

Abstract
The utilization of isolated Palladium Oxidative Addition Complexes (OACs) has had a significant impact on Pd-catalyzed and Pd-mediated cross-coupling reactions. Despite their importance, widespread utility of OACs has been limited by the instability of their precursor complexes. Herein, we report the use of Cámpora’s palladacycle as a new, more stable precursor to Pd OACs. Using this palladacycle, a series of biarylphosphine ligated OACs, including those with pharmaceutical-derived aryl halides and relevance to bioconjugation, were prepared.
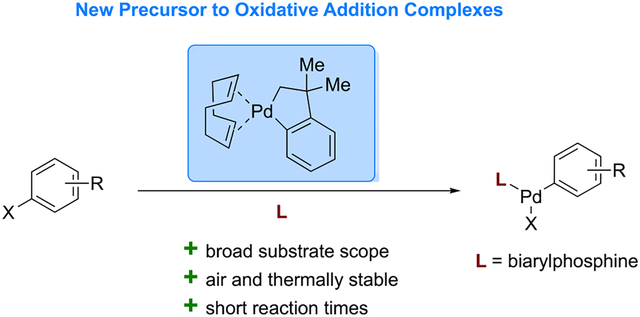
Graphical Abstract
Palladium-catalyzed cross-coupling reactions are important for the synthesis of complex molecules. They have become mainstays in natural product synthesis,1 materials science,2 agrochemical synthesis,1 and pharmaceutical development.3 Mechanistic studies utilizing stoichiometric Pd complexes have been valuable in expanding the generality of Pd catalysis by enabling each elementary step of the catalytic cycle to be probed (Figure 1A).4 Of these mechanistic inquiries, those focused on the generation and use of Pd oxidative addition complexes (OACs) are noteworthy. OACs have allowed for the study of different modes of catalyst deactivation,5 guided the synthesis of additional ligand scaffolds,6 been employed as an entry point to analyze other steps in the catalytic cycle,7 and been implemented as superior precatalysts for Pd-catalyzed carbon–heteroatom bond forming reactions.8
Figure 1.

(A) A general catalytic cycle for Pd0/PdII-catalyzed cross-coupling reactions highlighting the complexes of interest in this study. (B) Biarylphosphines utilized in this study.
The modularity and versatility of OACs have allowed them to be utilized stoichiometrically in Pd-mediated cross-coupling reactions as well.9 When catalytic cross-coupling methods are applied to complex aryl halides, they often fail to deliver any product.9a In the context of drug discovery, this can lead to fewer potential drug candidates, and first-pass reaction success outweighs the cost of using stoichiometric quantities of Pd.9a Additionally, Pd OACs have been applied to bioconjugation,9c–f radioisotope incorporation,9g and late-stage pharmaceutical diversification.9a,b
Despite the aforementioned successes, accessing OACs still remain a challenge. While OACs are, themselves, often air-, moisture-, and thermally stable, they necessitate the use of unstable precursors. In order to access OACs with a broad ligand and aryl (pseudo)halide scope, either (cod)Pd(CH2TMS)2 (cod = 1,5-cyclooctadiene),10 a thermally sensitive PdII complex, or (L·Pd)2(cod) (L = phosphine),11 a series of air-sensitive Pd0 complexes that require manipulation in a glovebox, is often employed. While these complexes can be used to access a wider range of OACs than the commercial and more stable Pd2(dba)3,5,12 their instability can limit their utility.
We sought to identify a more stable, user-friendly precursor to Pd OACs that would fulfill the following prerequisites: the new precursor should (1) display similar reactivity to (cod)Pd(CH2TMS)2, (2) be synthetically tractable, and (3) be stable at room temperature and under ambient atmosphere. With these goals in mind, we report the use of (cod)Pd(CH2CMe2C6H4) (P3), a five-membered palladacycle first reported by Cámpora,13 as a new precursor to OACs. Using P3, a series of OACs bearing a wide range of biarylphosphine ligands were accessible (Figure 1B). Palladacycle P3 was used to the generate (L·Pd)2(cod) complexes in situ, which allowed for the synthesis of pharmaceutical-derived OACs without the use of a glovebox.
