

Abstract
The application of next-generation sequencing to study congenital heart disease (CHD) is increasingly providing new insights into the causes and mechanisms of this prevalent birth anomaly. Whole-exome sequencing analysis identifies damaging gene variants altering single or contiguous nucleotides that are assigned pathogenicity based on statistical analyses of families and cohorts with CHD, high expression in the developing heart and depletion of damaging protein-coding variants in the general population. Gene classes fulfilling these criteria are enriched in patients with CHD and extracardiac abnormalities, evidencing shared pathways in organogenesis. Developmental single-cell transcriptomic data demonstrate the expression of CHD-associated genes in particular cell lineages, and emerging insights indicate that genetic variants perturb multicellular interactions that are crucial for cardiogenesis. Whole-genome sequencing analyses extend these observations, identifying non-coding variants that influence the expression of genes associated with CHD and contribute to the estimated ~55% of unexplained cases of CHD. These approaches combined with the assessment of common and mosaic genetic variants have provided a more complete knowledge of the causes and mechanisms of CHD. Such advances provide knowledge to inform the clinical care of patients with CHD or other birth defects and deepen our understanding of the complexity of human development. In this Review, we highlight known and candidate CHD-associated human genes and discuss how the integration of advances in developmental biology research can provide new insights into the genetic contributions to CHD.
Congenital heart disease (CHD) has long been recognized as the most common and often severe anomaly at birth, with a prevalence of 6–13 in 1,000 newborn babies1–5. CHD encompasses a broad spectrum of heart malformations that range from a solitary abnormality (atrial or ventricular septal defect or isolated dysplastic valve) to complex lesions consisting of multiple defects (tetralogy of Fallot or hypoplastic left heart syndrome) (Fig. 1). Advanced surgical and medical interventions, including palliation and definitive correction of critical (requiring early intervention for survival) malformations during infancy have significantly reduced CHD mortality6. Currently, >90% of patients with CHD survive to adulthood, accounting for the increased prevalence of CHD in the general population6. Moreover, the increased longevity of patients with CHD has led to a greater recognition of the associated comorbidities. Approximately 13% of newborn babies with CHD have extracardiac structural or functional defects and can develop neurodevelopmental delay7 throughout childhood. When these additional diagnoses are likely to have resulted from the same aetiology, CHD is classified as syndromic. Complex heart malformations most often occur in syndromic CHD, which can result in lifelong multisystem health issues. Understanding the aetiologies and mechanisms of CHD can therefore contribute insights into normal and aberrant developmental processes of multiple organ systems.
Fig. 1 |. Common types of congenital heart disease.
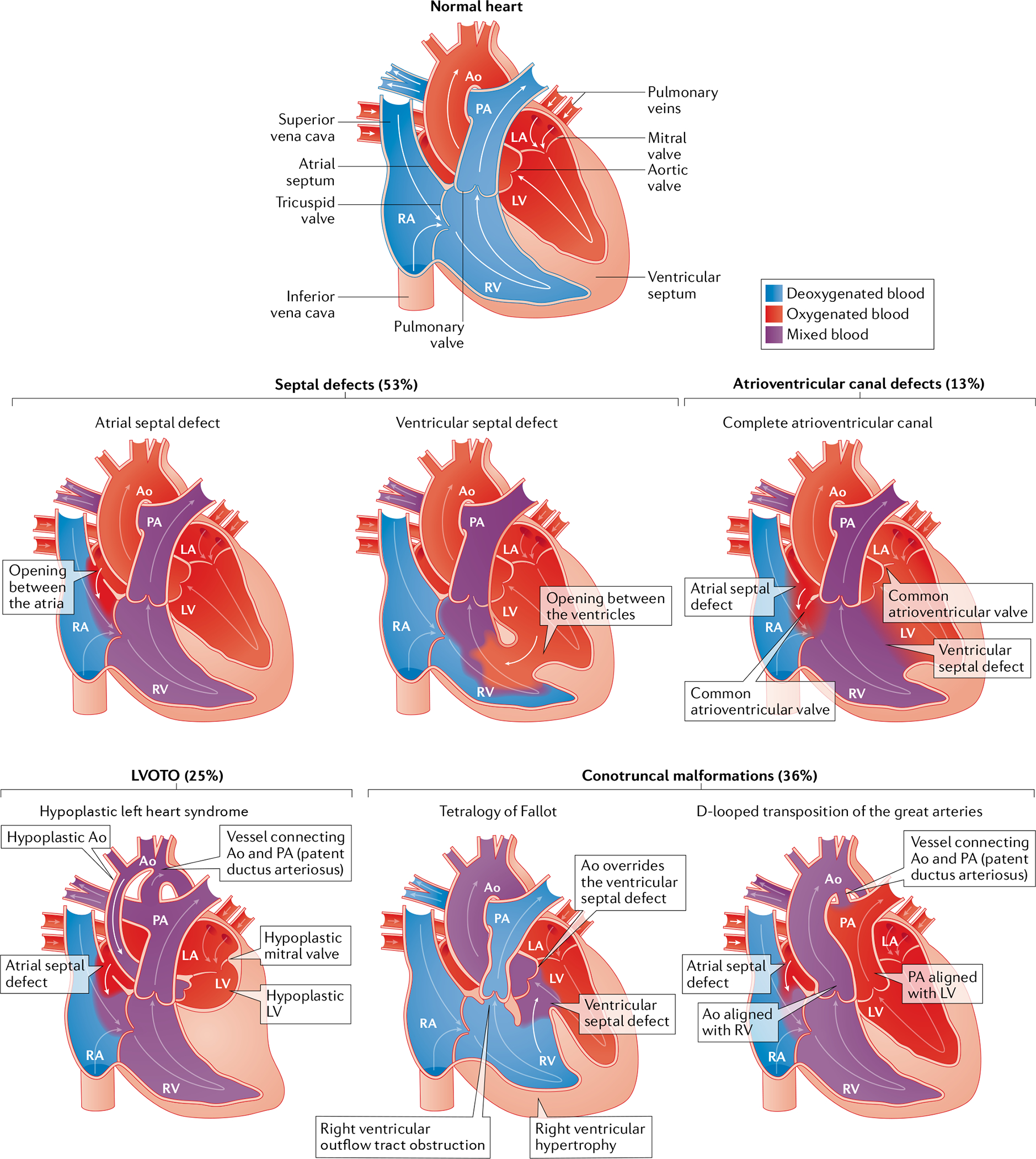
Schematic of congenital heart disease malformations grouped by anatomical subtype and showing the oxygen saturation of blood. Abnormal connections between the left and right chambers, such as atrial or ventricular septal defects or atrioventricular canal defects, are typically acyanotic owing to left-to-right shunting of oxygenated blood into the pulmonary circulation. Left ventricular outflow tract obstructions (LVOTOs) range from isolated aortic stenosis to a combination of anomalies (hypoplastic left heart syndrome with mitral and aortic stenosis) associated with cyanosis. Conotruncal malformations are frequently cyanotic, with shunting of deoxygenated blood into the systemic circulation, and include tetralogy of Fallot and transposition of the great arteries. The percentages denote the prevalence of malformation in the cohort of patients with congenital heart disease from the Paediatric Cardiac Genomics Consortium17,18. Note that the percentages exceed 100 due to coexisting structural cardiac abnormalities in the participants. Ao, aorta; LA, left atrium; LV, left ventricle; PA, pulmonary artery; RA, right atrium; RV, right ventricle.
Genetics has a central role in CHD pathogenesis, a discovery that has benefited from considerable technical advances in examining the human genome. Early evidence came from karyotyping patients with syndromic CHD that uncovered aneuploidies, most commonly trisomy 13, 18 or 21 (Down syndrome) and monosomy X (Turner syndrome), which occur in ~12% of patients with CHD8. Cytogenic analyses and genome arrays extended the molecular insights into subchromosomal structural rearrangements, revealing prevalent recurrent 3-Mb deletions at 22q11.2 in DiGeorge (velocardiofacial) syndrome9–12. Deletions at 22q11.2 are detected in approximately 2% of all patients with CHD13 and in 13% of patients with specific cardiac malformations, as assessed by fluorescence in situ hybridization and targeted amplification14. Karyotype-detected or microarray-detected abnormalities together account for almost 25% of cases of sporadic CHD15.
Genome-wide linkage analyses enabled the initial identification of monogenic causes of familial CHD, including variants in genes encoding transcription factors and transcriptional regulators of genes related to cardiac development16. A detailed knowledge of the human genome at base pair resolution and advances in sequencing technologies have substantially accelerated the discovery of genes associated with CHD. Contemporary whole-exome sequencing (WES) and whole-genome sequencing (WGS) platforms, bioinformatic analytical tools and large, aggregated, population-based sequencing datasets enable the identification of deleterious missense and loss-of-function (LOF) variants (together denoted as damaging variants), small insertions or deletions, copy number variants (CNVs) and structural variants. Application of these strategies has uncovered damaging variants that segregate in familial cases of CHD, reflect de novo mutations in CHD probands with unaffected parents, and occur at significantly higher rates in approximately 20% of patients with CHD in large cohort studies15–19.
Whole-exome sequencing.
(WES). Targeted sequencing of protein-encoding regions, which comprise 1% of the genome. WES can be used to determine single-nucleotide variants, small insertions and deletions, and copy number variants, but is less sensitive than WGS for the detection of structural variants.
Whole-genome sequencing.
(WGS). Sequencing of the entire genome (protein coding and non-coding regions) that can be used to determine single-nucleotide variants, small insertions and deletions, and structural variants.
Collectively, these approaches identify damaging coding variants in definitive and candidate genes for CHD in 45% of patients with CHD15–19. Genes with a definitive association with CHD harbour variants that significantly co-segregate in families with CHD, are significantly enriched in unrelated patients with CHD compared with controls or account for heart malformations in patients with syndromic CHD. Candidate genes for involvement in CHD have similar properties, but without sufficient statistical significance to warrant a definitive classification. Diverse types of variants (aneuploidies, CNVs, structural variants and LOF variants) alter the dosage of CHD-related genes, whereas deleterious missense variants can maintain physiological gene dosage but impair function of the encoded protein.
Genes linked to CHD provide new insights into the crucial molecules and pathways involved in cardiogenesis, complementing and extending the considerable information derived from exploring heart development in experimental models. Studies of spontaneous (de novo) gene variants in sporadic CHD can identify new genes involved in cardiogenesis, while detailed clinical evaluations allow the definition of a broader spectrum of cardiac and extracardiac phenotypes than studies in experimental models. Given that the dosage of most human variants is heterozygous, whereas studies in experimental models often explore homozygous variants, human CHD data are informative about subtler influences on heart formation. Additionally, the observation that the highest rates of de novo damaging variants in genes associated with CHD occur in patients with critical and complex malformations17–19 provides compelling evidence for profound deleterious effects on reproductive health and the evolutionary constraint on many genes linked to CHD.
Why is CHD the most common congenital anomaly? One hypothesis is that complex developmental processes of heart formation are exquisitely sensitive to changes in gene dosage among many essential genes and pathways. When gene dosage is altered, developmental errors can occur, many of which are readily detected by contemporary screening ultrasonography that is routinely performed at 18 weeks gestational age20. To highlight the exquisite molecular orchestration of heart formation, in this Review we briefly summarize cardiac development before discussing progress in the past 10 years in the genomic discoveries and strategies at the forefront of current CHD research. Several informative reviews have examined key signals that govern cardiac morphogenesis, and readers are referred to these publications for more details21–26. Throughout the Review, we highlight in bold those genes with damaging variants identified in patients with CHD.
Cardiac development
The heart forms during the first weeks of embryogenesis through the specification of cardiac progenitor fields that coalesce into a cardiac crescent. These progenitor fields evolve into a linear and then looped heart tube that grows, undergoes regional transformation and emerges as a nearly fully formed organ by 8 weeks of gestation in humans. Throughout cardiac morphogenesis, the interplay between differentiating progenitor cell populations generates specialized cardiac cells with precise spatial organization within constrained regions.
Overview of progenitor cell populations and morphogenesis.
Three major progenitor cell populations contribute to cardiac structures: the first heart field (FHF), derived from the lateral plate mesoderm, the second heart field (SHF), derived from the lateral plate splanchnic mesoderm, and migratory cardiac neural crest populating the III, IV and VI pharyngeal arches27–30 (Fig. 2). The symmetric FHF and SHF meet anteriorly to form the cardiac crescent, which then fuses into a linear heart tube with ventricular and atrial progenitors oriented on the anterior–posterior axis. The heart tube undergoes rightward looping with input from a morphogen gradient defining the left–right axis, producing the spatial arrangement of fetal cardiac chambers. Cardiac cells derived from the FHF give rise to the atria and left ventricle, whereas cells from the SHF become the right ventricle and outflow tract and also contribute to both atria. Ballooning of the outer curvature of the heart tube via asymmetric growth increases the size of the ventricular chamber31. Subsequent outflow tract septation and smooth muscle differentiation occur with contribution from the cardiac neural crest.
Fig. 2 |. Human heart development.
