

Abstract
In acute myeloid leukemia (AML), refractory disease is a major challenge and the leukemia microenvironment may harbor refractory disease. Human AML cell lines KG-1 and HL-60 expressed receptors also found on endothelial cells (ECs) such as VEGFRs, PDGFRs, and cKit. When human AML cells were co-cultured with human umbilical vein endothelial cells (HUVECs) and primary bone marrow endothelial cell (BMECs), the AML cells were more resistant to cytarabine chemotherapy, even in transwell co-culture suggesting angiocrine regulation. Primary BMECs secreted significantly increased levels of VEGF-A and PDGF-AB after exposure to cytarabine. Pazopanib, a receptor tyrosine kinase inhibitor (RTKI) of VEGFRs, PDGFRs, and cKit, removed EC protection of AML cells and enhanced AML cell sensitivity to cytarabine. Xenograft modeling showed significant regression of AML cells and abrogation of BM hypervascularity in RTKI treated cohorts. Together, these results show direct cytotoxicity of RTKIs on AML cells and reversal of EC protection. Combining RTKIs with chemotherapy may serve as promising therapeutic strategy for patients with AML.
1. Introduction
Although a majority of patients with acute myeloid leukemia (AML) achieve initial remission after standard induction chemotherapy, refractory disease remains a major challenge, especially in older individuals [1,2]. Treatment resistance may be due to leukemia cell-intrinsic mechanisms such as down-regulation of chemotherapy transport proteins and increase in multi-drug resistance receptors. However, targeting these cell-intrinsic resistance mechanisms has failed to show improved leukemia-free survival [3,4]. Therefore, increasing focus has been placed on leukemia cell-extrinsic regulation by the bone marrow (BM) microenvironment.
The BM microenvironment is a complex milieu of hematopoietic and stromal cell types that balances normal hematopoietic stem and progenitor cell (HSPC) self-renewal and differentiation [5]. A number of soluble and membrane-bound angiocrine factors produced by sinusoidal ECs and perivascular stromal cells have been identified as crucial signals that maintain quiescence and facilitate return to homeostasis following myeloablative chemotherapy including stem cell factor (SCF), Jagged-1, stromal cell derived factor 1 (SDF-1), and pleiotrophin [6–8]. Furthermore, we have demonstrated a developmental relationship between normal HSPCs and ECs by showing that adult HSPCs are capable of hemangioblast activity, generating both blood and blood vessels [9,10]. Given the close intercellular relationships between normal HSPCs and ECs in the bone marrow, it is plausible to implicate ECs in the establishment and progression of malignant HSPCs like AML.
A number of studies have suggested an intimate role between AML and blood vessels. The bone marrow of patients with AML exhibits increased vascularity and increased angiogenic factors that portend for worse prognosis independent of other factors such as cytogenetic abnormalities [11–15]. Recently, we demonstrated that AML cells can integrate within blood vessels, become quiescent, adopt EC-like features, and re-emerge as CD45+ leukemia cells [16]. Others have demonstrated that AML cells in proximity to ECs are more resistant to chemotherapy [17,18]. Together, these results indict the vascular niche as a major accomplice in AML, worthy of further study and targeting.
To test our hypothesis that increased bone marrow vascularity protects AML cells from cytotoxic therapy, we chose to use the multi-targeted receptor tyrosine kinase inhibitor (RTKI) pazopanib that has been shown to block tumor growth and inhibit angiogenesis by targeting VEGFRs, PDGFRs, and cKit (SDF-1 receptor) [19]. Pazopanib is approved for renal cell carcinoma and soft tissue sarcoma treatment, has a better health-related quality of life and is more cost effective than other RTKIs such as sunitinib [20,21].
2. Methods
2.1. Human AML cell lines
HL-60 and KG-1 cell lines were purchased from the American Tissue Culture Collection (Manassas, VA) and incubated with 5% CO2 in air at 37 °C. HL-60 and KG-1 cell lines were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM) (Invitrogen) supplemented with 20% heat-inactivated fetal bovine serum (FBS) (Atlanta Biologicals) and 1% penicillin/streptomycin (Cellgro). Human Umbilical Vein Endothelial Cells (HUVEC) were obtained from ATCC. The cell line was cultured in Endothelial Growth Medium-2 (EGM-2) (Lonza) supplemented with EGM-2 Bulletkit (Lonza) growth factors. GFP+ HL-60 cells were created using lentiviral transfection.
2.2. Therapeutic agents
Pazopanib (Votrient) was provided by GlaxoSmithKline, Inc. For in vitro experiments, pazopanib was dissolved in a l0 mM stock solution of dimethyl sulfoxide (DMSO) (Sigma–Aldrich). Further dilution to indicated concentrations was done with DMSO. Cytarabine (Ara-C) (Sigma–Aldrich) was dissolved in a 10 mM stock solution to indicated concentrations with DMSO. For in vivo experiments, pazopanib was suspended in 0.5% hydroxypropylmethyl cellulose (HPMC) (Colorcon) and 0.1% Tween-80 in water as a vehicle (pH 1.3–1.5) and given daily by oral gavage. Cytarabine was given intraperitoneally with 1 × Dulbecco’s Phosphate-Buffered Solution as a vehicle.
