

Abstract
Background
The time of survival in patients with amyotrophic lateral sclerosis (ALS) varies greatly, and the genetic factors that contribute to the survival of ALS are not well studied. There is a lack of a comprehensive study to elucidate the role of genetic factors in the survival of ALS.
Methods
The published studies were systematically searched and obtained from PubMed, EMBASE, and the Cochrane Library without any language restrictions from inception to Oct 27, 2021. A network meta-analysis for ALS causative/risk genes and a systematic review and pairwise meta-analysis for other genetic modifiers were conducted. The PROSPERO registration number: CRD42022311646.
Results
A total of 29,764 potentially relevant references were identified, and 71 papers were eligible for analysis based on pre-decided criteria, including 35 articles in network meta-analysis for 9 ALS causative/risk genes, 17 articles in pairwise meta-analysis for four genetic modifiers, and 19 articles described in the systematic review. Variants in three genes, including ATXN2 (HR: 3.6), C9orf72 (HR: 1.6), and FUS (HR:1.8), were associated with short survival of ALS, but such association was not identified in SOD1, TARDBP, TBK1, NEK1, UBQLN2, and CCNF. In addition, UNC13A rs12608932 CC genotype and ZNF521B rs2275294 C allele also caused a shorter survival of ALS; however, APOE ε4 allele and KIFAP3 rs1541160 did not be found to have any effect on the survival of ALS.
Conclusions
Our study summarized and contrasted evidence for prognostic genetic factors in ALS and would help to understand ALS pathogenesis and guide clinical trials and drug development.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12916-022-02411-3.
Keywords: Amyotrophic lateral sclerosis, Genes, Variants, Modifier, Survival
Background
Amyotrophic lateral sclerosis (ALS) is one of the most devastating neurodegenerative diseases, characterized by degeneration of the upper and lower motor neurons. It eventually results in progressive muscle atrophy and death in 3–5 years after disease onset [1]. About 5% to 10% of patients with ALS are present with a family history, called family ALS(FALS), while the remaining cases are sporadic (SALS) [2]. FALS always occurs due to a specific genetic mutation, but genetic causes also have been known to play an important role in SALS [1, 3]. The cumulative number of ALS-related genes has increased rapidly. To date, more than 130 genes/loci are reported to be associated with a risk of ALS [4]. Some of them also were reported to have a disease modification effect, which means they are always linked to a difference in the clinical phenotype of ALS, often survival time.
As we all know, aging, environmental and genetic factors play an essential role in the development of ALS. However, as a rare disease, we still don’t have an excellent strategy for preventing it from developing due to the limited knowledge of its etiology. Hence, more attention has been paid to the associated factors that affect the survival time. In our recent study, twenty-five non-genetic factors associated with ALS survival were identified, such as age at onset, onset site, the time between onset and diagnosis, et al. [5]. However, these non-genetic factors associated with ALS survival could be affected by confounding factors. Therefore, other unbiased methods exploring clinical outcomes are in the ascendant, such as the Mendelian Randomization study, which focuses on the actual causal effect on diseases or their phenotype by applying genetic variants. Till now, the genetic factors that contribute to the survival of ALS are not well studied and remain to be explored. Previous researches have reported that some potential loci may modify the survival of ALS, such as UNC13A rs12608932 and CAMTA1 rs2412208 [6–8]. In addition, the causative ALS genes (C9orf72, SOD1, FUS, TARDBP) might modify the disease course as well [9–13]. Yet, due to the limited sample size of patients with rare variants in ALS causative genes, the different genetic backgrounds, or other factors, there are inconsistencies in those results and a lack of a comprehensive review to elucidate the role of genetic factors in the survival of ALS. Consequently, our study tries to clarify the genetic factors that affect the survival of ALS by an integrated approach combining a network meta-analysis (NMA) on ALS causative/risk genes and a pairwise meta-analysis on other modified loci along with a systematic review.
Methods
Different genetic variants are considered as different interventions in this study, so we employed a NMA following the International Society for Pharmacoepidemiology and Outcomes Research (ISPOR) guidelines [14] and reported it using the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) extension for NMA [15]. In addition, the report of the pairwise meta-analysis for other modified loci was followed by the recommendations of the PRISMA (2009) guidelines [16]. The protocol for this study was registered with PROSPERO, registration number: CRD42022311646.
Search strategies and selection criteria
The published studies were systematically searched and obtained from PubMed, EMBASE, and the Cochrane Library without any language restrictions, by using the term: “(gene* OR geno* OR variant* OR mutation OR haplotyp* OR polymorphism* OR SNP OR Allel*) AND (prognosis* OR progress* OR survival OR outcome OR mortality OR death) AND ((amyotrophic lateral sclerosis) OR (motor neuron disease) OR (Lou Gehrig’s disease) OR (Gehrig Disease))”. Reference lists of full review articles were also reviewed to search for additional articles. All randomized clinical trials (RCTs), quasi-RCTs, and observational studies were eligible, but no RCTs were identified. The articles were updated to October 27, 2021.
According to previous survival research in ALS, survival was defined as the time between the onset of symptoms and noninvasive ventilation (NIV) for more than 23 hours per day, or tracheostomy or death [5, 17, 18]. The inclusion criteria are as follows: (1) assessed the association between present or absence with different genetic loci and survival time from the onset in patients with ALS; (2) had reported a hazard ratio (HR) and 95% confidential intervals (CI) for patients with genetic mutations or Kaplan-Meier plots from which we can estimate the HR and 95%CI; (3) abstract, reviews, letters without original study, secondary studies and studies that HR or 95%CI was unavailable in papers, excluded from the analysis; and (4) full papers published in English.
Data collection
First author’s name, year of publication or online, patients’ nation, number of patients, type of disease, diagnosis criteria, age at onset and gender distribution of patients, median survival time, and HR with their 95% CI were extracted. When HR was unavailable directly from the articles, the Kaplan-Meier curve was evaluated by Engauge Digitizer version 12.1, and HRs and 95%CI were estimated using Richard Steven’s excel workbook [19]. Data from multi-arm studies were extracted following the tutorial by B. S. Woods [20]. Extracted data from included studies by two independent reviewers to reduce bias and a third one verified the data to avoid repeat inclusion.
Appraisal of methodological quality
Only observational studies were included for analysis in this study, and their quality was appraised with the Newcastle-Ottawa Scale (NOS) [21]. As for publication bias, the assessment was conducted only when at least ten studies were available for the same factor by Begg’s test [22]. In addition, to overcome overestimation and mask funnel plot asymmetry induced by some multi-arm studies, we plotted data points corresponding to the study-specific basic parameters (different known ALS gene mutation comparisons with a common comparator). In each study, we used the control group (absence with known ALS gene mutation) as the common comparator, if this was unavailable, we used the same comparator (always, group with SOD1 mutation) against the remaining groups.
Synthesis of included studies
HR was used for each dichotomous outcome, and traditional pairwise meta-analyses were performed for studies, which directly compared different groups. And from known ALS causative/risk genes variants, outcome data were pooled using NMA models through the R 4.1.1 software. A network relationship diagram was drawn, among which parameters such as each node and line thickness respectively represent a certain gene’s variants and the researches sizes were considered from the included studies. The model fit was assessed using three criteria based on the deviance and node-based residuals. We evaluated the inconsistency, which means the difference between the pooled direct and indirect evidence of a particular comparison, using inconsistency factors based on a modified back-calculation approach [23]. In addition, we performed the rankogram plots to show the probability of each genetic mutation [24]. The remaining modification loci were pool analyzed under meta-analysis by random-effect model if analyzed in more than two studies (≥3 studies); otherwise they were conducted for systematic review if analyzed in less than three studies (< 3 studies) (including SMN2 deletion, SMN1 and SMN2 copy numbers, CX3CR1 V249I, CX3CR1 T280M, haplotype in CX3CR1, ABCC8 rs4148646, KCNJ11 rs5219, LXRs rs2279238, LXRs rs7120118, LXRs rs35463555, LXRs rs2695121, PRGN rs9897526, PRGN rs34424835, haplotype in PRGN, HTR2B rs10199752, STMN2 CA repeat, BDNF C270T, C7 gene rs3792646, PON1 rs854560, PON1 rs662, NIPA1 polyalanine repeat expansions, SLC11A2 rs407135, CAMTA1 rs2412208, GSTP1 rs1695, CNTF, HLA-DRA or HLA-DRB5 rs9268856, rs4623951, EPHA4 rs6436254, and IDE rs139550538). All computations were conducted on R (V4.1.1) package “gemtc,” “rjags,” and STATA/MP 16.0.
Results
Literature results
We identified 29,764 potentially relevant references from PubMed, EMBASE, and Cochrane Library and three additional records from reference lists (Fig. 1). Of these records, only 71 articles met the inclusion criteria. Finally, we included 35 articles using network meta-analysis for variants in nine ALS causative/risk genes (Additional file 1: Table S1) [9–13, 25–54], 17 articles using pairwise meta-analysis for four modification loci in ALS-related genes (Additional file 1: Table S2) [6–8, 55–68], and 19 articles using systematic review according to pre-decided criteria (Additional file 1: Table S3) [7, 69–87]. The characteristics of the 71 included trials are summarized in Additional file 1: Table S1-S3 and the quality of 52 articles conducted by network meta-analysis and pairwise meta-analysis was shown in Additional file 1: Table S4.
Fig. 1.

