

Abstract
Inhibition of the protein kinase CSNK2 with any of 30 specific and selective inhibitors representing different chemotypes, blocked replication of pathogenic human, bat, and murine β-coronaviruses. The potency of in-cell CSNK2A target engagement across the set of inhibitors correlated with antiviral activity and genetic knockdown confirmed the essential role of the CSNK2 holoenzyme in β-coronavirus replication. Spike protein endocytosis was blocked by CSNK2A inhibition, indicating that antiviral activity was due in part to a suppression of viral entry. CSNK2A inhibition may be a viable target for the development of anti-SARS-like β-coronavirus drugs.
Introduction
Coronaviruses (CoVs) are genetically diverse positive-sense RNA viruses that circulate in animals and humans.1 α-CoV and β-CoV can infect mammals, while γ-CoV and δ-CoV are restricted to birds. Three highly pathogenic human β-CoV of zoonotic origin that cause severe lower respiratory tract infection have emerged in recent years: severe acute respiratory syndrome CoV (SARS-CoV), Middle East respiratory syndrome (MERS)-CoV, and SARS-CoV-2, the causative agent of the COVID-19 pandemic. Despite the rapid development of effective vaccines and direct-acting antivirals, the perpetual evolution of β-CoV, the inevitable development of drug resistance, and the potential for the emergence of new zoonotic SARS-like β-CoVs have highlighted the need for effective broad-spectrum oral antiviral therapies to treat infections.2
CoVs are spherical enveloped viruses characterized by their crown-like surface projections composed of trimers of the viral spike protein.3 The CoV spike protein binds receptors on the surface of target host cells, allowing entry of the virus as the first step of infection. The CoV spike protein, which has adapted to target receptors of different hosts, determines the spectrum of infectivity of each virus. The spike proteins of SARS-CoV and SARS-CoV-2 bind to human angiotensin-converting enzyme 2 (ACE2) receptor,4 while MERS-CoV utilizes dipeptidyl peptidase 45 and the spike protein of mouse hepatitis virus (MHV) binds to mouse carcinoembryonic antigen-related cell adhesion molecule 1 receptor.6 While SARS-CoV, SARS-CoV-2, and MERS-CoV are all biosafety level 3 pathogens, the restriction of MHV infectivity to mice and its close phylogenetic relationship to other members of the β-CoV genus makes it a widely-accepted model system that can be studied within biosafety level 2 containment.7
β-CoV, including SARS-CoV-2 and MERS-CoV, enter cells primarily by clathrin-mediated endocytosis (CME).8,9 Cell surface fusion can provide an alternate pathway of cell entry, although its relative contribution is dependent on high expression levels of proteases such as TMPRSS2 and cathepsins.10 Endocytosis of the receptor-bound virus is followed by RNA release from the lumen of the endosome followed by uncoating of the CoV RNA genome.1 The virus encodes a replicase and an RNA-dependent RNA polymerase that transcribe the CoV mRNAs, which in turn are translated into the viral structural and nonstructural accessory proteins. Following the assembly of new virions, composed of the viral genomic RNA and structural proteins, the virus is translocated in vesicles to the host cell membrane and released by nonlytic exocytosis. The virus co-opts many host cell proteins through its life cycle to maintain efficient entry, replication, packaging, and exocytosis in addition to suppression of immune response pathways.11
Development of direct-acting antiviral agents has often been hampered by the potential for viruses to overcome negative selective pressure to generate drug-resistant mutants.12 Host cell proteins that are utilized by the virus during replication or for suppression of the immune response are less likely to be circumvented by viral escape mutants.13,14 Protein kinases are involved in almost all cell signaling processes and are often induced or suppressed by viruses during infection.15 Casein Kinase 2 (CSNK2) is a constitutively active serine/threonine kinase typically found as a tetramer consisting of two catalytic subunits and two regulatory subunits, forming either a homotetramer or heterotetramer depending on the identity of the catalytic subunit (Figure 1A).16 CSNK2 phosphorylates hundreds of physiological substrates and modulates the activity of many cell signaling pathways.16,17 The role of CSNK2 in-cell cycle regulation, cell growth, proliferation, and survival and its overexpression in numerous tumors has marked it as a potential anti-cancer target. The ATP-competitive CSNK2 inhibitor silmitasertib (Figure 1B) is being developed for the treatment of cholangiocarcinoma, while CIGB-300 (Figure S1), a 25-membered heterodetic cyclic peptide inhibitor of CSNK2 substrate phosphorylation, is under development for cervical cancer.17 Despite the ubiquitous role of CSNK2 in cell signaling, the safety and tolerability of these CSNK2 inhibitors has permitted on-going clinical development.18−20
Figure 1.

CSNK2 enzyme and ATP-competitive inhibitors. (A) CSNK2 is a homo- or heterotetramer composed of two copies of the catalytic 2A-subunit (A1 or A2) and two copies of the regulatory 2B-subunit. (B) Clinical candidate silmitasertib. (C) Prototypical CSNK2A inhibitors TBB and DMAT. Chemical probe SGC-CK2-1 and its negative control analogue SGC-CK2-1N.
A wide range of viruses have proteins that are phosphorylated by CSNK2.21 It remains unclear if all of these phosphorylation events are essential for virus replication or a manifestation of the broad range of CSNK2 substrate specificity. However, for human papillomaviruses, it appears that the phosphorylation of E1 protein by CSNK2 stabilizes ATP-dependent DNA helicase activity, which is a key step in their viral replication.22 Recently, a series of mass spectrometry proteomic and phosphoproteomic studies have mapped the interactions between β-CoV and host CSNK2 in infected cells.23−25 Both CSNK2A1 and CSNK2A2 were identified as participants in the SARS-CoV-2 interactome, specifically in a complex with the nucleocapsid protein.23 These observations were extended to SARS-CoV and MERS,24 suggesting that the interactions will be shared across other β-CoV members, such as MHV. Furthermore, phosphoproteomic profiling of cells following SARS-CoV-2 infection identified many CSNK2 substrates, consistent with upregulation of its kinase activity by the virus.25 These observations are indicative of β-CoV commandeering host cell CSNK2 to support its infectivity and replication and suggest that small molecule inhibitors may be promising antiviral compounds.
Chemogenomics is a method of drug target validation that utilizes selective and highly annotated small molecule inhibitors to link perturbation of a cell phenotype to a specific molecular target.26 For protein kinases, a robust chemogenomic strategy requires the use of multiple small molecule inhibitor chemotypes combined with inactive analogues to control for potential pleiotropic kinase inhibition and other off-target activity.27−29 Many examples of ATP-competitive inhibitors of the CSNK2A catalytic domain have been described over the past two decades, with the chronology of their development reviewed in detail.30 Among the early examples were the polybrominated benzotriazole TBB and benzimidazole DMAT (Figure 1B) which had modest potency and selectivity but were used as the initial tools to study CSNK2 biology.31 Structure-guided optimization subsequently led to a series of pyrazolo[1,5-a]pyrimidines with nanomolar potency as CSNK2 inhibitors.32,33 The pyrazolo[1,5-a]pyrimidine SGC-CK2-1 (Figure 1C) was recently described as a potent and exquisitely selective ATP-competitive CSNK2 inhibitor, which with its negative control analogue SGC-CK2-1N can be used as a high-quality chemical probe pair.34 Using a chemogenomic approach employing multiple ATP-competitive small molecule inhibitors, we now report that the potency of CSNK2A target engagement in cells, over a 3-log range, tracks with the suppression of murine, bat, and human β-CoV replication. The critical role of CSNK2 in β-CoV replication was further confirmed by the genetic knockdown of the individual catalytic and regulatory subunits. Finally, by studying the effect of CSNK2A inhibition on SARS-CoV-2 spike protein uptake, we provide evidence that antiviral activity may be due in part to inhibition of viral entry via CME.
Results and Discussion
Development of a β-coronavirus Reporter Assay
Mouse hepatitis virus (MHV) is a member of the β-CoV genus that has been widely used as a model to study the virulence of SARS-CoV and SARS-CoV-2.7 To develop a reporter virus to study the effect of compounds on β-CoV replication, the MHV-A59 G plasmid was engineered to replace most of the coding sequence for orf4a and orf4b with nanoluciferase (nLuc).35 The resulting virus, MHV-nLuc, replicated to high titer and efficiently expressed nLuc.
To determine the optimal titer and time point to analyze viral replication, mouse derived-from-brain-tumor (DBT) cells were inoculated with a range of multiplicities of infection (MOI) from 0.016 to 10 with MHV-nLuc and luciferase activity measured in cell lysates at multiple time points up to 24 h post infection (Figure 2A). The results indicated that inoculation of DBT cells by MHV-nLuc with an MOI of 0.1 and luciferase measurement at 10 h post infection were the optimal assay conditions, as viral replication was in the linear range and bioluminescence was within the dynamic range of the luminometer.
Figure 2.

β-CoV replication assay. (A) Optimization of MHV-nLuc assay. (B) Effect of CSNK2A inhibitors on replication of MHV-nLuc in DBT cells, n = 3 ± SE. No curve was fit to the SGC-CK2-1N data.
