

Abstract
A large body of evidence has demonstrated that cyclic-guanosine monophosphate (cGMP), signaling has anti-tumor effects that might be used for colon cancer prevention. The tumor-suppressive mechanism and the signaling components downstream of cGMP remain largely unknown. The present study has characterized the expression of cGMP-dependent protein kinases (PKG1, PKG2) in normal and cancerous tissue from human colon. PKG1 was detected in both normal and tumor tissue, where it localized exclusively to the lamina propria and stroma (respectively). In contrast, PKG2 localized specifically to the epithelium where its expression decreased markedly in tumors compared to matched normal tissue. Neither PKG isoform was detected at the RNA or protein level in established colon cancer cell lines. To test for a potential tumor-suppressor role of PKG2 in the colon epithelium, Prkg2 knockout (KO) mice were subjected to azoxymethane/dextran sulfate-sodium (AOM/DSS) treatment. PKG2 deficiency was associated with crypt hyperplasia (Ki67) and almost twice the number of polyps per mouse as wild-type (WT) siblings. In vitro culture of mouse colon epithelium as organoids confirmed that PKG2 was the only isoform expressed, and it was detected in both proliferating and differentiating epithelial compartments. Colon organoids derived from Prkg2 KO mice proliferated more rapidly and exhibited a reduced ability to differentiate compared to WT controls. Taken together our results highlight PKG2 as the central target of cGMP in the colon, where it suppresses carcinogenesis by controlling proliferation in an epithelial-cell intrinsic manner.
Cyclic-GMP is tumor suppressive in the colon but the downstream signaling is unknown. Here, we demonstrate that type 2 cGMP-dependent protein kinase is the central cGMP effector in the colon epithelium where it inhibits proliferation and carcinogenesis.
Graphical Abstract

Introduction
A large body of evidence supports the idea that increasing cyclic-guanosine monophosphate (cGMP) in the colon epithelium is a potential strategy for colon cancer chemoprevention (1). The concentration of cGMP in the gut is tightly controlled by guanylyl cyclase C (GC-C) receptors that convert guanosine triphosphate into cGMP upon stimulation by the endogenous peptide hormones guanylin and uroguanylin (2–4). While cGMP is well known to promote secretion in the small intestine (5,6), guanylin (GUCA2A) and GC-C (GUCY2C) knockout (KO) mice have shown that cGMP also regulates epithelial homeostasis in both intestine and colon (3,7). Epithelial renewal in the colon is maintained by stem cells at the crypt base that give rise to proliferative progenitors that differentiate as they move upward, into either absorptive colonocytes or secretory goblet and enteroendocrine cells (8). Mice that are deficient in cGMP signaling show increased proliferation and reduced differentiation of secretory lineage cells in the colon mucosa. GC-C KO mice also have a compromised epithelial barrier that is more susceptible to insult by chemicals, pathogens and ionizing radiation (9–12). These functions of cGMP-signaling are collectively thought to be tumor-suppressive because GC-C KO mice are also more susceptible to tumorigenesis in both azoxymethane (AOM) and ApcMin/+ mouse models of colon cancer (13,14). This idea is supported by the fact that guanylin and uroguanylin are commonly lost in colon cancers (15,16), and that increasing cGMP using exogenous GC-C agonist is tumor-suppressive in ApcMin/+ mice (17,18). In addition, amplifying endogenous cGMP signaling with phosphodiesterase 5 (PDE5) inhibitors is also tumor-suppressive in mouse models of colon cancer (18,19).
Despite the abundance of evidence for tumor-suppressive roles of cGMP in the intestine, the downstream signaling components remain poorly understood. Cyclic-GMP can directly regulate some ion channels and PDEs, but the cGMP-dependent protein kinases (PKG) are central effectors in most tissues (20). Mammals express three isoforms of PKG that are transcribed from two distinct genes, and their distribution and known functions have been extensively reviewed (21,22). The alternatively spliced α and β isoforms of type 1 PKG (PKG1) are widely distributed, but are most highly expressed in smooth muscle tissue where they mediate relaxation (20). The membrane-tethered type 2 PKG (PKG2) is also expressed in several tissues, including the intestinal epithelium (22). PKG2 mediates the effects of GC-C coupled secretagogues in the small intestine, but this effect is less pronounced in the colon where regulation of epithelial homeostasis may be its primary function (23–25).
The use of colon cancer cell lines to characterize the anti-tumor signaling downstream of cGMP has resulted in discrepant results. Many reports describe anti-proliferative and pro-apoptotic roles for the PKG1 isoform following activation by PDE inhibitors (26–29). Other studies suggest that PKG2 is more important because it suppresses growth and promotes differentiation (25,30). Adding to the confusion, it has been reported that colon cancer cell lines lose expression of all PKG isoforms compared to non-cancer cells (25,31). Which PKG isoforms are relevant to colon cancer in vivo is poorly understood. The present study has examined the expression of PKG isoforms in normal and neoplastic intestinal tissues from both mice and humans. PKG2 was determined to be the only isoform present in colon epithelial cells, where it has a cell-intrinsic anti-proliferative and anti-tumorigenic role.
Materials and methods
Animals and AOM/dextran sulfate-sodium treatment
All procedures involving animals were approved by the Augusta University Institutional Animal Care and Use Committee, which ensures compliance with the Animal Welfare Act, Public Health Service Policy on Humane Care and Use of Laboratory Animals. PKG2 KO mice in strain 129S1 genetic background (25) were crossed for 15 generations with C57BL/6J mice before experimentation (Jackson Laboratories). For stand-alone dextran sulfate-sodium (DSS) treatment, procedures were followed as detailed previously (32). For the AOM/DSS colon cancer model, we followed the procedure described previously with some modifications (19). Mice were given a single 7.5 mg/kg intraperitoneal dose of AOM (13.4M 98%, Sigma Aldrich) and subsequently subjected to three consecutive cycles of DSS treatment (2% w/v DSS for 5 days) at weeks 9, 12 and 15 of age. Recovery periods after the first and second cycles of DSS were for 14 days. After the third DSS cycle, mice were allowed to recover for 21 days prior to euthanasia. All mice were sacrificed at 18 weeks of age and the colorectums were processed for quantitation of polyp number and size. Briefly, colorectums were flushed with ice-cold phosphate-buffered saline (PBS) and cut longitudinally, flattened, contrast-stained with 1% Alcian blue dye, and placed on a light box for macroscopic high-resolution image capture. These images were enlarged for identification and marking of suspected polyps, and ImageJ software was used for enumeration and size quantification of the identified polyps.
Tissue and organoid culture
All cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in 5% CO2 in RPMI-1640 medium containing 10% fetal bovine serum, and supplemented with 2 mM L-glutamine, 10 IU/mL penicillin, 10 μg/mL streptomycin. ATTC cell lines are internally authenticated by short tandem-repeat profiling, and the lines used in the present studies were within four passages from purchase. De-identified human colectomy specimens were obtained from the Augusta University Biorepository, which obtained written consent from patient donors. The procedure was approved by the Augusta University Institutional Review Board. Colon tumor and grossly uninvolved (normal) tissues were classified by board-certified pathologists.
