

Abstract
Recent studies revealed that molecular events related with the physiology and pathology of αS might be regulated by specific sequence motifs in the primary sequence of αS. The importance of individual residues in these motifs remains an important open avenue of investigation. In this work, we have addressed the structural details related to the amyloid fibril assembly and lipid‐binding features of αS through the design of site‐directed mutants at position 39 of the protein and their study by in vitro and in vivo assays. We demonstrated that aromaticity at position 39 of αS primary sequence influences strongly the aggregation properties and the membrane‐bound conformations of the protein, molecular features that might have important repercussions for the function and dysfunction of αS. Considering that aggregation and membrane damage is an important driver of cellular toxicity in amyloid diseases, future work is needed to link our findings with studies based on toxicity and neuronal cell death.
Brief statement outlining significance
Modulation by distinct sequential motifs and specific residues of αS on its physiological and pathological states is an active area of research. Here, we demonstrated that aromaticity at position 39 of αS modulates the membrane‐bound conformations of the protein, whereas removal of aromatic functionality at position 39 reduces strongly the amyloid assembly in vitro and in vivo. Our study provides new evidence for the modulation of molecular events related with the physiology and pathology of αS.
Keywords: amyloid fibril, fluorescence and confocal microscopy, lipid interaction, NMR, sequence motifs, α‐synuclein
1. INTRODUCTION
Alpha‐synuclein (αS) is a disordered, neuronal cytoplasmic protein located predominantly at the presynaptic termini of the central nervous system. 1 Misfolding and aberrant aggregation of αS is associated with neuropathological disorders collectively referred to as synucleinopathies. 2 , 3 , 4 , 5 , 6 , 7 , 8 Although the physiological functions of αS remain elusive, it has been implicated in synaptic plasticity 9 and maintenance of synaptic vesicle pool size. 10 , 11 Strikingly, despite strong efforts over the years, the precise mechanisms leading to αS aggregation are unclear.
The primary structure of αS comprises 140 amino acids distributed in three regions: the amphipathic N‐terminus (residues 1–60), implicated in lipid binding 12 ; the central region, known as NAC (residues 61–95), which is highly hydrophobic and fibrillogenic 13 , 14 ; and the acidic C‐terminus (residues 96–140), critical for blocking rapid αS filament assembly. 15 , 16 , 17 Whereas in its free monomeric state, αS adopts an ensemble of disordered, aggregation‐autoinhibited conformations, 18 , 19 , 20 upon binding to lipid membranes, the protein undergoes a disorder‐to‐helix transition in the N‐terminal, encompassing the first 100 amino acids. 21
Systematic analysis of the role of distinct sequential motifs and specific residues of αS primary sequence on its physiological and pathological states remains an important open avenue of investigation. Studies focused on αS mutations, post‐translational modifications, and molecular interactions are emerging as of central importance to the normal function of the protein, as well as to its pathogenic role in Parkinson disease. 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 In this context, a segment of 12 residues at the amino‐terminus as well as a region surrounding Tyr‐39 were identified recently as canonical chaperone interaction motifs of αS in mammalian cells, through which molecular chaperones might prevent the transformation of αS towards pathological sates. 22 , 25 In parallel, other studies showed that the region consisting of residues 36–42 is important in promoting αS aggregation. 27 , 32 Added to that, post‐translational modifications at Tyr‐39 were suggested to play a role in the physiology and pathology of αS. 22 , 23 , 24 , 25 , 26 , 30 , 31 , 33 , 34
In the frame of our studies in the field of the structural biology of Parkinson disease, we recently showed that removing aromaticity at position 39 affects the compactness of the protein and impacts strongly on the inhibitory interaction of αS with amyloid‐blocking agents. 35 , 36 , 37 In this work, we have addressed structural details related to the aggregation and lipid‐binding features of αS and its site‐directed mutants Y39F, Y39L, and Y39A αS. By the characterization of the amyloid assembly of these species in vitro and in vivo, and the analysis of its conformations in the membrane‐bound state, we can draw the following conclusions: (i) removal of aromatic residues at position 39 impairs the in vitro amyloid fibril assembly of αS; (ii) the aggregation propensity of αS is also affected by the aromaticity at position 39 in cell‐based assays and animal models; and (iii) aromaticity at position 39 determines the membrane‐bound conformation of αS by modulating lipid interactions involving the central hydrophobic NAC domain.
2. RESULTS
2.1. Removal of aromaticity at position 39 impairs the in vitro amyloid fibril assembly of αS
We evaluated first the amyloid‐forming capability of the different αS species. As shown in Figure 1a, the time course of aggregation of wt αS monitored as monomer consumption by 1D 1H‐NMR spectroscopy was comparable to that measured for the Y39F mutant, whereas those species containing the Y39L and Y39A mutation showed a significant reduction in the rate and amount of aggregated protein (1A). Results consistent to those reported by NMR were independently obtained when circular dichroism (CD) and the amyloid‐sensitive Thioflavin T (ThT) fluorescence assay were used to determine the change in secondary structure content of αS species during fibril assembly (Figure S1a‐b).
FIGURE 1.
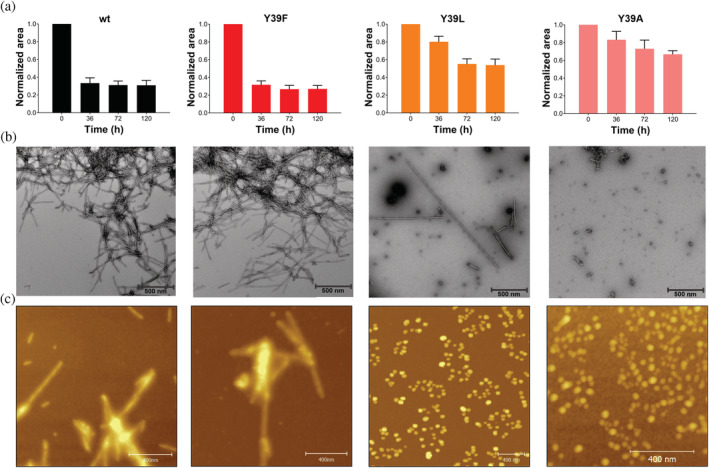
Effects of Y39 mutations on αS fibril formation. (a) Level of remaining soluble αS monomers determined by 1D 1H‐NMR as a function of aggregation time. (b) Representative negative‐stain electron microscopy images of αS aggregates (50 μM samples) taken at 120 hr. Scale bars, 500 nm. (c) AFM of αS aggregates (50 μM samples) after 120 hr incubation. Scale bars, 400 nm
The morphology of the aggregation products of the αS variants was then characterized by transmission electron microscopy (TEM). Ultrastructural visualization of the protein deposits obtained for the Y39F species revealed the presence of abundant amounts of typical amyloid fibrils that were morphologically indistinguishable from those formed for the wild‐type protein (Figure 1b). In contrast, the TEM images corresponding to the Y39L and Y39A species showed predominantly nonfibrillar protein structures (Figure 1b), with a minuscule amount of isolated fibrillar components detectable only for the Y39L variant (1C).