2.3. Co-culture studies
For direct contact co-culture studies in a 96-well format, HUVEC cells were seeded at 2 × 104 cells per well in a 100 uL of culture media and allowed to attach overnight. HUVEC cells were used in initial experiments as proof-of-principle, as well as their reliability and uniformity in cell culture propagation. Only early passage HUVECs were used for co-culture experiments. Subsequently KG-1 cells were seeded on direct contact at 1 × 105 cells per well in a 100 uL of culture media. Like wise, for a 24-well format, HUVEC cells were seeded at 5 × 104 cells per well in a 1 mL of culture media (EGM-2) and allowed to attach overnight. The next day, EGM-2 was aspirated and the KG-1 cells were seeded on direct contact at 5 × 104 cells per well in 1 mL of culture media. After treatment with cytarabine (AraC) alone, pazopanib alone, or a combination of both at indicated time and dose, assays were performed to determine cell viability or apoptosis.
For transwell co-culture studies HUVEC cells were seeded at 5 × 104 cells per well in a 1 mL of culture media (EGM-2) and allowed to attach overnight. KG-1 cells were seeded in transwell inserts the following day at 5 × 104 cells per well in 100 uL of culture media. Cell viability was assessed using flow cytometry by staining for PI and Annexin V (BD Biosciences).
2.4. Cell viability
Cell proliferation was measured using an XTT colorimetric assay (ATCC). Cells were seeded on a 96 well plate at 1 × 105 cells per well in 100 uL culture media. After a 24 h incubation with treatment at indicated doses, 50 μL of XTT reagent was added to each well and plates were incubated at 37 °C for 3 h. Plates were analyzed using an Infinite M200 Pro reader (Tecan). Prism (GraphPad) was used to calculate IC50 values. This same procedure was followed for cells seeded in co-culture as previously described.
The XTT assay was used to quantify cell growth in the drug sensitivity experiments. In brief, an absorbance microplate reader is used to measure the change in color that results when a tetrazolium dye is reduced to a soluble, highly pigmented derivative.
2.5. Apoptosis
To measure apoptosis, HL-60 or KG-1 cells were seeded at 2 × 105 cells/mL in culture medium and incubated with pazopanib and cytarabine for 48 h at the concentrations indicated. Briefly, cells were collected and stained using propidium iodide (PI) and Annexin V-APC (BD Biosciences) according to the manufacturer’s protocol. Analysis was performed using a Becton Dickinson FACSCanto II flow cytometer.
2.6. Human angiogenesis cytokine array
Media supernatants were assayed for the presence of angiogenic cytokines, after stimulating the cells with chemotherapy. The assay was performed using the Proteome Profiler™ Array from R&D Systems. HUVEC and KG-1 cells were seeded in co-culture using a transwell system as previously described. Media was collected after 48 h of treatment with increasing concentrations of cytarabine. Passage 3 primary human bone marrow endothelial cells (BMECs) were rinsed with 1X PBS and incubated with (unsupplemented) EBM-2 + GA1000 (antibiotic) and 20 uM cytarabine for 24 h. Supernatants were collected and then analyzed by cytokine array. Briefly, the membrane containing immobilized angiogenesis-related antibodies were blocked with bovine serum albumin for 1 h on a rocking platform at room temperature. Membranes were then incubated with cell culture supernatants along with a detection antibody cocktail overnight at 2 ◦C–8 ◦C on a rocking platform. The membrane was incubated with streptavidin–horseradish peroxidase conjugate followed by chemiluminescent detection reagent. The membrane was scanned and pixel density was presented by quantifying the mean spot densities. Density quantification was done using Image J software (NIH).
2.7. cDNA and PCR
RNA isolation from KG-1 and HL-60 cell lines was performed following the RNeasy Plus Mini Kit protocol (Qiagen) and analyzed with a SmartSpec Plus Spectrophotometer (BioRad). Reverse transcription for cDNA was done following the High Capacity cDNa Reverse Transcription Kit protocol (Applied Biosciences). PCR was done using human primers to SCFR, PDGFR-α and PDGFR-β attained from Invitrogen Oligos and New England BioLabs reagents. Analysis was done following agarose gel electrophoresis procedure.
2.8. Western blot
Cells were treated with various concentrations of pazopanib with or without the presence of recombinant human SCF or VEGF (R&D Systems). Cell extracts were isolated using RIPA buffer. For phosphorylation studies, samples were first immunoprecipitated using protein A/G-Agarose (Santa Cruz) in the presence of primary antibody against human SCFR and then separated on a 4–20% gel. Samples were then transferred onto a nitrocellulose membrane. The immunoprecipitated samples were probed with human anti pTYR antibody (Santa Cruz) and human anti SCF receptor antibody (Santa Cruz). For apoptosis studies whole cell lysates were separated on a 4–20% gel and subsequently blotted onto a nitrocellulose membrane. Horseradish peroxidase (HRP)-marked immunoglobulins from Santa Cruz were used as secondary antibodies. Antibody binding was detected using chemiluminescence reagent (ECL Western Blotting Analysis System, Amersham). Furthermore, in trying to elucidate a mechanism of action for the drug, protein lysates were immunoblotted for total and phosphorylated Src (Santa Cruz).
