

Abstract
Purpose:
PD-1/PD-L1 inhibitors are approved for multiple tumor types. However, resistance poses substantial clinical challenges.
Patients and Methods:
We conducted a phase I trial of CD40 agonist APX005M (sotigalimab) and CSF1R inhibitor cabiralizumab with or without nivolumab using a 3+3 dose-escalation design (NCT03502330). Patients were enrolled from June 2018 to April 2019. Eligibility included patients with biopsy-proven advanced melanoma, non–small cell lung cancer (NSCLC), or renal cell carcinoma (RCC) who progressed on anti-PD-1/PD-L1. APX005M was dose escalated (0.03, 0.1, or 0.3 mg/kg i.v.) with a fixed dose of cabiralizumab with or without nivolumab every 2 weeks until disease progression or intolerable toxicity.
Results:
Twenty-six patients (12 melanoma, 1 NSCLC, and 13 RCC) were enrolled in six cohorts, 17 on nivolumab-containing regimens. Median duration of follow-up was 21.3 months. The most common treatment-related adverse events were asymptomatic elevations of lactate dehydrogenase (n = 26), creatine kinase (n = 25), aspartate aminotransferase (n = 25), and alanine aminotransferase (n = 19); periorbital edema (n = 17); and fatigue (n = 13). One dose-limiting toxicity (acute respiratory distress syndrome) occurred in cohort 2. The recommended phase 2 dose was APX005M 0.3 mg/kg, cabiralizumab 4 mg/kg, and nivolumab 240 mg every 2 weeks. Median days on treatment were 66 (range, 23–443). Median cycles were 4.5 (range, 2–21). One patient had unconfirmed partial response (4%), 8 stable disease (31%), 16 disease progression (62%), and 1 unevaluable (4%). Pro-inflammatory cytokines were upregulated 4 hours post-infusion. CD40 and MCSF increased after therapy.
Conclusions:
This first in-human study of patients with anti-PD-1/PD-L1–resistant tumors treated with dual macrophage-polarizing therapy, with or without nivolumab demonstrated safety and pharmacodynamic activity. Optimization of the dosing frequency and sequence of this combination is warranted.
Introduction
PD-1/PD-L1 inhibitors are approved for several advanced cancers including melanoma, non–small cell lung cancer (NSCLC), and renal cell carcinoma (RCC; refs. 1-3). Anti-PD-1/PD-L1 can induce durable responses; however, not all patients respond and many develop resistance. New therapeutic strategies are needed to overcome this resistance.
While strategies testing anticancer drug combinations targeting adaptive immunity and T cells are common, there has been less focus on targeting innate immunity (4). Manipulation of innate immunity can enhance adaptive immunity through T-cell recruitment into “cold” tumors (5), induce direct tumoricidal activity (6) or antibody-dependent cellular cytotoxicity (7), and can impact the cytokine/chemokine balance in the tumor microenvironment (TME; ref. 8). Tumor-associated macrophages (TAM) comprise up to 30% of innate immune cells in the TME and are associated with anti-PD-1/PD-L1 resistance (9, 10). TAMs are comprised of negative (M1, pro-inflammatory) or positive (M2, anti-inflammatory) regulators of tumor growth (10). Although this nomenclature is evolving, M2 macrophages secrete immunosuppressive factors in the TME that negatively impact cytotoxic T-cell capabilities (11-14) and promote tumorigenesis, invasion, and metastasis (15, 16). Modulating macrophage function and phenotype is hypothesized to result in both T cell–dependent and independent antitumor effects (4, 17, 18) and could possibly overcome nonresponse or resistance to anti-PD-1 in a subset of patients.
Multiple emerging anticancer drugs inhibit or polarize macrophages toward a pro-inflammatory phenotype (19, 20). Macrophage colony-stimulating factor 1 (CSF1) is a chemotactic molecule that stimulates monocyte tumor infiltration and differentiation into M2 macrophages (11, 21) and interacts with its receptor CSF1R. Antibodies against CSF1R (αCSF1R) target M2 macrophages, where it is preferentially expressed (22).