To further characterize the structures observed in electron micrographs, atomic force microscopy (AFM) was performed. As shown in Figure 1c, samples of wild‐type and Y39F aggregates are comprised of typical amyloid fibrils. In contrast, AFM images of the final products of aggregation of Y39L and Y39A resulted mostly in the formation of nonfibrillar, spheroid‐like aggregates.
Altogether, these results demonstrate that mutations Y39L and Y39A impair the in vitro amyloid fibril assembly of αS.
2.2. The aggregation propensity of αS in biological models is affected by the aromaticity at position 39
The next question was whether the in vitro findings are corroborated by the behavior of the mutant proteins in the context of a mammalian cell model. To address this question, we assessed the effect of mutations at position 39 in a well‐established cell model that leads to positive intracellular protein inclusion formation for αS. 38 Treatment with αS promoted a significant number of transfected cells displaying αS inclusions (Figure 2a), while analysis of protein inclusion formation in the Y39F transfected cells showed a pattern comparable to that of wt αS (Figure 2b). In contrast, for the Y39L and Y39A variants, we observed a significant decrease in the percentage of cells with inclusions (Figure 2a‐b). To investigate whether these effects might be explained by differences in the levels of αS being expressed, we performed immunoblot analysis. There was no correlation between the expressed levels of the studied proteins and inclusions formation, indicating that the results were intimately correlated with the effects of the mutations on aggregation (Figure 2c).
FIGURE 2.
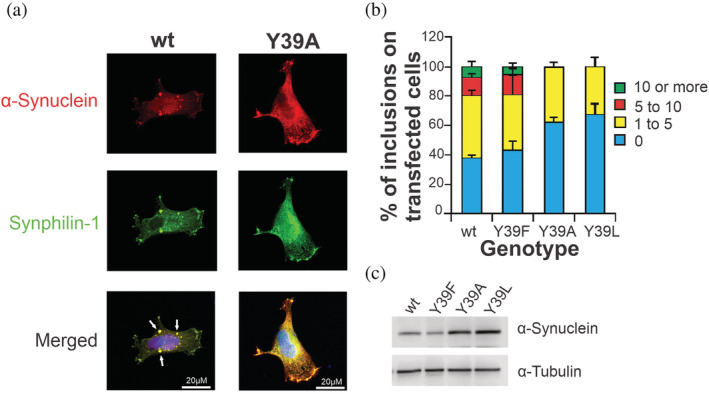
Modulation of αS inclusion formation in cultured cells. (a) Representative images of intracellular wt and Y39A αS inclusion formation in human cultured cells. Scale bars, 20 μm. (b) Quantification of αS inclusions. Transfected cells were classified in different groups: 1 to 4, 5 to 9, and equal to/more than 10 inclusions. Results were expressed as the percentage of the total number of transfected cells obtained from three independent experiments. At least 50 cells were scored per experiment (n = 3). (c) Immunoblot analysis of the expression levels of the αS variants studied in H4 cells
Next, to corroborate the results in postmitotic neurons, we used the nematode C. elegans as a multicellular model system to study the aggregation of αS variants at position 39. 39 When expressed in motoneurons as a C‐terminal fusion with mCherry, wt or Y39F αS mutant formed numerous large aggregates (Figure 3a‐b). By contrast, the number and size of these aggregates were reduced when Y39L or Y39A mutant forms of αS were expressed (Figure 3a‐b). Like in the cell model, the observed differences in aggregation were not due to differences in expression levels of αS as quantified by Western blot experiments (Figure 3c).
FIGURE 3.
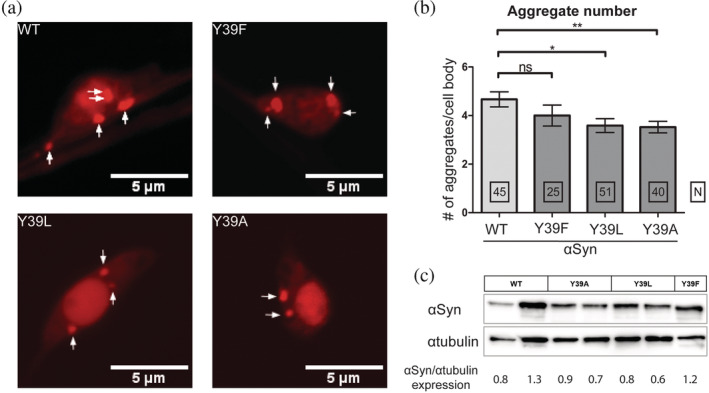
Aggregation of αS in C. elegans motoneurons is affected by the aromaticity at position 39. (a) Confocal images of one day adult motoneurons showing the formation of intracellular αS‐mCherry aggregates. Scale bars, 5 μm. (b) Quantification of intracellular αS aggregates. The values shown represent the mean ± SEM; for comparison an unpaired, two‐tailed t‐test was used; * and ** denote p < 0.05 and 0.01, respectively. (c) Total αS expression levels were quantified by Western blot experiments relative to αTubulin loading control
Overall, the results obtained in culture cells and C. elegans neurons demonstrate that intracellular inclusions of αS are significantly reduced when Y39L and Y39A αS variants were expressed as compared to wt or Y39F αS variants. This strongly suggests that like in vitro, the aggregation propensity of αS is also affected by the aromaticity at position 39 in vivo.
2.3. Aromaticity at position 39 is a determinant of the membrane‐bound conformation of αS
Since the association of αS with cellular membranes and changes in those interactions might have functional and pathological implications, we also investigated the effect of mutations at Y39 on the binding features of αS to lipid‐vesicles (small unilamellar vesicles: SUVs). Changes in αS structure associated with membrane binding were monitored by far‐UV CD, which reports on the increase in αS α‐helicity upon interaction with phospholipids. 28 The far‐UV CD spectra of αS is characterized by a minimum at 198 nm in the absence of SUVs, whereas a maximum at 192 nm and minima at 208 nm and 222 nm are typical of its membrane‐bound state. CD spectra for the wt and Y39A αS species in the presence of varying concentrations of SUVs are shown in Figure 4a‐b. To compare the binding affinities of αS and its Y39F, Y39L, and Y39A variants to SUVs, the mean residue molar ellipticity at 222 nm ([θ]MR,222) for all species was monitored as a function of lipid concentration (Figure 4c). In all cases, analysis of the titration data yielded K d values in the 0.1–0.2 μM range.