2.9. Mouse xenograft model
All animal studies were performed according to approved protocols from the University of Florida IACUC. To test the efficacy of pazopanib and cytarabine in systemic AML mouse models, human leukemia chimeras were established using NOD/scid/IL2Rγ−/−(NSG) mice (Jackson Laboratories, Bar Harbor, Maine). 6-week-old, female NSG mice were inoculated with 1 × 106 GFP-HL-60 or 2 × 106 KG1 cells via tail vein injection. We allowed for four weeks of engraftment for HL-60 cells and six weeks engraftment for KG1 cells. The mice were divided into five groups of 10 mice each: pazopanib, Ara-C, pazopanib+ Ara-C, vehicle, and untreated. Treatments were 500 mg/kg of body weight Ara-C (IP) on day 1 and day 2, 100 mg/kg of body weight pazopanib (oral gavage) daily for 8 days, and 100 uL of HPMC (vehicle control).
2.10. Tissue analysis
After the last treatment, the liver, kidney, spleen, lungs and femurs were harvested from all mice for the purpose of investigation. Engraftment was determined using flow cytometry staining for human CD45 and HLA (BD Biosciences), as well as Viaprobe to determine cell viability. CD45, also known as the leukocyte common antigen, is a highly specific marker for hematopoietic cells lines. This marker, together with human specific anti HLA, was used in order to label the human leukemia cells present in the mouse bone marrow. Femurs were sectioned and stained for MECA-32 (BD Biosciences) for the assessment of blood vessel formation.
2.11. Statistical analysis
Statistical analysis of the data was performed using Student’s t-test. P values of <0.05 were considered statistically significant.
3. Results
3.1. ECs establish a chemoprotective niche by angiocrine factor stimulation of AML cells
We initially examined the effect of ECs on AML cells exposed to cytotoxic chemotherapy. Utilizing a human AML cell line that expresses angiocrine factor receptors (Fig. 1A), we found that KG-1 cells grown in direct contact with ECs exhibited resistance to cytarabine chemotherapy compared to AML grown in suspension (Fig. 1B). Treatment of AML cells in co-culture with HUVECs with cytarabine for 48 h showed significantly decreased AML cell apoptosis compared to cells grown in the absence of an EC layer using Annexin V and PI staining analysis. To examine the role of direct contact between AML and HUVECs in chemoresistance, we utilized a co-culture system in which AML cells were seeded in a transwell in the presence or absence of an EC feeder layer (Fig. 1C). When AML cells that were not directly apposed to the EC feeder layer were exposed to cytarabine, the chemoprotective effect of the ECs was preserved indicating that soluble paracrine signals released from the EC feeder layer are at least partly responsible for the decreased apoptosis observed with cytarabine treatment.
Fig. 1.
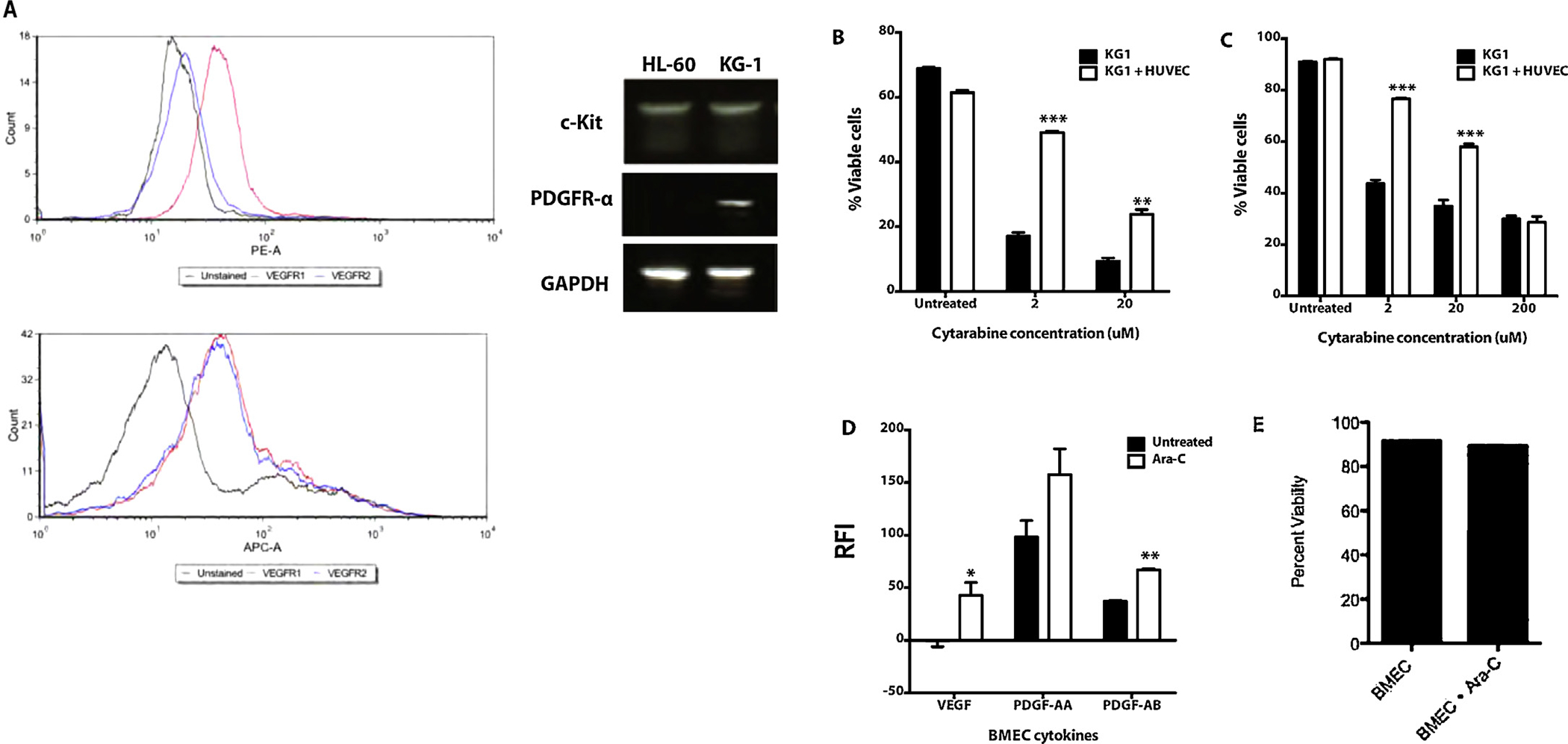
