

Abstract
Pleuropulmonary blastoma (PPB) is a primary embryonal malignancy of childhood that is characterized by distinct morphologic types: type Ir (regressed), type I (cystic), type II (cystic and solid), and type III (solid). Prognosis varies by PPB type. Most cases are associated with a germline pathogenic mutation in DICER1; however, there is limited data on the factor(s) at a cellular level that drive progression from type I to type III. In this study, we evaluated the expression of p53 and its prognostic implications. A total of 143 PPB cases were included in the study with the following distribution in PPB types: Ir (14%), I (23%), II (32%) and III (31%). P53 expression by immunohistochemistry (IHC) was recorded as four groups: 0%, 1%-25%, 26%-75% and 76%-100%. All type I PPBs showed 0-25% p53 expression compared to the higher p53 expression (>25%) in type III PPB (p<0.0001), to support the argument that p53 has a role in tumor progression. Additionally, type Ir with the architectural hallmarks of type I PPB, but lacking the primitive cell population, has negligible p53 expression. High p53 expression (staining observed in >25% of the tumor cells) was significantly associated with age over 1 year (p=0.0033), neoadjuvant therapy (p=0.0009), positive resection margin (p=0.0008) and anaplasia (p<0.0001). P53 expression was significantly associated with recurrence-free survival (p<0.0001) and overall survival (p=0.0350), with higher p53 expression associated with worse prognosis. Comparisons of concordance statistics showed no significant difference in prognostication when using morphologic types compared to p53 expression groups (p=0.647). TP53 sequence was performed in 16 cases; the most common variant identified was a missense variant (12 cases), and in one case a frameshift truncating variant was noted. Based on these findings, we recommend performing p53 IHC in all newly diagnosed cases of types II and III PPB to further aid in risk stratification.
Keywords: Pleuropulmonary blastoma, PPB, p53, p53 expression, TP53
INTRODUCTION
Pleuropulmonary blastoma (PPB), a primary embryonal type malignancy of lung of childhood, is one of the unique neoplasms in this age group whose morphologic features have some of the developmental hallmarks of the organ or site of origin. When PPB was first reported, it was described as a solid neoplasm arising in the lung and/or pleura with a mixed histopathologic pattern of embryonal type rhabdomyosarcoma, nodules of chondrosarcoma, islands of primitive tumor cells with blastemal features, spindle cell foci resembling infantile fibrosarcoma and individual and groups of large, pleomorphic and anaplastic cells(1). A neoplastic epithelial component was not identified in any of those original cases nor in the several hundred cases of PPB, which differentiates PPB from the biphasic epithelial-mesenchymal pulmonary blastoma, a rare primary lung neoplasm occurring almost exclusively in adults(2).
This initial period of our studies was followed by the recognition that the solid neoplasm was preceded by a purely cystic lesion, often diagnosed clinically in infancy as a “congenital lung cyst” and interpreted pathologically as a congenital pulmonary airway malformation, and yet another lesion of the lung whose cystic features resembled the so-called congenital lung cyst but had a solid component with the composite histologic features of the originally reported solid PPB(3). It was then proposed that there were three morphologic stages through which the PPB evolved from the purely cystic lesion (type I) to the mixed cystic and solid penultimate lesion (type II) to the ultimate stage of the solid PPB (type III). This clinicopathologic progression from type I to type III PPB was supported through the pathologic observations of cases previously diagnosed as congenital lung cyst of one type or another which was reviewed retrospectively to be an example of PPB type I at the time of a later recurrence as a PPB type II or type III. A second line of argument was the median age at diagnosis of PPB type I at 9 months, type II at 35 months, and type III at 41 months(4,5). There was a significant association with survival among the three pathologic types of PPB with an almost universally favorable outcome for type I, and 71% and 53% 5–year overall survival (OS) for types II and III, respectively(3,6).
It was recognized during the early phase of our studies of PPB, once established as a clinicopathologic entity, that there was an apparent familial predisposition as well as the occurrence of several extrapulmonary neoplasms including cystic nephroma and Sertoli-Leydig cell tumor of the ovary as just two examples. Several affected kindreds were later found to have a heterozygous germline mutation in DICER1 with five specific hotspot mutations(7). It is estimated that 70% of children with a PPB have a biallelic mutations in DICER1, one germline and the other a second hit somatic mutation(8).
We subsequently performed a comprehensive TP53 sequencing study of 15 paired tumor and non-tumor samples, and identified TP53 mutation as an early phenomenon in PPB with allele frequencies only slightly lower than the DICER1 RNase IIIb mutations(8). In addition, loss of TP53 was seen in 13 of 15 cases, of those 7 cases a deletion of one allele and a mutation in the remaining allele, five has a deletion of one allele, and one had a homozygous deletion. In this latter study the clinical implication and prognostication of TP53 status were not evaluated. Moreover, no comprehensive study to date has evaluated the significance of p53 expression in PPB cases.
Given the prior identification of TP53 as an early phenomenon in PPB and considering that p53 expression is a strong surrogate marker for TP53 missense mutations(9,10); we hypothesized that types I and Ir have a lower expression of p53 by immunohistochemistry compared to types II and III cases. Additionally, we have anecdotally observed among the cases submitted to the Registry an overexpression of p53 in cases of type II and III PPB. In light of this, the present study was undertaken to determine whether there is an association between p53 expression and the pathologic type of PPB. Given the significant differences in prognosis among these three types, we hypothesized that biallelic loss of p53 and p53 expression may be associated with PPB type and prognosis(8).
MATERIALS AND METHODS
All PPB patients included in this study were enrolled in the International Pleuropulmonary Blastoma/DICER1 Registry, which is located at Children’s Minnesota, Minneapolis and St. Paul, Minnesota, USA, with collaboration from Washington University School of Medicine, St. Louis, MO and the Children’s National Medical Center, Washington, DC. Registry procedures are approved by the relevant human subjects’ committees. The Registry welcomes cases of known or suspected PPB or other DICER1 related conditions from individuals worldwide and no case is vetted based on molecular or clinical behavior (https://www.ppbregistry.org/). All cases sent to the Registry are centrally reviewed by one of the two Registry pathologists (DAH and LPD) and once the diagnosis of PPB or other DICER1 related condition is confirmed the patient is enrolled in the Registry.
As mentioned above, all PPB Registry cases underwent central pathology review by one of the two Registry pathologists (DAH and LPD) to confirm the diagnosis, and only cases with confirmed diagnosis of PPB were included in the study. The cases in the current study were enrolled from 1994 to 2018. To aid in the evaluation of PPB, the majority of the cases have had ancillary immunohistochemistry (IHC) prospectively performed including vimentin, CD56, desmin, myogenin and p53.
