

Abstract
BACKGROUND
Although cardiomyopathy has emerged as a leading cause of death in Duchenne muscular dystrophy (DMD), limited studies and therapies have emerged for dystrophic heart failure.
OBJECTIVES
The purpose of this study was to model DMD cardiomyopathy using DMD patient-specific human induced pluripotent stem cell (hiPSC)-derived cardiomyocytes and to identify physiological changes and future drug therapies.
METHODS
To explore and define therapies for DMD cardiomyopathy, the authors used DMD patient-specific hiPSC-derived cardiomyocytes to examine the physiological response to adrenergic agonists and β-blocker treatment. The authors further examined these agents in vivo using wild-type and mdx mouse models.
RESULTS
At baseline and following adrenergic stimulation, DMD hiPSC-derived cardiomyocytes had a significant increase in arrhythmic calcium traces compared to isogenic controls. Furthermore, these arrhythmias were significantly decreased with propranolol treatment. Using telemetry monitoring, the authors observed that mdx mice, which lack dystrophin, had an arrhythmic death when stimulated with isoproterenol; the lethal arrhythmias were rescued, in part, by propranolol pre-treatment. Using single-cell and bulk RNA sequencing (RNA-seq), the authors compared DMD and control hiPSC-derived cardiomyocytes, mdx mice, and control mice (in the presence or absence of propranolol and isoproterenol) and defined pathways that were perturbed under baseline conditions and pathways that were normalized after propranolol treatment in the mdx model. The authors also undertook transcriptome analysis of human DMD left ventricle samples and found that DMD hiPSC-derived cardiomyocytes have dysregulated pathways similar to the human DMD heart. The authors further determined that relatively few patients with DMD see a cardiovascular specialist or receive β-blocker therapy.
CONCLUSIONS
The results highlight mechanisms and therapeutic interventions from human to animal and back to human in the dystrophic heart. These results may serve as a prelude for an adequately powered clinical study that examines the impact of β-blocker therapy in patients with dystrophinopathies.
Keywords: β-blockers, Duchenne disease modeling, muscular dystrophy cardiomyopathy, heart failure, human inducible pluripotent stem cells
Duchenne muscular dystrophy (DMD) is the most common form of muscular dystrophy, affecting 1 in 5,000 boys born in the United States (1). This genetic disease is caused by mutations of the DMD gene (2–4). Dystrophin is a component of the dystrophin glycoprotein complex (DGC), and the absence of dystrophin leads to muscle weakness (Figure 1A) (5). Ultimately, DMD is a lethal disease because of dilated cardiomyopathy (6,7), which is nearly ubiquitous in patients with DMD (7). Given the prevalence of cardiomyopathy in this patient population, an enhanced understanding of the pathophysiology of DMD cardiomyopathy is urgently needed.
FIGURE 1. The DGC in hiPSC-Derived CMs and Human Cardiac Samples in the Presence and Absence of Dystrophin.
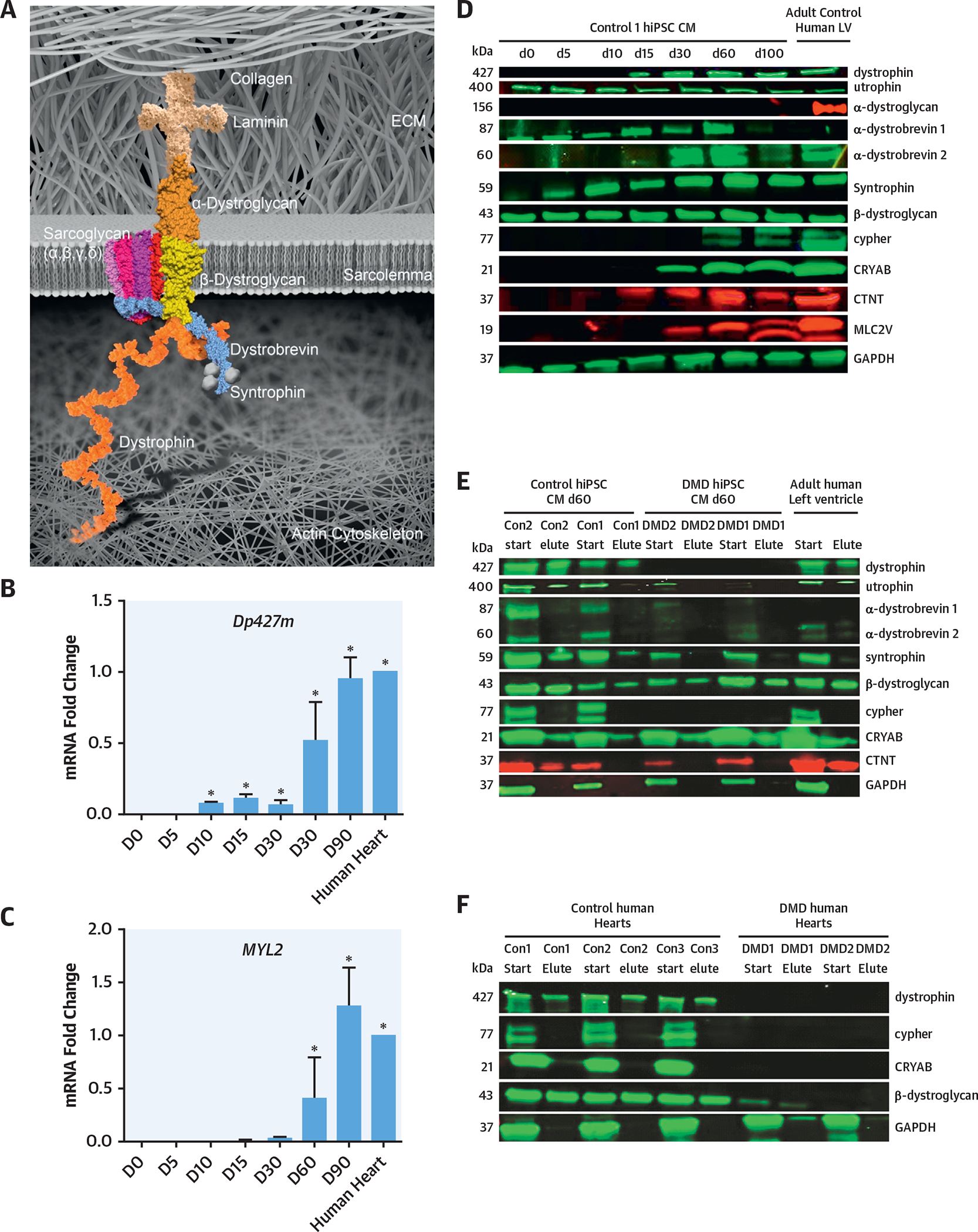
(A) Schematic of the DGC. (B, C) qRT-PCR time course of Dp427m (full-length muscle dystrophin gene) and MYL2 genes in control hiPSC-derived CMs (n = 3). (D) Western blot expression of the DGC components in hiPSC-derived CMs and adult human LV. These results showed that the day 60 hiPSC-derived CMs expressed the protein components of the DGC and sarcomeric proteins (n = 3). (E) WGA pulldown of control and DMD hiPSC-derived CM lines at day 60 differentiation demonstrated that the DGC components were all expressed and pulled down in hiPSC-derived CMs and adult human LV. In the DMD hiPSC-derived CMs at day 60, the absence of dystrophin leads to the lack of nucleation of the DGC. Cypher was also absent in DMD hiPSC-derived CMs (n = 3). (F) Control human LV samples showed the expression and pulldown of dystrophin, whereas an absence of dystrophin was seen in the 2 human DMD LV samples (n = 3). The DMD human LV samples have a lack of nucleation of the DGC, including the absence of cypher. CM = cardiomyocyte; Con = control; D = day; DGC = dystrophin-glycoprotein complex; DMD = Duchenne muscular dystrophy; GAPDH = glyceraldehyde 3-phsophate dehydrogenase; hiPSC = human induced pluripotent-derived cardiomyocytes; LV = left ventricle; qRT-PCR = quantitative real-time polymerase chain reaction; WGA = wheat germ agglutinin.
Our current understanding of the pathophysiology of DMD cardiomyopathy has been obtained from studies using the DMD mouse model (mdx), which lacks dystrophin and has a DMD phenotype (8–10). However, this research model has yielded limited clinical cardiovascular therapies for DMD cardiomyopathy, which underscores the need for new human-based research models.
In the present study, we hypothesized that cardiomyocytes (CMs) derived from human induced pluripotent stem cells (hiPSCs) from patients with DMD could be used to model DMD cardiomyopathy. We utilized DMD hiPSC lines isolated from patients with DMD having mutations in the DMD gene. The DMD hiPSCs and gene-edited DMD-null isogenic hiPSC-derived CMs also replicated the phenotype of increased arrhythmias due to irregular calcium transients, which were exacerbated by the addition of isoproterenol, a β-adrenergic agonist, and rescued with propranolol, a β-adrenergic antagonist. Based on these results, we undertook animal studies and observed that β-adrenergic receptor blockade rescued the adrenergic agonist–stimulated lethal phenotype of the mdx mouse model. We further undertook a genomics analysis to decipher possible mechanisms that promoted cardiomyopathic lethality in the mdx mouse model. We conclude that the DMD derived CMs are an important model for examining cardiomyopathic mechanisms and will serve as a platform for the development of therapies (Central Illustration).
