

Abstract
Parkinson’s Disease is a progressive neurodegenerative disorder that is characterized by pathological protein inclusions that form in the brains of patients, leading to neuron loss and the observed clinical symptoms. These inclusions, containing aggregates of the protein α-Synuclein, spread throughout the brain as the disease progresses. This spreading follows patterns that inform our understanding of the disease. One way to further our understanding of disease progression is to model the discrete steps from when a cell first encounters an aggregate to when those aggregates propagate to new cells. This review will serve to highlight the recent progress made in the effort to better understand the mechanistic steps that determine how this propagation happens at the cellular level.
Introduction
Parkinson’s Disease (PD), the second most prevalent neurodegenerative disorder worldwide [1] is commonly characterized by canonical motor symptoms such as resting tremor, as well as many non-motor symptoms [2]. The pathological hallmarks of PD are dense, proteinaceous inclusions in the brains of patients known as Lewy Bodies (LB) and Lewy Neurites (LN) that were first described over one hundred years ago [3]. The filamentous component of LB an LN has since been identified as aggregates of the protein α-Synuclein (αSyn). The implication of αSyn in LB pathology was established by both genetic [4] and histological [5,6] evidence, and has placed αSyn at the center of our understanding of PD. αSyn is an intrinsically disorder protein (IDP) that exhibits no persistent secondary structure in solution [7], however, upon binding to membranes it adopts an amphipathic α-helical conformation [8]. The helical conformation, which favors binding cellular vesicles, plays a key role in its cellular function. While the complete, native function of αSyn is still being elucidated, it participates in synaptic vesicle recycling and neurotransmitter release, among other roles [9, 10, 11, 12].
Monomers can also convert to β-sheet-rich amyloid multimers. These multimers grow through monomer addition to produce extended, fibrillar aggregates that are strikingly similar to the αSyn fibrils observed in LB [13,14]. αSyn aggregation is dynamic, involving one or more oligomeric species that serve as intermediates between monomers and fibrils. These oligomeric species can interconvert between conformations, elongate into fibrils or be liberated from the ends of mature aggregates [15]. The extent and rate of aggregation is also highly sensitive to local conditions, which further complicates the folding landscape of αSyn.
While the initial cause of aggregation remains uncertain, one unifying feature of PD that has been extensively described is the spread of αSyn pathology to specific brain regions during disease progression [16]. Staging of pathology in patient brains of increasing disease duration provides strong evidence that αSyn pathology spreads inter-neuronally from regions of initial aggregation to previously unaffected regions. This hypothesis of progressive spread is further supported by observations of pathology spreading in patients that received transplants of embryonic neuron grafts from surrounding tissue to the engrafted neurons [17]. There is also evidence that the spread of pathology can be initiated in the neurons of the enteric nervous system before spreading to the brain [18].
Given that spreading of pathology in the brain is a commonality in PD, it is imperative to understand the cellular consequences of neuronal exposure to exogenous fibrils and how this can lead to formation of new aggregates and neuronal dysfunction.
All model systems have strengths that make them uniquely suited to certain questions. Immortalized cells such as HEK-293 and SHSY-5Y do not replicate the complexity or αSyn expression of neurons but allow ease of manipulation of protein expression through genetic means that would be prohibitively difficult in complex systems. Primary rodent neurons are readily available and replicate many of the most critical hallmarks of disease such as insoluble pathological inclusions and human induced pluripotent stem cells (iPSC) derived neurons allow for comparison of underlying differences between patient and healthy genetic backgrounds, but both are only useful over a relatively short period of time. Animal models are the only reliable way to model disease progression on a lifetime scale and successfully replicate the progressive spreading nature of the real disease. While cellular models of αSyn pathology can not recapitulate the long time scales and complex intercellular connectivity of animal model systems, they are well suited for investigating the discrete steps that occur within the cell that can lead eventually to the formation of complex inclusions such as LBs. Being able to model the cellular fate of aggregated species may uncover pathways that can be targeted for interventional therapies. This review will highlight efforts to illuminate the key steps in the induction of αSyn pathology within the cell by exogenous aggregates, and the downstream consequences thereof, with an emphasis on recent progress.
Internalization of exogenous αSyn aggregates
Early cellular models of PD relied on overexpression of αSyn but did not lead to the formation of inclusions similar to those found in patients [19,20]. However, it was observed that aggregated forms of αSyn were selectively internalized in neuronal cell types by an active translocation mechanism [21]. It was later demonstrated that treating primary neurons with recombinant αSyn pre-formed fibrils (PFFs) lead to robust induction of intracellular inclusions [22] that were detergent-insoluble and positive for several common hallmarks of LB pathology such as αSyn phosphorylated at S129 (pS129), ubiquitin, and p62 [3]. Although the exact mechanism of internalization was yet unclear, this PFF treatment paradigm became the basis of many models for studying αSyn aggregation in cells.