Flow chart for literature selection. ALS, amyotrophic lateral sclerosis
Variants in nine ALS causative/risk genes by NMA
A total of 35 articles including 37 trials with nine known ALS causative/risk genes were involved in this network meta-analysis, which is shown in Fig. 2. C9orf72 was the most frequently investigated regimen with 40 comparisons (Additional file 1: Table S1). It was found that ALS patients carrying C9orf72 repeat expansion would have a poor prognosis compared to those without known ALS causative/risk genes variants and the HR was 1.6 (95%CI:1.4–1.9). In addition, patients with other two genes variants, ATXN2 and FUS, presented a short survival time compared to those without known ALS causative genes variants (HR:3.6 or 1.8, respectively). However, SOD1, TARDBP, TBK1, NEK1, UBQLN2, and CCNF did not affect the survival of ALS. The detailed features for each gene are shown in Table 1. Compared to C9orf72 repeats expansion, patients presented with SOD1 or TARDBP variants or without genetic variants seem to possess a better prognosis. However, no difference between C9orf72 repeats expansion and FUS variants and ATXN2 polyQ were identified. The detailed results are illustrated in Fig. 3.
Fig. 2.
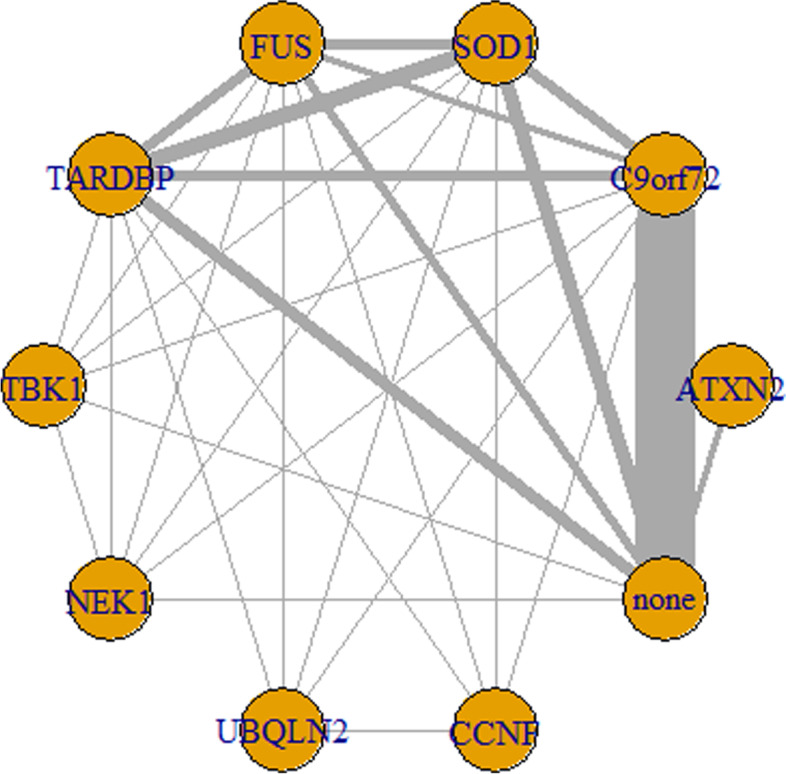
Network of analyzed comparisons. Each circle corresponds to a regimen included in the analysis. “none” means that there were no known ALS genetic variants, and the others mean that there were corresponding genetic variants shown in the circle. Each line represents comparisons between regimens, with the thickness corresponding to the number of within-trial comparisons
Table 1.
The characteristics of genes included in network meta-analysis
| Gene | Full name | Location | Category in ALSoD | Proportion of patients with ALS | Common mutations in ALS | Possible pathogenesis | Phenotype | Total HR (95% CI)a |
|---|---|---|---|---|---|---|---|---|
| ATXN2 | Ataxin 2 | 12q24.12 | Clinical modifier | <1% in northern European ancestry [88, 89], about 2% in French/French Canadian [90], about 2.5% in Italian [91], < 1% in Turkish [92] and in Chinese [93]. | CAG repeat size ≥ 31 | neurotoxicity caused by prion-like self-assembly and propagation of mutant or misfold protein [1, 94] | Mainly spinal onset | 3.6 (1.1, 12) |
| C9orf72 | C9orf72-SMCR8 complex subunit | 9p21.2 | Definitive ALS gene | FALS 33.7%, SALS 5.1% in European, FALS 2.3%, SALS 0.3% in Asian [95]. | GGGGCC hexanucleotide repeat expansion usually > 30, even > 1000 | RNA foci-mediated toxicity; dipeptide repeat protein (DPR)-mediated toxicity; and/or reduced levels of C9orf72 protein [96] | Mainly spinal onset, always with FTD | 1.6 (1.4, 1.9) |
| SOD1 | Superoxide dismutase 1 | 21q22.11 | Definitive ALS gene | FALS 14.8%, SALS 1.2% in European. FALS 30.0%, SALS 1.5% in Asian [95]. | A4V, I113T, G41A, A4T, G37R, D90A, D101N, E100G, A140A, D76Y, E21G, H46R, I149T, L106V, L144F, et al | autophagy, mitophagy, and neuroinflammation by mutant or misfold protein aggregates due to decreased SOD1 enzymatic activity [1, 97] | Mainly spinal onset | 0.85 (0.70, 1.0) |
| FUS | Fused in sarcoma | 16p11.2 | Definitive ALS gene | FALS 2.8%, SALS 0.3% in European. FALS 6.4%, SALS 0.9% in Asian [95]. | P525L, R495X, R521H, R521C, G504WfsX12, et al | a toxic gain-of-function due to FUS aggregation and/or a loss-of-function resulting from cytoplasmic mis-localization of FUS and subsequent loss of nuclear function [1, 98] | Mainly spinal onset, early-onset | 1.8 (1.1, 2.9) |
| TARDBP | TAR DNA binding protein | 1p36.22 | Definitive ALS gene | FALS 4.2%, SALS 0.8% in European. FALS 1.5%, SALS 0.2% in Asian [95]. | A382T, A382T, G294V, G295S, G348C, M337V, et al | neurotoxicity and motor neuron caused by degeneration dysregulation of transposable elements due to TDP-43 loss-of-function [1, 99] | Mainly spinal onset | 0.77 (0.61, 1.0) |
| TBK1 | TANK binding kinase 1 | 12q14.2 | Definitive ALS gene | About 2.8% of ALS patients [100] | T79del, G272_T331del, 690-713del, et al | autophagy, mitophagy, and neuroinflammation due to decrease Tbk1’s kinase activity [101] | Mainly spinal onset | 2.4 (0.39, 14.0) |
| NEK1 | NIMA related kinase 1 | 4q33 | definitive ALS gene | About 3% of ALS patients [102] | M545T, R261H, et al | impaired function for DNA damage repair [103] | NA | 1.2 (0.49, 2.9) |
| UBQLN2 | Ubiquilin 2 | Xp11.21 | Definitive ALS gene | rare [104] | P497H, P506S, T487T, T487I, et al | autophagy, mitophagy, and neuroinflammation by protein blocked protein degradation [105] | Mainly presence with FTD | 0.98 (0.40, 2.5) |
| CCNF | Cyclin F | 16p13.3 | Strong evidence | rare [106] | A74T, E528Q, E624K et al | neurotoxicity and motor neuron degeneration by defective protein degradation systems and the pathological accumulation of a protein involved in RNA processing and metabolism [107] | NA | 2.9 (0.19, 45.0) |
ALSoD Amyotrophic Lateral Sclerosis online Database [4], NA not Available
aTotal HR (95%CI) were calculated in this study
Fig. 3.

Forest plots of HR for pairwise comparison and rank probabilities for each regimen included in network meta-analysis. a Forest plot of HR for each genetic mutation when compared to absence with genetic mutation. b Forest plot of HR for absence with genetic mutation and other genetic mutation compared to C9orf72 repeat expansion. c The result of pairwise comparison among these genes. Bold: significative. d Rank probabilities among those genes for ALS survival based on the network meta-analysis. CI, confidence interval
The rankogram plots are shown in Fig. 3. According to the surface under the cumulative ranking curve (SUCRA), ATXN2 polyQ repeats (≥31) had the largest probability of being the worst genetic variants (SUCRA = 86.3%, data not shown). TBK1(70.0%), FUS (69.5%), CCNF (67.7%), and C9orf72 (63.4%) had a similar probability of being the worst. The result of pairwise comparison among these genes is shown in Fig. 3.
Furthermore, there is no significant difference between direct and indirect meta-analysis (Additional file 1: Fig. S1) and the heterogeneity between studies is also acceptable (Additional file 1: Fig. S2-S12), indicating the results of NMA are reliable. As for publication bias, we did not find significant funnel plot asymmetry in studies reporting C9orf72 expansion in patients with ALS compared to those without ALS-related mutation (Additional file 1: Fig. S13).
Four modification loci in ALS-related genes by pairwise meta-analysis
Four modification loci in ALS-related genes, APOE ε4 allele, KIFAP3 rs1541160, UNC13A rs12608932, and ZNF512B rs2275294, were available for pairwise meta-analysis (Additional file 1: Table S2). In this study, UNC13A rs12608932 CC (recessive model) and ZNF512B rs2275294 CC+CT (dominant model) could accelerate the death of ALS patients (HR 1.18 and 1.97, respectively, Fig. 4). However, APOE ε4 allele, KIFAP3 rs1541160 CC or CT (additive model) did not show any modificatory effect on ALS survival (Fig. 4). And no obvious heterogeneity was found.
