

Abstract
The BH-3 mimetic venetoclax overcomes apoptosis and therapy resistance caused by high expression of BCL2 or loss of BH3-only protein function. Although a promising therapy for hematologic malignancies, increased expression of anti-apoptotic MCL-1 or BCL-XL, as well as other resistance mechanisms prevent a durable response to venetoclax. Recent studies demonstrate that agents targeting epigenetic mechanisms such as DNA methyltransferase inhibitors, histone deacetylase (HDAC) inhibitors, histone methyltransferase EZH2 inhibitors, or bromodomain reader protein inhibitors may disable oncogenic gene expression signatures responsible for venetoclax resistance. Combination therapies including venetoclax and epigenetic therapies are effective in preclinical models and the subject of many current clinical trials. Here we review epigenetic strategies to overcome venetoclax resistance mechanisms in hematologic malignancies.
Keywords: Venetoclax, Azacytidine, Decitabine, EZH2, JQ1
1. Introduction
In the last decade remarkable progress has been made to improve our understanding of how altered expression of the apoptosis machinery may be inexorably linked to tumorigenesis. Cell life and death decisions are made largely dependent on the balance between pro-apoptotic and anti-apoptotic regulators. Malignancy may ensue when the balance is shifted to prevent normal cell death processes. This was highlighted by the identification of BCL-2 as an oncoprotein in lymphoma [1,2]. Elucidation of the mechanism by which BCL-2 and similar proteins prevent cell death have led to a new category of recently approved anticancer therapies. However, like many targeted therapies for malignancy, treatment can result in the induction of or selection for resistance, often through shifts in gene regulation mediated by changes in chromatin function. Thus, pharmacological agents that act upon epigenetic mechanisms and restore a more normal state of gene expression to cancer cells may be useful to counteract some resistance mechanisms of BCL-2 directed cancer therapies.
BCL-2 family members are categorized by their BCL-2 homology (BH) domains into three groups: anti-apoptotic proteins, pro-apoptotic BH3-only proteins and pro-apoptotic mitochondria pore-forming proteins. These BH domains facilitate interaction between family members to direct their anti- or pro-apoptotic function [3]. Patterns of gene expression, activation or subcellular localization of individual BCL-2 family members may vary depending on factors such as cell type, differentiation state, cellular stress stimuli or developmental stage. Anti-apoptotic proteins such as BCL-2, BCL-XL, and MCL-1 bind to and sequester mitochondria pore forming proteins BAX and BAK. Upon cellular stress, BH3-only proteins such as BIM (BCL2L11), BID, BAD, PUMA (BBC3), BIK and NOXA (PMAIP1) bind to anti-apoptotic proteins through their BH3 domain to release pore-forming proteins. These pore-forming proteins associate in the outer mitochondrial membrane to trigger membrane permeability and the release of cytochrome C from the mitochondria which activates caspases to initiate apoptosis.
More than 35 years ago, it was discovered that the t(14;18) chromosome translocation found in follicular lymphoma and B cell leukemia placed BCL2 from chromosome 18 under the control of the immunoglobulin heavy-chain promoter on chromosome 14, driving constitutive BCL2 expression and resistance to apoptosis [1,2]. This translocation is present in about 90% of follicular lymphoma and 10–30% of diffuse large B cell lymphoma (DLBCL), but is insufficient to cause lymphoma on its own [4–6]. High BCL2 expression is often observed in combination with other activated oncogenes. For example, approximately 5–15% of diffuse large B-cell lymphomas (DLBCL) harbor MYC, BCL2 and/or BCL6 translocations that have been characterized to drive proliferation and prevent apoptosis. These “double” or “triple hit” lymphomas demonstrate a high-grade histology and are difficult to treat with conventional chemotherapies leading to poor outcomes [7]. Genetic amplification of BCL2, aberrant activation of the NF-κB pathway, or downregulation of certain microRNAs can cause increased BCL-2 expression and resistance to therapy in a variety of human cancers [8,9]. The TCGA pan-cancer cBioPortal indicates that BCL2 amplification is found in about 4% of DLBCL, 2% of Sarcomas, 1% of Esophageal cancer and 1% of invasive breast carcinoma (Fig. 1) [10,11]. Furthermore, loss or inactivation of genes encoding BH3-only proteins is reported in many cancers and allows BCL-2 to persistently sequester pore-forming BAK and BAX, stalling apoptosis initiation. For instance, loss of BAD or BIK gene expression due to DNA methylation was reported in multiple myeloma (MM) and loss of function mutations in BAD have been reported in colon cancer [12–14]. The loss of the BH3-only protein tumor suppressor mechanism can lead to growth factor signaling independence [15]. Thus, developing a BH3-mimetic molecule to circumvent high BCL-2 or inactive BH3-only proteins has been an attractive approach to restore apoptosis.
Fig. 1.
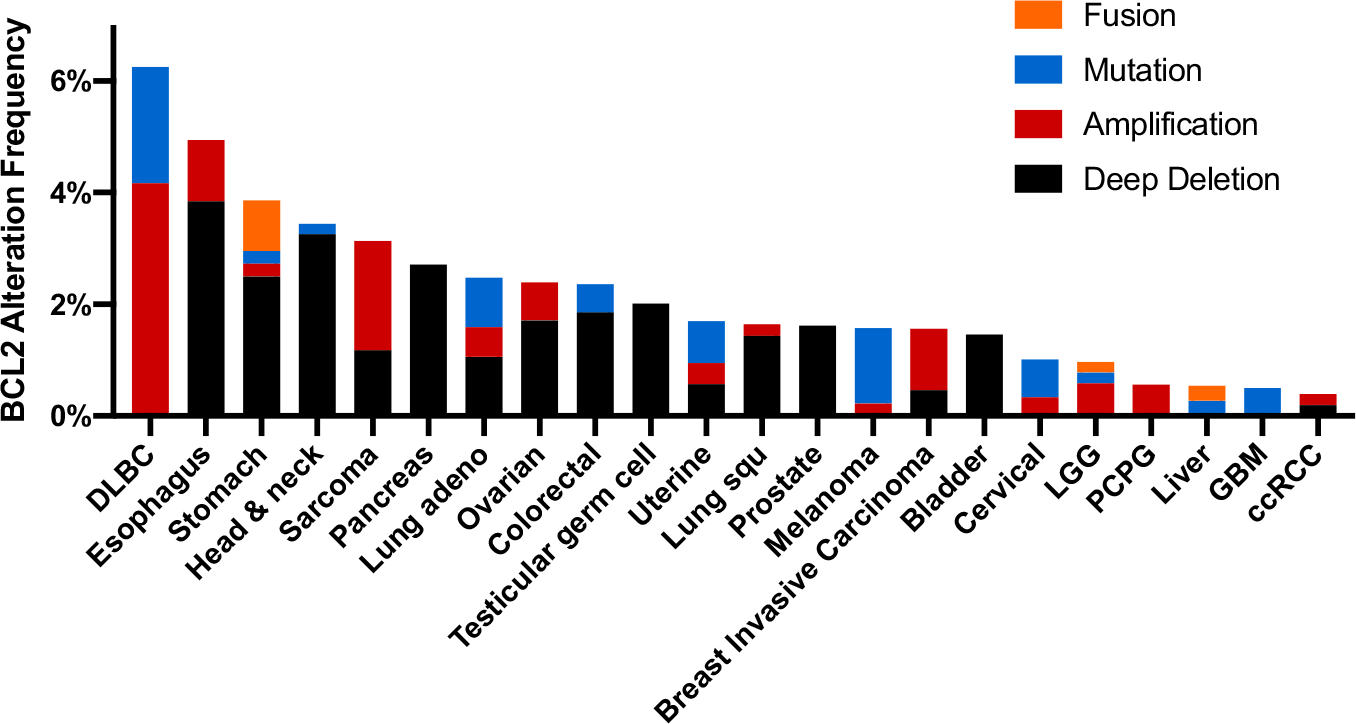
Frequency of BCL2 alterations in cancer. BCL2 alterations were identified by a search in cBioPortal of the TCGA Pan Cancer Atlas studies (10,967 samples from 10,953 patients) as of 12/2020. Abbreviations: DLBC, Diffuse large B-cell lymphoma; LGG, Low grade glioma; PCPG, Pheochromocytoma and paraganglioma; GBM, Glioblastoma multiforme; ccRCC, Clear cell renal cell carcinoma.
BH3 mimetics bind to and inactivate anti-apoptotic BCL-2 family members, allowing mitochondria pore forming proteins to initiate mitochondria permeabilization, cytochrome c release and activation of the apoptosis-driving caspase cascade. One of the first BH3 mimetics developed with high affinity to BCL-2 was ABT-737 [16]. In vitro studies demonstrated that this molecule could induce apoptosis of hematopoietic cancer cell lines and primary cancer cells. Furthermore, ABT-737 could synergize or cooperate with other chemotherapies such as bortezomib, melphalan or dexamethasone in a variety of cancers of hematologic origin [17–19]. However, clinical utility of ABT-737 was limited by both poor bioavailability and ineffectiveness in cancer cells with high MCL-1 expression [17,20]. A second generation BH3 mimetic, navitoclax (ABT-263), was developed with improved bioavailability, but it undermined platelet level due to BCL-XL inhibition [21]. Finally, a small molecule related to navitoclax was developed, venetoclax (ABT-199), that retained oral bioavailability. Venetoclax had improved binding selectivity for BCL-2 compared to BCL-XL and thus did not cause thrombocytopenia [22]. Venetoclax induced apoptosis of cells from chronic lymphocytic leukemia (CLL) patients in vitro and demonstrated substantial responses in patients with relapsed CLL, including those with poor prognostic features [23,24]. This led to the 2016 approval of venetoclax for the treatment of patients with relapsed and refractory CLL with 17p (site of TP53) deletion. In addition, acute myeloid leukemia (AML) myeloblasts have been reported to depend upon BCL-2 to a greater extent than normal hematopoietic stem cells, supporting the rationale for targeting BCL-2 in these patients [25]. Additional clinical trials demonstrated the efficacy of venetoclax as a monotherapy in patients of various hematologic malignancies including non-Hodgkin's lymphoma (NHL), MM, and AML. The results of these trials have spurred further investigation of combination strategies that include venetoclax [26–29].