CSNK2A Inhibitors Block β-CoV Replication
Silmitasertib (Figure 1B) is a modestly selective ATP-competitive CSNK2 inhibitor with a live cell CSNK2A1 target engagement of pIC50 = 6.5 (IC50 = 0.31 μM) as measured by nanoBRET assay.34,36 Silmitasertib was previously reported as demonstrating antiviral activity in African green monkey kidney epithelial Vero cells infected with SARS-CoV-2 (pIC50 = 5.6, IC50 = 2.5 μM)25 but was less potent at inhibiting SARS-CoV-2 infection of human lung epithelial A549-ACE2 cells (pIC50 < 5, IC50 > 10 μM). Silmitasertib demonstrated cell toxicity at micromolar doses in both cell lines,25 which may be due to its off-target inhibition of the kinases DYRK1A/1B37 and further complicated analysis of its anti-SARS-CoV-2 activity. However, when tested in our optimized MHV-nLuc assay in DBT cells, silmitasertib inhibited viral replication with pIC50 = 6.2 (IC50 = 0.63 μM) and with no effect on cell viability (Figures 2B and S2A). These data demonstrated for the first time that the anti-β-CoV activity of silmitasertib could be uncoupled from its effect on cell viability.
To provide additional evidence that host cell CSNK2 was required for coronavirus replication, we tested a second series of ATP-competitive CSNK2A inhibitors from the pyrazolo[1,5-a]pyrimidine chemotype, which is structurally and physiochemically distinct from silmitasertib (Table 1). We previously reported the identification of a series of 3-cyano-7-cyclopropylamino-pyrazolo[1,5-a]pyrimidines 1–7 as potent, selective, cell-active inhibitors with pIC50 = 6.6–8.9 (IC50 = 0.25–0.001 μM) in CSNK2A1/CSNK2A2 nanoBRET assays and low toxicity in many cell lines.34 When tested in the MHV-nLuc assay, pyrazolo[1,5-a]pyrimidines 1–6 demonstrated potent inhibition of viral replication. Pyrazolo[1,5-a]pyrimidines 1–4 demonstrated IC50 values < 10 nM with no effect on viability of DBT cells at concentrations up to 10 μM (Figure S2A). The N-benzyl pyrazolo[1,5-a]pyrimidine 7 was the only analogue with an IC50 above 1 μM. The lower potency of 7 in the MHV-nLuc assay was consistent with its weaker activity in the CSNK2A1/CSNK2A2 nanoBRET assays. Notably, compound 2, which was up to 100-fold more potent than silmitasertib as a CSNK2A inhibitor, also showed a 2-log improved potency in antiviral activity (Figure 2B).
Table 1. Structure–Activity Relationship of the N-(3-Aminophenyl)acetamide Series of 3-Cyano-7-cyclopropylamino-pyrazolo[1,5-a]pyrimidines 1–7a,b.
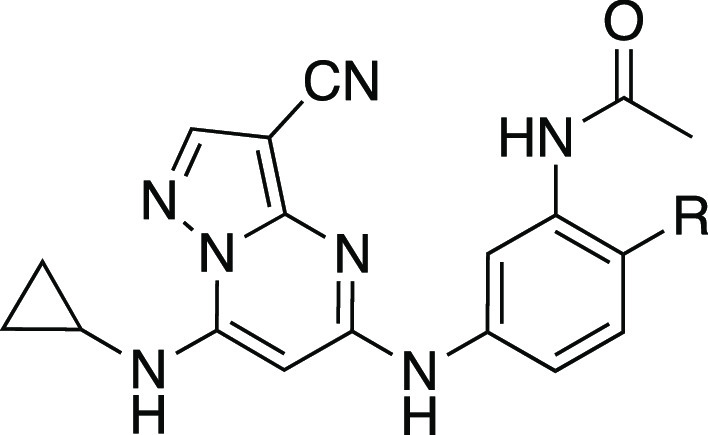

In-cell target engagement of CSNK2A-nLuc in HEK293 cells. Data from ref (24).
Inhibition of MHV-nLuc replication in DBT cells. Values are the mean of three assays with range ± 15%.
The kinome-wide selectivity of the 3-cyano-7-cyclopropylamino-pyrazolo[1,5-a]pyrimidine inhibitors is controlled in part by the para- and meta-aniline substituents.34SGC-CK2-1 (Figure 1B), which contains para-methyl and meta-propionamide aniline substituents, is the most selective of all known ATP-competitive small molecule CSNK2A inhibitors (3 kinases inhibited at 1 μM)34 and has been characterized as a high-quality chemical probe by the Structural Genomics Consortium.29 A close structural analogue, SGC-CK2-1N (Figure 1B), which lacks CSNK2A activity at concentrations up to 10 μM has been designated as a negative control compound. When tested in the MHV-nLuc assay, SGC-CK2-1 inhibited viral replication with pIC50 = 6.9 (IC50 = 0.21 μM) while negative control SGC-CK2-1N was inactive up to a concentration of 10 μM (Figure 2B). This result provided a third line of chemogenomic evidence that inhibition of host cell CSNK2 impeded replication of a murine β-CoV.
Relationship between CSNK2A Inhibitor Potency and anti-β-CoV Activity
To generate additional evidence that CSNK2 was required for β-CoV replication, two additional series of inhibitors based on the pyrazolo[1,5-a]pyrimidine chemotype were synthesized to strengthen the structure–activity relationship between kinase inhibition and viral replication (Scheme 1). The 3-cyano-7-cyclopropylamino-pyrazolo[1,5-a]pyrimidines, in particular, have demonstrated high cellular potency as CSNK2A inhibitors combined with good kinome-wide selectivity.34 A series of analogues (8–15, Table 2), where the aniline para-methyl group of the chemical probe SGC-CK2-1 was replaced by a basic side chain were synthesized by palladium-catalyzed cross-coupling of di-substituted aniline intermediates (i) and pyrazolo[1,5-a]pyrimidine building block (ii) (Scheme 1). The para-substituent on the aniline forces the propionamide to adopt an otherwise energetically disfavored cisoid configuration in the enzyme-active site that contributes to improved CSNK2A selectivity.34 Although the ATP-binding sites of CSNK2A1 and CSNK2A2 have high sequence identity (Figure S3), we opted to screen the new analogues 8–15 for cellular target engagement on both isozymes using nanoBRET assays. MHV replication tracked with nanoBRET activity, with the most potent dual CSNK2A1/CSNK2A2 inhibitors 8 and 9 showing the strongest MHV inhibition and the least effective inhibitors 12 and 14 showing the weakest inhibition of viral replication. Notably, analogues 10 and 15 showed modest selectivity for CSNK2A1 over CSNK2A2 (6–8 fold), but this did not translate into improved potency for MHV inhibition.
Scheme 1. Synthesis of 3-Cyano-pyrazolo[1,5-a]pyrimidines 8–33.

Reagents and conditions: (A) BINAP, Pd(OAc)2, t-BuOLi, 1,4-dioxane, microwave irradiation, 130 °C. R1 defined in Table 2. (B) Xantphos, Pd(OAc)2, Cs2CO3, 1,4-dioxane, microwave irradiation, 130 °C. R2 defined in Table 3.
Table 2. Structure–Activity Relationship of the N-(3-Aminophenyl)propionamide Series of 3-Cyano-7-cyclopropylamino-pyrazolo[1,5-a]pyrimidines 8–15a,b.


In-cell target engagement of CSNK2A2-nLuc in HEK293 cells.
Inhibition of MHV-nLuc replication in DBT cells. All values are the mean of three assays with range ± 15%.
The third series of potent CSNK2A inhibitors was based on pyrazolo[1,5-a]pyrimidine 16, which contained a 6-(acetamino)indole as its 5-substituent.34 Indole 16 also demonstrated potent anti-β-CoV activity with pIC50 = 7.4 (IC50 = 0.04 μM) in the MHV-nLuc assay (Table 3). Guided by the knowledge that the cyclopropylamine sits in the region of the kinase that is adjacent to the solvent-accessible channel, we explored a range of alternative 7-heterosubstituted analogues to define the structure–activity relationship for CSNK2A inhibition and anti-β-CoV activity (Table 3). Synthesis of the analogues 16–33 was achieved by palladium-catalyzed cross-coupling of 6-acetaminoindole (iii) and 7-substituted choloro-pyrazolo[1,5-a]pyrimidines (iv) (Scheme 1). In general, large modifications to the 7-cyclopropylamino group were found to be deleterious to in-cell CSNK2A1/CSNK2A2 target engagement, but smaller modifications retained activity on the enzyme (Table 3). Importantly, as was seen with the aniline-substituted pyrazolo[1,5-a]pyrimidines, anti-β-CoV activity of the 6-(acetamino)indoles tracked with their cellular potency on CSNK2A. Only the 7-cyclobutylamino analogue 17 demonstrated a pIC50 > 6.0 (IC50 < 1.0 μM) in the MHV-nLuc assay. However, several of the analogues that had modest potency in the CSNK2A1/CSNK2A2 nanoBRET assays demonstrated IC50 values in the 1–5 μM range in the MHV-nLuc assay. Importantly, the analogues 21 and 32 that were inactive at 10 μM on CSNK2A were unable to block β-CoV replication. As was seen before, analogues with modest CSNK2A1 over CSNK2A2 selectivity (e.g., 25 and 28) did not show improved antiviral potency. Thus, even though the compounds in the 6-(acetamino)indole series were generally less active as dual CSNK2A1/CSNK2A2 inhibitors, their potency tracked with antiviral activity.
Table 3. Structure–Activity Relationship of the N-(1H-indol-6-yl)acetamide Series of 3-Cyanopyrazolo[1,5-a]pyrimidines 16–33a,b.