Fig. 4.

Forest plots of HR for modification loci included in pairwise meta-analysis. a Forest plot of HR for APOE ε4. b Forest plot of HR for KIFAP3 rs1541160 CC. c Forest plot of HR for KIFAP3 rs1541160 CT. d Forest plot of HR for UNC13A rs12608932 CC. e Forest plot of HR for ZNF512B rs2275294 CC+TT. HR, hazard ratio; CI, confidence interval
Other loci in ALS-related genes by systematic review
The other loci in ALS-related genes, which were not suitable for pool analysis, were reviewed systematically and shown in Additional file 1: Table S3. We found the minor allele carriers of CX3CR1 V249I, KCNJ11 rs5219, LXRs rs2695121, PRGN rs34424835, HTR2B rs10199752, PON1 rs662, SLC11A2 rs407135, CAMTA1 s2412208, and IDE rs139550538 might have shorter survival than that without minor allele carriers, but ABCC8 rs4148646 GG, KCNJ11 rs5219 TT, HTR2B rs10199752 AA, and C7 gene rs3792646 AA might be related to more prolonged survival. Additionally, SMN1 and SMN2 copy numbers, and the remaining genes on display did not show any modificatory effect on ALS survival. However, there were not enough articles for meta-analysis.
Discussion
Genetic factors play a pivotal role in the pathogenesis and phenotypic modification of ALS. How the genetic factors affected the survival of ALS was largely unknown yet, especially for variants in the ALS causative genes, in consideration of the limited sample with genetic mutation. This study is the first systematic analysis for the effect of comprehensive genetic factors on the survival of ALS. Using NMA and pairwise meta-analysis, we found three genes, including ATXN2 polyQ, C9orf72 repeats, and FUS variants, and two genetic modifiers, ZNF521B rs2275294 C allele and UNC13A rs12608932 CC genotype, were associated with short survival of ALS.
This current study provided robust evidence that genetic factors affected the survival of ALS. For NMA, among the nine ALS causative/risk genes involved, we found C9orf72, ATXN2, and FUS were associated with much shorter survival. Ataxin 2 is a protein-coding gene and the N-terminal region of this protein contains a polyglutamine tract of 14-31 residues that can be expanded in the pathogenic state to 32-200 residues. The long trinucleotide repeats expansions (≥36) in ATXN2 have been known to be related to spinocerebellar ataxia type 2 (SCA2) [108], but the intermediate length expansions (27–33 repeats) were also discovered to increase susceptibility to ALS [88], not only in FALS but also in SALS [109]. Meanwhile, another study also reported that ATXN2 ployQ may render C9orf72 repeat expansion carriers more susceptible to ALS [110]. It was also associated with a more severe phenotype and a worse prognosis of ALS, causing a significantly shorter survival (1.2 vs. 4.2 years) [26]. In this study, we found it had the most significant probability of becoming the genetic background with the worst prognosis for ALS (HR=3.6). Ataxin-2 plays a critical role in the normal physiological functions of cells, including RNA processing, receptor endocytosis, the formation of stress granules, and induction of aberrant TDP-43 cleavage [93]. In ALS, it altered protein homeostasis and RNA metabolism, leading to neurotoxicity [1, 94]. Motor neurons in this condition may degenerate faster than those without ATXN2 ployQ expansion or with other genetic variants. Therefore, several gene therapies targeted for ATXN2 used in cell or ALS animal models to prevent or delay its progression were reported [111].
Thanks to advances in the next-generation technology, C9orf72 GGGGCC (G4C2) hexanucleotide repeat expansion was identified as the most common mutation in Europe ALS and the second most common mutation in Asia ALS [95]. Hence, whether the clinical studies are based on genetics and biomarkers, or the basic research based on pathogenies mechanisms or therapeutics, C9orf72 was the most studied gene in the ALS research field in the last ten years. Although healthy individuals can have from 2 to 25 G4C2 repeats, ALS or FTD patients harbor hundreds to even thousands of these repeats [112, 113]. Furthermore, it is involved with RNA foci-mediated toxicity, dipeptide repeat protein (DPR)-mediated toxicity and/or loss-of-function due to reduced levels of C9orf72 protein [96]. Here, after synthesizing a total of 40 comparisons, we found C9orf72 expansion was also associated with shorter survival (HR:1.6, 95%CI [1.4, 1.9]). In addition, cases with C9orf72 expansion may have a more rapid rate of cognitive decline and a higher risk of developing FTD [114], which were predictors for poor prognosis for ALS as well [5]. So far, gene therapy targeting C9orf72 has been carried out in preclinical studies [115, 116], and these methods are promising approaches for future in vivo studies.
SOD1, FUS, and TARDBP, the other three common ALS causative genes, usually present as missense variants. When all the reported mutation sites were analyzed together, we found only FUS would shorten the survival time of ALS. However, we could not ignore that different variants on these genes have different effects on ALS survival. Based on previous genetic studies, we summarized the characteristics of several common variants in these genes in Additional file 1: Table S5 [117–119]. The most significant heterogeneity may exist in SOD1 with more than 150 ALS-associated SOD1 variants described. SOD1 was believed to cause ALS through toxic gain of function caused by aggregation of misfolded SOD1 [1, 97]. Variants with different influences on survival are usually located in different domains and are more likely to have a strikingly different effect on protein structure and function. For example, SOD1A4V and SOD1H46R associated, with the shorter and significantly longer durations, respectively, were identified not only in Caucasians but also in Asians. Our recent cohort study has yielded similar results [13]. Thus, the results obtained in the current study may be due to a mixture of different variants in SOD1. There, it does not mean that SOD1 variants do not affect the survival of ALS, for individuals, specific variants should be identified. Further, we found a similar but slight difference from a meta-analysis of published FUS-ALS cases [119]. It was found to have a different duration from onset to the severe event among FUSR521, FUSP525L, frameshift/truncation, and the remaining variants in FUS [119]. However, there did not seem to be a mutation in FUS reported that would prolong the survival of ALS (Additional file 1: Table S5) [11–13, 119]. Taken together, it appears that genetic testing of FUS is indicated in patients with shorter disease duration. TARDBP gene encoded TDP-43, a DNA-/RNA-binding protein normally localized to the nucleus [120], which is also the most widespread and pathologic hallmark in the ALS/FTD spectrum [94]. Although TARDBP did not get a definitive positive result in this study, it seems tempting to suggest that TARDBP-ALS cases might have a better prognosis because the HR was 0.77 and 95% CI was 0.61 to 1. Moreover, there was no shortage of reports of TARDBP-ALS cases that have survived for decades [12]. Similarly, there are differences in the impact of different variants on the survival of ALS.
However. we did not find TBK1, NEK1, UBQLN2, and CCNF were associated with survival of ALS either. Maybe there were too few studies or limited patients with variants in these genes to reach the significant difference. The features for these genes were also shown in Table 1. It must be emphasized that those genes included in NMA have been regarded as ALS risk genes with strong evidence or above, but some of the variants in them might not be of demonstrated pathogenetic significance. For instance, most pathogenic variants identified in TBK1 are concentrated within the kinase and the coiled-coiled domains [121], which usually were loss-of-function types, while for some missense variants (such as p.K291E, p.I305T, p.L306I, p.H322Y, p.T3221I, p.R444Q, and p.A535T) may need more functional studies to research their pathogenicity [122]. And NEK1 missense variants confer risk of developing ALS is also a matter of debate [123], the similar condition exists in other genes as well. While repeat expansions in genes are more susceptible to reaching the statistical significance threshold. This limit due to our methods may have an impact on the results.
In addition, four genetic factors were studied by pairwise meta-analysis. Consistent with most previous studies [6, 8, 59, 60, 124], UNC13A rs12608932 CC was a predictor of poor survival in ALS. The rs12608932 in UNC13A is associated with ALS/FTD susceptibility [125] and may indicate poorer cognitive functioning, higher rates of behavioral impairment, and higher rates of FTD [60]. A previous meta-analysis for a single factor has already shown this effect [126]. Therefore, this genotype may function as a prognostic indicator and could be used to define patient endophenotypes in clinical trials. Although the mechanisms by which the CC genotype of rs12608932 in UNC13A significantly decreased the survival time of ALS patients was not entirely clear, the very recent studies found UNC13A variants exacerbate the effects of reduced TDP-43 function and TDP-43 can repress a cryptic exon-splicing event in UNC13A [127, 128]. Hence, it may provide a promising therapeutic target for TDP-43 proteopathies. We also detected that the ZNF521B rs2275294 C allele indicates a poor prognosis of ALS, meanwhile a meta-analysis suggested that the ZNF512B rs2275294 polymorphism is also associated with ALS risk [129]. The ZNF512B gene encodes a protein of 893 amino acids, which is expressed in the brain and spinal cord. Upregulation of ZNF512B activates TGF-β/Smad signaling, while downregulation inhibits this pathway and rs2275294C may reduce this neuroprotective TGF-β/Smad signaling [130]. Several other genes (such as EPHA4, CAMTA1, and HFE) may be associated with the prognosis of ALS, however, they cannot be performed by meta-analysis due to the limited studies.
