

Abstract
STAT4 plays a critical role in the generation of both innate and adaptive immune responses. In the absence of STAT4, Th1 responses, critical for resistance to fungal disease, do not occur. Infection with the dimorphic fungus, Coccidioides, is a major cause of community acquired pneumonia in the endemic regions of Arizona and California. In some people and often for unknown reasons, coccidioidal infection results in hematogenous dissemination and progressive disease rather than the typical self-limited pneumonia. Members of three generations in a family developed disseminated coccidioidomycosis, prompting genetic investigation. All affected family members had a single heterozygous base change in STAT4, c.1877A>G, causing substitution of glycine for glutamate at AA626 (STAT4E626G/+). A knock-in mouse, heterozygous for the substitution, developed more severe experimental coccidioidomycosis than WT mice. Stat4E626G/+ T cells were deficient in production of IFN-gamma after anti-CD3/CD28 stimulation. Spleen cells from Stat4E626G mice showed defective responses to IL-12/IL-18 stimulation in vitro. In vivo, early after infection, mutant Stat4E626G/+ mice failed to produce IFN-gamma and related cytokines in the lung and to accumulate activated adaptive immune cells in mediastinal lymph nodes. Therefore, defective early induction of IFN gamma and adaptive responses by STAT4 prevents normal control of coccidioidomycosis in both mice and humans.
Introduction
STAT4 is critical in the generation of the Th1 response, which is mediated in part by IL-12 and IL-18. Mice deficient in STAT4 do not make effective Th1 responses and are more susceptible to infections requiring effective IFN-gamma responses (1–7). In mice many polymorphisms of STAT4 are known but there is little data available on the impact of those substitutions (8). In humans, the IL-12 axis has been implicated in autoimmunity as well as infections, including paracoccidioidomycosis (4, 9–15).
Coccidioidomycosis (CM), also known as San Joaquin Valley fever, is caused by Coccidioides immitis and Coccidioides posadasii, soil-inhabiting dimorphic fungi in endemic regions of the Southwestern United States, Mexico, and other parts of the Western Hemisphere. Despite its relatively limited geographic distribution, at least 150,000 infections occur annually (16). Recent estimates from the CDC propose that the actual numbers could be several-fold higher (17). Economic estimates for the health-related cost of CM in California and Arizona total $1.4 billion/year (18, 19).
Most infections are either asymptomatic or self-limited, although the latter often cause significant illness for months. Within endemic areas of Arizona, CM accounts for a quarter of all community acquired pneumonias (20, 21). In approximately 0.5% - 1.0%, of all CM infections, extrapulmonary dissemination leads to progressive and destructive lesions in other organs, a complication known as disseminated CM (DCM). DCM is much more likely in patients with broadly depressed cellular immunity, either from co-morbidities or immunosuppressive therapies (22). However, most DCM patients do not have these risk factors (23, 24) leaving the immunologic basis for their progressive infections unclear.
We have previously investigated patients with DCM and found mutations in the IL-12/IFN-gamma pathway. These have been isolated patients or siblings with DCM, harboring deleterious mutations in IL12RB1, IFNGR1, STAT1 and STAT3 (25–29). Consistent with the above findings, IFN-gamma knockout mice have higher fungal burdens, especially in disseminated sites, than wild-type mice (30). Similarly, it is clear that transfer of immunity following vaccination requires IFN-gamma competent CD4+ T cells (30). Therefore, the IL-12/IL-18/STAT4/IFN-gamma axis appears to be essential for protection from DCM in both humans and experimental animals.
We have identified members of a three-generation family who developed DCM, all of whom possessed a shared, unique single amino acid missense mutation in STAT4. We investigated the consequences of that mutation in vitro, genetically engineered mice carrying the homologous mutation, experimentally infected those mice and examined the potency of a CM vaccine in them. The induced mutation impaired IFN-gamma production through STAT4-dependent agonists, leading to a failure to accumulate activated immune cells in the draining lymph nodes, shorter survival time, and increased Coccidioides fungal burden.
Materials and Methods
Sequencing
Peripheral blood was used for sequencing after written consent under National Institutes of Health (NIH) protocol 14-I-0146. Exome-enriched libraries were quantitated using the KAPA Library Quantification Kit and following the manufacturer’s protocol (KAPA Biosystems, Woburn, MA). Library quantifications were size adjusted based on the library peak size estimated from the Bioanlyzer profiles.
Exome-enriched libraries were clustered on the cBot using 10 pM and 11 pM of template and then sequenced on the Illumina HiSeq 2500 as 2 × 100-bp paired-end reads, following the manufacturer’s protocol (Illumina Inc, San Diego, CA).
Identification of SNPs and indels was performed using Strand NGS software. Strand NGS utilizes a modified Bayesian variant calling method adapted from the MAQ SNP calling algorithm which compares the nucleotides present on aligned reads against the reference at each position in the genome.
Mouse Production
CRISPR guide RNAs, used for mouse Stat4 gene E626G knock-in were designed using the CRISPOR web tool (31). Multiple guides were selected and the one successful for introducing the mutation was: CGACAGTCTCCCTTTGTTGTAGG (gRNA5)(PAM sequence in green). Synthetic single guide RNAs were made by Synthego. eSpCas9 recombinant protein was ordered from Millipore (ESPCAS9PRO-250UG). ssODNs with 30–70 bp homology to sequences on each side of each gRNA-mediated double-stranded break were designed and ordered from IDT. Silent mutations introducing RsaI restriction site for E626G knock-in was created in the corresponding oligoes for genotyping purposes.
Ribonucleoprotein complexes (RNP) were assembled by incubating recombinant Cas9 protein with sgRNA for 10 min at room temperature. Then the ss oligo was added to the mixture, followed by 10 minutes centrifugation at 10,000 rpm. The final concentrations used for the microinjections were 50/30/50 ng/ul respectively.
Fertilized eggs were collected from the oviducts of super ovulated BL6/NJ females. Microinjections were performed by continuous flow injection of the RNP/ssODN mixture into the pronucleus of 1-cell zygotes.
Tail-tipping of the newborn mice was utilized to purify DNA for genotyping by PCR, employing two screening primers: forward, 5’- CAGACTCCAGAAGGTCAGACG and reverse, 5’- TCAGAGGGTTTTCAGGGATG, producing 231bp band for the wild type and two additional bands of 141bp and 90bp in the positive mice when restricted with RsaI.
Fungal Strains
C. posadasii (Cp) strain RMSCC1038 (32) (Cp1038) was grown on 2X glucose-yeast extract agar (GYE) (2% glucose, 1% yeast extract, and 1.5% agar) at 30°C for 12 weeks. by the spin bar method, filtered through Pellon Thermolam Plus® to remove hyphal elements, and enumerated by hemocytometer Viability and enumeration was determined by growth of ten-fold serial dilutions on GYE at 37°C for 7 days. Viable suspensions for animal inoculation were diluted in isotonic saline. All manipulation of live fungus was performed at BSL3 with University of Arizona Institutional Biosafety Committee approval.
Mice
6–8 week old female C57BL/6NJ (Stock Number 005304) and C57BL-6J-Stat4em3Adiuj/J (Stat4 KO Stock Number 028526) mice were purchased from Jackson Laboratories. B6-Stat4em1Doe/em1Doe (B6-Stat4E626G) were maintained in house as homozygotes. Heterozygous B6-Stat4E626G/+ were produced by crossing with C57BL/6NJ mice. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Arizona.
Mouse infections
Mice were housed and manipulated at ABSL3 for all infection and post-infection procedures. Groups of 5–10 mice were infected intranasally under ketamine-xylazine anesthesia with ~50 coccidioidal spores in 30 μl of isotonic sterile saline. Animals were monitored by observation daily and weekly weight measurement for declining condition and were humanely euthanized when moribund. Fungal burden studies were sacrificed at specified time points and lung and spleen fungal burdens quantitated by serial dilution of organ homogenates on GYE plates at 35–37°C for up to 7 days as previously described (32). For survival studies, mice were observed for up to 6 weeks and moribund mice sacrificed as needed with quantitative culture of lungs and spleens. For immunologic studies, mice were sacrificed at pre-specified time points and lungs, spleens, lymph nodes and blood were collected for further analysis.
Cytokines
Lungs collected at time points ranging from 2 to 42 days p.i. were split and the right lung was thinly sliced and incubated for 24 hours in 2.5 mls of RPMI-1640 medium with 10% fetal calf serum at 37°C, 5% CO2, in 6-well tissue culture plates (33). Supernatant was centrifuged, filtered through a 0.8 μm filter to sterilize, and stored at −80°C until analysis. Cytokines were measured using a Luminex mouse 31-plex Panel (EMD Millipore, Billerica, MA) as previously described. Single cytokines secretion was confirmed using DuoSet ELISA (R&D Systems, Minneapolis, MN).
Preparation of cells for flow cytometry
Lungs were perfused with PBS to remove blood and then finely minced. Minced lung was then further processed using Miltenyi Lung Dissociation Kit and a gentleMACS OctoDissociator per manufacturers instruction (Miltenyi Biotec, Bergisch Gladbach, Germany). Spleens and lymph nodes were processed by mechanical separation of cells over 70um cell strainers as previously described (34). Red blood cells were lysed by using ammonium chloride–potassium carbonate lysis buffer. Viable cells were enumerated via Vi-Cell (Beckman Coulter, Indianapolis, IN).
Antibodies and Flow Cytometry
The following directly conjugated antibodies were utilized for flow cytometry analysis: B220 Clone RA3–6B2, CD3 Clone 17A2, CD4 Clone GK1.5, CD8 Clone 53–6.7, CD11b Clone M1/70, CD11c Clone N418, CD19 Clone 1D3, CD21 Clone HB5, CD23 Clone B3B4, CD24 Clone M1/69, CD25 Clone PC61, CD27 Clone LG.3A10, CD44 Clone IM7, CD45 Clone 104, CD62L Clone MEL-14, CD49a Clone HMα1, CD64 Clone 10.1, CD90.2 Clone 53–2.1, gamma delta TCR Clone UC7–13D5, IgD Clone 11–26c.2a, IgM Clone II/41, MHC Class II IA/IE Clone M5/114.15.2, NK1.1 Clone PK136, NKp46 Clone 29A1.4, Ly6G Clone RB6–8C5, ST2 Clone D1H4. All antibodies were titrated on normal B6 splenocytes prior to use. After preparation of single cell suspensions, cells were blocked for 30 min at 4°C with 24G2 supernatant to block Fc receptors. Cells were then stained with indicated antibodies for 30 min at 4°C in the dark. Cells were then washed three times with PBS + 2% BSA and analyzed on a Becton Dickinson LSRII flow cytometer. FlowJo (Treestar) was used for all flow cytometry analysis. Total cell numbers were calculated based on viable counts.
Fluorescence-linked immunosorbent assay (FLISA)
Coccidioides strain Δcps1 was incubated with mouse sera diluted 1:100 in PBS for 30 minutes at room temperature. Cells were then washed 3 times with PBS + 2% BSA. Labeled secondary antibodies were incubated for 30 minutes at room temperature in the dark. Cells were then washed three times with PBS + 2% BSA and analyzed on a Becton Dickinson LSRII flow cytometer. Results are expressed as ΔMFI compared to preinfection serum.
Results
Identification of STAT4 mutation.
Clinical Presentation
Three individuals across three generations within a family all developed DCM. The proband (Figure 1A) is an African American female who at age 48 years developed pulmonary CM, diagnosed by coccidioidal serology. A concurrent skin lesion was biopsied and showed spherules, verifying extrapulmonary dissemination. Fluconazole treatment resulted in healing of the lesion, however lesions recurred with fluconazole discontinuation; the patient has continued fluconazole for over two decades.
