

Abstract
Objective:
To describe the site of lesion responsible for the severe, bilateral, symmetrical, selective loss of vestibular function in Cerebellar Ataxia with Neuronopathy and Vestibular Areflexia Syndrome (CANVAS), an adult-onset recessively-inherited ataxia, characterized by progressive imbalance due to a combination of cerebellar, somatosensory, and selective vestibular impairment with normal hearing.
Methods:
Histologic examination of five temporal bones and the brainstems from four CANVAS patients and the brainstem only from one more, each diagnosed and followed from diagnosis to death by one of the clinician authors.
Results:
All five temporal bones showed severe loss of vestibular ganglion cells (cell counts 3–16% of normal), and atrophy of the vestibular nerves, whereas vestibular receptor hair cells and the vestibular nuclei were preserved. In contrast, auditory receptor hair cells, the auditory ganglia (cell counts 51–100% of normal), and the auditory nerves were relatively preserved. In addition, the cranial sensory ganglia (geniculate and trigeminal), present in two temporal bones, also showed severe degeneration.
Conclusions:
In CANVAS there is a severe cranial sensory ganglionopathy neuronopathy (ganglionopathy) involving the vestibular, facial, and trigeminal ganglia but sparing the auditory ganglia. These observations, when coupled with the known spinal dorsal root ganglionopathy in CANVAS, indicate a shared pathogenesis of its somatosensory and cranial nerve manifestations. This is the first published account of both the otopathology and neuropathology of CANVAS, a disease that involves the central as well as the peripheral nervous system.
Keywords: Bilateral vestibular impairment, CANVAS-ganglionopathy, Neuronopathy, Otopathology, Temporal bone
Cerebellar Ataxia with Neuronopathy and Vestibular Areflexia Syndrome (CANVAS) is an adult-onset neurodegenerative disease, recessively-inherited in familial cases (1,2), characterized by progressive bilateral vestibular impairment, somatosensory impairment, and cerebellar ataxia (3–10). Hearing is unaffected. Some patients have autonomic dysfunction (e.g., hypohidrosis and orthostatic hypotension) and chronic cough (8). Familial cases are recessively inherited with a biallelic intronic AAGGG repeat expansion in the replication factor C subunit 1 (RFC1) (1,2) Neuropathology shows severe, selective degeneration of vestibular, trigeminal and geniculate ganglia, spinal dorsal root ganglia, and cerebellar vermis Purkinje cells (5).
Previously, the temporal bones of a single patient with CANVAS (4) demonstrated severe degeneration of vestibular ganglion cells and vestibular nerves with preservation of receptor hair cells in all vestibular sensory regions. In contrast auditory ganglion cells, auditory nerves, and receptors hair cells in the cochlea were all preserved. In the temporal bones from this patient both the trigeminal and facial ganglia were severely degenerated, a finding that tied in with our subsequent report of brain and spine neuropathology in CANVAS, the only report so far of the neuropathology in this disorder (5). In this report, the temporal bones of three CANVAS patients displayed severe degeneration of all dorsal root ganglia, ascending spinal sensory pathways, and cerebellar Purkinje cells with preservation of brainstem structures. From this we surmised that the degeneration of the vestibular ganglia would explain the severe impairment of vestibular function in CANVAS. In other words, these patients have a true vestibular neuropathy, or more correctly, a vestibular neuronopathy (ganglionopathy).
To confirm these findings here, we describe the temporal bone pathology of three more CANVAS patients and review their brainstem neuropathology with particular attention to the vestibular nuclei.
METHODS
CANVAS patients, diagnosed and followed by one of the authors (D.J.S.), were recruited and enrolled in the MEEI temporal bone registry. Five temporal bones from four CANVAS patients were sent to our Otopathology Laboratory for histopathologic processing, including fixation in 10% neutral buffered formalin, decalcification in EDTA, embedding in celloidin, and serial sectioning in the axial plane at 20-μm intervals. Every tenth section was stained with hematoxylin and eosin and examined by light microscopy. Auditory and vestibular ganglion cell counts were done by 2D reconstruction, based on the method described by Schuknecht and Otte et al. (11–13).
Dorsal root ganglia, vestibular nuclei, and sural nerve obtained at postmortem examination were fixed in formalin and embedded in paraffin, sectioned at five microns and stained with hematoxylin and eosin.
The otopathology of two TBs from one CANVAS patient (case 1) has been previously reported (4).
RESULTS
Case 1
Clinical Findings
A 63-year-old woman presented with a 5-year history of slowly worsening stance and gait. She denied vertigo or hearing loss. There was no past history of aminoglycoside therapy and no family history of imbalance. Over subsequent years she developed dysarthria and dysphagia. At age 71, clinical examination revealed a broad-based ataxic gait, upper and lower limb ataxia, a positive Romberg’s test, absent tendon reflexes in the lower limbs, and cerebellar dysarthria. Oculomotor findings included bidirectionally saccadic horizontal and vertical visual pursuit with gaze-evoked nystagmus and bidirectionally abnormal horizontal and vertical vestibulo-ocular reflex (VOR) on the clinical head impulse test (HIT). As expected, the horizontal and vertical visually enhanced vestibule-ocular reflex (VVOR) was also bidirectionally impaired. Caloric testing revealed bilaterally absent responses to 0°C stimulation. There were no responses on rotational chair testing. The audiogram was normal for age. Brain magnetic resonance imaging (MRI) showed marked cerebellar atrophy (particularly evident in the vermis) but no brainstem abnormality. Nerve conduction studies showed absent upper and lower limb sensory nerve action potentials (SNAPs). Sural nerve biopsy revealed a severe loss of myelinated and unmyelinated fibers without evidence of vasculitis. Genetic testing for SCA1, 2, 3, 6, and 7 and for Friedreich’s ataxia was negative. Coeliac disease and thyroid antibodies were negative. Vitamin E levels were normal. The patient eventually became bed-bound and died of aspiration pneumonia at age 82. Both temporal bones were removed for histopathologic study.