Colon organoids were established by dissecting the colon from mice, rinsing in PBS, and then rocking for 30 min at 37°C in PBS containing 1% bovine serum albumin, 0.5 mM dithiothreitol and 2 mM ethylenediaminetetraacetic acid. Crypt fractions were obtained by repeated rounds of vortexing in PBS followed by passage through 70 µm filter and pelleting at 20 × g for 5 min. Fractions containing crypts were then suspended in a 1:1 mixture of matrigel (growth factor reduced; Corning) and stem medium, then plated in 20 μl domes in 12 well dishes. The procedures followed for organoid propagation were based on Wnt3a/R-spondin-1/Noggin conditioned medium as described previously (33). Stem medium contained 50% Advanced Dulbecco's modified Eagle's medium, 50% Wnt/R-spondin-1 and noggin (WRN) conditioned medium, 20% fetal bovine serum, 1 × Primocin (Invivogen, San Diego CA), 10 µM Y27632 (ATCC, Manassas, VA) and SB431542 (StemRD, Burlingame, CA). The medium was changed every 2 days, and organoids were passaged weekly by trypsinization. When comparing proliferating and differentiating organoids, the stem medium was changed daily to maintain proliferation, and was switched to differentiation medium containing 5% WRN without inhibitors for 3 days before harvest. In some cases, the γ-secretase inhibitor 5 µM DAPT (Tocris, Minneapolis, MN) was added to the differentiation medium.
Immunohistochemistry
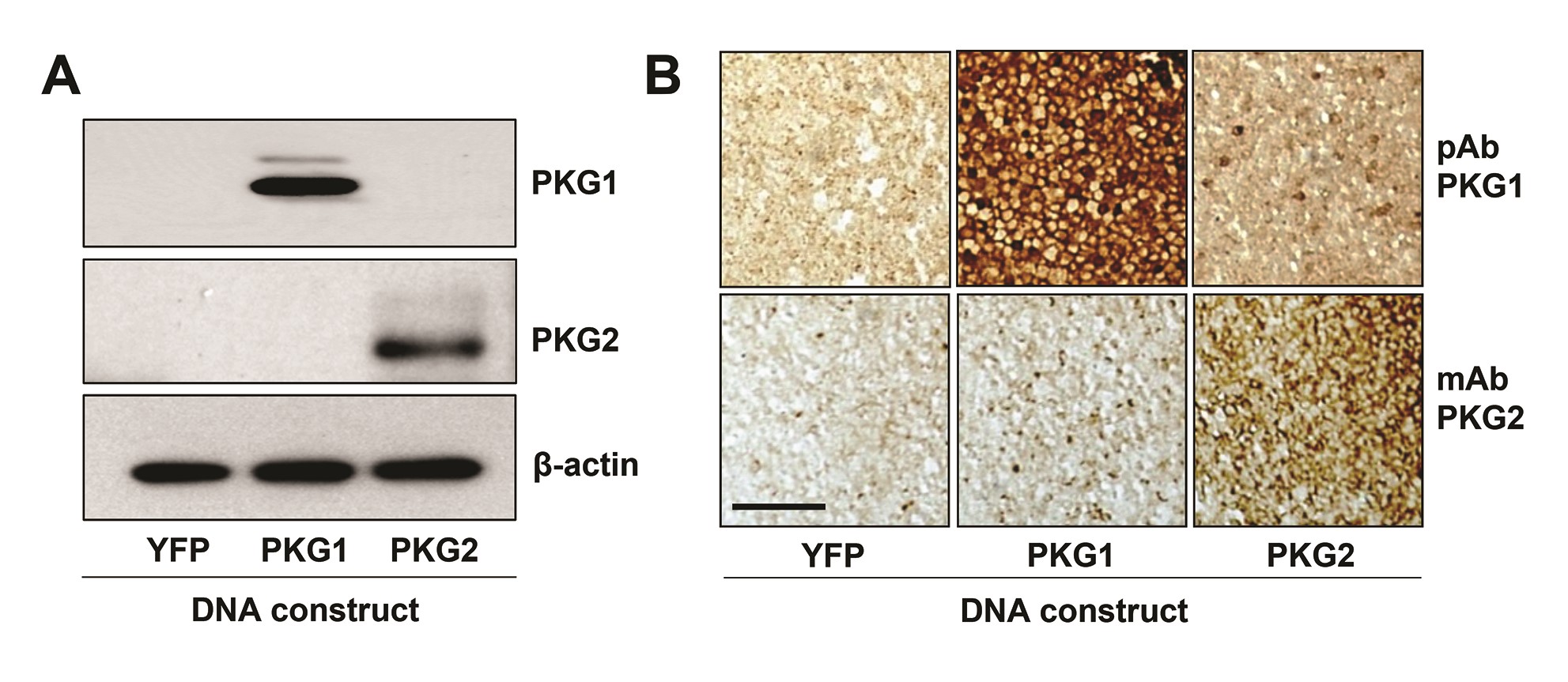
For antibody validation, HCT116 cells were transfected with plasmids encoding YFP, and human PRKG1 or PRKG2 complementary DNA (Supplementary Figure 1, available at Carcinogenesis Online). Transfected cells were pelleted by centrifugation, fixed in 10% formalin for 24 h, rinsed in PBS, and then mounted in 2% low melt agarose before dehydration and paraffin embedding. Paraffin blocks containing tissues or cell pellets were sectioned at a 5 μm thickness by the Augusta University Histology core. Sections were deparaffinized in xylene, rehydrated with decreasing concentrations of ethanol, and boiled in citrate-based buffer for antigen retrieval. Sections were stained antibodies as follows: PKG1 (1:15, gift from Robert Feil) and PKG2 (clone D3; 1:200, Santa Cruz), Vimentin (1:100, BD Pharmingen) and cytokeratin 8–18 (1 µg/mL, Epitomics). Antibody visualization of PKG1 used a biotinylated anti-rabbit antibody (Vector laboratories) and PKG2 used a biotinylated anti-mouse antibody (Vector laboratories). Amplification of biotinylated antibodies used ABC staining kit (Santa Cruz). Tissues were counterstained with hematoxylin, dehydrated in ethanol and mounted with coverslips using Acrymount (StatLab, Columbia, MD).