CLL and AML patients can become venetoclax resistant due to intrinsic or acquired mechanisms. Intrinsic venetoclax resistance has been primarily attributed to increased levels of MCL-1 or BCL-XL, which sequester the pro-apoptotic proteins that are released from BCL-2 upon venetoclax binding. Acquired resistance mechanisms include loss of BH3-only gene expression or BCL-2 mutations that alter its BH3 binding grove [30–33]. For instance, a glycine to valine missense mutation in the venetoclax binding site of BCL-2 at amino acid 101 has been reported to be acquired by CLL patients undergoing venetoclax therapy that reduced venetoclax binding ~180 fold [34]. In addition, molecular profiling of venetoclax-treated AML patient cells identified kinase activating mutations such as FLT3-ITD or biallelic TP53 mutation in leukemic clones upon relapse, illustrating the heterogenous mechanisms that may give rise to venetoclax-resistance [35]. In AML cell lines, xenografts and patient samples, MAPK activation has been reported to stabilize anti-apoptotic MCL-1 and inactivate pro-apoptotic BIM [36–38]. Moreover, venetoclax resistant monocytic AML has a distinct transcriptomic profile marked by loss of BCL-2 expression and dependence on MCL-1, resulting in a differential sensitivity that drives selective outgrowth of monocytic subpopulations [39]. Thus, mechanisms that alter the balance of BCL-2 family expression are key to understanding venetoclax resistance. Combination therapies that reduce expression of BCL-XL and MCL-1 or stimulate expression of BH3-only proteins may be useful to improve venetoclax efficacy.
In addition to primary DNA sequences directing transcription factors and other transcription regulators, gene expression is also regulated by epigenetic mechanisms. DNA methylation, histone post-translational modifications, and nucleosome positioning are epigenetic features that reversibly store and transmit heritable information marks governing gene expression. Proteins that catalyze the addition of an epigenetic modification onto either DNA or histones (usually on histone “tails” that extend from the nucleosome) can be thought of as “writers”. Epigenetic “readers” are effector proteins that recognize or are recruited to specific epigenetic modifications while epigenetic “erasers” remove these modifications. The inherent reversibility of epigenetic modifications makes the mechanisms that regulate these marks attractive therapeutic targets for reverting oncogenic gene expression signatures back to a more normal state (Fig. 2). In this review, we summarize current evidence that suggests therapies targeting epigenetic/gene expression mechanisms may be useful in combination with venetoclax to counteract de novo or acquired venetoclax resistance mechanisms in hematologic malignancies.
Fig. 2.
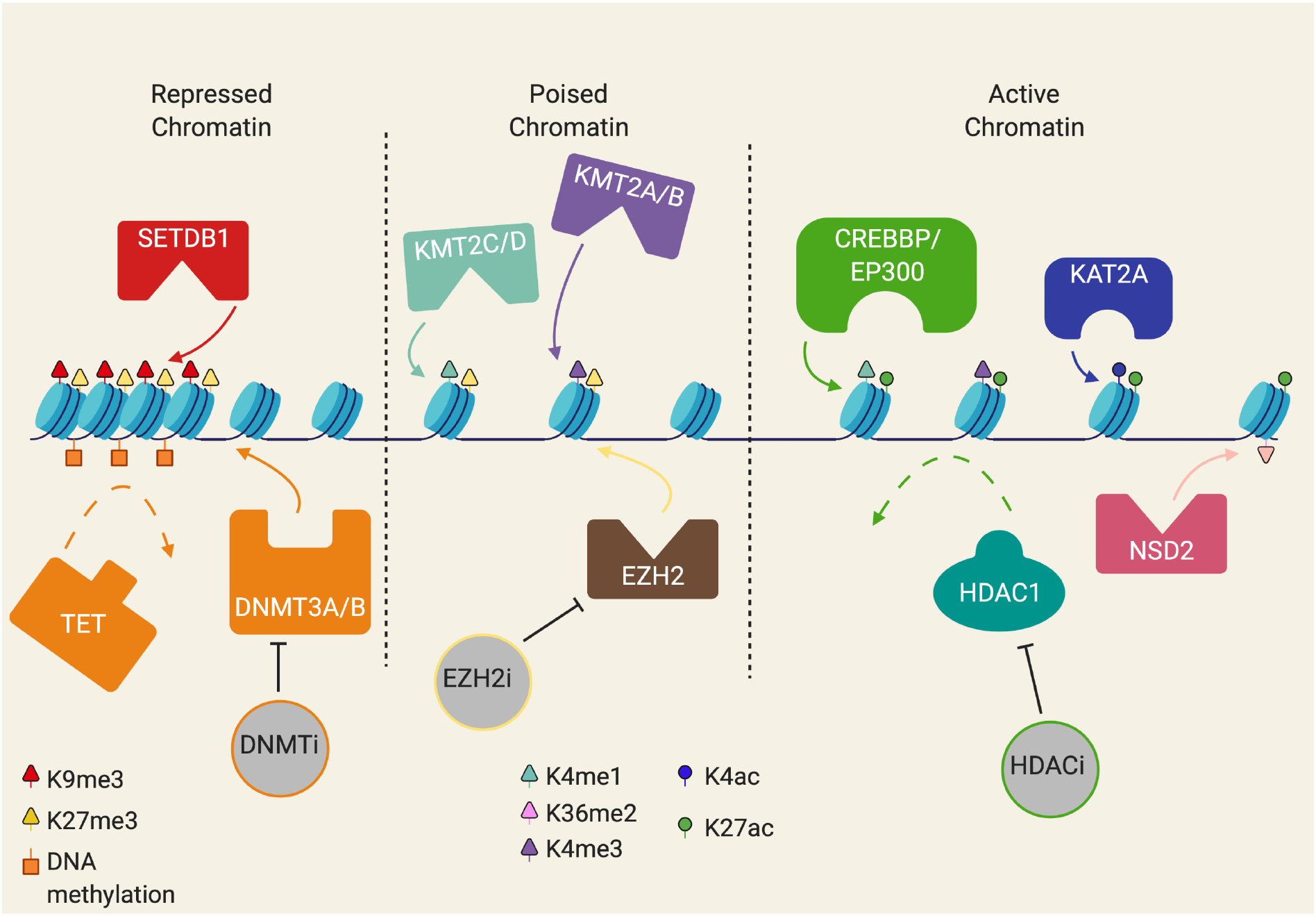
Schematic of chromatin states. Ranging from repressed heterochromatin to active euchromatin. Repressed chromatin is marked by modifications such as H3K9me3, H3K27me3, and DNA methylation of CpG islands. Poised chromatin is a state of inactive chromatin that has a combination of activating and repressing chromatin marks, such as H3K4me1/3 mixed with H3K27me3. Active chromatin is marked by histone acetylation marks, like H3K4ac and H3K27ac with certain activating methylation marks like H3K4me1/3.
2. DNA methylation inhibitors and venetoclax
DNA methyltransferases (DNMTs) methylate cytosines that precede guanine (CpG) to catalyze DNA methylation throughout the genome of mammals. The enzymes DNMT3A and DNMT3B establish de novo DNA methylation patterns while DNMT1 maintains daughter strand methylation after DNA replication [40,41]. Demethylation of DNA is facilitated by the TET enzymes that convert methylcytosine into hydroxymethylcytosine which is not recognized by DNMT1 [42,43]. Methylation of CpG-rich regions in cis-regulatory elements may repress expression of the corresponding gene by preventing binding of transcription factors or recruiting methyl-CpG binding proteins that interact with repressive histone modifying enzymes (Fig. 2). At the same time gene activation is associated with hydroxymethylation of DNA, particularly at enhancers and hence the activity of DNMTs and TET enzymes may be required for normal induction of gene expression [44]. Furthermore, DNA methylation within gene bodies helps maintain normal patterns of gene splicing, proper DNA repair, and prevents intragenic transcriptional initiation. AML is associated with many lesions that can upset DNA methylation patterns including loss of function mutations of TET2, gain of function mutations of IDH1 or IDH2, and loss of function mutations in the DNA methyltransferase DNMT3A [43,45–47].
The cytosine analogues 5′-azacytidine (azacitidine) and 5-aza-2′-deoxycytidine (decitabine) substitute nitrogen for carbon at the pyrimidine ring C-5 position and when incorporated into DNA irreversibly bind DNMT1, causing DNMT1 degradation and genome-wide hypomethylation [48,49]. The potential of DNA methyltransferase inhibitors (DNMTi) to reactivate tumor suppressor genes has spurred testing of these agents for hematologic malignancies and solid tumors. In 2004 (azacitidine) and in 2006 (decitabine) DNMTi were approved as therapies for myelodysplastic syndrome (MDS) [50,51]. In addition, active clinical trials using either DNMTi are currently ongoing for a variety of cancers both as single agents and in combination strategies (Table 1). Response to DNMTi treatment is correlated with a widespread loss of DNA methylation within leukemia cells, but how this may lead to a therapeutic response is uncertain. There is conflicting in vivo evidence of the activity of DNMTi as inducers of differentiation, DNA damage and re-expression of endogenous retroviruses [52–56].
Table 1.
Clinical trials evaluating combination of epigenetic inhibitors and venetoclax.
| Epigenetic drug combination with venetoclax | Cancer typea | Phase, trial ID |
|---|---|---|
|
| ||
| DNA methyltransferase inhibitors | ||
| Azacitidine | AML | II, NCT03466294 |
| AML | II, NCT04267081 | |
| AML | II, NCT03573024 | |
| AML | III, NCT04161885 | |
| AML | III, NCT04102020 | |
| AML | III, NCT02993523 | |
| AML | II, NCT04062266 | |
| AML | II, NCT04424147 | |
| AML | II, NCT04128501 | |
| MDS/CMML | I/II, NCT04160052 | |
| MDS | III, NCT04401748 | |
| MDS | I, NCT02966782 | |
| MDS | I, NCT02942290 | |
| MDS/CML | I/II, NCT04550442 | |
| Decitabine | AML | I, NCT03844815 |
| AML | II, NCT04476199 | |
| AML/MDS | II, NCT03404193 | |
| Azacitidine or decitabine | AML | III, NCT03941964 |
| AML | I/II, NCT02203773 | |
| Azacitidine and anti-PD-1 | AML | II, NCT04284787 |
| Azacitidine, decitabine and salsalate | MDS | II, NCT04146038 |
| Azacitidine and lintuzumab-Ac225 | AML | I/II, NCT03932318 |
| Azacitidine and APR-246 | TP53-mutant myeloid malignancies | I, NCT04214860 |
| Azacitidine and pevonedistat | AML | I, NCT04172844 |
| AML | II, NCT04266795 | |
| AML | II, NCT03862157 | |
| Azacitidine and gilteritinib | AML/MDS | I/II, NCT04140487 |
| Azactidine and trametinib | AML/MDS | II, NCT04487106 |
| Azacitidine and magrolimab | AML | I/II, NCT04435691 |
| Decitabine and nivolumab | AML | I, NCT04277442 |
| Decitabine and quizartinib | AML/MDS | I/II, NCT03661307 |
| Decitabine and ponatinib | AML/CML | II, NCT04188405 |
| ASTX727 (decitabine and cedazuridine) | AML | I, NCT04657081 |
| MDS/CML | I/II, NCT04655755 | |
| Azacitidine and ivosidenib | IDH1 mutant AML/MDS | Ib/II, NCT03471260 |
| Enasidenib | IDH2 mutant AML | Ib/II, NCT04092179 |
| Histone deacetylase inhibitors | ||
| Fimepinostat and rituximab | Lymphomas | I, NCT01742988 |
| BET protein family inhibitors | ||
| ABBV-075 | AML or solid tumors | I, NCT02391480 |
AML, Acute myeloid leukemia; CML, chronic myelogenous leukemia; CMML, chronic myelomonocytic leukemia; MDS, Myelodysplastic syndrome.