Otopathology
Both temporal bones showed similar findings and are described together. The organs of Corti appeared normal except a partial loss of outer hair cells in the extreme apex. The stria vascularis showed patchy atrophy but otherwise was intact. The spiral ganglion cells appeared normal (Fig. 1), and their total count in both cochleae corresponded to 70% of normal age-corrected spiral ganglion cell counts (11) (Table 1). Within the vestibular system, the most impressive change was severe atrophy of the superior and inferior vestibular nerves and of Scarpa’s ganglion (Fig. 2,A and B). The total Scarpa’s ganglion counts were the same in both temporal bones (16% of normal age-corrected Scarpa’s ganglion cells (ScGC) counts (12) [Table 1]). Dendrites and axons were also degenerated (Table 2).
FIG. 1.
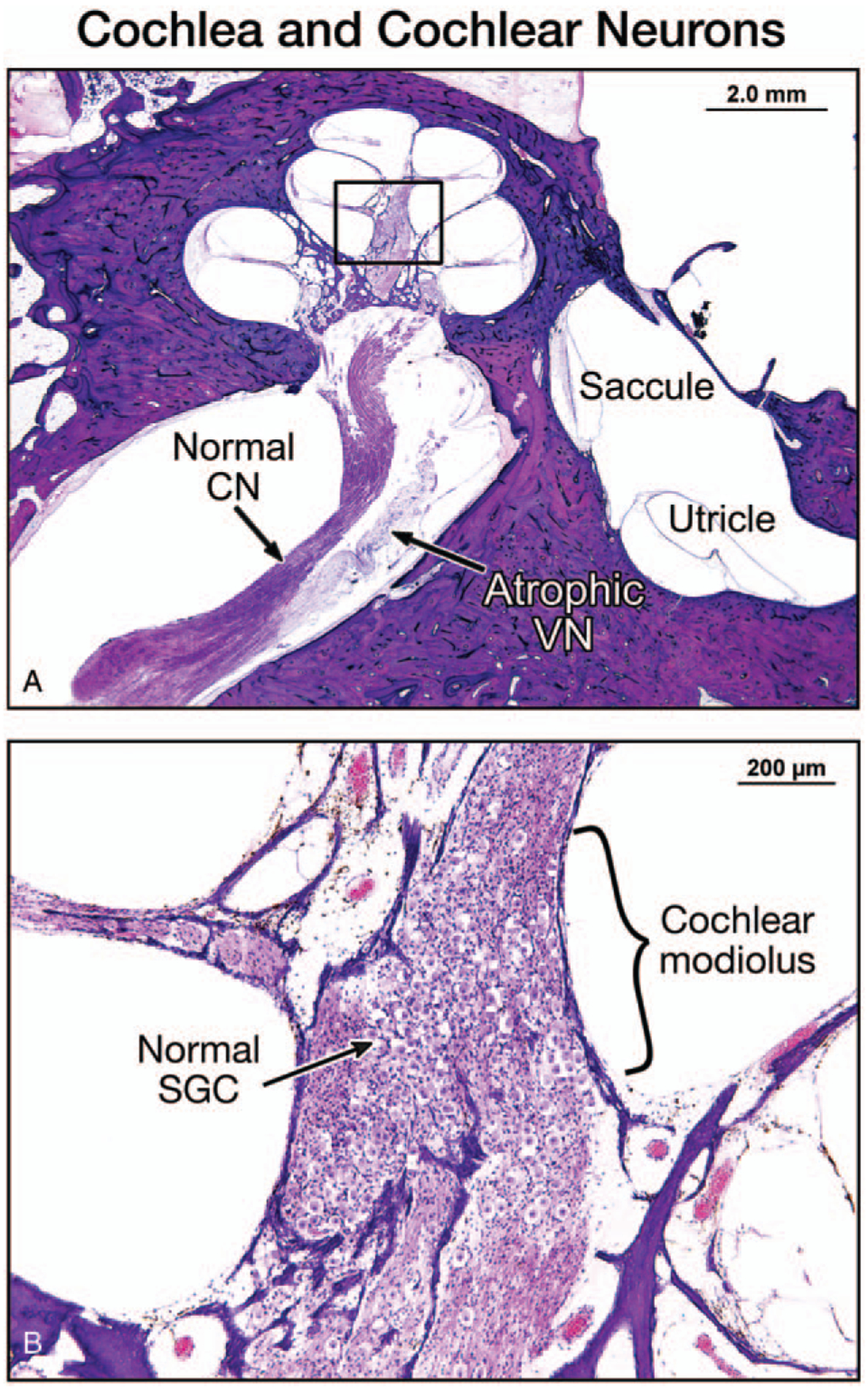
A, A low-power view of the right temporal bone of Patient 1 showed a normal cochlea and a normal cochlear nerve (CN) in a mid-modiolar section, and atrophy of the vestibular nerve (VN). The saccule and utricle were normal. B, A high-power view of the cochlear modiolus showed a normal population of spiral ganglion cells (SGC).
TABLE 1.
Ganglion cell histology
| Specimen | Patient/Side | Sex/Age | SGC, % nl. for Age (Absolute No.) | ScGC Count, % nl. for Age (Absolute No.) | GG | TG |
|---|---|---|---|---|---|---|
| 1 | 1R | 82/F | 70% (13008) | 16% (2596) | SA | N/A |
| 2 | 1L | 82/F | 70% (13102) | 16% (2587) | SA | SA |
| 3 | 2L | 90/M | 51% (9520) | 4% (735) | SA | SA |
| 4 | 3R | 81/F | 100% (19101) | 3% (581) | SA | N/A |
| 5a | 4L | 82/M | 88% (16598) | 3% (633) | SA | N/A |
Vestibular schwannoma in proximal IAC.
GG indicates geniculate ganglion; L, left; N/A, not available; nl., normal; No., number; R, right; SA, severe atrophy; ScGC, Scarpa’s ganglion cell counts; SGC, spiral ganglion cell counts; TG, trigeminal ganglion; IAC, internal auditory canal.
FIG. 2.