Fig. 1.
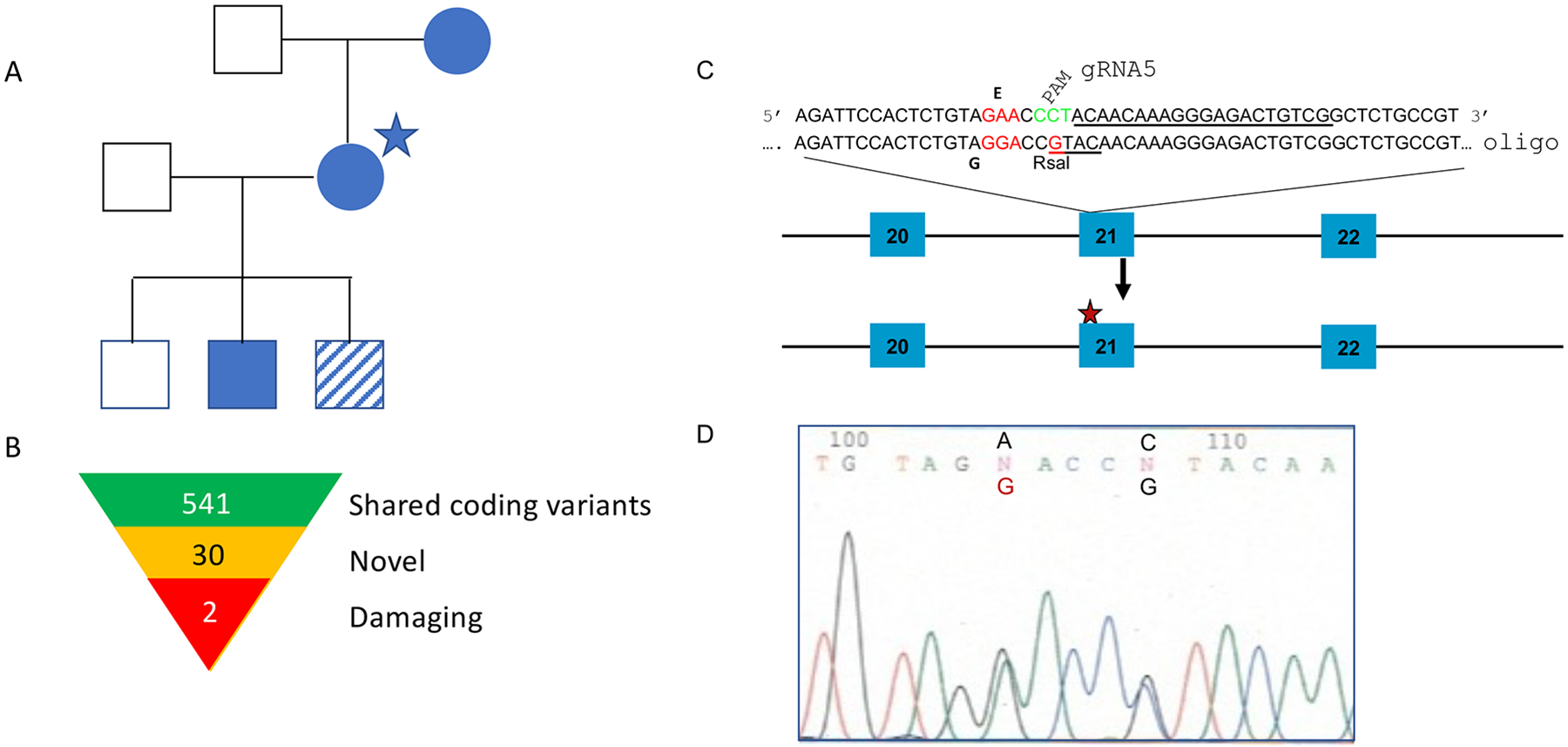
Family Pedigree (A) Proband is indicated with a star. All filled symbols were sequenced and had the E626G mutation in STAT4. Striped symbol was not available for sequencing but did develop DCM. (B) Summary of shared coding variants found through sequencing. (C) Sequence of the guide RNA, used to generate the E626G mutation in exon 21 of the mouse STAT4 gene. PAM sequence is in green. Oligo sequence shows the introduced SNPs, one for the intended change E626G, and one to destroy the PAM sequence and prevent re cutting, and also to introduce a RsaI restriction enzyme site, used for genotyping. (D) Actual sequence chromatogram result showing the introduced by CRISPR/Cas9 homology-directed repair SNPs in a heterozygous founder.
The proband’s mother, at age 37, developed severe coccidioidal pneumonia which was treated with amphotericin B. A nasal lesion was biopsied and demonstrated spherules, confirming DCM. She completed a course of amphotericin B with resolution of her illness and has not needed further therapy for the past five decades.
The proband’s son developed pulmonary symptoms at age 12, suffering a 12-pound weight loss before being diagnosed with coccidioidomycosis. He recovered without treatment. At 15 years he suffered a minimal trauma fracture of his left hand. Radiographs demonstrated a lytic lesion in one of the bones, which was subsequently biopsy-proven culture positive to be Coccidioides. He was treated for one year on a research protocol with either fluconazole or itraconazole (35), after which no further treatment has been needed for the subsequent 22 years.
Given the dominant inheritance pattern, the three family members underwent whole exome sequencing. We identified 541 shared coding variants, 30 of which were not present in dbSNPv141. Two of these novel variants, STAT4 c.1877A>G; p.E626G and ALKBH2 c.181C>T, p.R61W were predicted deleterious by multiple algorithms using dbNSFP (36, 37) (Figure 1B). In silico analysis of the STAT4 variant showed it within the SH2 domain, altering a residue within the highly conserved phospho-tyrosine binding pocket which is critical for signal transduction. The combined annotation dependent depletion (CADD) score for the STAT4 variant was 24.7. At the time of sequencing this mutation was not found in the Genome Aggregation Database (gnomAD) (38). Currently there is one variant in gnomAD, a frequency of 1/250864. Given the role of STAT4 in IL-12 signaling and previous demonstration of IL-12RB1 mutations leading to coccidioidomycosis, we pursued the STAT4 variant as the causative gene. ALKBH2 is a DNA-repair gene. To date there has been no link between DNA-repair and fungal susceptibility, however a different STAT4 mutation has been implicated in paracoccidioidomycosis (4).
Generation and characterization of B6-Stat4E626G mouse.
To test the impact of the Stat4E626G mutation in vivo, we used CRISPR/Cas9 to introduce the Stat4E626G mutation into B6/NJ embryos. A ssDNA oligonucleotide was synthesized to drive homology-directed repair to produce the desired substitution based on the published B6 sequence (plus a silent mutation to introduce a convenient restriction site for allele identification) using CRISPR/Cas9 mutagenesis by standard procedures (Figure 1C). (The oligo plus guide RNA used are shown in Supplemental Table 1.) Embryos were implanted into pseudopregnant females and the resulting pups were genotyped (Figure 1D). Mutation positive pups were bred back to wild-type B6 mice. Heterozygous offspring were then bred to establish a homozygous line. B6-Stat4em1Doe/em1Doe (noted hereafter as B6-Stat4E626G) mice were fertile and grossly normal. Both heterozygous B6-Stat4E626G/+ and homozygous B6-Stat4E626G/E626G mice showed similar numbers of immune cells and developmental markers compared to WT B6 mice, regardless of genotype (Supplemental Table 1). Western blot analysis of spleen cell lysates showed similar levels of STAT4 immunoreactivity in B6, B6-Stat4E626G/+, and B6-Stat4E626G/− mice, while B6-Stat4−/− had none (Supplemental Figure 1).
B6-Stat4E626G mice are highly sensitive to coccidioidal infection.
Given the isolated phenotype of DCM in the STAT4(E626G) patients without other notable infections, we sought to determine whether the B6-Stat4E626G mouse recapitulated the patients’ sensitivity to DCM. B6-Stat4E626G mice were intranasally infected with C. posadasii, Strain RMSCC1038 (Cp1038), which causes lethal infection in B6 mice, typically by 70 days (32). In a study comparing B6 control mice to B6-Stat4E626G heterozygous and homozygous mice, the mice carrying either one or two copies of the mutation had a median survival time (MST) of 38 (p=0.0072) and 35 (p=0.0013) days respectively, while control B6 mice survived to study endpoint day 42 (Figure 2A).
Fig. 2.
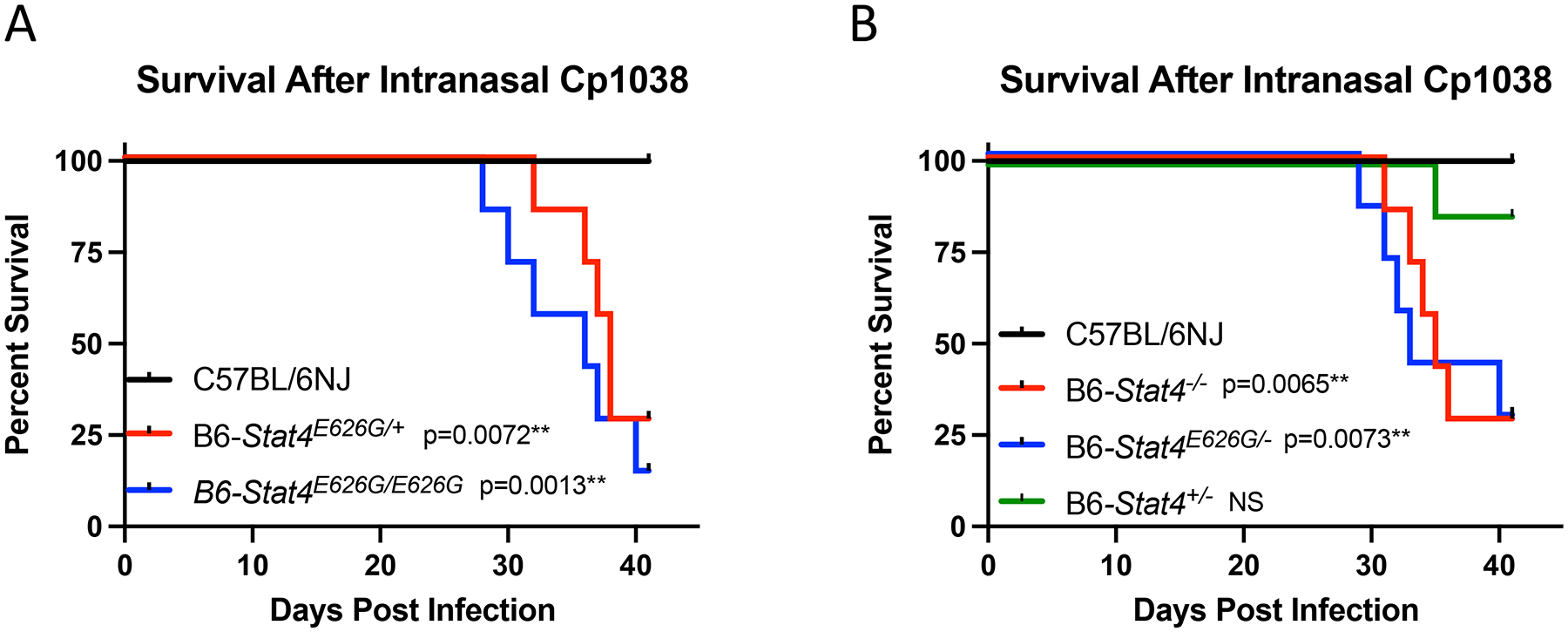
Mice carrying Stat4 mutations show increased susceptibility to Coccidioides infection. (A) C57BL/6NJ (black lines), B6-Stat4E626G/+ (red lines), or B6-Stat4E626G/E626G (blue lines) mice (N=7/group) were infected intranasally with ~50 CFU of Cp1038. Mice were followed for disease survival. (B) C57BL/6NJ (black lines), B6-Stat4−/− (red lines), B6-Stat4E626G/− (blue lines) or B6-Stat4+/− (green lines) mice (N=7/group) were infected intranasally with ~50 CFU of Cp1038. Mice were followed for disease survival. Graph is representative of 3 separate experiments of similar design. Significance was determined using a Mantel-Cox Log-rank test.