CENTRAL ILLUSTRATION. Duchenne Muscular Dystrophy Cardiomyopathy Disease Modeling From Bedside to Bench and Back Again.
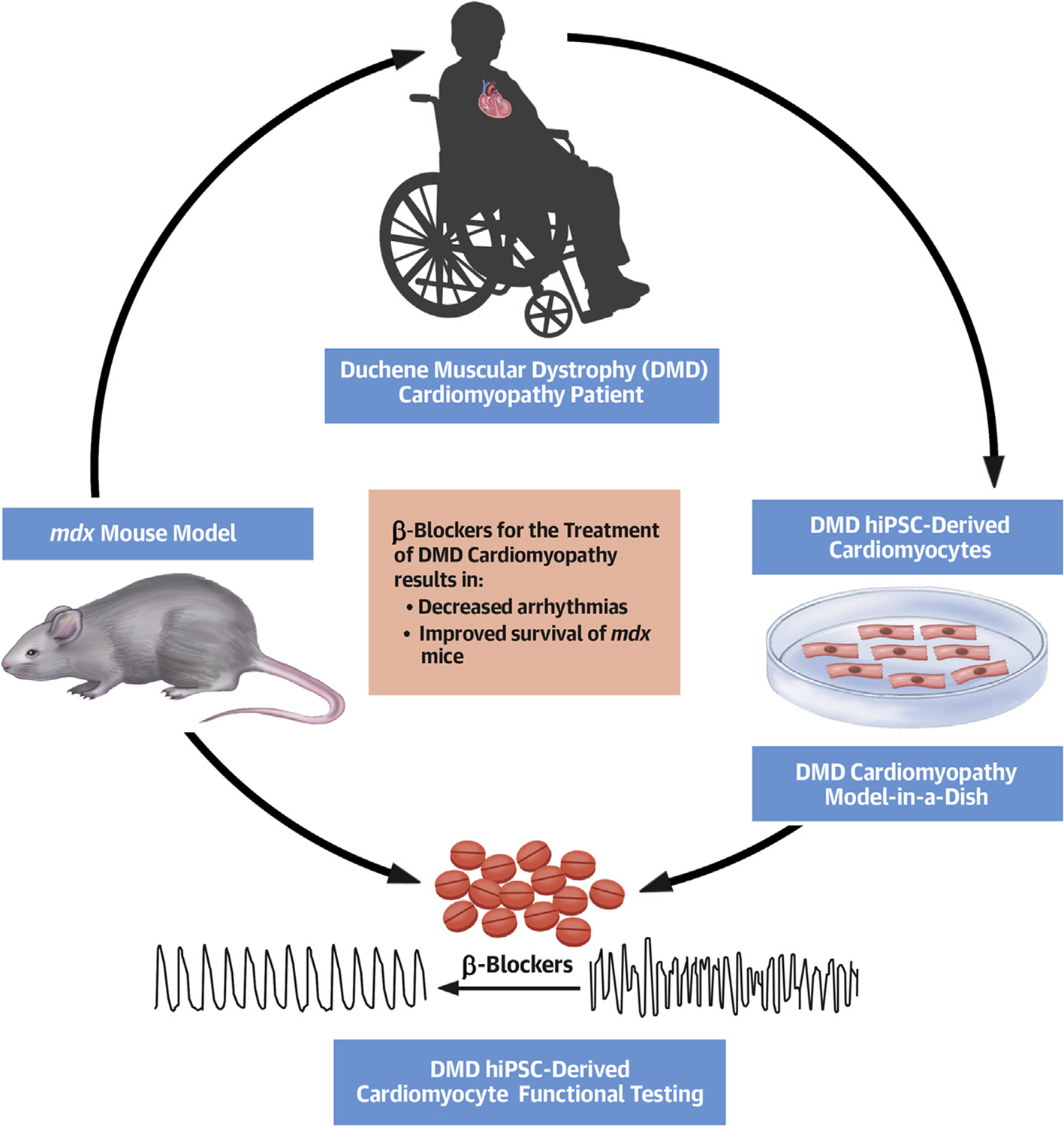
Schematic of DMD disease modeling from the patient with DMD cardiomyopathy to DMD hiPSC-derived CMs to the mdx mouse model and the role of β-blockers in rescuing the phenotype. DMD = Duchenne muscular dystrophy; hiPSC = human induced pluripotent stem cell; mdx = dystrophin-knockout mouse model.
METHODS
DERIVATION AND CULTURE OF hiPSC LINES.
DMD or unaffected hiPSC (control) lines were reprogrammed as previously reported (11) and differentiated to CMs (12,13) as outlined in the supplemental data section. The dystrophin gene editing for the generation of isogenic cell lines was performed as outlined in the supplemental data section. The electrophysiological analysis and immunoblot assays were performed as outlined in the Online Appendix.
CALCIUM IMAGING.
hiPSC-derived CMs were dissociated from a monolayer at day 60 of differentiation, loaded with 5 mmol/l Fluo-4 AM, spontaneous Ca2+ transients were recorded at 37°C by using standard whole-cell transient methods, and calcium transients were recorded under conditions as outlined in the Online Appendix.
SINGLE-CELL RNA-SEQ AND BULK RNA-SEQ ANALYSES.
For the single-cell RNA-seq studies, all libraries were sequenced using 75-base pair paired-end sequencing on MiSeq (Illumina, San Diego, California). The wild-type (WT) and DMD iPSC count matrices were merged based on Ensembl gene ID, resulting in 24,279 total genes. Genes with RNA-seq detection in 5 or fewer cells were removed, leaving 15,785 remaining genes. Clustering of genes was performed as described in the Online Appendix. Bulk RNA-seq of left ventricle (LV) samples was performed for C10 control (WT), C10 mice treated with isoproterenol, mdx control mice, mdx mice treated with isoproterenol, mdx mice treated with propranolol, and mdx mice treated with both isoproterenol and propranolol (n = 3 for each condition).
ANIMAL EXPERIMENTS.
WT (C57BL/10SnJ) and mdx (C57BL/10ScSn-Dmdmdx/J) mice were obtained from Jackson Laboratories (Bar Harbor, Maine). All animal procedures were approved by the University of Minnesota Institutional Animal Care and Use Committee, and studies were performed as outlined in the Online Appendix.
DYSTROPHINOPATHY PATIENT DATA.
We obtained patient data from the Muscular Dystrophy Association U.S. Neuromuscular Patient Registry. We identified those patients with both DMD and Becker Muscular Dystrophy (BMD) and subsequently identified which patients had received β-blocker therapy. Heart failure β-blockers were defined as carvedilol, metoprolol succinate, and bisoprolol. We also identified whether patients with dystrophinopathy had been seen by a cardiologist.
SUPPLEMENTAL AND STATISTICAL METHODS.
For additional detailed methods, please see the Online Appendix.
RESULTS
DMD PATIENT hiPSC CHARACTERIZATION.
To establish an in vitro model of DMD cardiomyopathy, hiPSC mutations were defined from male patients with DMD (Online Figure 1, Supplemental Data). Multiplex ligation–dependent probe amplification analyses were performed by using genomic DNA of all hiPSC lines. This analysis demonstrated that DMD1 had an out-of-frame deletion of exons 3 through 7 and that DMD2 had an out-of-frame deletion of exons 4 through 43 of the DMD gene. The DMD1, DMD2, and control lines were indistinguishable on morphological, karyotypic, pluripotency (OCT4 [octamer-binding transcription factor-4] and SSEA4 [stage specific embryonic antigen-4]) assays (Online Figures 2A and 2B).
FUNCTIONAL CARDIOMYOCYTES FROM CONTROL AND DMD-NULL hiPSC LINES.
hiPSCs were differentiated by using the small-molecule–directed differentiation protocol (Online Figure 2C, Online Videos 1 and 2). There was no difference in beating and expression of cardiac troponin T (CTNT) (Online Figures 2D, 2E, and 3B), which ranged from 75% to 90% without any further enrichment strategy (Online Figure 2F). Whole-cell patch-clamp electrophysiology indicated atrial, ventricular, and nodal-like cardiomyocytes (Online Figure 2G), as well as the expression of the sarcomeric proteins α-actinin and ventricular-specific myosin light chain 2V (MLC2V) (Online Figure 3A).