Several studies report that the efficacy of BH-3 mimetics is improved when administered in combination with DNMTi. For instance, it has been reported that azacitidine sensitizes AML cells to BH-3 mimetics by downregulating MCL-1 in a p53-independent manner, or by activating the integrated stress response which induces expression of the BH3-only protein NOXA, priming cells for apoptosis [57,58]. Retrospective cohort analyses using propensity score matching has suggested lower rate of relapse, 30-day mortality, and longer overall survival for patients receiving combination DNMTi and venetoclax compared to intensive chemotherapy [59–62]. Due to these results, combination of venetoclax with azacytidine, decitabine, or low dose cytarabine received regular approval from the United States Food and Drug Administration (FDA) in October of 2020 for treatment of newly diagnosed AML patients 75 years or older. However, the precise mechanism by which DNMTi promote sensitivity to venetoclax remains uncertain. In addition to causing changes in gene expression, other non-epigenetic mechanisms such as DNA damage may contribute to the synergy of DNMTi with venetoclax.
Mutations in the enzymes IDH1 and IDH2 that normally convert isocitrate to α-ketoglutarate are found in several tumors, including 10–20% of AML patients [63]. Mutant IDH1/2 reduces α-ketoglutarate to 2-hydroxygluterate (2-HG), a competitive inhibitor of the TET family of DNA methylcytosine dioxygenases that are key for DNA demethylation [64]. The metabolite 2-HG has been reported to inhibit α-ketoglutarate dependent Jumonji domain lysine demethylases and activate mTOR signaling [46,65,66]. In addition, increased intracellular 2-HG due to mutant IDH1/2 has been found to inhibit cytochrome c oxidase, causing cells with these mutations to be dependent upon BCL-2 as revealed by an RNA interference screen [67]. Furthermore, primary human AML cells with mutant IDH1/2 were more sensitive than wild-type IDH1/2 cells to venetoclax ex vivo and in xenograft models [67]. Clinical trials have demonstrated that relapsed/refractory AML patients with IDH-mutant AML have significantly improved outcomes when treated with venetoclax either as a single agent or in combination with DNMTi than the overall AML cohort [61,68].
Small molecule inhibitors of mutant IDH1 (ivosidenib or AG-120) or mutant IDH2 (enasidenib or AG-221) reduce 2-HG levels and induce differentiation of leukemic cells in vitro and in vivo. Their use as single agent therapies was recently approved by the FDA for the treatment of adults with IDH1 or IDH2-mutant AML [69]. Currently, clinical trials are underway that test the combination of venetoclax and mutant IDH1/2 inhibitors (Table 1). Given that the mechanism by which IDH mutations increase BCL-2 dependence is due to increased production of 2-HG, reduction of 2-HG by treatment with mutant IDH1/2 inhibitors may have been expected to antagonize venetoclax activity. Yet, evidence to date suggests this combination therapy is very effective. For instance, preliminary reports indicate that ivosidenib, venetoclax, and azacytidine may be a highly effective therapy for patients with IDH-mutated AML and this combination is being evaluated in early phase clinical trials [70]. Furthermore, in patient-derived xenograft (PDX) models of human IDH2-mutant AML, concurrent treatment of enasidenib and venetoclax resulted in the greatest reduction in leukemia engraftment compared to all other treatment combinations, including the sequential dosing of venetoclax and enasidenib [71]. Co-treatment with the mutant IDH-2 inhibitor enasidenib did not antagonize venetoclax activity even though enasidenib monotherapy resulted in a significant decrease in BCL2 gene expression [71]. Enasidenib treatment caused increased expression of myeloid differentiation markers [71], and enasidenib has been reported to enhance erythroid differentiation of hematopoietic progenitor cells independently of 2-HG [72]. Thus, although the mechanisms behind the effectiveness of venetoclax combination with mutant IDH1/2 inhibitors is unclear, myeloid differentiation may enable responsiveness to venetoclax and/or reduced BCL-2 may cause mitochondria priming leading to a cell state that is very sensitive to apoptosis induction, including by inhibition of the small remaining amount of BCL-2 [71].
While evidence is mounting that venetoclax combined with inhibitors targeting DNA methylation mechanisms represent an important new therapy combination especially for older AML patients, additional work is needed to determine the precise mechanisms by which DNMTi synergize with BH-3 mimetics to induce apoptosis. It remains unclear whether agents such as azacitidine promote venetoclax sensitivity due to any effect on DNA methylation-dependent gene expression or through some other mechanism such as activating DNA damage response pathways.
3. Histone deacetylase inhibitors and venetoclax
An essential means of gene regulation is through acetylation of histones by which acetyl groups are added or removed from lysine residues of the histone tails that extrude beyond the nucleosome. Addition and removal of these acetyl groups is a dynamic process, regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively (Fig. 2). Acetyl groups prevent the ionization and acquisition of positive charges by lysine residues of the histone “tail” residues. Thus, histone acetylation weakens the interaction between histones and the negative charge of the phospho-diester backbone of DNA, creating greater accessibility to the underlying DNA sequences. At the same time, acetylation of specific histone residues creates binding sites for chromatin regulators that activate gene expression and prevent the binding of repressive regulators. Typically, histone acetylation occurs at genomic regions that are either consistently active, therefore accessible, or at regions whose activity must be regulated between active and repressed transcription, e.g., promoter and enhancer regions [73–80]. As a hallmark of proper gene regulation essential to a healthy chromatin landscape, the distribution of acetyl marks is often deregulated in cancer.
In the context of tumor cells, promoters and enhancers of oncogenes are likely hyperacetylated (active transcription), while promoters and enhancers of tumor suppressor genes are typically hypoacetylated (repressed transcription) [81]. Studies in primary tumor tissue indicate that global hypoacetylation correlates with more aggressive cancers, increased risk of recurrence, and/or reduced survival [82–86]. In a survey of the cBioPortal database of cancer genomics, CREBBP and EP300, two essential HAT proteins, are significantly mutated in a variety of cancers (Fig. 3) [10,11]; in one study over 40% of basal cell carcinoma patients had a mutation in one of these two genes [87]. Somatic mutations in both of these genes are prevalent, and resulting loss of heterozygosity implicates them as tumor suppressor genes [81,88,89]. Interestingly, mutations in HDACs are rare, but HDAC overexpression is frequently observed in cancer patients [90], and this may contribute to histone hypoacetylation in many cancers.
Fig. 3.
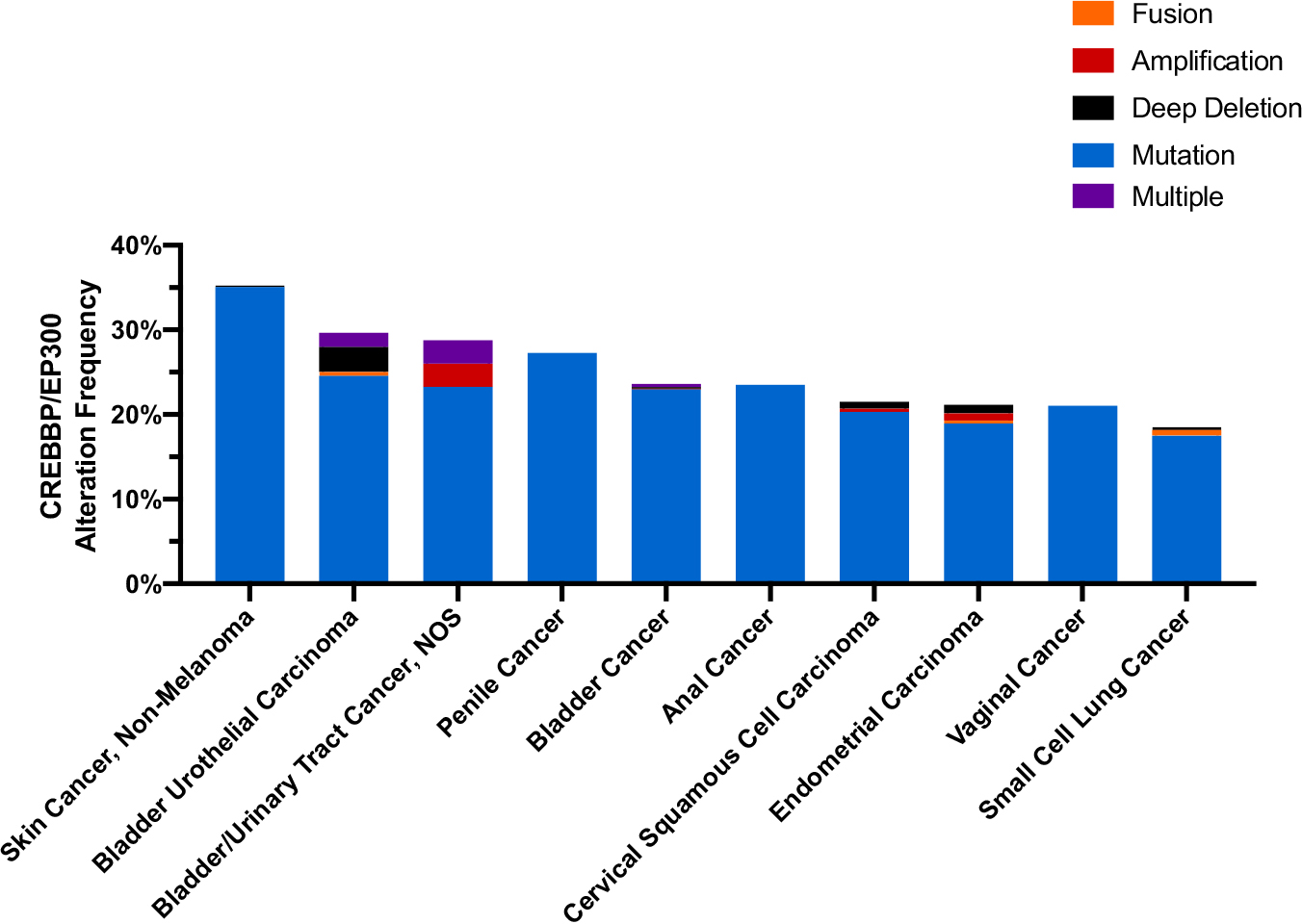
Alteration frequency of CREBBP/EP300 in the top 10 most frequently altered cancers. In non-melanoma skin cancer 35.26% of patients (183/519), 29.68% of bladder urothelial carcinoma patients (122/411), 28.77% of bladder/urinary tract cancer patients (21/73), 27.27% of penile cancer patients (3/11), 23.98% of bladder cancer patients (235/980), 23.53% of anal cancer patients (8/34), 21.51% of cervical squamous cell carcinoma patients (54/251), 21.16% of endometrial cancer patients (124/586), 21.05% of vaginal cancer patients (4/19), and 18.75% of small cell lung cancer patients (57/304).