FIGURE 4.
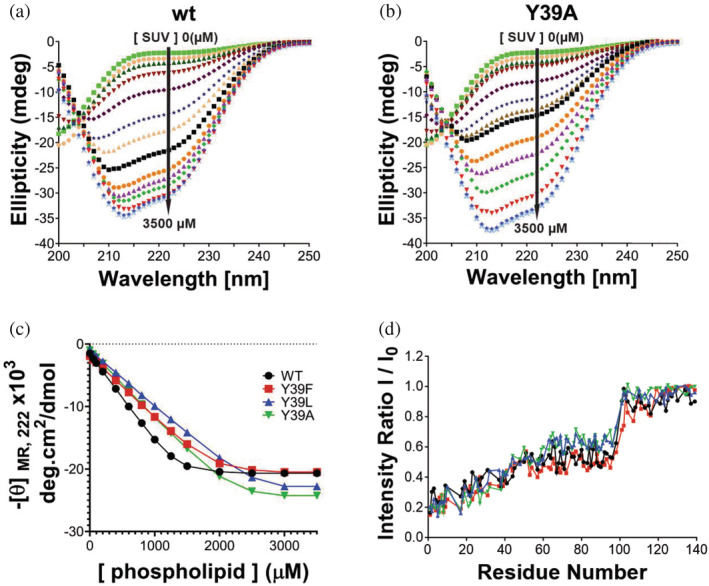
Effect of Y39 mutations on the membrane binding features of αS. (a‐b) cd spectra of wt (a) and Y39A αS (b) species with increasing concentrations of SUVs (0 to 3.5 mM). (c) Data from θMR,222 in wt αS and its Y39F, Y39L, and Y39A variants as a function of lipid concentration were fit to Equation 2 to calculate Kd values. (d) Graphs of I/I0 (where I and I0 are the peak intensities observed in the presence and absence of SUVs, respectively) plotted against residue number for wt αS and its Y39F, Y39L, and Y39A variants
To explore the structural details of the binding, we used NMR spectroscopy. The 15N‐labeled αS mutants were analyzed by 2D 1H–15N HSQC spectra recorded in the absence or presence of SUVs. This assay provides a residue‐by‐residue readout of lipid binding that is useful to evaluate which residues on the protein are in greatest contact with the vesicle. 40 NMR experiments correlate the normalized signal attenuation of resonances from specific residues, obtained in the presence of SUV, to the binding of these residues to the membrane. For each residue of αS, I/I0 is the proportion of molecules in an αS/SUV mixture in which the residue remains mobile and in solution, and the fractional attenuation (1 − I/I0) is the proportion of molecules in which the residue is tightly associated with the membrane. 40 We used this approach to characterize the αS–membrane interaction, in particular, to determine the fractional population of molecules bound to the membrane (“total bound population”) and whether the central hydrophobic (aggregation‐prone) region was either dissociated or associated in the case of conformers referred as “exposed” or “hidden,” respectively. 28 The fractional population of molecules bound to the membrane in either mode was determined from the mean attenuation of residues 3–9, based on the fact that the N‐terminal segment of αS has a higher membrane affinity compared to more C‐terminal regions 41 and is lipid‐bound in the case of both exposed and hidden conformers. 40 Regarding the conformation of the hydrophobic domain NAC at the membrane, which is described to have an important role in the regulation of biological properties of αS and on its oligomerization, 42 , 43 the fractional population of protein in the hidden state was determined by measuring the mean attenuation of residues 66–80, 28 a segment that plays a key role in αS self‐assembly. 14 On the other hand, the fractional population of protein with residues 66–80 in the exposed state was determined by subtracting the hidden population from the total bound population (residues 3‐9). 28
As shown in Table 1, the αS mutants exhibited nearly the same total populations for their membrane‐bound states. However, clear structural differences were observed among the vesicle‐bound forms of the αS variants at the level of the hydrophobic NAC domain. Our results indicate that the percentage of total bound molecules with the hydrophobic NAC in a membrane‐dissociated state was increased by a factor of 1.7 and 2.0 in the cases of the Y39L and Y39A variants compared to wt and Y39F. This means that aromaticity at position 39 regulates lipid membrane interactions involving the central hydrophobic NAC domain.
TABLE 1.
Fractional populations of membrane‐bound αS conformers determined by NMR
| Variant | Total bound a , b | Hidden a , c | Exposed a , d |
|---|---|---|---|
| wt | 0.72 | 0.57 | 0.15 |
| Y39F | 0.71 | 0.56 | 0.15 |
| Y39L | 0.74 | 0.49 | 0.25 |
| Y39A | 0.70 | 0.40 | 0.30 |
Values were determined from 2D 1H‐15N‐HSQC data in Figure 4 and are expressed as a fraction of the total protein. Each sample consisted of a mixture of αS and phospholipids at concentrations of 100 μM and 2.5 mM, respectively.
Determined from the mean attenuation of residues 3–9 (i.e., total bound = 1−{I/I0}mean,3−9). 28 , 44
3. DISCUSSION
Overall, the results presented here demonstrate that aromaticity at position 39 of αS might play a crucial role in molecular events that might represent functional or dysfunctional states of αS. In that direction, a recent study revealed that the aggregation of αS is regulated by specific sequence motifs flanking the NAC region. 27 , 32 The pre‐NAC segment 36GVLYVGS42 was suggested to be a critical requirement for αS aggregation in vivo, whereas removal of this motif prevented αS aggregation and also suppressed the aggregation‐induced toxicity in vivo. 27 , 32 However, certain questions remained unanswered, such as what is the significance of individual residues in that sequential motif in modulating the aggregation of αS. Our findings point to the significance of aromaticity in the 36GVLYVGS42 motif as a key factor determining the amyloid fibril assembly of αS. The substantial change of aggregation rate on passing from wt αS to the Y39A and the Y39L variants might be attributable to a loss of function of the aromatic side chain in the self‐assembly process and/or to a conformational stabilization of the native state.