Each case of PPB is assigned to one of four pathologic types: Ir, I, II, and III(3). Type I was defined as a multicystic lesion with a diffuse or focal group(s) of small primitive round cells with or without apparent rhabdomyoblastic differentiation on the basis of light and immunohistochemical findings with desmin and myogenin positivity or in the absence of the latter CD56 positivity in the small cell population; these small cells and rhabdomyoblasts were concentrated beneath the surface cuboidal epithelium with a cambium layer-like orientation (Figure 1). Type Ir (regressed) has the same multicystic architecture of type I, but is devoid of any small primitive round cells or rhabdomyoblasts by light microscopy or immunohistochemistry (Figure 1)(6). Type II consisted of any foci of a type I pattern and a grossly evident solid component whose microscopic composition was a variable collage of some or all the following: embryonal rhabdomyosarcoma, spindle cell sarcoma, nodules of atypical cartilage, islands of blastema with or without rhabdomyoblastic differentiation and large, highly pleomorphic anaplastic cells utilizing criteria for Wilms tumor(11) (Figure 2). The distribution and representation of these several primitive and sarcomatous patterns varied from one PPB type II to another, and any proportion of these patterns was regarded as a type II PPB. Type III lacked any identifiable foci of the type I pattern by gross or microscopic examination, but had the microscopic features of the solid component of type II PPB (Figure 3). As mentioned earlier anaplasia was defined using the criteria for Wilms tumor(11), and was evaluated prospectively in all the cases during enrollment in the Registry without quantification. Nonetheless, in our experience, when present anaplasia is usually diffuse, but patchy throughout the tumor.
Figure-1.
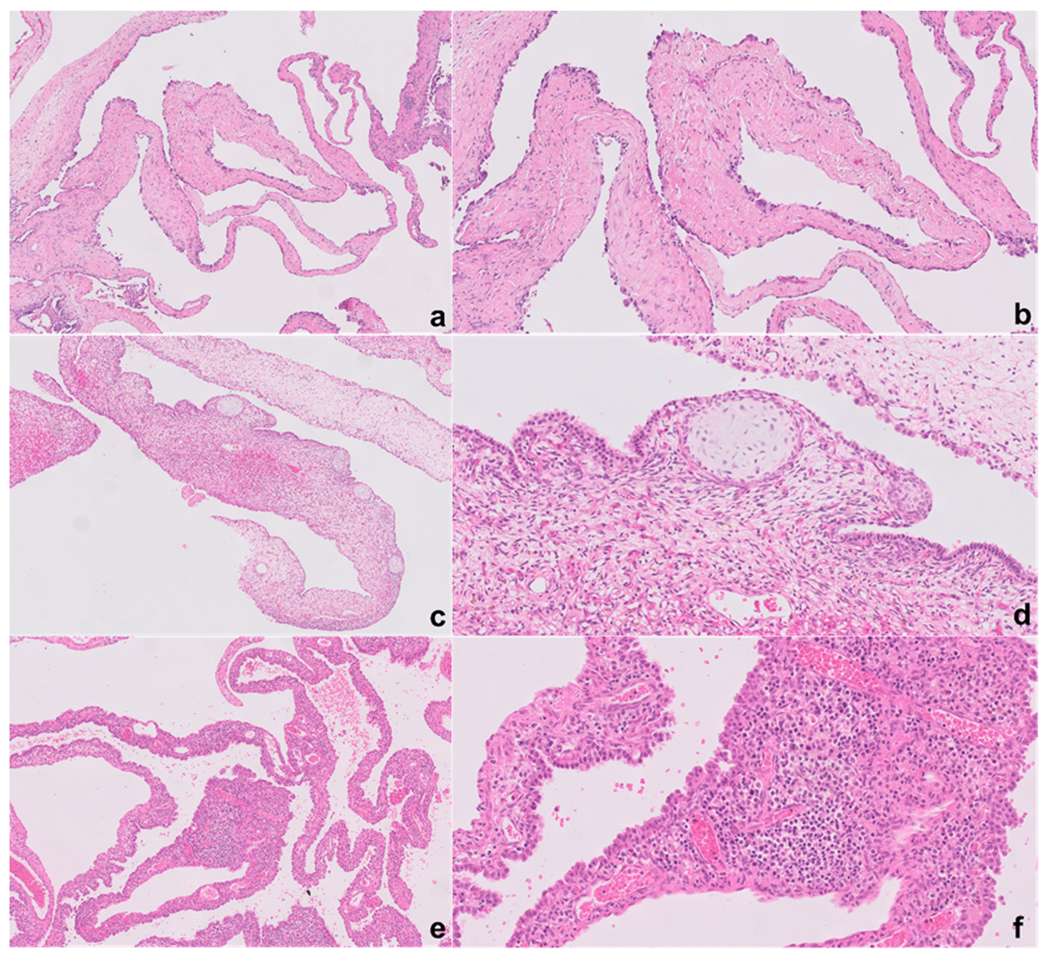
Type Ir PPB is characterized by multicystic structures where the septa lack the characteristic small primitive cells underneath the epithelium (a, b). In contrast, type I PPB is characterized by the presence of these small primitive cells which can either present as a continuous or discontinuous growth pattern (c, d, e, f). In more than half of the cases the primitive cells underneath the epithelium are rhabdomyoblasts, and a classic association with immature island of cartilage is seen in PPB regardless of type (c, d). Occasionally the cystic septa are expanded by the immature cells, a possible sign of early progression to type II (e, f).
Figure-2.