Soon after, heparan sulfate proteoglycans (HSPGs) were found to mediate PFF binding to, and uptake by, neuronal progenitor cells via macropinocytosis [23] (Figure 1a). This mode of binding was later shown to be selective for larger fibrils over smaller αSyn species for uptake by neuronal cells [24]. The binding was largely due to the presence of sufficient negative charge rather than any specific sulfation pattern. HSPGs above a certain length could bind well to αSyn fibrils and lead to their internalization, but binding was not specific to any one HSPG. In addition, one family of surface proteoglycans called syndecans, specifically the neuronally enriched syndecan-3, was found to selectively bind and promote the endocytosis of αSyn fibrils through a unique lipid-raft mechanism [25]. Both HSPGs and syndecans are also capable of binding aggregates of the Alzheimer’s Disease associated protein Tau [23. 25], which points to cell surface glycans as a general route for the uptake of large protein aggregates. However, while there are selective preference for binding between Tau aggregates and specific HSPG lengths and sulfation patterns, while αSyn is more promiscuous by comparison [26] which means therapeutic targeting of this binding would be difficult.
Figure 1.
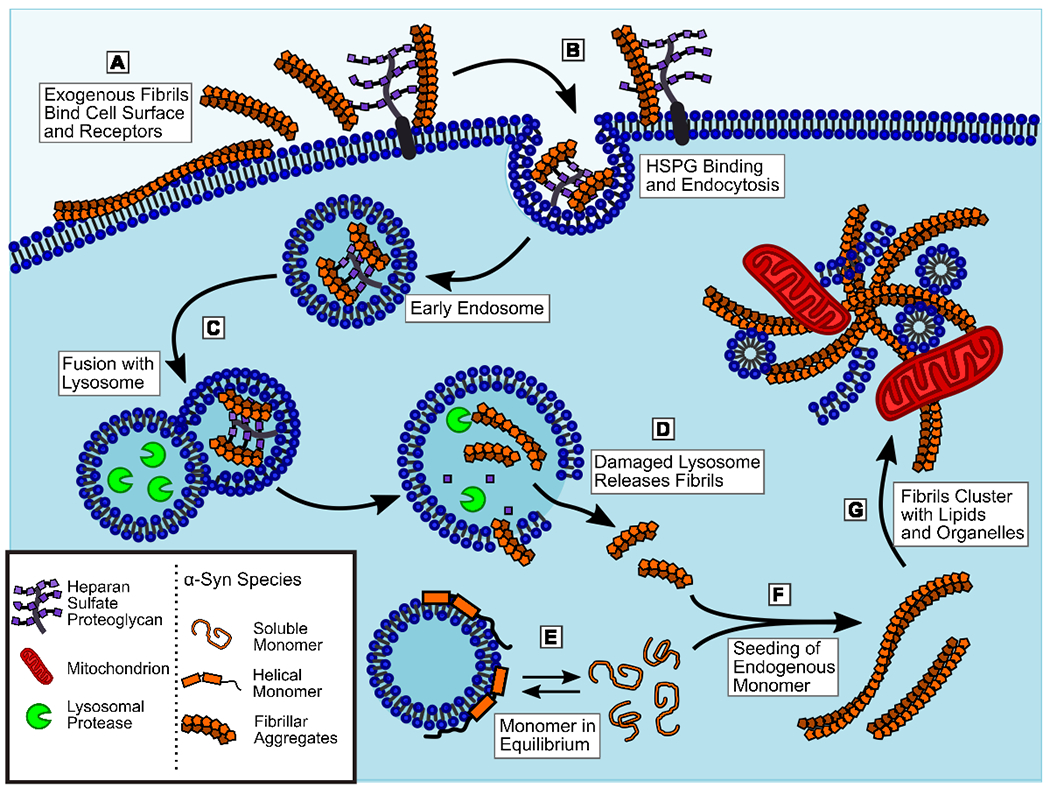
Exogenous αSyn fibrils are internalized into the cell and trigger aggregation of endogenous αSyn monomers. A) Extracellular fibrils are able to interact directly with cell membranes as well as bind to several classes of surface glycoproteins. B) Binding of heparan sulfate proteoglycans (HSPGs), syndecans, N-linked glycans, or receptor proteins can trigger endocytosis of fibrils by various mechanisms. C) Once internalized in endosomes, fibrils are delivered to the lysosome, however they are resistant to proteolytic degradation and persist in the lysosomal pathway. D) Failure to degrade fibrils in the lysosome leads to leakage, allowing some fibrils to escape into the cytosol. Some aggregate species may also be able to penetrate directly through the lysosomal membrane. E) Monomeric αSyn exists in equilibrium between the unstructured, soluble state and the α-helical, vesicle-bound state. The free monomer is more prone to aggregation, so factors that alter this equilibrium can promote or inhibit αSyn aggregation. F) Soluble monomers in the cytosol can interact with fibrils released from the lysosome, or otherwise delivered into the cell, to seed the generation of more fibrils and increase the aggregate burden in the cell. G) As fibrils accumulate in the cell, they can trap other cellular components such as lipids, vesicles, organelles, and other proteins. These fibrils will also become decorate by various post-translational modifications such as phosphorylation and ubiquitination. This process leads to the formation of a mature LB inclusions.