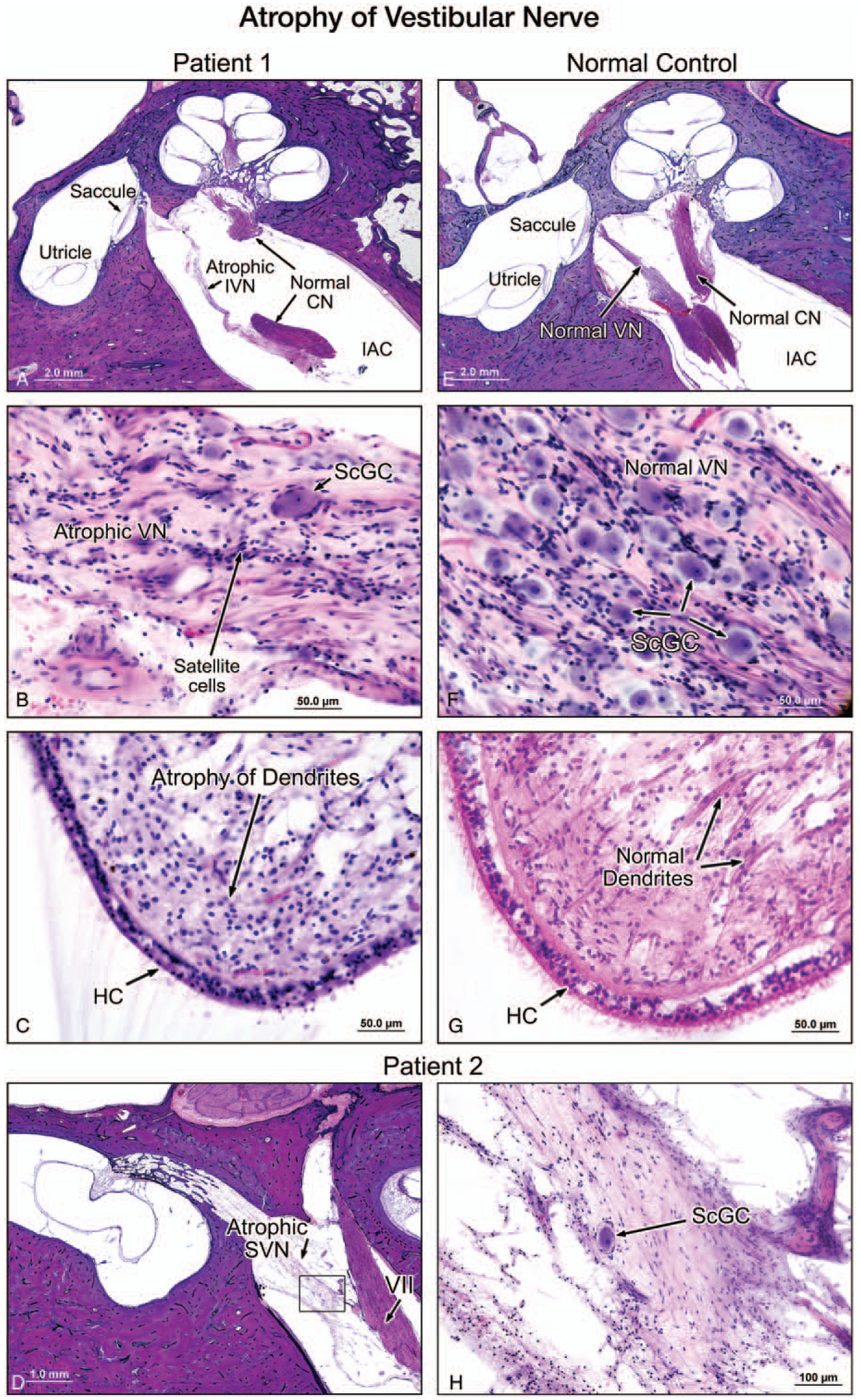
A, A low-power view of a midmodiolar section of the left temporal bone in Patient 1. In the internal auditory canal (IAC), the inferior vestibular nerve (VN) was atrophied and the cochlear nerve appeared normal. For comparison, the normal appearance of the vestibular nerve is demonstrated in a section from a normal temporal bone selected from our collection (E–G). B, A high-power view of the inferior vestibular nerve (VN) within the internal auditory canal. The nerve was severely degenerated, and there was severe loss of Scarpa’s ganglion cells (ScGC), compared with the inferior vestibular nerve in the normal control (F). No evidence of inflammation or vasculitis could be seen. C, The crista of the lateral semicircular canal in patient 1 showed a normal population of hair cells (HC) but a significantly reduced number of dendrites. Dendrites in a normal TB is shown for comparison (G). HC indicates vestibular hair cells. D, A low-power view (D) and a higher power view (H) of the superior vestibular nerve (SVN), of Patient 2 (left ear) showed severe atrophy and significant reduction of the number of Scarpa’s ganglion cells (ScGC). There was no evidence of inflammation or vasculitis.
TABLE 2.
Peripheral vestibular histology
| Specimen | Patient/Side | Vestibular Nerve | Peripheral Dendrites | ScGC % of nl. for Age | Vestibular Sense Organs |
|---|---|---|---|---|---|
| 1 | 1R | SA | Degenerated | 16% | Normala |
| 2 | 1L | SA | Degenerated | 16% | Normala |
| 3 | 2L | SA | Degenerated | 4% | Normala |
| 4 | 3R | SA | Degenerated | 3% | Normala |
| 5b | 4L | SA | Degenerated | 3% | Normala |
Hair cells present in both the cristae and maculae.
Vestibular schwannoma in proximal IAC.
L indicates left; nl., normal; R, right; SA, severe atrophy; ScGC, Scarpa’s ganglion cell counts; IAC, internal auditory canal.
Satellite cells were present. These small, star-like cells normally surround the body of neurones. Compact areas of satellite cells, named the nodules of Nageotte, were noted in clusters, reflecting ganglion cell loss in the vestibular (Scarpa’s) ganglion. The density of vestibular hair cells appeared normal despite a markedly reduced number of dendrites (Fig. 2C). There was no evidence of vasculitis or inflammation. The endolymphatic duct and sac were normal. Furthermore, there was severe atrophy of the sensory division of the facial nerve, most remarkable at the level of geniculate ganglion (Fig. 3,A and B). The trigeminal nerve and ganglion were present in the left temporal bone and showed severe atrophy of both the nerve fibers and ganglion cells (Fig. 4). The otopathology in this case has been previously reported (4).
FIG. 3.