To examine the possibility of STAT4 haploinsufficiency, we crossed WT B6 with B6-Stat4−/−,producing Stat4+/−, which have only a single functional copy of STAT4. As an additional control, we also bred Stat4E626G/E626G mice with B6-Stat4−/− to produce Stat4E626G/− mice. F1 mice along with control Stat4−/− and WT B6 mice were infected with Cp1038 and followed for disease progression. Both Stat4 E626G/− mice and Stat4 E626G/+ mice succumbed to infection (MST 35 days). In contrast, B6-Stat4+/− with a single WT Stat4 allele were indistinguishable from WT mice (MST >40 days). Therefore, Stat4E626G is dominant negative (Figure 2B) and Stat4 does not show haploinsufficiency in this challenge.
T cells from Stat4E626G mice are deficient in IFN-gamma production following in vitro stimulation.
Since resistance to fatal DCM infection is dependent on CD4+ T cell responses (30, 35, 36), we tested the ability of Stat4E626G T cells to respond to STAT4-dependent signals. To examine if the induced mutation had an effect on STAT4 signaling, purified splenic CD4+ T cells from naïve B6, B6-Stat4E626G/+, or B6-Stat4−/− mice were stimulated with anti-CD3/CD28 beads in the presence of IL-2. After 48 hours, media were supplemented with nothing, IL-12, IL-18 or IL-12+IL-18 for an additional 24 hours. Supernatants were collected and analyzed for IFN-gamma production. Addition of IL-18 or IL-12 + IL-18 increased the production of INF-gamma in WT-B6 T cells (Figure 3), whereas B6-Stat4−/− cells produced no IFN-gamma under any stimulation. B6-Stat4E626G/+ T cells produced less INF-gamma than WT-B6 when stimulated by IL-2+IL18, IL2+IL12+IL18 or PMA/Ionomycin, indicating a deficiency in STAT4 signaling downstream of the IL-12 and IL-18 receptors. Therefore, Stat4E626G protein dominantly inhibits STAT4 function, but does not completely abrogate it (Figure 3).
Fig. 3.
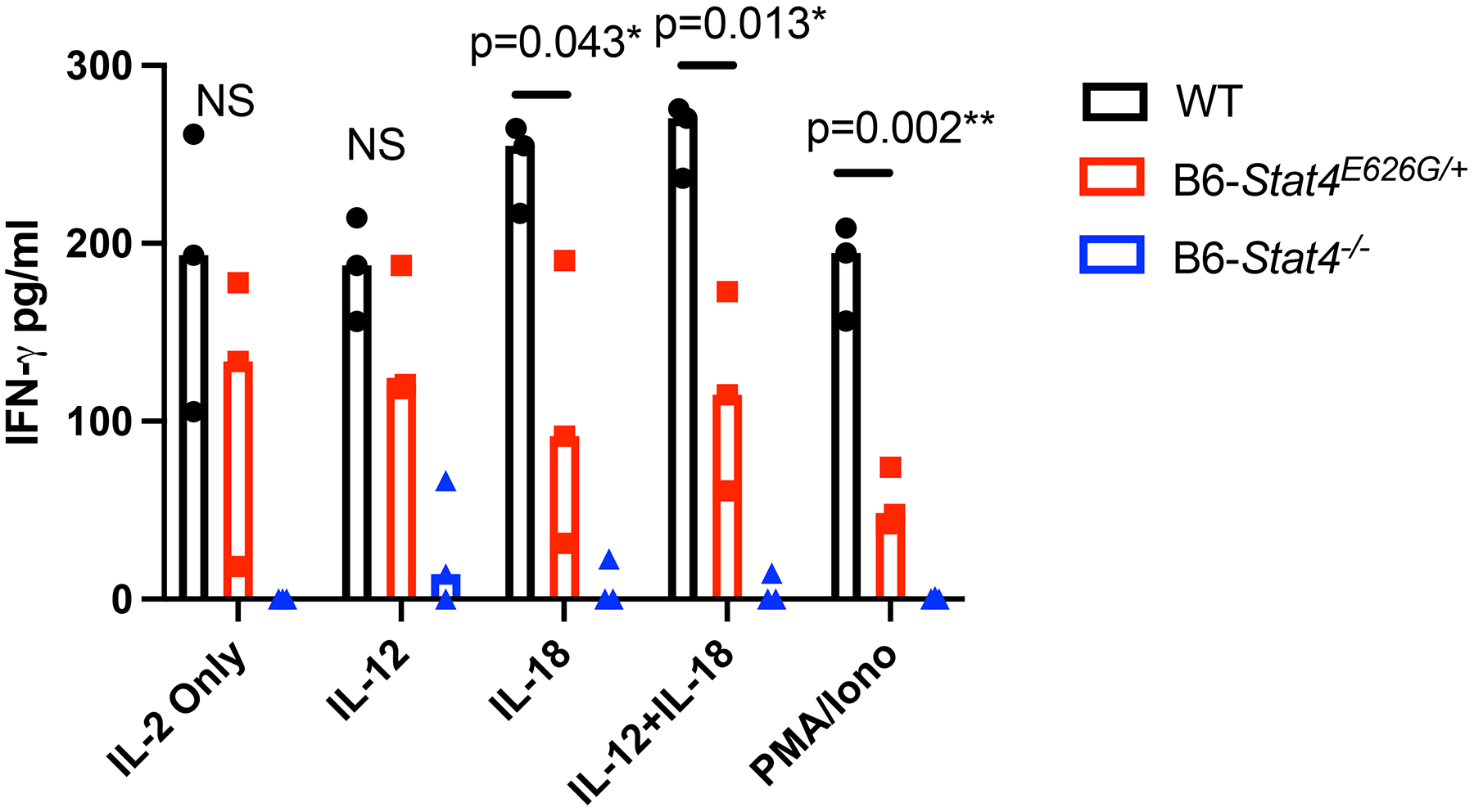
CD4+ T cells were purified from C57BL/6NJ (black circles), B6-Stat4E626G/+ (red squares), or B6-Stat4−/− (blue triangles) spleens. Purified cells were then plated with rmIL-2 and activated with CD3/28 Dynabeads according to the manufacturer’s instructions for 48 hours. At this point cells were supplemented with the indicated cytokine for 24 more hours. PMA/Ionomycin samples were treated for the last 4 hours of the experiment. Supernatants were then collected and analyzed for IFN-gamma production by ELISA. Graph is representative of 2 separate experiments of similar design. Significance was determined using a Mann-Whitney.
Cellularity and fungal burden following Cp1038 infection.
Since STAT4 signaling is depressed and mutant mice have increased mortality to Cp1038, we examined the underlying immune responses. We determined total cellularity in the lungs, mediastinal lymph nodes and spleens of B6-Stat4E626G/+ and WT-B6 mice following Cp1038 infection. Total lung cellularity after infection increased equivalently in B6-Stat4E626G/+ and WT-B6 mice (Figure 4A). The draining mediastinal lymph nodes (msLN) also showed increased cellularity over the course of the infection (Figure 4B). Notably, there was a delayed response in the B6-Stat4E626G/+ apparent at 2 weeks post infection (P=0.0218) suggesting a delayed adaptive immune response. However, by 4 weeks post infection this difference had normalized. Following this intranasal infection, the spleens showed very little increase in cellularity, indicating that most of the immune reactivity was in the thoracic cavity (Figure 4C).
Fig. 4.
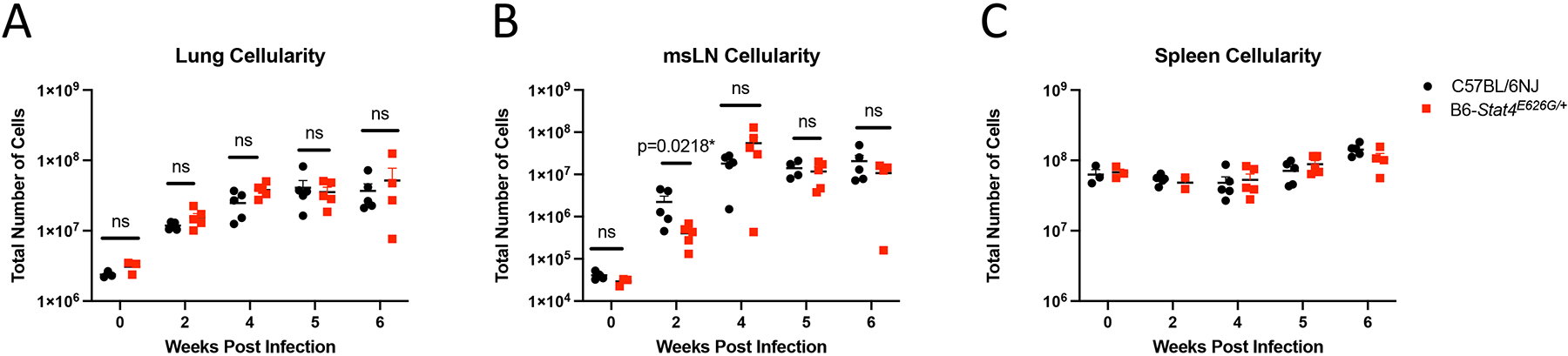
Stat4E626G/+ mice have similar cellularity in the lungs and mediastinal lymph nodes after Cp1038 infection. C57BL/6NJ (black circles), B6-Stat4E626G/+ (red squares) mice were intranasally infected with Cp1038. At the indicated time points mice were sacrificed (N=4–5/time point) and total cellularity was determined in the lungs (A) mediastinal lymph nodes (B) and spleen (C). Graph is representative of 2 separate experiments of similar design. Significance was determined using a Mann-Whitney on log transformed data.
Because of the decrease in total mediastinal lymph node cells in B6-Stat4E626G mice, the lung cellular composition at 2 weeks post infection was further characterized by multiparameter flow cytometry. The B6-Stat4E626G/+ lungs had a slight increase in total B cells as compared to WT-B6 mice (p=0.0213). This increase was seen in the IgM− IgD+ fractions, indicating that these are mature B Cells (p=0.0197) (Figure 5). Since there was a difference in mature B cells between the mouse strains we examined serum antibodies to Coccidioides. Fluorescence-linked immunosorbent assay (FLISA) examination of serum antibodies (IgG1, IgG2B, IgG2C and IgG3) to Coccidioides showed no differences between the two groups (Supplemental Figure 2). The T cell populations were not different between B6-Stat4E626G and WT-B6 mice in the lungs (Figure 5).
Fig. 5.