The first specific receptor protein identified that was capable of recognizing and internalizing αSyn aggregates was lymphocyte activation gene 3 (LAG3). This was identified through screening of a library of cell surface receptors expressed in human neuroblastoma cells (SHSY-5Y) for PFF binding. Deletion of LAG3 in neurons inhibited binding and internalization of αSyn PFF and reduced pathology formation [27]. However, the expression of LAG3 in neuronal cell types is not well supported, suggesting it’s role may be more indirect that initially thought [28]. αSyn endocytosis can also occur through binding to N-linked surface glycans [29], which leads to uptake via binding the neuronal glycoprotein neurexin 1β. Neurexin 1β was also identified as a hit in the same screen that identified LAG3. However, this N-linked glycan mechanism was shown to be highly selective for N-terminally acetylated αSyn aggregates. In vivo, αSyn is largely N-terminally acetylated, however, this acetylation predominately affects the dynamics of the α-helical conformation of αSyn [30], and thus many models utilizing recombinantly generated fibrils have not investigated it. The previously described results for HSPG and LAG3 binding would suggest that N-terminal acetylation is not strictly required for αSyn endocytosis. Nevertheless, the selective binding to N-linked glycans is likely to be a relevant route of uptake in the brain, where most αSyn should be the N-acetyl form.
More recently, a CRISPR-Cas9 knock-out screen in human derived HEK293 cells identified two novel genes that promote endocytosis of αSyn PFFs [31]. One gene coded for SLC35B2, a protein that imports the HSPG precursor PAPS (3’-phosphoadenosine-5’phosphosulfate) into the Golgi complex. Knock-out led to decreased HSPG synthesis and PFF binding. This agrees with previous findings suggesting the negative charge density of HSPGs is necessary for binding extracellular αSyn fibrils. The second gene identified coded for myosin-7B, which the authors of the study discovered was necessary for efficient clathrin-mediated endocytosis via maintenance of actin filament dynamics near clathrin-coated pits. While HEK293 are non-neuronal cells, they can still internalize aggregates and form αSyn inclusions, suggesting that some steps in the pathway necessary for αSyn pathology are not exclusive to neurons.
Taken together, these results illustrate that binding and uptake of αSyn fibrils can occur through multiple pathways, involving both specific receptor binding and non-specific charge-charge interactions, suggesting there is no single mode of entry for these aggregates (Figure 1b). While this means there is likely no single therapeutic target for totally inhibiting the uptake of αSyn fibrils by neurons in the brain, it remains to be determined which of the various modes of uptake are most important for disease progression, as it is not likely that they all contribute equally.
Another key consideration in modelling the cellular uptake of αSyn aggregates is the relative binding of various sized aggregates. While most of the experiments described above distinguish between monomeric and aggregated αSyn, it is not clear what size aggregates are being internalized. Generating fragmented fibrils of known size distribution led to the observation that essentially all aggregated αSyn species bind to SHSY-5Y cell surfaces, but there is a size cut-off of approximately 400 nm above which uptake is less efficient [32] (Figure 1a,b). Similarly, smaller pre-fibrillar oligomers of αSyn were shown to be able to enter cells more rapidly than longer fibrillar species in both SHSY-5Y and human iPSC-derived neurons [33] and this uptake correlated with acute cytotoxicity. This result suggests that uptake of oligomers derived from larger fibrils may be more important than the uptake for the fibrils themselves. However, this study did not determine if the oligomeric species and fibrils were being taken up by the same or different mechanisms, or if the internalized oligomers were also able to induce αSyn pathology in cells. It is possible that the uptake of oligomeric and fibril species occur by independent pathways and have discrete down-stream effects rather than converging on a single pathway.
Another uptake pathway that could by-pass endocytosis entirely is cell-to-cell transfer of αSyn species through tunneling nanotubes (TNTs). TNTs are F-actin containing, membranous extensions that allow transfer of cargoes between adjacent cells. While it remains to be determined how significant TNT formation is in vivo, primary mouse neurons and human iPSC-derived neurons are both capable of transferring αSyn species from cell to cell via this mechanism [34–36]. It also appears that αSyn aggregates are contained within or associated with lysosomal or mitochondrial membranes within the TNT rather than being transported freely [37], suggesting that αSyn may be internalized first by endocytosis or perhaps macroautophagy prior to TNT transport to recipient cells. These lipid-encapsulated species could then be processed similarly to other species internalized by endocytosis.