To date the FDA has approved four pan-HDAC inhibitors (HDACi), and they are primarily used in hematological malignancies, specifically T-cell lymphoma and myeloma [91]. Reports indicate that HDACi cause cell cycle arrest, leading to apoptosis in cultured cancer cells. Furthermore, these inhibitors increase transcription levels of p21, a prevalent cell-cycle regulator, and stimulate p53 acetylation [91], which enhances p53 stability that is required for checkpoint responses to DNA damage and to activate oncogenes [92]. HDACi also stimulate transcription of pro-apoptotic genes such as BAX and BAK [91]. However, in humans the anticancer efficacy of pan-HDAC inhibitors has been limited. These agents broadly target histone acetylation and can impact diverse mechanisms that lead to pleiotropic effects. For instance, HDACi non-specifically block angiogenesis that may hamper drug delivery to solid tumors and prevent inflammation, by inducing apoptosis of tumor-fighting immune cells [91]. In addition, HDACi have been reported to induce DNA damage and mitotic defects.
Altered patterns of histone acetylation are a common pan-cancer feature which can have significant consequences on the transcription of pro-apoptotic and other tumor suppressing genes. For example, histone hypoacetylation is associated with a more aggressive phenotype in adenocarcinoma of the lung and hypoacetylation is associated with breast cancer progression [93,94]. Reestablishing the appropriate balance of histone acetylation in cancer cells may be a promising approach to reengage apoptosis mechanisms. Treatment with HDACi can lead to the renewal of transcriptional profiles and cellular pathways that have been made dormant by oncogenesis, leading to upregulation of genes involved in tumor suppressing pathways, like apoptosis and cell-cycle arrest. Combination therapy of HDACi with venetoclax may serve as a novel method to overcome resistance mechanisms that limit the efficacy of each monotherapy. Recent data suggests that HDAC inhibition can act, at least in part, by shifting the balance of BCL-2 family proteins in favor of pro-apoptotic proteins, thus enhancing the ability of venetoclax to stimulate apoptosis. For example, treatment with pan-HDAC inhibitors has been reported to cause increased expression of pro-apoptotic factors, such as a 40-fold increase of pro-apoptotic BIM [95]. Furthermore, treatment of both AML cell lines and primary patient samples with panobinostat upregulated BIM expression and in combination with venetoclax synergistically induced cell death [96,97]. Panobinostat synergy with venetoclax depended upon BIM and shRNA knockdown of BIM in AML cell lines significantly attenuated apoptosis caused by the combination therapy [96,97]. In an ex vivo study of cells isolated from 25 patients with advanced CTCL, 93% of samples treated with venetoclax and vorinostat and 73% of samples treated with venetoclax and romidepsin displayed synergistic effects [98]. Another recent report demonstrated that synergy between venetoclax and panobinostat treatment was independent of TP53 status in patient derived AML xenograft models [99].
In addition, fimepinostat (CUDC-907), a dual PI3K and HDAC inhibitor, and venetoclax have been reported to synergistically induce apoptosis in AML cell lines, primary AML patient samples, and an AML xenograft mouse model. This combination is currently being tested in early-stage clinical trials (Table 1) [100]. Although the precise mechanism is unclear, fimepinostat downregulates expression of MCL1, CHK1, WEE1, RRM1, and c-MYC while upregulating BIM and inducing DNA damage to potentiate venetoclax-induced apoptosis [100]. Furthermore, venetoclax treatment enhanced fimepinostat-induced DNA damage potentially through inhibition of DNA repair [100]. This illustrates that pan-HDAC inhibitors may affect more than just gene expression of BCL-2 family members and their contribution to apoptosis induction may include other mechanisms such as inducing DNA damage [101].
4. Histone methyl transferase EZH2 inhibitors and venetoclax
Histone methylation at promoters and enhancers is crucial for modulating gene expression and when imbalanced can lead to oncogene activation or tumor suppressor silencing. The polycomb repressive complex 2 (PRC2) di- and tri-methylates lysine residue 27 on histone H3 (H3K27me2/3) to promote chromatin compaction and transcriptional silencing that is important for biological processes such as differentiation and maintaining cell identity [102]. PRC2 is comprised of core polycomb group (PcG) proteins SUZ12, EED, EZH2, and RbAp46/48, and the SET domain-containing histone methyltransferase EZH2 is the enzymatic subunit that facilitates methylation [102–105]. Removal of methyl groups from H3K27me2 and H3K27me3 is accomplished by histone demethylases KDM6A (UTX) and KDM6B (JMJD3) [106]. Inactivating mutations in KDM6A occur in many different cancers and loss of demethylase activity may be associated with local alterations in H3K27me3 levels similar to those observed in cells carrying gain-of-function EZH2 mutations [107,108]. Therefore, EZH2 inhibition presents an attractive target for restoring H3K27me3 balance and tumor suppressor gene expression in malignancies.
H3K27me3 levels are deregulated in numerous cancer types [109]. Major aberrations leading to increased H3K27me3 include overexpression of EZH2, gain of function mutations in EZH2 or loss of function mutations in chromatin regulators antagonizing EZH2 function, such as KDM6A [109]. Increased expression of EZH2 is associated with failure of differentiation, cell invasion and worse survival for patients with multiple myeloma or many solid cancers, including prostate, head and neck, breast, bladder, thyroid, and endometrial cancer [110–116]. Proliferation is attenuated in many EZH2-overexpressing cancer cell lines when cells are treated with pharmacological EZH2 inhibitors (EZH2i) or EZH2-targeting short hairpin RNA (shRNA) that deplete EZH2 protein expression [117]. Similarly, ectopic expression of EZH2 confers a growth advantage in primary mouse embryonic fibroblasts [118]. Significantly, 22% of germinal center B cell (GCB) DLBCL, 7%–12% of follicular lymphomas [119], and a small number of melanoma cases have a gain-of-function EZH2 mutant within the enzymatic SET domain at position Y641 [120]. In vitro biochemical enzymatic assays demonstrated that this mutation leads to a gain of function in EZH2 activity that subsequently results in increased levels of H3K27me3 [121]. This increase in H3K27me3 coincides with repression of Polycomb target genes that have recently been identified in DLBCL [122] and melanoma [123]. Point mutations at residues A677 and A687 have also been identified in non-Hodgkin's lymphomas (NHL), and likewise result in increased H3K27me3 levels [124,125]. Aside from EZH2, H3K27me3 levels can also be affected by aberrations in other PcG proteins, mutations in H3K27me3 reader proteins, histone mutations, or aberrations in related chromatin regulators [126].
Due to the common occurrence of gain-of-function EZH2 alterations in cancer, development of EZH2i has been an ongoing area of investigation. Diverse types of compounds have been developed, including a SAH hydrolase inhibitor, SAM-competitive inhibitors, hydrocarbon-stapled peptides that disrupt the protein interaction between EZH2 and EED or SUZ12, and compounds triggering EZH2 degradation (see Table 2 in review article [127]). Most of these compounds only show limited efficacy as monotherapy, but are promising therapeutics in combination with other chemotherapy agents.
EZH2 and BCL2 mutations commonly co-occur in DLBCL. Out of 1077 DLBCL cases from 5 different studies queried on the cBioPortal of cancer genomics in December 2020, 248 cases had EZH2 and/or BCL2 perturbations [10,11]. EZH2 and BCL2 mutations co-occurred in 15% of cases (37/248) and co-occurrence was associated with a significantly shorter survival rate (Fig. 4). This is in concordance with a recent study that determined EZH2 mutation and BCL2 translocation co-occur in 12% (69/574) of biopsy samples of which 88% belonged to the germinal center B-cell subtype [128]. The common co-occurrence of gain of function alterations in EZH2 and BCL2 in lymphoma justifies a rationale for combination treatment with EZH2 and BCL2 inhibitors.
Fig. 4.