RT-qPCR and western blotting
RNA extraction of human tissues, cells and organoids was done with TRIzol reagent (Life Technologies). RNA was DNAse treated (TURBO DNA-free kit, Life Technologies) and converted to complementary DNA using Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase (Invitrogen™). Quantitative PCR (qRT-PCR) analysis of the complementary DNA was done using SYBR Green PCR Master Mix (Applied Biosystems). Primers were designed using Primer Blast Software (NCBI; Supplementary Table 1, available at Carcinogenesis Online). Relative expression levels were determined by calculating ΔCT using β-actin (ACTB) as the reference, log2 transformed, and then normalized to mean expression in the colon cancer cell lines to determined fold changes. Amplification was performed in triplicate wells, and melt curve analysis was used done to confirm primer specificity.
All specimens were processed for Western blotting by extracting protein in lysis buffer containing 1% NP40, 0.1% sodium dodecyl sulfate, protease and phosphatase inhibitors (EMD Millipore). Human tissues were dissociated with a short pulse in a tissue grinder. Lysates were prepared clarified by centrifugation and boiled 10 min in 5× polyacrylamide gel electrophoresis buffer. Proteins were resolved on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis min-gels (BioRad), transferred to nitrocellulose membranes, and blocked with 5% bovine serum albumin in PBS containing 0.025% Tween 20. Antibodies used to detect PKG were as follows: human PKG2 (clone E7, Santa Cruz Biotechnology), mouse PKG2 (polyclonal H-120, Santa Cruz Biotechnology). The PKG1 antibody used was an affinity-purified polyclonal generated by Dr Feil laboratory. Antibodies recognizing proliferating cell nuclear antigen (PCNA), and both total and phospho-239 vasodilator-stimulated phosphoprotein (VASP) (Cell Signaling), β-actin (Sigma Chemical Co) and β-catenin (BD Transduction Laboratories).
Statistical analysis
All data were analyzed using GraphPad Prism 9 (San Diego, CA) and data are presented as the mean ± SD except where indicated. The qRT-PCR data were compared across tissue and PKG isoform using one-way ANOVA followed by Tukey’s multiple comparison test. Changes in PKG expression between tumor and cancer cell lines were done by paired t-tests for each isoform. Tumor multiplicity data were analyzed using the Mann–Whitney U test. P < 0.05 was considered to be significantly different.
Results
PKG isoforms are differentially expressed in colon tissues
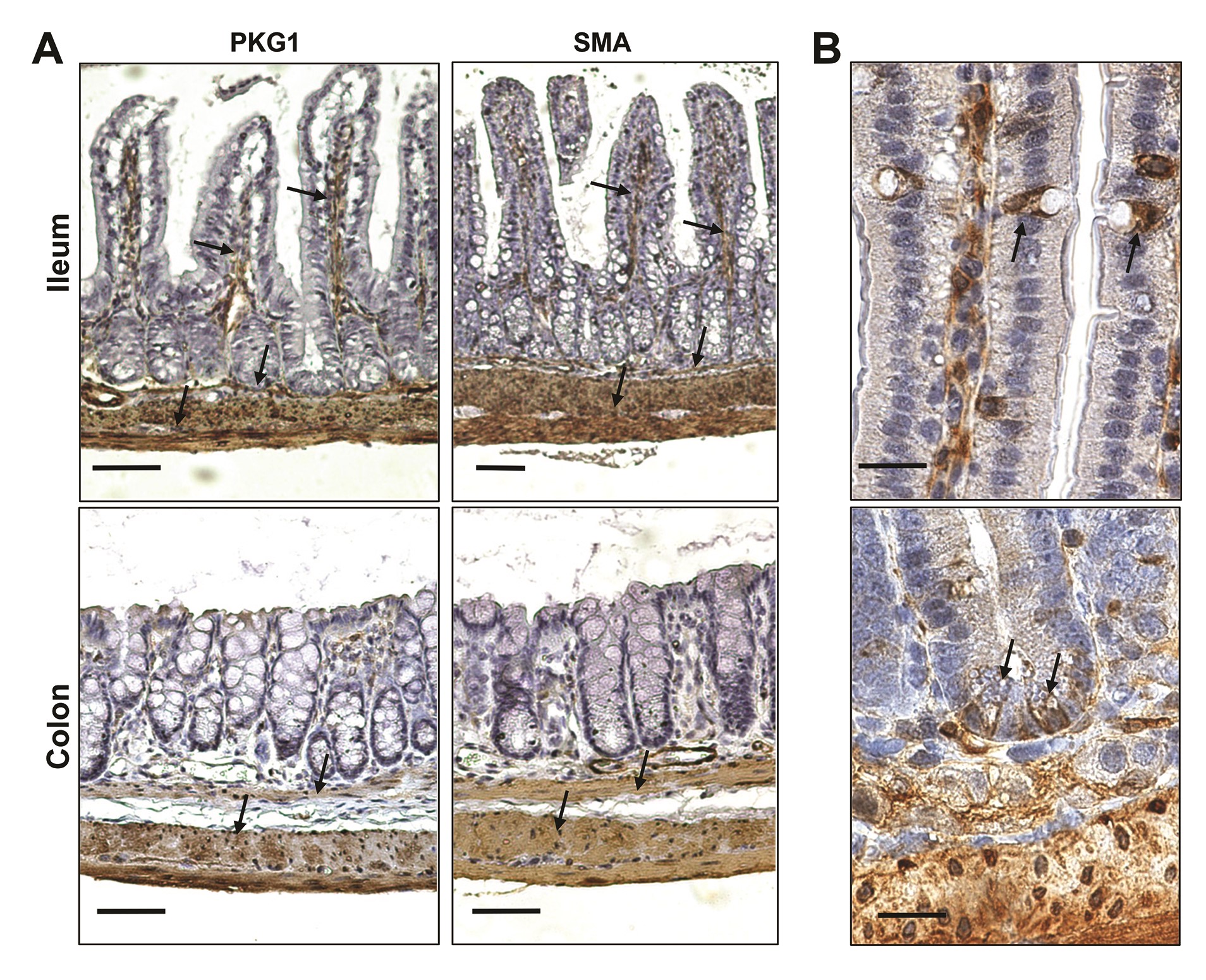
To gain some insight into which PKG isoforms might be involved in anti-cancer signaling, PKG expression in normal and tumor tissues from human colectomy specimens was examined, and compared to established colon cancer cell lines. All three PKG isoforms were detected at the RNA level in both normal and tumor tissues (Figure 1A). Only PKG2 showed reduced levels in tumor compared to normal tissue, but all three isoforms were lower in colon cancer cell lines compared to tumor tissue. Immunoblotting also showed a dramatic loss of PKG2 in tumors compared to normal tissue, in contrast with PKG1 that slightly increased, but neither isoform was detected in colon cancer cell lines (Figure 1B). Because tumor tissue is composed of numerous stromal cell types in addition to cancer epithelium, immunohistochemistry (IHC) was used to identify the cell-type localization of the PKG isoforms in human tumors (Figure 1C). These results showed that PKG1 colocalized with vimentin in the tumor stroma, but was not detected in the tumor epithelium. PKG2 expression was opposite that of PKG1, as it colocalized with cytokeratin 8 in the tumor epithelium but was undetectable in the stroma. IHC was also used to examine PKG isoform expression in normal intestinal epithelium (Figure 2). PKG1 was detected in supportive cells of the lamina propria, with strong expression in the muscularis and myofibroblasts in ileal and colon specimens from both human and mouse. PKG1 was not observed in the intestinal epithelium, but over-staining showed some signal in goblet and paneth cells of the ileum but not in the colon (Supplementary Figure 2, available at Carcinogenesis Online). In contrast, PKG2 was observed exclusively in the epithelium of both ileal and colon specimens. The antibody used to detect PKG2 by IHC only recognized human PKG2, and a similar antibody for mouse tissues is presently unavailable. To overcome this issue, we enriched the epithelial crypts from total mouse mucosa, and examined PKG expression by western blot (Figure 2C). These studies supported the IHC data from human tissues, as both isoforms were detected in whole mucosa, but only PKG2 was detected in purified crypts.