Endothelial cells protect leukemia cells from chemotherapy induced apoptosis in co-culture. (A) Flow cytometry shows HL-60 cells express VEGFR1 while KG1 cells express VEGFR1 and VEGFR2. PCR demonstrates HL-60 cells express c-Kit while KG-1 cells express c-Kit and PDGFR-a. (B) KG1 cells were seeded in direct co-culture with HUVECs and treated with varying concentrations of cytarabine. After 48 h of treatment the cells were stained with Annexin V and PI, and then analyzed by flow cytometry. KG1 AML cells were more resistant to cytarabine treatment when in co-culture with ECs (p < 0.001). (C) KG1 cells were seeded in transwell co-culture with HUVECs and treated with varying concentrations of cytarabine. After 48 h of treatment the cells were stained with Annexin V and PI, and then analyzed by flow cytometry. KG1 AML cells were more resistant to cytarabine treatment when in transwell co-culture with ECs (p < 0.001). (D) Angiogenic cytokine secretion from primary human BMECs. After cytarabine chemotherapy, BMECs secreted significantly increased VEGF-A (40-fold, p < 0.05) and PDGF-A/B (1.6-fold, p < 0.005). RFI = Relative Fluorescence Intensity. (E) BMEC viability after 24 h treatment with cytarabine chemotherapy at clinically relevant concentration, 20 μM. Flow cytometry analysis revealed no reduction in viability.
In order to identify angiocrine factors that contribute to the chemoprotective effect of HUVECs on AML cells, we analyzed the media of AML-EC co-cultures after treatment with cytarabine for soluble EC-derived paracrine factors. We found significantly increased levels of VEGF-A and PDGF-A/B in co-cultures treated with cytarabine compared to untreated (data not shown). To determine if these cytokines are elicited from primary human bone marrow ECs (BMECs) in response to chemotherapy, we cultured patient derived BMECs in EGM-2 until confluent. The primary BMECs were treated with cytarabine at 20 μM for 24 h. The supernatants were collected and analyzed for PDGF and VEGF-A. BMECs treated with cytarabine secreted 1.6-fold PDGF-AB (p < 0.005) and 40-fold the amount of VEGF (p < 0.05) compared to untreated BMECs (Fig. 1D). Cytarabine-treated BMECs at this time point showed no decrease in viability (Fig. 1E), suggesting that the cytokine release was associated with particular angiogenic/inflammatory response. These results are consistent with prior reports [22–24].
3.2. Pazopanib treatment results in an additive effect on apoptosis in combination with cytarabine in vitro
Given the evidence that ECs protect AML cells from chemotherapy and the specific angiogenic cytokines discovered in this protection (i.e., VEGF-A and PDGF-A/B), we selected pazopanib, a receptor tyrosine kinase inhibitor (RTKI) with specificity for VEGFRs, PDGFRs, and cKit (SCF receptor), as a potential anti-leukemia agent in combination with cytarabine. Treatment of HL-60 AML cells for 48 h with cytarabine alone or pazopanib alone resulted in a modest decrease in cell viability in vitro; however, when combined, the drugs had an additive effect on viability as measured by XTT assay (Fig. 2A). This effect of RTK inhibition in the absence of trophic soluble mediators secreted from ECs is not surprising given evidence that AML cells express endothelial-like receptors (Fig. 1A) and stimulate their own proliferation with angiocrine factors via autocrine signaling [25]. Next, we reasoned that in the presence of pazopanib RTK inhibition, the chemoprotective effect afforded by ECs would be abrogated. When AML cells were treated with combined therapy (pazopanib + cytarabine), there was an additive effect on apoptosis (Fig. 2B). Western blot of phosphorylated Src kinase, a downstream target of VEGFRs and PDGFRs, demonstrated a dose dependent response to pazopanib RTK inhibition suggesting a mechanism for direct leukemia cell cytotoxicity (Fig. 2C).
Fig. 2.