Based on immunohistochemical analysis, DMD hiPSC-derived CMs lacked dystrophin, whereas control hiPSC-derived CMs demonstrated co-staining with both dystrophin and CTNT (Online Figure 3B). These results were verified with Western blot analysis for dystrophin, β-dystroglycan, and CTNT at day 30 of differentiation (Online Figure 4). These results supported the conclusion that the DMD and control hiPSC lines could be efficiently differentiated into functional CMs and that the DMD hiPSC-derived CMs lacked dystrophin, consistent with the pathophysiology of DMD cardiomyopathy.
DGC TRANSCRIPTS AND PROTEINS WERE EXPRESSED AT DAY 60 OF DIFFERENTIATION, AND THE DGC IS NUCLEATED AT DAY 60 IN CONTROL hiPSC-DERIVED CMs AND ABSENT IN DMD PATIENT hiPSC-DERIVED CMs.
To evaluate the transcript and protein expression of dystrophin and DGC, a time course during CM differentiation compared to adult human LV was examined (Figures 1B to 1D, Online Tables 1 and 2) (14). The DGC and sarcomeric transcripts and proteins were expressed at day 60 of hiPSC-derived CM differentiation. To assess for dystrophin association with the DGC at day 60 hiPSC-derived CMs, a wheat germ agglutinin (WGA) pulldown assay was utilized. WGA is able to bind to α- and β-dystroglycan, and if bound to dystrophin, it will pull down both α- and β-dystroglycan as well as other DGC proteins, including dystrophin. The total lysates (without pulldown) and eluted lysates were immunoblotted and assayed. In the day 60 control hiPSC-derived CMs, the DGC was primarily nucleated, including the novel cardiac DGC-associated protein CRYAB (Figure 1E). The nucleation of the DGC in hiPSC-derived CMs was critical for the interrogation of the control and DMD hiPSC-derived CMs to further elucidate the pathophysiology of DMD cardiomyopathy. A WGA pulldown assay using male human control and human DMD LV was used to validate the day 60 hiPSC-derived CM results. As anticipated, all 3 human control LV samples expressed dystrophin, and dystrophin was pulled down, whereas the DMD human LV samples lacked dystrophin expression (Figure 1F). Similarly, β-dystroglycan was expressed and pulled down in control human LV samples, but there was limited β-dystroglycan expression in DMD human LV samples. Cypher was expressed in the control human LV samples, but the DMD human LV samples did not express cypher, further supporting our observation in day 60 DMD hiPSC-derived CMs (Figure 1F).
DMD hiPSC-DERIVED CMs HAVE IRREGULAR CALCIUM TRANSIENTS CORRESPONDING TO ARRHYTHMIAS.
Calcium transients have been determined to have a correlation with arrhythmias in hiPSC-derived CMs (15). In a blinded review of calcium transients from control and DMD hiPSC-derived CM lines, we determined that DMD hiPSC-derived CMs have markedly increased baseline arrhythmic calcium transients (Figures 2A to 2C). Control hiPSC-derived CMs at day 60 had 16% to 20% arrhythmic calcium transients, whereas the DMD hiPSC-derived CM lines at day 60 had 60% arrhythmic calcium transients at baseline (p < 0.05) (Figure 2C).
FIGURE 2. DMD hiPSC Derived-CMs and Isogenic DMD-Null hiPSC-Derived CMs Have Increased Arrhythmias at Baseline That Were Increased With the Addition of a β-Adrenergic Agonist and Rescued With Addition of a β-Adrenergic Antagonist.
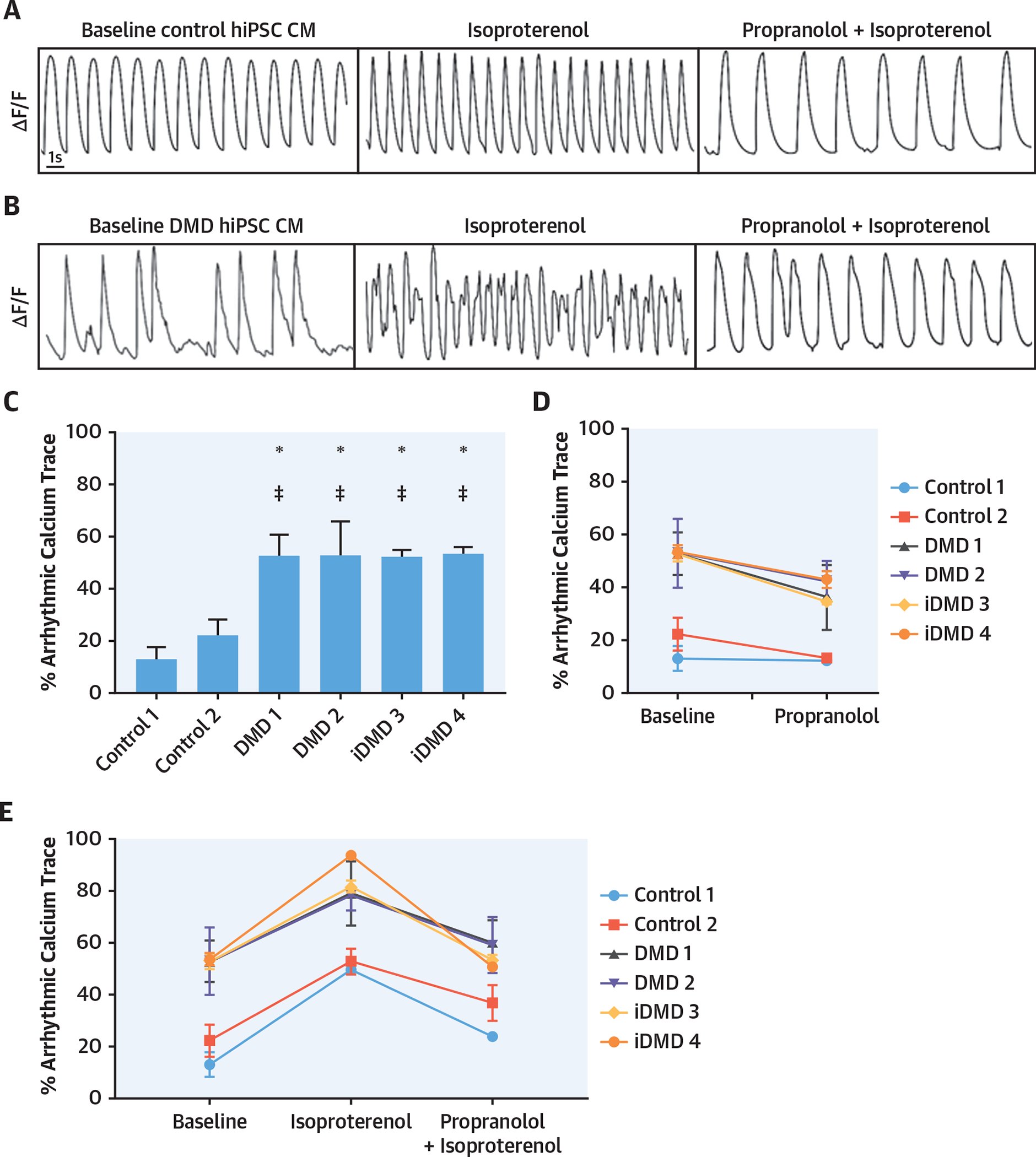
(A, B) Examples of calcium traces at baseline; isoproterenol, propranolol, and isoproterenol-treated control; and DMD hiPSC-derived CMs. (C) Arrhythmic calcium traces were significantly increased in DMD hiPSC-derived CMs at baseline versus control hiPSC-derived CMs (*p < 0.05 vs. control 1 and p < 0.05 vs. control 2; n = 120 cells/experiment repeated in triplicate). (D) Propranolol decreased arrhythmic calcium traces in all DMD hiPSC-derived CMs from baseline. (E) Isoproterenol increased arrhythmic calcium traces in DMD hiPSC-derived CMs from baseline while pre-treatment with propranolol followed by isoproterenol prevented isoproterenol-induced increase in arrhythmic calcium traces. Abbreviations as in Figure 1.
ISOPROTERENOL EXACERBATED ARRHYTHMIC CALCIUM TRANSIENTS IN DMD hiPSC-DERIVED CMs, AND PROPRANOLOL RESCUED THE PHENOTYPE.
β-adrenergic stress is increased in heart failure and can exacerbate ventricular arrhythmias (16). As expected, isoproterenol increased beating frequency in both control and DMD-null hiPSC-derived CMs (Figures 2A and 2B, Online Figure 5) and increased arrhythmic calcium traces in DMD-null hiPSC-derived CMs from a baseline of 60% to 80% (Figure 2E). The addition of propranolol led to a reduction of arrhythmic calcium traces below baseline in DMD-null hiPSC-derived CMs (Figure 2D). Additionally, when DMD-null hiPSC-derived CMs were pre-treated with propranolol followed by isoproterenol exposure, the arrhythmic calcium traces remained at baseline compared to the increased arrhythmias observed with isoproterenol alone (Figure 2E). Using clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9, we engineered isogenic control DMD-null hiPSC lines (iDMD3 and iDMD4). The DMD-null isogenic hiPSC-derived CMs demonstrated the absence of DMD gene or protein expression (Online Figure 6). Calcium imaging was repeated in the DMD-null isogenic hiPSC-derived CMs in the presence and absence of isoproterenol and propranolol and replicated the phenotype seen in DMD patient-specific hiPSC-derived CMs (Figures 2C to 2E).