Figure 1.

Expression of PKG isoforms in human colon-derived tissues and cancer cell lines. Comparison of PKG isoform expression in patient-derived colon matched normal/tumor specimens and colon cancer cell lines by (A) RT-qPCR and (B) Western blotting. (C) Colectomy specimens were processed for IHC to detect type 1 or 2 PKG as indicated. Serial sections were stained for vimentin and cytokeratin 8 as markers for stroma and tumor epithelium (respectively). Magnification is the same in all panels, scale bar = 100 μm. ∗P < 0.05 using ANOVA with Tukey’s multiple comparison test. PKG expression in the cell lines was less than tumor tissue using two-sided t-tests for each isoform (P < 0.05). IHC, immunohistochemistry.
Figure 2.

Expression of PKG in normal intestinal tissues. Normal colon and ileum tissues derived from human (A), and mouse (B) were processed for IHC and immunostained for PKG1 or PKG2 as indicated. Arrows in PKG1-stained tissues highlight non-epithelial supportive tissues including smooth muscle tissue and suspected myofibroblasts. Arrows in PKG2-stained tissues highlight epithelium. (C) Dissociated colon mucosa (M) or enriched crypts (C) were imaged by phase-contrast microscopy (left), and subjected to immunoblotting for PKG isoforms (right). The scale bar in upper panels = 50 μm, and in panel C = 100 μm. IHC, immunohistochemistry.
PKG2 suppresses proliferation and carcinogenesis in the mouse colon
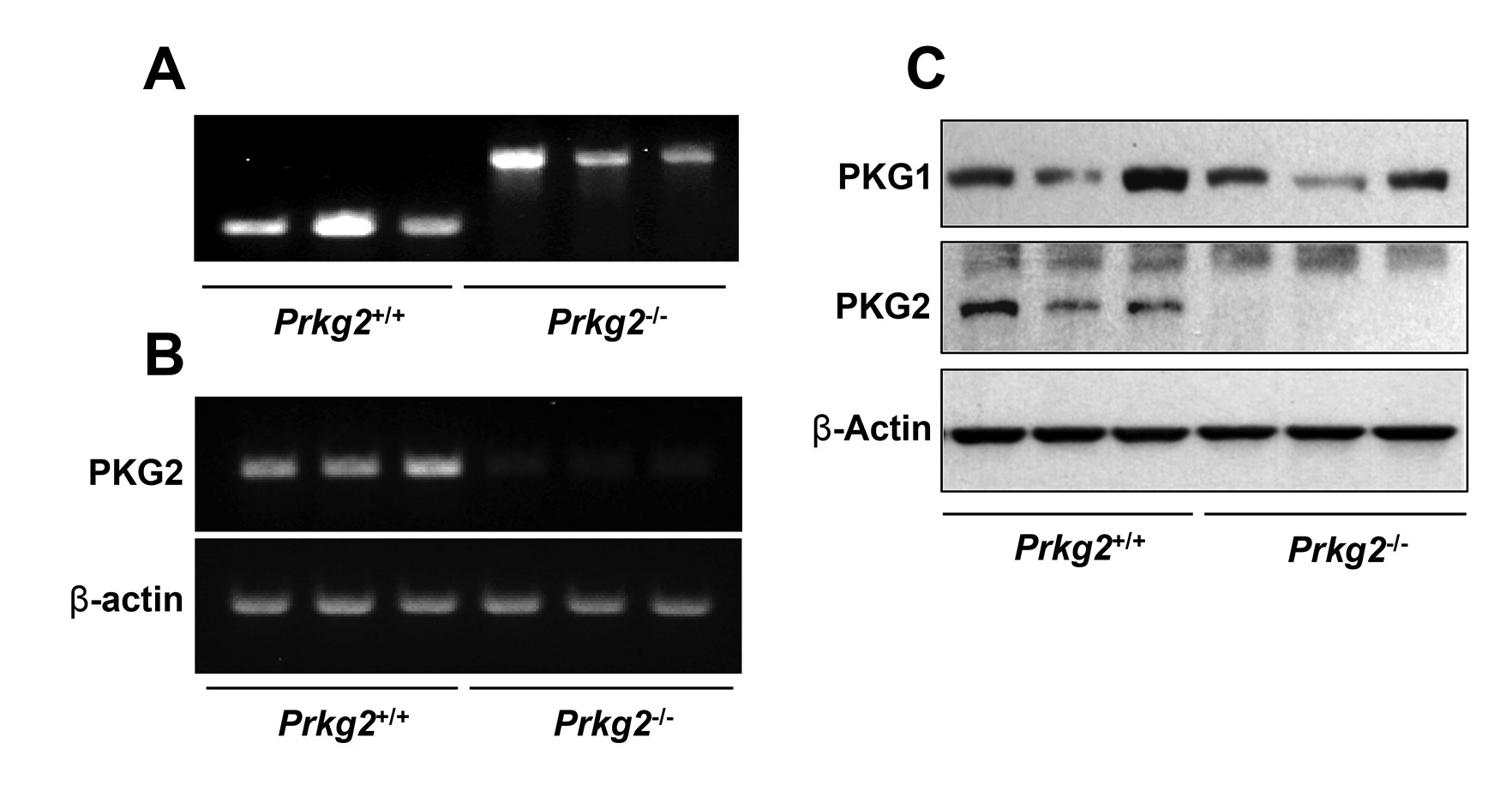
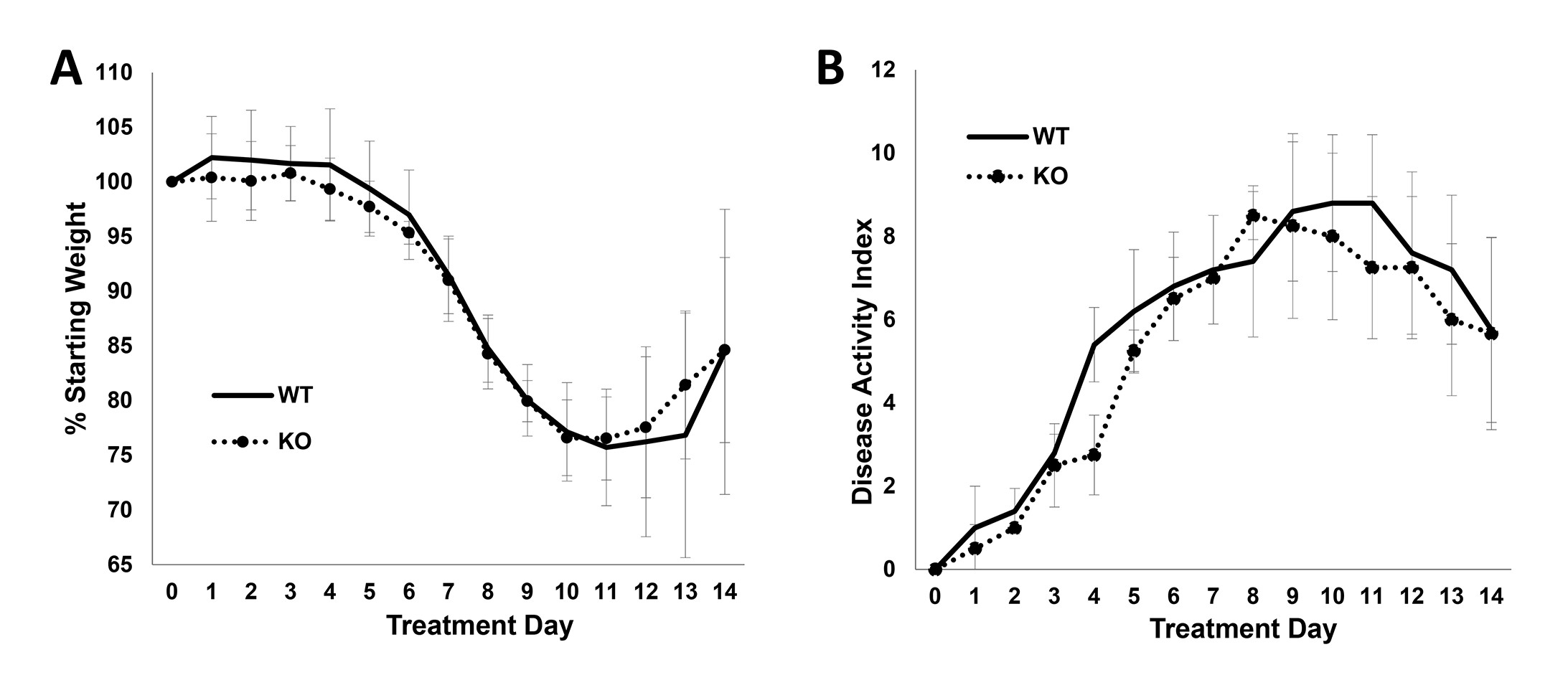
The IHC results clearly highlight PKG2 as the main effector of GC-C/cGMP in the colon epithelium, and could therefore mediate the anti-tumor effects of this pathway. To test this, we obtained PKG2 KO mice in the strain 129 background that have been previously reported to exhibit similar intestinal homeostatic changes as GC-C KO animals (25). These animals were backcrossed into C57Bl/6 mice that are well-characterized in colon cancer models (Supplementary Figure 3, available at Carcinogenesis Online). Colon homeostasis in the PKG2-deficient C57Bl/6 mice showed increased proliferation in the crypts, and increased apoptosis in the border epithelium (Figure 3A and B). The AOM/DSS colon carcinogenesis model is widely used to study colon cancer in mice because it recapitulates several aspects of human disease. When subjected to AOM/DSS treatment, the PKG2 KO mice produced almost twice the number of colonic polyps per mouse as wild-type (WT) siblings (Figure 3C and D). The mean size of polyps from the PKG2 KO mice was slightly lower than WT animals, but the difference did not reach significance in our study (Figure 3E). Similar to GC-C KO mice (34), PKG2 KO mice respond similarly to DSS as WT mice (Supplementary Figure 4, available at Carcinogenesis Online), indicating that the observed effect of PKG2 on polyp formation is unlikely to arise from differences in inflammation.