Endo-lysosomal trafficking and escape
Following the internalization of αSyn aggregates, it is critical to understand where within the cell they are transported, how long they persist, and how this may lead to the formation of cytoplasmic inclusions. It has been demonstrated, using fluorescently labelled PFFs, that aggregates could be internalized into endo-lysosomes in SHSY-5Y cells and recruit galectin-3 [38]. Normally diffuse in the cytosol, galectin-3 becomes concentrated at the site of ruptured endo-lysosomal vesicles. This was used to illustrate that PFFs can induce vesicle rupture within 24 hours of treatment which could lead to their release and eventual induction of endogenous aggregation (Figure 1c,d). However, this study did not look at later time points to see the long-term fate of the internalized fibrils or their ability to induce αSyn pathology.
Another study utilized PFFs generated with fluorescently labelled monomers to selectively track the internalized aggregates taken up by primary neurons [39]. Here the authors used either the pH sensitive dye pHRodo to selectively image PFFs within acidified lysosomes, or BODIPY to image the total internalized population in live cells. Extracellular BODIPY fluorescence was quenched with trypan blue, which is cell impermeable. This revealed that the majority of internalized PFFs are located in acidic lysosomal vesicles within 24 hours and remain there for several days (Figure 1c). Disruption of lysosomes with chloroquine, a weak lipophilic base, lead to increased pathology formation within treated neurons, and the authors were able to localize BODIPY-PFF seeds within endogenously formed αSyn pathology, further confirming that vesicle escape leads to induction of αSyn aggregation (Figure 1d,f).
The observation of lysosomal disruption involving galectin-3 reported above was corroborated by findings that upon PFF treatment of H4 neuroglioma cells, internalized fibrils lead to rupture of endo-lysosomal vesicles prior to the formation of pathological inclusions [40]. This rupture was indicated by recruitment of galectin-3 to LAMP1 positive vesicles containing HA-tagged PFF seeds. Galectin-3 recruitment was followed by the aggregation of endogenous αSyn, as indicated by association of split Venus YFP-N-or C-term fused to αSyn. Recently, an alternative approach to visualizing similar uptake dynamics was developed utilizing an αSyn-GFP fusion with an internal tobacco etch virus (TEV) protease cleavage site [41]. This allowed for selective cleavage of the GFP tag from any fibrils remaining outside the cell to quench without the need for the cytotoxic quencher trypan blue.
The ability of exogenous PFF to induce vesicle rupture was further confirmed in an analysis of fibril treated neurons by cryogenic electron tomography (cryo-ET). This technique allowed for the direct observation of fibrils formed in situ in neurons treated with PFF seeds labelled by gold nanoparticles [42]. The aggregates formed in the cytoplasm from endogenous αSyn could be seen extending from immunogold labelled seeds, in some cases with the PFF protruding directly from a vesicle (Figure 1d). This work offers evidence by an orthogonal technique to support the fluorescence-based studies referenced above.
For PFF treatment as in SH-SY5Y cells [38] and primary neurons [39], and co-treatment of neurons with the disaccharide trehalose greatly reduced the appearance of large, dilated lysosomes and reduced the amount of internalized PFFs [43]. Trehalose is an activator of autophagy, but it is unclear exactly how upregulation of autophagy would reduce the PFF-induced stress on lysosomes given that autophagosomes would require functional lysosomes to degrade their cargo. A similar observation was made in human iPSC derived neurons, where upregulation of autophagy by AMPK (5’-AMP-activated protein kinase) activation led to reduced αSyn inclusions following PFF treatment [44]. One possible explanation is that disruption of the autophagy-lysosome pathway (ALP) can drive multivesicular bodies to release their contents from the cell as exosomes [45]. By this logic, cells undergoing ALP stress due to PFF exposure could eject some of that material back into the extracellular space, reducing the amount of αSyn inclusions but potentially exacerbating the spread to other cells. Inhibition of glucocerebrosidase and induction of lysosomal stress in neurons causes a similar type of exosome release [46]. However, recent results investigating the pathology induced by exosome-contained αSyn in mice indicates such encapsulated aggregates may be less potent than naked fibrils [47].
Even if the lysosome remains functional after uptake of fibrils, it may still exacerbate the formation of pathology. This is because C-terminal truncation of α-synuclein species greatly increases their propensity to form new aggregates. Treatment of rat dopaminergic N27 cells lead to incomplete digestion of PFFs by lysosomal cathepsin proteases, resulting in aggregates that were more potent seeds than full-length controls [48].