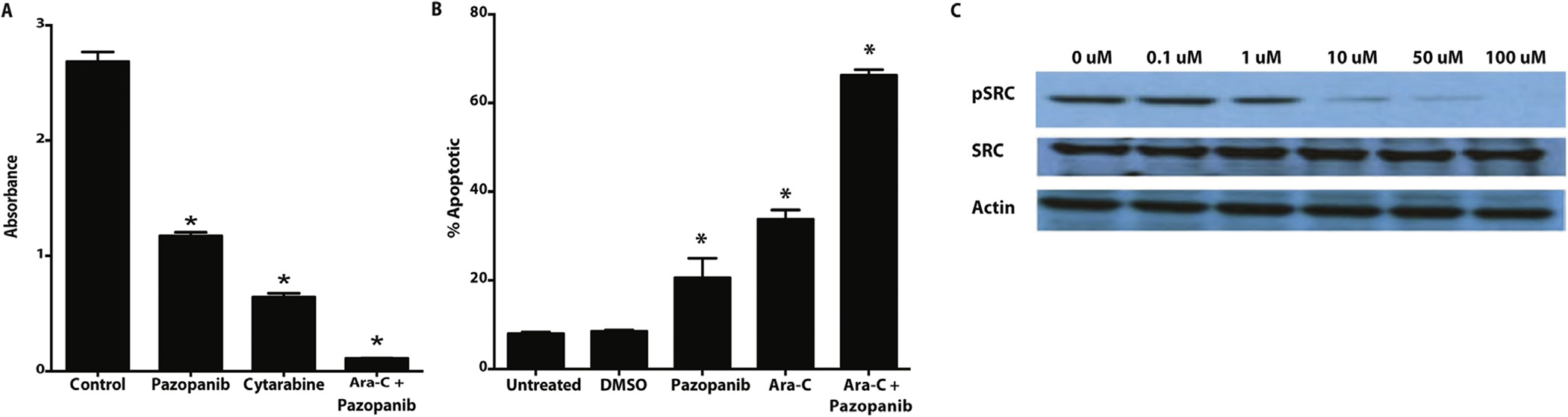
In vitro flow cytometry apoptosis analysis shows an additive effect of treatment with pazopanib and cytarabine. (A) KG-1 cells exposed to pazopanib and cytarabine demonstrate modest reduction in viability by XTT assay. In combination, the effect is additive, significantly decreasing viability of AML cells relative to controls. *p-values <0.05 for all treatment samples, compared to untreated control. (B) In the presence of an EC feeder layer, the addictive effect of pazopanib and cytarabine on AML cells persists. *p-values <0.05 for all treatment samples, compared to untreated control. (C) Western blot of phosphorylated SRC, a downstream target of RTK signaling, shows a dose-dependent decrease in phosphorylation with pazopanib exposure.
3.3. Pazopanib treatment in vivo results in decreased AML engraftment and abrogates cytarabine-induced angiogenesis
To test the effect of pazopanib RTK inhibition in vivo, NSG mice were injected with 1 × 106 GFP-tagged HL-60 cells and allowed to engraft. After 4 weeks, mice were treated with pazopanib, cytarabine, or combination for 8 days. The cohorts were sacked and engraftment was determined with flow cytometry. Pazopanib, cytarabine, and the combination showed similar levels of BM engraftment relative to the untreated cohort demonstrating that pazopanib alone is as effective as conventional cytotoxic chemotherapy (Fig. 3A). Examination of the bone marrow histology demonstrated a trend of increased vascularity in mice treated with cytarabine alone compared to treatment with pazopanib (Fig. 3B and C). To confirm reproducibility among cell lines, NSG mice were injected with 2 × 106 KG1 cells and allowed to engraft for six weeks. Treatment with cytarabine, pazopanib, or a combination reduced bone marrow AML engraftment comparable to the previous model (Fig. 3D). This angiogenic response to cytarabine is consistent with the increased angiocrine profile elicited from BMECs treated with cytarabine in vitro. The increased angiogenic effect of cytarabine was abrogated with addition of pazopanib (Fig. 3B and C).
Fig. 3.
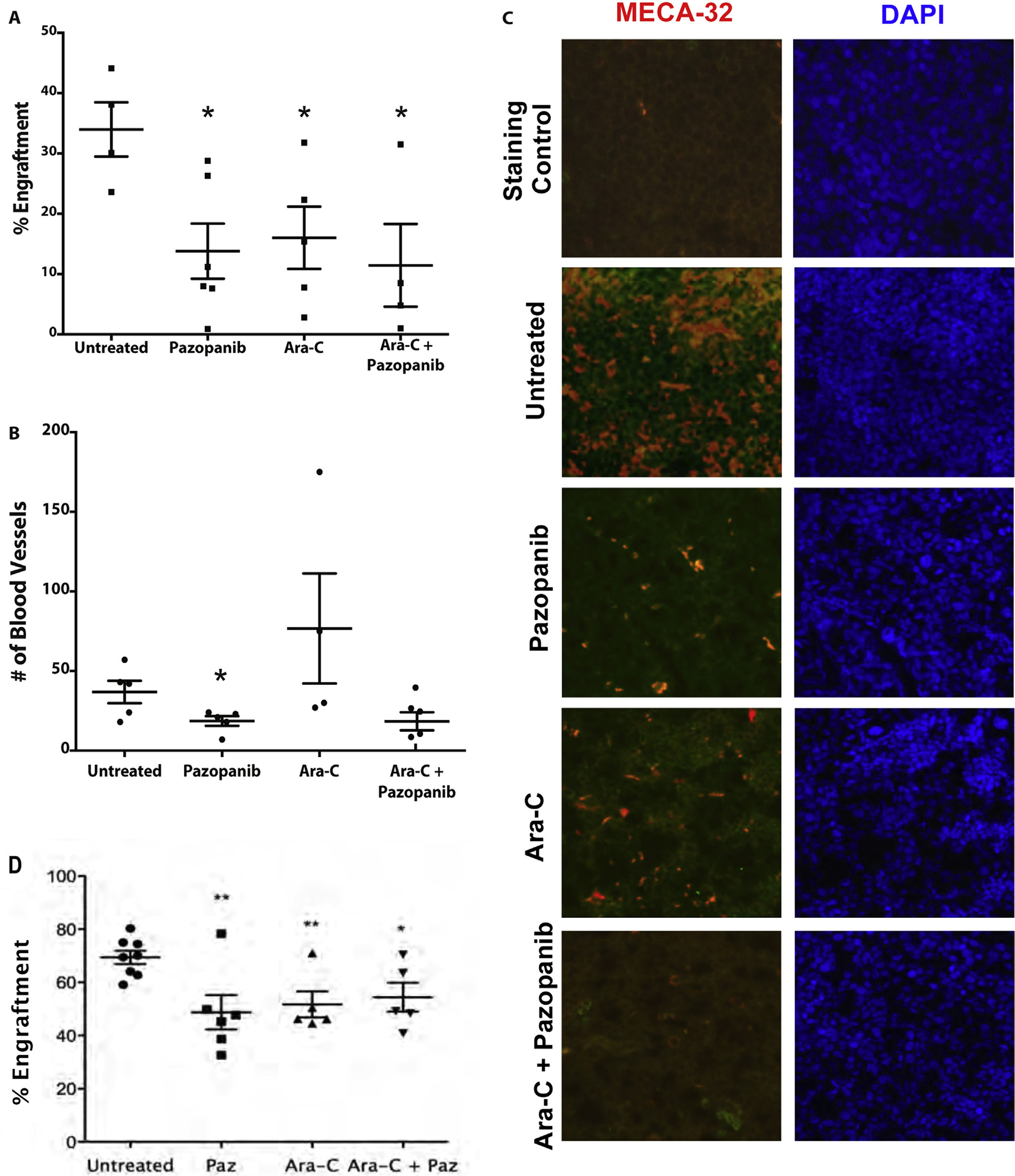
Pazopanib treatment decreases engraftment equivalent to cytarabine and disrupts microvessel formation. (A) Pazopanib and cytarabine decreased the level of HL-60 engraftment equally. The decrease in engraftment for all treatment groups was determined by flow cytometry analysis. HL-60 engraftment for untreated mice was approximately 34%. HL-60 engraftment for treated mice ranged 12–18%. *p-values <0.05 for all treatment samples compared to untreated control. (B) Pazopanib treatment leads to a reduction in the number of blood microvessels within leukemic cores compared to other treatment groups. Quantification of