It is controversial whether APOE polymorphism can change the course of ALS [56, 57]. However, APOE ε4 allele does not modify the clinical course of ALS as well under meta-analysis, so does KIFAP3 rs1541160. As for the remaining genetic factors, although SMN genes have been reported to be associated with ALS survival [69, 131], This conclusion was not supported based on the results of a recent and large study including new and all previously reported results on SMN1 and SMN2 copy number variation in ALS [87]. .The genetic mechanisms of SMN1 and SMN2 are implicated in motor neuron death in spinal muscular atrophy, but SMN expression levels in the physiological range may not modify the progression of ALS [87]. Additionally, a very recent large GWAS also looked at the role of some genetic factors mentioned above on modifying ALS survival and found the effect of common genetic risk factors for ALS susceptibility on disease progression was limited [132], suggesting the influence of a single SNP or gene on ALS survival cannot be magnified.
Although we find some potential genetic factors that affected the survival of ALS, these findings should be interpreted with caution, given some limitations. First and foremost, different variants in the same gene might have different effects on survival, it may result in information missing when a gene was analyzed as a whole factor. What is more, as we discussed above, whether some of the variants in these genes are ALS-associated mutations remains in doubt. Therefore, the results were not absolutely correct. Besides, some studies did not report the HR directly, when we get HR from the survival curve, there may be some slight gap with the accurate results. Some differences in follow-up time, the included population, and the definition of outcomes may lead to publication bias and the non-genetic factors related to ALS survival may also play a role [5]. Moreover, the lack of commonality of prognostic factors investigated in different cox PH models is also a limitation, and some studies are single-factor analyses and did not adjust for confounding factors. In addition, the small number of prospective studies was also a limitation. For example, ZNF521B rs2275294 were reported in only three articles, the results should be explained with caution. Therefore, more high-quality prospective studies are warranted. Finally, the rules of the review methodology of its restriction to articles published in English, and the low specificity of the search strategy, increase the risk of missing relevant studies.
Conclusions
The present meta-analysis summarized and contrasted evidence for genetic prognostic factors in patients with ALS and will help to understand ALS genetics. Genetic prognostic factors deserve attention and careful consideration as the field moves forward to combat and prevent this devastating disease.
Supplementary Information
Additional file 1: Table S1. Characteristics of the articles included in network meta-analysis for variants in ALS causative genes. Table S2. Characteristics of the articles included in pairwise meta-analysis for other modification loci. Table S3. Characteristics of the articles included in systematic review for other modification loci. Table S4. The quality of articles included in network meta-analysis and pairwise meta-analysis by NOS. Table S5. The feathers for different variants in SOD1, FUS, TARDBP. Figure S1. Forest plot for inconsistency test in network meta-analysis. Figure S2-S12. Heterogeneity analysis n network meta-analysis. Figure S13. publication bias for studies reporting C9orf72 expansion in patients with ALS compared to those without ALS-related mutation.
Acknowledgements
The authors would like to thank the ALS patients in the included studies and all the researchers of them.
Abbreviations
- ALS
Amyotrophic lateral sclerosis
- CI
Confidential intervals
- DPR
Dipeptide repeat protein
- FALS
Family amyotrophic lateral sclerosis
- FTD
Frontotemporal dementia
- GWAS
Genome-Wide Association Study
- HR
Hazard ratio
- ISPOR
International Society for Pharmacoepidemiology and Outcomes Research
- NIV
Noninvasive ventilation
- NMA
Network meta-analysis
- NOS
Newcastle-Ottawa Scale
- PH
Proportional hazard
- PRISMA
Preferred Reporting Items for Systematic Reviews and Meta-Analyses
- RCT
Randomized clinical trial
- SALS
Sporadic amyotrophic lateral sclerosis
- SCA2
Spinocerebellar ataxia type 2
- SNP
Single nucleotide polymorphism
- SUCRA
Surface under the cumulative ranking curve
Authors’ contributions
WMS and YPC conceived and designed the study. WMS, YPC, XJG, and ZJ selected the articles and extracted and cross-checked the data. WMS, YPC, XJG, and QQD contributed to the statistical analysis. WMS and YPC wrote the first draft of the manuscript. WMS, XG, YPC, and HFS revised and discussed the final edition. The authors read and approved the final manuscript.
Funding
This study was supported the National Natural Science Fund of China (Grant No. 81971188), the 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (Grant No. 2019HXFH046), and the Science and Technology Bureau Fund of Sichuan Province (No. 2019YFS0216 and No.2022ZDZX0023).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files. Effect size and study details were extracted from the original papers, which are available in the public domain.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Brown RH, Al-Chalabi A. Amyotrophic Lateral Sclerosis. N Engl J Med. 2017;377(2):162–172. doi: 10.1056/NEJMra1603471. [DOI] [PubMed] [Google Scholar]
- 2.Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D, Orozco G, Chinea A. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int. 2015;6(1):171. doi: 10.4103/2152-7806.169561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byrne S, Walsh C, Lynch C, Bede P, Elamin M, Kenna K, McLaughlin R, Hardiman O. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82(6):623–627. doi: 10.1136/jnnp.2010.224501. [DOI] [PubMed] [Google Scholar]
- 4.Abel O, Powell JF, Andersen PM, Al-Chalabi A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum Mutat. 2012;33(9):1345–1351. doi: 10.1002/humu.22157. [DOI] [PubMed] [Google Scholar]
- 5.Su WM, Cheng YF, Jiang Z, Duan QQ, Yang TM, Shang HF, Chen YP. Predictors of survival in patients with amyotrophic lateral sclerosis: A large meta-analysis. EBioMedicine. 2021;74:103732. doi: 10.1016/j.ebiom.2021.103732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diekstra FP, van Vught PW, van Rheenen W, Koppers M, Pasterkamp RJ, van Es MA, Schelhaas HJ, de Visser M, Robberecht W, Van Damme P, et al. UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33(3):630.e633–630.e638. doi: 10.1016/j.neurobiolaging.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 7.Fogh I, Lin K, Tiloca C, Rooney J, Gellera C, Diekstra FP, Ratti A, Shatunov A, van Es MA, Proitsi P, et al. Association of a Locus in the CAMTA1 Gene With Survival in Patients With Sporadic Amyotrophic Lateral Sclerosis. JAMA Neurol. 2016;73(7):812–820. doi: 10.1001/jamaneurol.2016.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiò A, Mora G, Restagno G, Brunetti M, Ossola I, Barberis M, Ferrucci L, Canosa A, Manera U, Moglia C, et al. UNC13A influences survival in Italian amyotrophic lateral sclerosis patients: a population-based study. Neurobiol Aging. 2013;34(1):357.e351–357.e355. doi: 10.1016/j.neurobiolaging.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabatelli M, Conforti FL, Zollino M, Mora G, Monsurrò MR, Volanti P, Marinou K, Salvi F, Corbo M, Giannini F, et al. C9ORF72 hexanucleotide repeat expansions in the Italian sporadic ALS population. Neurobiol Aging. 2012;33(8):1848.e1815–1848.e1820. doi: 10.1016/j.neurobiolaging.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miltenberger-Miltenyi G, Conceição VA, Gromicho M, Pronto-Laborinho AC, Pinto S, Andersen PM, de Carvalho M. C9orf72 expansion is associated with accelerated decline of respiratory function and decreased survival in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2019;90(1):118–120. doi: 10.1136/jnnp-2018-318032. [DOI] [PubMed] [Google Scholar]
- 11.Hübers A, Just W, Rosenbohm A, Müller K, Marroquin N, Goebel I, Högel J, Thiele H, Altmüller J, Nürnberg P, et al. De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. Neurobiol Aging. 2015;36(11):3117.e3111–3117.e3116. doi: 10.1016/j.neurobiolaging.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 12.Corcia P, Valdmanis P, Millecamps S, Lionnet C, Blasco H, Mouzat K, Daoud H, Belzil V, Morales R, Pageot N, et al. Phenotype and genotype analysis in amyotrophic lateral sclerosis with TARDBP gene mutations. Neurology. 2012;78(19):1519–1526. doi: 10.1212/WNL.0b013e3182553c88. [DOI] [PubMed] [Google Scholar]