Added to that, our findings also indicate that the aromatic character at position 39 of αS favors a NAC membrane‐anchored state for the lipid‐bound conformation of αS, supporting further a role for Tyr‐39 as a factor aiding in the folding of the amphiphilic region and promoting helix propagation. 29 Interestingly, it was proposed recently that the equilibrium between membrane‐bound and unbound states of the region 65–97, which overlaps with the NAC region, might play a role in the regulation of biological properties of αS. 43 Indeed, transition toward partly helical membrane‐bound states of αS has been associated with the control of vesicle docking and/or protein–protein interactions mediated by αS via a double‐anchor mechanism. 43 In line with this hypothesis, phosphorylation of αS at Tyr‐39 was shown to promote partly helical membrane‐bound states. 26 Accordingly, it was suggested that in the cellular context, alternation of αS between its two membrane‐bound states might be driven by phosphorylation at Tyr‐39, which would play then a role as a conformational switch. 26
Remarkably, the region encompassing residues 34–45 of αS has been identified as a ganglioside‐binding domain 45 which are specific of outer plasma membrane leaflets and are related to cellular uptake of αS monomers 46 and its pathological aggregates. 47 , 48 In that domain, Tyr‐39 was shown to be the most critical residue for the interaction. In line with our findings, the work concluded that it is not Y39 per se the residue responsible for the recognition of the lipid domain but the presence of an aromatic moiety at this position. 46
Interestingly, a lipid‐chaperone hypothesis was recently proposed, in which the amyloidogenic protein–lipid complex plays the role of the main actor in membrane damage. 49 In that context, the lipid–protein complexes rather than the bare proteins are considered the key players in membrane damage. Considering that aggregation and membrane damage is an important driver of cellular toxicity in amyloid diseases, future work is needed to link our findings with studies based on toxicity and neuronal cell death.
4. MATERIALS AND METHODS
4.1. Protein and reagents
Unlabeled and 15N isotopically enriched N‐terminally acetylated αS and its Y39F, Y39L, and Y39A variants were obtained by co‐transforming E. coli BL21 cells with the plasmid harboring the corresponding protein gene and a second one that encodes for the components of yeast NatB acetylase complex. 50 Both plasmids carried different antibiotic resistance, namely, Ampicillin and Chloramphenicol to select the doubly transformed E. coli colonies. Purification was carried out as previously reported, 15 with the exception that when required, both antibiotics were included in the growth flasks to avoid plasmid purge during growth and expression. The final purity of the protein samples was determined by SDS‐PAGE. Purified protein samples were dissolved in 20 mM MES buffer supplemented with 100 mM NaCl at pH 6.5 (Buffer A). 15N NH4Cl was purchased from Cambridge Isotope Laboratories, and MES buffer and D2O were purchased from Merck or Sigma. Protein concentrations were determined spectrophotometrically by measuring absorption at 274 nm and using an epsilon value of 5,600 M−1 cm−1.
4.2. Aggregation assay
Aggregation kinetics measurements were performed with 50 μM protein samples dissolved in Buffer A, which were incubated at 37°C under constant stirring. To perform the aggregation studies, we used an NMR‐based approach that measures the consumption of the monomeric state of the amyloid protein during the progression of the aggregation process. 51 , 52 The amount of soluble αS monomers at different time points of the aggregation assay was determined by integration of the protein NMR resonances in the aliphatic region of the one‐dimensional (1D) 1H NMR spectrum (0.7–1.0 ppm). Acquisition and processing of NMR spectra were performed using TOPSPIN 7.0 (Bruker Biospin). In addition, endpoint aliquots of aggregation assays were analyzed by the Thioflavin‐T (ThT) fluorescence assay 53 and CD spectroscopy. 42
4.3. Electron microscopy
Ten‐microliter aliquots withdrawn from aggregation reactions were adsorbed onto Formvar/carbon‐coated copper grids (Pella, Redding, CA) and negative stained with 2% (w/v) uranyl acetate. Images were obtained at various magnifications (1,000–90,000X) using a Philips CM120 transmission electron microscope.
4.4. AFM microscopy
Samples were prepared by spin‐coating. A volume 20 μL of each sample was deposited over Muscovite mica discs grade V1 (Ted Pella) of 10 mm diameter, which were previously cleaved. After promoting interactions with the substrate, mica discs were accelerated up to a constant rotation speed of 2000 rpm to ensure complete drying of the substrate. AFM imaging was performed on a Bruker Multimode 8 SPM (Santa Barbara, CA, USA) and a NanoScope V Controller (Santa Barbara, CA, USA) using a “J” type scanner. The AFM images were acquired in tapping mode using silicon tips NCHV (Bruker) with a spring constant of 40 N m−1 and a resonance frequency of ~320 kHz. Images analyses were performed using Gwyddion version 2.46 software (Brno, Czech Republic). 54
4.5. Preparation of SUVs
1,2‐Dioleoyl‐sn‐glycero‐3‐phosphoethanol‐amine (DOPE), 1,2‐dioleoyl‐sn‐glycero‐3‐phospho‐L‐serine (DOPS), and 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC) were purchased from Avanti Polar Lipids (Alabaster, AL), as pure DOPC and a DOPE/DOPS/DOPC mixture with a 5:3:2 weight ratio (Coagulation Reagent I). Phospholipid mixtures containing Coagulation Reagent I or 60% DOPC, 25% DOPE, and 15% DOPS (molar concentrations) were prepared by drying a mixture of the different lipids dissolved in chloroform under a stream of N2 gas and re‐suspending the lipid film in Buffer A. 55 SUVs were prepared by pulse‐sonicating the phospholipid suspensions in a bath sonicator (10 cycles of 2 min with separation of 2 min). The size of the resulting SUVs (hydrodynamic radii of 40–50 nm) was determined by dynamic light scattering using a Zetasizer Nano ZS instrument (Malvern Instruments, Worcestershire, U.K.).
4.6. Far‐UV circular dichroism (CD)
The content of α‐helix structure of αS monomer upon binding to SUVs was evaluated by far‐UV circular dichroism (CD). Solutions of 10 μM of αS monomer in the absence or presence of varying concentration of SUVs were analyzed at 25°C (SUV composition: 20% DOPC, 50% DOPE, and 30% DOPS). Far‐UV CD spectra were recorded from 200 to 250 nm in a J‐1500 CD spectrophotometer (JASCO, Inc.), using a 0.1 cm quartz cuvette (bandwith = 1 nm; scan rate = 50 nm/min; accumulation = 3).
The ellipticity at 222 nm was measured and the background associated with buffer or SUV solutions was subtracted. The mean residue molar ellipticity at 222 nm ([θ]MR,222) was calculated using Equation 1:
| (1) |
where θ222 is the measured ellipticity (millidegrees) at 222 nm, C is the protein concentration (molar), n = 140 (number of amino acid residues in the protein), and l is the path length of the cuvette in cm (0.1 cm). Lipid titration curves generated by plotting [θ]MR,222 versus the lipid concentration were analyzed by fitting to Equation 2, as previously described 44 :
| (2) |
where R is the measured θMR,222 at a given lipid concentration, R0 is the θMR,222 in the absence of lipid, Rf is the θMR,222 in the presence of saturating lipid, L is the total lipid concentration, C is the total protein concentration, K d is the apparent macroscopic dissociation equilibrium constant, and N is the binding stoichiometry (lipids/protein).