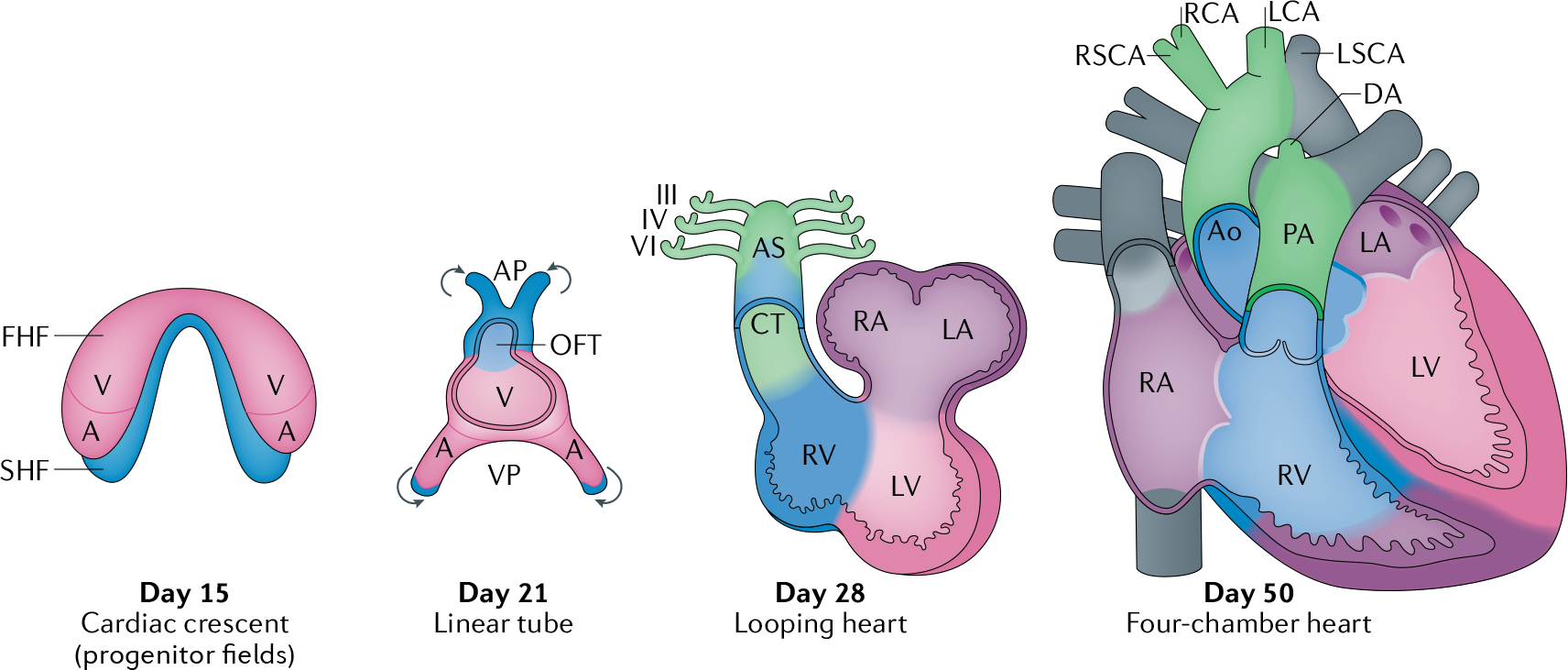
Human heart development involves the formation of the first heart field (FHF) or cardiac crescent at approximately embryonic day 15, with chamber-specific cardiac progenitors aligned in a specified spatial distribution. Concurrently, an additional population of cardiac progenitors, the second heart field (SHF), develops posterior to the FHF. By embryonic day 21, the midline fusion of the FHF forms a linear cardiac tube and SHF-derived cells migrate (arrows) into the arterial pole (AP) and venous pole (VP) of the tube. The linear cardiac tube undergoes looping by embryonic day 28 to form early ventricular chambers. Cardiac neural crest cells migrate to populate the III, IV and VI pharyngeal arches, the aortic sac (AS) and the conotruncal (CT) ridges. By embryonic day 50, the cardiac structures largely resemble those of the mature heart, with four main chambers and two outflow tracts (OFT). A, atrium, Ao, aorta; AV, atrioventricular; DA, ductus arteriosus; LA, left atrium; LCA, left carotid artery; LSCA, left subclavian artery; LV, left ventricle; PA, pulmonary artery; RA, right atrium; RCA, right carotid artery; RSCA, right subclavian artery; RV, right ventricle; V, ventricle.
Genetic regulators of cardiac morphogenesis.
Many crucial molecules participate in the differentiation of progenitor cell populations into cardiac cell lineages and in directing cardiac morphogenesis, some of which are encoded by genes that are associated with CHD. Formation of the FHF from cardiogenic mesoderm is induced by signalling pathways including bone morphogenetic protein (BMP) and fibroblast growth factor (FGF) from the adjacent endoderm32–35. In experimental model systems, ablation of genes encoding BMPs reduces the expression of crucial cardiac transcripts that are required for commitment to a cardiomyocyte lineage36. Sarcomere formation and contraction are initiated by the expression of genes such as GATA4, GATA6, NKX2–5 and TBX5 (REFS 21–26,37). Expression of IRX4 in both the FHF and SHF of the cardiac crescent promotes specification of ventricular cardiomyocytes38. Commitment to an atrial lineage is initiated by factors including retinoic acid within mesoderm progenitor fields39. Further specification results in cardiomyocytes destined to populate the left (IRX4 (REF.40), HAND1 (REF.41) and MSX1 (REF.42)) and right (IRX4 (REF.40) and GATA6 (REF.43)) ventricles and the outflow tract (GATA6 (REF.43), HAND2 (REF.41), ARID3B44 and TEAD45). SHF contributions to the outflow tract require FGF signalling35, which enables neural crest migration and is required to induce tbx1 expression in the SHF in zebrafish46. Despite stereotyped differentiation pathways, plasticity is maintained in the progenitor fields during cardiac development, because ablation of the FHF cells in zebrafish results in SHF compensation that regenerates ventricular cardiomyocytes47.
Signalling pathways and gene networks regulating cardiac neural crest differentiation have also been identified. Absence of retinoic acid signalling prevents normal differentiation of cardiac neural crest cells in mice48, which can be ameliorated by maternal vitamin A supplementation49, highlighting genotype and environmental interactions in CHD. By contrast, a GATA6 missense variant that is recurrently identified in patients with CHD and disrupts outflow track formation strikingly increases retinoic acid signalling43. Cardiac neural crest migration depends on FOXC2 expression50, which in turn mediates the activation of Sema3c expression in the SHF51, whereas Tbx1 expression results in counterbalance inhibitory signals51.
The heart also contains non-chamber cardiomyocytes within the atrioventricular canal and outflow tract as well as valvular endothelial cells and smooth muscle cells. Non-chamber myocardium is specified by the repression of chamber-specific genes by regulatory proteins such as those encoded by TBX2 and TBX3 (REF.52) and BMP2 and BMP4 (REF.53). A subset of valvular endothelial cells undergo endothelial-to-mesenchymal transition to populate the endocardial cushions under the guidance of signalling cascades that include vascular endothelial growth factor (VEGF)54 and transforming growth factor-β (TGFβ)55. Studies of outflow tract development in mouse models have also demonstrated the requirement for histone deacetylase 3 (encoded by Hdac3) for smooth muscle differentiation, whereas the pulmonary arteries and veins require FGF and Sonic hedgehog signalling56–58. Cardiomyocytes can also become non-chamber cells via transdifferentiation, and single-cell characterization of the mouse developing heart found a cardiomyocyte population that transdifferentiates into vascular smooth muscle cells of the great arteries59.
Establishment of a left–right signalling axis in the embryo occurs within the embryonic node, where signals mediated by motile and sensory cilia lead to the asymmetric expression of genes encoding downstream regulatory signals, including the genes coding for the TGFβ family members Nodal, Lefty1, Lefty2 and Zic3 (REFS60–62). These signals are crucial for appropriate rightward looping of the linear heart tube that properly arranges the atria, ventricles and outflow tract and establishes the sinoatrial node in the right atria. Perturbation of Sonic hedgehog or activin signalling in the chick embryo63 or knockout of Nodal, Lefty1 or Lefty2 (encoding left–right axis signalling proteins and transcriptional regulators) in mice lead to defects in cardiac looping and resultant congenital heart defects64,65.
Disruption of human cardiac morphogenesis
The anatomical study of human embryos has provided insights into human cardiogenesis and the aetiology of the cardiac malformations that result from the disruption of specific spatiotemporal processes. CHD lesions are often classified into anatomical subgroups that arise from disruption of shared embryonic processes. As such, the discovery of genes with pathogenic variants that elicit particular malformations provides clues into the downstream developmental and physiological consequences of genetic regulation in heart development.
Conotruncal defects.
Conotruncal defects, such as tetralogy of Fallot, persistent truncus arteriosus and transposition of the great arteries (Fig. 1), reflect maldevelopment of the ventricular septum in the outflow tract and/or the great arteries, or malalignment of these structures. Conotruncal defects frequently occur in autosomal dominant 22q11.2 deletion syndrome (which commonly involves deletion of the CHD-associated gene TBX1)66 and CHARGE syndrome (which in most cases involves deletions or variants in the CHD-associated gene CHD7)67. These syndromes are associated with abnormalities in the heart, face and head, which arise from neural crest cells, highlighting the effects of damaging gene variants on cell lineages that contribute to extracardiac malformations. Both of these CHD syndromes have variable penetrance and expressivity68,69 (defined in Table 1), which suggests that additional factors influence the clinical phenotype. Although these additional factors are largely unknown, a genetic modifier influencing conotruncal defects in 22q11.2 deletion syndrome has been reported70. Recurrent duplications and deletions at 1q21.1 — which spans the candidate genes for CHD GJA5 (encoding connexin 40) and CHD1L (a gene related to CHD7) — are associated with tetralogy of Fallot, often but not always with extracardiac abnormalities71,72.
Table 1 |.
Glossary of human genetics terms
| Domain | Term | Definition |
|---|---|---|
| Mode of inheritance | Autosomal dominant | A single copy of a gene variant on a non-sex chromosome is sufficient to cause disease |
| Autosomal recessive | A variant on each gene copy is required to cause disease | |
| X-linked | Gene variants encoded on the X chromosome can exhibit dominant or recessive transmission | |
| Hemizygous | Presence of a single gene copy in diploid cells, such as the X chromosome in males | |
| Mosaicism | The presence of at least two cell populations with differing genotypes, resulting in a gene variant being present in only a subset of cells | |
| De novo variant | A gene variant in an individual that is absent from both parents, and arises due to new mutation of parental gametes or after fertilization | |
| Monogenic inheritance | A single gene is causative for a trait | |
| Polygenic inheritance | Many genes contribute to a trait | |
| Oligogenic inheritance | A few genes influence the trait | |
| Variant types | Indel | An insertion or deletion of a sequence of 1–49 base pairs |
| Aneuploidy | An abnormal number of chromosomes | |
| Structural variant | A broad class of variants encompassing ≥50 base pairs of sequence that includes deletions, duplications, inversions, insertions or transversions | |
| Copy number variant | An unbalanced structural variant (deletion, duplication or insertion) that alters the copy number of a gene or genes | |
| Molecular consequences of genetic variants | Damaging | A general descriptive term indicating that a variant alters the expression level or function of a gene |
| Loss-of-function | A variant that extinguishes the expression of one gene copy or abrogates its function | |
| Gain-of-function | A variant that enhances gene function, produces a new function or inappropriately increases gene expression | |
| Frameshift | Insertion or deletion of nucleotides that shifts the triplet amino acid code used for translation of mRNA to protein | |
| Missense | A variant that substitutes one amino acid for another in the protein sequence | |
| Synonymous | A variant that alters a nucleotide but does not alter the amino acid sequence of the encoded protein | |
| Splicing | A gene variant that alters canonical nucleotide signals to excise introns or exclude exons from mature mRNAs; splicing variants can abrogate existing splice signals or create new splice signals | |
| Genotype–phenotype relationship | Variable expressivity | Variation in phenotype among individuals with the causal genetic variant |
| Penetrance | The proportion of individuals who manifest the phenotype associated with a genetic variant |
Non-syndromic conotruncal malformations are also mainly associated with monogenic LOF variants in genes related to two important signalling pathways orchestrating developmental programmes in the SHF and cardiac neural crest cells: the Notch pathway (NOTCH1) and the VEGF pathway (KDR, FLT4, VEGFA, FGD5, BCAR1, IQGAP1, FOXO1 and PRDM1)73,74. Why these progenitor fields and developing tissues have a unique susceptibility to alterations in the VEGF pathway remains an open question. Autosomal dominant damaging variants in ZFPM2 (encoding the transcription factor ZFPM2) and in the genes encoding its binding partners GATA4 and GATA6 also frequently result in conotruncal defects75–77. Both GATA genes encode transcription regulators that act as pioneer factors, binding and influencing closed chromatin regions to enable subsequent transcription factor binding, highlighting the importance of this transcriptional regulatory network in conotruncal development.
Atrial and ventricular septal defects.
Abnormal development of the endocardial cushions and/or the SHF-derived dorsal mesenchymal protrusion78,79 results in atrial and ventricular septation defects, atrioventricular valve abnormalities and atrioventricular canal defects that include septation and valve defects. Complete atrioventricular canal defects are found in only ~5% of individuals with CHD80, but notably occur in almost half of the ~40% of individuals with trisomy 21 who have CHD81,82. This remarkable association indicates that genes in chromosome 21, particularly those localized within a 0.96-Mb region83, are crucially involved in endocardial cushion development, but the responsible gene(s) remain unknown. As in other syndromes, CHD penetrance is incomplete in individuals with trisomy 21, and 60% of individuals have a structurally normal heart81,82.
Isolated atrial or ventricular septal defects are the most common congenital heart lesions; however, among patients with sporadic occurrence of these malformations in the absence of a genetic syndrome, the causal aetiology is rarely defined. Damaging variants in NKX2–5 and GATA4 were initially identified in patients with rare autosomal dominant familial septal defects, whereas TBX5 variants were found to cause Holt–Oram syndrome in families with septal defects and associated limb defects16. In model systems, the proteins coded by these genes have been shown to interact physically to promote proper atrial and ventricular septation84–86.