CD40 is expressed on macrophages and antigen-presenting cells (APC) and interacts with its ligand CD40L on T cells. CD40 agonists (CD40a) stimulate maturation and activation of APCs which enhance tumor-specific antigen presentation, resulting in cytotoxic T-cell recruitment. CD40a also enhance macrophage pro-inflammatory cytokine secretion and tumoricidal activity (18, 23). Our groups have demonstrated that combinations of αCSF1R and CD40a in preclinical models suppress growth of poorly T cell–infiltrated melanomas in a T cell–independent fashion. This combination alters TAM composition and increases type I inflammation and chemokine expression, CD8+ T-cell infiltration, and PD-1 expression (18), providing preclinical rationale for this study. Our groups have generated preclinical data in murine models demonstrating that the addition of anti-PD-1 to CD40a and αCSF1R improves survival in mouse tumor models compared with CD40a and αCSF1R alone (unpublished data).
Although CD40a+αCSF1R combinations in preclinical models show superiority compared with either alone (4, 17, 18, 24), human studies are limited (25). The anti-CSF1R antibody cabiralizumab (Five Prime Therapeutics; humanized IgG4 mAb against CSF1R) was previously studied with nivolumab and the recommended phase 2 dose (RP2D) was determined (NCT02526017). Clinical trials of CD40a like APX005M (sotigalimab; Apexigen, Inc.; humanized IgG1 agonistic mAb that binds CD40; ref. 26) have shown durable responses (27) including in patients with anti-PD-1–resistant melanoma (NCT03123783; refs. 27, 28).
We hypothesized that targeting innate immunity by combining αCSF1R and CD40a with anti-PD-1 will reprogram macrophages and dendritic cells, reverse T-cell suppression, and overcome anti-PD-1/PD-L1 resistance. We report results of the phase I portion of a trial of APX005M plus cabiralizumab with or without nivolumab in patients with advanced melanoma, NSCLC, or RCC whose disease has progressed on anti-PD-1/PD-L1 (NCT03502330).
Patients and Methods
Patients
Eligible patients had biopsy-proven advanced melanoma, NSCLC, or RCC with radiographic and/or clinical progression on anti-PD-1/PD-L1 without intervening therapy. Patients with melanoma were included irrespective of BRAF status. Patients with NSCLC with ALK rearrangement or EGFR-mutated tumors were eligible if previously treated with targeted therapy. Any number of prior therapies were allowed but excluded αCSF1R and/or CD40a. Other eligibility criteria included age ≥18 years, Eastern Cooperative Oncology Group (ECOG) performance status 0–1, life expectancy >6 months, and normal organ function. Other eligibility criteria are in the protocol (Supplementary Materials and Methods). Informed written consent was obtained from each subject. This study was approved by the Yale University Institutional Review Board and conducted in accordance with ethical guidelines as outlined by the U.S. Common Rule and with an assurance filed with and approved by the U.S. Department of Health and Human Services.
Study design
This single-institution phase I trial with 3+3 dose escalation performed at YCC determined the RP2D of APX005M with cabiralizumab and nivolumab. The RP2D of cabiralizumab and nivolumab was previously determined at 4 mg/kg and 240 mg i.v. every 2 weeks, respectively. Because APX005M had never been combined with cabiralizumab in humans, APX005M was escalated from 0.03 to 0.1 to 0.3 mg/kg in combination with cabiralizumab 4 mg/kg in cohorts 1, 3, and 5, to establish safety of this doublet. Nivolumab was sequentially added to the doublet for each escalating dose of APX005M (cohorts 2, 4, and 6). Two sets of cohorts were conducted concurrently: cohorts 2 and 3, followed by 4 and 5 (Fig. 1A).
Figure 1.

A, Study design: Patients in cohorts 1, 3, and 5 were treated with cabiralizumab and APX005M alone, as this doublet had not previously been administered to patients with cancer. Cohorts 2, 4, and 6 involved the addition of nivolumab to the other two drugs. Patients were enrolled concurrently in cohorts 2 and 3 and in cohorts 4 and 5. A standard 3+3 design was used. B–E, Example of tumor from a patient with melanoma treated on cohort 6 with increased CD4+ and CD8+ T-cell density in a right axillary lymph node at week 8 (C) compared with baseline (B). D shows baseline T-cell infiltration of tumor from a patient with RCC treated on cohort 1 in which there was a decrease in CD4+ and CD8+ T-cell density in a right flank mass at week 8 (E).