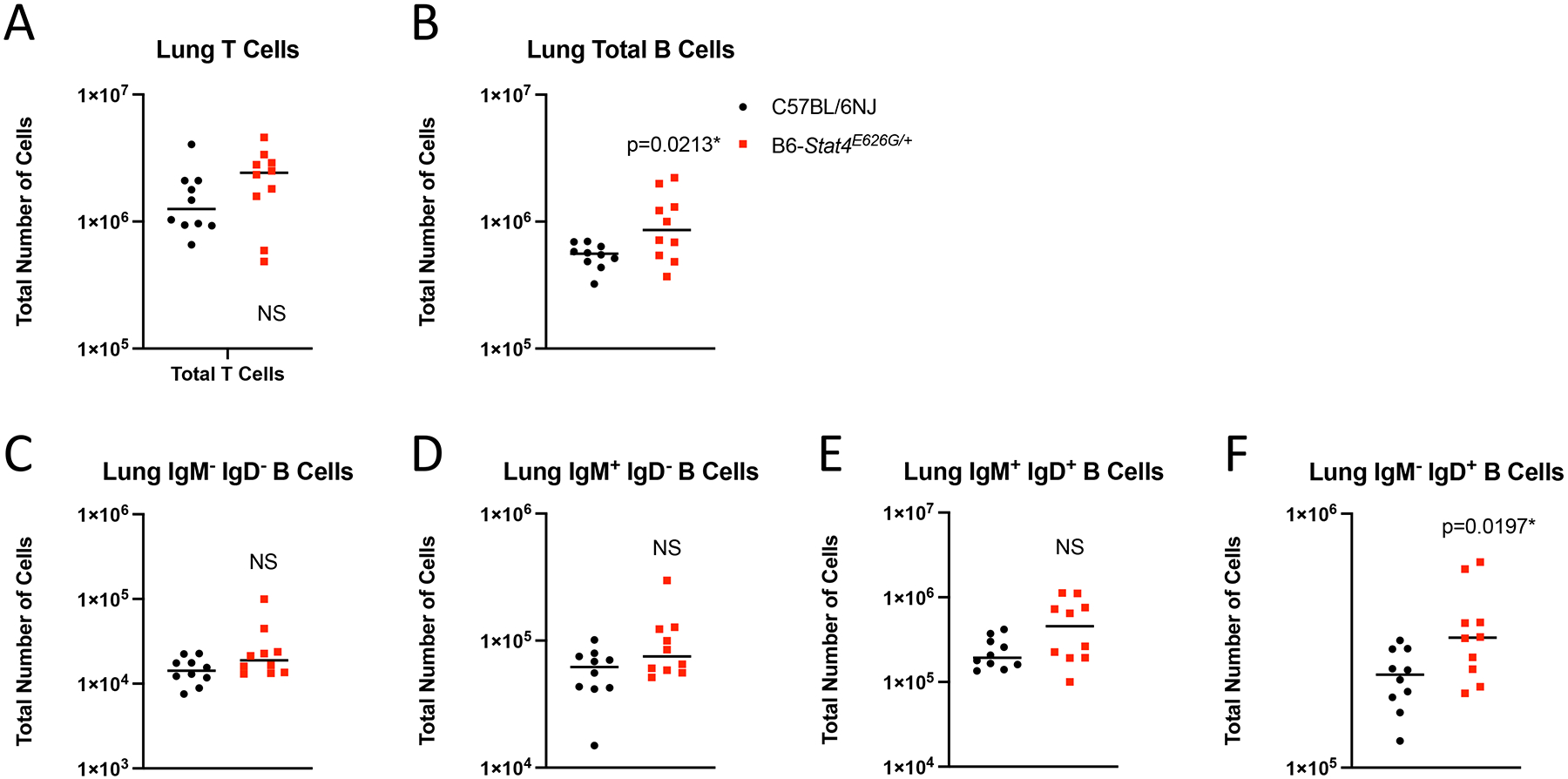
Stat4E626G/+ mice have slightly decreased accumulation of B cells the lungs after Cp1038 infection. C57BL/6NJ (black circles), B6-Stat4E626G/+ (red squares) mice were intranasally infected with Cp1038. At 2 weeks post infection mice were sacrificed (N=4–5/group) and total numbers of T (CD3+ CD19−)(A) and B (CD19+ B220+)(B) cells were determined in the lungs by flow cytometry. B cells were further subtyped by expression of IgM and IgD (C-F). Significance was determined by Mann-Whitney on log transformed data. Data is combined from 2 experiments of similar design.
Given that the msLN cellularity was decreased in B6-Stat4E626G/+ mice 2 weeks post-infection, we examined which cells were reduced. There was reduced accumulation of T-cells in the msLN of B6-Stat4E626G/+ compared to WT-B6 mice (Figure 6A). The defect in cell accumulation was seen in naïve, effector and central memory compartments of both CD4+ and CD8+ T cells (Figure 6B–G). Additionally, msLN from B6-Stat4E626G/+ failed to accumulate activated B cells as compared to WT-B6 mice (Figure 7).
Fig. 6.
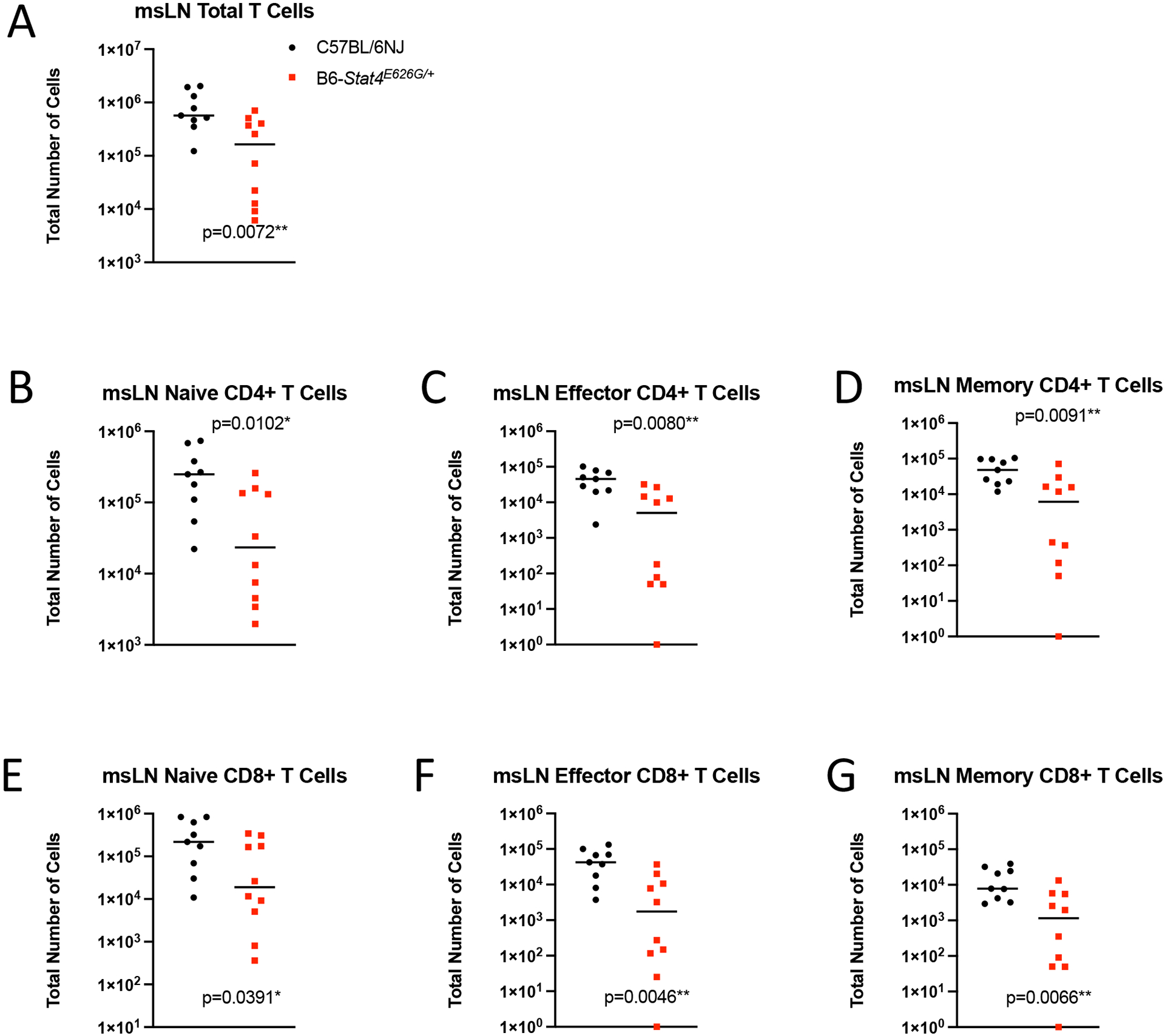
Stat4E626G/+ mice have decreased accumulation of total and activated T in the mediastinal lymph nodes 2 weeks after Cp1038 infection. C57BL/6NJ (black circles), B6-Stat4E626G/+ (red squares) mice were intranasally infected with Cp1038. Two weeks after infection mice were sacrificed (N=4–5/time point) and T cells were phenotyped in the mediastinal lymph nodes. (A) Total T Cells (CD3+ CD19−). (B,E) Naïve T cells (CD62L+ CD44var), (C,F) Effector T Cells (CD62L− CD44−), (D,G) Memory T Cells (CD62L− CD44+). Significance was determined by Mann-Whitney of log transformed data. Data is combined from 2 experiments of similar design.
Fig. 7.
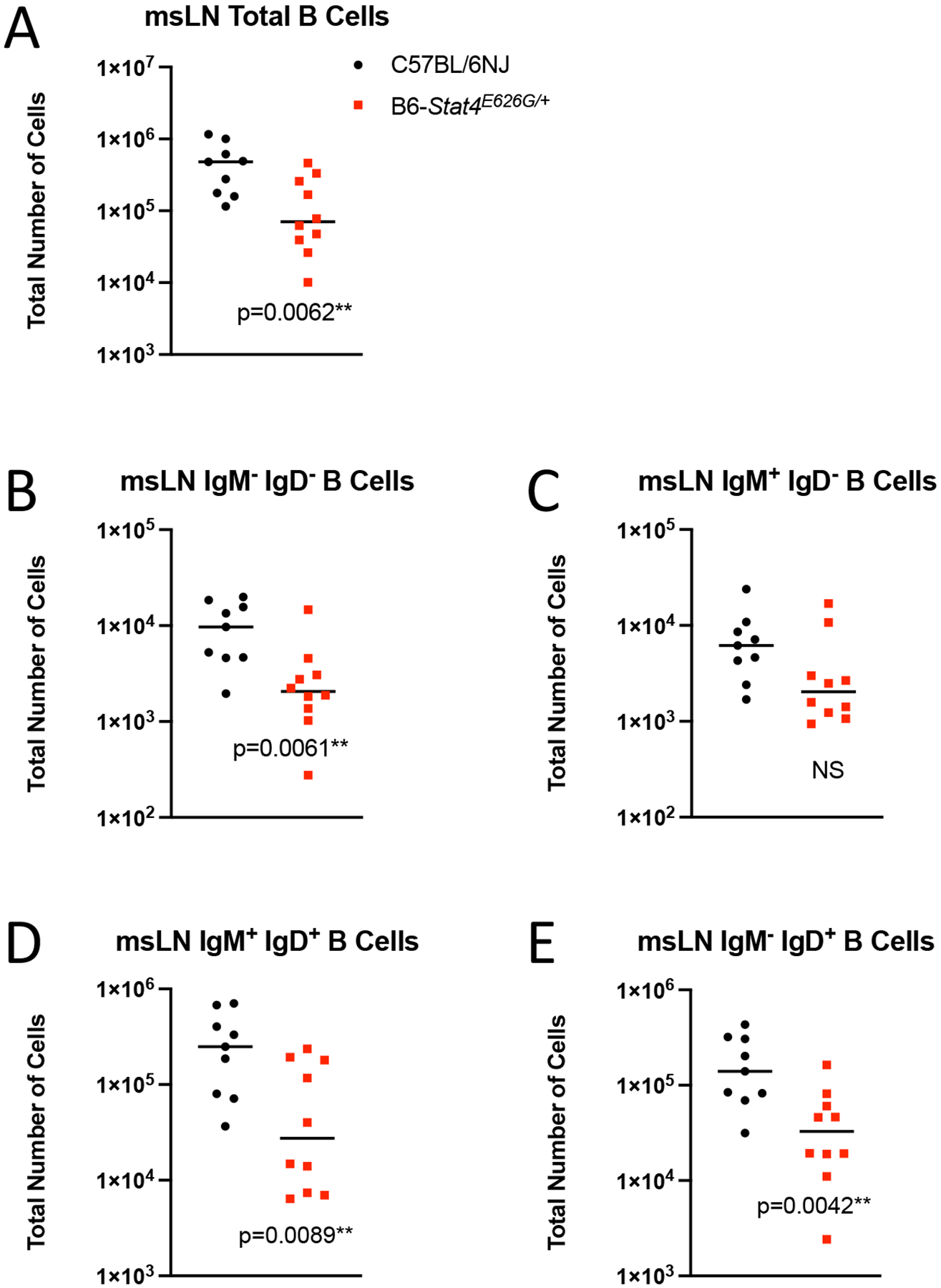
Stat4E626G/+ mice have decreased accumulation of total and activated B cells in the mediastinal lymph nodes 2 weeks after Cp1038 infection. C57BL/6NJ (black circles), B6-Stat4E626G/+ (red squares) mice were intranasally infected with Cp1038. Two weeks after infection mice were sacrificed (N=4–5/time point) and total B cells (CD19+ B220+) were enumerated in the mediastinal lymph nodes (A). B cell were further phenotyped for their expression of IgM and IgD (B-E). Significance was determined by Mann-Whitney of log transformed data. Data is combined from 2 experiments of similar design.