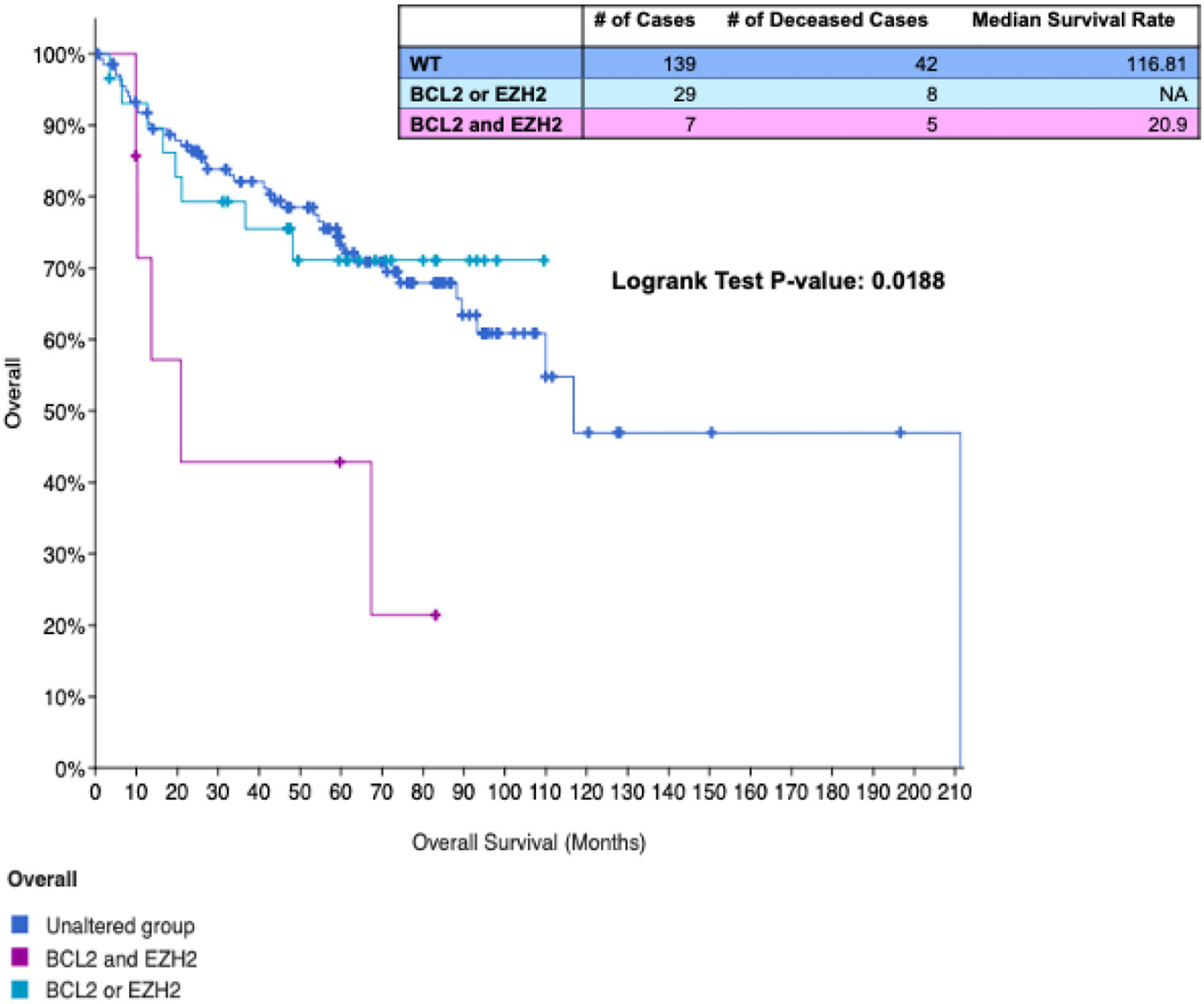
Co-occurrence of EZH2 and BCL2 alterations is associated with reduced overall survival in DLBCL. Overall survival rate from 4 datasets (cBioPortal) of patients with no BCL2 and EZH2 alterations (dark blue), an alteration in BCL2 or EZH2 (light blue), or alterations in both EZH2 and BCL2 (magenta). Table displays number of overall cases per group, number of deceased cases, and median survival rates.
To date, two studies have directly tested the efficacy of BCL2 inhibition and EZH2i combination therapy. In 2013, Beguelin et al. established that EZH2 cooperates with BCL2 to generate germinal center derived lymphomas [129]. When bone marrow from VavP-BCL2 mice was transduced with gain-of-function mutant EZH2Y641F, and transplanted into C57Bl6 recipient mice, the recipients developed splenomegaly and accelerated lymphoma that was accompanied by increased B-cell infiltration into other organs. Combination treatment with the BCL2 inhibitor Obatoclax and the EZH2i GSK503 attenuated cell growth in vitro and tumor xenograft growth in vivo. In addition, the synergy between EZH2i and BCL2 inhibitors has been confirmed in DLBCL cell lines, organoids and in vivo models [130]. Combination therapy of venetoclax and the EZH2i tazemetostat was synergistic in 6 of 6 DLBCLs with EZH2 mutation and BCL2 translocation, 2 of 4 DLBCLs with WT EZH2 and BCL2 translocation, and 0 of 5 DLBCLs without alterations in either EZH2 or BCL2. Synergy was confirmed in two EZH2 and BCL2 alteration-harboring organoid models, one derived from a cell line, the other from a patient-derived xenograft. In vivo, combination treatment attenuated tumor growth in DLBCL cell line xenografts and patient-derived xenografts with EZH2 mutations and BCL2 translocations. Both of these studies are promising pre-clinical models that support using the combination of EZH2i and venetoclax to eradicate tumor cells, and a clinical trial of this treatment strategy is currently being developed [130].
The mechanism of synergy between EZH2i and BCL2 inhibitors remains uncertain. Because EZH2 and BCL2 are both implicated in apoptosis evasion, amplifications or gain-of-function mutations of these genes may cooperate to overcome anti-apoptotic signaling in cancer cells. EZH2 may directly or indirectly alter gene expression of BCL2 family genes through H3K27me3 at promoters and enhancers of target genes. In addition, EZH2 regulates cell cycle genes such as CDKN1A and CDKN2A that also regulate p53, further implicating EZH2 in apoptosis evasion [109]. EZH2 has been reported to be downstream of the pRB-E2F pathway, and inhibition of EZH2 enhances E2F1 expression and consequently the expression of pro-apoptotic BIM [118,131]. Importantly, EZH2 inhibition may activate pro-apoptotic gene expression (such as BIM) to prime cells for subsequent BCL2 inhibition that now forces cancer cells to initiate apoptosis (Fig. 5). In support of this model, treatment of DLBCL cell lines with tazemetostat has been demonstrated to increase expression of pro-apoptotic BCL2 family proteins that may be direct or indirect target genes of EZH2 [130]. Although this combination therapy has only been tested in DLBCL, it may also be applicable to other cancers marked by increased EZH2 and BCL2 expression or activity.
Fig. 5.
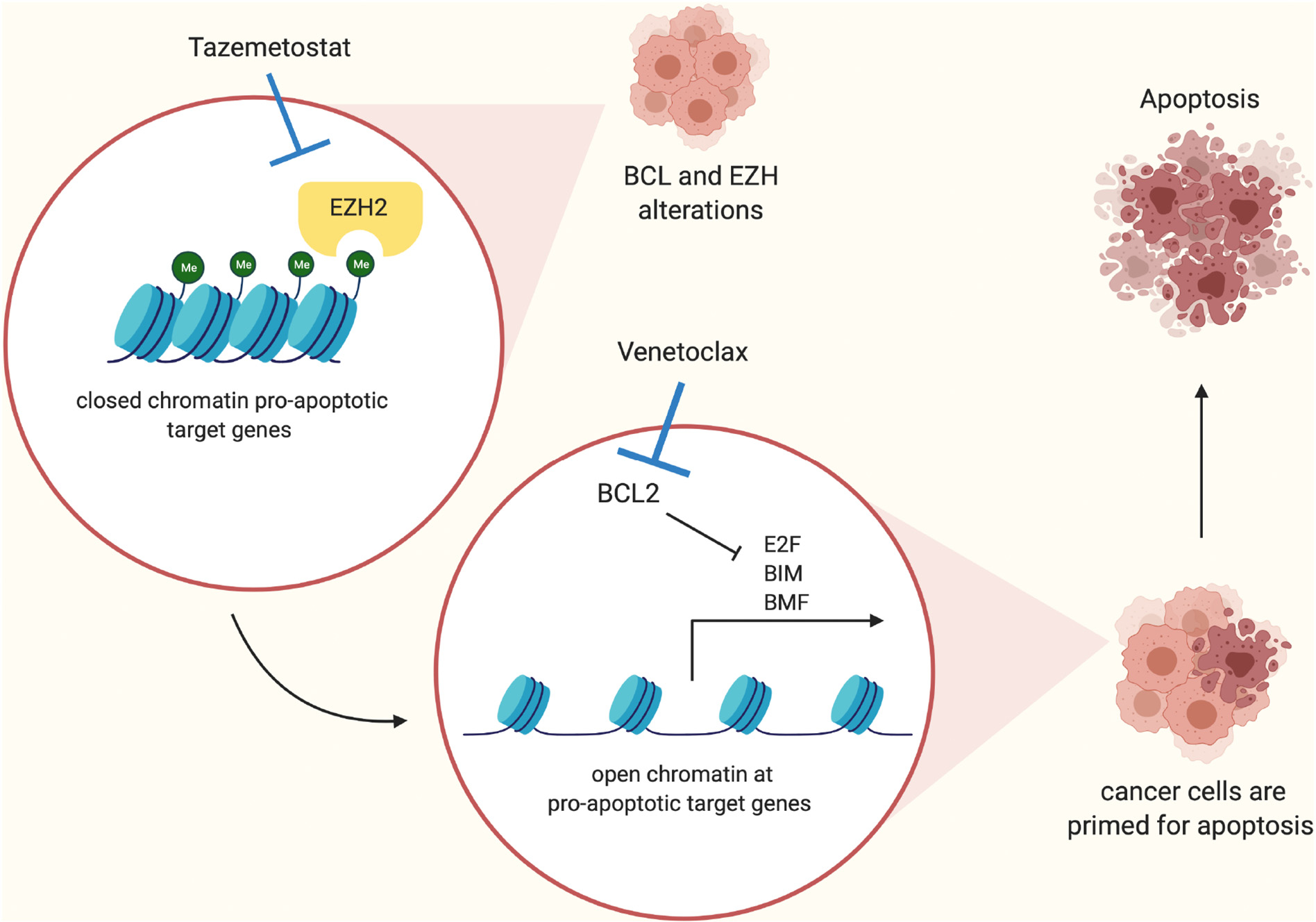
Proposed mechanism of cooperation between EZH2 and BCL2 inhibition to induce apoptosis. Cancer cells with BCL2 and EZH2 alterations have tightly compacted chromatin at pro-apoptotic target genes because of increased H3K27me3 levels. Inhibition of EZH2 through Tazemetostat treatment results in open chromatin at pro-apoptotic target genes such as BIM and BMF, activating their expression and priming cancer cells for apoptosis. Inhibition of BCL2 with venetoclax now releases pro-apoptotic proteins from BCL2 and cancer cells are forced to undergo apoptosis.