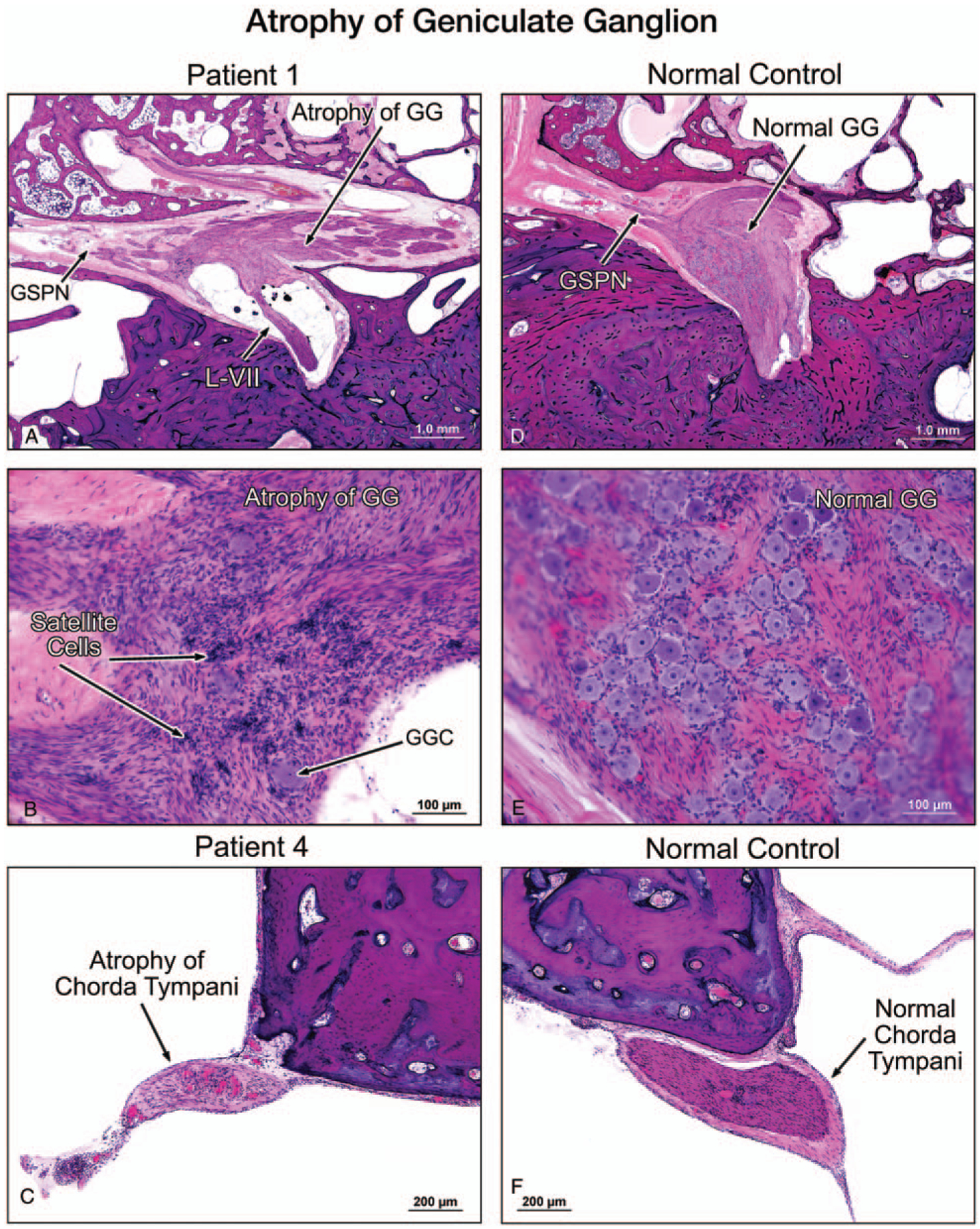
A low-power view (A) and a higher power view (B) of the facial nerve of the left temporal bone of Patient 1 showed severe atrophy of both the labyrinthine segment of the nerve (L-VII) and the geniculate ganglion (GG), as compared with a normal control which showed a normal population of GG cells, (D, E). There was no evidence of inflammation or vasculitis. GSPN indicates greater superficial petrosal nerve. A high-power view showed an atrophy of the chorda tympani nerve (C) in Patient 4, as compared with the chorda tympani nerve in a normal control (F).
FIG. 4.
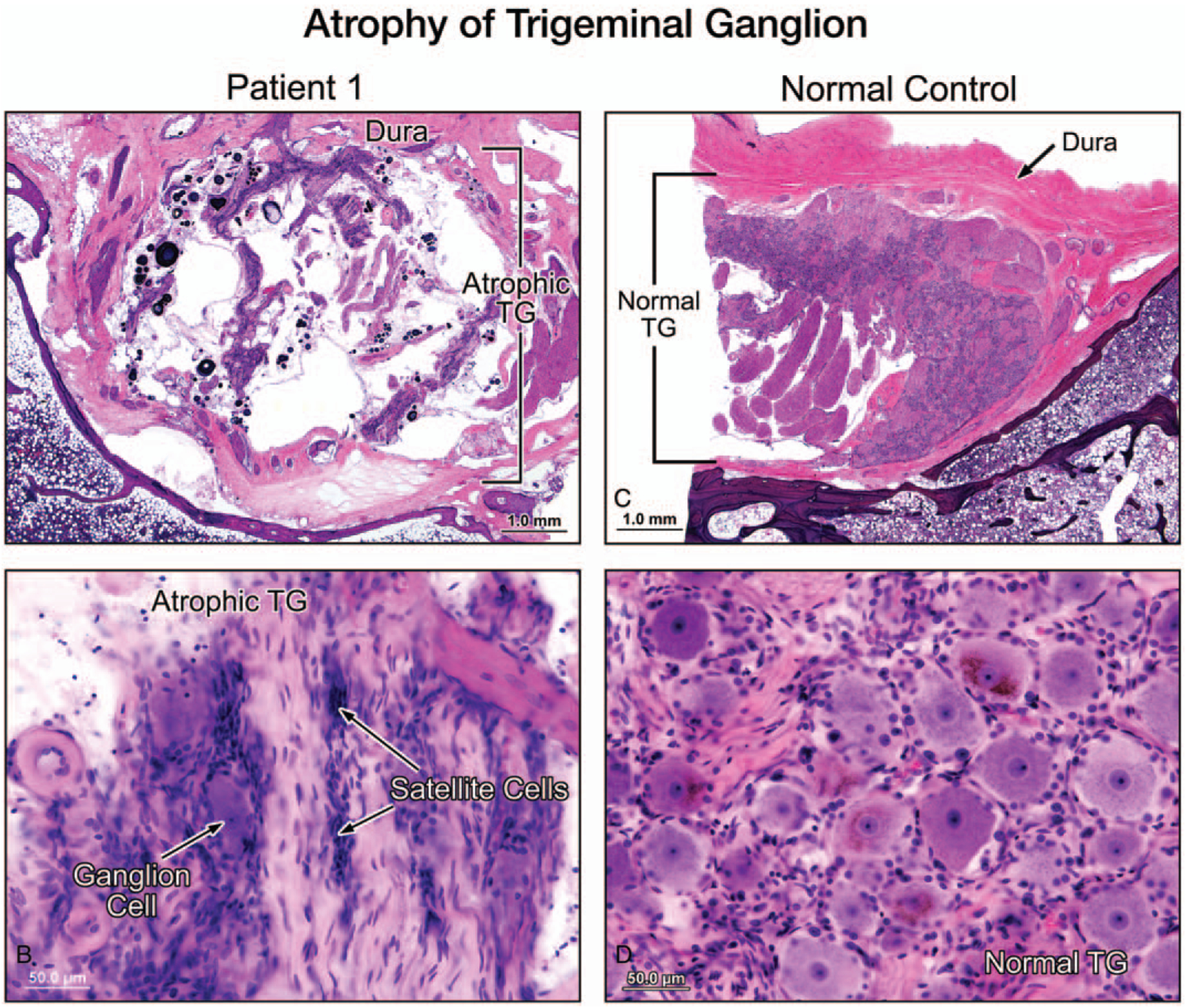
A low-power view (A) and a higher power view (B) of the trigeminal ganglion (TG) (left ear, Patient 1) showed severe atrophy and significant reduction of the number of TG cells, as compared with a normal temporal bone (C, D). There was no evidence of inflammation or vasculiti.
Neuropathology
The brainstem, including the vestibular nuclei, was unaffected. The cerebellum showed Purkinje cell loss more in the vermis than the hemispheres. Neuropathology was previously reported (5).
Case 2
Clinical Findings
A 68-year-old man presented with a 10-year history of progressive deterioration of stance and gait, on the background of a sensory neuropathy diagnosed 4 years earlier. There was a negative family history of ataxia. On examination he had a broad-based ataxic gait, a bidirectionally abnormal horizontal, and vertical HIT with saccadic visual pursuit, gaze-evoked nystagmus, saccadic hypermetria, and VVOR impairment; sensory impairment at the fingers and toes, absent ankle jerks, flexor plantae, four limb ataxia, and a positive Romberg’s test. In time, the distal proprioceptive sense was lost and progression to global tendon hyporeflexia (with flexor plantae) was noted. Caloric, rotational chair responses, and vestibular evoked myogenic muscle potentials testing showed marked bilateral semicircular canal and otolith impairment. An audiogram showed a moderate bilateral, symmetric, high-frequency sensorineural hearing loss. By age 82 he had developed dysarthria and his gait unsteadiness progressed to such a degree that despite preserved lower limb power he was confined to a wheel-chair. He suffered a painless burn of the forearm, and by age 86 he became housebound. An MRI confirmed cerebellar atrophy. Sural and tibial nerve conduction studies showed impaired neural conduction velocity. Upper and lower limb SNAPs were absent. He died aged 90 years of septicemia complicating a urinary tract infection. The brain and left temporal bone were available for study.