Maturation of aggregates in the cell
Once αSyn aggregates enter the cytosol, they can recruit endogenous monomer to propagate (Figure 1f). This accumulation leads to many of the neuronal phenotypes commonly associated with PD such as mitochondrial dysfunction. Treatment of iPSC derived neurons with PFFs lead to mitochondrial fragmentation and aberrant condensation in cells with cytoplasmic inclusion, but not those without [49].
Direct interaction between αSyn aggregates and mitochondrial membranes was found to impair cellular respiration [50] and cytoplasmic aggregates provided a source of αSyn oligomers that were shown to selectively bind cardiolipin-rich mitochondrial membranes and lead to leakage [51]. However, forming cytoplasmic fibrils is not the final step in the progression of αSyn pathology. LB pathology in the brain contains a variety of non-fibrillar components including ubiquitin, p62, cellular lipids, and fragments of organelles [3,52]. Ideally, cellular models of αSyn aggregation would demonstrate this complexity, but rarely do. Recent efforts have made great progress on this front, with cryo-ET [42] allowing the visualization of de novo aggregates among other components such as vesicles and mitochondria (Figure 1f,g). These results suggest that fibrils do not preferentially recruit cellular membranes to their location, based on the distribution of membranes and fibrils relative to untreated cells. Using a different imaging modality called correlative light and electron microscopy (CLEM), which involves the parallel imaging of PFF treated neurons by immunofluorescence microscopy and transmission electron microscopy, allowed for highly detailed identification of the cellular components surrounding pathological inclusions [53]. The authors demonstrated that some primary neurons treated with PFF develop dense αSyn inclusions with similar lipid components to LB (Figure 1g). The fraction of cells with these inclusions increases with time post-treatment and markers of mitochondrial stress and synaptic dysfunction also increase proportionally. This approach allows better characterization of the inclusions formed and allows more information to be extracted from existing experiments using fluorescent microscopy. Another study employing CLEM together with scanning electron microscopy and stimulated emission depletion microscopy (STED) analyzed LB from PD patient brain sections and found that many of them do not appear to contain significant amounts of αSyn fibrils, despite being positive for αSyn by immunostaining [54]. While the authors take this data to mean αSyn fibrils are not a major component of LB, the interpretation of this data is still a topic of debate in the field [55].
Concluding remarks
Neurodegenerative diseases like PD pose numerous challenges to our understanding of mechanisms of pathogenesis. The brain is a difficult organ to probe and disease progression can often take decades. The better modelling of this complex disease in more controllable systems becomes, the better our understanding of its mechanisms will be. The field must continue to dissect the details of the cellular processes that fall between what we can currently observe, shrinking the gap in our experimental knowledge steps by step with the goal of developing disease altering therapies. At this point, most of the mechanistic understanding of αSyn pathology formation is in broad strokes or in discontinuous fragments. The role of major pathways such as endocytosis or lysosomal maintenance are still not characterized to the level needed for targeted drug development, but new understanding of the underlying cell biology continues to be uncovered. The work featured here was selected to underline the usefulness of that approach and the progress it has made in recent years. New imaging modalities like super-resolution techniques and advances in electron microscopy are becoming more accessible and continue to uncover new information that couldn’t be captured before. However, this review also sought to highlight creative applications of the standard biochemical toolbox to ask insightful and productive questions. Lastly, it is crucial to be constantly assessing critically the current models in the field, both to reaffirm their utility and to confirm that they still reflect our changing understanding of PD and its etiology.
Highlights:
A central aspect of Parkinson’s Disease is the cell-to-cell spread of α-Synuclein pathology in the brain.
The spreading of pathology can be modelled by inducing aggregation in cultured cells
Cellular models allow the isolation of key steps in the pathway from aggregate uptake to propagation
Acknowledgements
This work was supported by the National Institutes of Health (grants NS110456 and U19-AG062418)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflict of interest.