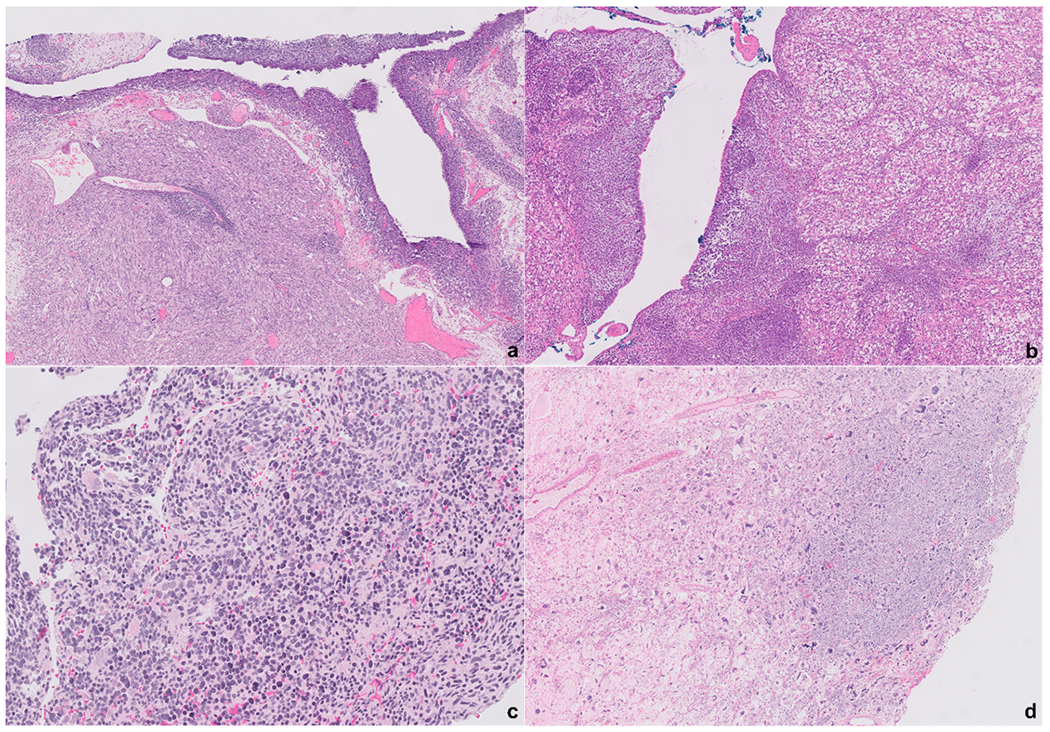
Type II PPB are characterized by the presence of both cystic and solid areas. Underneath the epithelium of the cystic structures a condensation of the immature cells can be noted in some cases forming a cambium-like layer (a). The solid components can have a wide range of morphology from a fibrosarcoma-like appearance (a) to a predominance of a rhabdomyoblasts proliferation (b). In some cases, the solid areas are composed of primitive small immature cells (c). The solid areas might also show frank anaplasia (d). All photomicrographs represent different cases.
Figure-3.
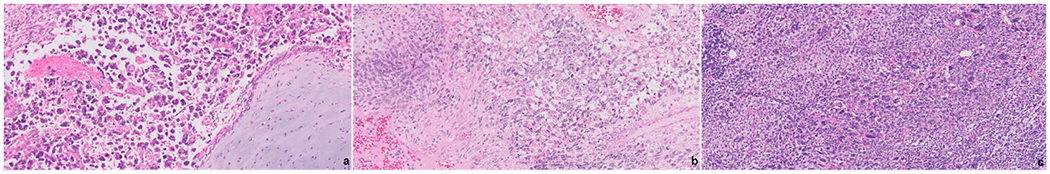
PPB composed exclusively of solid areas are consistent with a type III PPB. The solid component can have a broad morphology spectrum with the most classically seen being a rhabdomyosarcoma which is often associated with immature cartilaginous islands (a, b). In some cases, an undifferentiated sarcoma is seen with prominent anaplasia (c).
Inclusion of cases for the study was based upon representative H&E sections of the tumor and immunohistochemical stains to include p53 in each case. As stated earlier, p53 IHC was performed in all cases prospectively from 1994 to 2018 and in various platforms over this period, it is not possible to determine the exact protocol and antibody clone use in all cases. Given that all p53 IHC were performed prospectively the possibility of pre-analytical factors affecting the staining quality is unlikely. These sections were on file at the PPB/DICER1 Registry or the consultation files of the Lauren V. Ackerman Laboratory of Surgical Pathology, Barnes-Jewish and St. Louis Children’s Hospitals, St. Louis, MO. The archival slides were re-reviewed blinded from all clinical and follow-up information. The p53 immunostain score was obtained by assessing the entire slide and identifying the “hot-spot” areas; the percentage of tumor cells reactive for p53 was determined in these hot spot foci. Only nuclear staining was considered positive regardless of its staining intensity. In all cases, presence of internal control staining such as inflammatory cells, and normal epithelial and/or stromal cells if present were evaluated, especially in cases showing absence of p53 expression. All cases showed adequate internal controls. Depending on the percentage of positive tumor cells, the cases were classified into four groups: 0%, 1% – 25%, 26% – 75% and 76% – 100% (Figure 4); tumor staining intensity was not considered for grouping. A high p53 expression was considered for cases with >25% of tumor cells staining and low as <26%. Those cases showing absence of expression (0% group) were considered as abnormal complete loss of expression and not as a wild-type pattern of expression. In addition, in cases with a cystic component, namely types Ir, I and II, special attention was directed to the cystic lining epithelium, and p53 nuclear expression by immunohistochemistry was evaluated in these cells and noted as present or absent without quantification.
Figure-4.
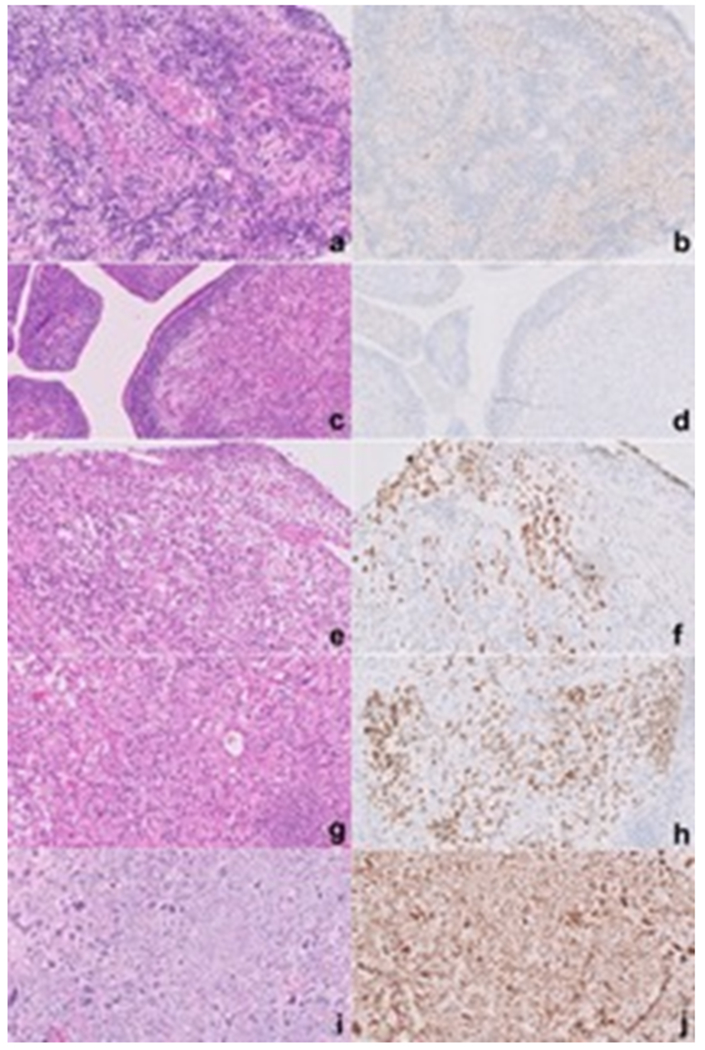
Separate cases of PPB paired with p53 immunostains showing various degrees of expression: 0%, group 0 (a, b); 10%, group 1 (1 – 25%) (c, d); 50% group 3 (26 – 75%) (e, f); 80% (g, h); and 100%, group 4 (76 – 100%) (i, j).