Each cycle was 14 days. Each cohort included 3 to 6 patients. Patients were evaluated for at least 4 weeks before dose escalation. If none of the first 3 evaluable patients in a dose cohort experienced a dose-limiting toxicity (DLT) in the first two cycles, then the next 3 patients would be treated in the next higher dose cohort. If 1 of 3 patients within a cohort experienced a DLT in the first 28 days, then 3 additional patients would have to be added to that cohort. If a second patient experienced a DLT at that dose level in the first 28 days, then the next cohort would not be initiated, the MTD would be exceeded, and the dose of one or more drugs would be de-escalated; however, this did not occur.
Treatment and assessments
Diphenhydramine, famotidine, ibuprofen, and acetaminophen were administered 30 minutes pre-treatment. Nivolumab, if applicable, was infused first over 30 minutes, followed by cabiralizumab which was administered 30 minutes after the end of the nivolumab infusion and infused over 30 minutes. Approximately 30 minutes later, APX005M was given last and infused over 60 minutes. Treatment was administered every 14 days until disease progression, intolerable toxicity, or consent withdrawal. Radiographic assessments were performed at baseline, every 8 weeks for 4 months, and every 12 weeks thereafter. MRI brain was performed at these timepoints for melanoma and NSCLC, and every 6 months for RCC. Treatment beyond progression was allowed if clinical benefit was derived.
Objectives
Primary objectives were safety, tolerability, and to determine the RP2D of APX005M with this regimen. Secondary objectives were treatment-related adverse event (TRAE) profiles. Exploratory objectives were to characterize the pharmacodynamic profile of the doublet and triplet.
Safety
AEs and serious AEs for all 26 patients were graded per the NCI Common Terminology Criteria for Adverse Events v5.0 from treatment initiation until 100 days after treatment or until a new anticancer therapy began. DLTs were defined as non-transient grade 3–5 AEs that occurred in the first 28 days, not attributable to other causes. Applicable DLT definitions for this study included: any grade ≥3 non-laboratory non-hematologic toxicity with several exceptions including grade 3 fatigue <1 week, grade 3 fever not associated with hemodynamic compromise, grade 3 endocrinopathy well controlled by hormone replacement, and grade 3 infusion reaction that returned to grade 1 in <6 hours. Any grade ≥3 non-hematologic laboratory value was defined as a DLT if: immunosuppression other than corticosteroids was required, the abnormality itself solely led to hospitalization or persisted for >1 week, it resulted in death not clearly due to underlying malignancy or extraneous causes, and any toxicity requiring dose reduction of APX005M or permanent discontinuation of any of the study drugs. For aspartate aminotransferase (AST)/alanine aminotransferase (ALT) >12× upper limit of normal (ULN), DLT was defined as elevations not improving after >7 days or if the value increased by more than 5× ULN over 72 hours. Serum creatine kinase (CK) elevation >15× ULN and ≤20× ULN that lasted for <7 days was not considered a DLT. Certain toxicities, even if already resolved, would make it unacceptable to advance to subsequent cohorts as planned, including but not limited to: grade 4 myocarditis, grade 3 neurotoxicity with the exception of peripheral neuropathy, grade 4 pneumonia, and bone marrow aplasia. Further details, including exceptions, are outlined in the protocol (Supplementary Materials and Methods). If DLTs were observed in 2 or more patients within a cohort, the MTD would have been exceeded and no further patients treated at that dose level.
Immunofluorescence staining
Melanoma and RCC tumor samples were cored and placed on a tissue microarray (TMA), as described previously (29). Biopsy specimens were cored perpendicular to the long axis and placed in the TMA master-block to maximize their use. The patient with NSCLC had no viable tissue. We stained for T-cell subsets (CD4, CD8) and macrophages (CD68) in available pre-treatment and on-treatment tumors collected at week 8 or during a necessary procedure. Staining methods and analysis were previously described (29-32). Details are provided in Supplemental Materials and Methods.
Cytokine/chemokine analysis
We hypothesized that pro-inflammatory cytokines/chemokines would increase with treatment and suggest pharmacodynamic activity of the study drug combinations. Serial plasma samples were collected before and during treatment and analyzed at six timepoints for cytokine/chemokine expression using the 65-plex Human Cytokine/Chemokine Discovery assay (catalog number: HD65, Eve Technologies Corp.): cycle 1 day 1 (C1D1) pre-treatment and 4 hours post-APX005M infusion, C1D2, and at the same timepoints for cycle 2. Twelve collections were missed for logistical reasons;144 samples were available for analysis. Single-plex analysis was conducted for IL34 and MCSF (ligands of CSF1R) and CD40, for a total of 68 analytes. A fluorescence intensity value was obtained for each analyte. Concentrations were calculated with a standard curve to yield an observed concentration (pg/mL).