5. Bromodomain inhibitors and venetoclax
Reader proteins recognize and bind specific epigenetic marks on chromatin or histones to either directly induce chromatin structural changes, recruit secondary chromatin modifiers or serve as structural support proteins for transcription, replication or repair complexes. Chromatin reader proteins can include several types of domains that recognize specific epigenetic marks and binding may depend on adjacent histone modifications [132]. The Bromodomain and Extra Terminal (BET) reader proteins BRD2, BRD3, BRD4, and BRDT share two conserved ~100 amino acid N-terminal bromodomains. These bromodomains contain a hydrophobic pocket which recognizes acetylated lysine residues. BET family proteins bind to acetylated lysine residues on chromatin and facilitate activation of transcription complexes to regulate gene expression. BRD4 is perhaps the most well characterized BET family protein. BRD4 binds to acetylated lysine of histone H3 at amino acid position 27 in transcriptionally active promoters and enhancer regions. Once bound, BRD4 interacts with the mediator complex, positive transcription elongation factor b (P-TEFb) and Jumonji domain containing 6 (JMJD6) to stimulate transcription elongation [133]. Oncogenic transcription factors such as MYC and E2F1 depend on BRD4 to drive gene expression [134,135].
BET family inhibitors (BETi) have been shown to cause downregulation of MYC and initiate apoptosis in cancer cells [134]. One of the first BET inhibitors developed, JQ1, can induce terminal differentiation of leukemic stem cells in primary AML samples and prevent ovarian carcinoma growth in xenografts [136,137]. In addition, dBET, a phthalimide-coupled JQ1 was demonstrated to induce BRD4 protein degradation and delay leukemia progression in mice [138]. Recently, next generation BET inhibitors have been developed with improved efficacy that are in clinical trials for hematologic malignancies [139].
Preclinical studies have demonstrated the usefulness of BETi to sensitize cells to venetoclax. For instance, BET inhibitors JQ1, ABBV-075, I-BET762, CPI-0610 have been reported to cooperate with venetoclax to initiate cell death in cutaneous T cell lymphoma and B cell lymphoma cell lines [140,141]. Furthermore, in vitro drug screening in primary T-cell acute lymphoblastic leukemia samples from high-risk patients and cell lines revealed synergistic activity between venetoclax and JQ1 that was confirmed in patient-derived xenograft models [142]. Significantly, this drug combination reduced expression of BCL2 and a miRNA negative regulator of pro-apoptotic BIM expression, leading to increased BIM expression and binding of BIM to BCL-2 [142]. JQ1 also increased venetoclax sensitivity in CLL cells, primary CD19(+) lymphocytes, and venetoclax-resistant CLL cell lines [143]. Similar results have been reported with other BETi such as CPI203 that decreases expression of MYC and BFL-1, a BCL-2-like protein, allowing venetoclax to now induce caspase-mediated apoptosis in venetoclax-resistant double hit lymphoma cultures and xenografts [144]. Although JQ1 has not been developed for clinical use due to its short half-life, next generation BETi, such as ABBV-075, have entered early phase clinical trials in combination with venetoclax. In AML blasts and patient derived CD34+ progenitor cells, treatment with ABBV-075 potentiates venetoclax efficacy by reducing MCL1 and BCL-XL levels, while inducing BIM, thereby allowing BAX and/or BAK to overcome venetoclax resistance mechanisms and initiate apoptosis [145]. In addition, two recently developed nonbenzodiazepine BETi, PLX51107 and PLX2853, activated BIM expression and BIM-dependent apoptosis by suppressing expression of miR-17–92 [146]. Consequently, administration of PLX51107 or PLX2853 with venetoclax initiated cell death in MYC-driven lymphoma cells with high BCL-2 expression [146].
6. Summary
The discovery of venetoclax has led to a significant improvement for the treatment of hematologic malignancies, particularly for older AML patients. Study of venetoclax resistance mechanisms remains an emerging field, but it is clear that at least some resistance mechanisms can be countered by combination therapies that include agents that target epigenetic mechanisms. Therapies that target epigenetic mechanisms may serve as counters to the activated growth signaling or chromosomal translocations that have led to increased BCL2 gene expression in cancer cells. Furthermore, mutations that affect epigenetic mechanisms are commonly found in cancer patients and many of these mutations may alter expression or function of apoptosis mechanisms, rendering cells resistant to venetoclax. For instance, in addition to the mechanisms described here, mutations in components of the SWI-SNF chromatin remodeling complex have been found to facilitate increased expression of BCL-XL [147]. Follicular lymphoma often harbors mutations in histone methyltransferases KMT2D or EZH2 that may cooperate with increased BCL2 gene expression caused by chromosome translocation to drive tumorigenesis [148]. Fortunately, epigenetic mechanisms of gene regulation are dynamic and often reversible, providing the opportunity for therapeutic intervention, and many ongoing preclinical studies and clinical trials are focused on testing whether epigenetic therapies may provide benefit when used in combination with venetoclax (Table 1).
In addition to affecting gene expression, evidence suggests that inhibitors of epigenetic mechanisms may affect other cell death mechanisms that could synergize with an apoptosis-inducing agent such as venetoclax. For instance, IDH1/2 are key enzymes in the tricarboxylic acid (TCA) cycle and mutant IDH1/2 alters metabolism in such a way to make cells more sensitive to BCL-2 inhibition. Increased 2-HG caused by mutant IDH1/2 suppresses cytochrome C oxidase activity in the mitochondria which in turn primes the mitochondria for apoptosis [71]. In addition to their effects on histone acetylation and DNA methylation, HDACi and DNMTi, respectively, have been reported to affect the DNA damage response. Furthermore, DNMTi and EZH2i have been reported to induce expression of normally silent endogenous retroviruses that induce the host-antiviral response. Therefore, the ability of epigenetic therapies to cooperate or synergize with BH3 mimetics like venetoclax may be due to their effect on diverse mechanistic pathways in addition to their classically regarded effects on gene expression. As our understanding for the role of epigenetic mechanisms in venetoclax resistance expands, new therapeutic strategies are expected to leverage this knowledge to improve patient outcomes in hematologic malignancies.
Acknowledgments
This work was funded by a LLS Specialized Center of Research grant, R01 R01CA195732 and R01 CA180475 (JDL). GP was supported by T32 DK074367. Figs. 2 and 5 were made using BioRender.com.
Footnotes
This article is part of a Special Issue entitled: Death mechanisms in cellular homeostasis edited by Prof. Peter Ruvolo and Prof. Geert Bultynck.
Declaration of competing interest
None.
CRediT authorship contribution statement
Gabriel Prado: Writing – original draft, Writing – review & editing. Charlotte L. Kaestner: Writing – original draft, Writing – review & editing. Jonathan D. Licht: Funding acquisition, Writing – review & editing. Richard L. Bennett: Conceptualization, Writing – original draft, Writing – review & editing, Supervision.
References
- [1].Bakhshi A, et al. , Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18, Cell 41 (3) (1985) 899–906. [DOI] [PubMed] [Google Scholar]
- [2].Tsujimoto Y, et al. , Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation, Science 226 (4678) (1984) 1097–1099. [DOI] [PubMed] [Google Scholar]
- [3].van Delft MF, Huang DC, How the Bcl-2 family of proteins interact to regulate apoptosis, Cell Res. 16 (2) (2006) 203–213. [DOI] [PubMed] [Google Scholar]
- [4].Chung C, Driving toward precision medicine for B cell lymphomas: targeting the molecular pathogenesis at the gene level, J. Oncol. Pharm. Pract. 26 (4) (2020) 943–966. [DOI] [PubMed] [Google Scholar]
- [5].Tsuyama N, et al. , BCL2 expression in DLBCL: reappraisal of immunohistochemistry with new criteria for therapeutic biomarker evaluation, Blood 130 (4) (2017) 489–500. [DOI] [PubMed] [Google Scholar]
- [6].McDonnell TJ, et al. , bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation, Cell 57 (1) (1989) 79–88. [DOI] [PubMed] [Google Scholar]
- [7].Johnson NA, et al. , Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone, J. Clin. Oncol. 30 (28) (2012) 3452–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cimmino A, et al. , miR-15 and miR-16 induce apoptosis by targeting BCL2, Proc. Natl. Acad. Sci. U. S. A. 102 (39) (2005) 13944–13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Warren CFA, Wong-Brown MW, Bowden NA, BCL-2 family isoforms in apoptosis and cancer, Cell Death Dis. 10 (3) (2019) 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cerami E, et al. , The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data, Cancer Discov. 2 (5) (2012) 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gao J, et al. , Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal, Sci. Signal. 6 (269) (2013) pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Pompeia C, et al. , Microarray analysis of epigenetic silencing of gene expression in the KAS-6/1 multiple myeloma cell line, Cancer Res. 64 (10) (2004) 3465–3473. [DOI] [PubMed] [Google Scholar]
- [13].Hatzimichael E, et al. , Bcl2-interacting killer CpG methylation in multiple myeloma: a potential predictor of relapsed/refractory disease with therapeutic implications, Leuk. Lymphoma 53 (9) (2012) 1709–1713. [DOI] [PubMed] [Google Scholar]
- [14].Lee JW, et al. , Inactivating mutations of proapoptotic Bad gene in human colon cancers, Carcinogenesis 25 (8) (2004) 1371–1376. [DOI] [PubMed] [Google Scholar]
- [15].Ranger AM, et al. , Bad-deficient mice develop diffuse large B cell lymphoma, Proc. Natl. Acad. Sci. U. S. A. 100 (16) (2003) 9324–9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Oltersdorf T, et al. , An inhibitor of Bcl-2 family proteins induces regression of solid tumours, Nature 435 (7042) (2005) 677–681. [DOI] [PubMed] [Google Scholar]
- [17].Konopleva M, et al. , Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia, Cancer Cell 10 (5) (2006) 375–388. [DOI] [PubMed] [Google Scholar]
- [18].Chauhan D, et al. , A novel Bcl-2/Bcl-X(L)/Bcl-w inhibitor ABT-737 as therapy in multiple myeloma, Oncogene 26 (16) (2007) 2374–2380. [DOI] [PubMed] [Google Scholar]
- [19].High LM, et al. , The Bcl-2 homology domain 3 mimetic ABT-737 targets the apoptotic machinery in acute lymphoblastic leukemia resulting in synergistic in vitro and in vivo interactions with established drugs, Mol. Pharmacol. 77 (3) (2010) 483–494. [DOI] [PubMed] [Google Scholar]
- [20].van Delft MF, et al. , The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized, Cancer Cell 10 (5) (2006) 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schoenwaelder SM, et al. , Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets, Blood 118 (6) (2011) 1663–1674. [DOI] [PubMed] [Google Scholar]
- [22].Souers AJ, et al. , ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets, Nat. Med. 19 (2) (2013) 202–208. [DOI] [PubMed] [Google Scholar]