TP53 Sequencing
TP53 sequencing was performed on formalin-fixed, paraffin embedded tumor tissue. Pure tumor areas were identified by H&E slide and 5mm cores were taken from the representative area for each tumor. DNA was extracted and prepared for sequencing as described in Pugh et al.(8) A custom multiplex PCR panel was used for the coding regions of TP53 with an average amplicon length less than 200 base pairs (Custom Ampliseq, Life Technologies). Sequencing was performed on a 318 chip (ION PGM Sequencing 200 kit V2, Life Technologies) with an average depth of coverage of 500 filtered reads. Variants were annotated with Alamut mutation HT software (Interactive Biosoftware, Rouen, France) and named using HUGO nomenclature for TP53 transcript NM_000546.5. For both DICER1 and TP53, nonsense, frameshift and canonical splice-site mutations were considered loss of function. SIFT was used to assess the potential significance of predicted novel missense amino acid substitutions(12). Allele frequencies from the common SNP c.215C>G; p.Pro72Arg were included, when present, as an assessment for loss of heterozygosity.
Statistical Analysis
The association of p53 expression groups and patient and tumor characteristics were assessed with chi-square and Kruskal-Wallis tests for categorical and numeric data, respectively. Recurrence-free survival (RFS) and overall survival (OS) were estimated using Kaplan-Meier curves, and the association with p53 expression groups was assessed via log rank test. The concordance statistic (c-index) was used to assess different scores ability to classify risk for RFS and OS(13). The c-index is a measure of discrimination with a value of 1 indicating perfect discrimination and a value of 0.5 indicating no predictive value (i.e., null value). To compare potential risk scores, differences between c-indices were used and standard errors were calculated using the infinitesimal jackknife method(14).
RESULTS
Patient and tumor characteristics
A total of 219 cases were identified in the archival materials of the Lauren V. Ackerman Laboratory of Surgical Pathology, 143 cases met all of the study inclusion criteria (Table 1). A subset of these cases were included in prior studies(3,6,8,15,16). An equal gender distribution was noted and the median age of presentation was 2.9 years (range: 0 – 19.6). Thirty patients (21.0%) presented with an associated pneumothorax and 69.2% had received neoadjuvant therapy. A positive surgical resection margin was present in 42.7% of the cases. Only 7 patients (4.9%) had evidence of metastatic disease at the time of presentation. The distribution of PPB types was the following: 14.0% type Ir, 23.1% type I, 32.2% type II, and 30.8% type III (Table 1). Anaplasia was identified in 40.6% of the cases.
Table-1.
Patient and tumor characteristics
| N = 143 |
|
|---|---|
| Gender, n (%) | |
| Male | 71 (49.7%) |
| Female | 72 (50.3%) |
| Age, median (range) (years) | 2.9 (0 – 19.6) |
| Procedure performed for initial diagnosis, n (%) | |
| Thoracoscopy/Thoracotomy | 90 (62.9%) |
| Needle biopsy | 22 (15.4%) |
| Unknown | 31 (21.7%) |
| Pneumothorax at time of presentation, n (%) | |
| Yes | 30 (21.0%) |
| No | 68 (47.6%) |
| Unknown | 45 (31.5%) |
| Neoadjuvant therapy, n (%) | |
| Yes | 99 (69.2%) |
| No | 40 (28.0%) |
| Unknown | 4 (2.8%) |
| Margin status for primary surgical resection, n (%) | |
| Positive | 61 (42.7%) |
| Negative | 49 (34.3%) |
| Unknown | 33 (23.1%) |
| Pleuropulmonary blastoma histologic type, n (%) | |
| Ir | 20 (14.0%) |
| I | 33 (23.1%) |
| II | 46 (32.2%) |
| III | 44 (30.8%) |
| Anaplasia, n (%) | |
| Yes | 58 (40.6%) |
| No | 85 (59.4%) |
| Metastatic disease at time of presentation, n (%) | |
| Yes | 7 (4.9%) |
| No | 135 (95.1%) |
Percentages might not add to 100% due to rounding
P53 Expression
The distribution of p53 expression included the following: 40 cases (28.0%) with 0% expression, 41 cases (28.7%) with 1-25% expression, 33 cases (23.1%) with 26-75% expression, and 29 cases (20.3%) with 76-100% expression (Table 2). A significant association was identified between the PPB type and p53 expression (p < 0.0001). All of the type Ir and type I PPBs had less than 25% p53 expression; the majority of the type II and type III PPBs had p53 expression greater than 25%. Anaplasia was significantly associated with higher p53 expression (groups 26 – 75% and 76 – 100%) (p < 0.0001).
Table-2.