CRedit author statement
Nicholas P Marotta: Conceptualization, Writing – original draft, Writing – reviewing and editing. Virginia M-Y Lee: Writing – reviewing and editing, Supervision, Funding
References
- 1.Lau LM de, Breteler MM: Epidemiology of Parkinson’s disease. Lancet Neurology 2006, 5:525–535. [DOI] [PubMed] [Google Scholar]
- 2.Marti MJ, Tolosa E, Campdelacreu J: Clinical overview of the synucleinopathies. Movement Disord 2003, 18:21–27. [DOI] [PubMed] [Google Scholar]
- 3.Goedert M, Spillantini MG, Tredici KD, Braak H: 100 years of Lewy pathology. Nat Rev Neurol 2012, 9:13–24. [DOI] [PubMed] [Google Scholar]
- 4.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. : Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276:2045–2047. [DOI] [PubMed] [Google Scholar]
- 5.Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M: α-Synuclein in Lewy bodies. Nature 1997, 388:839–840. [DOI] [PubMed] [Google Scholar]
- 6.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M: α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc National Acad Sci 1998, 95:6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT: NACP, A Protein Implicated in Alzheimer’s Disease and Learning, Is Natively Unfolded. Biochemistry-us 1996, 35:13709–13715. [DOI] [PubMed] [Google Scholar]
- 8.Chandra S, Chen X, Rizo J, Jahn R, Südhof TC: A Broken α-Helix in Folded ü-Synuclein. J Biol Chem 2003, 278:15313–15318. [DOI] [PubMed] [Google Scholar]
- 9.Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC: Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Sci New York N Y 2010, 329:1663–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaltieri M, Grigoletto J, Longhena F, Navarria L, Favero G, Castrezzati S, Colivicchi MA, Corte LD, Rezzani R, Pizzi M, et al. : α-synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J Cell Sci 2015, 128:2231–2243. [DOI] [PubMed] [Google Scholar]
- 11.Sun J, Wang L, Bao H, Premi S, Das U, Chapman ER, Roy S: Functional cooperation of α-synuclein and VAMP2 in synaptic vesicle recycling. Proc National Acad Sci 2019, 116: 11113–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Longhena F, Faustini G, Spillantini MG, Bellucci A: Living in Promiscuity: The Multiple Partners of Alpha-Synuclein at the Synapse in Physiology and Pathology. Int J Mol Sci 2019, 20:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giasson BI, Uryu K, Trojanowski JQ, Lee VM-Y: Mutant and Wild Type Human α-Synucleins Assemble into Elongated Filaments with Distinct Morphologies in Vitro. J Biol Chem 1999, 274:7619–7622. [DOI] [PubMed] [Google Scholar]
- 14.Conway KA, Harper JD, Lansbury PT: Fibrils Formed in Vitro from α-Synuclein and Two Mutant Forms Linked to Parkinson’s Disease are Typical Amyloid. Biochemistry-US 2000, 39:2552–2563. [DOI] [PubMed] [Google Scholar]
- 15.Nunilo Cremades, Cohen Samuel I A, Emma Deas, Abramov Andrey Y, Chen Allen Y, Orte Angel, Sandal Massimo, Clarke Richard W, Paul Dunne, Aprile Francesco A, et al. : Direct Observation of the Interconversion of Normal and Toxic Forms of α-Synuclein. Cell 2012, 149:1048–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This work was crucial in establishing the mechanism by which oligomeric aggregates can progress into fibrils. The authors established a link between the interconversion of oligomers and the lag phase of aggregation kinetics.
- 16.Braak H, Tredici KD, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rüb U: Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s Disease (preclinical and clinical stages). J Neurol 2002, 249:iii1–iii5. [DOI] [PubMed] [Google Scholar]
- 17.Kordower Jeffrey H, Yaping Chu, Hauser Robert A, Freeman Thomas B, Olanow C Warren: Lewy body–like pathology in long-term embryonic nigral transplants in Parkinson’s Disease. Nat Med 2008, 14:504. [DOI] [PubMed] [Google Scholar]
- 18.Kim S, Kwon S-H, Kam T-I, Panicker N, Karuppagounder SS, Lee S, Lee JH, Kim WR, Kook M, Foss CA, et al. : Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103:627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tabrizi SJ, Orth M, Wilkinson JM, Taanman J-W, Warner TT, Cooper JM, Schapira AHV: Expression of mutant α-synuclein causes increased susceptibility to dopamine toxicity. Hum Mol Genet 2000, 9:2683–2689. [DOI] [PubMed] [Google Scholar]
- 20.McLean PJ, Kawamata H, Hyman BT: α-Synuclein–enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience 2001, 104:901–912. [DOI] [PubMed] [Google Scholar]
- 21.Lee H-J, Suk J-E, Bae E-J, Lee J-H, Paik SR, Lee S-J: Assembly-dependent endocytosis and clearance of extracellular a-synuclein. Int J Biochem Cell Biology 2008, 40:1835–1849. [DOI] [PubMed] [Google Scholar]
- 22.Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM-Y: Exogenous α-Synuclein Fibrils Induce Lewy Body Pathology Leading to Synaptic Dysfunction and Neuron Death. Neuron 2011, 72:57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, et al. : Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc National Acad Sci 2013, 110:E3138–E3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ihse E, Yamakado H, Wijk XM, Lawrence R, Esko JD, Masliah E: Cellular internalization of alpha-synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci Reports 2017, 7:9008–9018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hudák A, Kusz E, Domonkos I, Jósvay K, Kodamullil AT, Szilák L, Hofmann-Apitius M, Letoha T: Contribution of syndecans to cellular uptake and fibrillation of α-synuclein and tau. Sci Reports 2019, 9:16543. doi: 10.1038/s41598-019-53038-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stopschinski BE, Holmes BB, Miller GM, Manon VA, Vaquer-Alicea J, Prueitt WL, Hsieh-Wilson LC, Diamond Ml: Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus α-synuclein and β-amyloid aggregates. J Biol Chem 2018, 293:10826–10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mao X, Ou MT, Karuppagounder SS, Kam T-I, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin J-H, et al. : Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353:aah3374 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emmenegger M, Cecco ED, Hruska-Plochan M, Eninger T, Schneider MM, Barth M, Tantardini E, Rossi P de, Bacioglu M, Langston RG, et al. : LAG3 is not expressed in human and murine neurons and does not modulate α-synucleinopathies. Embo Mol Med 2021, 13:e14745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Birol M, Wojcik SP, Miranker AD, Rhoades E: Identification of N-linked glycans as specific mediators of neuronal uptake of acetylated α-Synuclein. PloS Biol 2019, 17:e3000318. doi: 10.1371/journal.pbio.3000318 [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This work established a new pathway for internalization of fibrils while also reaffirming the importance of studying the native, N-terminally acetylated aSyn isoform in disease models more generally.