We sought to find a PdII complex that was air-, moisture-, and thermally stable with a labile ancillary ligand, such as cod, but could rapidly undergo reductive elimination upon ligand exchange with a phosphine ligand. This would generate a reactive L·Pd0 species that could subsequently undergo oxidative addition in the presence of an aryl (pseudo)halide to afford the desired OAC (Table 1). As a benchmark, (cod)Pd(CH2TMS)2 (P1) allowed for access to RuPhos-, XPhos-, and t-BuXPhos-ligated OACs in high yield in tetrahydrofuran (THF) at room temperature (entries 1–3). While stable dialkyl Pd complexes are underrepresented in the literature,10a,14 reportedly stable polyfluorinated diaryl Pd complexes have been thoroughly studied.15 These complexes seemed like potential viable alternatives, as they are stable when ligated to cod, but have been shown to undergo reductive elimination in the presence of biarylphosphine ligands catalytically.16 However, complete conversion of (cod)Pd(2,4,6-F3C6H2)2 (P2) was only observed in the presence of smaller dicyclohexylbiarylphosphines (entries 4–6). We, thus, ultimately concluded that diaryl Pd complexes are too stable to be used as general precursors to OACs.
Table 1.
Investigation of Pd Precursors for OAC Formationa

| |||||
|---|---|---|---|---|---|
| Entry | Pd Precursor | L | Temp (°C) | Yield A (%) | Yield B (%) |
| 1b | P1 | RuPhos | rt | n/a | 99 |
| 2b | P1 | XPhos | rt | n/a | 97 |
| 3b | P1 | t-BuXPhos | rt | n/a | 99 |
| 4b | P2 | RuPhos | rt | n/a | 98 |
| 5b | P2 | XPhos | rt | n/a | 99 |
| 6b | P2 | t-BuXPhos | rt | n/a | 40 |
| 7 | P3 | RuPhos | rt | 99 | 0 |
| 8 | P3 | XPhos | rt | 70 | 0 |
| 9 | P3 | t-BuXPhos | rt | 0 | 10 |
| 10 | P3 | t-BuBrettPhos | rt | 0 | 0 |
| 11 | P3 | RuPhos | 60 | 41 | 59 |
| 12 | P3 | XPhos | 60 | 0 | 99 |
| 13 | P3 | t-BuXPhos | 60 | 0 | 82 |
| 14 | P3 | t-BuBrettPhos | 60 | 0 | 24 |

| |||||
Yields determined by 1H NMR using 0.5 equiv of 1,3,5-trimethoxybenzene as an internal standard. Reaction conditions: Pd precursor (0.05 mmol), L (0.055 mmol), aryl bromide (0.075 mmol), and THF (0.50 mL, 0.10 M).
Reactions run for 16 h.
Since the dialkyl Pd complex P1 afforded desired reactivity at the expense of stability and the diaryl Pd complex P2 had the desired stability at the cost of reactivity, we wondered whether the use of a mixed alkyl,aryl-Pd complex could combine the favorable aspects of each. While stable acyclic alkyl,aryl-Pd complexes without strong electron-withdrawing groups7b remain rare, a series of air-stable 5-membered alkyl,aryl-palladacycles have been reported by Catellani,17 Echavarren,18 and Cámpora.13 Not only have these palladacycles been frequently invoked as intermediates in catalytic transformations19 and commonly utilized as mechanistic tools,13,20 two reports have shown that they can undergo thermal reductive elimination from PdII complexes to generate Pd0 intermediates and the corresponding benzocyclobutene.21 In these cases, this reductive elimination was not used as a means to generate a Pd0 species. We, however, anticipated that the L·Pd0 intermediates generated in situ by this method could subsequently react with aryl halides to afford OACs.