Statistical methods
We used summary statistics for continuous variables and numbers and percentages for categorical variables. Best overall responses (BOR) using RECIST v1.1 were documented as confirmed if the same response was observed on ≥2 consecutive scans; otherwise the response was unconfirmed. Progression-free survival (PFS) was defined as time from treatment initiation to date of first progression (PD) by RECIST or investigator’s clinical determination.
Data analysis was performed with R v3.4.3 for cytokine/chemokine analyses. Observed concentrations for the 68 cytokines/chemokines were ranked, with the smallest and largest observed concentrations assigned the lowest and highest ranks, respectively. Ranked values were used for analysis of differentially expressed cytokines/chemokines with the Mann–Whitney U test and selected for significance if P < 0.001 to correct for multiple comparisons.
Fold change was applied to the observed concentration and ranked to evaluate change in cytokine/chemokine levels at pre-specified timepoints compared with baseline. We performed exploratory analyses to determine whether change in cytokine chemokine levels at C2D2 compared with baseline were associated with duration of time on treatment in days using 200 days as the cut-off point between short and long duration of therapy. We also assessed whether pre-treatment CD4+ and CD8+ T-cell density or CD68+ macrophage density correlated with time on treatment in days.
Results
We enrolled 26 patients (June 2018–April 2019). Table 1 shows demographic and disease characteristics.
Table 1.
Baseline patient characteristics.
| Characteristics Nivolumab Cabiralizumab APX005M |
Cohort 1 (n = 3) — 4 mg/kg 0.03 mg/kg |
Cohort 2 (n = 6) 240 mg 4 mg/kg 0.03 mg/kg |
Cohort 3 (n = 3) — 4 mg/kg 0.1 mg/kg |
Cohort 4 (n = 5) 240 mg 4 mg/kg 0.1 mg/kg |
Cohort 5 (n = 3) — 4 mg/kg 0.3 mg/kg |
Cohort 6 (n = 6) 240 mg 4 mg/kg 0.3 mg/kg |
Total (n = 26) |
|---|---|---|---|---|---|---|---|
| Age | |||||||
| Median | 61 | 58 | 69 | 58 | 61 | 61 | 61 |
| Range | 59–66 | 40–71 | 69–73 | 40–63 | 60–67 | 44–84 | 40–84 |
| Sex | |||||||
| Male (%) | 3 (100) | 4 (67) | 1 (33) | 5 (100) | 3 (100) | 4 (67) | 20 (77) |
| Female (%) | 0 (0) | 2 (33) | 2 (67) | 0 (0) | 0 (0) | 2 (33) | 6 (23) |
| Cancer type | |||||||
| Melanoma (%) | 0 (0) | 2 (33) | 2 (67) | 3 (60) | 0 (0) | 5 (83) | 12 (46) |
| RCC (%) | 3 (100) | 4 (67) | 1 (33) | 2 (40) | 2 (67) | 1 (17) | 13 (50) |
| NSCLC (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (33) | 0 (0) | 1 (4) |
| ECOG PS | |||||||
| 0 | 3 (100) | 4 (67) | 2 (67) | 5 (100) | 1 (33) | 4 (67) | 19 (73) |
| 1 | 0 (0) | 2 (33) | 1 (33) | 0 (0) | 2 (67) | 2 (33) | 7 (27) |
| All lines of prior treatment | |||||||
| Median | 5 | 3 | 1 | 3 | 3 | 2 | 3 |
| Range | 3–5 | 1–5 | 1–5 | 1–6 | 1–6 | 1–4 | 1–6 |
| Lines of prior immunotherapy-based treatment only | |||||||
| Median | 4 | 2 | 1 | 3 | 3 | 2 | 2 |
| Range | 2–5 | 1–3 | 1–3 | 1–5 | 1–3 | 1–4 | 1–5 |
| Response to prior immune checkpoint inhibitor(s) | |||||||
| Clinical benefit | 3 (100) | 1 (17) | 2 (67) | 3 (60) | 2 (67) | 4 (67) | 15 (58) |
| 1° resistance | 0 (0) | 5 (83) | 1 (33) | 2 (40) | 1 (33) | 2 (33) | 11 (42) |
Abbreviation: ECOG PS, Eastern Cooperative Oncology Group performance status.