Otopathology
There was severe atrophy of the vestibular (Scarpa’s) ganglion cell count (4% of normal age-corrected ScGC count), the peripheral dendrites, and the superior (Fig. 2D) and inferior vestibular nerves (Tables 1 and 2). The vestibular sense organs and the endolymphatic duct and sac were unremarkable. There was no inflammation or vasculitis. In the cochlea, there was some loss of outer hair cells.
7The spiral ligament, stria vascularis, and organ of Corti were normal. Neuronal loss was noted in the spiral ganglion (cell count corresponding to 51% of normal for age (Table 1)). The trigeminal and facial nerves were atrophic, and satellite cells were demonstrated in the ganglia of both nerves.
Neuropathology
There was minor generalized atrophy of the cerebrum and cerebellar cortical atrophy with relative sparing of the inferior vermis. Microscopic examination showed severe loss of Purkinje cells in the hemispheres and the anterior/superior vermis. The brainstem showed only age-related changes. The vestibular nuclei showed no qualitative neuronal loss or gliosis. As previously reported (5) (patient 3) there was atrophy of the spinal dorsal root ganglia with subtotal neuronal loss and secondary atrophy of the dorsal roots and posterior columns and subtotal loss of myelinated axons.
As previously published4 there was atrophy of the spinal dorsal root ganglia with subtotal neuronal loss and secondary atrophy of the dorsal roots and posterior columns and subtotal loss of myelinated axons.
Case 3
Clinical History
A 67-year-old woman presented with a 30-year history of dysesthesia affecting her feet and fingertips, and progressive deterioration of stance and gait, particularly in the dark. Speech and swallowing were unaffected. There was no family history of neuropathy or ataxia. On examination, she had ataxia of gait and of all four limbs, a positive Romberg’s test and signs of a severe somatosensory impairment with absent ankle jerks, impairment of light touch, proprioception, and vibration distally with global loss of pin-prick sensation. Visual pursuit was markedly saccadic, with reduced velocity of saccades to target. The HIT and VVOR were bilaterally abnormal, with normal vestibulo-ocular reflex suppression and bilateral horizontal gaze-evoked nystagmus was noted. She had finger-to-nose ataxia and severe heel-to-shin ataxia. She also displayed a “glove and stocking” pattern of sensory loss and mild loss of distal vibration and proprioceptive perception. Caloric responses were absent to ice water stimulation.