Clinicopathologic characteristics and p53 expression
| p53 expression | |||||
|---|---|---|---|---|---|
| 0% n: 40 |
1 – 25% n: 41 |
26 – 75% n: 33 |
76 – 100% n: 29 |
P | |
|
|
|||||
| Gender, n (%) | |||||
| Male | 21 (52.5%) | 23 (46.9%) | 15 (60.0%) | 12 (41.4%) | 0.5441 |
| Female | 19 (47.5%) | 26 (53.1%) | 10 (40.0%) | 17 (58.6%) | |
| Age, median (range) (years) | 1.5 (0.2 – 11.5) | 2.1 (0 – 19.6) | 2.9 (1.5 – 15.3) | 3.1 (0 – 7.7) | 0.0033 |
| Procedure performed for initial diagnosis, n (%) | |||||
| Thoracoscopy/Thoracotomy | 27 (67.5%) | 29 (70.7%) | 19 (57.6%) | 15 (51.7%) | 0.0081 |
| Needle biopsy | 2 (5.0%) | 2 (4.9) | 8 (24.2%) | 10 (34.5%) | |
| Unknown | 11 (27.5%) | 10 (24.4%) | 6 (18.2%) | 4 (13.8%) | |
| Pneumothorax at time of presentation, n (%) | |||||
| Yes | 6 (15.0%) | 9 (21.9%) | 7 (21.2%) | 8 (27.6%) | 0.0577 |
| No | 28 (70.0%) | 17 (41.5%) | 13 (39.4%) | 10 (34.5%) | |
| Unknown | 6 (15.0%) | 15 (36.6%) | 13 (39.4%) | 11 (37.9%) | |
| Neoadjuvant therapy, n (%) | |||||
| Yes | 7 (17.5%) | 6 (14.6%) | 10 (30.3%) | 17 (58.6%) | 0.0009 |
| No | 33 (82.5%) | 34 (82.9%) | 21 (63.6%) | 11 (37.9%) | |
| Unknown | 0 | 1 (2.4%) | 2 (6.1%) | 1 (3.5%) | |
| Margin status for primary surgical resection, n (%) | |||||
| Positive | 8 (20.0%) | 14 (34.2%) | 18 (54.6%) | 21 (72.4%) | 0.0008 |
| Negative | 20 (50.0%) | 17 (41.5%) | 9 (27.3%) | 3 (10.3%) | |
| Unknown | 12 (30.0%) | 10 (34.4%) | 6 (18.2%) | 5 (17.2%) | |
| Pleuropulmonary blastoma histologic type, n (%) | |||||
| Ir | 15 (37.5%) | 5 (12.2%) | 0 | 0 | < 0.0001 |
| I | 15 (37.5%) | 18 (43.9%) | 0 | 0 | |
| II | 4 (10.0%) | 11 (26.8%) | 20 (60.6%) | 11 (37.9%) | |
| III | 6 (15.0%) | 7 (17.1%) | 13 (39.4%) | 18 (62.1%) | |
| Anaplasia, n (%) | |||||
| Yes | 9 (22.5%) | 9 (22.0%) | 15 (45.5%) | 25 (86.2%) | < 0.0001 |
| No | 31 (77.5%) | 32 (78.1%) | 18 (54.6%) | 4 (13.8%) | |
| Metastatic disease at time of presentation, n (%) | |||||
| Yes | 0 | 1 (2.4%) | 3 (9.1%) | 3 (10.7%) | 0.1199 |
| No | 40 (100%) | 40 (97.6%) | 30 (90.9%) | 25 (89.3%) | |
Percentages might not add to 100% due to rounding
In cases with a cystic component (i.e., all except type III PPBs), attention was directed to the epithelial cystic lining to determine if p53 was expressed in the epithelium. Epithelial expression of p53 was associated with PPB type and was observed in 31 (75.6%) cases of type II compared to 15 (45.5%) cases of type I PPB (p < 0.0001). Of note, in 5 type II PPB cases the information on p53 expression in the epithelium lining was not available due to the p53 IHC being performed in a section without a cystic component. Only 3 cases (15.0%) of PPB type Ir showed any expression of p53 in the lining epithelium with only very patchy positivity compared to types I and II.
Given the strong association of p53 expression with PPB type, it is not surprising that p53 expression was also associated with several other factors known to be associated with PPB type. For example, median age at diagnosis trended upward with higher p53 expression regarded as >25% (p = 0.0033). Similarly, although in a substantial proportion of the patients (31 cases, 21.7%), the procedure performed during initial diagnosis was unknown, a significant association was noted with a p53 expression greater than 25% in those having a needle biopsy compared to those in which a thoracoscopy/thoracotomy was performed (p = 0.0081). Also, a p53 expression greater than 25% was noted in cases treated with neoadjuvant therapy (p = 0.0009). Cases with p53 expression less than 26% were associated with a negative surgical resection while those with a p53 expression higher than 25% which were associated with a positive margin (p = 0.0008).
Recurrence-Free Survival and Overall Survival
PPB type was significantly associated with RFS (p = 0.0002) and OS (p = 0.0022) with type Ir and type III having the most and least favorable survivals, respectively (Figure 5). Expression of p53 was also significantly associated with RFS (p < 0.0001) and OS (p = 0.0350), with lower (and higher) levels of expressions associated with a more (and less) favorable outcomes (Figure 6). In this cohort, 5-year RFS in type Ir was 100%; type I, 86.2%, type II, 52.0%, and type III, 46.5%. The differences in 5-year RFS did not reach statistical significance for types II and III, likely due to small sample size. With the proposed p53 groups, three groups can be identified with best prognosis among patients with 0% p53 expression (5-year RFS 83.3%), intermediate prognosis with 1-75% p53 expression (5-year RFS 65.2% and 64.8% for the 1-25% and 26-75% groups, respectively), and worst prognosis when p53 expression is greater than 75% (5-year RFS 33.3%).
Figure-5.
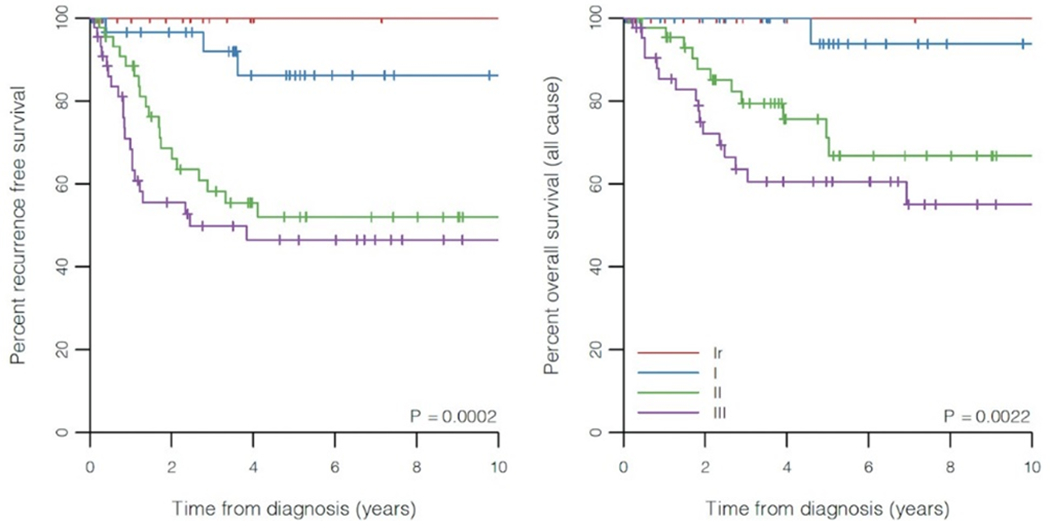
Kaplan-Meier plots of recurrence-free and overall survival for PPB types.
Figure-6.