4.7. NMR spectroscopy of the interaction with lipid vesicles
NMR spectra were recorded on a Bruker 600 MHz HD Advance III spectrometer, equipped with a cryogenically cooled triple resonance 1H (13C/15N) TCI probe. Two‐dimensional 2D 1H–15N HSQC experiments were recorded at 15°C using standard pulse sequences form the Topspin suite (Bruker) library. Sequence‐specific assignments for the backbone of the intrinsically unfolded αS were obtained from our previous works. In the case of the Y39A, Y39F, and Y39L mutants, assignment of backbone resonances in the vicinity of the mutation was confirmed by 3D TOCSY‐HSQC and NOESY‐HSQC experiments. Binding of monomers of αS and its Y39F, Y39L, and Y39A variants to SUVs was determined via 2D 1H−15N HSQC experiments in which 100 μM 15N‐labeled αS species in Buffer A, 10% D2O, was incubated in the absence or the presence of 2.5 mM SUVs (SUV composition: 60% DOPC, 25% DOPE, and 15% DOPS). To monitor residue‐specific interactions of αS variants with SUVs, the fractional signal attenuation of each residue peak was determined by comparing their intensities in the presence (I) and absence of SUVs (I0), as previously described. 40 The I/I0 ratios of nonoverlapping cross‐peaks were plotted as a function of the protein sequence to obtain the protein–lipid interaction profiles. The fractional population of molecules bound to the membrane (“total bound population”) was determined from the mean attenuation of residues 3–9 28 , 44 (Table 1). Regarding the conformation of the hydrophobic domain NAC at the membrane surface, in the fractional population of protein in the associated state (referred to here as “hidden”), we determined the mean attenuation of residues 66–80 28 , 44 (Table 1). The fractional population of protein with residues 66–80 in the dissociated state (referred to here as “exposed”) was determined by subtracting the hidden population from the total bound population (residues 3–9). 28 , 44 (Table 1). Acquisition and processing of NMR spectra were performed using TOPSPIN 7.0 (Bruker Biospin). 2D spectra analysis and visualization were performed with CCPN.
4.8. Cell‐based assay
4.8.1. Cell culture and transfection
For the studies with cell‐based models of αS aggregation, H4 neuroglioma cells were maintained at 5% CO2 and 37°C in Dulbecco's Modified Eagle's Medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum and 1% Penicillin Streptomycin. One day before the transfection, cells were seeded in 12‐well plates. Cells were co‐transfected with plasmids encoding a C‐terminally modified variant of αS or its point mutants (SynT construct) and synphilin‐1 38 using FuGene (Promega), according to the manufacturer's instructions. Inclusion formation was assessed 48 hr post‐transfection.
4.8.2. Immunocytochemistry
Cells were fixed with 4% paraformaldehyde in PBS 48 hr after transfection. For permeabilization, cells were treated with 0.1% Triton X‐100 and blocked with 1.5% bovine serum albumin (BSA) in PBS. Cells were then incubated with anti‐αS primary antibody (BD 610787, 1:1000) and anti‐V5 tag (Abcam ab9116, 1:100), either 4 hr at room temperature or overnight at 4°C, and 1 hr in Alexa Fluor 488 donkey anti‐rabbit and Alexa Fluor 555 donkey anti‐mouse as secondary antibodies (Invitrogen A21202 and A31570, respectively, 1:1000). Cells were further stained with DAPI (Sigma, D9542) for 5–10 min. Images were captured using a Nikon C2 Plus confocal microscope.
4.8.3. Quantification of αS intracellular inclusions
Transfected cells were detected and scored based on the pattern of αS intracellular inclusions, by classifying them into four groups: cells without inclusions, less than five inclusions (<5 inclusions), between five to nine inclusions (5–9 inclusions), and equal/more than ten inclusions (≥10 inclusions). 38 Results were expressed as the percentage of the total number of transfected cells obtained from three independent experiments. At least 50–100 cells were counted per condition.
4.8.4. Western blot analysis
H4 cells were lysed with radio‐immunoprecipitation assay (RIPA) lysis buffer (50 mM Tris pH 8.0, 0.15 M NaCl, 0.1% SDS, 1% NP40, 0.5% Na‐deoxycholate) and 2 mM EDTA and supplemented with a Protease Inhibitor Cocktail (Roche Diagnostics, Mannheim, Germany). Protein concentration was determined using the Bradford assay (BioRad Laboratories, Hercules, CA, USA) and the gels were loaded with 40 μg protein after denaturation for 5 min at 100°C in SDS‐PAGE protein sample buffer. The samples were separated on 12% SDS‐polyacrylamide gels (SDS‐PAGE). The transfer was carried out to nitrocellulose membrane (Amersham™ Hybond®, GE Healthcare Life Sciences) for 90 min with constant current at 0.3 A and 4°C, using Tris‐Glycine transfer buffer. Membranes were fixed in 8% para‐formaldehide and blocked with 5% (w/v) skim milk in 1xTBS‐Tween (50 mM Tris, 150 mM NaCl, 0.05% Tween, pH 7.5) for 60 min at room temperature. Membranes were further incubated with the primary antibody, mouse anti‐ASYN (1:1000, 2B2D1 BD Biosciences, San Jose, CA, USA), and mouse anti‐αtubulin (T6199, Sigma‐Aldrich) overnight at 4°C. After washing three times in TBS‐Tween for 5 min, membranes were incubated for 1 hr with secondary antibody, anti‐mouse IgG (A4416, Sigma‐Aldrich). Detection was carried out by chemiluminescence (Bio‐Lumina; Kalium Technologies, Argentina) using a Licor C‐Digit Blot Scanner (LI‐COR Biosciences) according to the manufacturer instructions.