Left-sided obstructive lesions.
This group of CHDs includes the spectrum of stenotic and atretic anomalies of the left-side chambers and valves that occur in isolation (mitral stenosis, bicommissural aortic valve, aortic stenosis and coarctation of the aorta) or in combination, such as Shone complex (annulo-leaflet mitral ring, parachute mitral valve, subaortic stenosis and coarctation of the aorta) and hypoplastic left heart syndrome (mitral and aortic stenosis or atresia, and hypoplastic left ventricle). Left-sided obstructive lesions have higher rates of heritability than other CHDs, and a range of malformations is observed within families, indicating that the development of the involved structures shares genetic pathways87.
Patients with monosomy X or Turner syndrome (45,XO karyotype) often have bicommissural aortic valve (15–30%), aortic coarctation (7–20%) and, less commonly, hypoplastic left heart syndrome88,89. The increased risk of left-sided obstruction lesions in patients with Turner syndrome relates to altered dosages of genes on chromosome X and copy number polymorphisms at the 12q13.31 locus90. However, the mechanisms by which these genotypes influence the development of left-sided structures is unknown. The autosomal dominant Jacobsen syndrome (11q23 deletion), which causes diverse and severe left-sided obstructive lesions in 33% of the patients91, is mediated in part by a requirement for protein C-ets-1 (encoded by ETS1) function in the developing endocardium to support cardiomyocyte proliferation and extracellular matrix organization92. Monogenic damaging variants in GATA5 cause bicuspid aortic valve93,94 and in RBFOX2 cause hypoplastic left heart syndrome95. Rare recessive missense variants in MYH6 (encoding the cardiac α-myosin heavy chain) cause Shone complex17 and hypoplastic left heart syndrome96. Given that cardiac α-myosin heavy chain is a contractile protein in the sarcomere that is highly expressed during cardiogenesis, MYH6 is one of a very few CHD-associated genes that is not related to transcription or translation.
Right-sided obstructive lesions.
Obstruction to the flow of systemic venous blood as it travels from the right side of the heart to the pulmonary capillary beds can occur in tricuspid atresia, pulmonary atresia with intact ventricular septum, isolated pulmonary valve stenosis and supravalvular and branch pulmonary artery stenosis, and reflects a wide range of embryological abnormalities. Tricuspid atresia results from agenesis of the right-sided atrioventricular valve and is associated with chromosomal trisomy in humans97 and Zfpm2 LOF in mice98. Pulmonary valve stenosis is present in >50% of children with Noonan syndrome and is more frequently associated with variants in the PTPN11 gene99. The pulmonary valve stenosis associated with PTPN11 variants results from increased activation of mitogen-activated protein kinases (MAPKs) in endocardial cells that leads to excess endothelial-to-mesenchymal transition in the developing pulmonary valve100. Pulmonary valve stenosis is a common feature of other syndromes (such as Costello and cardiofaciocutaneous syndromes) that result from excessive activation of the RAS–MAPK signalling pathway101. Autosomal dominant and recessive forms of Robinow syndrome are associated with isolated pulmonary valvular stenosis and conotruncal lesions, such as tetralogy of Fallot102,103, and arise from pathogenic variants in genes encoding receptors and ligands controlling planar cell polarity (WNT5A, ROR2, DVL1 and DVL3)102,104–106, demonstrating the importance of the planar cell polarity pathway in the development of the SHF-derived right ventricular infundibulum and the outflow tract cushions. Individuals with autosomal dominant Alagille syndrome (JAG1 and NOTCH2) can have tetralogy of Fallot, pulmonary valve stenosis and/or stenosis of the peripheral pulmonary arterial branches, demonstrating the dose sensitivity of Notch signalling in right ventricular outflow tract development, pulmonary valve formation and vasculogenesis that contributes to the pulmonary arterial tree107,108.
Left–right patterning defects.
Defects in left–right axis patterning cause heterotaxy syndrome, resulting in abnormal positioning of the viscera as well as CHD that includes structural malformations and aberrant arterial and venous connections. Normal cilia function is crucial for left–right axis patterning, and a subset of patients with primary ciliary dyskinesia have heterotaxy syndrome with CHD. Patients with non-syndromic CHD and damaging variants in cilia-related genes generally have transposition of the great arteries109. Damaging variants in Nodal and TGFβ signalling pathway genes that participate in laterality (NODAL, FOXH1, ZIC3, ACVR2B and SMAD2) are also frequent among patients with cardiac left–right patterning defects109,110. Variants in cilia-related genes that are responsible for laterality defects cause CHD by autosomal dominant or autosomal recessive mechanisms. CHD-associated variants in genes encoding Nodal and TGFβ signalling proteins are dominant, whereas ZIC3 variants cause X-linked heterotaxy syndrome in hemizygous males111,112.
Emerging CHD genes from cohort studies
Genes linked to CHD are increasingly being identified from analyses of CHD cohorts, recruited from single centres or by national and international collaborations17,19,113. Most studies enrol patients with severe or critical CHD, under-represent patients with prevalent simple CHD lesions and exclude patients with clinically established genetic syndromes114. Variants are typically discovered by WES and analyses of case–control comparisons. These studies show that patients with CHD have the same rates of total de novo variants but significantly more rare (allele frequency ≤ 1 × 10−5 in the general population) damaging variants compared with the general population, including 9% excess of de novo damaging variants, 7% excess of dominant inherited variants and 1% recessive inherited variants17–19. Among patients with unexplained sporadic CHD, damaging variants are identified in approximately 8–10% of patients17,19. The rate of de novo damaging variants is highest among patients with syndromic CHD, whereas the rate of inherited LOF variants is highest among patients with isolated CHD19.
Cohort studies have confirmed the importance of previously identified biological pathways in CHD pathogenesis, including genes related to the Notch115 and VEGF115 signalling cascades. However, the most prominent functional class of newly identified CHD-associated genes is chromatin remodelling18,116. Damaging variants in these genes encoding chromatin remodelling proteins occurred most often in patients with CHD who had extracardiac anomalies and neurodevelopmental delay. The broad expression pattern of these genes indicates that shared epigenetic and transcriptional regulatory pathways are employed across organ development.
WES data from patients with CHD supports previous evidence indicating the importance of ciliary function in cardiogenesis. As has been identified in other recessive disorders, damaging variants in cilia-related genes are often inherited from an unaffected parent117. By contrast, damaging variants in genes related to chromatin modification are more often de novo variants117, indicating the substantial deleteriousness of even one mutant allele in this gene class.
Definitive genes for CHD.
Analyses of WES data from two cohorts of patients with CHD, totalling 2,056 patients with isolated CHD, 2,994 with syndromic CHD and 808 with CHD and unknown extracardiac status, were of sufficient statistical power to detect a significant excess of damaging variants in patients with CHD compared with matched controls, with correction for the analyses of all protein-coding genes (P ≤ 2 × 10−6)17,19,118. The identified genes from these cohorts have a definitive causal association with CHD (FiGS 3,4, genes shown in white font), as are the genes associated with rare familial CHD, but the genes discovered in large study cohorts have greater contribution to the overall population prevalence of CHD. A total of 18 genes were significantly enriched in these two WES cohorts: ADNP, ANKRD11, CDK13, CHD4, CHD7, DDX3X, DYRK1A, FLT4, KMT2A, KMT2D, NOTCH1, NSD1, PACS1, PRKD1, PTPN11, RBFOX2, SMAD6 and TAB2 (REFS17, 19, 73). Notably, despite exclusion of patients with syndromic CHD at enrolment, the analysis identified variants in CHD7 (CHARGE syndrome), KMT2D (Kabuki syndrome), NSD1 (Sotos syndrome) and PTPN11 (Noonan syndrome), highlighting the possibility that some variants associated with syndromic CHD result in formes frustes of syndromic phenotypes that escape clinical recognition. Variants in CDK13, CHD4 and PRKD1 are enriched in syndromic CHD19, whereas SMAD6 variants are more prevalent in isolated CHD119.
Fig. 3 |. Definitive and candidate genes for congenital heart disease: genes with LOF variants.
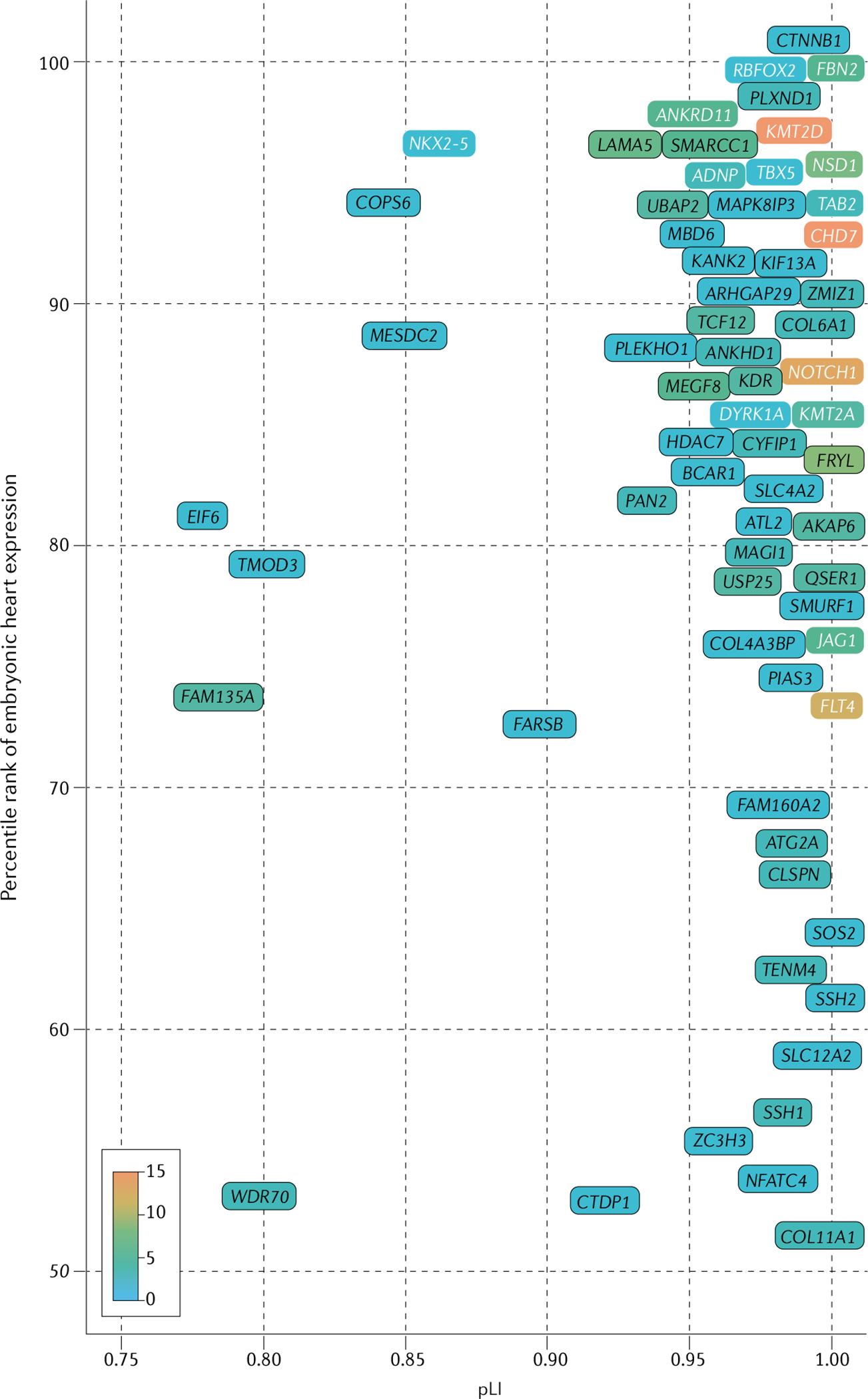
Genes with a definitive association with congenital heart disease (CHD) identified in whole-exome sequencing analyses of patients with CHD17,19 are significantly enriched for damaging variants in patients with CHD compared with controls (binomial P ≤ 2 × 10−6) after Bonferroni correction (for 20,440 human genes). Candidate genes for CHD (P < 0.05) were identified by binomial comparison of per-gene de novo loss-of-function (LOF) variants identified in combined data from 4,891 CHD trios17,19,118 (patients and unaffected parents) and 1,789 unaffected trios (from the Simons Simplex Collection cohort128). Enrichment for rare, transmitted LOF variants was determined by comparing 4,443 patients with CHD with 141,456 individuals without CHD included in the Genome Aggregation Database cohort129. Candidate genes were restricted by genetic constraint (probability of LOF intolerance (pLI) >0.75), high expression (>50th percentile) in the developing heart18,130, and identification of three or more LOF variants in CHD cohorts17,19,118 (accounting for differences in variant frequencies). Definitive (white font) and candidate (black font) genes with a nominal increase in LOF variants (scale 0–15 variants per gene; listed in Supplementary Table 1) among patients with CHD compared with participants without CHD are shown, ordered by percentile rank for expression in the mouse developing heart and by genetic constraint (pLI). Genes without expression data are not shown. See the Online Mendelian Inheritance in Man database for the clinical phenotypes associated with the genetic variants.
Fig. 4 |. Definitive and candidate genes for congenital heart disease: genes with damaging missense variants.