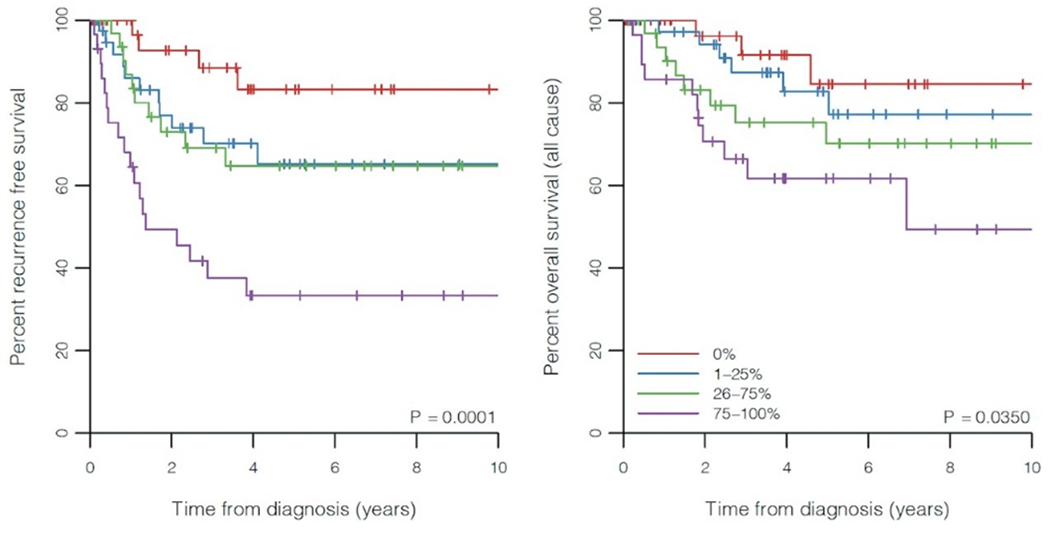
Kaplan-Meier plots of recurrence-free and overall survival for p53 expression groups.
The c-indices for RFS were 0.685 for the four p53 expression groups and 0.704 for p53 as a (numeric ordinal) percentage (Table 3). These values were comparable to using PPB type as a risk score, which had a c-index of 0.702 (p = 0.647 and 0.964 when compared to p53 groups and percentage, respectively). When stratified by type, p53 expression still showed some predictive ability (average c-index of 0.594 for p53 percentage), which was best among type III patients (c-index of 0.653 for p53 percentage) but essentially random for type II patients (c-index of 0.528 for p53 percentage). For OS, the c-indices were 0.669 for the four p53 expression groups and 0.680 for p53 as a (numeric ordinal) percentage (Table 4). PPB type had a c-index of 0.717, which was not significantly different from c-indices of p53 groups and p53 percentage (p = 0.374 and 0.475, respectively). When stratified by type, p53 expression did not differentiate patients well for OS (average c-index of 0.540 for p53 percentage). In particular, among type I patients, the c-index of 0.200 was due to only one overall death, which occurred in a patient with 0% p53 expression.
Table-3.
Concordance statistics (c-index) of various risk scores for RFS
| Risk Factor | C-index (SE) | Difference (SE) | P |
|---|---|---|---|
| PPB type | 0.702 (± 0.035) | ||
|
| |||
| p53 group | 0.685 (± 0.039) | −0.017 (± 0.038) | 0.647 |
| Stratified by type | 0.581 (± 0.045) | ||
| Type I | 0.611 (± 0.112) | ||
| Type II | 0.503 (± 0.068) | ||
| Type III | 0.651 (± 0.056) | ||
|
| |||
| p53 percent | 0.704 (± 0.038) | 0.002 (± 0.037) | 0.964 |
| Stratified by type | 0.594 (± 0.046) | ||
| Type I | 0.627 (± 0.143) | ||
| Type II | 0.528 (± 0.066) | ||
| Type III | 0.653 (± 0.063) | ||
Abbreviations: RFS – recurrence-free survival; SE – standard error; PPB – pleuropulmonary blastoma.
Table-4.
Concordance statistics (c-index) of various risk scores for OS
| Risk Factor | C-index (SE) | Difference (SE) | P |
|---|---|---|---|
| PPB type | 0.717 (± 0.038) | ||
|
| |||
| p53 group | 0.669 (± 0.047) | −0.048 (± 0.054) | 0.374 |
| Stratified by type | 0.543 (± 0.054) | ||
| Type I | 0.200 (± 0.063) | ||
| Type II | 0.559 (± 0.085) | ||
| Type III | 0.542 (± 0.070) | ||
|
| |||
| p53 percent | 0.680 (± 0.046) | −0.037 (± 0.051) | 0.475 |
| Stratified by type | 0.540 (± 0.056) | ||
| Type I | 0.200 (± 0.063) | ||
| Type II | 0.571 (± 0.086) | ||
| Type III | 0.529 (± 0.074) | ||
Abbreviations: OS – overall survival; SE – standard error; PPB – pleuropulmonary blastoma.
TP53 Sequencing
TP53 sequencing was performed in 16 PPB cases (6 type II and 10 type III PPB) (Table 5). All of the sequenced cases showed TP53 mutations with the most common being a missense mutation (12 cases, 75%) followed by a frameshift mutation (1 case, 6%), a nonsense mutation (1 case, 6%); in two cases (13%) the mutation type was unknown. These latter two unknown variants consisted of a c.673-1G>C and a c.314G>T; p.G105V per International Agency for Research on Cancer (IARC) nomenclature. According to the IARC TP53 database, c.673-1G>C results in a splice site on intron 6, and c.314G>T in a deleterious sorting intolerant from tolerant (SIFT) on exon 4. Six of the missense mutations were predicted to be deleterious by SIFT and the remainder were considered to represent single nucleotide polymorphism (SNP). Of the 16 cases, 13 cases the TP53 expression by immunohistochemistry matched the genotype. In two cases, both showing anaplasia, a missense and a nonsense mutation were identified but these cases did not show p53 expression.
Table-5.