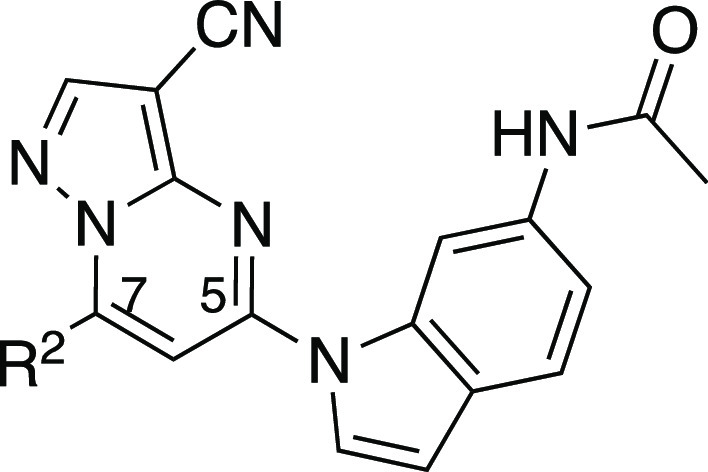

In-cell target engagement of CSNK2A2-nLuc in HEK293 cells.
Inhibition of MHV-nLuc replication in DBT cells. All values are the mean of three assays with range ± 15%. i.a. inactive.
Relationship between CSNK2A Potency and β-CoV Replication
Over the three series of 3-cyano-pyrazolo[1,5-a]pyrimidines (Tables 1–3) with a wide range of 5- and 7-substituents, anti-β-CoV activity shadowed their potency in the live cell CSNK2A1/CSNK2A2 target engagement assays (Figure 3). Within each series, the most potent kinase inhibitors were the most potent in the antiviral assay, and the least active CSNK2A inhibitors were unable to block viral replication. The relationship was maintained over more than a 3-log range in activity with an R2 = 0.68 when the lower value for inhibition of CSNK2A1 or CSNK2A2 was compared to antiviral potency (Figure 3). The relationship was also maintained with R2 > 0.6 when either CSNK2A1 or CSNK2A2 alone were used in the analysis (Figure S3). However, the improved correlation obtained using target engagement data from both catalytic isoforms suggests that the heterotetramer form of the holoenzyme (Figure 1A) is the active complex in cells and that dual CSNK2A1/CSNK2A2 inhibition translates into improved antiviral potency. The modest potency of silmitasertib, which belongs to a different chemotype of CSNK2A inhibitors, was also consistent with the relationship between CSNK2A and anti-β-CoV activity. The N-(3-aminophenyl) acetamide series (Table 1) contained the most potent inhibitors of CSNK2A and MHV-nLuc. Several analogues in the N-(3-aminophenyl)propionamide series (Table 2) maintained potent CSNK2A activity with many analogues showing activity < 1 μM, but no single analogue as potent in the β-CoV replication assay as members of the N-acetamide series. Thus, while paired analogues had equivalent activity in the CSNK2A nanoBRET assays, the propionamide series was generally less potent in the antiviral assay (see 1 vs SGC-CK2-1, 2 vs 8, and 3 vs 9). These nuances in the structure–activity relationship are unlikely to be due to species differences between the human kinase and the murine viral assay, since both human and mouse CSNK2A1 are identical in the kinase domain, and CSNK2A2 differs by only a single amino acid E253D at the base of the C-lobe at > 30 Å from the ATP-binding pocket (Figure S3). Physiochemical properties can also contribute to nonenzymatic viral inhibition mechanisms such as phospholipidosis.38 This nonspecific activity is unlikely to confound our results due to the strong correlation between CSNK2A activity and MHV inhibition across a wide dose range and the nanomolar potency of many of the CSNK2A inhibitors. However, we cannot rule out some potential nonspecific mechanisms with the two weakest CSNK2A inhibitors (pIC50 < 6, IC50 > 1 μM) that lie the furthest from the trendline (Figure 3).
Figure 3.

Correlation of potency for CSNK2A target engagement with inhibition of β-CoV replication across three subseries of pyrazolo[1,5-a]pyrimidines and silmitasertib. The analysis uses the lower value of CSNK2A1 or CSNK2A2 pIC50 from Tables 1–3 for each active analogue. Inactive analogues SGC-CK2-1N, 20, 21, 22, 31, and 32 were not included in the analysis. The dashed line represents the logarithmic trendline with R2 = 0.68.
Targeted Knockdown of CSNK2 Blocks β-CoV Replication
CSNK2 is a serine/threonine kinase that is expressed endogenously as a tetramer of two catalytic subunits and two regulatory subunits, forming either a homotetramer or heterotetramer depending on the identity of the catalytic subunit (Figure 1A).16 Transcripts for each of the three subunits (CSNK2A1, CSNK2A2, CSNK2B) were detected in uninfected DBT cells by qRT-PCR. To study the effect of MHV infection, DBT cells were inoculated at an MOI of 0.1, and the transcript abundance of each CSNK2 subunit was determined through the time course of infection. By qRT-PCR, MHV infection did not change the abundance of any CSNK2 subunit transcripts in DBT cells over 12 h (Figure 4A).
Figure 4.

CSNK2 target validation. (A) Relative expression after MHV infection of the CSNK2 subunit mRNAs in DBT cells by qRT-PCR. (B) Expression of CSNK2 subunits by Western blot after siRNA targeting. DBT, untreated cells. NT, nontargeting control siRNA. (C) Inhibition of MHV replication by siRNA knockdown of CSNK2 subunits. *p < 0.05, ***p < 0.001.
To further validate the role of CSNK2 in supporting β-CoV replication, targeted knockdown of the individual subunits of the enzyme was performed with siRNA specific to CSNK2A1, CSNK2A2, or CSNK2B, respectively. Effective knockdown of each subunit was confirmed by Western blot using a well-characterized antibody (Figure 4B).34 Following knockdown, the DBT cells were infected with MHV-nLuc at an MOI of 0.1 to determine the role of each subunit in viral replication. Knockdown of CSNK2A1 inhibited MHV replication by 40% compared to a nontargeting control siRNA, whereas CSNK2A2 knockdown had no significant effect on MHV replication. Knockdown of CSNK2B inhibited MHV replication by 85% compared to the control siRNA (Figure 4C). These results support the model (Figure 1A) in which a functional CSNK2 tetramer can be assembled using two copies of either CSNK2A1 or CSNK2A2 but must always contain two CSNK2B subunits. Thus, while the two catalytic subunits can be any mix of CSNK2A1 and CSNK2A2 subunits, the absence of CSNK2B yields a nonfunctional enzyme and loss of MHV replication in the DBT cells. To confirm our interpretation of the results, both CSNKA1 and CSNKA2 were depleted simultaneously. Dual knockdown of CSNK2A1 and CSNK2A2 inhibited MHV replication by 90% compared to the control siRNA (Figure 4B), further supporting the critical role of both isoforms of the catalytic unit during β-CoV replication. Evidence that the 2A1:2B:2B:2A2 heterotetramer is likely to be the primary form of the CSNK2 holoenzyme in cells was also provided by the chemogenomic analysis, which showed a stronger correlation with inhibition of β-CoV replication using target engagement data from both the CSNK2A1 and CSNK2A2 catalytic units (Figures 3 and S4) and the observation that inhibitors with modest selectivity for CSNK2A1 over CSNK2A2 did not show improved antiviral potency.
CSNK2 Inhibition Blocks SARS-like β-CoV Replication
To extend these findings to SARS-like β-CoV of pandemic potential,39 including the clinically relevant SARS-CoV-2 that is the cause of the COVID-19 pandemic, we studied the effect of CSNK2 inhibition on bat and human β-CoV replication in both continuous cell lines and in primary human cells. The severe contagion risk of SARS-like bat and human β-CoV mandates the use of high containment biosafety laboratory 3 containment for these assays. Given the resource-intensive nature of this work, we restricted this analysis to the potent pyrazolo[1,5-a]pyrimidine CSNK2A inhibitor 2, the chemical probe SGC-CK2-1, and its negative control SGC-CK2-1N (Figure 2B and Table 1). When tested in A549-ACE2 cells infected with our well-characerized SARS-CoV-2 reporter virus expressing nLuc,40−42 inhibition of viral replication was observed with both inhibitor 2 and SGC-CK2-1 but not the control SGC-CK2-1N (Figure 5A). To provide additional evidence that CSNK2 inhibition blocks replication of other SARS-like β-CoV, we measured the effect of the CSNK2 inhibitors on two bat viruses, SHC014-CoV and WIV1-CoV, which are poised for human emergence.39,43 In A549-ACE2 cells inoculated with either bat virus, inhibitor 2 and SGC-CK2-1 produced dose-dependent decreases in viral replication while the negative control SGC-CK2-1N remained inactive (Figure 5A). Viability of the A549-ACE2 cells was not affected at doses below 10 μM (Figure S2B), possibly reflecting the improved CSNK2 selectivity of the pyrazolo[1,5-a]pyrimidine-based inhibitors.34 To further explore the potential therapeutic utility of CSNK2 inhibition for the treatment of COVID-19, we studied the effect of the most potent CSNK2 inhibitor compound 2 on the replication of SARS-CoV-2 in primary human airway epithelial cells (HAE) grown in culture on an air–liquid interface. These primary lung cells model the architecture and cellular complexity of the conducting airway and are readily infected by zoonotic CoV, including SARS-CoV-2.44−46 At a dose of 10 μM, CSNK2A inhibitor 2 caused a 1.5 to 2-log reduction in the level of SARS-CoV-2 in HAE derived from three different donors after 18 h without affecting cell viability (Figure 5B). The efficacy of 2 was equivalent to remdesivir dosed at a 2.5 μM concentration. A dose–response assay in the HAE cells indicated that 2 had an IC50 in the 200–300 nM range for inhibition of SARS-CoV-2 replication without affecting cell viability at doses up to 10 μM (Figure 5C). Combined with the results from A549-ACE2 cells inoculated with human and bat viruses, these data provide strong evidence of the efficacy of host cell CSNK2A inhibitors in preventing the replication of SARS-like β-CoV.