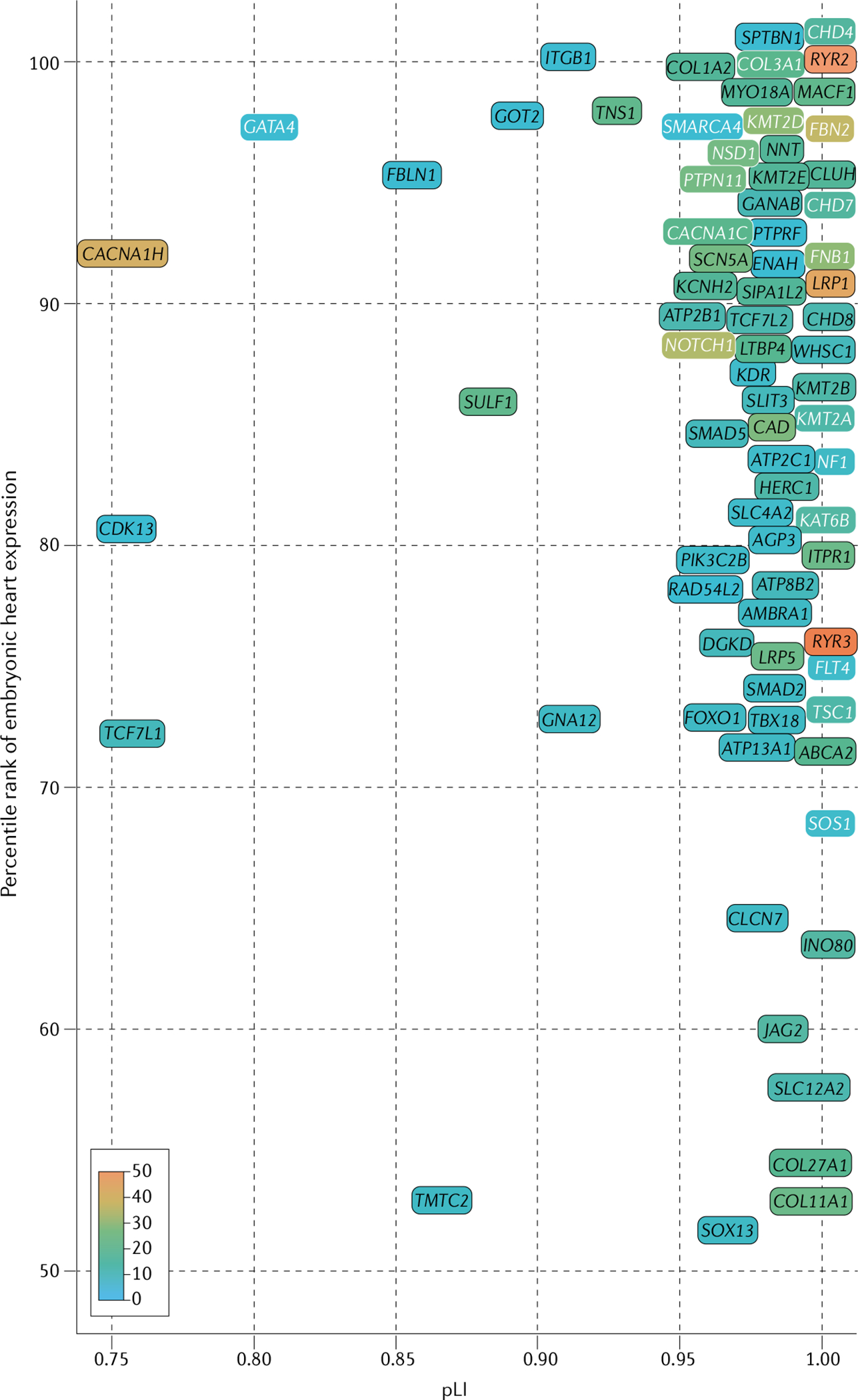
Genes with a definitive association with congenital heart disease (CHD) identified in whole-exome sequencing analyses of patients with CHD17,19 are significantly enriched for damaging variants in patients with CHD compared with controls (binomial P ≤ 2 × 10−6) after Bonferroni correction (for 20,440 human genes). Candidate genes for CHD (P < 0.05) were identified by binomial comparison of per-gene de novo deleterious missense variants identified in combined data from 4,891 CHD trios17,19,118 (patients and unaffected parents) and 1,789 unaffected trios (from the Simons Simplex Collection cohort128). Enrichment for rare, transmitted damaging missense variants was determined by comparing 4,443 patients with CHD with 141,456 individuals without CHD included in the Genome Aggregation Database cohort129. Candidate genes were restricted by genetic constraint (probability of loss-of-function intolerance (pLI) >0.75), high expression (>50th percentile) in the developing heart18,130, and identification of eight or more damaging missense variants in patients with CHD17,19,118 (accounting for differences in variant frequencies). Definitive (white font) and candidate (black font) genes with a nominal increase in damaging missense variants (scale 0–50 variants per gene, except RYR3, with 51 variants identified; all genes listed in Supplementary Table 1) among patients with CHD compared with participants without CHD are shown, ordered by percentile rank for expression in the mouse developing heart and by genetic constraint (pLI). Genes without expression data are not shown. See the Online Mendelian Inheritance in Man database for the clinical phenotypes associated with the genetic variants.
Previous studies highlight the biological relevance of some definitive CHD-associated genes. Null mutations of the murine homologues of SMAD6 and NOTCH1 cause heart malformations in mice120,121. The levels of RBFOX2 transcripts are reduced in some tissues from patients with CHD compared with control individuals122. Finally, the protein encoded by DYRK1A is a therapeutic target for neurodevelopmental phenotypes in individuals with trisomy 21 (REF123). Recurrent variants in other genes have yielded new phenotype correlations. NOTCH1 and FLT4 variants cause tetralogy of Fallot, accounting for 7% of patients in one study73. Genes previously associated with intellectual disability, including ADNP, ANKRD11 (KBG syndrome), CDK13 and DDX3X (REFS124–127), are now recognized also to be associated with CHD124–127. However, the roles of many definitive CHD-associated genes in cardiac development remain unknown.
Candidate CHD-associated genes with damaging variants.
WES data predict that approximately 400 genes contribute to CHD116, far more than are known today. Some of these genes probably harbour damaging variants in patients with CHD, but without statistical significance for classification as a definitive CHD-associated gene. To address the limitations in statistical power due to small sample sizes, we performed meta-analyses of the WES data from the two large cohorts of patients with CHD described in the previous section17,19,73,118. Patients with aneuploidies or 22q11 deletions were excluded from the analysis. WES data were jointly analysed and compared with WES data from a cohort of individuals without CHD128 and the Genome Aggregation Database cohort129. We identified 132 candidate genes for CHD (FiGS 3,4), including 66 enriched for LOF variants and 78 enriched for deleterious missense variants.
The findings of this meta-analysis provide further support for the roles of CHD-associated genes in transcriptional regulation (TABLE 2; Supplementary Tables 1, 2). Many of the candidate genes for CHD with LOF variants have functional annotations as chromatin modification, transcription factors or RNA processing, and some have been previously implicated in CHD or in organogenesis, accounting for associated extracardiac phenotypes. In the group of definitive and candidate genes for CHD related to the regulation of gene expression, the presence of extracardiac phenotypes was more common in patients with LOF variants than in those with damaging missense variants17,19 (Fig. 5a). Notably, these analyses confirmed that in genes related to chromatin modification, de novo LOF variants were enriched in patients with CHD with extracardiac abnormalities, whereas transmitted LOF variants were enriched in patients with isolated CHD (Fig. 5b). These observations might imply cardiac-specific functions for some chromatin-modifying proteins.
Table 2 |.
Functional assignment of 132 definitive and candidate genes for CHD
| Category | Functional class | CHD-associated genes enriched for variant type (%) | Example genes associated with syndromic and non-syndromic CHD | ||
|---|---|---|---|---|---|
| Loss of function | Damaging missense | Gene | Phenotypea | ||
| Gene regulation | Chromatin modification | 15 | 19 | KMT2D | Kabuki syndrome |
| CHD7 | CHARGE syndrome | ||||
| KAT6B | Genitopatellar syndrome | ||||
| Transcription factor | 8 | 8 | GATA4 | Isolated atrial, ventricular or atrioventricular septal defect, tetralogy of Fallot | |
| TBX5 | Holt–Oram syndrome | ||||
| mRNA processing | 8 | 3 | CDK13 | Distinctive faces, intellectual disability | |
| DNA binding | 3 | 1 | CLSPN | Xeroderma pigmentosa | |
| Signalling | Extracellular matrix | 8 | 12 | COL3A1 | Polymicrogyria |
| Cilia | 0 | 1 | AMBRA1 | Corpus callosum agenesis, seizures, cataracts, pigmentation defects, cardiomyopathy, variable immune defects | |
| Cytoskeleton | 12 | 13 | NF1 | Neurofibromatosis | |
| Signal transduction | 29 | 23 | PTPN11 | Noonan syndrome | |
| JAG1 | Alagille syndrome | ||||
| Other | Ion binding, transmembrane transport, oxidoreductase activity | 18 | 21 | CLCN7 | Hypopigmentation, organomegaly, delayed myelination, developmental delays |
Genes associated with congenital heart disease (CHD) identified in the analysis shown in FIGS 3 and 4 were mapped to gene ontology terms found in the Gene Ontology database. The gene ontology categorization of 132 genes and the identified variants are provided in Supplementary Table 1.
Phenotypes are detailed at Online Mendelian Inheritance in Man.
Fig. 5 |. Relationship between extracardiac features, genotypes and functions of CHD-associated genes.
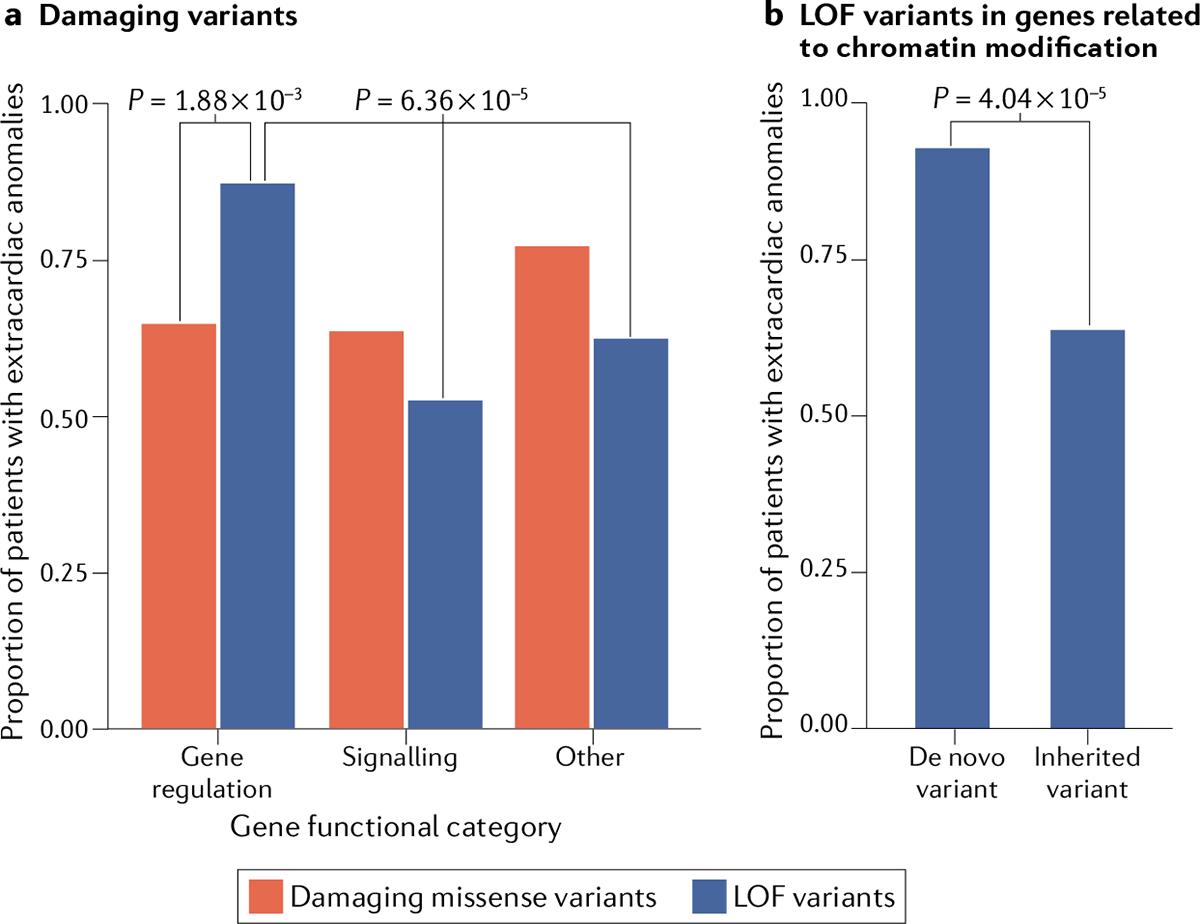
a | In a cohort of 2,871 patients with congenital heart disease (CHD) for whom whole-exome sequencing and clinical data were available17, extracardiac abnormalities are more common among patients with CHD with loss-of-function (LOF) variants (blue bars) in genes encoding proteins with gene-regulatory functions than among patients with LOF variants in genes related to signalling or other categories (P=6.36 × 10−5) or among patients with damaging missense variants (red bars) in regulatory genes (P=1.88 × 10−3)17,19. b | For LOF variants in genes encoding a protein involved in chromatin modification, extracardiac abnormalities are more common among patients with CHD with de novo variants than in patients with an inherited variant (P=4.04 × 10−5).