TP53 sequence information
| Case | PPB type | IARC TP53 nomenclature | Variant type | IARC TP53 database effects | Predicted alleles frequency | Genotype / phenotype match |
|---|---|---|---|---|---|---|
| 1 | II | c.673-1G>C | Unknown | Splice site, intron 6 | 74% | Yes |
| 2 | III | c.518T>C; p.V173A | Missense | SIFT deleterious, exon 5 | 83% | Yes |
| 3 | II | c.215C>G; p.P72R | Missense | SNP | Heterozygous | Yes |
| 4 | II | c.215C>G; p.P72R | Missense | SNP | LOH | Yes |
| 5 | III | c.215C>G; p.P72R | Missense | SNP | LOH | No |
| 6 | II | c.1009C>T; p.R337C | Missense | SIFT deleterious, exon 10 | 71% | Yes |
| 7 | III | c.215C>G; p.P72R | Missense | SNP | LOH | Yes |
| 8 | III | c.215C>G; p.P72R | Missense | SNP | LOH | Yes |
| 9 | III | c.513_514insA | Frameshift | Frameshift truncating | 50% | Yes |
| 10 | II | c.215C>G; p.P72R | Missense | SNP | Heterozygous | Yes |
| 11 | III | c.314G>T; p.G105V | Unknown | SIFT deleterious, exon 4 | 5% | No |
| 12 | III | c.415A>T; p.K139* | Nonsense | Nonsense | 76% | No |
| 13 | II | c.556G>A; p.D186N | Missense | SIFT deleterious, exon 5 | 4% | Yes |
| 14 | III | c.481G>A; p.A161T | Missense | SIFT deleterious, exon 5 | 7% | Yes |
| 15 | III | c.580C>T; p.L194F | Missense | SIFT deleterious, exon 6 | 95% | Yes |
| 16 | III | c.391A>T; p.N131Y | Missense | SIFT deleterious, exon 5 | 36% | Yes |
Abbreviations: PPB – pleuropulmonary blastoma; IARC – International Agency for Research on Cancer; SIFT – sorting intolerant from tolerant; SNP – single nucleotide polymorphism; LOH – loss of heterozygosity.
DISCUSSION
Pleuropulmonary blastoma is one of the few pediatric malignancies that follows a temporal progression from an indolent to a highly aggressive, potentially lethal neoplasm. This progression follows a chronologic line based upon the median age at diagnosis from the type I PPB at 9 months to type III PPB at 44 months. This unique characteristic of PPB allows for the opportunity to characterize the genetic and cellular factors that may promote the progression of this neoplasm. The stepwise abnormalities that result in this morphologic spectrum with its prognostic implications remain unclear. In this report, some insight has been gained upon the suggested role of p53 in the progression of PPB type I to types II and III. Type I PPB was significantly associated with a lower expression of p53 compared to types II and III, demonstrating an increase in protein expression with tumor progression (p < 0.0001). Moreover, a significant expression of p53 in the cystic epithelial cells was identified from type Ir (3% of cases), type I (45.5% of cases) and type II (75.6%). We hypothesized based on these findings that TP53 mutations in the cystic epithelial cells also have an important role in PPB type progression. The lack of expression of p53 in PPB type Ir may provide a partial explanation for the lower risk for these lesions to progress to type II or III PPB.
In this cohort of cases, a higher p53 expression was significantly associated with the use of neoadjuvant treatment (p = 0.0009) and with the specimen being a needle biopsy (p = 0.0081). These associations are somewhat expected given the strong association with p53 expression groups and PPB types. Since PPB types II and III are commonly treated with neoadjuvant therapy which is rarely given in types Ir and I(3,16). Similarly, there is an increase tendency to perform needle biopsies in larger PPBs, which are more commonly types II and III, and a thoracoscopy/thoracotomy approach for purely cystic lesions(3).
One of the first studies to evaluate p53 expression in PPB was conducted by Pacinda et al., who evaluated three PPBs; only one case showed p53 expression in more than 10% of the neoplastic cells. The PPB type and follow-up information were not provided in this study(17). The expression of p53 in our cohort was shown to be a significant prognostic factor. Those cases with higher p53 expression were found in children over 1 year of age (p = 0.0033), which further validates the concept of tumor progression over time. However, the evidence is still equivocal as to whether p53 expression alone drives the process of tumor progression; other possible factors are under investigation.
The p53 expression groups were significantly associated with both RFS and OS. Moreover, utilizing this grouping for stratification, three distinct groups were identified: favorable (0%), intermediate (1%-75%) and poor (>75%) prognosis. As both PPB types and p53 expression groups were significantly associated with survival, the c-index was used to assess if PPB type or p53 expression better stratifies patients’ outcome. The c-index for both RFS was comparable between p53 expression and PPB type. Based on the latter finding, at least in this cohort of cases there is not enough evidence to suggest which model is better for prognostic stratification. Moreover, when stratified by type, the c-index for RFS suggests p53 still has prognostic value, in particular among type III PPB patients.
The expression of p53 by immunohistochemistry has been shown to be a strong surrogate marker for TP53 missense mutations(9,10,18). TP53 is a well-established tumor suppressor gene and its mutations, particularly missense mutations, have been implicated in multiple neoplasms(19–21). Of note, nonsense or frameshift pathogenic variants or loss of TP53 would not be predicted to result in protein misfolding and resistance to degradation leading to p53 overexpression, and in contrast leads to an absence of p53 expression(18). One of the first descriptions of TP53 mutations in PPB was reported by Kusafuka et al(22). In their report of three PPB cases, two showed TP53 mutation; one was a Val - Leu substitution at codon 173, and the second case was an ArgArg to TrpCys substitution at codon 282 and 283. Both children whose tumors had TP53 mutations in their study died of their disease and the case without evidence of TP53 mutations did not have evidence of disease progression(22). A subsequent study by Vargas et al. analyzed two PPB cases, both type II, and in both cases expression of p53 was noted in the epithelium and the stroma but no mutations in TP53 were identified(23).
One of the most comprehensive TP53 sequence studies in PPB was performed by Pugh et al(8). A total of 15 paired tumor and non-tumor samples underwent whole exome sequencing. In all cases a somatic DICER1 mutation was detected; in 12 cases a germline loss of function DICER1 pathogenic variant was identified. Additionally, 13 cases had loss of TP53 and 7 cases a TP53 mutation including a frameshift mutation in 3 and a missense mutation in 4 cases. More importantly, this study identified TP53 mutation or loss as an early phenomenon in PPB with alleles frequencies only slightly lower than the DICER1 RNase IIIb mutations. In this study, clinical correlation with TP53 status was not available. In our experience with 16 PPB cases, all had a mutation in TP53; the most common was a missense mutation (12 cases). The other mutations included a nonsense and a frameshift mutation (1 case each), and in two cases the mutation was unknown. Most of the missense mutations were considered to be deleterious (58%) and the remainder were deemed to represent SNP. One limitation of our study is that only type II and III PPB cases underwent TP53 sequencing; sequencing was not available in Types I and Ir PPB. Further studies are needed to evaluate those cases and correlate the genetic TP53 information with the pattern expression given that some of these cases showed no expression of p53.