Figure 5.

SARS-like β-CoV replication assays. (A) A549-ACE2 cells infected with SARS-CoV-2-nLuc, SHC012-CoV-nLuc, or WIV1-CoV-nLuc. Dose–response effect of CSNK2A inhibitor 2 (open triangles) and SGC-CK2-1 (closed squares) on inhibition of viral replication. No curve was fit to the SGC-CK2-1N data (closed triangles). Remdesivir (*, 2.5 μM) was included as a comparator. Assays performed in triplicate with data ranges shown by error bars. (B) Primary HAE cells from three donors (black, gray, while bars) infected with SARS-CoV-2-nLuc or unifected (U/I). CSNK2A inhibitor 2 (10 μM) produced a 1.5–2.0 log reduction in virus compared with the vehicle control (Veh). Remdesivir (Rem, 2.5 μM) was included as a comparator. (C) Dose–response effect of CSNK2A inhibitor 2 in primary HAE cells infected with SARS-CoV-2-nLuc (open circles) without affecting cell viability determined by LDH assay (closed circles). Remdesivir (*, 2.5 μM) was included as a comparator. Assay performed in triplicate with data ranges shown by error bars.
CSNK2A Inhibitors Block SARS-CoV-2 Spike Protein Uptake
The broad substrate specificity of CSNK2 provides little guidance on the mechanism of antiviral activity of CSNK2A inhibitors.47 There are several key steps (endocytic entry, replication, packaging, and egress), where CSNK2A inhibition may impact the virus life cycle1 and over 40 different viral proteins have been shown to be CSNK2 substrates.48 Notably, several other host kinases have been implicated in the regulation of virus entry into cells.49 β-CoVs infect cells following the attachment of their spike glycoprotein to receptors on the cell surface membrane.10 A primary mechanism by which the β-CoV spike-membrane complex enters cells is the process of CME.9 The internalized β-CoV accumulates in endosomes until the action of host cell proteases leads to the release of the virus mRNA from the endosome lumen into the cytosol. To study the effect of CSNK2 inhibition on CME, we utilized an assay that measures the uptake of the SARS-CoV-2 spike protein trimer into cells.8 His6-tagged spike protein was incubated with HEK293T-ACE2 cells for 30 min at 4 °C to allow complex formation between the spike protein and ACE2, and at 37 °C for 30 min to promote internalization by CME. The cells were then acid-washed to remove extracellular spike protein and fixed. The intracellular spike protein was visualized using a His6 antibody and quantified by imaging (Figure 6). This spike protein uptake assay phenocopies the CME of lentivirus pseudotyped with spike glycoprotein, a common model of SARS-CoV-2 infectivity.8 Vehicle-treated cells have efficient uptake of the His6-tagged SARS-CoV-2 spike protein. Treatment of the HEK293T-ACE2 cells with 1 μM of the CSNK2A chemical probe SGC-CK2-1 or CSNK2A inhibitor 2 resulted in a 70–80% decrease in spike protein uptake (Figure 6). Notably, the negative control analogue SGC-CK2-1N had no effect on spike protein uptake.
Figure 6.

Effect of CSNK2A inhibition on His6-SARS-CoV-2 spike protein uptake into HEK293T-ACE2 cells. (A) Cell nuclei stained with DAPI. Spike protein detected using a His6 antibody. Cells were treated with 1 μM of CSNK2A inhibitors (SGC-CK2-1 or Compound 2), negative control (SGC-CK2-1N), or vehicle control (DMSO). (B) Quantification of the His6-SARS-CoV-2 spike protein uptake. The data is from three separate experiments, with n = 9 for each sample. The total spike-His-tag fluorescence was divided by the number of cells to control for the number of cells per frame. AU, arbitrary units; ns, not significant; and ****p < 0.0001.
Since high levels of surface protease expression can support an alternative membrane fusion pathway of viral entry, the spike protein uptake studies were repeated in Caco-2 cells (high ACE2 expression)50 and Calu-3 cells (high TMPRSS2 and ACE2 expression).51 In both cell lines, inhibitor 2 and SGC-CK2-1 produced a > 75% decrease in spike protein uptake (Figure S5) despite the different levels of TMPRSS2 protease expression51 and consistent with our prior study demonstrating that CME is the primary pathway for SARS-CoV-2 uptake into cells.8 Finally, we utilized both chemical inhibition and genetic knockdown to demonstrate that CME is the primary mechanism for entry of MHV-nLuc into DBT cells. CME is dependent on the GTPase dynamin, and the selective dynamin inhibitors dynasore52 and hydroxy-dynasore53 produced dose-dependent inhibition of MHV replication (Figure S6A). In addition, siRNA knockdown of the clathrin heavy chain, an essential protein in CME, also blocked MHV replication (Figure S6B). Together, these data demonstrate that, despite differences in their cell surface receptors, MHV and SARS-CoV-2 utilize a common endocytosis pathway for cell entry. The dramatic decrease in spike protein uptake by CSNK2 inhibitors in multiple cell lines when combined with the observation of their antiviral activity on SARS-like β-CoV suggests that inhibition of CME may be a common mechanism that is targeted by these compounds.
The proteomic and phosphoproteomic studies that identified a key role of CSNK2 in SARS-CoV-2 infection had focused on the role of the kinase in virus egress through the remodeling of the extracellular matrix.25 Our data demonstrate that CSNK2 may also be involved in virus entry through CME (Figure 6), suggesting that β-CoVs utilize a common kinase for multiple steps in viral trafficking during their life cycle. The Numb-associated kinases AAK1 and GAK have also been implicated in the regulation of virus entry by CME.49,54 However, inhibitors of these kinases generally demonstrate antiviral activity at only micromolar concentrations,15 and the antiviral activity often does not track with kinase inhibition.55,56 Furthermore, AAK1 inhibition was recently reported to block SARS-CoV-2 virus uptake only in a subset of cells that lack the ACE2 receptor,57 implicating a mechanism independent of CME. In our assays, selective chemical probes for AAK158 or GAK59 failed to prevent β-CoV replication when used at their recommended 1 μM dose (Table S1). In contrast, we saw antiviral activity that tracked with CSNK2A potency (Figure 3) and robust inhibition of SARS-CoV-2 spike protein uptake by selective CSNK2A inhibitors (Figure 5).
CSNK2 is a Host Target for Inhibition of SARS-like β-CoV
Multiple observations argue in favor of CSNK2A inhibition as an antiviral mechanism in the MHV replication assay. First, the high-quality chemical probe SGC-CK2-1 showed inhibition of virus replication at doses where it has remarkably high kinase selectivity.34 Second, the structurally related negative control analogue SGC-CK2-1N had no effect on virus replication at doses up to 100-fold higher in concentration. Third, silmitasertib, a chemotype of CSNK2A inhibitor with different chemical and physical properties, also inhibited virus replication. Fourth, across three series of ATP-competitive 3-cyano-pyrazolo[1,5-a]pyrimidine CSNK2A inhibitors with substitutions at multiple sites on the heterocyclic core, the structure–activity relationship for virus inhibition matched the potency of kinase inhibition (Figure 3). The combined chemogenomic evidence strongly implicates CSNK2A inhibition as the molecular mechanism of action of antiviral activity. Confirmation that CSNK2 is a host cell kinase required for β-CoV replication was provided by genetic knockdown of the essential regulatory subunit CSNK2B or dual knockdown of the catalytic subunits CSNK2A1 and CSNK2A2 (Figure 4C). While further studies will be required to dissect the molecular details of the signaling pathway that requires CSNK2 for virus uptake and its relative contribution to SARS-like β-CoV replication, the potent anti-β-CoV activity of CSNK2A inhibition suggests that it may be a viable broad-spectrum antiviral therapy for current and future SARS-like β-CoVs. Although silmitasertib has progressed to clinical studies as an oncology drug, it may be challenging to repurpose it as an antiviral drug given its relatively weak potency (Figure 3). We have shown that more potent CSNK2 inhibitors can be identified with > 100-fold improvement in antiviral activity in cells, which portends that further optimization for anti-β-CoV activity and in vivo pharmacokinetic properties could lead to drugs with efficacy at doses that would be attainable in a clinical setting.
Materials and Methods
Cell Culture
DBT cells were cultured at 37 °C in Dulbecco’s modified Eagle’s medium (DMEM; Sigma) supplemented with 10% fetal bovine serum (Gibco) and penicillin and streptomycin (Sigma). Primary human airway epithelial (HAE) cells were cultured according to standard protocol.60 Briefly, HAE cells were expanded on plates coated with Bovine Collagen Type I/III (Advanced BioMatrix) and cultured in BEGM media. HAE cells were seeded onto transwells coated with HPC Collagen IV (Sigma) and cultured with ALI media. Cells were visually inspected for hallmarks of differentiation and used in studies between days 28–35 post seeding into transwells. HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). Cells were incubated in 5% CO2 at 37 °C. Cells lines were passaged every 72 h with trypsin and not allowed to reach confluency.