Analyses of larger cohorts of patients with CHD and reference cohorts composed of individuals of diverse races and ethnicities and studied using newer techniques17,19,130 and with functional assessment of deleterious missense variants are expected to reclassify some candidates genes for CHD as definitive, improve genotype-phenotype correlations and increase our knowledge of their precise roles in cardiac development.
Beyond rare monogenic coding variants
Aneuploidy, CNVs, indels and damaging coding variants have been identified in only 45% of patients with CHD. These data, combined with the recurrence rates of 3–7% for CHD in the offspring of patients with CHD131–135, indicate that monogenic variants cannot fully account for human CHD17,18,116. Other genotypes, including common and mosaic variants as well as variants in non-coding regulatory regions, are have been predicted to cause or contribute to CHD.
Common variants.
Polymorphic loci across the genome are analysed in genome-wide association studies to define risk alleles for traits of interest with the use of imputation of genomic sequences from low-cost, single-nucleotide polymorphism (SNP) arrays to provide genome-wide data or directly by WGS. Genome-wide association studies in CHD have identified loci associated with tetralogy of Fallot136, left-sided CHD lesions137–139 and atrial septal defects140. Protection from CHD is associated with a variant in the FIGN locus141 (encoding fidgetin), perhaps reflecting the reported association between increased fidgetin expression and increased folate transport and utilization and that folate deficiency is associated with developmental defects. Moreover, SNPs in GPR98 (encoding an adhesion G protein-coupled receptor) are enriched in patients with tetralogy of Fallot with the 22q11 deletion142, indicating that common variants might modify the malformations caused by pathogenic genotypes.
Mosaic variants.
Damaging de novo variants that arise after initial zygote formation result in mosaicism, with the variant genotype present in only a subset of cells. Mosaic variants are identified by an alternative allele fraction significantly lower than the 50% expected for a heterozygous genotype143. Interrogation of data from WES of saliva or blood-derived samples have identified likely pathogenic mosaic variants in <1% of patients with CHD (25 of 2,530), indicating a fairly small role for mosaic variants in CHD144,145. Similar rates of mosaicism occur in autism spectrum disorder and neurodegenerative disorders144. Emerging strategies to better define CHD-associated damaging mosaic variants include high-depth WGS, which provides more uniform genome coverage than WES, single-cell sequencing to detect low-level mosaicism, particularly when combined with phasing of nearby SNPs to exclude polymerase-generated errors146,147, and analyses of CHD tissues.
Probability of LOF intolerance.
(pLI). A measure of evolutionary constraint estimated by the ratio of observed LOF variant alleles in the gnomAD cohort compared with the expected number of LOF variants on the basis of mutation rate, cohort size and sequence context.
Genome-wide association studies.
A statistical genetic analysis approach that associates common genetic variants, often with a minor allele frequency of ≥5%, with quantitative and qualitative traits. Given the number of genomic loci, a commonly accepted significance threshold of P< 5 × 10−8 is used regardless of how many loci are included in the analysis.
Alternative allele fraction.
The proportion of total nucleotide reads at a particular genomic position that represent a non-reference allele. For example, in a sequencing library containing ten reads of the reference allele A and ten reads of the alternative allele T, the alternative allele fraction would be 0.5 (10/20).
Whole-genome sequencing
Contemporary WGS provides technical advantages over WES, including avoiding the need for PCR amplification, a more uniform coverage of the genome and superior validation rates for SNPs and CNVs148–149. In addition to exome sequences, WGS allows the interrogation of rare or common mitochondrial sequences, microRNAs, non-coding RNAs, and promoter and regulatory sequences. Successful WGS analyses of patients with CHD include the identification of the genome-wide association of a MYH6 variant with aortic coarctation150 and the identification of increased rates of damaging variants among VEGF pathway genes in adults with tetralogy of Fallot with a pulmonary valve absent compared with adults with tetralogy of Fallot with a pulmonary valve present74.
Non-coding sequence variation via WGS.
Approximately 1% of the genome encodes protein and the remaining 99% is non-coding sequences that comprise promoters, enhancers, elements that influence chromatin topology or binding site accessibility for RNA or protein factors151–154 and other unknown functions. Damaging variants in some of these non-coding sequences might cause or influence the expression of CHD-associated genes. This hypothesis is based on the topological association between non-coding regions and gene promoters during cardiomyocyte differentiation in vitro155–157 and evidence that assessing non-coding sequences within CNVs can help define crucial regions involved in CHD158. Indeed, focused analyses of non-coding sequences flanking TBX5 identified three putative enhancers, one of which harboured a homozygous variant in a patient with CHD159. Compared with the normal sequence of the putative enhancer, the presence of the homozygous non-coding variant abrogated cardiac expression in model organisms159.
A genome-wide analysis of de novo variants in non-coding sequences in CHD focused on variants in non-coding elements that were likely to participate in the regulation of gene expression with the use of two related approaches160. WGS was used to identify variants by scores derived from machine learning analysis of diverse genomic annotations or by location within regions of active transcription during human induced pluripotent stem cell (hiPSC) differentiation into cardiomyocytes (hiPSC-CMs). Functional assays validated the candidate non-coding variants prioritized by both strategies. Additional analyses showed that non-coding variants within binding sites of RNA-binding proteins were increased among patients with CHD compared with control individuals. Collectively, these data indicate the potential for de novo non-coding variants, particularly within enhancers and RNA-binding protein sites, to contribute to 4–12% of CHD. Although a first step, the landscape of enhancer elements and RNA-binding protein sites across the genome is huge and considerably more analyses of larger cohorts will be needed to validate and extend these initial observations.
Structural variation analyses using WGS.
WGS enables the identification of structural variants with greater resolution than that of previous methodologies and expands the repertoire of identified LOF variants in human disease129,161–164. Previous studies have shown that small structural variants can cause CHD, including an 8-kb deletion in FLT4 that has been associated with tetralogy of Fallot74 and a 10-kb deletion mediated by a retrotrans-poson mobile element (Alu element) insertion in CHD7 that has been associated with CHARGE syndrome165. WGS can also detect genomic inversions and complex rearrangements. These structural variants were identified in patients with unexplained Alagille syndrome and affected the expression of the JAG1 and NOTCH2 genes166. Advances in structural variant algorithms to interrogate WGS are expected to increase substantially the identification of deletions, insertions, translocations and other genomic rearrangements in CHD.
Future genomic applications.
Unexplored genome features provide ongoing opportunities to improve our understanding of CHD genetics and to explain the 55% of unexplained cases of CHD. WGS affords the opportunity to genotype more completely an individual’s SNPs and structural variants and, therefore, the investigation of oligogenic causes of CHD (encompassing both rare and common damaging variants) is now possible. Expanded genome-wide variant analyses might contribute insights into CHD penetrance and phenotypic diversity, both in familial cases and among unrelated patients with shared CHD genotypes. How exposures influence the consequences of a CHD genotype remains an untapped and fruitful area for future research, given the prominence of CHD-associated genes related to chromatin remodelling and the evidence for environmental influences on chromatin structure167,168. Exploring these complexities might provide information on enigmatic features of CHD, including epidemiological evidence that the recurrence rates of CHD in families (3–7%)131–135 are lower than those predicted from the current knowledge of CHD genotypes, which would anticipate 25% and 50% recurrence for autosomal recessive and dominant disorders, respectively
Integrating biology to infer causality
The discovery of genes and variants with a potential causal link to CHD combined with analyses of gene expression during development and functional studies in model systems provide crucial evidence for assigning causality in the clinical setting169,170.
Insights from single-cell transcriptomics.
The advent of single-nuclear RNA sequencing and single-cell RNA sequencing (scRNA-seq) has enabled a detailed characterization of the many cells that populate the mouse developing heart171 and human mature heart172,173. Data from these studies have identified transcriptional heterogeneity between different cell lineages and distinct cell states within each lineage that were unappreciated in analyses of bulk RNA sequencing. For example, the human healthy adult heart contains four subsets of ventricular cardiomyocytes and five subsets of atrial cardiomyocytes172. These data raise new questions regarding the developmental origins, plasticity and physiological functions of distinct cell subsets, which will propel ongoing studies. scRNA-seq studies of human fetal hearts, although more limited in scope than studies on adult hearts, have identified transcriptional signatures of cardiac muscle cells (TNNI3 and TNNT2), fibroblast-like cells (COL1A1, COL1A2 and POSTN), endothelial cells (PECAM1 and KDR) and valvular cells (SOX9)174. Genes of the Notch signalling pathway are highly enriched in endocardial cells at gestational week 7, when compaction of the myocardium occurs, whereas genes related to the BMP pathway are expressed in endocardial and fibroblast-like cells from gestational week 5 to week 25, reflecting periods of endocardial-to-mesenchymal transition174.
Analyses of scRNA-seq from the developing mouse embryo complement and extend the limited datasets available from human tissues. These datasets revealed the cellular complexity and transcriptional profiles of lineages derived from three germ layers: early lateral plate mesoderm, paraxial mesoderm and neural crest cells171. Expression of Mesp1 in mouse cells at embryonic day (E) 6.75 to E7.5 indicates a loss of pluripotency and the acquisition of cardiac lineage commitment175. Hand2 expression initially demarcates the SFF but sub-sequently specifies cells of the outflow fract41. At E9.5 and beyond, cardiomyocytes exit the cell cycle and begin to show chamber-specific gene expression patterns174,176.
These datasets also demonstrate the considerable crosstalk between cell lineages that promotes cardiac differentiation and morphogenesis. The use of these resources to interrogate the temporal and lineage-specific expression of genes that harbour damaging variants linked to CHD provides more granular detail than is feasible from transcriptional analyses of bulk cardiac tissues. FIGURE 6 depicts scRNA-seq data from embryonic germ-layer derivatives that contribute to heart development in mice171 (FIG. 6a), showing the temporal emergence of cardiomyocytes, endothelial cells and neural crest cells (FIG. 6b). Definitive and candidate genes for CHD identified in the meta-analysis described in the previous sections show diverse lineage-specific and temporal expression in embryonic cells that are crucial for normal cardiovascular development (FIG. 6c). As expected, genes with damaging variants identified in patients with CHD with tetralogy of Fallot74, including Kdr, Flt1 and Flt4, have high expression in E8.25–E8.5 endothelial cells, consistent with the known functions of the encoded proteins in the VEGF pathway. By contrast, three genes, Rpl19, Rps24 and Rps26, that have been associated with Diamond–Blackfan anaemia and CHD177 have variable cellular and temporal expression in the mouse developing heart. Rpl19 and Rps26 are expressed in mesenchyme at E7.5–E7.75, and Rps24 is enriched in cardiomyocytes at E8.5; therefore, these genes might have differing roles in cardiac development. Finally, genes that encode chromatin-modifying proteins with integral functions in cardiac development and that have been show to harbour damaging mutations in patients with CHD (Chd4, Chd7, Kat6a, Kat6b, Kmt2 and Kmt2d) show diverse cellular and temporal expression profiles in the mouse developing heart, implying that altered transcriptional dynamics in many cell types probably results in CHD. We anticipate that the resolution provided by scRNA-seq datasets will provide the context to understand the role of poorly studied genes that are enriched for damaging variants in patients with CHD and will aid in identifying the pathways that are perturbed in CHD. These insights are expected to improve our understanding of the morphological consequences of damaging variants and the cellular deficits that contribute to lifelong adverse events in patients with CHD.
Fig. 6 |. Cardiac lineage-specific and temporal expression of definitive and candidate genes for CHD.