microvessels based on MECA-32+. Values represent mean ± SEM. p < .05. (C) Representative bone marrow micrographs showing microvessel density (MECA-32 endothelial stain, red) and nuclear staining (DAPI, blue) of mouse femurs after transplant with human AML cells (HL-60) and treatment with pazopanib and cytarabine. Untreated animals with robust AML engraftment showed high microvessel density in the bone marrow. Pazopanib alone markedly decreased microvessel density in the bone marrow. Cytarabine also decreased microvessel density. The combination of pazopanib and cytarabine also caused marked reduction in blood vessels within bone marrow. (D) Pazopanib and cytarabine decreased levels of bone marrow KG1 engraftment equally. The decrease in engraftment for all treatment groups was determined by flow cytometry analysis. KG1 engraftment for untreated mice ranged 59–80%. KG1 engraftment for treated mice ranged 33–78%. **p-values <0.005, *p-value <0.05 for all treatment samples compared to untreated control.
4. Discussion
Whereas progress in genetics has greatly advanced our understanding of AML as a multi-genetic and oligoclonal disease [26,27], targeting recurrent genetic mutations and intracellular resistance mechanisms has not improved the 60–80% relapse rate in AML patients. Therefore, leukemia cell-extrinsic factors in the AML microenvironment are gaining greater interest. In this study we demonstrate that ECs provide protection to AML cells from cytarabine chemotherapy, and that this protection is partly due to paracrine communication. Our results confirm previous reports on the broad role of ECs in AML [28], but also extend our understanding of AML pathobiology by showing bone marrow relevance through the use of primary BMECs as protective reservoirs. Recently we demonstrated that AML cells functionally integrate within endovascular linings and exit cell cycle. We postulated that EC-associated AML cells would be more resistant to cell cycle active agents [16]. In this study, we addressed this hypothesis by showing resistance to cytarabine, an S-phase agent, when leukemia cells were associated with ECs.
In search of a cell biology explanation, we found that BMECs secrete angiogenic/inflammatory cytokines, such as VEGF-A and PDGFs, in response to cytarabine. When considering that AML cells can also express endothelial-like receptors such as VEGFRs and PDGFRs (Fig. 1 and [29]), the paracrine or angiocrine release from ECs may contribute to pathologic protection and promotion of leukemia. Therefore, we applied an FDA-approved TKI, pazopanib, with known inhibition of VEGFRs, PDGFRs, and cKit (SCF receptor) and found three outcomes: (1) direct killing of AML cells, (2) reversal of EC protection of AML cells, and (3) abrogation of BM angiogenesis.