By the age of 77, she had developed cerebellar dysarthria and dysphagia, hypometric saccades to target, and global loss of somatosensory pinprick sensation. Her bedside hearing test was described as “satisfactory.” MRI showed moderate cerebellar atrophy and moderate atrophy of the brainstem and upper cervical cord. Nerve conduction studies showed absent SNAPs with only mild slowing of motor conduction velocities. Genetic testing for SCA 1, 2, 3, 6, 7, and FA was normal. She died at age 81 of an unknown cause.
Otopathology
The right temporal bone was available for study. In the cochlea, the organ of Corti, stria vascularis, and spiral ligament appeared normal for age. The spiral ganglion cell count was 100% of normal for age. Similar to the previous cases, the most significant findings were found in the vestibular system (Tables 1 and 2). There was marked degeneration of both superior and inferior vestibular nerves, and Scarpa’s ganglion. The ScGC count was 3% of normal for age. Although the cristae and maculae of the sensory end organs (semicircular canals, saccule, and utricle) appeared normal, their dendrites were markedly degenerated.
The geniculate ganglion and facial nerve were also markedly atrophied. Nodules of Nageotte were seen in both geniculate and vestibular ganglia. The trigeminal ganglion was not present in this specimen. There was no evidence of inflammation or vasculitis in the nerves. The endolymphatic duct and sac appeared normal.
Neuropathology
The cerebellum was found to be atrophic in a pattern which predominantly affected the superior and inferior aspects of the vermis and greater than that observed in the cerebellar hemispheres. Spinal cord examination revealed significantly atrophic DRG and posterior column atrophy. Microscopically, the cerebellar vermis revealed marked neuronal loss in the Purkinje cell layer and a minor degree of atrophy of the granular cell layer with secondary loss of white matter tracts. These findings were largely mirrored in the cerebellar hemispheres. The cerebellar dentate, pontine, and cranial nerve (including the eighth cranial nerve) nuclei were all unaffected. There was no evidence to support retrograde transneuronal degeneration in the vestibular system. Whilst the corticospinal tracts appeared unaffected, the posterior columns displayed extensive atrophy and demyelination, with very few residual neurons in the dorsal root ganglia. The findings were previously reported (5).
Case 4
Clinical History
A 69-year-old man presented with a 16-year history of progressive impairment of stance and gait. There was no family history of imbalance or neurological disease. Examination revealed an ataxic gait, an abnormal Romberg’s test, and distal impairment of vibration sense to the level of the ankles with absent ankle reflexes. Oculomotor examination revealed saccadic horizontal and vertical pursuit eye movements and bidirectional VOR impairment on HIT, normal vestibulo-ocular reflex suppression, and abnormal VVOR. Bilateral upper and lower limb ataxia was present. The Romberg test was positive. In addition, there was a distal impairment of vibration sense at the ankles and absent ankle jerks. Both caloric and rotational chair testing confirmed severe bilateral vestibular impairment and there was reduced VVOR gain on rotational testing. He had symmetrical high-frequency hearing loss consistent with presbycusis and noise-induced injury. Nerve conduction studies demonstrated reduced amplitude SNAPs in all four limbs and preserved motor conduction. Brain MRI showed moderate cerebellar atrophy, particularly of the superior vermis. Genetic testing for SCA 1, 2, 3, 6, 7, and FA was negative, as were tests for coeliac disease and thyroid antibodies. By the age of 82, he was chair-bound with severe cerebellar dysarthria and dysphagia causing him difficulty in managing his own secretions. Limb power was preserved with normal upper limb and absent lower limb tendon reflexes (plantae were flexor). Perception of proprioception and pinprick was normal in the upper limbs and absent in the toes. There was marked upper and lower limb ataxia. He died at aged 83 years of bronchopneumonia.
Otopathology
The organ of Corti appeared normal with the exception of postmortem artifact and a missing area in the hook region of the cochlea. There was mild to moderate atrophy of the stria vascularis and spiral ligament. The spiral ganglion cell count corresponded to 88% of normal for his age (Table 1). As seen in previous cases, there was striking degeneration of vestibular axons, dendrites, and ganglion cells (3% of normal for his age) (Tables 1 and 2). The vestibular end-organs appeared normal. In addition, there was severe neuronal loss in the geniculate ganglion, and nodules of Nageotte were present in both vestibular and facial ganglia. There was atrophy of the chorda tympani nerve (Fig. 3C). There was no inflammation or vasculitis in the nerves. The endolymphatic duct and sac were unremarkable. Notably, an incidental vestibular schwannoma was present in the proximal portion of the internal auditory canal in contact with the facial, auditory, and vestibular nerves.
Neuropathology
Marked cerebellar atrophy, principally affecting the vermis (superior greater than inferior) and the hemispheres, was seen with near total loss of the Purkinje cells. The vestibular nerves were atrophic with loss of myelinated fibers. In the dorsal pons and medulla there was marked microglial activation in the vestibular nuclear complex; possibly reflecting Wallerian degeneration in the vestibular nerves. There was moderate atrophy of the dorsal root ganglia with severe neuronal loss and secondary atrophy with loss of myelinated axons in the dorsal columns.
Case 5
In an attempt to confirm that the vestibular nuclei are unaffected in CANVAS, a histopathological sample was acquired from the stored postmortem brainstem of a CANVAS patient who died aged 75 years of sepsis in the context of aspiration pneumonia and a small bowel obstruction. He presented with a 15-year history of slowly progressive gait ataxia and loss of sensory perception in the lower limbs. He had bilaterally reduced VOR gain on caloric and rotational chair testing, a saccadic VVOR and electrophysiologically absent upper and lower limb SNAPS. The vestibular nuclei displayed a normal population of neurons with no evidence of gliosis or reactive changes. Otopathological examination was not carried out on this patient.
DISCUSSION
The hallmark of CANVAS is the slowly progressive triad of vestibular, cerebellar, and somatosensory impairment (8). The abnormality of the VVOR is the characteristic clinical sign of combined vestibular and cerebellar dysfunction (10). The VVOR relies on three compensatory oculomotor reflexes, namely, the VOR, the optokinetic reflex, and smooth pursuit. In addition to the impaired (saccadic) VVOR, there is sensory impairment which may involve the perception of light touch, vibration, or proprioception. A diagnostic protocol has been suggested, which aids in the exclusion of other causes of ataxia, including other inherited spinocerebellar ataxias and late-onset Friedreich’s ataxia (7).