ISOGENIC DMD-NULL hiPSC-DERIVED CMs HAVE AN INCREASE IN FIBROTIC GENES BASED ON SINGLE-CELL RNA-SEQ.
High-quality DMD (iDMD3, iDMD4) and control single hiPSC-derived CMs (n = 176) from days 30 and 60 were visualized using the t-distributed stochastic neighbor embedding (t-SNE) algorithm. Because of cellular heterogeneity, we used the principal curve on the 2-dimensional t-SNE subspace to show the pseudo-temporal expression pattern of the single cells (Figure 3A) (17). The single cells were grouped into 5 clusters according to their projections on the principal curve (P1 to P5). We found that mature CM markers (TNNT2 and MYL2) were highly expressed in P1 to P4 cell groups and that known cardiofibroblast (CF) markers (CDH11) were highly enriched in the P5 cell group. The pathway analysis of genes that were significantly upregulated in P1 to P4 versus P5 cell groups suggested that these genes were associated with normal CM functions such as striated muscle tissue development (Figure 3B). Moreover, we found that the genes related to calcium/channel transportation (CDH2) and G protein-coupled receptor signaling and metabolism (ACAT1, ATP5J) were highly expressed at the P1 to P4 cells and that the genes related to fibrosis were enriched in the P5 cells (Figure 3C). These results suggested that the heterogeneous expression patterns of the DMD and WT single cells and the pseudo-time reflected the spectrum of CM and CF cells. Notably, we observed a larger proportion of P5 cells from DMD samples (Figure 3A). Given the heterogeneity between single cells from both groups that may have been a result of cell culture heterogeneity or other factors, we focused the analysis on the P1 to P4 CM population between DMD and control hiPSC CMs. The pathway analysis of highly expressed genes in the day 60 cells in DMD-null hiPSC-derived CMs (compared with the remaining cells), after excluding the CDH11-expressing CF cells, also suggested an increased fibrosis program (Figure 3D) as DMD-null hiPSC-derived CMs (Figure 3E) and had an induction of known fibrosis genes (COL1A1, ADAMTS2, COL6A1, and THY1) compared with control CMs (day 60) (Wilcoxon rank sum test, p < 0.001) (Figures 3E to 3I). In summary, the single-cell RNA-seq analysis showed that the fibrosis program was significantly more activated specifically in the DMD hiPSC-derived CM population (P1 to P4) at days 30 and 60.
FIGURE 3. Single-Cell Genomics of Isogenic DMD hiPSC-Derived CMs.
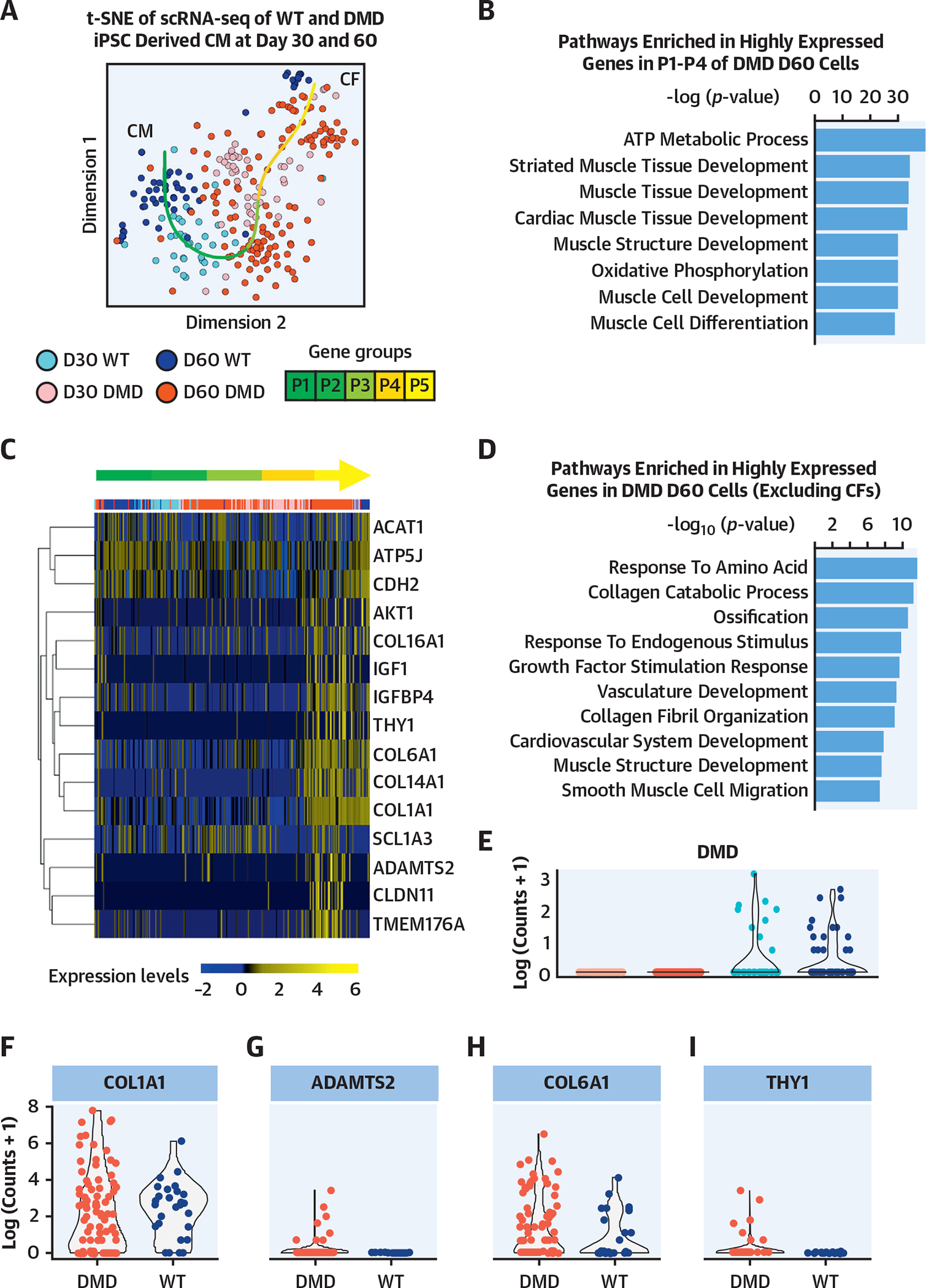
(A) The small conditional RNA-seq data matrix is normalized and treated for batch effects using ComBat (Surrogate Variable Analysis R package, JHU Bloomberg School of Public Health, Baltimore, Maryland). The t-SNE plot shows the clustering of the resulting matrix and indicates grouping of CMs and CFs. The dark red points represent DMD cells at day 60 (n = 151), pink points represent DMD cells at day 30 (n = 52), dark blue points represent WT cells at day 60 (n = 54), and light blue points represent WT cells at day 30 (n = 41). Using R package princurve (AT&T Bell Laboratories, Murray Hill, New Jersey), a principal curve is fit to the t-SNE data matrix, and the curve is split into 5 segments, shown in the green gradient labeled P1 to P5. (B) Highly expressed gene groups within each segment determined pathways enriched in segments P1 to P4. These segments indicate the subset of cells that are cardiomyocytes. According to the top 8 hits shown in the bar plot of the ToppGene pathway analysis (Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio), based on −log(p value), the genes highly expressed in P1 to P4 are linked to cardiac function. (C) The heatmap highlights genes that are related to fibrosis, calcium-channel transportation issues, and GPCR signaling and metabolism. Expression is calculated by getting log-transformed, normalized counts of the reads mapped to the gene. Columns are in the same order as principal curve segments P1 to P5, indicated by the same green gradient color as in A. (D) The pathway analysis indicated the biological processes enriched in highly expressed genes in DMD day 60 cells, after excluding the 62 CDH11-expressing CF cells. (E) The violin plot shows the log-transformed, normalized expression of the reads mapped to the deleted DMD sequence. The day 30 and day 60 DMD cells do not show the presence of this sequence, confirming that the DMD sequence is present only in the WT cells. (F to I) The violin plots show the log-transformed, normalized expression of genes related to fibrosis and indicate that there is higher representation of these genes in the DMD day 60 samples compared with WT day 60. CF = cardiofibroblast; GPCR = G protein-coupled receptor; t-SNE = t-stochastic neighbor embedding; WT = wild type; other abbreviations as in Figure 1.
ISOPROTERENOL ADMINISTRATION WAS LETHAL IN MDX MICE, AND PROPRANOLOL RESCUED THE LETHALITY IN MDX MICE.