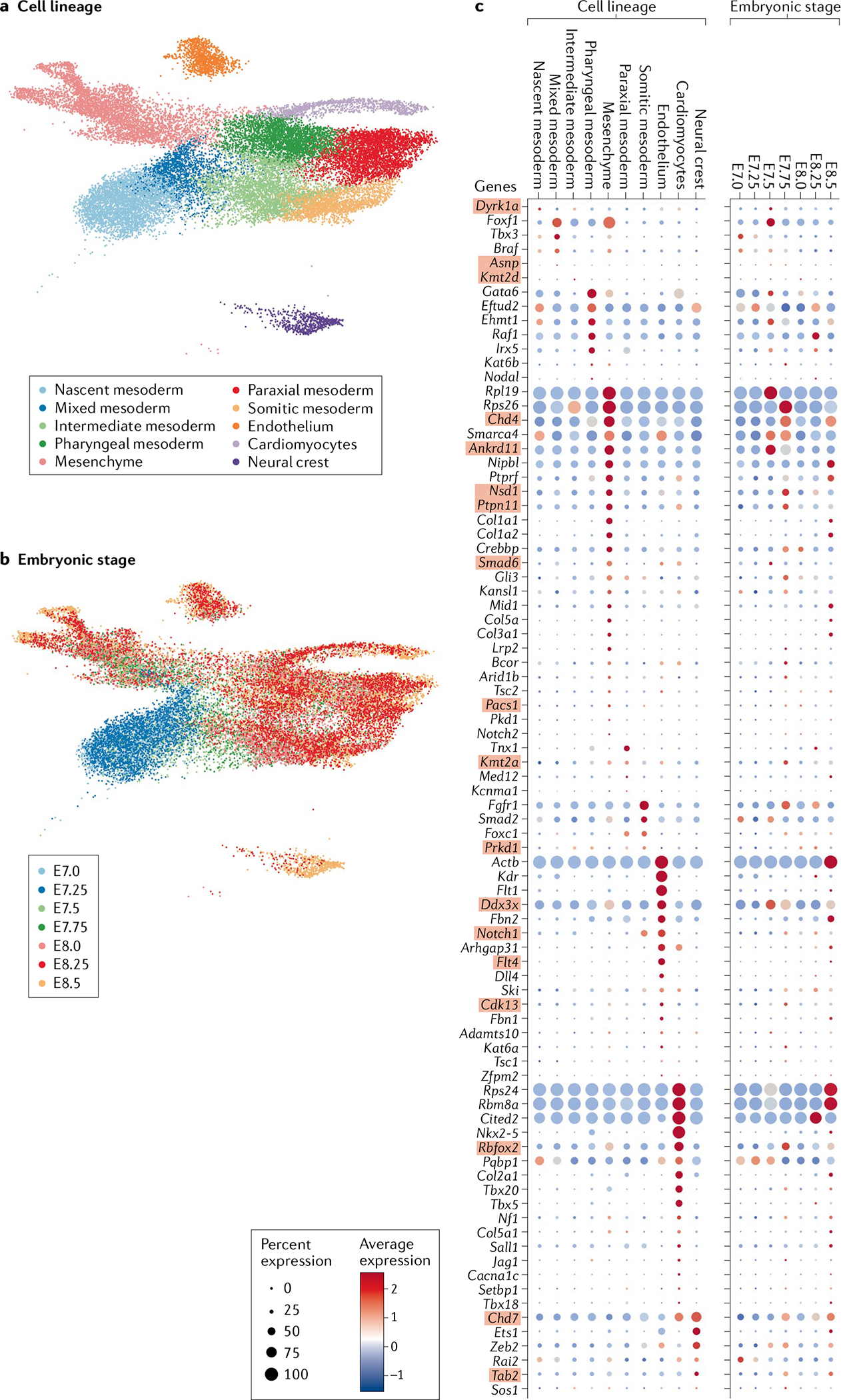
2D (UMAP) representations of transcriptional profiles of ceils involved in cardiogenesis labelled by cell lineage (part a) and mouse embryonic day (E7.0–E8.5; part b). Expression profile in the mouse embryos of definitive and candidate genes for congenital heart disease (CHD) that were identified in a meta-analysis of whole-exome sequencing data from patients with CHD17,19 (part c), classified by lineage and mouse embryonic stage. Definitive and candidate genes for CHD with a probability of loss-of-function intolerance (pLI) of >0.5 and the highest enrichment for loss-of-function or damaging missense variants in patients with CHD versus the Genome Aggregation Database are shown (84 genes in total). Definitive genes identified by statistical enrichment in case–control studies are shown in red boxes. Data adapted from a study on single-cell RNA sequencing of whole mouse embryos during organogenesis171.
Insights from induced pluripotent stem cell models of CHD.
hiPSCs, derived from either patients or CRIPPR–Cas9 editing of isogenic cell lines, provide an experimental approach in human cells to ascertain the functional consequences of damaging variants identified in patients with CHD. The differentiation process of hiPSC-CMs demonstrates properties consistent with the FHF178,179 or SHF, cardiac neural crest or pre-valvular endocardial cells180–182 that enable analyses of early developmental stages, such as the differentiation of mesoderm progenitor cells into early cardiomyocytes178. When combined with epigenetic and transcriptional profiling, differentiating hiPSC-CMs provide a window into early cardiogenesis that is difficult to achieve with model organisms43,180–182.
The molecular characterization of mutant hiPSCs can reveal the functional consequences of CHD-associated variants on gene expression, chromatin status and differentiation states. Although these in vitro assays lack the influences of in vivo morphology and haemodynamics, hiPSC-CMs have provided insights into CHD pathogenesis. hiPSCs with GATA6 LOF variants show disruption of crucial downstream regulators associated with outflow tract development (KDR and HAND2), accounting for the association between GATA6 LOF variants and outflow tract malformations in patients43. By contrast, hiPSCs with a GATA6 missense variant that is recurrently identified in patients with both CHD and pancreatic agenesis have a broadly disrupted epigenetic landscape and augmented retinoic acid signalling43. Similarly, transcriptomic analyses of hiPSCs with TBX5 haploinsufficiency have revealed that gene regulatory networks controlling cardiomyocyte differentiation are sensitive to TBX5 dosage and elucidated novel genetic interactions that might influence CHD phenotypes183. hiPSCs with a GATA4 missense variant that is associated with septal defects and pulmonary stenosis show loss of GATA4–TBX5 interactions and inappropriate expression of endothelial cell genes in iPSC-CMs84.
The use of hiPSCs also shows promise for the assessment of non-coding variants in CHD, overcoming the barriers of the limited conservation of non-coding sequences between species that impede analyses in experimental models. Massively parallel assays designed for hiPSCs can distinguish probable contributory non-coding variants from the vast numbers of benign polymorphisms and can help identify the associated genes184. Moreover, epigenetic and transcriptional profiling of hiPSCs during cell lineage differentiation provides a window into early cardiogenesis that is difficult to achieve in model organisms156.
Translating genomics into the clinic
The landscape of clinical genetic testing in patients with CHD is rapidly evolving due to technical advances in testing methodologies and identified associations between genotypes and longitudinal clinical phenotypes. Traditional genetic testing strategies have largely focused on syndromic CHD or selected CHD phenotypes that are strongly associated with a particular genotype (such as the 22q11 deletion)185. However, with the increasing availability and cost-effectiveness of clinical WES, gene-based diagnosis of both syndromic and non-syndromic CHD is emerging. Given that WES provides comprehensive analyses that provide information on the causes of CHD as well as detect incidental findings that are clinically actionable and/or of unclear significance, informed consent and guidance of genetic counsellors or experienced clinicians is essential. Moreover, with ongoing discoveries, the evidence for pathogenicity of damaging variants in genes associated with CHD requires criteria similar to those proposed by ClinGen and the American College of Medical Genetics and Genomics169,170,186,187.
The identification of definitive causal CHD variants influences patient care. Both the family of a newborn baby with CHD and adults with repaired or palliated CHD benefit when the genotype helps to inform the risk of recurrence, prenatal genetic counselling and pre-implantation genotyping188. Increasingly, the genotype can indicate the risk of associated extracardiac anomalies or comorbidities such as neurodevelopmental delay18 and highlight the need for surveillance for deleterious postoperative outcomes189 and the future development of cancers190–192.
CHD associated with extracardiac anomalies or neurodevelopmental delay.
The presence of an extracardiac anomaly or neurodevelopmental delay increases the likelihood of detecting a pathogenic CHD genotype from 8% to 22%18,19, particularly in genes encoding proteins involved in modifying chromatin17. WES studies of cohorts with neurocognitive disorders have also identified enrichment of damaging variants in chromatin-modifying genes18,193, indicating a strong genetic contribution to the association between CHD and neurodevelopmental delay. Therefore, the clinical identification of chromatin-related damaging genotypes in a newborn baby with CHD can inform pre-emptive interventions that might improve the baby’s learning and social skills.
De novo CNVs influence perioperative outcomes.
CHD genotypes can help identify high-risk patients for cardiac surgical interventions. Patients with CHD with 22q11.2 deletions have longer cardiopulmonary bypass times, more re-operations and longer intensive care stays than patients with CHD without this deletion194,195. Patients with CHD with other large CNVs (>300 kb) have a higher risk of death or need for transplantation than patients with CHD without large CNVs196. WES studies are extending these observations. Patients with syndromic or non-syndromic CHD with de novo CNVs or damaging single-nucleotide variants have increased time to extubation and decreased transplantation-free survival189. These adverse outcomes are most pronounced among patients with CHD without extracardiac anomalies. Given that some of these risk-associated genomic regions are also linked to cardiac trabeculation and myocardial performance197, the relationships between genotype and morphological structure might underlie the risk of heart failure and transplantation in some patients with CHD.
Risk of cancer in patients with CHD.
Adults with CHD have a higher cancer prevalence than the general population (1.4-fold to 2-fold)190–192. In addition to increased radiation exposure from therapeutic interventions198, certain CHD genotypes might also increase the risk of cancer118, because many genes with damaging variants associated with CHD are also recognized as genes associated with the risk of cancer. This association might be intertwined with the fundamental and broad developmental role of some CHD-associated genes, because patients with CHD with damaging variants in genes linked to cancer risk often have extracardiac manifestations.
Future clinical applications.
Investigation of the relationship between causal CHD-associated variants and clinical outcomes remains an area of enormous opportunity. How do variants in specific genes or defects in signalling pathways relate to the variability observed in CHD phenotypes? Do variants in genes linked to CHD or other genetic variants affect survival after surgical palliation or repair in patients with CHD? How do damaging variants that result in structural heart disease relate to measures of long-term cardiovascular function? To address these areas of inquiry, large genome-wide sequencing datasets harmonized with comprehensive, high-quality clinical data will be necessary. Given the expense and magnitude of the datasets necessary to address these questions, multicentre collaborative efforts are likely to be crucial in accelerating these areas of research.
Conclusions
Technological advances have propelled the discovery of the genetic architecture of CHD, which encompasses damaging germline and mosaic variants, copy number and structural variants that encompass many genes and variants in non-coding sequences. The influence of these discoveries extends well beyond the elucidation of the molecular basis for this common birth defect. These insights define crucial developmental genes and pathways that participate in the formation and function of the heart and many other organs. CHD-associated variants also identify genetic variants that promote neurocognitive dysfunction and cancer and confer distinct risks on patients who need surgical intervention for CHD.
Despite our current understanding of the genetic architecture of CHD, there is still much to learn. Aetiologies remain unknown in >50% of patients with CHD. New directions are needed to address this challenge. The identification of aberrant gene expression from direct analyses of human CHD tissues, comprehensive assessments of non-coding sequences and oligogenic variants, and deciphering the effect of environmental exposures might be productive. The success of these and other strategies holds great opportunities not only to improve the diagnosis, classification and clinical care of patients with CHD, but also to enrich the fundamental knowledge in developmental and genome biology.
Supplementary Material
Key points.
A genetic risk contributes substantially to congenital heart disease (CHD), and variantsin >400 genes are estimated to cause human CHD.
Analyses of large cohorts of patients with CHD have empowered the identification of novel genes associated with human CHD.
Non-coding variants contribute to CHD, in part by altering the activity of cardiac developmental enhancers.
Despite the genetic insights gained so far, a definitive cause is not identified in half of the cases of CHD.
Integration of genomic data with developmental biology promises to increase our understanding of the pathogenesis of CHD.
Acknowledgements
The authors’ work is supported in part by grants from the Harvard Medical School Epigenetics & Gene Dynamics Award, AHA postdoctoral Fellowship, and Boston Children’s Hospital Office of Faculty Development Career Development Fellowship Award (S.U.M.), the John S. LaDue Memorial Fellowship at Harvard Medical School (D.Q.), National Institutes of Health (J.G.S.: UM1 HL098166, HL151257; C.E.S.: UM1 HL0981479, 3U01HL131003), and the Howard Hughes Medical Institute (C.E.S.).
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information
The online version contains supplementary material available at https://doi.org/10.1038/s41569-021-00587-4.