To chronicle adaptive immune cell accumulation early in the response, mediastinal lymph nodes and lungs were harvested at 2, 9 and 14 days post infection. At 2 days post infection there was no increase in lung or mediastinal lymph node cellularity in WT or mutant mice compared to uninfected mice. This lack of very early immune activation is consistent with the fact that Coccidioides spherule rupture with Cp1038 occurs 5 days after infection (34). However, at 9 days post infection the mediastinal lymph nodes had significantly fewer CD4+ (Figure 8) and CD8+ (Figure 9) T cells in the B6-Stat4E626G/+ mice than in WT-B6 mice, consistent with an impaired adaptive immune response. At 14 days post infection the T cell numbers were not significantly different in this infection though the B6-Stat4E626G/+ mice were trending towards lower accumulation similar to earlier experiments. Additionally we examined the accumulation of Innate Lymphoid Cells (ILC) in the lung and mediastinal lymph nodes on days 2, 7, 10 and 14 post infection. There were no differences amongst mouse strains in these cell types in the lungs. In the msLN, as with other cell types examined, no differences were found between B6 and B6-Stat4E626G/+on day 2 post infection. There was a decrease in Natural Killer (NK) and the IFN-γ producing ILC1s between day 7 and 10 in both mouse strains but the loss was more severe in the B6-Stat4E626G/+ mice. The NK and ILC1 cell numbers recovered in both mouse strains by day 14 (Figure 10). There was no differences observed in ILC2 and ILC3.
Fig. 8.
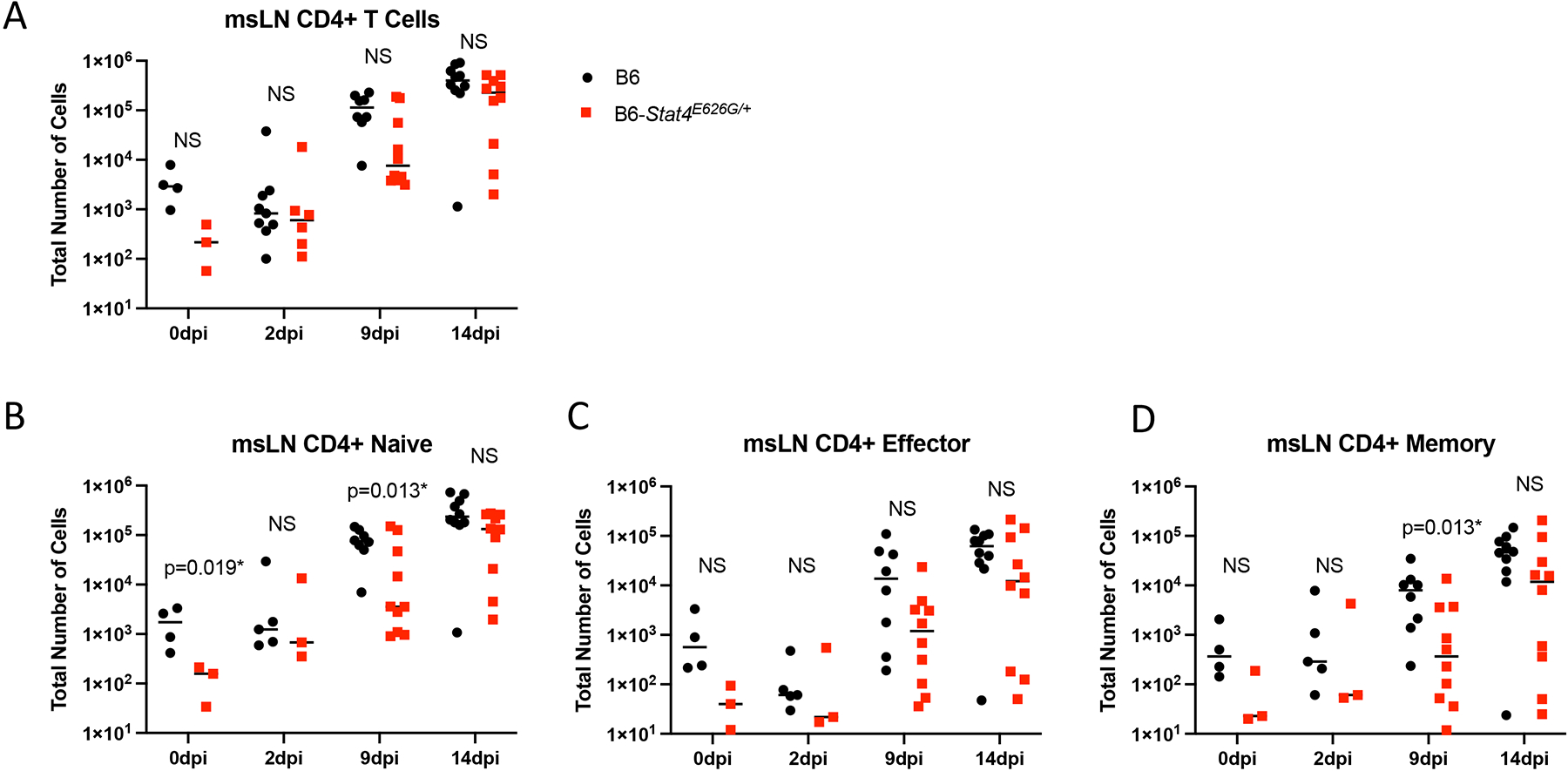
Stat4E626G/+ mice have decreased accumulation of total and activated CD4+ T cells in the mediastinal lymph nodes early after Cp1038 infection. C57BL/6NJ (black circles), B6-Stat4E626G/+ (red squares) mice were intranasally infected with Cp1038. At the indicated time point after infection mice were sacrificed (N=4–5/time point) and total CD4+ T cells were enumerated in the mediastinal lymph nodes (A). Cells were further divided into naïve (B) (CD62L+, CD44var), Effector (C) (CD62L− CD44−), and Memory (D) (CD62L− CD44+) subtypes. Significance was determined by Mann-Whitney of log transformed data. Data is combined from 2 experiments of similar design.
Fig. 9.
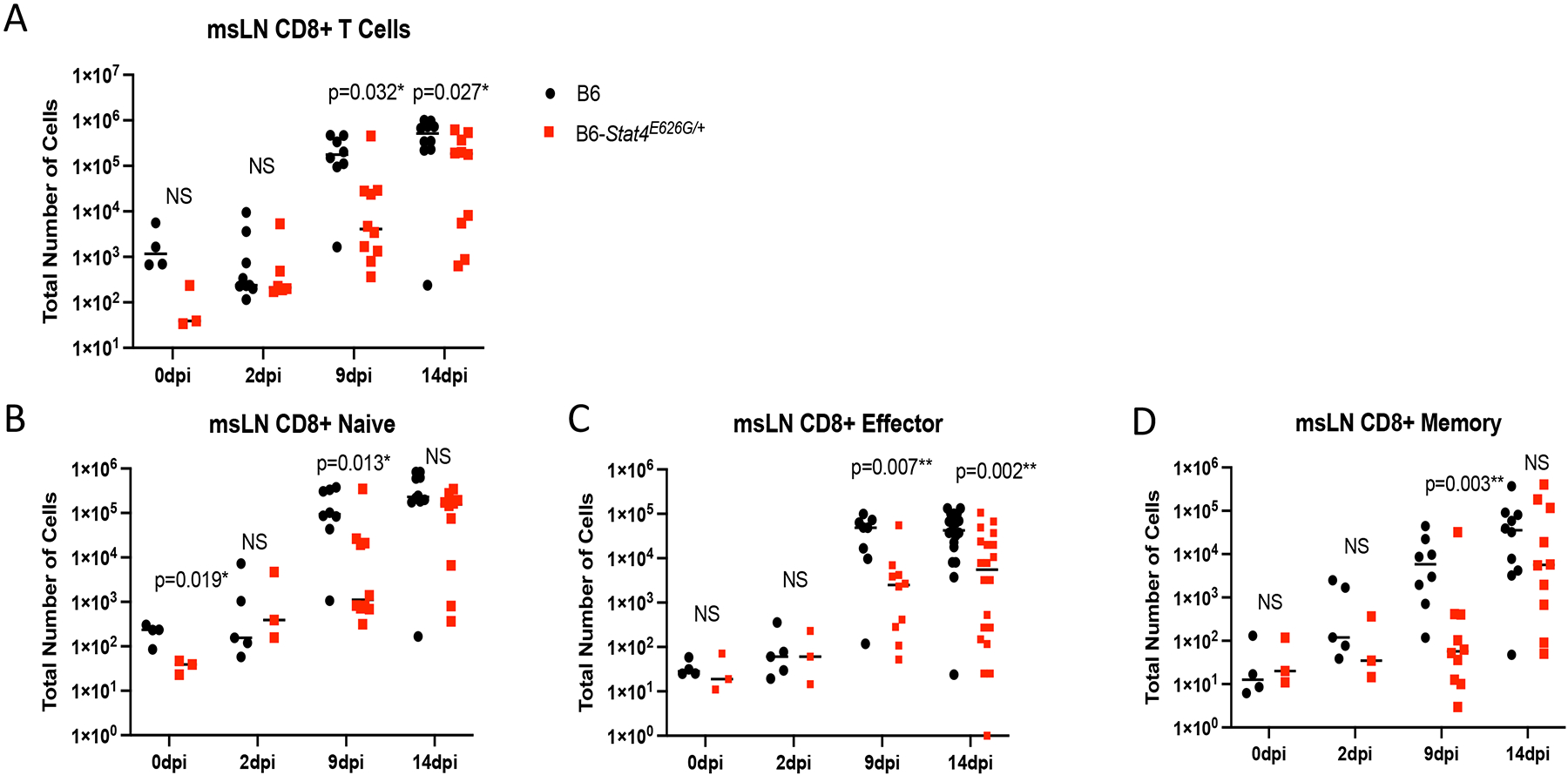
Stat4E626G/+ mice have decreased accumulation of total and activated CD8+ T cells in the mediastinal lymph nodes early after Cp1038 infection. C57BL/6NJ (black circles), B6-Stat4E626G/+ (red squares) mice were intranasally infected with Cp1038. At the indicated time point after infection mice were sacrificed (N=4–5/time point) and total CD8+ cells were enumerated in the mediastinal lymph nodes (A). Cells were further divided into naïve (B) (CD62L+, CD44var), Effector (C) (CD62L− CD44−), and Memory (D) (CD62L− CD44+) subtypes. Significance was determined by Mann-Whitney of log transformed data. Data is combined from 2 experiments of similar design.