The findings here are of five CANVAS TBs (which consisted of three previously unpublished and two previously published TBs. The latter from a single patient (4) (Tables 1 and 2)). The otopathology in all five TBs is that of marked loss of vestibular (Scarpa’s) ganglion cells (3– 16% of normal for age) resulting in severe atrophy of vestibular nerves (axons) and dendrites, with preservation of vestibular receptor hair cells. In addition, a detailed focused review of all five brainstems confirmed our previous finding (6) of preservation of the vestibular nuclei. In other words, we can be confident that the severe impairment of vestibular function in CANVAS patients is entirely due to degeneration of vestibular ganglion cells; a vestibular neuronopathy. This selective degeneration of vestibular ganglion cells is pathologically the same as occurs in other cranial nerve (facial, trigeminal) and spinal (dorsal root) sensory ganglion cells in CANVAS (6). In contrast, with the absence of degenerative changes in the auditory system; not only are cochlear receptor hair cells, but also auditory ganglion cells (spiral ganglion) and auditory nerves are intact, which would explain the normal (for age) hearing in CANVAS patients. All five temporal bones showed collections of satellite cells and nodules of Nageotte, in the ganglia of degenerated nerves (vestibular, facial, and trigeminal). These cells are consistent with neuronophagia and ganglion cell degeneration and appear in other disorders associated with sensory neuronopathy (e.g., Friedreich ataxia and HIV infection) (14–18). Furthermore, preservation of vestibular hair cells and the absence of cochlear/saccular hydrops, excludes other, potentially, comorbid peripheral vestibular disorders such as Meniere’s disease (18).
It might be asked why were the vestibular nuclei normal, when there was such severe degeneration of the vestibular nerves? Why was there no evidence of trans-synaptic degeneration? The reason might be that vestibular nuclei receive many neural inputs apart from primary vestibular afferent inputs: from the visual system, the somatosensory system, and the cerebellum (19) and these are enough to prevent the development of trans-synaptic degeneration from vestibular deafferentation.
Our study is unique in that our patients had: 1) a definite phenotype with now an identified genotype (1,2), 2) detailed vestibular and auditory testing, and 3) focused pathologic examination of both the peripheral and central vestibular system. We were able to find only one vaguely similar study: Spoendlin (20) and van Bogaert and Martin (21) in 1974 reported a group of patients who would today be considered to have an unidentified hereditary ataxia with auditory and vestibular involvement (21) or possibly Friedreich’s Ataxia (gene not yet identified) (20). Their patients had both a neuropathological and otopathological examination which showed, as in our cases, preservation of the receptor hair cells. In contrast to our cases degeneration of both vestibular and cochlear nerves, and the vestibular and cochlear nuclei, was reported.
Clinically, the differential diagnosis of CANVAS includes both dominant (e.g., SCA3) and a recessive (Friedreich’s) hereditary ataxia, both of which can display the CANVAS phenotype (22–24). In SCA3 the vestibular nuclei are atrophic with secondary demyelination (19) but there are no otopathological reports. In Friedreich’s ataxia (FA) there is atrophy of vestibular ganglia, which as in CANVAS results in vestibular nerve atrophy, and the vestibular end-organs themselves are unaffected (20,25). There is however some loss of vestibular nuclei neurons in some FA patients (26). A limitation of these “FA” reports is that they preceded the availability of the FA diagnostic gene test (26) so these results could be confounded by misdiagnosis. In CANVAS, the facial nerve is atrophic secondary to geniculate ganglion cell loss (Fig. 3).
In our cohort of CANVAS patients, the facial and trigeminal sensory neuronopathies, unlike the vestibular neuronopathy, were not only asymptomatic but clinically inapparent. This might be due to inadequate relevant clinical data in this retrospective study or to minor facial and trigeminal nerve sensory deficits, despite marked histologic degeneration of these nerves. In 80 CANVAS patients, trigeminal or facial sensory impairments were not recorded on routine office examinations (8), and in another study only 2/26 CANVAS patients had loss of trigeminal sensation (27). While clinically these deficits were largely unapparent, there was electrophysiological evidence of facial and trigeminal sensory neuronopathies (9).
From this confirmatory evidence we report here that one can be confident that the pathology responsible for the severe impairment of vestibular function in CANVAS is entirely peripheral, i.e., in the vestibular ganglia, and is not central, i.e., in the vestibular nuclei.
Potential Limitations of This Study
The temporal bone findings from Patient 4 are potentially confounded by an incidentally identified vestibular schwannoma in the proximal internal auditory canal. Although vestibular and facial nerve degeneration may be due to the vestibular schwannoma, this is usually related to the size of the tumor; small in this case. The presence of marked neuronal loss in the vestibular ganglion suggests that CANVAS was more likely the cause of the neural degeneration in this case, as well. Genotyping for RFC1, the gene responsible for CANVAS (1,2), had not been undertaken as the discovery of this gene, which was only recently identified, postdated the death of the subjects in this article.
CONCLUSIONS
In this study, the histopathology of five temporal bones in four CANVAS patients demonstrates a severe selective sensory neuronopathy (ganglionopathy) that involved the vestibular (Scarpa) ganglia with cell counts of 3 to 16% of normal individuals, in addition to severe facial and trigeminal ganglia atrophy. However, the cochlear ganglion and auditory nerve were preserved. This selective sensory neuronopathy is likely to have a shared pathogenesis with the spinal dorsal root ganglia pathology also seen in CANVAS. Additionally, the finding of normal vestibular nuclei demonstrates that the vestibular impairment in CANVAS is not of a central etiology (i.e., in the vestibular nuclei) but purely related to the peripheral vestibular pathology, which involves vestibular ganglion cell loss, that is, a vestibular neuronopathy.
Acknowledgments:
The authors also thank and acknowledge Barbara Burgess, Diane Jones, Jennifer O’Malley, and Meng Yu Zhu for histologic preparation of the specimens, and Garyfallia Pagonis for assistance in preparing the figures.
Footnotes
The authors disclose no conflicts of interest.