Following a slightly modified literature procedure, the cod-bound Cámpora palladacycle P3 could be accessed in 79% isolated yield on 10 mmol scale and in a 65% average isolated yield on 25 mmol scale (Scheme 1). This synthesis employs a commercially available Grignard reagent, proceeds at ambient temperature, and has a predictable C–H activation/cyclization step. To determine the viability of P3 as a precursor to OACs, we treated it with a series of biarylphosphine ligands and 2-(trimethylsilyl)ethyl 4-bromobenzoate. In the presence of RuPhos at room temperature, we observed complete consumption of P3; however, no desired OAC was formed. Instead, formation of the RuPhos-ligated palladacycle was observed by LC/MS and 1H NMR spectroscopy (Table 1, entry 7). When XPhos was utilized, partial conversion to the XPhos-ligated palladacycle was detected (entry 8). However, the analogous reaction with t-BuXPhos or t-BuBrettPhos resulted in trace or no OAC formation, respectively (entries 9–10). By heating each reaction mixture to 60 °C for 1 h, the RuPhos- and XPhos-ligated OACs could be formed in 59% and 100% yield, respectively (entries 11–12). In general, for dicyclohexylbiarylphosphines, the ligand exchange process is facile at room temperature, but the subsequent reductive elimination requires heating. In the case of di-tert-butylbiaryl-phosphines, the t-BuXPhos-ligated OAC was formed in 82% yield (entry 13), while the formation of the t-BuBrettPhos OAC was slower and resulted in 24% yield after 1 h at 60 °C (entry 14). Improved yields for the latter were obtained by heating the reaction mixture to 80 °C. Changing the reaction solvent from THF to n-hexane and heating each reaction mixture for 2 h allowed for isolation of the corresponding OAC by precipitation in excellent yield.
Scheme 1.

Synthesis of Palladacycle P3
aAverage of 3 runs.
To expand on these preliminary studies, we investigated the scope of ligands that could form OACs using P3 (Figure 2A). Both the t-BuXPhos-bound and XPhos-bound OACs of 2-(trimethylsilyl)ethyl 4-bromobenzoate (1 and 2) were prepared in 90% and 68% isolated yield, respectively. Notably, the use of P3 that had been stored under ambient conditions for 1 year allowed for the isolation of 1 in 80% yield.22 By employing the same aryl halide, OACs bearing t-BuBrettPhos (3), RuPhos (4), Ad(Cy)BrettPhos (5), and GPhos (6) could be obtained. OACs ligated to the extremely bulky dialkylter-arylphosphine AlPhos were accessible, as both the bromide complex 7 and triflate complex 8 could be formed from P3. Additionally, OACs bearing electron-neutral (L = BrettPhos, 9) and electron-rich arenes (L = CPhos 10) could be generated in excellent yields. While some of these OACs have been utilized as precatalysts,8 we determined that P3 itself can also be used as a precatalyst (see SI for details).
Figure 2.

OAC scope from P3. Isolated yields shown. Reaction conditions: (A) P3 (0.50 mmol), L (0.53 mmol), aryl halide (0.60 mmol), and n-hexane (5.0 mL, 0.10 M); (B) P3 (0.60 mmol), L (0.72 mmol), aryl halide (0.60 mmol), 2-MeTHF (2.5 mL), and THF (5.0 mL). aIsolated yield using P3 stored under ambient conditions for 1 y. bCyclohexane (5.0 mL), 80 °C. c1.2 equiv of P3 and 1.0 equiv of aryl halide. d1.0 equiv of P3, 3.0 equiv of aryl halide, and 2-MeTHF (7.5 mL). See Supporting Information for details.
Much of the utility of Pd OACs arises from their versatility, with respect to both the ancillary ligand and the aryl halide. We were interested in determining if P3 could form OACs from more complex aryl halides, including pharmaceuticals. Formation of OACs from pharmaceutical-like aryl halides has a tremendous impact on first-pass reaction success.9a However, accessing these OACs is often particularly challenging. Attempting to prepare OACs from (cod)Pd(CH2TMS)2 and pharmaceutical-derived aryl halides often leads to incomplete conversion and low yield of the product. Although preligating the desired ancillary ligand to the palladium center through the use of (L·Pd)2(cod) complexes can lead to improved yields, these Pd0 complexes are oxygen sensitive and require storage and manipulation in a glovebox.