Figure 3.

PKG2-deficient mice exhibit increased crypt hyperplasia and carcinogenesis in the colon. (A) Paraffin sections of colon from WT and PKG2 KO mice were stained for proliferation (Ki-67) and apoptosis (cleaved caspase 3; CC3), and quantitated (B). Arrows in the Ki-67 panel indicate positive nuclei in the lower crypt, and in the CC3 panel they indicate apoptotic cells at the luminal surface. Data shown are means and error bars show SD. ∗P < 0.01, ∗∗P < 0.005 two tailed t-test (n = 3 mice per group). (C) Experimental outline for treating WT (Prkg2+/+, WT) and PKG2 KO (Prkg2−/−, KO) mice with the AOM/DSS model of colon cancer (upper panel). Images of distal colon and rectum of representative WT and PKG2 knock-out mice following AOM/DSS treatment (lower panel). Arrows indicate examples of polyps counted. (D) Enumeration of colorectal polyps per mouse in AOM/DSS treated animals. The line shows median value, and ∗∗P < 0.005 using a Mann–Whitney U test. (E) Mean colon polyp size in AOM/DSS treated mice. Error bars show SD, and NS indicates P > 0.05 from two-sided t-test (n = 8). KO, knockout; WT, wild type.
PKG2 suppresses colon proliferation in an epithelial cell-autonomous manner
The results shown here indicate that PKG2 suppresses proliferation and carcinogenesis in the colon epithelium of mice. However, mesenchymal cells in the lamina propria also play important roles in epithelial homeostasis and carcinogenesis in the colon (35). Indeed, it has been suggested that the epithelial hyperplasia observed in GC-C KO mice is indirect, involving hepatocyte growth factor secretion from fibroblasts in the lamina propria (36). Colon organoids are composed of pure-epithelium that are cultured in the absence of a mesenchymal niche (33,37). This model was therefore used to determine the importance of epithelial cell-intrinsic PKG2 signaling to the crypt hyperplasia observed in KO mice. Murine colon organoids were cultured as proliferating epithelium and compared to differentiated epithelium by reducing WRN levels in the medium. Upon differentiation, the proliferation (Ki67) and T-cell factor-target genes (cMyc, LGR5, SOX9) were dramatically reduced (Figure 4A and B), concomitantly with increased expression of differentiation genes (Muc2, Bmp2, Villin) (Figure 4C). As described previously (38), inhibiting notch signaling skewed differentiation toward secretory lineage cells resulting in a further increase in secretory gene expression.
Figure 4.