REFERENCES
- 1.Cortese A, Simone R, Sullivan R, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet 2019;51:649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rafehi H, Szmulewicz DJ, Bennett MF, et al. Bioinformatics-based identification of expanded repeats: A non-reference intronic pentamer expansion in RFC1 causes CANVAS. Am J Hum Genet 2019;105:151–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szmulewicz DJ, Waterston JA, Halmagyi GM, et al. Sensory neuropathy as part of the cerebellar ataxia neuropathy vestibular areflexia syndrome. Neurology 2011;76:1903–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Szmulewicz DJ, Merchant SN, Halmagyi GM. Cerebellar ataxia with neuropathy and bilateral vestibular areflexia syndrome: A histopathologic case report. Otol Neurotol 2011;32:63–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szmulewicz DJ, McLean CA, Rodriguez ML, et al. Dorsal root ganglionopathy is responsible for the sensory impairment in CANVAS. Neurology 2014;82:. 1410–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szmulewicz DJ, Roberts L, McLean CA, et al. Proposed diagnostic criteria for cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS). Neurol Clin Pract 2016;6:61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szmulewicz DJ, McLean CA, MacDougall HG, Roberts L, Storey E, Halmagyi GM. CANVAS an update: Clinical presentation, investigation and management. J Vestib Res 2014;24:465–74. [DOI] [PubMed] [Google Scholar]
- 8.Szmulewicz DJ, Seiderer L, Halmagyi GM, Storey E, Roberts L. Neurophysiological evidence for generalized sensory neuronopathy in cerebellar ataxia with neuropathy and bilateral vestibular areflexia syndrome. Muscle Nerve 2015;51:600–3. [DOI] [PubMed] [Google Scholar]
- 9.Szmulewicz DJ, Waterston JA, MacDougall HG, et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS): A review of the clinical features and video-oculographic diagnosis. Ann N Y Acad Sci 2011;1233:139–47. [DOI] [PubMed] [Google Scholar]
- 10.Petersen JA, Wichmann WW, Weber KP. The pivotal sign of CANVAS. Neurology 2013;81:1642–3. [DOI] [PubMed] [Google Scholar]
- 11.Otte J, Schunknecht HF, Kerr AG. Ganglion cell populations in normal and pathological human cochleae. Implications for cochlear implantation. Laryngoscope 1978;88 (8 pt 1):1231–46. [DOI] [PubMed] [Google Scholar]
- 12.Velázquez-Villaseñor L, Merchant SN, Tsuji K, Glynn RJ, Wall C 3rd, Rauch SD. Temporal bone studies of the human peripheral vestibular system. Normative Scarpa’s ganglion cell data. Ann Otol Rhinol Laryngol Suppl 2000;181:14–9. [DOI] [PubMed] [Google Scholar]
- 13.Schuknecht HF. Techniques for study of cochlear function and pathology in experimental animals; development of the anatomical frequency scale for the cat. ANA Arch Otolaryngol 1953;58: 377–97. [DOI] [PubMed] [Google Scholar]
- 14.Keswani SC, Pardo CA, Cherry CL, Hoke A, McArthur JC. HIV-associated sensory neuropathies. AIDS 2002;16:2105–17. [DOI] [PubMed] [Google Scholar]
- 15.Spencer PS, Weinberg HJ, Raine CS, Prineas JW. The perineurial window—a new model of focal demyelination and remyelination. Brain Res 1975;96:323–9. [DOI] [PubMed] [Google Scholar]
- 16.Adle-Biassette H, Bell JE, Creange A, et al. DNA breaks detected by in situ end-labelling in dorsal root ganglia of patients with AIDS. Neuropathol Appl Neurobiol 1998;24:373–80. [DOI] [PubMed] [Google Scholar]
- 17.Koeppen AH, Mazurkiewicz JE. Friedreich Ataxia: Neuropathology Revised. J Neuronpathol Exp Neurol. 2013;72:78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nadol JB. Disorders of unknown or multiple causes. Shelton: Merchant SN, Nadol JB Schuknecht’s Pathology of the Ear. 3rd ed.; 2010. pp. 572–615. [Google Scholar]
- 19.Highstein SM, Holstein GR. The anatomy of the vestibular nuclei. Prog Brain Res 2006;151:157–203. [DOI] [PubMed] [Google Scholar]
- 20.Spoendlin H. Optic and cochleovestibular degenerations in hereditary ataxias: II. Temporal bone pathology in two cases of Friedreich’s ataxia with vestibulo-cochlear disorders. Brain 1974;97: 41–8. [DOI] [PubMed] [Google Scholar]
- 21.van Bogaert L, Martin L. Optic and cochleovestibular degenerations in the hereditary ataxias. I. Clinico-pathological and genetic aspects. Brain 1974;97:15–40. [DOI] [PubMed] [Google Scholar]
- 22.Gordon CR, Joffe V, Vainstein G, Gadoth N. Vestibulo-ocular areflexia in families with spinocerebellar ataxia type 3 (Machado-Joseph disease). J Neurol Neurosurg Psychiatry 2003;74:1403–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Linnemann C, Tezenas du Montcel S, Rakowicz M, et al. Peripheral neuropathy in spinocerebellar ataxia Type 1, 2, 3, and 6. Cerebellum 2016;15:165–73. [DOI] [PubMed] [Google Scholar]
- 24.Rüb U, Brunt ER, de Vos RAI, et al. Degeneration of the central vestibular system in spinocerebellar ataxia type 3 (SCA3) patients and its possible clinical significance. Neuropathol Appl Neurobiol 2004;30:402–14. [DOI] [PubMed] [Google Scholar]
- 25.Oppenheimer DR. Brain lesions in Friedreich’s ataxia. Can J Neurol Sci 1979;6:173–6. [DOI] [PubMed] [Google Scholar]
- 26.Lamarche JB, Lemieux B, Lieu HB. The Neuropathology of “Typical” Friedreich’s Ataxia in Quebec. Can J Neurol Sci 1984;11 (4 Suppl):592–600. [DOI] [PubMed] [Google Scholar]
- 27.Wu TY, Taylor JM, Kilfoyle DH, et al. Autonomic dysfunction is a major feature of cerebellar ataxia, neuropathy, vestibular areflexia ‘CANVAS’ syndrome. Brain 2014;137:2649–56. [DOI] [PubMed] [Google Scholar]