By simultaneously heating P3 with the desired ligand and aryl halide (conditions A), the protocol utilized with simple aryl halides, the t-BuXPhos-bound OAC derived from the “informer compound” X43b 11 was formed in 63% isolated yield (Figure 2B). However, these conditions were not general for other complex aryl halides, as they failed to deliver either the rivaroxaban-derived OAC 12 or the gefitinib-derived OAC 13. We postulated that by allowing P3 to react with t-BuXPhos prior to the addition of the aryl halide, we could generate [(t-BuXPhos)Pd]2(cod) in situ. This may, in turn, increase the yield of OACs prepared from pharmaceutical-like aryl halides. By generating (L·Pd)2(cod) before adding the desired aryl halide (conditions B), t-BuXPhos-bound 12 and 13 could be obtained in 80% and 88% isolated yield, respectively. Furthermore, this stepwise approach allowed for access to the XPhos-ligated 14 and BrettPhos-ligated 15, both derived from rivaroxaban.
Another application of Pd OACs involves their use as reagents for the selective delivery of arenes into densely functionalized molecules under mild conditions. These reagents have recently emerged as useful tools for the incorporation of radioisotopes9g and for the functionalization of biomolecules (Figure 2C).9c–f In this regard, we were able to synthesize the cationic RuPhos complex 16.9c By allowing 4,4′-dibromobiphenyl to react with 2 equiv of P3, we were able to access the bis-Pd OAC 17, which has been used as a peptide stapling reagent.9d While aryl iodides are not converted to product under conditions A,23 we found that the BrettPhos-ligated Pd OAC of 1,4-diiodobenzene 18 could be prepared using conditions B with excess aryl halide. Additionally, OACs bearing orthogonal functional handles, including N-hydroxy-succinimide (19) and pentafluorophenoxy esters (20 and 21), could be prepared. In order to demonstrate that the chemistry was scalable, we also performed the synthesis of 19 on a 10 mmol scale, which resulted in 99% isolated yield. Complex 20 is also noteworthy, as amphiphilic sulfonated phosphine ligands have allowed for cross-coupling in aqueous conditions9e and site-selective functionalization through ion-pairing interactions.24
Prior to OAC formation, intermediate L·Pd0 and L·PdII complexes are generated in situ that are interesting to access in their own right (Figure 3). The t-BuXPhos-ligated 22 and BrettPhos-ligated 23 formed in the absence of aryl halides have been previously prepared from P1, although the protocol employed long reaction times, typically 48 h.11
Figure 3.

Scope of other Pd0 and PdII complexes accessible via P3. Isolated yields shown. Reaction conditions: P3 (0.50 mmol), L (0.53 mmol), and CyH (5.0 mL, 0.10 M). Yields in parentheses are determined by 1H NMR spectroscopy of the crude reaction mixture using 1,3,5-trimethoxybenzene as the internal standard. See Supporting Information for details. aL (0.50 mmol), Et2O (5.0 mL, 0.05 M), 1 h. bL (0.50 mmol), pentane (5.0 mL, 0.05 M), 1 h.
With P3, each complex could be produced in 2 h. XPhos-bound 24 can also be accessed. Additionally, by allowing P3 to react with each ligand at room temperature, palladacycles bearing XPhos (25) and RuPhos (26) can be prepared. Although these complexes undergo reductive elimination at elevated temperatures, we found that samples of each showed no evidence of decomposition after 3 months in the solid state under ambient conditions.