Fig. 10.
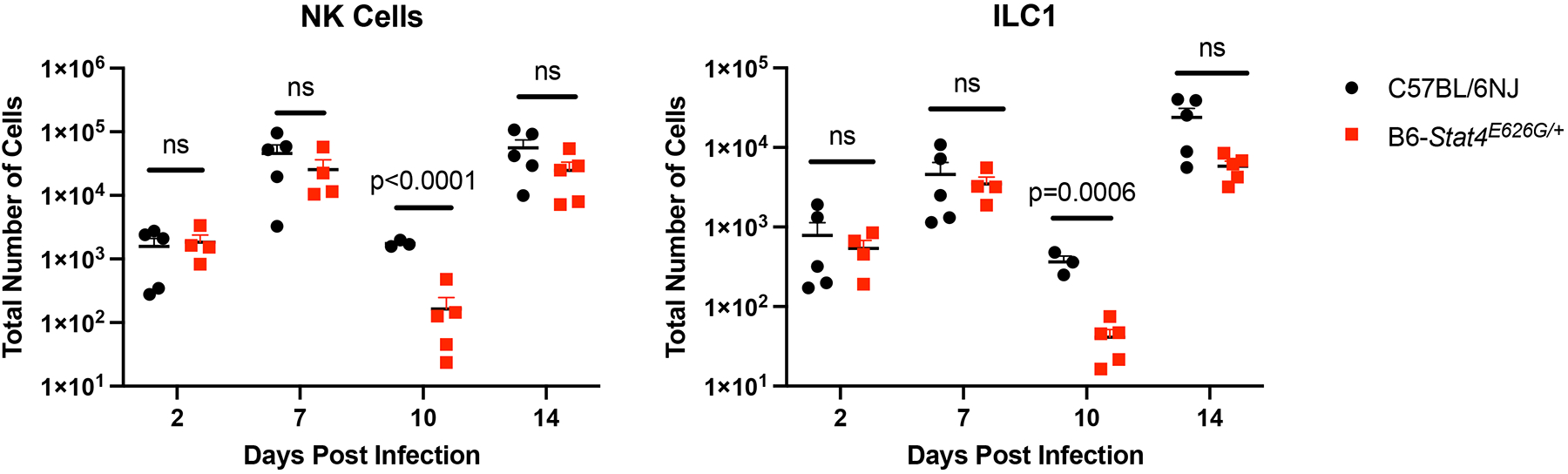
Stat4E626G/+ mice have decreased accumulation of NK and ILC1 cells in the mediastinal lymph nodes early after Cp1038 infection. C57BL/6NJ (black circles), B6-Stat4E626G/+ (red squares) mice were intranasally infected with Cp1038. At the indicated time point after infection mice were sacrificed (N=3–5/time point) and total NK cells (NK1.1+, NKp46+, CD49a−) (A) and Type 1 ILCs (NK1.1+, NKp46+, CD49a+) were enumerated in the mediastinal lymph nodes. Significance was determined by Mann-Whitney of log transformed data.
Innate cell recruitment to the lungs was not different between the WT and mutant mouse strains indicating that Stat4E626G mutation did not affect initial recognition of Coccidioides, innate recruitment, or retention (Supplemental Figure 3).
B6-Stat4E626G mice have lower levels of IFN-gamma related cytokines after Coccidioides infection
IFN-gamma responses are crucial to recovery from CM in humans and mice (37). As expected, STAT4 is also critical as it functions in both CD4 and CD8 T cells as an regulator of the IFN-gamma production. We hypothesized that the decreased STAT4 signaling observed in vitro would also result in lowered IFN-gamma responses in vivo. To test this, we characterized the differences in secretion of cytokines following CM in B6-Stat4E626G and WT-B6 mice infected intranasally with Cp1038 and sacrificed 2, 4, 5 and 6 weeks later. Lungs were collected to determine both cellular composition and cytokine production. Spleens, peripheral blood, and msLN were analyzed for total cell number and cellular composition. Both B6-Stat4E626G and WT-B6 mice showed similar increases in lung weights, a general measure of disease progression (39), beginning 4 weeks post infection (Supplemental Figure 3). Lung fungal burdens were similar in both B6-Stat4E626G and WT-B6, except at 5 weeks post infection, when it was greater in B6-Stat4E626G/+ mice (Figure 11A). Splenic fungal burden was absent at 2 weeks post infection but identical thereafter in both B6-Stat4E626G and WT-B6 (Figure 11B).
Fig. 11.
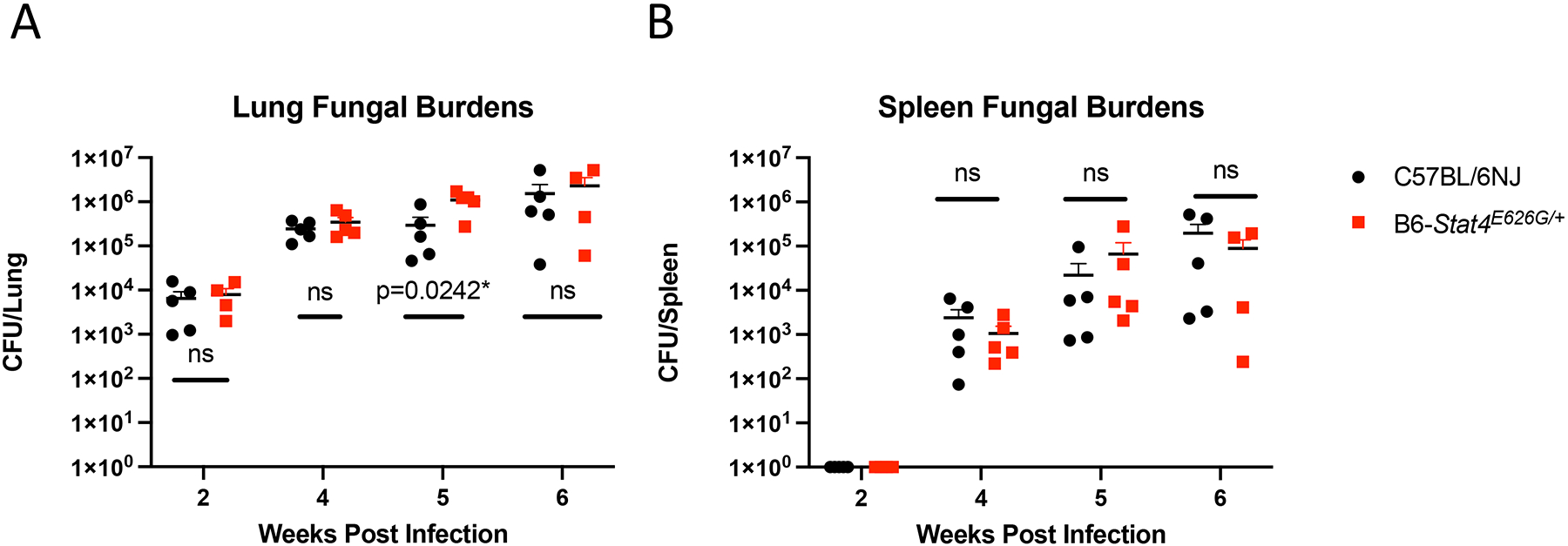
Stat4E626G/+ mice have similar fungal burdens after Cp1038 infection as compared to B6 mice. C57BL/6NJ (black circles), B6-Stat4E626G/+ (red squares) mice were intranasally infected with Cp1038 at the indicated time point after infection mice were sacrificed (N=4–5/time point) and fungal burdens were determined in the lungs (A) and spleen (B). Significance was determined by Mann-Whitney of log transformed data. Data in representative of 2 experiments of similar design.
To examine the cytokine milieu in the lungs directly, thinly sliced lung sections were cultured over-night and cytokine secretion was measured in the supernatants by Luminex. Differences in cytokine levels were reanalyzed by targeted ELISA to confirm the results of the screening assay. Lung slices from WT-B6 mice demonstrated upregulation of IFN-gamma (Figure 12A) and IFN-gamma related cytokines, IP-10 (Figure 12B) and MIG (Figure 12C) post infection. In contrast, B6-Stat4E626G/+ mice failed to increase production of these IFN-gamma-dependent cytokines and chemokines. These in vitro results correlated with serum cytokine levels (Figure 12D–F).
Fig. 12.