Safety and tolerability
All patients had at least one TRAE (Table 2), which most commonly represented laboratory abnormalities anticipated with cabiralizumab: elevations of lactate dehydrogenase (LDH; 100%), AST (96%), CK (96%), and ALT (73%). None of these laboratory abnormalities were associated with clinical symptoms and all resolved once study drugs were discontinued. Most clinical TRAEs were grade 1–2 and most commonly periorbital edema (62%) secondary to cabiralizumab, and fatigue (50%).
Table 2.
TRAEs.
| Grade 1–2 |
Grade 3–5 |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Laboratory abnormalities | Cohort 1 (n = 3) |
Cohort 2 (n = 6) |
Cohort 3 (n = 3) |
Cohort 4 (n = 5) |
Cohort 5 (n = 3) |
Cohort 6 (n = 6) |
Cohort 1 (n = 3) |
Cohort 2 (n = 6) |
Cohort 3 (n = 3) |
Cohort 4 (n = 5) |
Cohort 5 (n = 3) |
Cohort 6 (n = 6) |
Total (%) (n = 26) |
| LDH elevation | 3 | 6 | 3 | 5 | 3 | 6 | — | — | — | — | — | — | 26 (100) |
| AST elevation | 3 | 3 | 2 | 2 | 2 | 2 | — | 2 | 1 | 3 | 1 | 4 | 25 (96) |
| CK elevation | 1 | 3 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 4 | 1 | 4 | 25 (96) |
| ALT elevation | 1 | 3 | 2 | 2 | 3 | 6 | — | — | 1 | 1 | — | — | 19 (73) |
| Alkaline phosphatase elevation | 1 | 1 | — | — | 1 | 1 | — | — | — | — | — | — | 4 (15) |
| Grade 1–2 |
Grade 3–5 |
||||||||||||
| Clinical events | Cohort 1 (n = 3) |
Cohort 2 (n = 6) |
Cohort 3 (n = 3) |
Cohort 4 (n = 5) |
Cohort 5 (n = 3) |
Cohort 6 (n = 6) |
Cohort 1 (n = 3) |
Cohort 2 (n = 6) |
Cohort 3 (n = 3) |
Cohort 4 (n = 5) |
Cohort 5 (n = 3) |
Cohort 6 (n = 6) |
Total (%) (n = 26) |
| Periorbital edemaa | 2 | 4 | 3 | 2 | 2 | 3 | — | — | — | — | — | — | 16 (62) |
| Fatigue | 2 | 2 | 2 | 1 | 2 | 4 | — | — | — | — | — | — | 13 (50) |
| Cytokine release syndromeb | 2 | 1 | — | 1 | 3 | 4 | — | — | — | — | — | — | 11 (42) |
| Rash – maculopapular | — | 3 | 2 | 2 | — | 2 | — | — | — | — | — | — | 9 (35) |
| Pruritis | 1 | — | 2 | 2 | — | 1 | — | — | — | — | — | — | 6 (23) |
| Infusion-related reactionc | — | 1 | — | — | 1 | 2 | — | — | — | — | — | — | 4 (15) |
| Myalgia | 2 | 2 | — | — | 1 | — | — | — | — | — | — | — | 5 (19) |
| Nausea | 1 | — | 1 | — | 1 | 1 | — | — | — | — | — | — | 4 (15) |
| Headache | 1 | — | 1 | 1 | — | 1 | — | — | — | — | — | — | 4 (15) |
| Diarrhea | — | — | 2 | — | 1 | 1 | — | — | — | — | — | — | 4 (15) |
| Vomiting | 1 | — | — | — | 1 | 1 | — | — | — | — | — | — | 3 (12) |
| Anorexia | — | — | — | 1 | 1 | — | — | — | — | — | — | — | 2 (8) |
| Edema – limbs | — | 1 | — | — | — | 1 | — | — | — | — | — | — | 2 (8) |
| Hoarseness | 1 | — | — | — | — | — | — | — | — | — | — | — | 1 (4) |
| Acute respiratory distress syndrome | — | — | — | — | — | — | — | 1 | — | — | — | — | 1 (4) |
| Pain – back | 1 | — | — | — | — | — | — | — | — | — | — | — | 1 (4) |
| Erythema multiforme | — | — | 1 | — | — | — | — | — | — | — | — | — | 1 (4) |
| Dry mouth | — | — | — | 1 | — | — | — | — | — | — | — | — | 1 (4) |
| Sleep apnea | — | — | — | 1 | — | — | — | — | — | — | — | — | 1 (4) |
| Joint pain | — | — | — | — | 1 | — | — | — | — | — | — | — | 1 (4) |
| Uveitis | — | — | — | — | 1 | — | — | — | — | — | — | — | 1 (4) |
| Peripheral sensory neuropathy | — | — | — | — | — | 1 | — | — | — | — | — | — | 1 (4) |
| Clogged ears | — | — | — | — | 1 | — | — | — | — | — | — | — | 1 (4) |
| Paresthesia | — | — | — | — | — | 1 | — | — | — | — | — | — | 1 (4) |
| Muscle weakness | — | — | — | — | — | 1 | — | — | — | — | — | — | 1 (4) |
Note: Patients reporting more than one AE of the same term were counted once at the highest grade reported. AEs were deemed “treatment-related” if they were possibly, probably, or definitely related to one or more of the study drugs.