In conclusion, we demonstrated that palladacycle P3 can be utilized as an air-, moisture-, and thermally stable precursor to a broad range of Pd OACs. Even after storing P3 under ambient conditions for a year, it showed no evidence of decomposition and allowed for access to a diverse array of biarylphosphine ligated OACs, including densely functionalized OACs relevant to pharmaceutical diversification and OACs bearing orthogonal functional handles for selective incorporation of PdII into biomolecules. OAC formation from P3 was found to be scalable, as P3 allowed for access to OACs on 10 mmol scale. We believe that the use of P3 will greatly expand the availability of OACs in both academic and industrial settings, which in turn will allow for the investigation of new chemical transformations and increase the accessible chemical space.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA and the National Institute of Health (R35-GM122483). The content of this communication solely reflects the research and opinion of the authors and does not necessarily represent the official views of the NIH. We are grateful to Millipore-Sigma for the generous donation of t-BuXPhos, RuPhos, SPhos, XPhos, and BrettPhos used in this work. We thank Dr. Alexander Schuppe, Dr. Veronika Kottisch, and Dr. Christine Nguyen (MIT) for assistance in the preparation of this manuscript.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.1c02307.
Experimental details and characterization data (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.orglett.1c02307
The authors declare the following competing financial interest(s): MIT has patents on some ligands and precatalysts that are described in this manuscript, from which S.L.B. and former co-workers receive royalty payments.
Contributor Information
Ryan P. King, Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States.
Shane W. Krska, Merck & Co., Inc., Kenilworth, New Jersey 07033, United States.
Stephen L. Buchwald, Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States.
REFERENCES
- (1).Ruiz-Castillo P; Buchwald SL Applications of Palladium-Catalyzed C-N Cross-Coupling Reactions. Chem. Rev 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Leone AK; Mueller EA; McNeil AJ The History of Palladium-Catalyzed Cross-Couplings Should Inspire the Future of Catalyst-Transfer Polymerization. J. Am. Chem. Soc 2018, 140, 15126–15139. [DOI] [PubMed] [Google Scholar]
- (3).(a) Brown DG; Boström J Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem 2016, 59, 4443–4458. [DOI] [PubMed] [Google Scholar]; (b) Kutchukian PS; Dropinski JF; Dykstra KD; Li B; DiRocco DA; Streckfuss EC; Campeau L-C; Cernak T; Vachal P; Davies IW; Krska SW; Dreher SD Chemistry informer libraries: a chemoinformatics enabled approach to evaluate and advance synthetic methods. Chem. Sci 2016, 7, 2604–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Sather AC; Lee HG; De La Rosa VY; Yang Y; Müller P; Buchwald SL A Fluorinated Ligand Enables Room-Temperature and Regioselective Pd-Catalyzed Fluorination of Aryl Triflates and Bromides. J. Am. Chem. Soc 2015, 137, 13433–13438. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maleckis A; Sanford MS Catalytic Cycle for Palladium-Catalyzed Decarbonylative Trifiuoromethylation using Trifiuoroacetic Esters as a CF3 Source. Organometallics 2014, 33, 2653–2660. [Google Scholar]
- (5).Widenhoefer RA; Zhong HA; Buchwald SL Synthesis and Solution Structure of Palladium Tris(o-tolyl)phosphine Mono(amine) Complexes. Organometallics 1996, 15, 2745–2754. [Google Scholar]