- 13.Chen YP, Yu SH, Wei QQ, Cao B, Gu XJ, Chen XP, et al. Role of genetics in amyotrophic lateral sclerosis: a large cohort study in Chinese mainland population. J Med Genet. 2021. 10.1136/jmedgenet-2021-107965. [DOI] [PMC free article] [PubMed]
- 14.Jansen JP, Trikalinos T, Cappelleri JC, Daw J, Andes S, Eldessouki R, Salanti G. Indirect treatment comparison/network meta-analysis study questionnaire to assess relevance and credibility to inform health care decision making: an ISPOR-AMCP-NPC Good Practice Task Force report. Value Health. 2014;17(2):157–173. doi: 10.1016/j.jval.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 15.Hutton B, Salanti G, Caldwell DM, Chaimani A, Schmid CH, Cameron C, Ioannidis JP, Straus S, Thorlund K, Jansen JP, et al. The PRISMA extension statement for reporting of systematic reviews incorporating network meta-analyses of health care interventions: checklist and explanations. Ann Intern Med. 2015;162(11):777–784. doi: 10.7326/M14-2385. [DOI] [PubMed] [Google Scholar]
- 16.Moher D, Liberati A, Tetzlaff J, Altman DG, Group P Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. 2009;339:b2535. doi: 10.1136/bmj.b2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cudkowicz ME, van den Berg LH, Shefner JM, Mitsumoto H, Mora JS, Ludolph A, Hardiman O, Bozik ME, Ingersoll EW, Archibald D, et al. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): a randomised, double-blind, phase 3 trial. Lancet Neurol. 2013;12(11):1059–1067. doi: 10.1016/S1474-4422(13)70221-7. [DOI] [PubMed] [Google Scholar]
- 18.Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012;3:CD001447. [DOI] [PMC free article] [PubMed]
- 19.Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time-to-event data into meta-analysis. Trials. 2007;8:16. doi: 10.1186/1745-6215-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woods BS, Hawkins N, Scott DA. Network meta-analysis on the log-hazard scale, combining count and hazard ratio statistics accounting for multi-arm trials: a tutorial. BMC Med Res Methodol. 2010;10:54. doi: 10.1186/1471-2288-10-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol. 2010;25(9):603–605. doi: 10.1007/s10654-010-9491-z. [DOI] [PubMed] [Google Scholar]
- 22.Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics. 1994;50(4):1088–1101. doi: 10.2307/2533446. [DOI] [PubMed] [Google Scholar]
- 23.Dias S, Welton NJ, Caldwell DM, Ades AE. Checking consistency in mixed treatment comparison meta-analysis. Stat Med. 2010;29(7-8):932–944. doi: 10.1002/sim.3767. [DOI] [PubMed] [Google Scholar]
- 24.Salanti G, Ades AE, Ioannidis JP. Graphical methods and numerical summaries for presenting results from multiple-treatment meta-analysis: an overview and tutorial. J Clin Epidemiol. 2011;64(2):163–171. doi: 10.1016/j.jclinepi.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 25.Borghero G, Pugliatti M, Marrosu F, Marrosu MG, Murru MR, Floris G, Cannas A, Parish LD, Cau TB, Loi D, et al. ATXN2 is a modifier of phenotype in ALS patients of Sardinian ancestry. Neurobiol Aging. 2015;36(10):2906.e2901–2906.e2905. doi: 10.1016/j.neurobiolaging.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiò A, Calvo A, Moglia C, Canosa A, Brunetti M, Barberis M, Restagno G, Conte A, Bisogni G, Marangi G, et al. ATXN2 polyQ intermediate repeats are a modifier of ALS survival. Neurology. 2015;84(3):251–258. doi: 10.1212/WNL.0000000000001159. [DOI] [PubMed] [Google Scholar]
- 27.Byrne S, Elamin M, Bede P, Shatunov A, Walsh C, Corr B, Heverin M, Jordan N, Kenna K, Lynch C, et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. 2012;11(3):232–240. doi: 10.1016/S1474-4422(12)70014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ratti A, Corrado L, Castellotti B, Del Bo R, Fogh I, Cereda C, Tiloca C, D'Ascenzo C, Bagarotti A, Pensato V, et al. C9ORF72 repeat expansion in a large Italian ALS cohort: evidence of a founder effect. Neurobiol Aging. 2012;33(10):2528.e2527–2528.e2514. doi: 10.1016/j.neurobiolaging.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 29.van Rheenen W, van Blitterswijk M, Huisman MH, Vlam L, van Doormaal PT, Seelen M, Medic J, Dooijes D, de Visser M, van der Kooi AJ, et al. Hexanucleotide repeat expansions in C9ORF72 in the spectrum of motor neuron diseases. Neurology. 2012;79(9):878–882. doi: 10.1212/WNL.0b013e3182661d14. [DOI] [PubMed] [Google Scholar]
- 30.Millecamps S, Boillee S, Le Ber I, Seilhean D, Teyssou E, Giraudeau M, Moigneu C, Vandenberghe N, Danel-Brunaud V, Corcia P, et al. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J Med Genet. 2012;49(4):258–263. doi: 10.1136/jmedgenet-2011-100699. [DOI] [PubMed] [Google Scholar]
- 31.Debray S, Race V, Crabbé V, Herdewyn S, Matthijs G, Goris A, Dubois B, Thijs V, Robberecht W, Van Damme P. Frequency of C9orf72 repeat expansions in amyotrophic lateral sclerosis: a Belgian cohort study. Neurobiol Aging. 2013;34(12):2890.e2897–2890.e2812. doi: 10.1016/j.neurobiolaging.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 32.García-Redondo A, Dols-Icardo O, Rojas-García R, Esteban-Pérez J, Cordero-Vázquez P, Muñoz-Blanco JL, Catalina I, González-Muñoz M, Varona L, Sarasola E, et al. Analysis of the C9orf72 gene in patients with amyotrophic lateral sclerosis in Spain and different populations worldwide. Hum Mutat. 2013;34(1):79–82. doi: 10.1002/humu.22211. [DOI] [PubMed] [Google Scholar]
- 33.Irwin DJ, McMillan CT, Brettschneider J, Libon DJ, Powers J, Rascovsky K, Toledo JB, Boller A, Bekisz J, Chandrasekaran K, et al. Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2013;84(2):163–169. doi: 10.1136/jnnp-2012-303507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Laere K, Vanhee A, Verschueren J, De Coster L, Driesen A, Dupont P, Robberecht W, Van Damme P. Value of 18fluorodeoxyglucose-positron-emission tomography in amyotrophic lateral sclerosis: a prospective study. JAMA Neurol. 2014;71(5):553–561. doi: 10.1001/jamaneurol.2014.62. [DOI] [PubMed] [Google Scholar]
- 35.Calvo A, Canosa A, Bertuzzo D, Cugnasco P, Solero L, Clerico M, De Mercanti S, Bersano E, Cammarosano S, Ilardi A, et al. Influence of cigarette smoking on ALS outcome: a population-based study. J Neurol Neurosurg Psychiatry. 2016;87(11):1229–1233. doi: 10.1136/jnnp-2016-313793. [DOI] [PubMed] [Google Scholar]
- 36.Umoh ME, Fournier C, Li Y, Polak M, Shaw L, Landers JE, Hu W, Gearing M, Glass JD. Comparative analysis of C9orf72 and sporadic disease in an ALS clinic population. Neurology. 2016;87(10):1024–1030. doi: 10.1212/WNL.0000000000003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gendron TF, Daughrity LM, Heckman MG, Diehl NN, Wuu J, Miller TM, Pastor P, Trojanowski JQ, Grossman M, Berry JD, et al. Phosphorylated neurofilament heavy chain: A biomarker of survival for C9ORF72-associated amyotrophic lateral sclerosis. Ann Neurol. 2017;82(1):139–146. doi: 10.1002/ana.24980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reniers W, Schrooten M, Claeys KG, Tilkin P, D'Hondt A, Van Reijen D, Couwelier G, Lamaire N, Robberecht W, Fieuws S, et al. Prognostic value of clinical and electrodiagnostic parameters at time of diagnosis in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(5-6):341–350. doi: 10.1080/21678421.2017.1288254. [DOI] [PubMed] [Google Scholar]
- 39.Cammack AJ, Atassi N, Hyman T, van den Berg LH, Harms M, Baloh RH, Brown RH, van Es MA, Veldink JH, de Vries BS, et al. Prospective natural history study of C9orf72 ALS clinical characteristics and biomarkers. Neurology. 2019;93(17):e1605–e1617. doi: 10.1212/WNL.0000000000008359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rooney J, Murray D, Campion A, Moloney H, Tattersall R, Doherty M, Hammond M, Heverin M, McLaughlin R, Hardiman O. The C9orf72 expansion is associated with accelerated respiratory function decline in a large Amyotrophic Lateral Sclerosis cohort. HRB Open Res. 2019;2:23. doi: 10.12688/hrbopenres.12940.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trojsi F, Siciliano M, Femiano C, Santangelo G, Lunetta C, Calvo A, Moglia C, Marinou K, Ticozzi N, Ferro C, et al. Comparative Analysis of C9orf72 and Sporadic Disease in a Large Multicenter ALS Population: The Effect of Male Sex on Survival of C9orf72 Positive Patients. Front Neurosci. 2019;13:485. doi: 10.3389/fnins.2019.00485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benatar M, Zhang L, Wang L, Granit V, Statland J, Barohn R, Swenson A, Ravits J, Jackson C, Burns TM, et al. Validation of serum neurofilaments as prognostic and potential pharmacodynamic biomarkers for ALS. Neurology. 2020;95(1):e59–e69. doi: 10.1212/WNL.0000000000009559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Schaepdryver M, Lunetta C, Tarlarini C, Mosca L, Chio A, Van Damme P, Poesen K. Neurofilament light chain and C reactive protein explored as predictors of survival in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2020;91(4):436–437. doi: 10.1136/jnnp-2019-322309. [DOI] [PubMed] [Google Scholar]