NanoBRET Assay
Assays were run as previously described.34 Briefly, a 10 μg/mL solution of DNA in Opti-MEM without serum was made containing 9 μg/mL of Carrier DNA (Promega) and 1 μg/mL of NL-CSNK2A1 or CSNK2A2-NL (Promega) for a total volume of 1.05 mL. Then, 31.5 μL of FuGENE HD (Promega) was added to form a lipid:DNA complex. The solution was then mixed by inversion eight times and incubated at room temperature for 20 min. The transfection complex (1.082 mL) was then gently mixed with 21 mL of HEK293 cells (ATCC) suspended at a density of 2 ×105 cells/mL in DMEM (Gibco) + 10% FBS (Corning). Briefly, 100 μL was dispensed into 96-well tissue culture treated plates (Corning #3917) and incubated at 37 °C in 5% CO2 for 24 h. The media was removed and replaced with 85 μL of Opti-MEM without phenol red. A total of 5 μL per well of 20 μM nanoBRET Tracer K10 (CSNK2A1) or K5 (CSNK2A2) in Tracer Dilution Buffer (Promega N291B) was added to all wells, except the “no tracer” control wells. Test compounds (10 mM in DMSO) were diluted in Opti-MEM media (99%) to prepare 1% DMSO stock solutions and evaluated at 11 concentrations. A total of 10 μL per well of the 10-fold test compound stock solutions (final assay concentration of 0.1% DMSO) were added. For “no compound” and “no tracer” control wells, a total of 10 μL per well of Opti-MEM plus DMSO (9 μL Opti-MEM with 1 μL DMSO) was added for a final concentration of 1% DMSO. Then, 96-well plates containing cells with nanoBRET Tracer K5 and test compounds (100 μL total volume per well) were equilibrated (37 °C/5% CO2) for 2 h. The plates were cooled to room temperature for 15 min. nanoBRET Nano-Glo substrate (Promega) at a ratio of 1:166 to Opti-MEM media in combination with extracellular NanoLuc Inhibitor (Promega) diluted 1:500 (10 μL of 30 mM stock per 5 mL Opti-MEM plus substrate) were combined to create a 3X stock solution. A total of 50 μL of the 3X substrate/extracellular NL inhibitor was added to each well. The plates were read within 10 min on a GloMax Discover luminometer (Promega) equipped with 450 nm BP filter (donor) and 600 nm LP filter (acceptor) using 0.3 s integration time. Raw milliBRET (mBRET) values were obtained by dividing the acceptor emission values (600 nm) by the donor emission values (450 nm) and multiplying by 1000. Averaged control values were used to represent complete inhibition (no tracer control: Opti-MEM + DMSO only) and no inhibition (tracer only control: no compound, Opti-MEM + DMSO + Tracer K5 only) and were plotted alongside the raw mBRET values. The data with n = 3 biological replicates was first normalized and then fitted using Sigmoidal, 4PL binding curve in Prism Software to determine IC50 values.
Viruses
MHV-nLuc
The MHV-A59 G plasmid was engineered to replace most of the coding sequence for orf4a and 4b with nLuc. Briefly, nucleotides 27,983 to 28,267 were removed and replaced with SalI and SacII restriction sites; approximately 111 bp of the 3′ end of orf4B was left to maintain the TRS for orf5. nLuc was PCR-amplified with primers 5′nLuc SalI (5′-NNNNNNGTCGACATGGTCTTCACACTCGAAGATTTC-3′) and 3′nLuc SacII (5′-NNNNNNCCGCGGTTACGCCAGAATGCGTTCGCAC-3′), digested with SalI and SacII, and then cloned into the G plasmid which had been similarly digested. A sequence-verified G-nLuc plasmid was used with MHV-A59 wild type A, B, C, D, E, and F plasmids to recover virus expressing nLuc and the recombinant virus sequence-verified. MHV-nLuc virus stocks were grown on DBT cells, and their titers were determined using the 50% tissue culture infectious dose (TCID50) assay.
SARS-CoV-2-nLuc, SHC014-CoV-nLuc, and WIV1-CoV-nLuc
A549-ACE2 cells (85–95% confluent) were infected at MOI of 0.01 with SARS-CoV-2-nLuc,42 WIV1-nLuc,61 or SHC014-nLuc61 in DMEM containing 5% heat-inactivated serum. Infected monolayers were incubated at 37 °C with 5% CO2 until CPE involved approximately 50% of the monolayer (generally between 66 and 72 h). The infected cell culture supernatant was recovered and clarified by centrifugation, and aliquots of the clarified supernatant were frozen at −80 °C until use.
MHV Assay
DBT cells were plated in 96-well plates to be 80% confluent at the start of the assay. Test compounds were diluted to 15 μM in DMEM. Serial 4-fold dilutions were made in DMEM, providing a concentration range of 15–0.22 μM. Media was aspirated from the DBT cells, and 100 μL of the diluted test compounds were added to the cells for 1 h at 37 °C. After 1 h, MHV-nLuc was added at an MOI of 0.1 in 50 μL DMEM so that the final concentration of the first dilution of the compound was 10 μM (T = 0). After 10 h, the media was aspirated, and the cells were washed with phosphate-buffered saline (PBS) and lysed with passive lysis buffer (Promega) for 20 min at room temperature. Relative light units (RLUs) were measured using a luminometer (Promega; GloMax). Triplicate data was analyzed in Prism GraphPad to generate IC50 values. For experiments employing dynasore and hydroxy-dynasore, the compounds were resuspended in serum-free DMEM. DBT cells were plated in 96-well plates to be 80% confluent at the start of the assay in DMEM supplemented with 10% FBS. The following morning, the cells were washed four times with PBS and serum-starved for 2 h before adding 100 μL of diluted compounds to cells, and incubated at 37 °C for 1 h. MHV-nLuc was diluted in serum-free DMEM and added to cells at an MOI of 0.1 (T = 0) and incubated at 37 °C. After 1 h, media was aspirated from the cells and replaced with DMEM with 10% FBS. At 10 hpi, the media was aspirated, and cells were washed with PBS, lysed, and RLUs measured, as described above.
A549-ACE2 Assay
Human lung epithelial A549-ACE2 cells were cultured in DMEM containing 10% heat-inactivated FBS, nonessential amino acids, and pen strep. A549-ACE2 cells were seeded at 20,000 cells per well in a 96-well solid black plate 1 day prior to infection. To assay drug effect, cells were pretreated with drug for 1 h and then infected with virus, with drug maintained during the infection. Then, 2 h after infection, the supernatant was removed, monolayers were rinsed with PBS, and media containing drug was added to each well. At 48 h post infection start, Nano-glo was added to each well as per the manufacturer’s protocol (Promega), and RLUs were measured using a Promega GloMax.
HAE Assay
HAE cultures were washed 3 times with prewarmed PBS (20 min each wash) to remove mucus from the apical surface. After the last apical wash, the spent ALI media was removed and replaced with media containing drug, DMSO, or media only, as needed. Immediately after the media was replaced, 100 μL of ic-SARS-CoV-2-nLuc42 was added to the apical side of the HAE cultures to achieve MOI = 0.5. Cultures were returned to the incubator and allowed to infect for 2 h. The inoculum was removed, and the apical surface was washed 3 times with PBS to remove the unbound virus before the cells were returned to the incubator. Typically, 24 h post infection, the cells were washed by adding 100 μL of prewarmed PBS to the apical surface and incubated at 37 °C for 20 min. The apical wash was removed, the inserts were transferred to a new 12-well plate, and 150 μL of Passive Lysis Buffer (Promega) was added to each well. After 10 min of incubation at room temperature, the inserts were scraped with a pipette tip, and the lysed cell mixture was recovered. Fifty microliter aliquots of lysed cell mixture were transferred to a clear bottom, black-walled plate and mixed with 50 μL of Nano-Glo reagent (Promega). Luminescence was read on a GloMax instrument (Promega). For calculations, wells containing passive lysis buffer mixed with Nano-Glo reagent were used as background luminescence, and this background was subtracted from the RLU of each sample. RLUs (background adjusted) were graphed directly in the bar graphs. For dose–response curves, the percent inhibition was calculated as follows: (1-(Sample RLU-background)/(virus only RLU-background adjusted)) x 100 with range normalized from 0–100 and IC50 calculated using GraphPad Prism.