- (6).(a) Lee HG; Milner PJ; Buchwald SL Pd-Catalyzed Nucleophilic Fiuorination of Aryl Bromides. J. Am. Chem. Soc 2014, 136, 3792–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McCann SD; Reichert EC; Arrechea PL; Buchwald SL Development of an Aryl Amination Catalyst with Broad Scope Guided by Consideration of Catalyst Stability. J. Am. Chem. Soc 2020, 142, 15027–15037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Dennis JM; White NA; Liu RY; Buchwald SL Pd-Catalyzed C - N Coupling Reactions Facilitated by Organic Bases: Mechanistic Investigation Leads to Enhanced Reactivity in the Arylation of Weakly Binding Amines. ACS Catal 2019, 9, 3822–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Culkin DA; Hartwig JF Carbon-Carbon Bond-Forming Reductive Elimination from Arylpalladium Complexes Containing Functionalized Alkyl Groups. Infiuence of Ligand Steric and Electronic Properties on Structure, Stability, and Reactivity. Organometallics 2004, 23, 3398–3416. [Google Scholar]
- (8).Ingoglia BT; Buchwald SL Oxidative Addition Complexes as Precatalysts for Cross-Coupling Reactions Requiring Extremely Bulky Biarylphosphine Ligands. Org. Lett 2017, 19, 2853–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Uehling MR; King RP; Krska SW; Cernak T; Buchwald SL Pharmaceutical diversification via palladium oxidative addition complexes. Science 2019, 363, 405–408. [DOI] [PubMed] [Google Scholar]; (b) Roque JB; Kuroda Y; Jurczyk J; Xu L-P; Ham JS; Göttemann LT; Roberts CA; Adpressa D; Saurí J; Joyce LA; Musaev DG; Yeung CS; Sarpong R C-C Cleavage Approach to C-H Functionalization of Saturated Aza-Cycles. ACS Catal 2020, 10, 2929–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Vinogradova EV; Zhang C; Spokoyny AM; Pentelute BL; Buchwald SL Organometallic palladium reagents for cysteine bioconjugation. Nature 2015, 526, 687–691. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rojas AJ; Zhang C; Vinogradova EV; Buchwald NH; Reilly J; Pentelute BL; Buchwald SL Divergent unprotected peptide macrocyclisation by palladium-mediated cysteine arylation. Chem. Sci 2017, 8, 4257–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Rojas AJ; Pentelute BL; Buchwald SL Water-Soluble Palladium Reagents for Cysteine S-Arylation under Ambient Aqueous Conditions. Org. Lett 2017, 19, 4263–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Dhanjee HH; Buslov I; Windsor IW; Raines RT; Pentelute BL; Buchwald SL Palladium-Protein Oxidative Addition Complexes by Amine-Selective Acylation. J. Am. Chem. Soc 2020, 142, 21237–21242. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Lee HG; Milner PJ; Placzek MS; Buchwald SL; Hooker JM Virtually Instantaneous, Room-Temperature [11C]-Cyanation Using Biaryl Phosphine Pd(0) Complexes. J. Am. Chem. Soc 2015, 137, 648–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Pan Y; Young GB Syntheses and spectroscopic characteristics of dialkylpalladium(II) complexes; PdR2(cod) as precursors for derivatives with N- or P-donor ligands. J. Organomet. Chem 1999, 577, 257–264. [Google Scholar]; (b) (cod)Pd(CH2TMS)2 shows significant decomposition even when stored at –35 °C in a glovebox.
- (11).Lee HG; Milner PJ; Colvin MT; Andreas L; Buchwald SL Structure and reactivity of [(L·Pd)n·(1,5-cyclooctadiene)] (n = 1–2) complexes bearing biaryl phosphine ligands. Inorg. Chim. Acta 2014, 422, 188–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Wallow TI; Goodson FE; Novak BM New Methods for the Synthesis of ArPdL2I (L = Tertiary Phosphine) Complexes. Organometallics 1996, 15, 3708–3716. [Google Scholar]; (b) Zalesskiy SS; Ananikov VP Pd2(dba)3 as a Precursor of Soluble Metal Complexes and Nanoparticles: Determination of Palladium Active Species for Catalysis and Synthesis. Organometallics 2012, 31, 2302–2309. [Google Scholar]