- 44.Kläppe U, Chamoun S, Shen Q, Finn A, Evertsson B, Zetterberg H, et al. Cardiac troponin T is elevated and increases longitudinally in ALS patients. Amyotroph Lateral Scler Frontotemporal Degener. 2022;23(1-2):58–65. [DOI] [PubMed]
- 45.Puentes F, Lombardi V, Lu CH, Yildiz O, Fratta P, Isaacs A, Bobeva Y, Wuu J, Benatar M, Malaspina A. Humoral response to neurofilaments and dipeptide repeats in ALS progression. Ann Clin Transl Neurol. 2021;8(9):1831–1844. doi: 10.1002/acn3.51428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cudkowicz ME, McKenna-Yasek D, Sapp PE, Chin W, Geller B, Hayden DL, Schoenfeld DA, Hosler BA, Horvitz HR, Brown RH. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann Neurol. 1997;41(2):210–221. doi: 10.1002/ana.410410212. [DOI] [PubMed] [Google Scholar]
- 47.Liu W, Li X, Sun Y, Yu X, Wang Y, Liu N, et al. Genotype-phenotype correlations in a chinese population with familial amyotrophic lateral sclerosis. Neurol Res. 2022;44(3):206–16. [DOI] [PubMed]
- 48.Lattante S, Conte A, Zollino M, Luigetti M, Del Grande A, Marangi G, Romano A, Marcaccio A, Meleo E, Bisogni G, et al. Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology. 2012;79(1):66–72. doi: 10.1212/WNL.0b013e31825dceca. [DOI] [PubMed] [Google Scholar]
- 49.Borghero G, Pugliatti M, Marrosu F, Marrosu MG, Murru MR, Floris G, Cannas A, Parish LD, Occhineri P, Cau TB, et al. Genetic architecture of ALS in Sardinia. Neurobiol Aging. 2014;35(12):2882.e2887–2882.e2812. doi: 10.1016/j.neurobiolaging.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Millecamps S, Salachas F, Cazeneuve C, Gordon P, Bricka B, Camuzat A, Guillot-Noël L, Russaouen O, Bruneteau G, Pradat PF, et al. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J Med Genet. 2010;47(8):554–560. doi: 10.1136/jmg.2010.077180. [DOI] [PubMed] [Google Scholar]
- 51.Chiò A, Borghero G, Restagno G, Mora G, Drepper C, Traynor BJ, Sendtner M, Brunetti M, Ossola I, Calvo A, et al. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain. 2012;135(Pt 3):784–793. doi: 10.1093/brain/awr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCann EP, Williams KL, Fifita JA, Tarr IS, O'Connor J, Rowe DB, Nicholson GA, Blair IP. The genotype-phenotype landscape of familial amyotrophic lateral sclerosis in Australia. Clin Genet. 2017;92(3):259–266. doi: 10.1111/cge.12973. [DOI] [PubMed] [Google Scholar]
- 53.Govaarts R, Beeldman E, Kampelmacher MJ, van Tol MJ, van den Berg LH, van der Kooi AJ, Wijkstra PJ, Zijnen-Suyker M, Cobben NA, Schmand BA, et al. The frontotemporal syndrome of ALS is associated with poor survival. J Neurol. 2016;263(12):2476–2483. doi: 10.1007/s00415-016-8290-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brand D, Polak M, Glass JD, Fournier CN. Comparison of Phenotypic Characteristics and Prognosis Between Black and White Patients in a Tertiary ALS Clinic. Neurology. 2021;96(6):e840–e844. doi: 10.1212/WNL.0000000000011396. [DOI] [PubMed] [Google Scholar]
- 55.Ammar Al-Chalabi ZEE, Bakker MC, et al. Association of apolipoprotein E4 allele with bulbar-onset motor neuron disease. Lancet. 1996;347:159–160. doi: 10.1016/S0140-6736(96)90343-8. [DOI] [PubMed] [Google Scholar]
- 56.Drory VE, Birnbaum M, Korczyn AD, Chapman J. Association of APOE epsilon4 allele with survival in amyotrophic lateral sclerosis. J Neurol Sci. 2001;190(1-2):17–20. doi: 10.1016/S0022-510X(01)00569-X. [DOI] [PubMed] [Google Scholar]
- 57.Zetterberg H, Jacobsson J, Rosengren L, Blennow K, Andersen PM. Association of APOE with age at onset of sporadic amyotrophic lateral sclerosis. J Neurol Sci. 2008;273(1-2):67–69. doi: 10.1016/j.jns.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 58.Steenland K, MacNeil J, Seals R, Levey A. Factors affecting survival of patients with neurodegenerative disease. Neuroepidemiology. 2010;35(1):28–35. doi: 10.1159/000306055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vidal-Taboada JM, Lopez-Lopez A, Salvado M, Lorenzo L, Garcia C, Mahy N, Rodríguez MJ, Gamez J. UNC13A confers risk for sporadic ALS and influences survival in a Spanish cohort. J Neurol. 2015;262(10):2285–2292. doi: 10.1007/s00415-015-7843-z. [DOI] [PubMed] [Google Scholar]
- 60.Tan HHG, Westeneng HJ, van der Burgh HK, van Es MA, Bakker LA, van Veenhuijzen K, van Eijk KR, van Eijk RPA, Veldink JH, van den Berg LH. The Distinct Traits of the UNC13A Polymorphism in Amyotrophic Lateral Sclerosis. Ann Neurol. 2020;88(4):796–806. doi: 10.1002/ana.25841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tetsuka S, Morita M, Iida A, Ikegawa S, Nakano I. ZNF512B gene serves as a prognostic factor in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13:122. doi: 10.1016/j.jns.2012.10.029. [DOI] [PubMed] [Google Scholar]
- 62.Yu CJ, Wang L, Mao SL, Zhang Y, Song LL, Cai LY, Tao Y. The clinical assessment of amyotrophic lateral sclerosis patients' prognosis by ZNF512B gene, neck flexor muscle power score and body mass index (BMI) BMC Neurol. 2018;18(1):211. doi: 10.1186/s12883-018-1219-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang H, Yang B, Wang F, Li K, Zhu Y, Liu B, Ren H, Tian S, Xu Y, Pang A, et al. Association of Single Nucleotide Polymorphism at rs2275294 in the ZNF512B Gene with Prognosis in Amyotrophic Lateral Sclerosis. Neuromolecular Med. 2021;23(2):242–246. doi: 10.1007/s12017-020-08634-y. [DOI] [PubMed] [Google Scholar]
- 64.Landers JE, Melki J, Meininger V, Glass JD, van den Berg LH, van Es MA, Sapp PC, van Vught PW, McKenna-Yasek DM, Blauw HM, et al. Reduced expression of the Kinesin-Associated Protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2009;106(22):9004–9009. doi: 10.1073/pnas.0812937106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Traynor BJ, Nalls M, Lai SL, Gibbs RJ, Schymick JC, Arepalli S, Hernandez D, van der Brug MP, Johnson JO, Dillman A, et al. Kinesin-associated protein 3 (KIFAP3) has no effect on survival in a population-based cohort of ALS patients. Proc Natl Acad Sci U S A. 2010;107(27):12335–12338. doi: 10.1073/pnas.0914079107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Orsetti V, Pegoraro E, Cima V, D'Ascenzo C, Palmieri A, Querin G, Volpe M, Ermani M, Angelini C, Sorarù G. Genetic variation in KIFAP3 is associated with an upper motor neuron-predominant phenotype in amyotrophic lateral sclerosis. Neurodegener Dis. 2011;8(6):491–495. doi: 10.1159/000327755. [DOI] [PubMed] [Google Scholar]
- 67.van Doormaal PT, Ticozzi N, Gellera C, Ratti A, Taroni F, Chiò A, Calvo A, Mora G, Restagno G, Traynor BJ, et al. Analysis of the KIFAP3 gene in amyotrophic lateral sclerosis: a multicenter survival study. Neurobiol Aging. 2014;35(10):2420.e2413–2420.e2424. doi: 10.1016/j.neurobiolaging.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Czell D, Sapp PC, Neuwirth C, Weber M, Andersen PM, Brown RH. Further analysis of KIFAP3 gene in ALS patients from Switzerland and Sweden. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(3-4):302–304. doi: 10.1080/21678421.2017.1280509. [DOI] [PubMed] [Google Scholar]
- 69.Gamez J, Barceló MJ, Muñoz X, Carmona F, Cuscó I, Baiget M, Cervera C, Tizzano EF. Survival and respiratory decline are not related to homozygous SMN2 deletions in ALS patients. Neurology. 2002;59(9):1456–1460. doi: 10.1212/01.WNL.0000032496.64510.4E. [DOI] [PubMed] [Google Scholar]
- 70.Lopez-Lopez A, Gamez J, Syriani E, Morales M, Salvado M, Rodríguez MJ, Mahy N, Vidal-Taboada JM. CX3CR1 is a modifying gene of survival and progression in amyotrophic lateral sclerosis. PLoS One. 2014;9(5):e96528. doi: 10.1371/journal.pone.0096528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Calvo A, Moglia C, Canosa A, Cammarosano S, Ilardi A, Bertuzzo D, Traynor BJ, Brunetti M, Barberis M, Mora G, et al. Common polymorphisms of chemokine (C-X3-C motif) receptor 1 gene modify amyotrophic lateral sclerosis outcome: A population-based study. Muscle Nerve. 2018;57(2):212–216. doi: 10.1002/mus.25653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vidal-Taboada JM, Pugliese M, Salvadó M, Gámez J, Mahy N, Rodríguez MJ. K(ATP) Channel Expression and Genetic Polymorphisms Associated with Progression and Survival in Amyotrophic Lateral Sclerosis. Mol Neurobiol. 2018;55(10):7962–7972. doi: 10.1007/s12035-018-0970-7. [DOI] [PubMed] [Google Scholar]