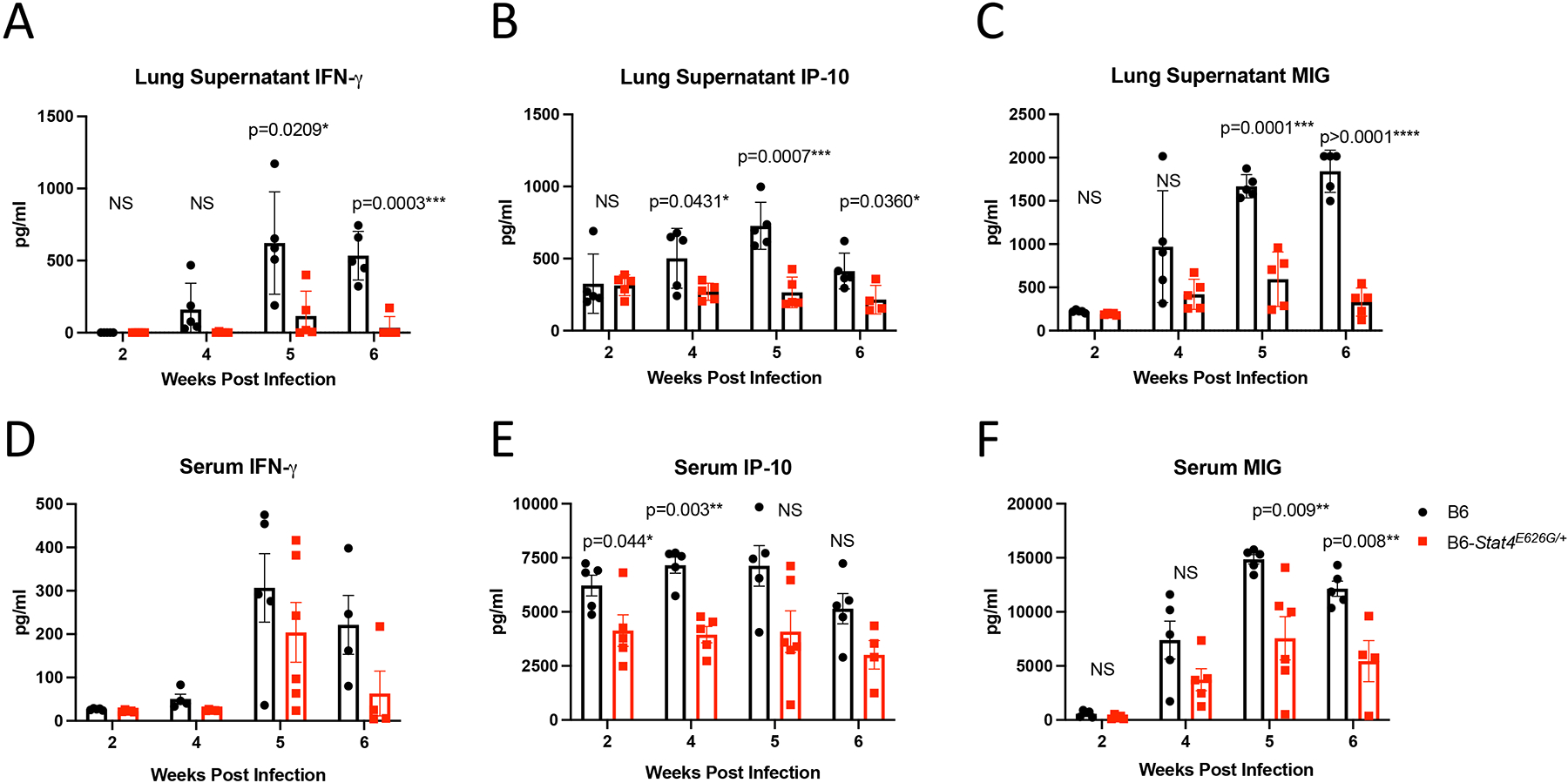
Stat4E626G/+ mice have decreased production of IFN-gamma and related cytokines after Cp1038 infection as compared to B6 mice. C57BL/6NJ (black circles), B6-Stat4E626G/+ (red squares) mice were intranasally infected with Cp1038 at the indicated time point after infection mice were sacrificed (N=4–5/time point). Lungs were finely sliced and placed in complete RPMI for 24 hours. Supernatant was then collected and analyzed for IFN-gamma (A), IP-10 (B), and MIG (C) cytokine production by ELISA. Serum was harvested and analyzed for IFN-gamma (D), IP-10 (E), and MIG (F) by ELISA. Significance was determined by Mann-Whitney.
These results demonstrate a dominant negative effect of STAT4E626G on the function of STAT4 both in vivo and in vitro in mice, modeling the effect in humans. Mice carrying the mutation associated with human DCM had significantly more rapid disease progression than did WT mice. Similarly, the apparently normal lymphoid system of the E626 mice is similar to the apparently normal immune system in the patients.
Discussion
We identified a multi-generational family with DCM associated with a novel heterozygous mutation, STAT4 E626G. As with the patients, mice heterozygous for Stat-4E626G were more susceptible to coccidioidal infection. These mice showed normal development of immune cells in both primary and secondary lymphoid compartments, indicating this mutation did not lead to a global defect in immune system development. This is not surprising since Stat4 knockout mice are also grossly normal (2, 40).
STAT4 regulates the IL-12-IFN-gamma pathway in both humans and mice. A variety of mutations in the IL-12/IFN-gamma pathways have been described predisposing to a variety of fungal diseases (28, 41–43). Although a complete loss of STAT4 function in knockout mice dramatically ablates the IFN-gamma response and results in the loss of Th1 function, little information is available on specific missense mutations. STAT4 E651V mutation was reported to reduce in vitro STAT4 phosphorylation and nuclear translocation in a family with Paracoccidioides brasiliensis (4). Although Stat4E626G is expressed at levels similar to WT Stat4, CD4+ T cells were deficient in making IFN-gamma in response to IL-12+IL-18. Mice either heterozygous or homozygous for Stat4E626G were similarly susceptible to infection with Coccidioides, indicating that one copy of mutant Stat4 was sufficient for infection sensitivity while mice heterozygous for a null allele (Stat4+/−) were phenotypically wild type, confirming Stat4E626G as a dominant negative allele.
The immune response after intranasal Coccidioides infection of B6-Stat4E626G mice showed relatively few global differences compared to those in B6 controls. Spleens showed no increase in total cellularity, indicating that most of the immune response was limited to the lungs and draining mediastinal lymph nodes. The cellular accumulation into the lungs showed few differences. The proliferation and homing of T cells was unaffected, although B6-Stat4E626G/+ mice did have a slightly elevated number of B cells compared to controls. In contrast to the relatively normal cell numbers, supernatants from lung homogenates from infected B6-Stat4E626G/+ mice made significantly less IFN-gamma and IFN-gamma driven chemokines than controls. The IFN-gamma that was detected 4–5 weeks after infection was most likely produced as a result of adaptive immune responses. These defects in IFN-gamma and its downstream induced genes are overall consistent with impaired expression of T-bet and reduced polarization of CD4 cells towards Th1 responses, as well as IFN-gamma production of CD8 T cells.
Many groups have shown a dependence of IFN-gamma for both survival of Coccidioides infection as well as development of protective immunity by experimental vaccination (30, 44–47). Of note, there was no detectable IL-12p40 or IL-12p70 in the lung supernatants of either WT-B6 or B6-Stat4E626G, consistent with these low-abundance IFN-gamma inducing cytokines being consumed in a paracrine manner and not detectable in culture supernatants.
The most significant alterations in the immune responses were found in the draining mediastinal lymph nodes. B6-Stat4E626G/+ mice failed to accumulate activated T cells and B cells during the early phase of infection, < 2 weeks post infection. At 2 days post infection, before the first rupture of Coccidioides endospores, there was no increase in cellularity in the mediastinal lymph nodes in either WT-B6 or B6-Stat4E626G. This indicates little widespread immune response or activation to the initial infection, although it is possible that there may have been some small local responses in the lung.
At 9- and 14-days post infection B6-Stat4E626G/+ mice had significantly fewer cells in their mediastinal lymph nodes. At 2–6 weeks post infection, while there was no difference between WT-B6 or B6-Stat4E626G total lung cellularity, there was significant and persistent reduction in cellularity in the mediastinal lymph nodes. Whether this cellular accumulation is recruitment from the periphery or local expansion remains to be explored. While this defect in cellularity is subtle, it has a profound effect on disease severity and time to death. Interestingly, the fungal burdens in lungs and spleen as well as lung weights were similar between WT-B6 or B6-Stat4E626G. Therefore, the early death seen in B6-Stat4E626G was not due to fungal proliferation or lung vascular permeability but correlated most closely with the impaired IFN-gamma responses. It is important to note that STAT4 is also involved in IL-23 signaling, another cytokine critically important to mycobacterial and fungal responses (48–52). Improperly high IL-4 production has been seen in other fungal infections including Histoplasma, Aspergillus, and Candida (53–55), and we detected elevated IL-4 in the serum and lungs of some of infected B6-Stat4E626G/+mice, but as a group the difference was not significantly different from B6. Investigation of the effective cause of early mortality in the B6-Stat4E626G mice is ongoing.
Dominant negative mutation in STAT4 impairs the production of IFN-gamma and its downstream effectors. It is noteworthy that our patients were apparently not subject to other severe viral, bacterial or fungal infections, suggesting that STAT4 is redundant for the majority of immune responses required in everyday life. However, this mutation predisposed them, so far apparently uniquely, to disseminated coccidioidomycosis, highlighting the importance of host genetics in human coccidioidal infections.
Supplementary Material
References
- 1.Deng JC, Zeng X, Newstead M, Moore TA, Tsai WC, Thannickal VJ, and Standiford TJ. 2004. STAT4 is a critical mediator of early innate immune responses against pulmonary Klebsiella infection. J Immunol 173: 4075–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elsegeiny W, Zheng M, Eddens T, Gallo RL, Dai G, Trevejo-Nunez G, Castillo P, Kracinovsky K, Cleveland H, Horne W, Franks J, Pociask D, Pilarski M, Alcorn JF, Chen K, and Kolls JK. 2018. Murine models of Pneumocystis infection recapitulate human primary immune disorders. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, Kung AL, Cereb N, Yao TP, Yang SY, and Reiner SL. 2001. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science 292: 1907–1910. [DOI] [PubMed] [Google Scholar]
- 4.Schimke LF, Hibbard J, Martinez-Barricarte R, Khan TA, de Souza Cavalcante R, de Oliveira E. Borges Junior, Franca T. Takahashi, Iqbal A, Yamamoto G, Arslanian C, Feriotti C, Costa TA, Bustamante J, Boisson-Dupuis S, Casanova JL, Marzagao Barbuto JA, Zatz M, Mendes R. Poncio, Garcia Calich VL, Ochs HD, Torgerson TR, Cabral-Marques O, and Condino-Neto A. 2017. Paracoccidioidomycosis Associated With a Heterozygous STAT4 Mutation and Impaired IFN-gamma Immunity. J Infect Dis 216: 1623–1634. [DOI] [PubMed] [Google Scholar]
- 5.Thieu VT, Yu Q, Chang HC, Yeh N, Nguyen ET, Sehra S, and Kaplan MH. 2008. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity 29: 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinstein JS, Laidlaw BJ, Lu Y, Wang JK, Schulz VP, Li N, Herman EI, Kaech SM, Gallagher PG, and Craft J. 2018. STAT4 and T-bet control follicular helper T cell development in viral infections. J Exp Med 215: 337–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu J, Jankovic D, Oler AJ, Wei G, Sharma S, Hu G, Guo L, Yagi R, Yamane H, Punkosdy G, Feigenbaum L, Zhao K, and Paul WE. 2012. The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity 37: 660–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blake JA, Eppig JT, Kadin JA, Richardson JE, Smith CL, Bult CJ, and G. the Mouse Genome Database. 2017. Mouse Genome Database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucleic Acids Res 45: D723–D729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balashov KE, Smith DR, Khoury SJ, Hafler DA, and Weiner HL. 1997. Increased interleukin 12 production in progressive multiple sclerosis: induction by activated CD4+ T cells via CD40 ligand. Proc Natl Acad Sci U S A 94: 599–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caragol I, Raspall M, Fieschi C, Feinberg J, Larrosa MN, Hernandez M, Figueras C, Bertran JM, Casanova JL, and Espanol T. 2003. Clinical tuberculosis in 2 of 3 siblings with interleukin-12 receptor beta1 deficiency. Clin Infect Dis 37: 302–306. [DOI] [PubMed] [Google Scholar]
- 11.Comabella M, Balashov K, Issazadeh S, Smith D, Weiner HL, and Khoury SJ. 1998. Elevated interleukin-12 in progressive multiple sclerosis correlates with disease activity and is normalized by pulse cyclophosphamide therapy. J Clin Invest 102: 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.el-Shabrawi Y, Livir-Rallatos C, Christen W, Baltatzis S, and Foster CS. 1998. High levels of interleukin-12 in the aqueous humor and vitreous of patients with uveitis. Ophthalmology 105: 1659–1663. [DOI] [PubMed] [Google Scholar]
- 13.MacLennan C, Fieschi C, Lammas DA, Picard C, Dorman SE, Sanal O, MacLennan JM, Holland SM, Ottenhoff TH, Casanova JL, and Kumararatne DS. 2004. Interleukin (IL)-12 and IL-23 are key cytokines for immunity against Salmonella in humans. J Infect Dis 190: 1755–1757. [DOI] [PubMed] [Google Scholar]
- 14.Ottenhoff TH, Verreck FA, Hoeve MA, and van de Vosse E. 2005. Control of human host immunity to mycobacteria. Tuberculosis (Edinb) 85: 53–64. [DOI] [PubMed] [Google Scholar]