VEGF has been a target of interest for many years in AML [25,30]. Although monoclonal antibody therapy directed against VEGF (i.e., bevacizumab) in AML patients reduces BM VEGF levels, minimal improvements in clinical outcomes are seen [31,32]. Anti-angiogenic TKIs have also been tested as monotherapies in AML and have shown mixed responses in early phase clinical trials [33,34]. In the clinical setting of AML where the disease is rapidly hyperproliferative, cytoreductive chemotherapy is often required immediately. From a clinical practical perspective, any viable microenvironmental targeting strategy for AML, especially refractory/relapsed disease, will require combination therapy with a cytoreductive reagent. Therefore, in this study we tested an anti-angiogenic RTKI in combination with cytarabine chemotherapy and demonstrate an enhanced additive effect. Together, these results support the combination of anti-angiogenic RTKI in combination with cytarabine for patients with AML. Preliminary results from a phase I clinical study of sunitinib (RTKI with activity against FLT3, VEGFRs, PDGFRs, cKit, and others) in combination with standard AML induction chemotherapy presented by Fiedler et al. showed impressive disease response (70% CR/CRi) despite myelosuppression [35]. Optimizing a combination strategy of targeting the AML microenvironment plus chemotherapy for AML will require adjusting the dose of cytoreductive chemotherapy, intentional selection of patients with AML myeloblasts expressing relevant EC-like receptors, and/or rationally designing a timed-sequential therapy based on angiogenic cytokine release as Karp et al. previously piloted [31].
When considering our study, several considerations should be made. First, our initial experiments utilized HUVECs, which may differ from ECs that interact with leukemia cells. To address this, we harvested primary BM specimens from patients, established BMECs, and used these to confirm EC-AML protection. In follow-up experiments, we will test whether there are differences between healthy volunteer BMECs and leukemia BM-derived BMECs. It may also be interesting to test EC-AML interactions in serum free conditions, such as with adenovirus E4+ ECs [6]. Second, we used AML cell lines rather than primary AML specimens. The main reason for cell line usage was because the focus of our experiments was on targeting known RTKs, rather than discovering new receptors in AML. Prior investigators have done an excellent job demonstrating EC-like expression on primary AML myeloblasts, and we found no cause to repeat these experiments [30,36]. Finally, the xenograft model showed significant BM regression of leukemia in the RTKI cohorts, but no improved BM regression when combining RTKI with cytarabine. Reasons for this may include the dosing and schedule of the treatment agents. We approximated clinically relevant dosing; however, drug dose and schedule are difficult to establish and extrapolate in mouse models, especially NSG xenografts [37]. At a minimum, our results show strong proof-of-principle that pazopanib RTK inhibition has in vivo activity in AML. Moreover, abrogation of BM hypervascularity may show its greatest benefit in terms of time to relapse, which can take several months to years in human patients. By reducing the number of vascular niches that serve as protective reservoirs of disease, anti-angiogenic RTKIs may winnow niches for minimal residual disease.
Acknowledgements
The Leukemia & Lymphoma Society supported CRC with a Scholar in Clinical Research award (2400–13). This work was supported by the Gatorade Trust, which is administered by the University of Florida Department of Medicine.
Footnotes
Conflict of interest statement
The authors declare no conflict of interest.
References
- [1].Lowenberg B, Downing JR, Burnett A, Acute myeloid leukemia, New Engl. J. Med. 341 (14) (1999) 1051–1062. [DOI] [PubMed] [Google Scholar]
- [2].Appelbaum FR, et al. , Age and acute myeloid leukemia, Blood 107 (9) (2006) 3481–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chauncey TR, et al. , Sequential phase II Southwest Oncology Group studies (S0112 and S0301) of daunorubicin and cytarabine by continuous infusion, without and with ciclosporin, in older patients with previously untreated acute myeloid leukaemia, Br. J. Haematol. 148 (1) (2010) 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Roboz GJ, et al. , Phase I trial of SAR103168, a novel multi-kinase inhibitor, in patients with refractory/relapsed acute leukemia or high-risk myelodysplastic syndrome, Leuk. Lymphoma 56 (2) (2015) 395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Morrison SJ, Scadden DT, The bone marrow niche for haematopoietic stem cells, Nature 505 (7483) (2014) 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Butler JM, et al. , Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells, Cell Stem Cell 6 (3) (2010) 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kobayashi H, et al. , Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells, Nat. Cell Biol. 12 (11) (2010) 1046–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Poulos MG, et al. , Endothelial Jagged-1 is necessary for homeostatic and regenerative hematopoiesis, Cell Rep. 4 (5) (2013) 1022–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Grant MB, et al. , Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization, Nat. Med. 8 (6) (2002) 607–612. [DOI] [PubMed] [Google Scholar]
- [10].Cogle CR, et al. , Adult human hematopoietic cells provide functional hemangioblast activity, Blood 103 (1) (2004) 133–135. [DOI] [PubMed] [Google Scholar]