- 73.Mouzat K, Molinari N, Kantar J, Polge A, Corcia P, Couratier P, Clavelou P, Juntas-Morales R, Pageot N, Lobaccaro JA, et al. Liver X Receptor Genes Variants Modulate ALS Phenotype. Mol Neurobiol. 2018;55(3):1959–1965. doi: 10.1007/s12035-017-0453-2. [DOI] [PubMed] [Google Scholar]
- 74.Sleegers K, Brouwers N, Maurer-Stroh S, van Es MA, Van Damme P, van Vught PW, van der Zee J, Serneels S, De Pooter T, Van den Broeck M, et al. Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology. 2008;71(4):253–259. doi: 10.1212/01.wnl.0000289191.54852.75. [DOI] [PubMed] [Google Scholar]
- 75.El Oussini H, Bayer H, Scekic-Zahirovic J, Vercruysse P, Sinniger J, Dirrig-Grosch S, Dieterlé S, Echaniz-Laguna A, Larmet Y, Müller K, et al. Serotonin 2B receptor slows disease progression and prevents degeneration of spinal cord mononuclear phagocytes in amyotrophic lateral sclerosis. Acta Neuropathol. 2016;131(3):465–480. doi: 10.1007/s00401-016-1534-4. [DOI] [PubMed] [Google Scholar]
- 76.Theunissen F, Anderton RS, Mastaglia FL, Flynn LL, Winter SJ, James I, Bedlack R, Hodgetts S, Fletcher S, Wilton SD, et al. Novel STMN2 Variant Linked to Amyotrophic Lateral Sclerosis Risk and Clinical Phenotype. Front Aging Neurosci. 2021;13:658226. doi: 10.3389/fnagi.2021.658226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu L, Tian D, Li J, Chen L, Tang L, Fan D. The Analysis of Two BDNF Polymorphisms G196A/C270T in Chinese Sporadic Amyotrophic Lateral Sclerosis. Front Aging Neurosci. 2017;9:135. doi: 10.3389/fnagi.2017.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.He J, Fu J, Fan D. The complement C7 variant rs3792646 is associated with amyotrophic lateral sclerosis in a Han Chinese population. Neurobiol Aging. 2021;99:103.e101–103.e107. doi: 10.1016/j.neurobiolaging.2020.10.012. [DOI] [PubMed] [Google Scholar]
- 79.Diekstra FP, Beleza-Meireles A, Leigh NP, Shaw CE, Al-Chalabi A. Interaction between PON1 and population density in amyotrophic lateral sclerosis. NeuroReport. 2009;20(2):186–190. doi: 10.1097/WNR.0b013e32831af220. [DOI] [PubMed] [Google Scholar]
- 80.Verde F, Tiloca C, Morelli C, Doretti A, Poletti B, Maderna L, Messina S, Gentilini D, Fogh I, Ratti A, et al. PON1 is a disease modifier gene in amyotrophic lateral sclerosis: association of the Q192R polymorphism with bulbar onset and reduced survival. Neurol Sci. 2019;40(7):1469–1473. doi: 10.1007/s10072-019-03834-2. [DOI] [PubMed] [Google Scholar]
- 81.Blauw HM, van Rheenen W, Koppers M, Van Damme P, Waibel S, Lemmens R, van Vught PW, Meyer T, Schulte C, Gasser T, et al. NIPA1 polyalanine repeat expansions are associated with amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21(11):2497–2502. doi: 10.1093/hmg/dds064. [DOI] [PubMed] [Google Scholar]
- 82.Blasco H, Vourc'h P, Nadjar Y, Ribourtout B, Gordon PH, Guettard YO, Camu W, Praline J, Meininger V, Andres CR, et al. Association between divalent metal transport 1 encoding gene (SLC11A2) and disease duration in amyotrophic lateral sclerosis. J Neurol Sci. 2011;303(1-2):124–127. doi: 10.1016/j.jns.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 83.de Sousa Barros JB, de Faria SK, da Cruz Pereira Bento D, Prado Assunção LD, da Silva Santos R, da Silva Reis AA. Influence of GSTP1 rs1695 polymorphism on survival in male patients' amyotrophic lateral sclerosis: a genetic association study in Brazilian population. Mol Biol Rep. 2022;49(2):1655–9. [DOI] [PubMed]
- 84.Al-Chalabi A, Scheffler MD, Smith BN, Parton MJ, Cudkowicz ME, Andersen PM, Hayden DL, Hansen VK, Turner MR, Shaw CE, et al. Ciliary neurotrophic factor genotype does not influence clinical phenotype in amyotrophic lateral sclerosis. Ann Neurol. 2003;54(1):130–134. doi: 10.1002/ana.10638. [DOI] [PubMed] [Google Scholar]
- 85.Van Vught PW, Van Wijk J, Bradley TE, Plasmans D, Jakobs ME, Veldink JH, de Jong JM, Van den Berg LH, Baas F. Ciliary neurotrophic factor null alleles are not a risk factor for Charcot-Marie-Tooth disease, hereditary neuropathy with pressure palsies and amyotrophic lateral sclerosis. Neuromuscul Disord. 2007;17(11-12):964–967. doi: 10.1016/j.nmd.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 86.Yang X, Zheng J, Tian S, Chen Y, An R, Zhao Q, Xu Y. HLA-DRA/HLA-DRB5 polymorphism affects risk of sporadic ALS and survival in a southwest Chinese cohort. J Neurol Sci. 2017;373:124–128. doi: 10.1016/j.jns.2016.12.055. [DOI] [PubMed] [Google Scholar]
- 87.Moisse M, Zwamborn RAJ, van Vugt J, van der Spek R, van Rheenen W, Kenna B, Van Eijk K, Kenna K, Corcia P, Couratier P, et al. The Effect of SMN Gene Dosage on ALS Risk and Disease Severity. Ann Neurol. 2021;89(4):686–697. doi: 10.1002/ana.26009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee T, Li YR, Ingre C, Weber M, Grehl T, Gredal O, de Carvalho M, Meyer T, Tysnes OB, Auburger G, et al. Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum Mol Genet. 2011;20(9):1697–1700. doi: 10.1093/hmg/ddr045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Van Damme P, Veldink JH, van Blitterswijk M, Corveleyn A, van Vught PW, Thijs V, Dubois B, Matthijs G, van den Berg LH, Robberecht W. Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology. 2011;76(24):2066–2072. doi: 10.1212/WNL.0b013e31821f445b. [DOI] [PubMed] [Google Scholar]
- 90.Daoud H, Belzil V, Martins S, Sabbagh M, Provencher P, Lacomblez L, Meininger V, Camu W, Dupre N, Dion PA, et al. Association of long ATXN2 CAG repeat sizes with increased risk of amyotrophic lateral sclerosis. Arch Neurol. 2011;68(6):739–742. doi: 10.1001/archneurol.2011.111. [DOI] [PubMed] [Google Scholar]
- 91.Corrado L, Mazzini L, Oggioni GD, Luciano B, Godi M, Brusco A, D'Alfonso S. ATXN-2 CAG repeat expansions are interrupted in ALS patients. Hum Genet. 2011;130(4):575–580. doi: 10.1007/s00439-011-1000-2. [DOI] [PubMed] [Google Scholar]
- 92.Lahut S, Omur O, Uyan O, Agim ZS, Ozoguz A, Parman Y, Deymeer F, Oflazer P, Koc F, Ozcelik H, et al. ATXN2 and its neighbouring gene SH2B3 are associated with increased ALS risk in the Turkish population. PLoS One. 2012;7(8):e42956. doi: 10.1371/journal.pone.0042956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen Y, Huang R, Yang Y, Chen K, Song W, Pan P, Li J, Shang HF. Ataxin-2 intermediate-length polyglutamine: a possible risk factor for Chinese patients with amyotrophic lateral sclerosis. Neurobiol Aging. 2011;32(10):1925 e1921–1925 e1925. doi: 10.1016/j.neurobiolaging.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 94.Kim G, Gautier O, Tassoni-Tsuchida E, Ma XR, Gitler AD. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron. 2020;108(5):822–842. doi: 10.1016/j.neuron.2020.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017;88(7):540–549. doi: 10.1136/jnnp-2016-315018. [DOI] [PubMed] [Google Scholar]
- 96.Freibaum BD, Taylor JP. The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front Mol Neurosci. 2017;10:35. doi: 10.3389/fnmol.2017.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ekhtiari Bidhendi E, Bergh J, Zetterstrom P, Forsberg K, Pakkenberg B, Andersen PM, Marklund SL, Brannstrom T. Mutant superoxide dismutase aggregates from human spinal cord transmit amyotrophic lateral sclerosis. Acta Neuropathol. 2018;136(6):939–953. doi: 10.1007/s00401-018-1915-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, Shneider NA. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun. 2016;7:10465. doi: 10.1038/ncomms10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7(5):409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cui R, Tuo M, Li P, Zhou C. Association between TBK1 mutations and risk of amyotrophic lateral sclerosis/frontotemporal dementia spectrum: a meta-analysis. Neurol Sci. 2018;39(5):811–820. doi: 10.1007/s10072-018-3246-0. [DOI] [PubMed] [Google Scholar]
- 101.Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain. 2017;10(1):5. doi: 10.1186/s13041-017-0287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yao L, He X, Cui B, Zhao F, Zhou C. NEK1 mutations and the risk of amyotrophic lateral sclerosis (ALS): a meta-analysis. Neurol Sci. 2021;42(4):1277–1285. doi: 10.1007/s10072-020-05037-6. [DOI] [PubMed] [Google Scholar]
- 103.Higelin J, Catanese A, Semelink-Sedlacek LL, Oeztuerk S, Lutz AK, Bausinger J, Barbi G, Speit G, Andersen PM, Ludolph AC, et al. NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res. 2018;30:150–162. doi: 10.1016/j.scr.2018.06.005. [DOI] [PubMed] [Google Scholar]