LDH Assay
DBT cells were plated to be 80% confluent at the start of the assay. Compounds were diluted as done for the MHV assay and incubated with cells at 37 °C for 1 h. After 1 h, 50 μL of DMEM was added to the cells (T = 0); 45 min before harvest, lysis buffer was added to positive wells. LDH activity in cell-free supernatants was measured at 10 h after infection using the Sigma Tox7 kit as per the manufacturer’s directions. A549-ACE2 cells were seeded at 20,000 cells per well 1 day prior to infection in 96-well plates. Cells were pretreated for 1 h and then mock-infected. Then, 2 h post-mock infection, the media was removed, the monolayer was rinsed one time with PBS, and media containing drug was added to each well. Typically,48 h after mock infection, plates were centrifuged, and an aliquot of the cell culture supernatant was removed. For LDH assays using Sigma Tox7 kit, the clarified supernatant was transferred to a clean plate and assayed following the manufacturer’s protocol.
siRNA Knockdown
SMARTPool ON-TARGETplus mouse siRNAs were purchased for Csnk2a1 (L-058653-00-0005), Csnk2a2 (L-051582-00-0005), Csnk2b (L-049417-00-0005), Clathrin heavy polypeptide (L-063954-00-0005), or nontargeting (D-001810-10-05) genes (Horizon). Then, 200 μL of transfection master mix (12.5 nmol siRNA, RNAi Max, Opti-MEM) was reverse-transfected with DBT cells and incubated at 37 °C for 48 h. Cells were either collected for Western blot analysis or trypsinized and replated with fresh siRNA transfection master mix. Replated cells were 80% confluent and used for MHV assay experiments.
qRT-PCR
Cells were scraped, pelleted, and stored at −80 °C until the time of analysis. RNA was extracted from cell pellets using TRIzol (Thermo Fisher Scientific) and chloroform. After a 10 min spin, an equal volume of isopropanol was added to the aqueous layer and RNA was precipitated overnight at −20 °C. RNA was washed with ethanol and DNAse-treated (TURBO DNase, Thermo Fisher Scientific). RNA was quantified by NanoDrop (Thermo Fisher Scientific), and 2 μg of RNA was used to make cDNA (High-Capacity cDNA Reverse Transcription kit, Thermo Fisher Scientific). For real-time PCR, 0.5 μM of gene-specific primers (csnk2a1: Fwd GGTGAGGATAGCCAAGGTTCTG, Rev TCACTGTGGACAAAGCGTTCCC; csn2a2: Fwd GGATTACTGCCACAGCAAGGGA, Rev GGATGATAGAACTCTGCCAGACC; csnk2b: Fwd CAGAGCGACTTGATCGAACAGG, Rev CGAGGACAGTAGCCAAAGTCTC), and 1X SYBR green master mix were added to 2 μL of cDNA. RNA abundance was quantified using a standard curve generated from 10-fold serial dilutions of a DNA standard specific for each primer pair. The relative expression at t = 4, 8, and 12 h post infection was determined by dividing the RNA abundance at each time point by the value determined following mock infection (t = 0).
Western Blot Analysis
Cells were scraped and pelleted for western blot analysis and stored at −80 °C until the time of analysis. Pellets were thawed on ice and lysed for 10 min in radioimmunoprecipitation assay buffer (RIPA: 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1% sodium deoxycholate) supplemented with 1x cOmplete protease inhibitor cocktail (Roche). Cells were spun at 4 °C to pellet debris, and the protein concentration was determined via Bradford assay (VWR). Equal amounts of protein were resolved on a 10% SDS-PAGE gel and transferred to nitrocellulose membranes (Amersham). Membranes were blocked for 1 h at room temperature with 5% nonfat milk in TBS-T (20 mM Tris-HCl [pH 7.6], 140 mM NaCl, 0.1% Tween 20). Membranes were washed with TBS-T prior to incubation with a primary antibody. Rabbit polyclonal antibodies were dissolved in 5% bovine serium albumin (BSA) in TBS-T and incubated overnight at 4 °C. Blots were washed twice in TBS-T for 10 min prior to incubation with secondary horseradish peroxidase-conjugated rabbit antibody for 1 h at room temperature. Blots were imaged using a chemiluminescent digital imager (Bio-Rad). Antibodies were provided by Dr. David Litchfield (Western University) and have been described previously:62−64 anti-CSNK2A1 (KLH-CK2α; 1:5000), anti-CSNK2B antibody (KLH-CK2β; 1:10 000), and anti-CSNK2A1/CSNK2A2 antibody (1:2000). Antibody for clathrin heavy chain was purchased from Cell Signaling Technology #4796 (1:500).
Spike Uptake Assay
The protocol used for the uptake of spike protein has been described previously.8 Briefly, HEK293T-ACE2 cells were seeded onto poly-l-lysine-treated coverslips 24 h prior to experimentation. Calu-3 cells and Caco-2 cells were seeded as single cells on poly-d-lysine-treated coverslips and allowed to adhere for 36 h at 37 °C. Seeding at single cells allowed for reduction in clustering and cyst formation in Caco-2 and clear separation between cells for Calu-3 cells.One hour prior to the addition of spike protein, cell media were changed to starvation media (lack of serum) along with 1 μM test compounds and DMSO (vehicle control). Spike protein (5 μg per well) was added to each coverslip, and cells were incubated on ice for 30 min. Cells were then washed with PBS, and the media was replaced with fresh starvation media supplemented with the same test compound at 1 μM. Cells were then incubated for 30 min at 37 °C. Prior to fixation, cells were acid-washed for 60 s, followed by an acid-rinse to remove any extracellular spike protein. This was followed by PBS wash and fixation for 10 min with PFA at 4 °C. Cells were then permeabilized and blocked with 5% bovine serum albumin. His-tag antibody (HIS.H8) conjugated with Dylight 550 (Thermo Fisher) was used to identify spike protein uptake. Cells were then mounted and imaged using Leica SP8 microscope. Quantification was done with Leica LAS X software, with statistical calculations and graphs produced using Prism GraphPad software.
Acknowledgments
Constructs for nanoBRET measurements of CSNK2A1 and CSNK2A2, were provided by Promega. CSNK2 antibodies were generously provided by D. Litchfield (University of Western Ontario). The authors thank A. Edwards (SGC) and K. Saikatendu Singh (Takeda) for facilitating collaborative interactions, constructive criticism throughout the project, and critical reading of the manuscript. WuXi AppTec (Shanghai) and ChemSpace LLC provided chemical synthesis support. The authors acknowledge the support of the Neuro Microscopy Imaging Centre and Advanced BioImaging Facility at McGill University. The authors thank J. Bloom (University of Washington) for the HEK293T-ACE2 stable cell line.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00378.
Chemical structure of CIGB-300, cell viability data, human and mouse CSNK2A sequence comparison, correlation of CSNK2 isozyme potency on MHV inhibition, SARS-CoV-2 spike protein uptake in Calu-3 and Caco-2 cells, effect of CME inhibition on MHV replication, the effect of kinase Chemical Probes on MHV replication, and chemical synthesis and characterization of CSNK2 inhibitors (PDF)
Author Contributions
T.M.W., C.I.W., A.D.A., R.S.B., M.T.H., P.S.M., and N.J.M. conceived of the study. X.Y., J.W.B., E.C., and A.D.A. designed compounds and performed the synthesis. R.J.D., A.B., S.A.T.-B., J.L.S., E.A.M., E.M.L., and B.L.Y. performed biological studies. T.M.W., P.S.M., and N.J.M. wrote the manuscript. All authors read and approved the manuscript.
The Structural Genomics Consortium (SGC) is a registered charity (No: 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada through Ontario Genomics Institute [OGI-196], EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking [EUbOPEN Grant 875510], Janssen, Merck KGaA (aka EMD in Canada and US), Pfizer, and Takeda. Research reported in this publication was supported in part by the NC Biotech Center Institutional Support Grant 2018-IDG-1030 by the NIH Illuminating the Druggable Genome 1U24DK116204-01 and Department of Defense ALSRP Award AL190107. This project was supported by the North Carolina Policy Collaboratory at the University of North Carolina at Chapel Hill with funding from the North Carolina Coronavirus Relief Fund established and appropriated by the North Carolina General Assembly and by a grant from Takeda. This work was additionally supported by a grant from the Natural Sciences and Engineering Research Council to P.S.M. A.B. is supported by Fonds de recherche du Québec doctoral award and a studentship from the Parkinson Society of Canada. P.S.M. is a Distinguished James McGill Professor and a Fellow of the Royal Society of Canada. E.M. was supported by the Carol and Edward Smithwick Dissertation Completion Fellowship awarded by the UNC Graduate School.
The authors declare no competing financial interest.