- 30.Dikiy I, Eliezer D: N-terminal Acetylation Stabilizes N-terminal Helicity in Lipid- and Micelle-bound α-Synuclein and Increases Its Affinity for Physiological Membranes. J Biol Chem 2014, 289:3652–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Q, Xu Y, Lee J, Jarnik M, Wu X, Bonifacino JS, Shen J, Ye Y: A myosin-7B–dependent endocytosis pathway mediates cellular entry of α-synuclein fibrils and polycation-bearing cargos. Proc National Acad Sci 2020, 117:10865–10875 [DOI] [PMC free article] [PubMed] [Google Scholar]; *This work is an excellent mechanistic study of the consequences of two novel genes implicated in fibril uptake, demonstrating that the variety of pathways involved in αSyn aggregate propagation remains underexplored.
- 32.Zhang X, Wesén E, Kumar R, Bernson D, Gallud A, Paul A, Wittung-Stafshede P, Esbjörner EK: Correlation between Cellular Uptake and Cytotoxicity of Fragmented α-Synuclein Amyloid Fibrils Suggests Intracellular Basis for Toxicity. Acs Chem Neurosci 2020, 11:233–241. [DOI] [PubMed] [Google Scholar]
- 33.Cascella R, Chen SW, Bigi A, Camino JD, Xu CK, Dobson CM, Chiti F, Cremades N, Cecchi C: The release of toxic oligomers from α-synuclein fibrils induces dysfunction in neuronal cells. Nature Commun 2021, 12:1814. doi: 10.1038/s41467-021-21937-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study highlights the importance of considering the role of smaller aggregate and oligomeric species in disease propagation, with a special emphasis on the release of smaller oligomers from fibrils at the cell surface.
- 34.Abounit S, Bousset L, Loria F, Zhu S, Chaumont F de, Pieri L, Olivo-Marin J, Melki R, Zurzolo C: Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. Embo J 2016, 35:2120–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Senol AD, Samarani M, Syan S, Guardia CM, Nonaka T, Liv N, Latour-Lambert P, Hasegawa M, Klumperman J, Bonifacino JS, et al. : α-Synuclein fibrils subvert lysosome structure and function for the propagation of protein misfolding between cells through tunneling nanotubes. Plos Biol 2021, 19:e3001287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grudina C, Kouroupi G, Nonaka T, Hasegawa M, Matsas R, Zurzolo C: Human NPCs can degrade α–syn fibrils and transfer them preferentially in a cell contact-dependent manner possibly through TNT-like structures. Neurobiol Dis 2019, 132:104609. [DOI] [PubMed] [Google Scholar]
- 37.Valdinocci D, Kovarova J, Neuzil J, Pountney DL: Alpha-Synuclein Aggregates Associated with Mitochondria in Tunnelling Nanotubes. Neurotox Res 2021, 39:429–443. [DOI] [PubMed] [Google Scholar]
- 38.Flavin WP, Bousset L, Green ZC, Chu Y, Skarpathiotis S, Chaney MJ, Kordower JH, Melki R, Campbell EM: Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol 2017, 134:629–653. [DOI] [PubMed] [Google Scholar]
- 39.Karpowicz RJ, Haney CM, Mihaila TS, Sandler RM, Petersson EJ, Lee VM-Y: Selective imaging of internalized proteopathic α-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J Biol Chem 2017, 292:13482–13497. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This work gives the most complete timeline, to date, of the progression of fibrils within the cell as they traffic the endo-lysosomal pathway and, eventually, induce pathology.