WT (strain C10) and mdx mice were exposed to isoproterenol. All (100%) of the mdx mice died by day 2 of isoproterenol treatment (Figure 4A), whereas 15 out of 16 (94%) WT mice survived the entire isoproterenol treatment. Additionally, those mdx mice that were pre-treated with propranolol before and during isoproterenol exposure had a 54% survival compared with those mdx mice that were not treated with propranolol (Figure 4A). Continuous telemetry monitoring demonstrated that mdx mice exposed to isoproterenol had significantly more ventricular arrhythmias compared with WT mice (Figures 4B and 4C). Baseline electrocardiographic and echocardiographic data were comparable between control and mdx mice (Online Figure 7). Quantification of the arrhythmias over a 24-h period before death or termination of the experiment showed a significant increase in arrhythmias in the mdx versus WT mice exposed to isoproterenol (Figure 4D), and additionally mdx mice pre-treated with propranolol had a significant reduction in arrhythmias. All of the mdx mice exposed to isoproterenol had an arrhythmic death (Figure 4E). Cardiac samples taken at the time of death or termination of the experiment showed that mdx mice exposed to isoproterenol had an immunoglobulin (Ig) G infiltration, suggestive of severe acute myocardial injury and fibrosis, compared with control mice (Figures 4F and 4G). The propranolol-pre-treated mdx mice, which survived the following isoproterenol challenge, did not have significant acute injury (Figures 4F and 4G). These results established that isoproterenol administration resulted in acute myocardial injury and necrosis in the mdx heart, leading to death, whereas propranolol pre-treatment had a protective effect through the modulation of the fibrotic response. Survival, arrhythmia, and histological data of additional WT and mdx groups are presented in Online Figure 8.
FIGURE 4. β-Adrenergic Antagonist Propranolol Rescued mdx Mice From Isoproterenol-Induced Death.
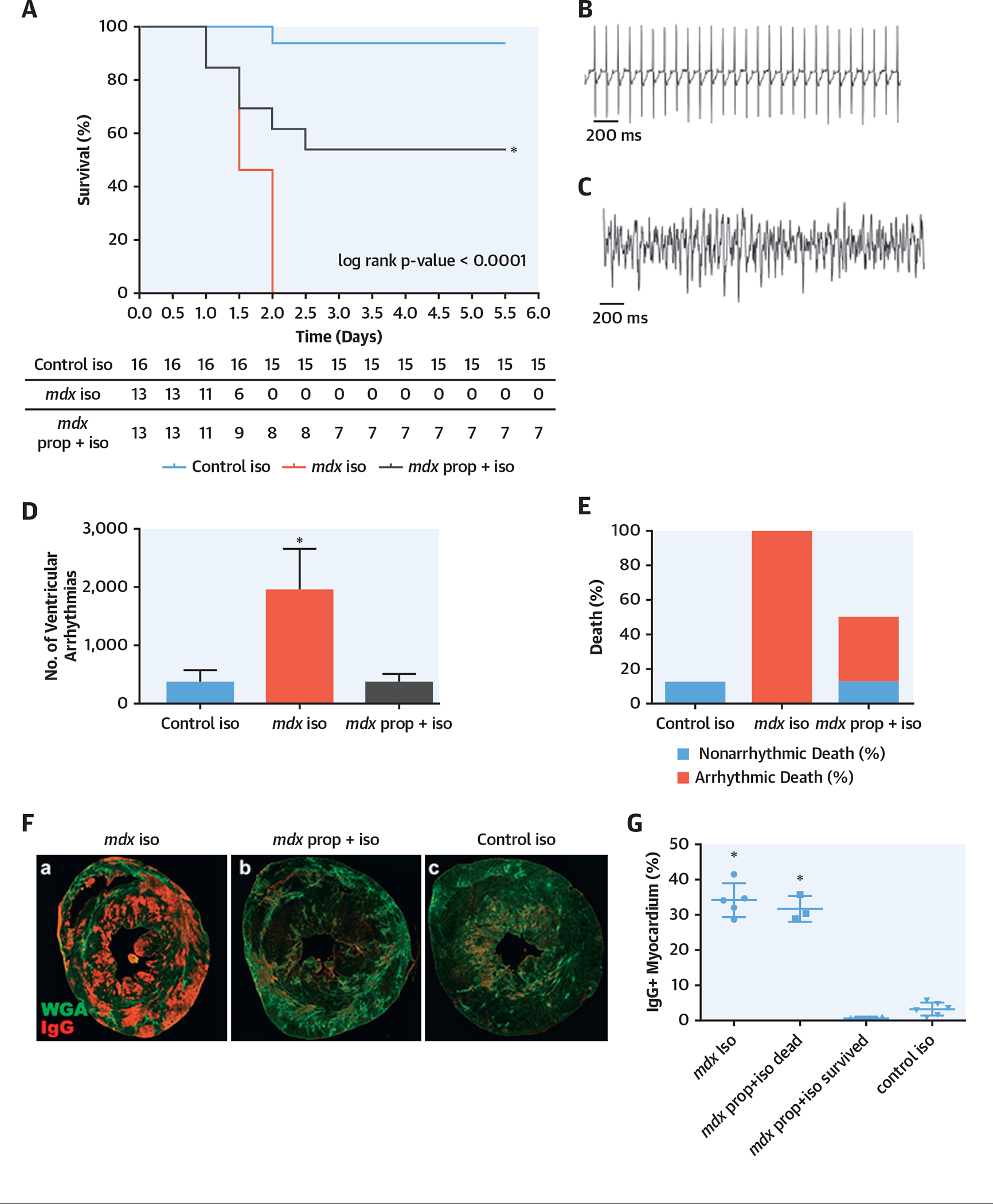
(A) Kaplan-Meier survival curve demonstrated normal survival for male control (C10) mice exposed to isoproterenol (blue line) (n = 16), 100% death in male mdx mice after isoproterenol exposure (red line) (n = 13), and 54% survival in mdx mice pre-treated with propranolol and then isoproterenol (green line) (n = 13) (mdx: all male animals; mean age: 5.3 months; range: 3.4 to 5.6 months; control: all male animals; mean age: 5.3 months; range: 3.3 to 5.6). (B) Normal electrocardiograph telemetry tracing of a representative mdx mouse before treatment. (C) Sustained ventricular arrhythmias on electrocardiograph telemetry tracing of an mdx mouse after isoproterenol treatment. (D) The mdx mice treated with isoproterenol have a significantly increased number of ventricular arrhythmias compared with C10 control mice, and mdx mice pre-treated with propranolol have an incidence of ventricular arrhythmias comparable to control mice treated with isoproterenol (p < 0.05). (E) All mdx mice treated with isoproterenol died due to an arrhythmic death. (F) Representative histology sections from (a) mdx mouse exposed to isoproterenol showing high IgG (red) infiltration, suggestive of severe acute injury and fibrosis (WGA); (b) mdx mouse exposed to propranolol and isoproterenol, showing minimal acute injury; and (c) control mouse exposed to isoproterenol, showing very mild acute injury. (G) Quantification of IgG in the 2 groups. Ig = immunoglobulin; iso = isoproterenol; mdx mouse = dystrophin-knockout mouse model; prop = propranolol.
PROPRANOLOL EXPOSURE NORMALIZED THE ABNORMAL MOLECULAR PROGRAM IN THE MDX HEART.
First, we examined the molecular programs that were perturbed in the mdx heart compared with the control (WT) mouse heart. These analyses showed a marked induction of transcripts related to inflammation, epithelial-mesenchymal transition, cancer pathways, and transporters/channels (Online Figure 9). These results showed a baseline dystrophic cardiomyopathy in the mdx heart. Then, we performed and compared the bulk RNA-seq datasets from the mdx and WT (C10) hearts after pre-treatment and adrenergic stimulation conditions (Online Table 3). In the mdx heart, propranolol treatment resulted in both the upregulation of mitochondrial/metabolic and signaling-associated genes and downregulation of specific genes involved in ion binding (Dsg2, Atp6v0d2, Cdh3) and channel-activity genes. An antifibrotic pattern was observed in the propranolol-treated mdx mice with a decrease in the expression of fibrotic genes (Lgals3, Col12a1, Tnc, F10, Galnt6) (Online Figure 10). These results demonstrated a molecular basis for the beneficial effect of propranolol in the dystrophic heart.