Supplementary Material
References
- V’Kovski P.; Kratzel A.; Steiner S.; Stalder H.; Thiel V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders J. M.; Monogue M. L.; Jodlowski T. Z.; Cutrell J. B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 323, 1824–1836. 10.1001/jama.2020.6019. [DOI] [PubMed] [Google Scholar]
- Li F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. 10.1146/annurev-virology-110615-042301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J.; Ge J.; Yu J.; Shan S.; Zhou H.; Fan S.; Zhang Q.; Shi X.; Wang Q.; Zhang L.; Wang X. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Raj V. S.; Mou H.; Smits S. L.; Dekkers D. H.; Muller M. A.; Dijkman R.; Muth D.; Demmers J. A.; Zaki A.; Fouchier R. A.; et al. Dipeptidyl Peptidase 4 Is a Functional Receptor for the Emerging Human Coronavirus-EMC. Nature 2013, 495, 251–254. 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J.; Wan Y.; Liu C.; Yount B.; Gully K.; Yang Y.; Auerbach A.; Peng G.; Baric R.; Li F. Structure of Mouse Coronavirus Spike Protein Complexed with Receptor Reveals Mechanism for Viral Entry. PLoS Pathog. 2020, 16, e1008392 10.1371/journal.ppat.1008392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Körner R.; Majjouti M.; Alcazar M. A. A.; Mahabir E. Of Mice and Men: The Coronavirus Mhv and Mouse Models as a Translational Approach to Understand SARS-CoV-2. Viruses 2020, 12, 880. 10.3390/v12080880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayati A.; Kumar R.; Francis V.; McPherson P. S. SARS-CoV-2 Infects Cells after Viral Entry Via Clathrin-Mediated Endocytosis. J. Biol. Chem. 2021, 296, 100306 10.1016/j.jbc.2021.100306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkard C.; Verheije M. H.; Wicht O.; van Kasteren S. I.; van Kuppeveld F. J.; Haagmans B. L.; Pelkmans L.; Rottier P. J.; Bosch B. J.; de Haan C. A. Coronavirus Cell Entry Occurs through the Endo-/Lysosomal Pathway in a Proteolysis-Dependent Manner. PLoS Pathog. 2014, 10, e1004502 10.1371/journal.ppat.1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson C. B.; Farzan M.; Chen B.; Choe H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. 10.1038/s41580-021-00418-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin-Cocon L.; Diaz O.; Jacquemin C.; Barthel V.; Ogire E.; Ramiere C.; Andre P.; Lotteau V.; Vidalain P. O. The Current Landscape of Coronavirus-Host Protein-Protein Interactions. J. Transl. Med. 2020, 18, 319 10.1186/s12967-020-02480-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin K. K.; Renzette N.; Kowalik T. F.; Jensen J. D. Antiviral Drug Resistance as an Adaptive Process. Virus Evol. 2016, 2, vew014 10.1093/ve/vew014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N.; Sharma S.; Kumar R.; Tripathi B. N.; Barua S.; Ly H.; Rouse B. T. Host-Directed Antiviral Therapy. Clin. Microbiol. Rev. 2020, 33, e00168-19 10.1128/CMR.00168-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitalia V. C.; Munawar A. H. A Painful Lesson from the COVID-19 Pandemic: The Need for Broad-Spectrum, Host-Directed Antivirals. J. Transl. Med. 2020, 18, 390 10.1186/s12967-020-02476-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Cárceles J.; Caballero E.; Gil C.; Martinez A. Kinase Inhibitors as Underexplored Antiviral Agents. J. Med. Chem. 2022, 65, 935–954. 10.1021/acs.jmedchem.1c00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litchfield D. W. Protein Kinase CK2: Structure, Regulation and Role in Cellular Decisions of Life and Death. Biochem J. 2003, 369, 1–15. 10.1042/bj20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgo C.; D’Amore C.; Sarno S.; Salvi M.; Ruzzene M. Protein Kinase CK2: A Potential Therapeutic Target for Diverse Human Diseases. Signal Transduction Targeted Ther. 2021, 6, 183 10.1038/s41392-021-00567-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borad M. J.; Bai L.-Y.; Chen M.-H.; Hubbard J. M.; Mody K.; Rha S. Y.; Richards D. A.; Davis S. L.; Soong J.; Huang C.-E. C.-E.; et al. Silmitasertib (CX-4945) in Combination with Gemcitabine and Cisplatin as First-Line Treatment for Patients with Locally Advanced or Metastatic Cholangiocarcinoma: A Phase Ib/II Study. J. Clin. Oncol. 2021, 39, 312. 10.1200/JCO.2021.39.3_suppl.312. [DOI] [Google Scholar]
- Perea S. E.; Baladron I.; Valenzuela C.; Perera Y. CIGB-300: A Peptide-Based Drug That Impairs the Protein Kinase CK2-Mediated Phosphorylation. Semin. Oncol. 2018, 45, 58–67. 10.1053/j.seminoncol.2018.04.006. [DOI] [PubMed] [Google Scholar]
- Marschke R. F.; Borad M. J.; McFarland R. W.; Alvarez R. H.; Lim J. K.; Padgett C. S.; Hoff D. D. V.; O’Brien S. E.; Northfelt D. W. Findings from the Phase I Clinical Trials of CX-4945, an Orally Available Inhibitor of CK2. J. Clin. Oncol. 2011, 29, 3087. 10.1200/jco.2011.29.15_suppl.3087. [DOI] [Google Scholar]
- Keating J. A.; Striker R. Phosphorylation Events During Viral Infections Provide Potential Therapeutic Targets. Rev. Med. Virol. 2012, 22, 166–181. 10.1002/rmv.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piirsoo A.; Piirsoo M.; Kala M.; Sankovski E.; Lototskaja E.; Levin V.; Salvi M.; Ustav M. Activity of CK2α Protein Kinase Is Required for Efficient Replication of Some HPV Types. PLoS Pathog. 2019, 15, e1007788 10.1371/journal.ppat.1007788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D. E.; Jang G. M.; Bouhaddou M.; Xu J.; Obernier K.; White K. M.; O’Meara M. J.; Rezelj V. V.; Guo J. Z.; Swaney D. L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D. E.; Hiatt J.; Bouhaddou M.; Rezelj V. V.; Ulferts S.; Braberg H.; Jureka A. S.; Obernier K.; Guo J. Z.; Batra J.; et al. Comparative Host-Coronavirus Protein Interaction Networks Reveal Pan-Viral Disease Mechanisms. Science 2020, 370, eabe9403 10.1126/science.abe9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhaddou M.; Memon D.; Meyer B.; White K. M.; Rezelj V. V.; Correa Marrero M.; Polacco B. J.; Melnyk J. E.; Ulferts S.; Kaake R. M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712e619. 10.1016/j.cell.2020.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L. H.; Bunnage M. E. Applications of Chemogenomic Library Screening in Drug Discovery. Nat. Rev. Drug Discovery 2017, 16, 285–296. 10.1038/nrd.2016.244. [DOI] [PubMed] [Google Scholar]
- Wells C. I.; Al-Ali H.; Andrews D. M.; Asquith C. R. M.; Axtman A. D.; Dikic I.; Ebner D.; Ettmayer P.; Fischer C.; Frederiksen M.; et al. The Kinase Chemogenomic Set (KCGS): An Open Science Resource for Kinase Vulnerability Identification. Int. J. Mol. Sci. 2021, 22, 566. 10.3390/ijms22020566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Schapira M. The Promise and Peril of Chemical Probe Negative Controls. ACS Chem. Biol. 2021, 16, 579–585. 10.1021/acschembio.1c00036. [DOI] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Audia J. E.; Austin C.; Baell J.; Bennett J.; Blagg J.; Bountra C.; Brennan P. E.; Brown P. J.; Bunnage M. E.; et al. The Promise and Peril of Chemical Probes. Nat. Chem. Biol. 2015, 11, 536–541. 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson E. L.; Iegre J.; Brear P. D.; Zhabina E. A.; Hyvonen M.; Spring D. R. Downfalls of Chemical Probes Acting at the Kinase Atp-Site: CK2 as a Case Study. Molecules 2021, 26, 1977 10.3390/molecules26071977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano M. A.; Andrzejewska M.; Ruzzene M.; Sarno S.; Cesaro L.; Bain J.; Elliott M.; Meggio F.; Kazimierczuk Z.; Pinna L. A. Optimization of Protein Kinase CK2 Inhibitors Derived from 4,5,6,7-Tetrabromobenzimidazole. J. Med. Chem. 2004, 47, 6239–6247. 10.1021/jm049854a. [DOI] [PubMed] [Google Scholar]
- Dowling J. E.; Chuaqui C.; Pontz T. W.; Lyne P. D.; Larsen N. A.; Block M. H.; Chen H.; Su N.; Wu A.; Russell D.; et al. Potent and Selective Inhibitors of CK2 Kinase Identified through Structure-Guided Hybridization. ACS Med. Chem. Lett. 2012, 3, 278–283. 10.1021/ml200257n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z.; Perretta C.; Erickson P.; Margosiak S.; Almassy R.; Lu J.; Averill A.; Yager K. M.; Chu S. Structure-Based Design, Synthesis, and Study of Pyrazolo[1,5-a][1,3,5]Triazine Derivatives as Potent Inhibitors of Protein Kinase CK2. Bioorg. Med. Chem. Lett. 2007, 17, 4191–4195. 10.1016/j.bmcl.2007.05.041. [DOI] [PubMed] [Google Scholar]