4.9. C. elegans study
4.9.1. Strains
All strains were grown on Agar plates containing OP50 bacteria at 20°C as described previously. 56 Transgenic animals expressing αS from an extra‐chromosomal array were generated by injecting wild‐type Bristol N2 worms with plasmids expressing different forms of human αS with mCherry fused to its C‐terminus. Mutations in αS (Y39F, Y39L, Y39A) were introduced by mutagenesis PCR and validated by sequencing. The protein αS was expressed in neuronal cells of C. elegans under the control of the pan‐neuronal rab‐3 promoter (rab‐3p:αS‐mCherry) using the expression vector pD115.62 as a backbone. To generate transgenic lines, the gonads of young adult N2 worms have been injected with a plasmid mix containing 50 ng/μl rab‐3p:αS‐mCherry, 5 ng/μl of a co‐injection marker (myo‐2p::mTFP), and 45 ng/μl pBluescript KSII (Stratagene) to reach a final DNA concentration of 100 ng/μl, as described previously. 57
4.9.2. Fluorescent imaging and analysis
Young adult C. elegans were mounted on a 2% agarose pad and immobilized by 50 mM sodium azide in M9 buffer (17.2 mM KH2PO4, 42.3 mM Na2HPO4, 85.6 mM NaCl, 1 mM MgSO4). Z‐stacks of the neuronal cell bodies were taken on a Zeiss LSM 780 confocal microscope with a 63x/1.40 NA oil immersion objective. Projections of the stacks and statistical analysis have been carried out with ImageJ 2.1.0 and Prism Version 5.03.
4.9.3. Western blotting
About 100 αS expressing worms were washed twice in ice cold PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4) with phosphatase and protease inhibitors (PhosphoStop Phosphatase Inhibitor Cocktail Tablets (Roche), complete EDTA‐free Protease Inhibitor Cocktail Tablets (Roche)) and lysed in 60 μL lysis buffer (150 mM NaCl, 1 mM EDTA, 25 mM Tris pH 7.5, 10% (v/v) glycerol, 1% (v/v) Triton X and with freshly added 100 mM DTT, 4 mM Pefabloc SC‐Protease Inhibitor (A 154.1, Roth), Phosphostop Phosphatase Inhibitor Cocktail Tablets (Roche), and complete EDTA‐free Protease Inhibitor Cocktail Tablets (Roche)) prior to shock‐freezing in liquid nitrogen. Samples were directly transferred to −80°C. After thawing, SDS‐PAGE protein sample buffer was added. Subsequently, the samples were sonicated on ice for 3 min (20 sec on, 10 sec off, 70% amplitude) with an SonoPlus mini20 Sonicator (3,665, Bandelin, Sonotrode Type MS2.5). After boiling at 100°C for 10 min, the lysate was centrifuged at 17000 g for 10 min. About 30 μL of the lysate has been separated on 12% SDS‐polyacrylamide gels and subsequently transferred to a PVDF membrane (10,600,023, Amersham Hybond) for 90 min at 220 mA under cooling conditions. Membranes were blocked with 1% semi‐skimmed milk in 1% TBS‐T buffer (20 mM Tris, 150 mM NaCl, 0.1% (v/v) Tween 20, pH 7.6) for 1 hr. Primary antibody incubation was performed in blocking solution with mouse anti‐αS (1:1000, 610,787, BD Transduction Laboratories) or mouse anti‐αtubulin (1:2000, T6199, Sigma‐Aldrich) overnight, with agitation at 4°C. Next, membranes were washed 3 times in TBS‐T buffer for 7 min and incubated at room temperature with agitation in (goat) anti‐mouse HRP‐conjugated secondary antibody in blocking solution (1:5000, 1,721,011, Bio‐RAD Laboratories, Inc.). For the detection of the chemiluminescence, the ECL Kit Prime Western Blotting Detection Reagents (RPN2232, Amersham) and an Intas ChemoCam Imager were used according to the manufacturer's instructions.
AUTHOR CONTRIBUTIONS
Fiamma A. Buratti: Conceptualization (equal); data curation (equal); formal analysis (equal); investigation (equal); writing – original draft (equal); writing – review and editing (equal). Nicola Boeffinger: Formal analysis (equal); investigation (equal). Hugo A. Garro: Formal analysis (equal); investigation (equal). Jesica S. Flores: Formal analysis (equal); investigation (equal). Francisco J. Hita: Formal analysis (equal); investigation (equal). Phelippe do Carmo Gonçalves: Formal analysis (equal); investigation (equal). Federico dos Reis Copello: Formal analysis (equal); investigation (equal). Leonardo Lizarraga: Formal analysis (equal); investigation (equal). Giulia Rossetti: Formal analysis (equal); investigation (equal). Paolo Carloni: Formal analysis (equal); supervision (equal); writing – original draft (equal). Markus Zweckstetter: Formal analysis (equal); supervision (equal); writing – original draft (equal). Tiago F. Outeiro: Formal analysis (equal); writing – original draft (equal). Stefan Eimer: Formal analysis (equal); supervision (equal); writing – original draft (equal). Christian Griesinger: Formal analysis (equal); supervision (equal); writing – original draft (equal). Claudio Oscar Fernandez: Conceptualization (lead); formal analysis (lead); funding acquisition (lead); investigation (lead); methodology (lead); project administration (lead); resources (lead); supervision (lead); validation (lead); writing – original draft (lead); writing – review and editing (lead).
CONFLICTS OF INTEREST
There are no conflicts to declare.
Supporting information
Figure S1 Final products of aggregation of αS and its Y39F, Y39L, and Y39A mutants. (A) FarUV CD spectra corresponding to the supernatant (red) and pellet (green) obtained from the final product of aggregation (t = 120 hr) of the studied αS species. Black curves correspond to the CD spectra of the monomeric states (t = 0 hr). (A) ThT fluorescence intensities corresponding to the final state of aggregation of the studied αS species (t = 120 hr) and their monomeric states (t = 0 hr)
ACKNOWLEDGMENTS
C.O.F. thanks Universidad Nacional de Rosario (UNR) and ANPCyT‐FONCyT (PICT 2014‐3704 and PICT 2017‐4665) for financial support. C.O.F. and C.G. thank the Max Planck Society (P10390) for support. F.A.B. thanks CONICET for fellowship. TFO is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy ‐ EXC 2067/1‐ 390729940, and SFB1286 (Project B8). M.Z. was supported by the EU Horizon 2020 research and innovation program (grant agreement No. 787679) and by The Michael J. Fox Foundation for Parkinson's Research (Grant ID: MJFF‐019033). Open Access funding enabled and organized by Projekt DEAL.