- 104.McLaughlin RL, Kenna KP, Vajda A, Byrne S, Bradley DG, Hardiman O. UBQLN2 mutations are not a frequent cause of amyotrophic lateral sclerosis in Ireland. Neurobiol Aging. 2014;35(1):267 e269–267 e211. doi: 10.1016/j.neurobiolaging.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 105.Wu JJ, Cai A, Greenslade JE, Higgins NR, Fan C, Le NTT, Tatman M, Whiteley AM, Prado MA, Dieriks BV, et al. ALS/FTD mutations in UBQLN2 impede autophagy by reducing autophagosome acidification through loss of function. Proc Natl Acad Sci U S A. 2020;117(26):15230–15241. doi: 10.1073/pnas.1917371117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pan C, Jiao B, Xiao T, Hou L, Zhang W, Liu X, Xu J, Tang B, Shen L. Mutations of CCNF gene is rare in patients with amyotrophic lateral sclerosis and frontotemporal dementia from Mainland China. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(3-4):265–268. doi: 10.1080/21678421.2017.1293111. [DOI] [PubMed] [Google Scholar]
- 107.Williams KL, Topp S, Yang S, Smith B, Fifita JA, Warraich ST, Zhang KY, Farrawell N, Vance C, Hu X, et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat Commun. 2016;7:11253. doi: 10.1038/ncomms11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fernandez M, McClain ME, Martinez RA, Snow K, Lipe H, Ravits J, Bird TD, La Spada AR. Late-onset SCA2: 33 CAG repeats are sufficient to cause disease. Neurology. 2000;55(4):569–572. doi: 10.1212/WNL.55.4.569. [DOI] [PubMed] [Google Scholar]
- 109.Wang MD, Gomes J, Cashman NR, Little J, Krewski D. Intermediate CAG repeat expansion in the ATXN2 gene is a unique genetic risk factor for ALS--a systematic review and meta-analysis of observational studies. PLoS One. 2014;9(8):e105534. doi: 10.1371/journal.pone.0105534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.van Blitterswijk M, Mullen B, Heckman MG, Baker MC, DeJesus-Hernandez M, Brown PH, Murray ME, Hsiung GY, Stewart H, Karydas AM, et al. Ataxin-2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol Aging. 2014;35(10):2421.e2413–2421.e2427. doi: 10.1016/j.neurobiolaging.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, Messing J, Kim HJ, Soriano A, Auburger G, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544(7650):367–371. doi: 10.1038/nature22038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rohrer JD, Isaacs AM, Mizielinska S, Mead S, Lashley T, Wray S, Sidle K, Fratta P, Orrell RW, Hardy J, et al. C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol. 2015;14(3):291–301. doi: 10.1016/S1474-4422(14)70233-9. [DOI] [PubMed] [Google Scholar]
- 113.Chen Y, Lin Z, Chen X, Cao B, Wei Q, Ou R, Zhao B, Song W, Wu Y, Shang HF. Large C9orf72 repeat expansions are seen in Chinese patients with sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2016;38:217 e215–217 e222. doi: 10.1016/j.neurobiolaging.2015.10.021. [DOI] [PubMed] [Google Scholar]
- 114.Yang T, Hou Y, Li C, Cao B, Cheng Y, Wei Q, Zhang L, Shang H. Risk factors for cognitive impairment in amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2021;92(7):688–693. doi: 10.1136/jnnp-2020-325701. [DOI] [PubMed] [Google Scholar]
- 115.Hu J, Rigo F, Prakash TP, Corey DR. Recognition of c9orf72 Mutant RNA by Single-Stranded Silencing RNAs. Nucleic Acid Ther. 2017;27(2):87–94. doi: 10.1089/nat.2016.0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Martier R, Liefhebber JM, Miniarikova J, van der Zon T, Snapper J, Kolder I, Petry H, van Deventer SJ, Evers MM, Konstantinova P. Artificial MicroRNAs Targeting C9orf72 Can Reduce Accumulation of Intra-nuclear Transcripts in ALS and FTD Patients. Mol Ther Nucleic Acids. 2019;14:593–608. doi: 10.1016/j.omtn.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bali T, Self W, Liu J, Siddique T, Wang LH, Bird TD, Ratti E, Atassi N, Boylan KB, Glass JD, et al. Defining SOD1 ALS natural history to guide therapeutic clinical trial design. J Neurol Neurosurg Psychiatry. 2017;88(2):99–105. doi: 10.1136/jnnp-2016-313521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Arisato T, Okubo R, Arata H, Abe K, Fukada K, Sakoda S, Shimizu A, Qin XH, Izumo S, Osame M, et al. Clinical and pathological studies of familial amyotrophic lateral sclerosis (FALS) with SOD1 H46R mutation in large Japanese families. Acta Neuropathol. 2003;106(6):561–568. doi: 10.1007/s00401-003-0763-5. [DOI] [PubMed] [Google Scholar]
- 119.Naumann M, Peikert K, Günther R, van der Kooi AJ, Aronica E, Hübers A, Danel V, Corcia P, Pan-Montojo F, Cirak S, et al. Phenotypes and malignancy risk of different FUS mutations in genetic amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2019;6(12):2384–2394. doi: 10.1002/acn3.50930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, Marroquin N, Nordin F, Hübers A, Weydt P, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18(5):631–636. doi: 10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]
- 122.Chia R, Chio A, Traynor BJ. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 2018;17(1):94–102. doi: 10.1016/S1474-4422(17)30401-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lattante S, Doronzio PN, Conte A, Marangi G, Martello F, Bisogni G, Meleo E, Colavito D, Del Giudice E, Patanella AK, et al. Novel variants and cellular studies on patients' primary fibroblasts support a role for NEK1 missense variants in ALS pathogenesis. Hum Mol Genet. 2021;30(1):65–71. doi: 10.1093/hmg/ddab015. [DOI] [PubMed] [Google Scholar]
- 124.Calvo A, Restagno G, Brunetti M, Ossola I, Majounie E, Renton AE, Canosa A, Manera U, Bersano E, Moglia C, et al. UNC13A influences survival in an Italian population-based series. Eur J Neurol. 2012;19:262. [Google Scholar]
- 125.Diekstra FP, Van Deerlin VM, van Swieten JC, Al-Chalabi A, Ludolph AC, Weishaupt JH, Hardiman O, Landers JE, Brown RH, Jr, van Es MA, et al. C9orf72 and UNC13A are shared risk loci for amyotrophic lateral sclerosis and frontotemporal dementia: a genome-wide meta-analysis. Ann Neurol. 2014;76(1):120–133. doi: 10.1002/ana.24198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yang B, Jiang H, Wang F, Li S, Wu C, Bao J, Zhu Y, Xu Z, Liu B, Ren H, et al. UNC13A variant rs12608932 is associated with increased risk of amyotrophic lateral sclerosis and reduced patient survival: a meta-analysis. Neurol Sci. 2019;40(11):2293–2302. doi: 10.1007/s10072-019-03951-y. [DOI] [PubMed] [Google Scholar]
- 127.Brown AL, Wilkins OG, Keuss MJ, Hill SE, Zanovello M, Lee WC, Bampton A, Lee FCY, Masino L, Qi YA, et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature. 2022;603:131–137. doi: 10.1038/s41586-022-04436-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, Briner A, Rodriguez CM, Guo C, Akiyama T, et al. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature. 2022;603:124–130. doi: 10.1038/s41586-022-04424-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ning P, Yang X, Yang B, Zhao Q, Huang H, An R, Chen Y, Hu F, Xu Z, Xu Y. Meta-analysis of the association between ZNF512B polymorphism rs2275294 and risk of amyotrophic lateral sclerosis. Neurol Sci. 2018;39(7):1261–1266. doi: 10.1007/s10072-018-3411-5. [DOI] [PubMed] [Google Scholar]
- 130.Schober A, Peterziel H, von Bartheld CS, Simon H, Krieglstein K, Unsicker K. GDNF applied to the MPTP-lesioned nigrostriatal system requires TGF-beta for its neuroprotective action. Neurobiol Dis. 2007;25(2):378–391. doi: 10.1016/j.nbd.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 131.Veldink JH, van den Berg LH, Cobben JM, Stulp RP, De Jong JM, Vogels OJ, Baas F, Wokke JH, Scheffer H. Homozygous deletion of the survival motor neuron 2 gene is a prognostic factor in sporadic ALS. Neurology. 2001;56(6):749–752. doi: 10.1212/WNL.56.6.749. [DOI] [PubMed] [Google Scholar]
- 132.van Rheenen W, van der Spek RAA, Bakker MK, van Vugt J, Hop PJ, Zwamborn RAJ, de Klein N, Westra HJ, Bakker OB, Deelen P, et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat Genet. 2021;53(12):1636–1648. doi: 10.1038/s41588-021-00973-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Characteristics of the articles included in network meta-analysis for variants in ALS causative genes. Table S2. Characteristics of the articles included in pairwise meta-analysis for other modification loci. Table S3. Characteristics of the articles included in systematic review for other modification loci. Table S4. The quality of articles included in network meta-analysis and pairwise meta-analysis by NOS. Table S5. The feathers for different variants in SOD1, FUS, TARDBP. Figure S1. Forest plot for inconsistency test in network meta-analysis. Figure S2-S12. Heterogeneity analysis n network meta-analysis. Figure S13. publication bias for studies reporting C9orf72 expansion in patients with ALS compared to those without ALS-related mutation.
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files. Effect size and study details were extracted from the original papers, which are available in the public domain.