- 15.van de Vosse E, and Ottenhoff TH. 2006. Human host genetic factors in mycobacterial and Salmonella infection: lessons from single gene disorders in IL-12/IL-23-dependent signaling that affect innate and adaptive immunity. Microbes Infect 8: 1167–1173. [DOI] [PubMed] [Google Scholar]
- 16.Galgiani JN, Ampel NM, Blair JE, Catanzaro A, Geertsma F, Hoover SE, Johnson RH, Kusne S, Lisse J, MacDonald JD, Meyerson SL, Raksin PB, Siever J, Stevens DA, Sunenshine R, and Theodore N. 2016. 2016 Infectious Diseases Society of America (IDSA) Clinical Practice Guideline for the Treatment of Coccidioidomycosis. Clin Infect Dis 63: e112–146. [DOI] [PubMed] [Google Scholar]
- 17.Freedman M, Jackson BR, McCotter O, and Benedict K. 2018. Coccidioidomycosis Outbreaks, United States and Worldwide, 1940–2015. Emerg Infect Dis 24: 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grizzle AJ, Wilson L, Nix DE, and Galgiani JN. 2021. Clinical and Economic Burden of Valley Fever in Arizona: An Incidence-Based Cost-of-Illness Analysis. Open Forum Infect Dis 8: ofaa623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson L, Ting J, Lin H, Shah R, MacLean M, Peterson MW, Stockamp N, Libke R, and Brown P. 2019. The Rise of Valley Fever: Prevalence and Cost Burden of Coccidioidomycosis Infection in California. Int J Environ Res Public Health 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim MM, Blair JE, Carey EJ, Wu Q, and Smilack JD. 2009. Coccidioidal pneumonia, Phoenix, Arizona, USA, 2000–2004. Emerg Infect Dis 15: 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valdivia L, Nix D, Wright M, Lindberg E, Fagan T, Lieberman D, Stoffer T, Ampel NM, and Galgiani JN. 2006. Coccidioidomycosis as a common cause of community-acquired pneumonia. Emerg Infect Dis 12: 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blair JE, Ampel NM, and Hoover SE. 2019. Coccidioidomycosis in selected immunosuppressed hosts. Med Mycol 57: S56–S63. [DOI] [PubMed] [Google Scholar]
- 23.Adam RD, Elliott SP, and Taljanovic MS. 2009. The spectrum and presentation of disseminated coccidioidomycosis. Am J Med 122: 770–777. [DOI] [PubMed] [Google Scholar]
- 24.Seitz AE, Prevots DR, and Holland SM. 2012. Hospitalizations associated with disseminated coccidioidomycosis, Arizona and California, USA. Emerg Infect Dis 18: 1476–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Odio CD, Marciano BE, Galgiani JN, and Holland SM. 2017. Risk Factors for Disseminated Coccidioidomycosis, United States. Emerg Infect Dis 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Odio CD, Milligan KL, McGowan K, Spergel A. K. Rudman, Bishop R, Boris L, Urban A, Welch P, Heller T, Kleiner D, Jackson MA, Holland SM, and Freeman AF. 2015. Endemic mycoses in patients with STAT3-mutated hyper-IgE (Job) syndrome. J Allergy Clin Immunol 136: 1411–1413 e1411–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sampaio EP, Hsu AP, Pechacek J, Bax HI, Dias DL, Paulson ML, Chandrasekaran P, Rosen LB, Carvalho DS, Ding L, Vinh DC, Browne SK, Datta S, Milner JD, Kuhns DB, Priel D. A. Long, Sadat MA, Shiloh M, Marco B. De, Alvares M, Gillman JW, Ramarathnam V, de la Morena M, Bezrodnik L, Moreira I, Uzel G, Johnson D, Spalding C, Zerbe CS, Wiley H, Greenberg DE, Hoover SE, Rosenzweig SD, Galgiani JN, and Holland SM. 2013. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J Allergy Clin Immunol 131: 1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vinh DC, Masannat F, Dzioba RB, Galgiani JN, and Holland SM. 2009. Refractory disseminated coccidioidomycosis and mycobacteriosis in interferon-gamma receptor 1 deficiency. Clin Infect Dis 49: e62–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vinh DC, Schwartz B, Hsu AP, Miranda DJ, Valdez PA, Fink D, Lau KP, Long-Priel D, Kuhns DB, Uzel G, Pittaluga S, Hoover S, Galgiani JN, and Holland SM. 2011. Interleukin-12 receptor beta1 deficiency predisposing to disseminated Coccidioidomycosis. Clin Infect Dis 52: e99–e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shubitz LF, Powell DA, Trinh HT, Lewis ML, Orbach MJ, Frelinger JA, and Galgiani JN. 2018. Viable spores of Coccidioides posadasii Deltacps1 are required for vaccination and provide long lasting immunity. Vaccine 36: 3375–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Concordet JP, and Haeussler M. 2018. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res 46: W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shubitz LF, Powell DA, Butkiewicz CD, Lewis ML, Trinh HT, Frelinger JA, Orbach MJ, and Galgiani JN. 2021. A Chronic Murine Disease Model of Coccidioidomycosis Using Coccidioides posadasii, Strain 1038. J Infect Dis 223: 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roberts LM, Davies JS, Sempowski GD, and Frelinger JA. 2014. IFN-gamma, but not IL-17A, is required for survival during secondary pulmonary Francisella tularensis Live Vaccine Stain infection. Vaccine 32: 3595–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Powell DA, and Frelinger JA. 2017. Efficacy of Resistance to Francisella Imparted by ITY/NRAMP/SLC11A1 Depends on Route of Infection. Front Immunol 8: 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galgiani JN, Catanzaro A, Cloud GA, Johnson RH, Williams PL, Mirels LF, Nassar F, Lutz JE, Stevens DA, Sharkey PK, Singh VR, Larsen RA, Delgado KL, Flanigan C, and Rinaldi MG. 2000. Comparison of oral fluconazole and itraconazole for progressive, nonmeningeal coccidioidomycosis. A randomized, double-blind trial. Mycoses Study Group. Ann Intern Med 133: 676–686. [DOI] [PubMed] [Google Scholar]
- 36.Liu X, Jian X, and Boerwinkle E. 2011. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat 32: 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X, Wu C, Li C, and Boerwinkle E. 2016. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum Mutat 37: 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, C. Genome Aggregation Database, Neale BM, Daly MJ, and MacArthur DG. 2020. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581: 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huppert M, Sun SH, Gleason-Jordon I, and Vukovich KR. 1976. Lung weight parallels disease severity in experimental coccidioidomycosis. Infect Immun 14: 1356–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ren M, Kazemian M, Zheng M, He J, Li P, Oh J, Liao W, Li J, Rajaseelan J, Kelsall BL, Peltz G, and Leonard WJ. 2020. Transcription factor p73 regulates Th1 differentiation. Nat Commun 11: 1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bustamante J, Boisson-Dupuis S, Jouanguy E, Picard C, Puel A, Abel L, and Casanova JL. 2008. Novel primary immunodeficiencies revealed by the investigation of paediatric infectious diseases. Curr Opin Immunol 20: 39–48. [DOI] [PubMed] [Google Scholar]
- 42.Ouederni M, Sanal O, Ikinciogullari A, Tezcan I, Dogu F, Sologuren I, Pedraza-Sanchez S, Keser M, Tanir G, Nieuwhof C, Colino E, Kumararatne D, Levy J, Kutukculer N, Aytekin C, Herrera-Ramos E, Bhatti M, Karaca N, Barbouche R, Broides A, Goudouris E, Franco JL, Parvaneh N, Reisli I, Strickler A, Shcherbina A, Somer A, Segal A, Angel-Moreno A, Lezana-Fernandez JL, Bejaoui M, Bobadilla-Del Valle M, Kachboura S, Sentongo T, Ben-Mustapha I, Bustamante J, Picard C, Puel A, Boisson-Dupuis S, Abel L, Casanova JL, and Rodriguez-Gallego C. 2014. Clinical features of Candidiasis in patients with inherited interleukin 12 receptor beta1 deficiency. Clin Infect Dis 58: 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zerbe CS, and Holland SM. 2005. Disseminated histoplasmosis in persons with interferon-gamma receptor 1 deficiency. Clin Infect Dis 41: e38–41. [DOI] [PubMed] [Google Scholar]
- 44.Cole GT, Hung CY, Sanderson SD, Hurtgen BJ, Wuthrich M, Klein BS, Deepe GS, Ostroff GR, and Levitz SM. 2013. Novel strategies to enhance vaccine immunity against coccidioidomycosis. PLoS Pathog 9: e1003768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Donovan FM, Shubitz L, Powell D, Orbach M, Frelinger J, and Galgiani JN. 2019. Early Events in Coccidioidomycosis. Clin Microbiol Rev 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Viriyakosol S, Mdel P. Jimenez, Gurney MA, Ashbaugh ME, and Fierer J. 2013. Dectin-1 is required for resistance to coccidioidomycosis in mice. mBio 4: e00597–00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xue J, Chen X, Selby D, Hung CY, Yu JJ, and Cole GT. 2009. A genetically engineered live attenuated vaccine of Coccidioides posadasii protects BALB/c mice against coccidioidomycosis. Infect Immun 77: 3196–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kagami S, Rizzo HL, Kurtz SE, Miller LS, and Blauvelt A. 2010. IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans. J Immunol 185: 5453–5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khader SA, Guglani L, Rangel-Moreno J, Gopal R, Junecko BA, Fountain JJ, Martino C, Pearl JE, Tighe M, Lin YY, Slight S, Kolls JK, Reinhart TA, Randall TD, and Cooper AM. 2011. IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung. J Immunol 187: 5402–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee MP, Wu KK, Lee EB, and Wu JJ. 2020. Risk for deep fungal infections during IL-17 and IL-23 inhibitor therapy for psoriasis. Cutis 106: 199–205. [DOI] [PubMed] [Google Scholar]
- 51.Nur S, Sparber F, Lemberg C, Guiducci E, Schweizer TA, Zwicky P, Becher B, and LeibundGut-Landmann S. 2019. IL-23 supports host defense against systemic Candida albicans infection by ensuring myeloid cell survival. PLoS Pathog 15: e1008115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wozniak TM, Ryan AA, and Britton WJ. 2006. Interleukin-23 restores immunity to Mycobacterium tuberculosis infection in IL-12p40-deficient mice and is not required for the development of IL-17-secreting T cell responses. J Immunol 177: 8684–8692. [DOI] [PubMed] [Google Scholar]
- 53.Cenci E, Mencacci A, Sero G. Del, Bacci A, Montagnoli C, d’Ostiani CF, Mosci P, Bachmann M, Bistoni F, Kopf M, and Romani L. 1999. Interleukin-4 causes susceptibility to invasive pulmonary aspergillosis through suppression of protective type I responses. J Infect Dis 180: 1957–1968. [DOI] [PubMed] [Google Scholar]
- 54.Gildea LA, Gibbons R, Finkelman FD, and Deepe GS Jr. 2003. Overexpression of interleukin-4 in lungs of mice impairs elimination of Histoplasma capsulatum. Infect Immun 71: 3787–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tonnetti L, Spaccapelo R, Cenci E, Mencacci A, Puccetti P, Coffman RL, Bistoni F, and Romani L. 1995. Interleukin-4 and -10 exacerbate candidiasis in mice. Eur J Immunol 25: 1559–1565. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.