- [11].Hussong JW, Rodgers GM, Shami PJ, Evidence of increased angiogenesis in patients with acute myeloid leukemia, Blood 95 (1) (2000) 309–313. [PubMed] [Google Scholar]
- [12].Padro T, et al. , Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia, Blood 95 (8) (2000) 2637–2644. [PubMed] [Google Scholar]
- [13].Loges S, et al. , Analysis of concerted expression of angiogenic growth factors in acute myeloid leukemia: expression of angiopoietin-2 represents an independent prognostic factor for overall survival, J. Clin. Oncol. 23 (6) (2005) 1109–1117. [DOI] [PubMed] [Google Scholar]
- [14].Lee CY, et al. , Marrow angiogenesis-associated factors as prognostic biomarkers in patients with acute myelogenous leukaemia, Br. J. Cancer 97 (7) (2007) 877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shih TT, et al. , Bone marrow angiogenesis magnetic resonance imaging in patients with acute myeloid leukemia: peak enhancement ratio is an independent predictor for overall survival, Blood 113 (14) (2009) 3161–3167. [DOI] [PubMed] [Google Scholar]
- [16].Cogle CR, et al. , Functional integration of acute myeloid leukemia into the vascular niche, Leukemia 28 (10) (2014) 1978–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pezeshkian B, et al. , Leukemia mediated endothelial cell activation modulates leukemia cell susceptibility to chemotherapy through a positive feedback loop mechanism, PLOS ONE 8 (4) (2013) pe60823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Poulos MG, et al. , Activation of the vascular niche supports leukemic progression and resistance to chemotherapy, Exp. Hematol. 42 (11) (2014) 976–986, el-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li H, et al. , Pazopanib, a receptor tyrosine kinase inhibitor, suppresses tumor growth through angiogenesis in dedifferentiated liposarcoma xenograft models, Transl. Oncol. 7 (6) (2014) 665–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hansen RN, et al. , Health care costs among renal cancer patients using pazopanib and sunitinib, J. Manag. Care Spec. Pharm. 21 (1) (2015) 37–44, 44a-d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Delea TE, et al. , Cost-effectiveness of pazopanib versus sunitinib for renal cancer in the United States, J. Manag. Care Spec. Pharm. 21 (1) (2015) 46–54, 54a-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chervontseva AM, Romanov Iu A, Savchenko VG, [Effect of cytarabine on expression of cell adhesion molecules and on endothelium–leukocyte interaction in vitro], Ter. Arkh. 78 (7) (2006) 67–72. [PubMed] [Google Scholar]
- [23].Romanov YA, et al. , Vascular endothelium: target or victim of cytostatic therapy? Can. J. Physiol. Pharmacol. 85 (3–4) (2007) 396–403. [DOI] [PubMed] [Google Scholar]
- [24].Liersch R, et al. , Expression of VEGF-C and its receptor VEGFR-3 in the bone marrow of patients with acute myeloid leukaemia, Leuk. Res. 32 (6) (2008) 954–961. [DOI] [PubMed] [Google Scholar]
- [25].Dias S, et al. , Inhibition of both paracrine and autocrine VEGF/VEGFR-2 signaling pathways is essential to induce long-term remission of xenotransplanted human leukemias, Proc. Natl. Acad. Sci. U. S. A. 98 (19) (2001) 10857–10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Walter MJ, et al. , Clonal architecture of secondary acute myeloid leukemia, N. Engl. J. Med. 366 (12) (2012) 1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Welch JS, et al. , The origin and evolution of mutations in acute myeloid leukemia, Cell 150 (2) (2012) 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hatfield K, et al. , Primary human acute myeloid leukaemia cells increase the proliferation of microvascular endothelial cells through the release of soluble mediators, Br. J. Haematol. 144 (1) (2009) 53–68. [DOI] [PubMed] [Google Scholar]
- [29].Padro T, et al. , Overexpression of vascular endothelial growth factor (VEGF) and its cellular receptor KDR (VEGFR-2) in the bone marrow of patients with acute myeloid leukemia, Leukemia 16 (7) (2002) 1302–1310. [DOI] [PubMed] [Google Scholar]
- [30].Fiedler W, et al. , Vascular endothelial growth factor, a possible paracrine growth factor in human acute myeloid leukemia, Blood 89 (6) (1997) 1870–1875. [PubMed] [Google Scholar]
- [31].Karp JE, et al. , Targeting vascular endothelial growth factor for relapsed and refractory adult acute myelogenous leukemias: therapy with sequential 1-beta-D-arabinofuranosylcytosine, mitoxantrone, and bevacizumab, Clin. Cancer Res. 10 (11) (2004) 3577–3585. [DOI] [PubMed] [Google Scholar]
- [32].Zahiragic L, et al. , Bevacizumab reduces VEGF expression in patients with relapsed and refractory acute myeloid leukemia without clinical antileukemic activity, Leukemia (2007). [DOI] [PubMed] [Google Scholar]
- [33].Fiedler W, et al. , A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease, Blood 105 (3) (2005) 986–993. [DOI] [PubMed] [Google Scholar]
- [34].Fiedler W, et al. , An open-label, phase I study of cediranib (RECENTIN) in patients with acute myeloid leukemia, Leuk. Res. 34 (2) (2010) 196–202. [DOI] [PubMed] [Google Scholar]
- [35].Fiedler W, et al. , A phase I/II study combining sunitinib with standard ara-C/daunorubicin chemotherapy in patients 60 years or older with FLT3 mutated AML, ASH Ann. Meet. Abstr. 116 (21) (2010) 3285-. [Google Scholar]
- [36].Dias S, et al. , Vascular endothelial growth factor (VEGF)-C signaling through FLT-4 (VEGFR-3) mediates leukemic cell proliferation, survival, and resistance to chemotherapy, Blood 99 (6) (2002) 2179–2184. [DOI] [PubMed] [Google Scholar]
- [37].Wunderlich M, et al. , AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model, Blood 121 (12) (2013) e90–e97. [DOI] [PMC free article] [PubMed] [Google Scholar]