Colon organoids recapitulate the crypt-villus axis in vitro. Mouse colon organoids were maintained under proliferating and differentiation conditions as described in Materials and Methods. (A) Proliferating and differentiating organoids were imaged live using phase contrast microscopy (upper panels, scale bar = 100 µm), or processed for staining for Ki67 (proliferation; middle panels, bar = 25 µm), or alcian blue/periodic acid-Schiff (AB/PAS; bottom panels, bar = 50 µm). AB/PAS identifies goblet cells by staining mucopolysaccharides. (B) RT-qPCR analysis of β-catenin/TCF target gene expression in proliferating (Pro), differentiating (Diff) or Notch inhibitor-treated differentiating (DAPT) colon organoids. (C) RT-qPCR analysis of differentiation genes in colon organoids. The data show mean with SD error bars, n > 3. Data were analyzed using one-way ANOVA with Dunnetts multiple comparisons test for each gene relative to proliferation values. ∗P < 0.05, ∗∗P < 0.005, ∗∗∗P < 0.0005.
PKG2 was detected in both proliferating and differentiating organoids, but PKG1 was undetectable at either the mRNA or protein level (Figure 5). The notion that PKG2 is the only isoform expressed in colon epithelium was confirmed using the well-established PKG substrate, VASP (Figure 5C). VASP was phosphorylated at the PKG-specific site (Ser239) in response to treatment of WT organoids with the membrane permeable cGMP analog 8Br-cGMP, but not in PKG2 KO organoids. Results with RT-qPCR, immunoblotting and PKG activity all demonstrated reduced expression of PKG2 when the organoids were pushed to differentiate into secretory cells using a notch inhibitor, suggesting that PKG2 is more prominent in colonocytes.
Figure 5.

Expression of PKG isoforms in mouse colon organoids. Mouse colon organoids were maintained under proliferating (Pro) and differentiation (Diff) conditions with or without the Notch inhibitor (DAPT). PKG1 and PKG2 expression was measured by (A) RT-qPCR (error bars show SD, n = 3), (B) Immunoblotting. Mouse colon mucosa (Col) and HEK293 cells (293) were positive controls. (C) PKG activity was measured by immunoblotting to detect phosphorylation of Vasodilator-Stimulated Phosphoprotein (VASP) on Ser239. Organoids from WT or PKG2 KO mice were either untreated (−), or treated (+) with the membrane-permeable cGMP analog 8-Br-cGMP. KO, knockout; WT, wild type.
The colon organoid model was further used to determine the effect of PKG2-deficiency on epithelial growth and differentiation. The WT and PKG2 KO organoids increased in size at a similar rate initially, but the PKG2 KO organoids subsequently grew faster and were double the size of WT organoids by three days after seeding (Figure 6A and B). The higher proliferation of PKG2 KO relative to WT organoids was supported by higher PCNA and β-catenin expression levels (Figure 6C). When cultured in reduced WRN medium to promote differentiation, the PCNA expression decreased dramatically in both organoids, but remained higher in the PKG2 KOs. In addition, the differentiation genes Bmp2, Vil1 and Muc2 either failed to increase (Vil1) or were at much-reduced levels in the PKG2 KO organoids compared to controls (Bmp2, Muc2) in the reduced WRN medium (Figure 6D). The level of Bmp2 RNA in the PKG2 KO organoids was notably less than WT even under proliferating conditions.
Figure 6.

PKG2-deficient organoids show higher proliferation than WT controls. Colon organoids from WT and PKG2 KO mice were seeded in proliferation medium. (A) When spheres were visible, the cultures were imaged daily and mean organoid volumes were quantitated. At day three ∗∗ P < 0.01 using two tailed t-test (n = 3). (B) Bright field image of WT and PKG2 KO organoid cultures at 3 days. Bar = 300 µm. (C) Organoid extracts were subjected to immunoblotting for proliferation (PCNA and β-catenin) using β-actin as a loading control. (D) WT and PKG2 KO organoids were maintained in proliferating (Pro) or differentiating medium without (Diff) or with DAPT (DAPT), and the expression of differentiation genes was quantitated by RT-qPCR. The data are means with error bars showing SD. ∗P < 0.05, ∗∗P < 0.005 relative to proliferating organoids (paired t-test, n = 3). KO, knockout; WT, wild type.
Discussion
Signaling through cGMP has anti-carcinogenic effects in the colon but the downstream effectors remain poorly understood. The epithelium is the main site of cGMP production in the colon due to activation of GC-C by guanylin, and PKG is the main effector of cGMP in most cells. It was shown here that all PKG isoforms are detectable in normal colon mucosa and in colon tumors, but not in established colon cancer cell lines. We have previously reported the lack of PKG in colon cancer cell lines (25,31,39) but this is the first time it has been directly compared with normal and tumor tissues. The lack of PKG protein expression in colon cancer cell lines is ostensibly incongruous with reports of growth-inhibitory effects of PDE inhibitors in colon cancer cell lines (26,40,41). However, a recent report indicates that the anti-proliferative effects are cGMP-independent and do not require PKG (42). An important finding of the present study is that PKG1 was primarily expressed in supportive cells of the lamina-propria and the tumor stroma. This explains its absence in established colon cancer cell lines and in colon organoids that do not contain stromal cells. The localization of PKG2 contrasted greatly with PKG1 as it was exclusive to the epithelium in both normal and tumor tissues. This observation is consistent with previous reports of PKG2 in normal intestinal tissues (6,43,44). The reduced expression of PKG2 at both protein and RNA levels in colon cancer tissue and its further loss in colon cancer cell lines suggests that silencing of the Prkg2 gene occurs during the normal to adenocarcinoma progression. While the underlying mechanism warrants further investigation, the results clearly highlight PKG2 as the central cGMP effector in both normal and tumor epithelia.
To determine whether PKG2 has a tumor-suppressive role in the colon, we subjected Prkg2−/− mice to AOM/DSS treatment. PKG2 KO mice showed a significantly higher polyp multiplicity compared to WT siblings, but there was no difference in polyp size. Because WT and PKG2 KO mice responded similarly to DSS treatment, these observations suggest that PKG2 suppresses polyp initiation rather than progression. This interpretation is consistent with previous studies showing that cGMP is effective at suppressing polyp initiation (during AOM treatment), but not promotion (during DSS treatment) (19). The lack of effect of PKG2 expression on initiated tumors is not surprising as the cGMP-stimulating hormone guanylin is silenced very early during neoplastic transformation (16,45). It was shown here that proliferation was significantly higher in PKG2- KO mice compared to WT, which is consistent with previous work implicating PKG2 as a mediator of cGMP-dependent inhibition of proliferation in the colon (25,32). The larger proliferative compartment in the colons of PKG2 KO mice would increase susceptibility to genotoxic stress such as AOM. In addition, it has been reported that the amount of proliferation in a tissue is a determinant of cancer risk due to random mutations (46–48). Increasing cGMP with either GC-C agonist or with PDE5 inhibitors also suppresses intestinal polyp formation in ApcMIN mice (18,19). Tumorigenesis in this model does not require exogenous mutagens or cycles of inflammation like the AOM/DSS model, but depends upon random mutations that lead to loss of heterozygosity at the Apc locus. The inhibition of epithelial proliferation by cGMP/PKG2 is therefore likely to be the central tumor-suppressive mechanism of this pathway.