We then analyzed the differential expression of transcripts in response to isoproterenol in mdx and WT (C10) mice, because we observed that the mdx mice failed to induce the fibrotic (Col5a3, Col16a1, Col5a1, Fyb1, PCOLCE, Fyb1, MMP12, Bgn, etc.) and pro-survival (Cln8, Sk38, CIAPIN1, etc.) mechanisms in response to isoproterenol-induced stress (Online Figure 11). The C10 mice also had elevated levels of inflammatory immune-response genes and channel/transporter genes in response to isoproterenol, whereas the mdx mice did not have a similar response to isoproterenol (Online Figure 11). Interestingly, several transcriptional repressors, genes regulating voltage-gated channel activity or transcripts associated with Golgi-mediated cargo transport and G protein-coupled receptor–mediated exocytosis/endocytosis genes were downregulated in C10 mice in response to isoproterenol, whereas their expression remained mostly unchanged in mdx mice (Online Figure 12). Next, we examined the transcripts upregulated (innate immunity) or downregulated (metabolism and iron homeostasis) in the mdx heart in response to isoproterenol but that remained unchanged in C10 hearts after isoproterenol challenge and in the mdx mice that received propranolol pre-treatment (Online Figures 13 and 14).
We further hypothesized that isoproterenol treatment would perturb distinct cardiac gene programs in the mdx heart and lead to their demise. To test this hypothesis, we identified the transcripts that met 3 criteria: 1) the transcripts that were significantly dysregulated in mdx plus isoproterenol versus mdx hearts (DESeq [European Molecular Biology Laboratory, Heidelberg, Germany], p < 0.001), 2) the transcripts that were significantly dysregulated in control plus isoproterenol versus control hearts (DESeq, p < 0.001), and 3) those transcripts having a similar expression pattern between C10 and mdx plus propranolol plus isoproterenol samples (DESeq, p > 0.1). Thus, the expression patterns of these transcripts were significantly perturbed by the isoproterenol treatment and were successfully normalized after propranolol treatment. The normalized expression of a subset of these transcripts that fulfilled the 3 criteria are outlined in Figure 5A. The pathway analysis of showed that the perturbation of these recovered genes leads to abnormal myocardial functions, abnormal heart morphology, or cardiac fibrosis, suggesting the beneficial role of propranolol treatment (Figure 5B). These results supported the notion that the isoproterenol treatment of mdx mice increased the expression pattern of genes that are associated with cardiac dysfunction and fibrosis.
FIGURE 5. The mdx Mouse Hearts and Human DMD iPSC-Derived CMs Showed Similar Dysregulated Biological Processes.
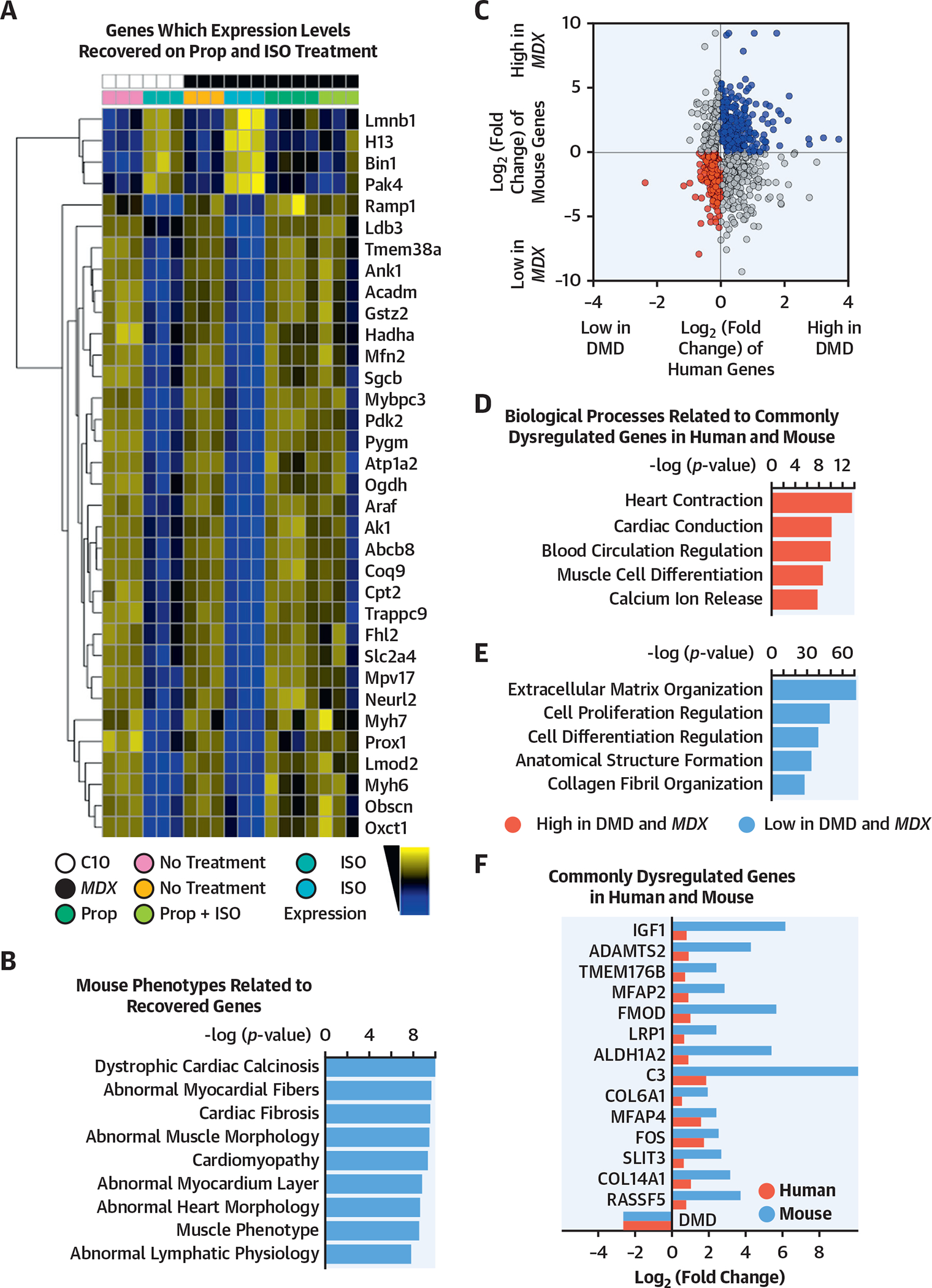
(A) The heatmap shows log transformed, normalized counts of the reads mapped to the genes, where the color blue indicates low expression, and the color yellow indicates high expression. The heatmap indicates a subset of all the transcripts dysregulated in the respective mouse models with or without treatment (mdx: all male animals; mean age: 5.3 months; range: 3.4 to 5.6 months; control: all male animals; mean age: 5.3 months; range: 3.3 to 5.6 months). Transcripts were gathered from (B) ToppGene pathway analysis of the transcripts that fit these criteria, ordered by their −log(p value). (C) Merged data from mouse and human samples. After differential expression analysis with DESeq2, differentially expressed transcripts (p < 0.05) were plotted based on fold change. Upregulated (blue) and downregulated (red) transcripts in DMD conditions were used as queries for pathway analysis in ToppGene. (D, E) The bar plots showed the ToppGene results of the biological processes that were related to commonly dysregulated genes in human and mouse, ordered by −log(p value). (F) Transcripts involved in the 5 biological processes of upregulated and downregulated genes were shown. These genes were commonly dysregulated in human (black) and mouse (gray). C10 = control mouse model; mdx mouse = dystrophin-knockout mouse model; other abbreviations as in Figures 1 and 4.
THE MDX HEARTS AND ISOGENIC HUMAN DMD hiPSC-DERIVED CMs HAVE SIMILAR DYSREGULATED MOLECULAR PROGRAMS AND BIOLOGICAL PROCESSES.
We combined the DMD hiPSC-derived CM small conditional RNA-seq datasets and the mouse mdx bulk RNA-seq dataset to define the commonly dysregulated molecular programs in the dystrophic (i.e., DMD) cardiomyocytes and mouse heart. Among the 16,060 homologous genes found between human and mouse, 332 and 556 genes were commonly upregulated or downregulated in DMD (mdx) heart samples (Figure 5C, Online Table 4). Interestingly, the pathway analysis showed that the genes upregulated in both DMD hiPSC-derived CMs and mdx samples were significantly associated with extracellular matrix (ECM) organization, cellular proliferation, and fibrosis, whereas the transcripts that were highly downregulated were associated with heart contraction and calcium function (Figure 5D to 5F). These results suggest that mdx hearts and human DMD hiPSC-derived CMs shared conserved biological programs that were perturbed by the DMD mutations.
DMD hiPSC-DERIVED CMs AND HUMAN DMD LV SAMPLES HAVE SIMILAR DYSREGULATED MOLECULAR PROGRAMS AND BIOLOGICAL PROCESSES.
We compared bulk RNA-seq of human DMD patient LVs to isogenic control DMD hiPSC-derived CMs (days 45 to 60) to underscore the utility of DMD hiPSC-derived CMs as a model for studying dystrophic cardiomyopathies. Our analysis showed that DMD hiPSC-derived CMs and left human DMD patient LV samples have similar dysregulated biological processes (Online Figure 15). After correcting for the batch effects between the iPSC-derived samples and heart tissue samples, we identified 922 and 1,147 genes that were significantly upregulated and downregulated between control and DMD samples, both in the iPSC and heart samples (Online Figure 15). The pathway analysis indicated that fibrosis, ossification, and ECM organization were commonly upregulated in DMD samples, whereas metabolic and cardiac muscle cell development were commonly downregulated in DMD samples. These results suggested that DMD hiPSC-derived CMs exhibited similar perturbations in molecular pathways as human DMD heart samples and supported the use of DMD hiPSC-derived CMs as a model of DMD cardiomyopathy.