- Wells C. I.; Drewry D. H.; Pickett J. E.; Tjaden A.; Kramer A.; Muller S.; Gyenis L.; Menyhart D.; Litchfield D. W.; Knapp S.; Axtman A. D. Development of a Potent and Selective Chemical Probe for the Pleiotropic Kinase CK2. Cell Chem. Biol. 2021, 28, 546–558 e10. 10.1016/j.chembiol.2020.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M. P.; Unch J.; Binkowski B. F.; Valley M. P.; Butler B. L.; Wood M. G.; Otto P.; Zimmerman K.; Vidugiris G.; Machleidt T.; et al. Engineered Luciferase Reporter from a Deep Sea Shrimp Utilizing a Novel Imidazopyrazinone Substrate. ACS Chem. Biol. 2012, 7, 1848–1857. 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasta J. D.; Corona C. R.; Wilkinson J.; Zimprich C. A.; Hartnett J. R.; Ingold M. R.; Zimmerman K.; Machleidt T.; Kirkland T. A.; Huwiler K. G.; et al. Quantitative, Wide-Spectrum Kinase Profiling in Live Cells for Assessing the Effect of Cellular ATP on Target Engagement. Cell Chem. Biol. 2018, 25, 206–214 e211. 10.1016/j.chembiol.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iegre J.; Atkinson E. L.; Brear P. D.; Cooper B. M.; Hyvonen M.; Spring D. R. Chemical Probes Targeting the Kinase CK2: A Journey Outside the Catalytic Box. Org. Biomol. Chem. 2021, 19, 4380–4396. 10.1039/D1OB00257K. [DOI] [PubMed] [Google Scholar]
- Tummino T. A.; Rezelj V. V.; Fischer B.; Fischer A.; O’Meara M. J.; Monel B.; Vallet T.; White K. M.; Zhang Z.; Alon A.; et al. Drug-Induced Phospholipidosis Confounds Drug Repurposing for SARS-CoV-2. Science 2021, 373, 541–547. 10.1126/science.abi4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V. D.; Yount B. L. Jr.; Debbink K.; Agnihothram S.; Gralinski L. E.; Plante J. A.; Graham R. L.; Scobey T.; Ge X. Y.; Donaldson E. F.; et al. A SARS-Like Cluster of Circulating Bat Coronaviruses Shows Potential for Human Emergence. Nat. Med. 2015, 21, 1508–1513. 10.1038/nm.3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen M.; Ren J.; Liu Q.; Glassman C. R.; Sheahan T. P.; Picton L. K.; Moreira F. R.; Rustagi A.; Jude K. M.; Zhao X.; et al. Facile Discovery of Surrogate Cytokine Agonists. Cell 2022, 185, 1414–1430 e1419. 10.1016/j.cell.2022.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan T. P.; Sims A. C.; Zhou S.; Graham R. L.; Pruijssers A. J.; Agostini M. L.; Leist S. R.; Schafer A.; Dinnon K. H. 3rd; Stevens L. J.; et al. An Orally Bioavailable Broad-Spectrum Antiviral Inhibits SARS-CoV-2 in Human Airway Epithelial Cell Cultures and Multiple Coronaviruses in Mice. Sci. Transl. Med. 2020, 12, eabb5883 10.1126/scitranslmed.abb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y. J.; Okuda K.; Edwards C. E.; Martinez D. R.; Asakura T.; Dinnon K. H. 3rd; Kato T.; Lee R. E.; Yount B. L.; Mascenik T. M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446 e414. 10.1016/j.cell.2020.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V. D.; Yount B. L. Jr.; Sims A. C.; Debbink K.; Agnihothram S. S.; Gralinski L. E.; Graham R. L.; Scobey T.; Plante J. A.; Royal S. R.; et al. SARS-Like WIV1-CoV Poised for Human Emergence. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 3048–3053. 10.1073/pnas.1517719113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y. J.; Chiba S.; Halfmann P.; Ehre C.; Kuroda M.; Dinnon K. H. 3rd; Leist S. R.; Schafer A.; Nakajima N.; Takahashi K.; et al. SARS-CoV-2 D614G Variant Exhibits Efficient Replication Ex Vivo and Transmission in Vivo. Science 2020, 370, 1464–1468. 10.1126/science.abe8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruijssers A. J.; George A. S.; Schafer A.; Leist S. R.; Gralinksi L. E.; Dinnon K. H. 3rd; Yount B. L.; Agostini M. L.; Stevens L. J.; Chappell J. D.; et al. Remdesivir Inhibits SARS-CoV-2 in Human Lung Cells and Chimeric SARS-CoV Expressing the SARS-CoV-2 RNA Polymerase in Mice. Cell Rep. 2020, 32, 107940 10.1016/j.celrep.2020.107940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan T. P.; Sims A. C.; Graham R. L.; Menachery V. D.; Gralinski L. E.; Case J. B.; Leist S. R.; Pyrc K.; Feng J. Y.; Trantcheva I.; et al. Broad-Spectrum Antiviral GS-5734 Inhibits Both Epidemic and Zoonotic Coronaviruses. Sci. Transl. Med. 2017, 9, eaal3653 10.1126/scitranslmed.aal3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Denis N. A.; Litchfield D. W. Protein Kinase Ck2 in Health and Disease: From Birth to Death: The Role of Protein Kinase CK2 in the Regulation of Cell Proliferation and Survival. Cell Mol. Life Sci. 2009, 66, 1817–1829. 10.1007/s00018-009-9150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggio F.; Pinna L. A. One-Thousand-and-One Substrates of Protein Kinase CK2?. FASEB J. 2003, 17, 349–368. 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- Bekerman E.; Neveu G.; Shulla A.; Brannan J.; Pu S. Y.; Wang S.; Xiao F.; Barouch-Bentov R.; Bakken R. R.; Mateo R.; et al. Anticancer Kinase Inhibitors Impair Intracellular Viral Trafficking and Exert Broad-Spectrum Antiviral Effects. J. Clin. Invest. 2017, 127, 1338–1352. 10.1172/JCI89857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossel E. C.; Huang C.; Narayanan K.; Makino S.; Tesh R. B.; Peters C. J. Exogenous Ace2 Expression Allows Refractory Cell Lines to Support Severe Acute Respiratory Syndrome Coronavirus Replication. J. Virol. 2005, 79, 3846–3850. 10.1128/JVI.79.6.3846-3850.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch J.; Uckeley Z. M.; Doldan P.; Stanifer M.; Boulant S.; Lozach P. Y. TMPRSS2 Expression Dictates the Entry Route Used by SARS-CoV-2 to Infect Host Cells. EMBO J. 2021, 40, e107821 10.15252/embj.2021107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen T.; Macia E.; Pelish H. E. Use of Dynasore, the Small Molecule Inhibitor of Dynamin, in the Regulation of Endocytosis. Methods Enzymol. 2008, 438, 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCluskey A.; Daniel J. A.; Hadzic G.; Chau N.; Clayton E. L.; Mariana A.; Whiting A.; Gorgani N. N.; Lloyd J.; Quan A.; et al. Building a Better Dynasore: The Dyngo Compounds Potently Inhibit Dynamin and Endocytosis. Traffic 2013, 14, 1272–1289. 10.1111/tra.12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdonck S.; Pu S. Y.; Sorrell F. J.; Elkins J. M.; Froeyen M.; Gao L. J.; Prugar L. I.; Dorosky D. E.; Brannan J. M.; Barouch-Bentov R.; et al. Synthesis and Structure-Activity Relationships of 3,5-Disubstituted-Pyrrolo[2,3-b]Pyridines as Inhibitors of Adaptor-Associated Kinase 1 with Antiviral Activity. J. Med. Chem. 2019, 62, 5810–5831. 10.1021/acs.jmedchem.9b00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saul S.; Huang P. T.; Einav S.; Asquith C. R. M. Identification and Evaluation of 4-Anilinoquin(az)olines as Potent Inhibitors of Both Dengue Virus (DENV) and Venezuelan Equine Encephalitis Virus (VEEV). Bioorg. Med. Chem. Lett. 2021, 52, 128407 10.1016/j.bmcl.2021.128407. [DOI] [PubMed] [Google Scholar]
- Saul S.; Pu S. Y.; Zuercher W. J.; Einav S.; Asquith C. R. M. Potent Antiviral Activity of Novel Multi-Substituted 4-Anilinoquin(az)olines. Bioorg. Med. Chem. Lett. 2020, 30, 127284 10.1016/j.bmcl.2020.127284. [DOI] [PubMed] [Google Scholar]
- Puray-Chavez M.; LaPak K. M.; Schrank T. P.; Elliott J. L.; Bhatt D. P.; Agajanian M. J.; Jasuja R.; Lawson D. Q.; Davis K.; Rothlauf P. W.; et al. Systematic Analysis of SARS-CoV-2 Infection of an ACE2-Negative Human Airway Cell. Cell Rep. 2021, 36, 109364 10.1016/j.celrep.2021.109364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells C.; Counago R. M.; Limas J. C.; Almeida T. L.; Cook J. G.; Drewry D. H.; Elkins J. M.; Gileadi O.; Kapadia N. R.; Lorente-Macias A.; et al. SGC-AAK1-1: A Chemical Probe Targeting AAK1 and BMP2k. ACS Med. Chem. Lett. 2020, 11, 340–345. 10.1021/acsmedchemlett.9b00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asquith C. R. M.; Berger B. T.; Wan J.; Bennett J. M.; Capuzzi S. J.; Crona D. J.; Drewry D. H.; East M. P.; Elkins J. M.; Fedorov O.; et al. SGC-GAK-1: A Chemical Probe for Cyclin G Associated Kinase (GAK). J. Med. Chem. 2019, 62, 2830–2836. 10.1021/acs.jmedchem.8b01213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulcher M. L.; Gabriel S.; Burns K. A.; Yankaskas J. R.; Randell S. H. Well-Differentiated Human Airway Epithelial Cell Cultures. Methods Mol. Med. 2005, 107, 183–206. [DOI] [PubMed] [Google Scholar]
- Rappazzo C. G.; Tse L. V.; Kaku C. I.; Wrapp D.; Sakharkar M.; Huang D.; Deveau L. M.; Yockachonis T. J.; Herbert A. S.; Battles M. B.; et al. Broad and Potent Activity against SARS-Like Viruses by an Engineered Human Monoclonal Antibody. Science 2021, 371, 823–829. 10.1126/science.abf4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosc D. G.; Slominski E.; Sichler C.; Litchfield D. W. Phosphorylation of Casein Kinase II by p34cdc2. Identification of Phosphorylation Sites Using Phosphorylation Site Mutants in Vitro. J. Biol. Chem. 1995, 270, 25872–25878. 10.1074/jbc.270.43.25872. [DOI] [PubMed] [Google Scholar]
- Litchfield D. W.; Luscher B.; Lozeman F. J.; Eisenman R. N.; Krebs E. G. Phosphorylation of Casein Kinase Ii by p34cdc2 in Vitro and at Mitosis. J. Biol. Chem. 1992, 267, 13943–13951. 10.1016/S0021-9258(19)49661-0. [DOI] [PubMed] [Google Scholar]
- Litchfield D. W.; Lozeman F. J.; Cicirelli M. F.; Harrylock M.; Ericsson L. H.; Piening C. J.; Krebs E. G. Phosphorylation of the Beta Subunit of Casein Kinase II in Human A431 Cells. Identification of the Autophosphorylation Site and a Site Phosphorylated by p34cdc2. J. Biol. Chem. 1991, 266, 20380–20389. 10.1016/S0021-9258(18)54934-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.