In conclusion, the expression of p53 in PPB is significantly associated with PPB type, suggesting that p53 has an important role in PPB progression. This hypothesis is further supported by the expression of p53 in the epithelial lining of the cystic component of PPB types with expression in 15%, 45.5% and 75.6% of types Ir, I and II, respectively. Further studies are needed to validate this hypothesis. More importantly, p53 expression groups are significantly associated with both RFS and OS with higher expression associated with worse outcome. In addition, in this cohort of cases there is no significant difference in the stratification of PPB cases when comparing the traditional method of PPB pathologic types to the p53 expression groups. Based on these results the authors suggest evaluating p53 expression in PPB cases with special emphasis in PPB types II and III to further aid in prognostic stratification.
Acknowledgments
This work is supported by National Institutes of Health grants NCI R01CA143167 (DAH, KS) and NCI R37CA244940 (KS, DAH), and the Parson’s Foundation (DAH, YM). The International Pleuropulmonary Blastoma (PPB)/DICER1 Registry is supported by Pine Tree Apple Classic Fund, Children’s Minnesota Foundation, Pediatric Cancer Research Foundation and the Randy Shaver Community Cancer Fund.
Footnotes
Disclosure
D. Ashley Hill is the owner of ResourcePath, a private research and clinical laboratory. ResourcePath has no financial interests or products described in or promoted by this manuscript.
REFERENCES
- 1.Manivel JC, Priest JR, Watterson J, Steiner M, Woods WG, Wick MR, et al. Pleuropulmonay Blastoma. The So-Called Pulmonary Blastoma of Childhood. Cancer. 1988;62:1516–26. [DOI] [PubMed] [Google Scholar]
- 2.Dehner LP. Pleuropulmonary Blastoma is THE Pulmonary Blastoma of Childhood. Semin Diagn Pathol. 1994;11:144–51. [PubMed] [Google Scholar]
- 3.Messinger YH, Stewart DR, Priest JR, Williams GM, Harris AK, Schultz KAP, et al. Pleuropulmonary blastoma: A report on 350 central pathology-confirmed pleuropulmonary blastoma cases by the international pleuropulmonary blastoma registry. Cancer. 2015;121:276–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Priest JR, Watterson J, Strong L, Huff V, Woods WG, Byrd RL, et al. Pleuropulmonary blastoma: A marker for familial disease. J Pediatr. 1996;128:220–4. [DOI] [PubMed] [Google Scholar]
- 5.Schultz KAP, Stewart DR, Kamihara J, Bauer AJ, Merideth MA, Stratton P, et al. DICER1 Tumor Predisposition. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. , editors. Seattle (WA); 2014. [Google Scholar]
- 6.Hill DA, Jarzembowski JA, Priest JR, Williams G, Schoettler P, Dehner LP. Type I Pleuropulmonary Blastoma: Pathology and Biology Study of 51 Cases From the International Pleuropulmonary Blastoma Registry. Am J Surg Pathol. 2008;32:282–95. [DOI] [PubMed] [Google Scholar]
- 7.Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science (80- ). 2009;325:965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pugh TJ, Yu W, Yang J, Field AL, Ambrogio L, Carter SL, et al. Exome sequencing of pleuropulmonary blastoma reveals frequent biallelic loss of TP53 and two hits in DICER1 resulting in retention of 5p-derived miRNA hairpin loop sequences. Oncogene. 2014;33:5295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guedes LB, Almutairi F, Haffner MC, Rajoria G, Liu Z, Klimek S, et al. Analytic, Preanalytic, and Clinical Validation of p53 IHC for Detection of TP53 Missense Mutation in Prostate Cancer. Clin Cancer Res. 2017;23:4693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iggo R, Gatter K, Bartek J, Lane D, L HA. Increased expression of mutant forms of p53 oncogene in primary lung cancer. Lancet. 1990;335:675–9. [DOI] [PubMed] [Google Scholar]
- 11.Zuppan CW, Beckwith JB, Luckey DW. Anaplasia in unilateral Wilms’ tumor: A report from the National Wilms’ Tumor Study Pathology Center. Hum Pathol. 1988;19:1199–209. [DOI] [PubMed] [Google Scholar]
- 12.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrell FE Jr, Califf RM, Pryor DB, Lee KL, Rosati RA. Evaluating the Yield of Medical Tests. JAMA. 1982;247:2543–6. [PubMed] [Google Scholar]
- 14.Therneau TM, Watson DA. The Concordance Statistic and the Cox Mode. Tech Rep 85. 2017; [Google Scholar]
- 15.Priest JR, McDermott MB, Bhatia S, Watterson J, Manivel JC, Dehner LP. Pleuropulmonary Blastoma: A Clinicopathologic Study of 50 Cases . Cancer. 1997;80:147–61. [PubMed] [Google Scholar]
- 16.Priest JR, Hill DA, Williams GM, Moertel CL, Messinger Y, Finkelstein MJ, et al. Type I Pleuropulmonary Blastoma: A Report From the International Pleuropulmonary Blastoma Registry. J Clin Oncol. 2006;24:4492–8. [DOI] [PubMed] [Google Scholar]
- 17.Pacinda SJ, Ledet SC, Gondo MM, Langston C, Brown RW, Carmona PA, et al. p53 and MDM2 Immunostaining in Pulmonary Blastomas and Bronchogenic Carcinomas. Hum Pathol. 1996;27:542–6. [DOI] [PubMed] [Google Scholar]
- 18.Murnyak B, Hortobagyi T. Immunohistochemical correlates of TP53 somatic mutations in cancer. Oncotarget. 2016;7:64910–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lane DP. p53, Guardian of the Genome. Nature. 1992;358:15–6. [DOI] [PubMed] [Google Scholar]
- 20.Levine AJ, Perry ME, Chang A, Silver A, Dittmer D, Wu M, et al. The 1993 Walter Hubert Lecture: The role of the p53 tumour-suppressor gene in tumorigenesis. Br J Cancer. 1994;69:409–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, et al. p14(ARF) links the tumour suppressors RB and p53 [5]. Nature. 1998;395:124–5. [DOI] [PubMed] [Google Scholar]
- 22.Kusafuka T, Kuroda S, Inoue M, Ara T, Yoneda A, Oue T, et al. P53 Gene Mutations in Pleuropulmonary Blastoma. Pediatr Hematol Oncol. 2002;19:117–28. [DOI] [PubMed] [Google Scholar]
- 23.Vargas SO, Korpershoek E, Kozakewich HPW, De Kruger RR, Fletcher JA, Perez-Atayde AR. Cytogenetic and p53 profiles in congenital cystic adenomatoid malformation: Insights into its relationship with pleuropulmonary blastoma. Pediatr Dev Pathol. 2006;9:190–5. [DOI] [PubMed] [Google Scholar]