PATIENTS WITH DYSTROPHINOPATHIES WERE RARELY TREATED WITH β-BLOCKERS.
Finally, we examined the care that patients with DMD and BMD with dystrophinopathies received because the most common cause of death for the population of patients with DMD or BMD was cardiomyopathy (7). Using the Muscular Dystrophy Association U.S. Neuromuscular Registry, we identified 945 patients with DMD and BMD and determined that there was only a 10% utilization of β-blockers (Figure 6A). Among these patients, the use of heart failure–specific β-blockers was even lower, at 7.8% (Figure 6B). The percentage of β-blocker medications used by age was highest in the 16- to 20-year-old age range (Figure 6C). Additionally, we determined that the majority of patients with dystrophinopathy (77%) had not been evaluated by a cardiologist (Figure 6D). We further determined that only 11.8% of the patients with DMD were taking β-blockers and only 8.8% were taking heart failure–specific β-blockers (Figures 6E and 6F).
FIGURE 6. Muscular Dystrophy Association Dystrophinopathy Registry Data Demonstrated a Low Utilization of β-Adrenergic Blockers in Patients With Duchenne and Becker Muscular Dystrophy.
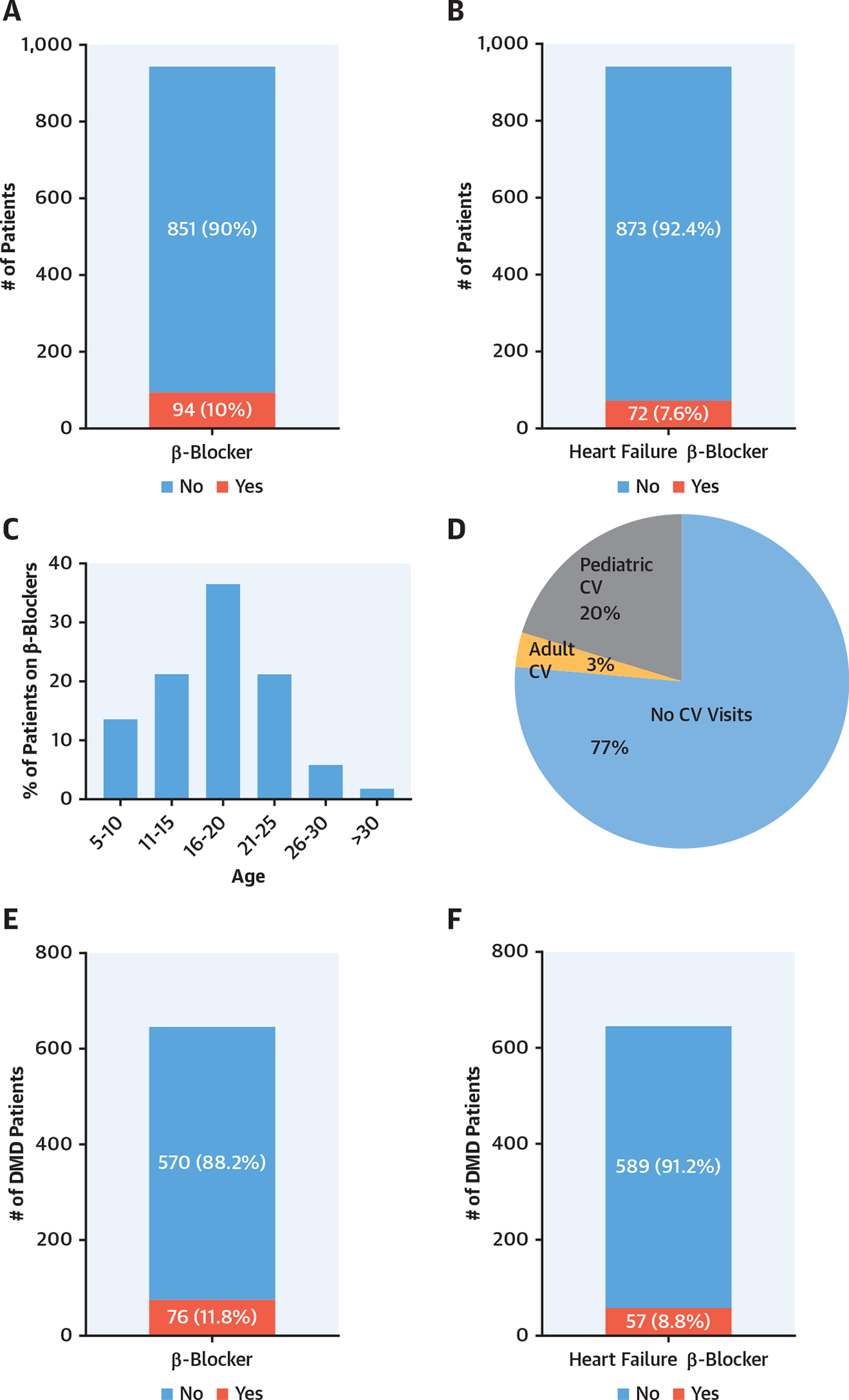
(A) β-Blocker use was 10% in male patients with DMD and BMD (n = 945). (B) Heart failure β-blocker use was <8% in patients with DMD and BMD. (C) Age histogram based on β-blocker use. (D) The majority of patients with DMD and BMD were not seen by cardiologists. (E) β-blocker use was approximately 12% in patients with DMD only (n = 646). (F) Heart failure β-blocker use was approximately 9% in patients with DMD. BMD = Becker muscular dystrophy; CV = cardiovascular; DMD = Duchenne muscular dystrophy.
DISCUSSION
DMD is a lethal disease, and essentially all the patients develop a cardiomyopathy (7). Relatively few patients with DMD receive therapies for their dystrophic cardiomyopathy, nor have these patients seen a cardiovascular specialist. Here, we describe the use of a DMD hiPSC-derived CM model to define and examine the response to pharmacological agents. We complement these studies by using a well-described mouse model to make 4 fundamental discoveries.
Our first discovery used hiPSC-derived CMs (18–21) and human LV samples to define the DGC in the human control and dystrophic heart. In the present study, using hiPSC-derived CMs from patients with DMD, we defined the expression of the DGC components in human cardiomyocyte cell lines and showed that dystrophin is localized to the DGC in hiPSC-derived CMs at day 30, which were primarily nucleated by day 60, supporting the notion that the dystrophin-DGC complex was functional and localized to the cell membrane. Although hiPSC-derived CMs are not fully mature, these human cardiomyocytes serve as a clinically relevant model for DMD cardiomyopathy. In the present study, the hiPSC-derived CM model has allowed for identification and validation of novel cardiac DGC proteins, such as cypher and CRYAB, which are implicated in the development of human cardiomyopathies (14,22,23). The absence of cytoskeletal proteins such as cypher may play an important role in the development of DMD cardiomyopathy and warrants further study in the future. Thus, the DMD hiPSC cardiac model allows for identification and testing of novel mechanisms underlying the development of DMD cardiomyopathy. Importantly, we replicated the DMD phenotype in vitro using DMD hiPSC-derived CMs and used these models to examine potential therapies for dystrophic cardiomyopathies.
Our second discovery demonstrated that DMD hiPSC-derived CMs replicated the phenotype observed in patients with DMD cardiomyopathy. We determined that DMD hiPSC-derived CMs have a markedly increased incidence of arrhythmic calcium traces, even under baseline conditions, compared with control hiPSC-derived CMs. These findings corroborated previously published results showing that patients with DMD cardiomyopathy had an increased incidence of arrhythmias (24,25). We further demonstrated that β-blocker therapies in vitro and in vivo decreased the incidence of arrhythmogenesis and rescued lethality in mdx mice after β-adrenergic stimulation. Although previous studies and guideline therapies have emphasized the structural, functional, and survival benefits of β-blocker therapies in nondystrophic cardiomyopathy, the results of the present study provide a platform for future studies that will examine the short- and long-term impact of β-blocker agents in DMD and BMD cardiomyopathy.