- [23].Anderson MA, et al. , The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism, Blood 127 (25) (2016) 3215–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Roberts AW, et al. , Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia, N. Engl. J. Med. 374 (4) (2016) 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vo TT, et al. , Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML, Cell 151 (2) (2012) 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Davids MS, et al. , Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-Hodgkin lymphoma, J. Clin. Oncol. 35 (8) (2017) 826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kumar S, et al. , Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma, Blood 130 (22) (2017) 2401–2409. [DOI] [PubMed] [Google Scholar]
- [28].Konopleva M, et al. , Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia, Cancer Discov. 6 (10) (2016) 1106–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Guerra VA, DiNardo C, Konopleva M, Venetoclax-based therapies for acute myeloid leukemia, Best Pract. Res. Clin. Haematol. 32 (2) (2019) 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Punnoose EA, et al. , Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist venetoclax in multiple myeloma models, Mol. Cancer Ther. 15 (5) (2016) 1132–1144. [DOI] [PubMed] [Google Scholar]
- [31].Bodo J, et al. , Acquired resistance to venetoclax (ABT-199) in t(14;18) positive lymphoma cells, Oncotarget 7 (43) (2016) 70000–70010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bose P, Gandhi V, Konopleva M, Pathways and mechanisms of venetoclax resistance, Leuk. Lymphoma 58 (9) (2017) 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Birkinshaw RW, et al. , Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations, Nat. Commun. 10 (1) (2019) 2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Blombery P, et al. , Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia, Cancer Discov. 9 (3) (2019) 342–353. [DOI] [PubMed] [Google Scholar]
- [35].DiNardo CD, et al. , Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML, Blood 135 (11) (2020) 791–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yeh YY, et al. , Up-regulation of CDK9 kinase activity and Mcl-1 stability contributes to the acquired resistance to cyclin-dependent kinase inhibitors in leukemia, Oncotarget 6 (5) (2015) 2667–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Konopleva M, et al. , MEK inhibition enhances ABT-737-induced leukemia cell apoptosis via prevention of ERK-activated MCL-1 induction and modulation of MCL-1/BIM complex, Leukemia 26 (4) (2012) 778–787. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [38].Pei XY, et al. , MEK1/2 inhibitors potentiate UCN-01 lethality in human multiple myeloma cells through a Bim-dependent mechanism, Blood 110 (6) (2007) 2092–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pei S, et al. , Monocytic subclones confer resistance to venetoclax-based therapy in patients with acute myeloid leukemia, Cancer Discov. 10 (4) (2020) 536–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Okano M, et al. , DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development, Cell 99 (3) (1999) 247–257. [DOI] [PubMed] [Google Scholar]
- [41].Bostick M, et al. , UHRF1 plays a role in maintaining DNA methylation in mammalian cells, Science 317 (5845) (2007) 1760–1764. [DOI] [PubMed] [Google Scholar]
- [42].Tahiliani M, et al. , Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1, Science 324 (5929) (2009) 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Figueroa ME, et al. , Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation, Cancer Cell 18 (6) (2010) 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Greenberg MVC, Bourc'his D, The diverse roles of DNA methylation in mammalian development and disease, Nat. Rev. Mol. Cell Biol. 20 (10) (2019) 590–607. [DOI] [PubMed] [Google Scholar]
- [45].Hanada M, et al. , bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia, Blood 82 (6) (1993) 1820–1828. [PubMed] [Google Scholar]
- [46].Xu W, et al. , Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases, Cancer Cell 19 (1) (2011) 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Russler-Germain DA, et al. , The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers, Cancer Cell 25 (4) (2014) 442–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Santi DV, Norment A, Garrett CE, Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine, Proc. Natl. Acad. Sci. U. S. A. 81 (22) (1984) 6993–6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ghoshal K, et al. , 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal, Mol. Cell. Biol. 25 (11) (2005) 4727–4741. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [50].Silverman LR, et al. , Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B, J. Clin. Oncol. 20 (10) (2002) 2429–2440. [DOI] [PubMed] [Google Scholar]
- [51].Kantarjian H, et al. , Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study, Cancer 106 (8) (2006) 1794–1803. [DOI] [PubMed] [Google Scholar]
- [52].Pinto A, et al. , 5-Aza-2'-deoxycytidine induces terminal differentiation of leukemic blasts from patients with acute myeloid leukemias, Blood 64 (4) (1984) 922–929. [PubMed] [Google Scholar]
- [53].Figueroa ME, et al. , MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation, Blood 114 (16) (2009) 3448–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chiappinelli KB, et al. , Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses, Cell 162 (5) (2015) 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Roulois D, et al. , DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts, Cell 162 (5) (2015) 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jiemjit A, et al. , p21(WAF1/CIP1) induction by 5-azacytosine nucleosides requires DNA damage, Oncogene 27 (25) (2008) 3615–3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tsao T, et al. , Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells, Ann. Hematol. 91 (12) (2012) 1861–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Jin S, et al. , 5-Azacitidine induces NOXA to prime AML cells for venetoclax-mediated apoptosis, Clin. Cancer Res. 26 (13) (2020) 3371–3383. [DOI] [PubMed] [Google Scholar]
- [59].Maiti A, et al. , Venetoclax with decitabine versus intensive chemotherapy in acute myeloid leukemia: a propensity score matched analysis stratified by risk of treatment-related mortality, Am. J. Hematol. 96 (3) (2020) 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].DiNardo CD, et al. , 10-Day decitabine with venetoclax for newly diagnosed intensive chemotherapy ineligible, and relapsed or refractory acute myeloid leukaemia: a single-centre, phase 2 trial, Lancet Haematol. 7 (10) (2020) e724–e736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].DiNardo CD, et al. , Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study, Lancet Oncol. 19 (2) (2018) 216–228. [DOI] [PubMed] [Google Scholar]
- [62].DiNardo CD, et al. , Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia, Blood 133 (1) (2019) 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tefferi A, et al. , IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis, Leukemia 24 (7) (2010) 1302–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ward PS, et al. , The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate, Cancer Cell 17 (3) (2010) 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lu C, et al. , IDH mutation impairs histone demethylation and results in a block to cell differentiation, Nature 483 (7390) (2012) 474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Carbonneau M, et al. , The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway, Nat. Commun. 7 (2016) 12700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chan SM, et al. , Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia, Nat. Med. 21 (2) (2015) 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].DiNardo CD, et al. , Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies, Am. J. Hematol. 93 (3) (2018) 401–407. [DOI] [PubMed] [Google Scholar]
- [69].Stein EM, et al. , Safety and efficacy of AG-221, a potent inhibitor of mutant IDH2 that promotes differentiation of myeloid cells in patients with advanced hematologic malignancies: results of a phase 1/2 trial, Blood 126 (23) (2015) 323. [Google Scholar]
- [70].Lachowiez CA, et al. , Phase Ib/II study of the IDH1-mutant inhibitor ivosidenib with the BCL2 inhibitor venetoclax +/− azacitidine in IDH1-mutated hematologic malignancies, J. Clin. Oncol. 38 (15_suppl) (2020) 7500. [Google Scholar]
- [71].Cathleen S, et al. , Combination of enasidenib and venetoclax shows superior antileukemic activity against IDH2 mutated AML in patient-derived xenograft models, Blood 132 (2018) 562. [Google Scholar]
- [72].Dutta R, et al. , Enasidenib drives human erythroid differentiation independently of isocitrate dehydrogenase 2, J. Clin. Invest. 130 (4) (2020) 1843–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Allis CD, et al. , Epigenetics, 2 ed., Cold Spring Harbor Laboratory Press, 2015, p. 984. [Google Scholar]
- [74].Allis CD, Jenuwein T, The molecular hallmarks of epigenetic control, Nat. Rev. Genet. 17 (8) (2016) 487–500. [DOI] [PubMed] [Google Scholar]
- [75].Alberts B, Molecular biology of the cell, in: 1 volume (various pagings), Sixth ed., Garland Science, Taylor and Francis Group, New York, NY, 2015. [Google Scholar]
- [76].Chen T, Dent SY, Chromatin modifiers and remodellers: regulators of cellular differentiation, Nat. Rev. Genet. 15 (2) (2014) 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Klemm SL, Shipony Z, Greenleaf WJ, Chromatin accessibility and the regulatory epigenome, Nat. Rev. Genet. 20 (4) (2019) 207–220. [DOI] [PubMed] [Google Scholar]
- [78].Eberharter A, Becker PB, Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics, EMBO Rep. 3 (3) (2002) 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kouzarides T, Chromatin modifications and their function, Cell 128 (4) (2007) 693–705. [DOI] [PubMed] [Google Scholar]
- [80].Audia JE, Campbell RM, Histone modifications and cancer, Cold Spring Harb. Perspect. Biol. 8 (4) (2016), a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Baylin SB, Jones PA, Epigenetic determinants of cancer, Cold Spring Harb. Perspect. Biol. 8 (9) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Elsheikh SE, et al. , Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome, Cancer Res. 69 (9) (2009) 3802–3809. [DOI] [PubMed] [Google Scholar]
- [83].Fraga MF, et al. , Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer, Nat. Genet. 37 (4) (2005) 391–400. [DOI] [PubMed] [Google Scholar]
- [84].Manuyakorn A, et al. , Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: results from RTOG 9704, J. Clin. Oncol. 28 (8) (2010) 1358–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Seligson DB, et al. , Global histone modification patterns predict risk of prostate cancer recurrence, Nature 435 (7046) (2005) 1262–1266. [DOI] [PubMed] [Google Scholar]
- [86].Seligson DB, et al. , Global levels of histone modifications predict prognosis in different cancers, Am. J. Pathol. 174 (5) (2009) 1619–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Bonilla X, et al. , Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma, Nat. Genet. 48 (4) (2016) 398–406. [DOI] [PubMed] [Google Scholar]