- (13).Cámpora J; López JA; Palma P; del Rio D; Carmona E; Valerga P; Graiff C; Tiripicchio A Synthesis and Insertion of the Cyclometalated Palladium-Alkyl Complexes Pd(CH2CMe2-o-C6H4)L2. Observation of a Pentacoordinated Intermediate in the Insertion of SO2. Inorg. Chem 2001, 40, 4116–4126. [DOI] [PubMed] [Google Scholar]
- (14).Shultz LH; Tempel DJ; Brookhart M Palladium(II) β-Agostic Alkyl Cations and Alkyl Ethylene Complexes: Investigation of Polymer Chain Isomerization Mechanisms. J. Am. Chem. Soc 2001, 123, 11539–11555. [DOI] [PubMed] [Google Scholar]
- (15).López G; Garcia G; Santana MD; Sánchez G; Ruiz J; Hermoso JA; Vegas A; Martínez-Ripoll M Synthesis and structural study of neutral mononuclear and anionic binuclear 2,4,6-trifiuorophenyl derivatives of palladium(II). Crystal structure of [P(CH2Ph)Ph3]2[(C6F3H2)2Pd(μ-SCN)(μ-NCS)Pd(C6F3H2)2]. J. Chem. Soc., Dalton Trans 1990, 1621–1626. [Google Scholar]
- (16).Yang Y; Oldenhuis NJ; Buchwald SL Mild and General Conditions for Negishi Cross-Coupling Enabled by the Use of Palladacycle Precatalysts. Angew. Chem., Int. Ed 2013, 52, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Catellani M; Chiusoli GP Palladium-(II) and -(IV) complexes as intermediates in C-C bond-forming reactions. J. Organomet. Chem 1988, 346, C27–C30. [Google Scholar]
- (18).Cárdenas DJ; Mateo C; Echavarren AM Synthesis of Oxaand Azapalladacycles from Organostannanes. Angew. Chem., Int. Ed. Engl 1995, 33, 2445–2447. [Google Scholar]
- (19).(a) Chaumontet M; Piccardi R; Audic N; Hitce J; Peglion J-L; Clot E; Baudoin O Synthesis of Benzocyclobutenes by Palladium-Catalyzed C-H Activation of Methyl Groups: Method and Mechanistic Study. J. Am. Chem. Soc 2008, 130, 15157–15166. [DOI] [PubMed] [Google Scholar]; (b) Catellani M Catalytic Multistep Reactions via Palladacycles. Synlett 2003, 3, 298–313. [Google Scholar]; (c) Wang J; Dong G Palladium/Norbornene Cooperative Catalysis. Chem. Rev 2019, 119, 7478–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ye J; Lautens M Palladium-catalysed norbornene-mediated C-H functionalization of arenes. Nat. Chem 2015, 7, 863–870. [DOI] [PubMed] [Google Scholar]
- (20).(a) Cámpora J; Palma P; del Río D; López JA;Álvarez E; Connelly NG Redox Behavior of an Organometallic Palladium(II)/Palladium(IV) System. A New Method for the Synthesis of Cationic Palladium(IV) Complexes. Organometallics 2005, 24, 3624–3628. [Google Scholar]; (b) Pérez-Temprano MH; Racowski JM; Kampf JW; Sanford MS Competition between sp3-C-N vs. sp3-C-F Reductive Elimination from PdIV Complexes. J. Am. Chem. Soc 2014, 136, 4097–4100. [DOI] [PubMed] [Google Scholar]; (c) Qu F; Khusnutdinova JR; Rath NP; Mirica LM Dioxygen activation by an organometallic Pd(II) precursor: formation of a Pd(IV)-OH complex and its C-O bond formation reactivity. Chem. Commun 2014, 50, 3036–3039. [DOI] [PubMed] [Google Scholar]
- (21).(a) Gutiérrez-Bonet Á; Juliá-Hernández F; de Luis B; Martin R Pd-Catalyzed C(sp3)-H Functionalization/Carbenoid Insertion: All-Carbon Quaternary Centers via Multiple C-C Bond Formation. J. Am. Chem. Soc 2016, 138, 6384–6387. [DOI] [PubMed] [Google Scholar]; (b) Clemenceau A; Thesmar P; Gicquel M; Le Fiohic A; Baudoin O Direct Synthesis of Cyclopropanes from gem-Dialkyl Groups Through Double C-H Activation. J. Am. Chem. Soc 2020, 142, 15355–15361. [DOI] [PubMed] [Google Scholar]
- (22).See SI for studies of the stability of P3.
- (23).Under conditions A, the reaction with aryl iodides led to a complex mixture of products.
- (24).Golding WA; Pearce-Higgins R; Phipps RJ Site-Selective Cross-Coupling of Remote Chlorides Enabled by Electrostatically Directed Palladium Catalysis. J. Am. Chem. Soc 2018, 140, 13570–13574. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.