Our third discovery defined mechanisms in human and rodent DMDs that contributed to dystrophic cardiomyopathy and lethality. Previous studies have emphasized the pathophysiology of DMD skeletal muscle and heart, including the increased permeability of the cellular membrane, due to the loss of dystrophin, which resulted in increased intracellular calcium leading to fibrosis, structural dysfunction, and impaired function. In the present study, we used single-cell and bulk RNA-seq in human and rodent models of DMD to further define molecular mechanisms that were perturbed in DMD cardiomyopathy and the benefits of β-blocker therapies. These studies highlighted the perturbed expression of transcripts associated with fibrosis, reactive oxygen species, channels, calcium signaling, and metabolism in DMD cardiomyopathy. Importantly, these studies demonstrated the normalization of these molecular responses in the dystrophic background after treatment with β-blocker agents such as propranolol. This normalization of transcript expression in the dystrophic heart provided by β-blocker treatment further supports the multifactorial benefits associated with reverse remodeling of the cardiomyopathic heart in response to such medical interventions. In addition, these molecular results support a mechanism whereby β-blocker agents promote viability in the adrenergic stimulated mdx mouse model.
Our fourth discovery was the similar dysregulated molecular and biological pathways in DMD hiPSC-derived CM and human DMD patient LV transcriptomes. Fibrosis, ossification, and ECM pathways were upregulated in both DMD hiPSC-derived CM and human DMD LV samples, whereas cardiac muscle and metabolic pathways were downregulated. These findings suggest that the use of DMD hiPSC-derived CMs, although immature, can serve as a model for human DMD cardiomyopathy. However, because >1,000 mutations have been identified in the DMD gene and the most frequent deletions are observed in the mid-distal hotspot (exons 42 to 52), several more DMD heart samples and iPSC models from those patients, or gene-edited models mimicking those mutations, are needed to better understand the diverse and complex mechanisms involved in DMD cardiomyopathy.
Based on our results and the results of others showing the near universal development of cardiomyopathy in patients with DMD, we examined the only known registry of patients with DMD that assembles longitudinal data from multiple centers across the United States. These registry results highlight the lack of cardiovascular care and therapies initiated in patients with DMD. We recognize that the MDA registry reflects only a subpopulation of patients with DMD. It is possible that this subpopulation of patients included in the MDA registry are those patients who have regular follow-up clinical visits and are treated more aggressively and, therefore, represent an overestimation of the patients with DMD seen by a cardiovascular specialist. Nevertheless, these results support the need for aggressive cardiovascular care for patients with DMD and the need to establish a larger and more comprehensive registry that will allow for the longitudinal analysis of this patient population.
STUDY LIMITATIONS.
DMD patient–derived hiPSC CMs are an excellent model for DMD cardiomyopathy; however, they are less mature than adult human heart tissue. We assessed 2 patient samples and 2 gene-edited samples, but future studies should interrogate a larger number of DMD lines and mutations. Likewise, the transcriptomic data analyses need to be expanded to include biopsy samples from living patients with DMD with varying degrees of dystrophic cardiomyopathy to complement our results and further enhance our understanding of the molecular mechanisms of DMD cardiomyopathy.
CONCLUSION
In summary, these studies validate the use of hiPSC-derived CMs as an in vitro model for studying DMD cardiomyopathy. Furthermore, the results of these experiments highlight important mechanisms and therapeutic interventions from human to animal and back to human in the dystrophic heart. Importantly, these results and discoveries may serve as a prelude for an adequately powered clinical study that examines the impact of β-blocker therapy in patients with dystrophinopathies.
Supplementary Material
PERSPECTIVES.
COMPETENCY IN MEDICAL KNOWLEDGE:
DMD can be associated with a refractory, arrhythmogenic cardiomyopathy. In a mouse model, patient-specific hiPSC-derived cardiomyocytes identified a phenotype of arrhythmias caused by abnormal calcium handling and ameliorated by β-blockers.
TRANSLATIONAL OUTLOOK:
Further investigation is warranted to understand the mechanism by which β-blockers reduce the risk of arrhythmias in patients with DMD-associated cardiomyopathy.
ACKNOWLEDGMENTS
The authors acknowledge the Duke Cardiac Repository for providing the human control LV samples and the University of Minnesota Muscular Dystrophy Center for providing the DMD LV samples. The authors acknowledge the Muscular Dystrophy Association for providing data from the Muscular Dystrophy Association U.S. Neuromuscular Patient Registry. The authors acknowledge the technical support of Debra Kulhanek, Daniel Mickelson, and Cynthia Faraday. The authors acknowledge the help of Grafika Labs in creating the graphic illustrations.
Funding is acknowledged from the National Institutes of Health (R01HL122576 and U01HL100407). Dr. Kamp has been a consultant for Cellular Dynamics, Inc. Dr. Wu reports a relationship with Khloris. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
ABBREVIATIONS AND ACRONYMS
- BMD
Becker muscular dystrophy
- CF
cardiofibroblast
- CM
cardiomyocyte
- CTNT
cardiac troponin T
- DGC
dystrophin glycoprotein complex
- DMD
Duchenne muscular dystrophy
- hiPSC
human induced pluripotent stem cell
- LV
left ventricle
- mdx mouse
dystrophin knock-out mouse model
- RNA-seq
RNA sequencing
- WGA
wheat herm agglutinin
- WT
wild type
Footnotes
APPENDIX For an expanded Methods section as well as supplemental figures, tables, and videos, please see the online version of this paper.
REFERENCES
- 1.Mendell JR, Lloyd-Puryear M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve 2013;48:21–6. [DOI] [PubMed] [Google Scholar]
- 2.Emery AE. The muscular dystrophies. Lancet 2002;359:687–95. [DOI] [PubMed] [Google Scholar]
- 3.Cox GF, Kunkel LM. Dystrophies and heart disease. Curr Opin Cardiol 1997;12:329–43. [PubMed] [Google Scholar]
- 4.Kamdar F, Garry DJ. Dystrophin-deficient cardiomyopathy. J Am Coll Cardiol 2016;67:2533–46. [DOI] [PubMed] [Google Scholar]
- 5.Rybakova IN, Patel JR, Ervasti JM. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol 2000;150:1209–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord 2002; 12:926–9. [DOI] [PubMed] [Google Scholar]
- 7.Nigro G, Comi LI, Politano L, Bain RJ. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol 1990; 26:271–7. [DOI] [PubMed] [Google Scholar]
- 8.Coulton GR, Curtin NA, Morgan JE, Partridge TA. The mdx mouse skeletal muscle myopathy: II. contractile properties. Neuropathol Appl Neurobiol 1988;14:299–314. [DOI] [PubMed] [Google Scholar]
- 9.Kamogawa Y, Biro S, Maeda M, et al. Dystrophin-deficient myocardium is vulnerable to pressure overload in vivo. Cardiovasc Res 2001;50: 509–15. [DOI] [PubMed] [Google Scholar]
- 10.Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS, Levin LS. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord 2004;14:491–6. [DOI] [PubMed] [Google Scholar]
- 11.Lowry WE, Richter L, Yachechko R, et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci U S A 2008;105:2883–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Klos M, Wilson GF, et al. Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: the matrix sandwich method. Circ Res 2012;111:1125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lian X, Zhang J, Azarin SM, et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat Protoc 2013;8:162–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson EK, Zhang L, Adams ME, et al. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS One 2012;7: e43515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spencer CI, Baba S, Nakamura K, et al. Calcium transients closely reflect prolonged action potentials in iPSC models of inherited cardiac arrhythmia. Stem Cell Rep 2014;3:269–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bristow MR. The adrenergic nervous system in heart failure. N Engl J Med 1984;311:850–1. [DOI] [PubMed] [Google Scholar]
- 17.Petropoulos S, Edsgard D, Reinius B, et al. Single-cell RNA-seq reveals lineage and X chromosome dynamics in human preimplantation embryos. Cell 2016;167:1012–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moretti A, Bellin M, Welling A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med 2010;363: 1397–409. [DOI] [PubMed] [Google Scholar]
- 19.Kamdar F, Klaassen Kamdar A, Koyano-Nakagawa N, Garry MG, Garry DJ. Cardiomyopathy in a dish: using human inducible pluripotent stem cells to model inherited cardiomyopathies. J Card Fail 2015;21:761–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lan F, Lee AS, Liang P, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 2013;12:101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsa E, Rajamohan D, Dick E, et al. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J 2011;32: 952–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vatta M, Mohapatra B, Jimenez S, et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol 2003;42:2014–27. [DOI] [PubMed] [Google Scholar]
- 23.Sacconi S, Feasson L, Antoine JC, et al. A novel CRYAB mutation resulting in multisystemic disease. Neuromuscul Disord 2012;22:66–72. [DOI] [PubMed] [Google Scholar]
- 24.Chenard AA, Becane HM, Tertrain F, de Kermadec JM, Weiss YA. Ventricular arrhythmia in Duchenne muscular dystrophy: prevalence, significance and prognosis. Neuromusc Disord 1993; 3:201–6. [DOI] [PubMed] [Google Scholar]
- 25.Corrado G, Lissoni A, Beretta S, et al. Prognostic value of electrocardiograms, ventricular late potentials, ventricular arrhythmias, and left ventricular systolic dysfunction in patients with Duchenne muscular dystrophy. Am J Cardiol 2002;89:838–41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.