- [88].Muraoka M, et al. , p300 gene alterations in colorectal and gastric carcinomas, Oncogene 12 (7) (1996) 1565–1569. [PubMed] [Google Scholar]
- [89].Gayther SA, et al. , Mutations truncating the EP300 acetylase in human cancers, Nat. Genet. 24 (3) (2000) 300–303. [DOI] [PubMed] [Google Scholar]
- [90].Dell’Aversana C, Lepore I, Altucci L, HDAC modulation and cell death in the clinic, Exp. Cell Res. 318 (11) (2012) 1229–1244. [DOI] [PubMed] [Google Scholar]
- [91].Yoon S, Eom GH, HDAC and HDAC inhibitor: from cancer to cardiovascular diseases, Chonnam Med. J 52 (1) (2016) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Reed SM, Quelle DE, p53 acetylation: regulation and consequences, Cancers 7 (1) (2014) 30–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Minamiya Y, et al. , Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung, Lung Cancer 74 (2) (2011) 300–304. [DOI] [PubMed] [Google Scholar]
- [94].Suzuki J, et al. , Protein acetylation and histone deacetylase expression associated with malignant breast cancer progression, Clin. Cancer Res. 15 (9) (2009) 3163–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Luo S, Rubinsztein DC, BCL2L11/BIM: a novel molecular link between autophagy and apoptosis, Autophagy 9 (1) (2013) 104–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Niu X, et al. , Binding of released Bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells, Clin. Cancer Res. 22 (17) (2016) 4440–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Schwartz J, et al. , Synergistic anti-leukemic interactions between ABT-199 and panobinostat in acute myeloid leukemia ex vivo, Am. J. Transl. Res. 8 (9) (2016) 3893–3902. [PMC free article] [PubMed] [Google Scholar]
- [98].Cyrenne BM, et al. , Synergy of BCL2 and histone deacetylase inhibition against leukemic cells from cutaneous T-cell lymphoma patients, Blood 130 (19) (2017) 2073–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Salmon JM, et al. , Combined BCL-2 and HDAC targeting has potent and TP53 independent activity in AML, Blood 132 (2018) 1426.30068506 [Google Scholar]
- [100].Li X, et al. , The HDAC and PI3K dual inhibitor CUDC-907 synergistically enhances the antileukemic activity of venetoclax in preclinical models of acute myeloid leukemia, Haematologica 106 (5) (2021) 1262–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Lee JH, et al. , Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair, Proc. Natl. Acad. Sci. U. S. A. 107 (33) (2010) 14639–14644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Margueron R, Reinberg D, The Polycomb complex PRC2 and its mark in life, Nature 469 (7330) (2011) 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Laugesen A, Helin K, Chromatin repressive complexes in stem cells, development, and cancer, Cell Stem Cell 14 (6) (2014) 735–751. [DOI] [PubMed] [Google Scholar]
- [104].Cao R, et al. , Role of histone H3 lysine 27 methylation in Polycomb-group silencing, Science 298 (5595) (2002) 1039–1043. [DOI] [PubMed] [Google Scholar]
- [105].Czermin B, et al. , Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites, Cell 111 (2) (2002) 185–196. [DOI] [PubMed] [Google Scholar]
- [106].Hong S, et al. , Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases, Proc. Natl. Acad. Sci. U. S. A. 104 (47) (2007) 18439–18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Jankowska AM, et al. , Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A, Blood 118 (14) (2011) 3932–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].van Haaften G, et al. , Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer, Nat. Genet. 41 (5) (2009) 521–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Kim KH, Roberts CW, Targeting EZH2 in cancer, Nat. Med. 22 (2) (2016) 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Masudo K, et al. , EZH2 overexpression as a useful prognostic marker for aggressive behaviour in thyroid cancer, In Vivo 32 (1) (2018) 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Mochizuki D, et al. , Aberrant epigenetic regulation in head and neck cancer due to distinct EZH2 overexpression and DNA hypermethylation, Int. J. Mol. Sci. 19 (12) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Pawlyn C, et al. , Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control, Blood Cancer J. 7 (3) (2017), e549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Raman JD, et al. , Increased expression of the polycomb group gene, EZH2, in transitional cell carcinoma of the bladder, Clin. Cancer Res. 11 (24 Pt 1) (2005) 8570–8576. [DOI] [PubMed] [Google Scholar]
- [114].Roh JW, et al. , Clinical and biological significance of EZH2 expression in endometrial cancer, Cancer Biol. Ther. 21 (2) (2020) 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Varambally S, et al. , The polycomb group protein EZH2 is involved in progression of prostate cancer, Nature 419 (6907) (2002) 624–629. [DOI] [PubMed] [Google Scholar]
- [116].Wang X, et al. , Clinical and prognostic relevance of EZH2 in breast cancer: a meta-analysis, Biomed. Pharmacother. 75 (2015) 218–225. [DOI] [PubMed] [Google Scholar]
- [117].Han Li C, Chen Y, Targeting EZH2 for cancer therapy: progress and perspective, Curr. Protein Pept. Sci. 16 (6) (2015) 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Bracken AP, et al. , EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer, EMBO J. 22 (20) (2003) 5323–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Morin RD, et al. , Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin, Nat. Genet. 42 (2) (2010) 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Tiffen J, et al. , Somatic copy number amplification and hyperactivating somatic mutations of EZH2 correlate with DNA methylation and drive epigenetic silencing of genes involved in tumor suppression and immune responses in melanoma, Neoplasia 18 (2) (2016) 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Yap DB, et al. , Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation, Blood 117 (8) (2011) 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Hao B, et al. , Global characterization of proteome and lysine methylome features in EZH2 wild-type and mutant lymphoma cell lines, J. Proteome 213 (2020), 103614. [DOI] [PubMed] [Google Scholar]
- [123].Tiffen J, et al. , EZH2 cooperates with DNA methylation to downregulate key tumor suppressors and IFN gene signatures in melanoma, J. Invest. Dermatol. 140 (12) (2020) 2442–2454, e5. [DOI] [PubMed] [Google Scholar]
- [124].Majer CR, et al. , A687V EZH2 is a gain-of-function mutation found in lymphoma patients, FEBS Lett. 586 (19) (2012) 3448–3451. [DOI] [PubMed] [Google Scholar]
- [125].McCabe MT, et al. , Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27), Proc. Natl. Acad. Sci. U. S. A. 109 (8) (2012) 2989–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Ezponda T, Licht JD, Molecular pathways: deregulation of histone h3 lysine 27 methylation in cancer-different paths, same destination, Clin. Cancer Res. 20 (19) (2014) 5001–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Duan R, Du W, Guo W, EZH2: a novel target for cancer treatment, J. Hematol. Oncol. 13 (1) (2020) 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Schmitz R, et al. , Genetics and pathogenesis of diffuse large B-cell lymphoma, N. Engl. J. Med. 378 (15) (2018) 1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Beguelin W, et al. , EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation, Cancer Cell 23 (5) (2013) 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Scholze H, et al. , Combined EZH2 and Bcl-2 inhibitors as precision therapy for genetically defined DLBCL subtypes, Blood Adv. 4 (20) (2020) 5226–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Wu ZL, et al. , Polycomb protein EZH2 regulates E2F1-dependent apoptosis through epigenetically modulating Bim expression, Cell Death Differ. 17 (5) (2010) 801–810. [DOI] [PubMed] [Google Scholar]
- [132].Ruthenburg AJ, et al. , Multivalent engagement of chromatin modifications by linked binding modules, Nat. Rev. Mol. Cell Biol. 8 (12) (2007) 983–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Yang Z, He N, Zhou Q, Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression, Mol. Cell. Biol. 28 (3) (2008) 967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Delmore JE, et al. , BET bromodomain inhibition as a therapeutic strategy to target c-Myc, Cell 146 (6) (2011) 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Xu L, et al. , Targetable BET proteins- and E2F1-dependent transcriptional program maintains the malignancy of glioblastoma, Proc. Natl. Acad. Sci. U. S. A. 115 (22) (2018) E5086–E5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Zuber J, et al. , RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia, Nature 478 (7370) (2011) 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Baratta MG, et al. , An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma, Proc. Natl. Acad. Sci. U. S. A. 112 (1) (2015) 232–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Winter GE, et al. , Drug development. Phthalimide conjugation as a strategy for in vivo target protein degradation, Science 348 (6241) (2015) 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Chaidos A, et al. , Potent antimyeloma activity of the novel bromodomain inhibitors I-BET151 and I-BET762, Blood 123 (5) (2014) 697–705. [DOI] [PubMed] [Google Scholar]
- [140].Kim SR, et al. , BET inhibition in advanced cutaneous T cell lymphoma is synergistically potentiated by BCL2 inhibition or HDAC inhibition, Oncotarget 9 (49) (2018) 29193–29207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Hogg SJ, et al. , BET inhibition induces apoptosis in aggressive B-cell lymphoma via epigenetic regulation of BCL-2 family members, Mol. Cancer Ther. 15 (9) (2016) 2030–2041. [DOI] [PubMed] [Google Scholar]
- [142].Peirs S, et al. , Targeting BET proteins improves the therapeutic efficacy of BCL-2 inhibition in T-cell acute lymphoblastic leukemia, Leukemia 31 (10) (2017) 2037–2047. [DOI] [PubMed] [Google Scholar]
- [143].Carra G, et al. , Inhibition of bromodomain and extra-terminal proteins increases sensitivity to venetoclax in chronic lymphocytic leukaemia, J. Cell. Mol. Med. 24 (2) (2020) 1650–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Esteve-Arenys A, et al. , The BET bromodomain inhibitor CPI203 overcomes resistance to ABT-199 (venetoclax) by downregulation of BFL-1/A1 in in vitro and in vivo models of MYC+/BCL2+ double hit lymphoma, Oncogene 37 (14) (2018) 1830–1844. [DOI] [PubMed] [Google Scholar]
- [145].Fiskus W, et al. , Superior efficacy of cotreatment with BET protein inhibitor and BCL2 or MCL1 inhibitor against AML blast progenitor cells, Blood Cancer J. 9 (2) (2019) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Cummin TEC, et al. , BET inhibitors synergize with venetoclax to induce apoptosis in MYC-driven lymphomas with high BCL-2 expression, Blood Adv. 4 (14) (2020) 3316–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Agarwal R, et al. , Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma, Nat. Med. 25 (1) (2019) 119–129. [DOI] [PubMed] [Google Scholar]
- [148].Green MR, Chromatin modifying gene mutations in follicular lymphoma, Blood 131 (6) (2018) 595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]