- 40.Jiang P, Gan M, Yen S-H, McLean PJ, Dickson DW: Impaired endo-lysosomal membrane integrity accelerates the seeding progression of α-synuclein aggregates. Sci Reports 2017, 7:7690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jarvela TS, Chaplot K, Lindberg I: A protease protection assay for the detection of internalized alpha-synuclein pre-formed fibrils. Plos One 2021, 16:e0241161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trinkaus VA, Riera-Tur I, Martínez-Sánchez A, Bäuerlein FJB, Guo Q, Arzberger T, Baumeister W, Dudanova I, Hipp MS, Hartl FU, et al. : In situ architecture of neuronal α-Synuclein inclusions. Nature Commun 2021, 12:2110. doi: 10.1038/s41467-021-22108-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; **The authors not only demonstrate with great detail the accumulation of cellular components in LB-like inclusions, but also establish a quantitative framework to evaluate the specificity of these accumulations.
- 43.Hoffmann A-C, Minakaki G, Menges S, Salvi R, Savitskiy S, Kazman A, Miranda HV, Mielenz D, Klucken J, Winkler J, et al. : Extracellular aggregated alpha synuclein primarily triggers lysosomal dysfunction in neural cells prevented by trehalose. Sci Reports 2019, 9:544. doi: 10.1038/s41598-018-35811-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao J, Perera G, Bhadbhade M, Halliday GM, Dzamko N: Autophagy activation promotes clearance of α-synuclein inclusions in fibril-seeded human neural cells. J Biol Chem 2019, 294:14241–14256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Minakaki G, Menges S, Kittel A, Emmanouilidou E, Schaeffner I, Barkovits K, Bergmann A, Rockenstein E, Adame A, Marxreiter F, et al. : Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 2018, 14:1–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gegg ME, Verona G, Schapira AHV: Glucocerebrosidase deficiency promotes release of α-synuclein fibrils from cultured neurons. Hum Mol Genet 2020, 29:1716–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karampetsou M, Sykioti VS, Leandrou E, Melachroinou K, Lambiris A, Giannelos A, Emmanouilidou E, Vekrellis K: Intrastriatal Administration of Exosome-Associated Pathological Alpha-Synuclein Is Not Sufficient by Itself to Cause Pathology Transmission. Front Neurosci 2020, 14:246. doi: 10.3389/fnins.2020.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McGlinchey RP, Lacy SM, Huffer KE, Tayebi N, Sidransky E, Lee JC: C-terminal α-synuclein truncations are linked to cysteine cathepsin activity in Parkinson’s disease. J Biol Chem 2019, 294:9973–9984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gribaudo S, Tixador P, Bousset L, Fenyi A, Lino P, Melki R, Peyrin J-M, Perrier AL: Propagation of α-Synuclein Strains within Human Reconstructed Neuronal Network. Stem Cell Rep 2019, 12:230–244. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This work is an excellent example of using a novel culture system to answer complex question about pathological aSyn spreading in a cell-based assay.
- 50.Wang X, Becker K, Levine N, Zhang M, Lieberman AP, Moore DJ, Ma J: Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathologica Commun 2019, 7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghio S, Camilleri A, Caruana M, Ruf VC, Schmidt F, Leonov A, Ryazanov S, Griesinger C, Cauchi RJ, Kamp F, et al. : Cardiolipin Promotes Pore-Forming Activity of Alpha-Synuclein Oligomers in Mitochondrial Membranes. Acs Chem Neurosci 2019, 10:3815–3829. [DOI] [PubMed] [Google Scholar]
- 52.McCormack A, Keating DJ, Chegeni N, Colella A, Wang JJ, Chataway T: Abundance of Synaptic Vesicle-Related Proteins in Alpha-Synuclein-Containing Protein Inclusions Suggests a Targeted Formation Mechanism. Neurotox Res 2019, 35:883–897. [DOI] [PubMed] [Google Scholar]
- 53.Mahul-Mellier A-L, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, Leleu M, Knott GW, Lashuel HA: The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc National Acad Sci 2020, 117:4971–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This is one of the best demonstrations to date that complex, LB-like inclusions can be systematically induced and studied in cultured cells and offers a platform for future investigation.
- 54.Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, Castaño-Díez D, Schweighauser G, Graff-Meyer A, Goldie KN, et al. : Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci 2019, 22:1099–1109. [DOI] [PubMed] [Google Scholar]; *While there is still some debate about the interpretation of the results, this study is significant for the level of detail revealed about the pathological inclusions analyzed and illustrates the wealth of information such approaches can yield.
- 55.Lashuel HA: Do Lewy bodies contain alpha-synuclein fibrils? and Does it matter? A brief history and critical analysis of recent reports. Neurobiol Dis 2020, 141:104876. [DOI] [PubMed] [Google Scholar]