Related links
American College of Medical Genetics and Genomics: https://www.acmg.net/
ClinGen: https://clinicalgenome.org
Gene Ontology: http://geneontology.org/
Genome Aggregation Database: https://gnomad.broadinstitute.org/
Online Mendelian inheritance in Man: https://www.omim.org/
References
- 1.Reller MD, Strickland MJ, Riehle-Colarusso T, Mahle WT & Correa A Prevalence of congenital heart defects in metropolitan Atlanta, 1998–2005. J. Pediatr. 153, 807–813 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffman JIE & Kaplan S The incidence of congenital heart disease. J. Am. Coll. Cardiol. 39, 1890–1900 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Leirgul E et al. Birth prevalence of congenital heart defects in Norway 1994–2009-A nationwide study. Am. Heart J. 168, 956–964 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Liu Y et al. Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies. Int. J. Epidemiol 48, 455–463 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bakker MK et al. Prenatal diagnosis and prevalence of critical congenital heart defects: an international retrospective cohort study. BMJ Open 9, e028139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mccracken C et al. Mortality following pediatric congenital heart surgery: an analysis of the causes of death derived from the national death index. J. Am. Heart Assoc. 7, e010624 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egbe A et al. Prevalence of congenital anomalies in newborns with congenital heart disease diagnosis. Ann. Pediatr. Cardiol. 7, 86–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartman RJ et al. The contribution of chromosomal abnormalities to congenital heart defects: a population-based study. Pediatr. Cardiol. 32, 1147–1157 (2011). [DOI] [PubMed] [Google Scholar]
- 9.de la Chapelle A, Herva R, Koivisto M & Aula P A deletion in chromosome 22 can cause DiGeorge syndrome. Hum. Genet. 57, 253–256 (1981). [DOI] [PubMed] [Google Scholar]
- 10.Greenberg F, Elder FFB, Haffner P, Northrup H & Ledbetter DH Cytogenetic findings in a prospective series of patients with DiGeorge anomaly. Am. J. Hum. Genet. 43, 605–611 (1988). [PMC free article] [PubMed] [Google Scholar]
- 11.Jalali GR et al. Detailed analysis of 22q11.2 with a high density MLPA probe set. Hum. Mutat. 29, 433–440 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thienpont B et al. Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur. Heart J. 28, 2778–2784 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Agergaard P, Olesen C, Østergaard JR, Christiansen M & Sørensen KM The prevalence of chromosome 22q11.2 deletions in 2,478 children with cardiovascular malformations. A population-based study. Am. J. Med. Genet. Part. A 158A, 498–508 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Peyvandi S et al. 22q11.2 deletions in patients with conotruncal defects: data from 1,610 consecutive cases. Pediatr. Cardiol. 34, 1687–1694 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pierpont ME et al. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation 115, 3015–3038 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Fahed AC, Gelb BD, Seidman JG & Seidman CE Genetics of congenital heart disease: the glass half empty. Circ. Res. 112, 707–720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin SC et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 49, 1593–1601 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Homsy J et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350, 1262–1266 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sifrim A et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 48, 1060–1065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.International Society of Ultrasound in Obstetrics & Gynecology. Cardiac screening examination of the fetus: guidelines for performing the ‘basic’ and ‘extended basic’ cardiac scan. Ultrasound Obstet. Gynecol. 27, 107–113 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Meilhac SM & Buckingham ME The deployment of cell lineages that form the mammalian heart. Nat. Rev. Cardiol. 15, 705–724 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Kathiriya IS, Nora EP & Bruneau BG Investigating the transcriptional control of cardiovascular development. Circ. Res. 116, 700–714 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Günthel M, Barnett P & Christoffels VM Development, proliferation, and growth of the mammalian heart. Mol. Ther. 26, 1599–1609 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui M, Wang Z, Bassel-Duby R & Olson EN Genetic and epigenetic regulation of cardiomyocytes in development, regeneration and disease. Development 145, dev171983 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Weerd JH & Christoffels VM The formation and function of the cardiac conduction system. Development 143, 197–210 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Jain R & Epstein JA Competent for commitment: you’ve got to have heart! Genes. Dev. 32, 4–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mjaatvedt CH et al. The outflow tract of the heart is recruited from a novel heart-forming field. Dev. Biol. 238, 97–109 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Cai C-L et al. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 5, 877–889 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hutson MR & Kirby ML Model systems for the study of heart development and disease. Cardiac neural crest and conotruncal malformations. Semin. Cell Dev. Biol. 18, 101–110 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin C-J, Lin C-Y, Chen C-H, Zhou B & Chang C-P Partitioning the heart: mechanisms of cardiac septation and valve development. Development 139, 3277–3299 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christoffels VM et al. Chamber formation and morphogenesis in the developing mammalian heart. Dev. Biol. 223, 266–278 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Schultheiss TM, Burch JB & Lassar AB A role for bone morphogenetic proteins in the induction of cardiac myogenesis. Genes Dev. 11,451–462 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Alsan BH & Schultheiss TM Regulation of avian cardiogenesis by Fgf8 signaling. Development 129, 1935–1943 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Schultheiss TM, Xydas S & Lassar AB Induction of avian cardiac myogenesis by anterior endoderm. Development 121, 4203–4214 (1995). [DOI] [PubMed] [Google Scholar]
- 35.Itoh N, Ohta H, Nakayama Y & Konishi M Roles of FGF signals in heart development, health, and disease. Front. Cell Dev. Biol. 4, 110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marques SR & Yelon D Differential requirement for BMP signaling in atrial and ventricular lineages establishes cardiac chamber proportionality. Dev. Biol. 328, 472–482 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Targoff KL, Schell T & Yelon D Nkx genes regulate heart tube extension and exert differential effects on ventricular and atrial cell number. Dev. Biol. 322, 314–321 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nelson DO, Jin DX, Downs KM, Kamp TJ & Lyons GE Irx4 identifies a chamber-specific cell population that contributes to ventricular myocardium development. Dev. Dyn. 243, 381–392 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JH, Protze SI, Laksman Z, Backx PH & Keller GM Human pluripotent stem cell-derived atrial and ventricular cardiomyocytes develop from distinct mesoderm populations. Cell Stem Cell 21, 179–194.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Cheng Z et al. Two novel mutations of the IRX4 gene in patients with congenital heart disease. Hum. Genet. 130, 657–662 (2011). [DOI] [PubMed] [Google Scholar]
- 41.de Soysa TY et al. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 572, 120–124 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen YH, Ishii M, Sun J, Sucov HM & Maxson RE Msx1 and Msx2 regulate survival of secondary heart field precursors and post-migratory proliferation of cardiac neural crest in the outflow tract. Dev. Biol. 308, 421–437 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Sharma A et al. GATA6 mutations in hiPSCs inform mechanisms for maldevelopment of the heart, pancreas, and diaphragm. eLife 9, e53278 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uribe V et al. Arid3b is essential for second heart field cell deployment and heart patterning. Development 141,4168–4181 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Creemers EE, Sutherland LB, McAnally J, Richardson JA & Olson EN Myocardin is a direct transcriptional target of Mef2, Tead and Foxo proteins during cardiovascular development. Development 133, 4245–4256 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Felker A et al. Continuous addition of progenitors forms the cardiac ventricle in zebrafish. Nat. Commun. 9, 2001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sánchez-Iranzo H et al. Tbx5a lineage tracing shows cardiomyocyte plasticity during zebrafish heart regeneration. Nat. Commun. 9, 428 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang X et al. Normal fate and altered function of the cardiac neural crest cell lineage in retinoic acid receptor mutant embryos. Mech. Dev. 117, 115–122 (2002). [DOI] [PubMed] [Google Scholar]
- 49.El Robrini N et al. Cardiac outflow morphogenesis depends on effects of retinoic acid signaling on multiple cell lineages. Dev. Dyn. 245, 388–401 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Inman KE et al. Foxc2 is required for proper cardiac neural crest cell migration, outflow tract septation, and ventricle expansion. Dev. Dyn. 247, 1286–1296 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kodo K et al. Regulation of Sema3c and the interaction between cardiac neural crest and second heart field during outflow tract development. Sci. Rep. 7, 6771 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ribeiro I et al. Tbx2 and Tbx3 regulate the dynamics of cell proliferation during heart remodeling. PLoS ONE 2, e398 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Niessen K & Karsan A Notch signaling in cardiac development. Circ. Res. 102, 1169–1181 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Dor Y et al. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development 128, 1531–1538 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Bischoff J Endothelial-to-mesenchymal transition. Circulation Res. 124, 1163–1165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh N et al. Histone deacetylase 3 regulates smooth muscle differentiation in neural crest cells and development of the cardiac outflow tract. Circ. Res. 109, 1240–1249 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marguerie A et al. Congenital heart defects in Fgfr2-IIIb and Fgf10 mutant mice. Cardiovasc. Res. 71,50–60 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Peng T et al. Coordination of heart and lung co-development by a multipotent cardiopulmonary progenitor. Nature 500, 589–592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu X et al. Single-Cell RNA-seq of the developing cardiac outflow tract reveals convergent development of the vascular smooth muscle cells. Cell Rep. 28, 1346–1361.e4 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Montague TG, Gagnon JA & Schier AF Conserved regulation of nodal-mediated left-right patterning in zebrafish and mouse. Development 145, dev171090 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weninger WJ et al. Cited2 is required both for heart morphogenesis and establishment of the left-right axis in mouse development. Development 132, 1337–1348 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Sutherland MJ, Wang S, Quinn ME, Haaning A & Ware SM Zic3 is required in the migrating primitive streak for node morphogenesis and left-right patterning. Hum. Mol. Genet. 22, 1913–1923 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levin M, Johnson RL, Stern CD, Kuehn M & Tabin C A molecular pathway determining left-right asymmetry in chick embryogenesis. Cell 82, 803–814 (1995). [DOI] [PubMed] [Google Scholar]
- 64.Meno C et al. Diffusion of nodal signaling activity in the absence of the feedback inhibitor Lefty2. Dev. Cell 1, 127–138 (2001). [DOI] [PubMed] [Google Scholar]
- 65.Meno C et al. lefty-1 is required for left-right determination as a regulator of lefty-2 and nodal. Cell 94, 287–297 (1998). [DOI] [PubMed] [Google Scholar]
- 66.Goldmuntz E et al. Frequency of 22q11 deletions in patients with conotruncal defects. J. Am. Coll. Cardiol. 32, 492–498 (1998). [DOI] [PubMed] [Google Scholar]
- 67.Corsten-Janssen N et al. The cardiac phenotype in patients with a CHD7 mutation. Circ. Cardiovasc. Genet. 6, 248–254 (2013). [DOI] [PubMed] [Google Scholar]
- 68.Layman WS, Hurd EA & Martin DM Chromodomain proteins in development: lessons from CHARGE syndrome. Clin. Genet. 78, 11–20 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rozas MF, Benavides F, León L & Repetto GM Association between phenotype and deletion size in 22q11.2 microdeletion syndrome: systematic review and meta-analysis. Orphanet. J. RareDis. 14, 195 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao Y et al. Complete sequence of the 22q11.2 allele in 1,053 subjects with 22q11.2 deletion syndrome reveals modifiers of conotruncal heart defects. Am. J. Hum. Genet. 106, 26–40 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Greenway SC et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat. Genet. 41, 931–935 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mefford HC et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 359, 1685–1699 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Page DJ et al. Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of Fallot. Circ. Res. 124, 553–563 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reuter MS et al. Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of Fallot. Genet. Med. 21, 1001–1007 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.De Luca A et al. New mutations in ZFPM2/FOG2 gene in tetralogy of Fallot and double outlet right ventricle. Clin. Genet. 80, 184–190 (2011). [DOI] [PubMed] [Google Scholar]
- 76.Yang YQ et al. GATA4 loss-of-function mutations underlie familial tetralogy of Fallot. Hum. Mutat. 34, 1662–1671 (2013). [DOI] [PubMed] [Google Scholar]
- 77.Kodo K et al. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorinplexin signaling. Proc. Natl Acad. Sci. USA 106, 13933–13938 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Burns T, Yang Y, Hiriart E & Wessels A The dorsal mesenchymal protrusion and the pathogenesis of atrioventricular septal defects. J. Cardiovasc. Dev. Dis. 3, 29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lana-Elola E et al. Genetic dissection of Down syndrome-associated congenital heart defects using a new mouse mapping panel. eLife 5, e11614 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Calabrò R & Limongelli G Complete atrioventricular canal. Orphanet J. Rare Dis. 1, 8 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Freeman SB et al. Population-based study of congenital heart defects in Down syndrome. Am. J. Med. Genet. 80, 213–217 (1998). [PubMed] [Google Scholar]
- 82.Bergström S et al. Trends in congenital heart defects in infants with Down syndrome. Pediatrics 138, e20160123 (2016). [DOI] [PubMed] [Google Scholar]
- 83.Pelleri MC et al. Genotype-phenotype correlation for congenital heart disease in Down syndrome through analysis of partial trisomy 21 cases. Genomics 109, 391–400 (2017). [DOI] [PubMed] [Google Scholar]
- 84.Ang Y-S et al. Disease model of GATA4 mutation reveals transcription factor cooperativity in human cardiogenesis. Cell 167, 1734–1749.e22 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Garg V et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424, 443–447 (2003). [DOI] [PubMed] [Google Scholar]
- 86.Durocher D, Charron F, Warren R, Schwartz RJ & Nemer M The cardiac transcription factors nkx2–5 and GATA-4 are mutual cofactors. EMBO J. 16, 5687–5696 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McBride KL et al. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: segregation, multiplex relative risk, and heritability. Am. J. Med. Genet. 134A, 180–186 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Silberbach M et al. Cardiovascular health in Turner syndrome: a scientific statement from the American Heart Association. Circulation. Genomic Precis. Med. 11, e000048 (2018). [DOI] [PubMed] [Google Scholar]
- 89.Lara DA, Ethen MK, Canfield MA, Nembhard WN & Morris SA A population-based analysis of mortality in patients with Turner syndrome and hypoplastic left heart syndrome using the Texas Birth Defects Registry. Congenit. Heart Dis. 12, 105–112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Prakash SK et al. Autosomal and X chromosome structural variants are associated with congenital heart defects in Turner syndrome: the NHLBI GenTAC registry. Am. J. Med. Genet. A 170, 3157–3164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Grossfeld PD et al. The 11q terminal deletion disorder: a prospective study of 110 cases. Am. J. Med. Genet. 129A, 51–61 (2004). [DOI] [PubMed] [Google Scholar]
- 92.Miao Y et al. Intrinsic endocardial defects contribute to hypoplastic left heart syndrome. Cell Stem Cell 27, 574–589.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shi LM et al. GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int. J. Mol. Med. 33, 1219–1226 (2014). [DOI] [PubMed] [Google Scholar]
- 94.Bonachea EM et al. Rare GATA5 sequence variants identified in individuals with bicuspid aortic valve. Pediatr. Res. 76, 211–216 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Verma SK et al. Rbfox2 function in RNA metabolism is impaired in hypoplastic left heart syndrome patient hearts. Sci. Rep. 6, 30896 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Theis JL et al. Recessive MYH6 mutations in hypoplastic left heart with reduced ejection fraction. Circ. Cardiovasc. Genet. 8, 564–571 (2015). [DOI] [PubMed] [Google Scholar]