Buratti FA, Boeffinger N, Garro HA, Flores JS, Hita FJ, Gonçalves PC, et al. Aromaticity at position 39 in α‐synuclein: A modulator of amyloid fibril assembly and membrane‐bound conformations. Protein Science. 2022;31(7):e4360. 10.1002/pro.4360
Review Editor: Hideo Akutsu
Funding information Michael J. Fox Foundation, Grant/Award Number: 019033; EU Horizon 2020; Deutsche Forschungsgemeinschaft, Grant/Award Number: 2067/1‐390729940; Max Planck Society, Grant/Award Number: P10390; Universidad Nacional de Rosario; ANPCyT‐FONCyT PICT, Grant/Award Number: 2017‐4665; Alexander von Humboldt Foundation
REFERENCES
- 1. Maroteaux L, Campanelli JT, Scheller RH. Synuclein: A neuron‐specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8(8):2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fanciulli A, Wenning GK. Multiple‐system atrophy. N Engl J Med. 2015;372(3):249–263. [DOI] [PubMed] [Google Scholar]
- 3. Gómez‐Tortosa E, Newell K, Irizarry MC, Sanders JL, Hyman BT. α‐Synuclein Immunoreactivity in dementia with Lewy bodies: Morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol. 2000;99(4):352–357. [DOI] [PubMed] [Google Scholar]
- 4. Irwin DJ, Lee VM, Trojanowski JQ. Amyloid Beta‐peptide and the dementia of Parkinson's disease. Nat Rev Neurosci. 2013;14(9):626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Luk KC, Lee VMY. Modeling Lewy pathology propagation in Parkinson's disease. Parkinsonism Relat Disord. 2014;20(01):S85–S87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McCann H, Stevens CH, Cartwright H, Halliday GM. α‐Synucleinopathy phenotypes. Parkinsonism Relat Disord. 2014;20(1):S62–S67. [DOI] [PubMed] [Google Scholar]
- 7. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α‐Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A. 1998;95(11):6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. α‐Synuclein in Lewy Bodies. Nature. 1997;388(6645):839–840. [DOI] [PubMed] [Google Scholar]
- 9. Watson JB, Hatami A, David H, et al. Alterations in Corticostriatal synaptic plasticity in mice overexpressing human α‐Synuclein. Neuroscience. 2009;159(2):501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cooper AA, Gitler AD, Cashikar A, et al. α‐Synuclein blocks ER‐Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313(5785):324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci. 2014;34(28):9364–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bussell R, Eliezer D. A structural and functional role for 11‐Mer repeats in α‐Synuclein and other exchangeable lipid binding proteins. J Mol Biol. 2003;329(4):763–778. [DOI] [PubMed] [Google Scholar]
- 13. Du HN, Tang L, Luo XY, et al. A peptide motif consisting of glycine, alanine, and valine is required for the fibrillization and cytotoxicity of human α‐Synuclein. Biochemistry. 2003;42(29):8870–8878. [DOI] [PubMed] [Google Scholar]
- 14. Giasson BI, Murray IVJ, Trojanowski JQ, Lee VMY. A hydrophobic stretch of 12 amino acid residues in the middle of α‐Synuclein is essential for filament assembly. J Biol Chem. 2001;276(4):2380–2386. [DOI] [PubMed] [Google Scholar]
- 15. Hoyer W, Cherny D, Subramaniam V, Jovin TM. Impact of the acidic C‐terminal region comprising amino acids 109−140 on α‐Synuclein aggregation in vitro. Biochemistry. 2004;43(51):16233–16242. [DOI] [PubMed] [Google Scholar]
- 16. Kim TD, Paik SR, Yang CH. Structural and functional implications of C‐terminal regions of α‐Synuclein. Biochemistry. 2002;41(46):13782–13790. [DOI] [PubMed] [Google Scholar]
- 17. Sang MP, Han YJ, Kim TD, Jeon HP, Yang CH, Kim J. Distinct roles of the N‐terminal‐binding domain and the C‐terminal‐solubilizing domain of α‐Synuclein, a molecular chaperone. J Biol Chem. 2002;277(32):28512–28520. [DOI] [PubMed] [Google Scholar]
- 18. Bertoncini CW, Jung YS, Fernandez CO, et al. Release of Long‐range tertiary interactions potentiates aggregation of natively unstructured α‐Synuclein. Proc Natl Acad Sci U S A. 2005;102(5):1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cho MK, Nodet G, Kim HY, et al. Structural characterization of α‐Synuclein in an aggregation prone state. Protein Sci. 2009;18(9):1840–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dedmon MM, Lindorff‐Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. Mapping Long‐range interactions in α‐Synuclein using spin‐label NMR and ensemble molecular dynamics simulations. J Am Chem Soc. 2005;127(2):476–477. [DOI] [PubMed] [Google Scholar]
- 21. Eliezer D, Kutluay E, Bussell R, Browne G. Conformational properties of α‐Synuclein in its free and lipid‐associated states. J Mol Biol. 2001;307(4):1061–1073. [DOI] [PubMed] [Google Scholar]
- 22. Aspholm EE, Matečko‐Burmann I, Burmann BM. Keeping α‐Synuclein at bay: A more active role of molecular chaperones in preventing mitochondrial interactions and transition to pathological states? Life. 2020;10(11):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brahmachari S, Karuppagounder SS, Ge P, et al. C‐Abl and Parkinson's disease: Mechanisms and therapeutic potential. J Parkinsons Dis. 2017;7(4):589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burai R, Ait‐Bouziad N, Chiki A, Lashuel HÁ. Elucidating the role of site‐specific nitration of α‐Synuclein in the pathogenesis of Parkinson's disease via protein Semisynthesis and mutagenesis. J Am Chem Soc. 2015;137(15):5041–5052. [DOI] [PubMed] [Google Scholar]
- 25. Burmann BM, Gerez JA, Matečko‐Burmann I, et al. Regulation of α‐Synuclein by chaperones in mammalian cells. Nature. 2020;577(7788):127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dikiy I, Fauvet B, Jovičić A, et al. Semisynthetic and in vitro phosphorylation of alpha‐Synuclein at Y39 promotes functional partly helical membrane‐bound states resembling those induced by PD mutations. ACS Chem Biol. 2016;11(9):2428–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Doherty CPA, Ulamec SM, Maya‐Martinez R, et al. A short motif in the N‐terminal region of α‐Synuclein is critical for both aggregation and function. Nat Struct Mol Biol. 2020;27:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lima VDA, Do Nascimento LA, Eliezer D, Follmer C. Role of Parkinson's disease‐linked mutations and N‐terminal acetylation on the Oligomerization of α‐Synuclein induced by 3,4‐Dihydroxyphenylacetaldehyde. ACS Chem Nerosci. 2019;10(1):690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lokappa SB, Suk JE, Balasubramanian A, Samanta S, Situ AJ, Ulmer TS. Sequence and membrane determinants of the random coil‐helix transition of α‐Synuclein. J Mol Biol. 2014;426(10):2130–2144. [DOI] [PubMed] [Google Scholar]