The signaling events downstream of PKG2 that inhibit proliferation in the colon are not understood. Inhibition of epithelial proliferation signaling (e.g. AKT/FoxO3a) has been implicated as important, suggesting that cGMP inhibits proliferation in an epithelial cell-intrinsic manner (12,14,49). However, a more complex model has been proposed in which epithelial cGMP activates transforming growth factor β production, which in turn suppresses growth-promoting hepatocyte growth factor secretion by fibroblasts in the lamina propria (36). The present study has tested this model by examining proliferation of colon epithelium grown as organoids. The lack of detectable PKG1 in the colon organoids confirmed that PKG2 is the only isoform expressed in the epithelium, and validated the absence of contaminating cells from the lamina propria. Importantly, the PKG2-deficient colon organoids grew more rapidly than WT based upon both organoid size and PCNA expression. This clearly demonstrates that PKG2 suppresses colon epithelial cell proliferation in a cell-autonomous manner that does not require mesenchymal participation. It is possible that PKG2 inhibits proliferation signaling indirectly by promoting differentiation, which was shown to be deficient in the PKG2 KO organoids. The pro-differentiation gene BMP2 was at lower levels in PKG2 KO organoids under both proliferating and differentiating conditions. BMP2 is well-established to suppress proliferation and promote differentiation in normal colon epithelium, in part by blocking β-catenin expression to antagonize Wnt signaling (50,51). It follows that PKG2 might facilitate the proliferation-differentiation transition by increasing BMP2 levels. This idea was supported by the higher β-catenin levels observed in the PKG2 KO organoids compared to WT, and that PKG2 is required for efficient differentiation of secretory lineage cells in vivo (25).
Taken together, the results show that PKG2 is the central effector kinase of cGMP in the intestinal epithelium, where it has an anti-proliferative and anti-carcinogenic role. While a detailed signaling mechanism downstream of PKG2 in the colon epithelium remains to be determined, the colon organoid culture system has shown here to be a useful model.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Dr Robert Feil, Interfakultäres Institut für Biochemie, University of Tübingen, Germany for providing the PKG1-specific antibody.
Glossary
Abbreviations
- AOM
azoxymethane
- DSS
dextran sulfate-sodium
- GC-C
guanylyl cyclase C
- IHC
immunohistochemistry
- KO
knockout
- PBS
phosphate-buffered saline
- PCNA
proliferating cell nuclear antigen
- PKG
protein kinase G
- VASP
Vasodilator-Stimulated Phosphoprotein
Contributor Information
Bianca N Islam, Department of Internal Medicine, Case Western Reserve University, Cleveland, OH, USA.
Sarah K Sharman, Department of Biochemistry and Molecular Biology, Medical College of Georgia at Augusta University, Augusta, GA, USA.
Yali Hou, Department of Biochemistry and Molecular Biology, Medical College of Georgia at Augusta University, Augusta, GA, USA.
Rui Wang, Department of Surgery, Case Western Reserve University, Cleveland, OH, USA.
Justin Ashby, Department of Biochemistry and Molecular Biology, Medical College of Georgia at Augusta University, Augusta, GA, USA.
Honglin Li, Department of Biochemistry and Molecular Biology, Medical College of Georgia at Augusta University, Augusta, GA, USA.
Kebin Liu, Department of Biochemistry and Molecular Biology, Medical College of Georgia at Augusta University, Augusta, GA, USA.
Kenneth J Vega, Department of Medicine, Section of Gastroenterology and Hepatology, Augusta University, Augusta, GA, USA.
Darren D Browning, Department of Biochemistry and Molecular Biology, Medical College of Georgia at Augusta University, Augusta, GA, USA.
Funding
National Institutes of Health (CA172627-01A1 to D.D.B.); Augusta University Research Institute (IGPB00001 to D.D.B.).
Conflict of Interest Statement
None declared.
References
- 1. Islam, B.N., et al. (2018) Phosphodiesterase-5 inhibitors for colon cancer chemoprevention. Aging (Albany NY), 10, 2216–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Forte, L.R., et al. (2004) Uroguanylin and guanylin peptides: pharmacology and experimental therapeutics. Pharmacol. Ther., 104, 137–162. [DOI] [PubMed] [Google Scholar]
- 3. Steinbrecher, K.A., et al. (2002) Targeted inactivation of the mouse guanylin gene results in altered dynamics of colonic epithelial proliferation. Am. J. Pathol., 161, 2169–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fritsch, R.M., et al. (2004) InsP3R-associated cGMP kinase substrate (IRAG) is essential for nitric oxide-induced inhibition of calcium signaling in human colonic smooth muscle. J. Biol. Chem., 279, 12551–12559. [DOI] [PubMed] [Google Scholar]
- 5. Geiselhoringer, A., et al. (2004) IRAG is essential for relaxation of receptor-triggered smooth muscle contraction by cGMP kinase. EMBO J., 23, 4222–4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Geiselhoringer, A., et al. (2004) Distribution of IRAG and cGKI-isoforms in murine tissues. FEBS Lett., 575, 19–22. [DOI] [PubMed] [Google Scholar]
- 7. Li, P., et al. (2007) Homeostatic control of the crypt-villus axis by the bacterial enterotoxin receptor guanylyl cyclase C restricts the proliferating compartment in intestine. Am. J. Pathol., 171, 1847–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gehart, H., et al. (2019) Tales from the crypt: new insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol., 16, 19–34. [DOI] [PubMed] [Google Scholar]
- 9. Garin-Laflam, M.P., et al. (2009) Activation of guanylate cyclase C signaling pathway protects intestinal epithelial cells from acute radiation-induced apoptosis. Am. J. Physiol. Gastrointest. Liver Physiol., 296, G740–G749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Han, X., et al. (2011) Loss of guanylyl cyclase C (GCC) signaling leads to dysfunctional intestinal barrier. PLoS One, 6, e16139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harmel-Laws, E., et al. (2013) Guanylate cyclase C deficiency causes severe inflammation in a murine model of spontaneous colitis. PLoS One, 8, e79180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lin, J.E., et al. (2012) GUCY2C opposes systemic genotoxic tumorigenesis by regulating AKT-dependent intestinal barrier integrity. PLoS One, 7, e31686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li, P., et al. (2007) Guanylyl cyclase C suppresses intestinal tumorigenesis by restricting proliferation and maintaining genomic integrity. Gastroenterology, 133, 599–607. [DOI] [PubMed] [Google Scholar]
- 14. Lin, J.E., et al. (2010) The hormone receptor GUCY2C suppresses intestinal tumor formation by inhibiting AKT signaling. Gastroenterology, 138, 241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Steinbrecher, K.A., et al. (2000) Expression of guanylin is downregulated in mouse and human intestinal adenomas. Biochem. Biophys. Res. Commun., 273, 225–230. [DOI] [PubMed] [Google Scholar]