- 97.Wald RM et al. Outcome after prenatal diagnosis of tricuspid atresia: a multicenter experience. Am. Heart J. 153, 772–778 (2007). [DOI] [PubMed] [Google Scholar]
- 98.Svensson EC et al. A syndrome of tricuspid atresia in mice with a targeted mutation of the gene encoding Fog-2. Nat. Genet. 25, 353–356 (2000). [DOI] [PubMed] [Google Scholar]
- 99.Prendiville TW et al. Cardiovascular disease in Noonan syndrome. Arch. Dis. Child. 99, 629–634 (2014). [DOI] [PubMed] [Google Scholar]
- 100.Gelb BD & Tartaglia M Noonan syndrome and related disorders: dysregulated RAS-mitogen activated protein kinase signal transduction. Hum. Mol. Genet. 15, R220–R226 (2006). [DOI] [PubMed] [Google Scholar]
- 101.Roberts A et al. The cardiofaciocutaneous syndrome. J. Med. Genet. 43, 833–842 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Danyel M, Kortüm F, Dathe K, Kutsche K & Horn D Autosomal dominant Robinow syndrome associated with a novel DVL3 splice mutation. Am. J. Med. Genet. Partt. A 176, 992–996 (2018). [DOI] [PubMed] [Google Scholar]
- 103.Atalay S et al. Congenital heart disease and Robinow syndrome. Clin. Dysmorphol. 2, 208–210 (1993). [PubMed] [Google Scholar]
- 104.Afzal AR et al. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat. Genet. 25, 419–422 (2000). [DOI] [PubMed] [Google Scholar]
- 105.Person AD et al. WNT5A mutations in patients with autosomal dominant Robinow syndrome. Dev. Dyn. 239, 327–337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.White J et al. DVL1 frameshift mutations clustering in the penultimate exon cause autosomal-dominant Robinow syndrome. Am. J. Hum. Genet. 96, 612–622 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Penton AL, Leonard LD & Spinner NB Notch signaling in human development and disease. Semin. Cell Dev. Biol. 23, 450–457 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McElhinney DB et al. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation 106, 2567–2574 (2002). [DOI] [PubMed] [Google Scholar]
- 109.Liu X et al. Exome-based case-control analysis highlights the pathogenic role of ciliary genes in transposition of the great arteries. Circ. Res. 126, 811–821 (2020). [DOI] [PubMed] [Google Scholar]
- 110.Li AH et al. Genetic architecture of laterality defects revealed by whole exome sequencing. Eur. J. Hum. Genet. 27, 563–573 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mohapatra B et al. Identification and functional characterization of NODAL rare variants in heterotaxy and isolated cardiovascular malformations. Hum. Mol. Genet. 18, 861–871 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ware SM et al. Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am. J. Hum. Genet. 74, 93–105 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lahaye S et al. Utilization of whole exome sequencing to identify causative mutations in familial congenital heart disease. Circ. Cardiovasc. Genet. 9, 320–329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hoang TT et al. The congenital heart disease genetic network study: cohort description. PLoS ONE 13, e0191319 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Preuss C et al. Family based whole exome sequencing reveals the multifaceted role of Notch signaling in congenital heart disease. PLoS Genet. 12, e1006335 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zaidi S et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Watkins WS et al. De novo and recessive forms of congenital heart disease have distinct genetic and phenotypic landscapes. Nat. Commun. 10, 4722 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Morton SU et al. Association of damaging variants in genes with increased cancer risk among patients with congenital heart disease. JAMA Cardiol. 6, 457–462 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tan HL et al. Nonsynonymous variants in the SMAD6 gene predispose to congenital cardiovascular malformation. Hum. Mutat. 33, 720–727 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Krebs LT et al. Notch signaling regulates left-right asymmetry determination by inducing Nodal expression. Genes Dev. 17, 1207–1212 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Galvin KM et al. A role for Smad6 in development and homeostasis of the cardiovascular system. Nat. Genet. 24, 171–174 (2000). [DOI] [PubMed] [Google Scholar]
- 122.McKean DM et al. Loss of RNA expression and allele-specific expression associated with congenital heart disease. Nat. Commun. 7, 12824 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Duchon A & Herault Y DYRK1A, a dosage-sensitive gene involved in neurodevelopmental disorders, is a target for drug development in down syndrome. Front. Behav. Neurosci 10, 104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Helsmoortel C et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 46, 380–384 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sirmaci A et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet. 89, 289–294 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bostwick BL et al. Phenotypic and molecular characterisation of CDK13-related congenital heart defects, dysmorphic facial features and intellectual developmental disorders. Genome Med. 9, 73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang X et al. Phenotypic expansion in DDX3X - a common cause of intellectual disability in females. Ann. Clin. Trans!. Neurol 5, 1277–1285 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fischbach GD & Lord C The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron 68, 192–195 (2010). [DOI] [PubMed] [Google Scholar]
- 129.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581,434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yokouchi-Konishi T et al. Recurrent congenital heart diseases among neonates born to mothers with congenital heart diseases. Pediatr. Cardiol. 40, 865–870 (2019). [DOI] [PubMed] [Google Scholar]
- 132.Ellesøe SG et al. Familial co-occurrence of congenital heart defects follows distinct patterns. Eur. Heart J. 39, 1015–1022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gill HK, Splitt M, Sharland GK & Simpson JM Patterns of recurrence of congenital heart disease: an analysis of 6,640 consecutive pregnancies evaluated by detailed fetal echocardiography. J. Am. Coll. Cardiol. 42, 923–929 (2003). [DOI] [PubMed] [Google Scholar]
- 134.Øyen N et al. Recurrence of congenital heart defects in families. Circulation 120, 295–301 (2009). [DOI] [PubMed] [Google Scholar]
- 135.Burn J et al. Recurrence risks in offspring of adults with major heart defects: results from first cohort of British collaborative study. Lancet 351,311–316 (1998). [DOI] [PubMed] [Google Scholar]
- 136.Cordell HJ et al. Genome-wide association study identifies loci on 12q24 and 13q32 associated with Tetralogy of Fallot. Hum. Mol. Genet. 22, 1473–1481 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hanchard NA et al. A genome-wide association study of congenital cardiovascular left-sided lesions shows association with a locus on chromosome 20. Hum. Mol. Genet. 25, 2331–2341 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hu Z et al. A genome-wide association study identifies two risk loci for congenital heart malformations in Han Chinese populations. Nat. Genet. 45, 818–821 (2013). [DOI] [PubMed] [Google Scholar]
- 139.Lin Y et al. Association analysis identifies new risk loci for congenital heart disease in Chinese populations. Nat. Commun. 6, 8082 (2015). [DOI] [PubMed] [Google Scholar]
- 140.Cordell HJ et al. Genome-wide association study of multiple congenital heart disease phenotypes identifies a susceptibility locus for atrial septal defect at chromosome 4p16. Nat. Genet. 45, 822–824 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang D et al. A genetic variant in FIGN gene reduces the risk of congenital heart disease in Han Chinese populations. Pediatr. Cardiol. 38, 1169–1174 (2017). [DOI] [PubMed] [Google Scholar]
- 142.Guo T et al. Genome-wide association study to find modifiers for tetralogy of Fallot in the 22q11.2 deletion syndrome identifies variants in the GPR98 locus on 5q14.3. Circ. Cardiovasc. Genet. 10, e001690 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Huang AY et al. MosaicHunter: accurate detection of postzygotic single-nucleotide mosaicism through next-generation sequencing of unpaired, trio, and paired samples. Nucleic Acids Res. 45, e76 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Manheimer KB et al. Robust identification of mosaic variants in congenital heart disease. Hum. Genet. 137, 183–193 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hsieh A et al. EM-mosaic detects mosaic point mutations that contribute to congenital heart disease. Genome Med. 12, 42 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.King DA et al. Detection of structural mosaicism from targeted and whole-genome sequencing data. Genome Res. 27, 1704–1714 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wei W et al. Frequency and signature of somatic variants in 1461 human brain exomes. Genet. Med. 21, 904–912 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Belkadi A et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl Acad. Sci. USA 112, 5473–5478 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Noll AC et al. Clinical detection of deletion structural variants in whole-genome sequences. NPJ Genomic Med. 1, 16026 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bjornsson T et al. A rare missense mutation in MYH6 associates with non-syndromic coarctation of the aorta. Eur. Hear: J 39, 3243–3249 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wright CF et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 385, 1305–1314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Turner TN & Eichler EE The role of de novo noncoding regulatory mutations in neurodevelopmental disorders. Trends Neurosci. 42, 115–127 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Montefiori LE et al. A promoter interaction map for cardiovascular disease genetics. eLife 7, e35788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hoelscher SC et al. MicroRNAs: pleiotropic players in congenital heart disease and regeneration. J. Thorac. Dis. 9, S64–S81 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Melnikov A et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 30, 271–277 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Akerberg BN et al. A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat. Commun. 10, 4907 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Vanoudenhove J, Yankee TN, Wilderman A & Cotney J Epigenomic and transcriptomic dynamics during human heart organogenesis. Circ. Res. 127, E184–E209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Thorsson T et al. Chromosomal imbalances in patients with congenital cardiac defects: a meta-analysis reveals novel potential critical regions involved in heart development. Congenit. Heart Dis. 10, 193–208 (2015). [DOI] [PubMed] [Google Scholar]
- 159.Smemo S et al. Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum. Mol. Genet. 21, 3255–3263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Richter F et al. Genomic analyses implicate noncoding denovo variants in congenital heart disease. Nat. Genet. 52, 769–777 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Audano PA et al. Characterizing the major structural variant alleles of the human genome. Cell 176, 663–675.e19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sudmant PH et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Collins RL et al. A structural variation reference for medical and population genetics. Nature 581, 444–451 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Turner TN et al. Genomic patterns of de novo mutation in simplex autism. Cell 171, 710–722.e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Udaka T et al. An Alu retrotransposition-mediated deletion of CHD7 in a patient with CHARGE syndrome. Am. J. Med. Genet. A 143, 721–726 (2007). [DOI] [PubMed] [Google Scholar]
- 166.Rajagopalan R et al. Genome sequencing increases diagnostic yield in clinically diagnosed Alagille syndrome patients with previously negative test results. Genet. Med. 23, 323–330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Legoff L, D’Cruz SC, Tevosian S, Primig M & Smagulova F Transgenerational inheritance of environmentally induced epigenetic alterations during mammalian development. Cells 8, 1559 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Barua S & Junaid MA Lifestyle, pregnancy and epigenetic effects. Epigenomics 7, 85–102 (2015). [DOI] [PubMed] [Google Scholar]
- 169.Strande NT et al. Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the clinical genome resource. Am. J. Hum. Genet. 100, 895–906 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Richards S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Pijuan-Sala B et al. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566, 490–495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Litviňuková M et al. Cells of the adult human heart. Nature 588, 466–472 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cao J et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Cui Y et al. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 26, 1934–1950.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 175.Lescroart F et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 359, 1177–1181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.DeLaughter DM et al. Single-cell resolution of temporal gene expression during heart development. Dev. Cell 39, 480–490 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ulirsch JC et al. The genetic landscape of Diamond-Blackfan anemia. Am. J. Hum. Genet. 103, 930–947 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Robertson C, Tran DD & George SC Concise review: maturation phases of human pluripotent stem cell-derived cardiomyocytes. Stem Cell 31, 829–837 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Takahashi K et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007). [DOI] [PubMed] [Google Scholar]
- 180.Zhang J et al. Functional cardiac fibroblasts derived from human pluripotent stem cells via second heart field progenitors. Nat. Commun. 10, 2238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kreitzer FR et al. A robust method to derive functional neural crest cells from human pluripotent stem cells. Am. J. Stem Cell 2, 119–131 (2013). [PMC free article] [PubMed] [Google Scholar]
- 182.Neri T et al. Human pre-valvular endocardial cells derived from pluripotent stem cells recapitulate cardiac pathophysiological valvulogenesis. Nat. Commun. 10, 1929 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kathiriya IS et al. Modeling human TBX5 haploinsufficiency predicts regulatory networks for congenital heart disease. Dev. Cell 56, 292–309.e9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hamdan FF et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am. J. Hum. Genet. 101, 664–685 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Pierpont ME et al. Genetic basis for congenital heart disease: revisited: a scientific statement from the American Heart Association. Circulation 138, e653–e711 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Philippakis AA et al. The matchmaker exchange: a platform for rare disease gene discovery. Hum. Mutat. 36, 915–921 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.MacArthur DG et al. Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–476 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yu Y et al. Functional mutant GATA4 identification and potential application in preimplantation diagnosis of congenital heart diseases. Gene 641, 349–354 (2018). [DOI] [PubMed] [Google Scholar]
- 189.Boskovski MT et al. De novo damaging variants, clinical phenotypes and post-operative outcomes in congenital heart disease. Circ. Genomic Precis. Med. 13, e002836 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gurvitz M et al. Prevalence of cancer in adults with congenital heart disease compared with the general population. Am. J. Cardiol. 118, 1742–1750 (2016). [DOI] [PubMed] [Google Scholar]
- 191.Mandalenakis Z et al. Risk of cancer among children and young adults with congenital heart disease compared with healthy controls. JAMA Netw. Open 2, e196762 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lee YS et al. The risk of cancer in patients with congenital heart disease: a nationwide population-based cohort study in Taiwan. PLoS ONE 10, 1–13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Krupp DR et al. Exonic mosaic mutations contribute risk for autism spectrum disorder. Am. J. Hum. Genet. 101, 369–390 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Mercer-Rosa L, Pinto N, Yang W, Tanel R & Goldmuntz E 22q11.2 deletion syndrome is associated with perioperative outcome in tetralogy of Fallot. J. Thorac. Cardiovasc. Surg. 146, 868–873 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.O’Byrne ML et al. 22q11.2 deletion syndrome is associated with increased perioperative events and more complicated postoperative course in infants undergoing infant operative correction of truncus arteriosus communis or interrupted aortic arch. J. Thorac. Cardiovasc. Surg. 148, 1597–1605 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kim DS et al. Burden of potentially pathologic copy number variants is higher in children with isolated congenital heart disease and significantly impairs covariate-adjusted transplant-free survival. J. Thorac. Cardiovasc. Surg. 151, 1147–1151.e4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Meyer HV et al. Genetic and functional insights into the fractal structure of the heart. Nature 584, 589–594 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Beauséjour Ladouceur V et al. Exposure to low-dose ionizing radiation from cardiac procedures in patients with congenital heart disease. Circulation 133, 12–20 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.