- 30. Mahul‐Mellier AL, Fauvet B, Gysbers A, et al. C‐Abl phosphorylates α‐Synuclein and regulates its degradation: Implication for α‐Synuclein clearance and contribution to the pathogenesis of Parkinson's disease. Hum Mol Genet. 2014;23(11):2858–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sevcsik E, Trexler AJ, Dunn JM, Rhoades E. Allostery in a disordered protein: Oxidative modifications to α‐Synuclein act distally to regulate membrane binding. J Am Chem Soc. 2011;18:7152–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tripathi TA. Master regulator of α‐Synuclein aggregation. ACS Chem Nerosci. 2020;11(10):1376–1378. [DOI] [PubMed] [Google Scholar]
- 33. Uversky VN, Yamin G, Munishkina LA, et al. Effects of nitration on the structure and aggregation of α‐Synuclein. Mol Brain Res. 2005;134(1):84–102. [DOI] [PubMed] [Google Scholar]
- 34. Zhao K, Lim YJ, Liu Z, et al. Parkinson's disease‐related phosphorylation at Tyr39 rearranges α‐Synuclein amyloid fibril structure revealed by Cryo‐EM. Proc Natl Acad Sci U S A. 2020;117(33):20305–20315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lamberto GR, Binolfi A, Orcellet ML, et al. Structural and mechanistic basis behind the inhibitory interaction of PcTS on α‐Synuclein amyloid fibril formation. Proc Natl Acad Sci U S A. 2009;106(50):21057–21062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lamberto GR, Torres‐Monserrat V, Bertoncini CW, et al. Toward the discovery of effective polycyclic inhibitors of α‐Synuclein amyloid assembly. J Biol Chem. 2011;286(37):32036–32044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Palomino‐Hernandez O, Buratti FA, Sacco PS, Rossetti G, Carloni P, Fernandez CO. Role of Tyr‐39 for the structural features of α‐Synuclein and for the interaction with a strong modulator of its amyloid assembly. Int J Mol Sci. 2020;21(14):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lázaro DF, Rodrigues EF, Langohr R, et al. Systematic comparison of the effects of alpha‐Synuclein mutations on its Oligomerization and aggregation. PLoS Genet. 2014;10(11):e1004741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Karpinar DP, Balija MBG, Kügler S, et al. Pre‐Fibrillar α‐Synuclein variants with impaired Β‐structure increase neurotoxicity in Parkinson's disease models. EMBO J. 2009;28(20):3256–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bodner CR, Maltsev AS, Dobson CM, Bax A. Differential phospholipid binding of α‐synuclein variants implicated in Parkinson's disease revealed by solution NMR spectroscopy. Biochemistry. 2010;49:862–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bartels T, Ahlstrom LS, Leftin A, et al. The N‐terminus of the intrinsically disordered protein α‐synuclein triggers membrane binding and helix folding. Biophys J. 2010;99:2116–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fonseca‐Ornelas L, Eisbach SE, Paulat M, et al. Small molecule‐mediated stabilization of vesicle‐associated helical α‐Synuclein inhibits pathogenic Misfolding and aggregation. Nat Commun. 2014;5:5857. [DOI] [PubMed] [Google Scholar]
- 43. Man WK, Tahirbegi B, Vrettas MD, et al. The docking of synaptic vesicles on the presynaptic membrane induced by α‐Synuclein is modulated by lipid composition. Nat Commun. 2021;12(1):927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ysselstein D, Joshi M, Mishra V, et al. Effects of impaired membrane interactions on α‐Synuclein aggregation and neurotoxicity. Neurobiol Dis. 2015;79:150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fantini J, Yahi N. Molecular basis for the glycosphingolipid‐binding specificity of α‐Synuclein: Key role of tyrosine 39 in membrane insertion. J Mol Biol. 2011;408(4):654–669. [DOI] [PubMed] [Google Scholar]
- 46. Park JY, Kim KS, Lee SB, et al. On the mechanism of internalization of α‐Synuclein into microglia: Roles of ganglioside GM1 and lipid raft. J Neurochem. 2009;110(1):400–411. [DOI] [PubMed] [Google Scholar]
- 47. Fusco G, Chen SW, Williamson PTF, et al. Structural basis of membrane disruption and cellular toxicity by A‐Synuclein oligomers. Science. 2017;358(6369):1440–1443. [DOI] [PubMed] [Google Scholar]
- 48. Masaracchia C, Hnida M, Gerhardt E, et al. Membrane binding, internalization, and sorting of alpha‐Synuclein in the cell. Acta Neuropathol Commun. 2018;6(1):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sciacca MF, Lolicato F, Tempra C, et al. Lipid‐chaperone hypothesis: A common molecular mechanism of membrane disruption by intrinsically disordered proteins. ACS Chem Nerosci. 2020;11(24):4336–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miotto MC, Rodriguez EE, Valiente‐Gabioud AA, et al. Site‐specific copper‐catalyzed oxidation of α‐Synuclein: Tightening the link between metal binding and protein oxidative damage in Parkinson's disease. Inorg Chem. 2014;53(9):4350–4358. [DOI] [PubMed] [Google Scholar]
- 51. Binolfi A, Rodriguez EE, Valensin D, et al. Bioinorganic chemistry of Parkinson's disease: Structural determinants for the copper‐mediated amyloid formation of alpha‐Synuclein. Inorg Chem. 2010;49(22):10668–10679. [DOI] [PubMed] [Google Scholar]
- 52. Valiente‐Gabioud AA, Riedel D, Outeiro TF, Menacho‐Márquez MA, Griesinger C, Fernández CO. Binding modes of Phthalocyanines to amyloid β peptide and their effects on amyloid fibril formation. Biophys J. 2018;114(5):1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fernández CO, Hoyer W, Zweckstetter M, et al. NMR of α‐Synuclein‐polyamine complexes elucidates the mechanism and kinetics of induced aggregation. EMBO J. 2004;23(10):2039–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nečas D, Klapetek P. Gwyddion: An open‐source software for SPM data analysis. Centr Eur JPhys. 2012;10(1):181–188. [Google Scholar]
- 55. Maltsev AS, Ying J, Bax A. Impact of N‐terminal acetylation of α‐Synuclein on its random coil and lipid binding properties. Biochemistry. 2012;51(25):5004–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brenner S. The genetics of Caenorhabditis Elegans. Genetics. 1974;77(1):71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C.Elegans: Extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10(12):3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Final products of aggregation of αS and its Y39F, Y39L, and Y39A mutants. (A) FarUV CD spectra corresponding to the supernatant (red) and pellet (green) obtained from the final product of aggregation (t = 120 hr) of the studied αS species. Black curves correspond to the CD spectra of the monomeric states (t = 0 hr). (A) ThT fluorescence intensities corresponding to the final state of aggregation of the studied αS species (t = 120 hr) and their monomeric states (t = 0 hr)