- 16. Wilson, C., et al. (2014) The paracrine hormone for the GUCY2C tumor suppressor, guanylin, is universally lost in colorectal cancer. Cancer Epidemiol. Biomarkers Prev., 23, 2328–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shailubhai, K., et al. (2000) Uroguanylin treatment suppresses polyp formation in the ApcMin/+ mouse and induces apoptosis in human colon adenocarcinoma cells via cyclic GMP. Cancer Res., 60, 5151–5157. [PubMed] [Google Scholar]
- 18. Sharman, S.K., et al. (2018) Cyclic-GMP-elevating agents suppress polyposis in ApcMin mice by targeting the preneoplastic epithelium. Cancer Prev. Res. (Phila), 11, 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Islam, B.N., et al. (2017) Sildenafil suppresses inflammation-driven colorectal cancer in mice. Cancer Prev. Res. (Phila), 10, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Francis, S.H., et al. (2010) cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev., 62, 525–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hofmann, F., et al. (2009) cGMP regulated protein kinases (cGK). Handb. Exp. Pharmacol., 13, 7–62. [DOI] [PubMed] [Google Scholar]
- 22. Hofmann, F., et al. (2006) Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol. Rev., 86, 1–23. [DOI] [PubMed] [Google Scholar]
- 23. Pfeifer, A., et al. (1996) Intestinal secretory defects and dwarfism in mice lacking cGMP-dependent protein kinase II. Science, 274, 2082–2086. [DOI] [PubMed] [Google Scholar]
- 24. Vaandrager, A.B., et al. (2000) Differential role of cyclic GMP-dependent protein kinase II in ion transport in murine small intestine and colon. Gastroenterology, 118, 108–114. [DOI] [PubMed] [Google Scholar]
- 25. Wang, R., et al. (2012) Type 2 cGMP-dependent protein kinase regulates proliferation and differentiation in the colonic mucosa. Am. J. Physiol. Gastrointest. Liver Physiol., 303, G209–G219. [DOI] [PubMed] [Google Scholar]
- 26. Thompson, W.J., et al. (2000) Exisulind induction of apoptosis involves guanosine 3ʹ,5ʹ -cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated beta-catenin. Cancer Res., 60, 3338–3342. [PubMed] [Google Scholar]
- 27. Tinsley, H.N., et al. (2011) Inhibition of PDE5 by sulindac sulfide selectively induces apoptosis and attenuates oncogenic Wnt/beta-catenin-mediated transcription in human breast tumor cells. Cancer Prev. Res. (Phila), 4, 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li, N., et al. (2013) Sulindac selectively inhibits colon tumor cell growth by activating the cGMP/PKG pathway to suppress Wnt/beta-catenin signaling. Mol. Cancer Ther., 12, 1848–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee, K., et al. (2016) β-Catenin nuclear translocation in colorectal cancer cells is suppressed by PDE10A inhibition, cGMP elevation, and activation of PKG. Oncotarget, 7, 5353–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Basu, N., et al. (2014) Intestinal cell proliferation and senescence are regulated by receptor guanylyl cyclase C and p21. J. Biol. Chem., 289, 581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hou, Y., et al. (2006) An anti-tumor role for cGMP-dependent protein kinase. Cancer Lett., 240, 60–68. [DOI] [PubMed] [Google Scholar]
- 32. Wang, R., et al. (2014) Type 2 cGMP-dependent protein kinase regulates homeostasis by blocking c-Jun N-terminal kinase in the colon epithelium. Cell Death Differ., 21, 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miyoshi, H., et al. (2013) In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc, 8, 2471–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Steinbrecher, K.A., et al. (2011) Murine guanylate cyclase C regulates colonic injury and inflammation. J. Immunol., 186, 7205–7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koliaraki, V., et al. (2017) Mesenchymal cells in colon cancer. Gastroenterology, 152, 964–979. [DOI] [PubMed] [Google Scholar]
- 36. Gibbons, A.V., et al. (2013) Intestinal GUCY2C prevents TGF-β secretion coordinating desmoplasia and hyperproliferation in colorectal cancer. Cancer Res., 73, 66541–66666. doi: 10.1158/0008-5472.CAN-13-0887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sato, T., et al. (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature, 459, 262–265. [DOI] [PubMed] [Google Scholar]
- 38. VanDussen, K.L., et al. (2015) Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut, 64, 911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hou, Y., et al. (2006) A role for cyclic-GMP dependent protein kinase in anoikis. Cell. Signal., 18, 882–888. [DOI] [PubMed] [Google Scholar]
- 40. Whitt, J.D., et al. (2012) A novel sulindac derivative that potently suppresses colon tumor cell growth by inhibiting cGMP phosphodiesterase and beta-catenin transcriptional activity. Cancer Prev. Res. (Phila), 5, 822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li, N., et al. (2015) Suppression of β-catenin/TCF transcriptional activity and colon tumor cell growth by dual inhibition of PDE5 and 10. Oncotarget, 6, 27403–27415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hou, Y., et al. (2022) Inhibition of colon cancer cell growth by phosphodiesterase inhibitors is independent of cGMP signaling. J Pharmacol. Exp. Ther. doi: 10.1124/jpet.121.001075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Golin-Bisello, F., et al. (2005) STa and cGMP stimulate CFTR translocation to the surface of villus enterocytes in rat jejunum and is regulated by protein kinase G. Am. J. Physiol. Cell Physiol., 289, C708–C716. [DOI] [PubMed] [Google Scholar]
- 44. Markert, T., et al. (1995) Endogenous expression of type II cGMP-dependent protein kinase mRNA and protein in rat intestine. Implications for cystic fibrosis transmembrane conductance regulator. J. Clin. Invest., 96, 822–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pattison, A.M., et al. (2020) Silencing the intestinal GUCY2C tumor suppressor axis requires APC loss of heterozygosity. Cancer Biol. Ther., 21, 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tomasetti, C., et al. (2017) Role of stem-cell divisions in cancer risk. Nature, 548, E13–E14. [DOI] [PubMed] [Google Scholar]
- 47. Tomasetti, C., et al. (2017) Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science, 355, 1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tomasetti, C., et al. (2015) Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science, 347, 78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang, R., et al. (2017) cGMP Signaling increases antioxidant gene expression by activating forkhead box O3A in the colon epithelium. Am. J. Pathol., 187, 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Reynolds, A., et al. (2014) Canonical Wnt signals combined with suppressed TGFβ/BMP pathways promote renewal of the native human colonic epithelium. Gut, 63, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Freeman, T.J., et al. (2012) Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of beta-catenin. Gastroenterology, 142, 562–571 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.