“Periorbital edema” also includes the term “facial swelling.”
“Cytokine release syndrome” includes the terms “fever,” “chills,” “sweats,” and “night sweats” and the symptoms occurred after drug dosing.
“Infusion-related reaction” includes the terms “chills” and “flushing” and the symptoms occurred during drug dosing.
One asymptomatic grade 4 CK elevation occurred, and asymptomatic grade 3 elevations of CK (n = 14), AST (n = 11), and ALT (n = 2) also occurred. Per protocol, in the absence of myocarditis or myositis, cabiralizumab is held for CK > 15× ULN and other drugs continued, but this never occurred. The only clinically significant grade ≥3 laboratory TRAE was an AST elevation that was non-transient and required steroids.
Grade 1 APX005M infusion-related reactions occurred in 4 patients (15%) with chills or flushing. Grade 1 post-infusion cytokine release syndrome secondary to APX005M consisting of fever, chills, and/or sweats occurred in 11 patients (42%), 7 at the highest APX005M dose. All were managed with anti-pyretics and additional premedications at subsequent cycles.
Immune-related AEs (irAEs) were moderate, low grade, and did not require systemic corticosteroids: rash (n = 9), diarrhea (n = 4), joint pain (n = 1), and uveitis (n = 1). There were no recurrences of grade ≥3 irAEs in patients who had already experienced specific irAEs prior to enrollment on this study.
The only DLT occurred in the first patient in triplet cohort 2. After two cycles, the patient developed grade 4 acute respiratory distress syndrome and came off trial. Cohort 2 was expanded by 3 patients due to the possibility that this DLT was related to any of the study drugs or their combination. There were no additional DLTs. Cohort 5 was expanded to 5 patients to gain additional experience with the doublet at the highest APX005M dose. Cohort 6 was expanded to 6 patients to confirm the RP2D.
The most common reason for drug discontinuation was PD: 20 (92%) radiographic PD, 4 (15%) with clinical deterioration. One patient discontinued therapy due to headaches related to a dural arteriovenous malformation complicated by a stroke. Another discontinued for inability to comply with study procedures. The determined RP2D was cohort 6 dosing: nivolumab 240 mg, cabiralizumab 4 mg/kg, and APX005M 0.3 mg/kg i.v. every 2 weeks.
Antitumor activity
As of August 6, 2020, all 26 patients were off trial. Median days on treatment was 66 (23–443). Median duration of follow-up was 21.3 months. The BORs were 1 unconfirmed partial response (PR; 4%), 8 stable disease (SD; 31%), 16 PD (62%), and 1 (4%) unevaluable. BOR was numerically similar for patients with melanoma and RCC (Table 3). There were no clear differences in response by number of prior therapies. The patient with melanoma with an unconfirmed PR had primary resistance to ipilimumab and nivolumab which was the only prior treatment. Median PFS was 72 days [95% confidence interval (CI): 54–182 days]. Waterfall plots demonstrating the percent change in RECIST v1.1 target lesions from baseline are provided as Supplementary Fig. S1A-S1C.
Table 3.
BOR.
| Disease type | ||||
|---|---|---|---|---|
| Best overall response |
Melanoma (n = 12) |
NSCLC (n = 1) |
RCC (n = 13) |
TOTAL (n = 26) |
| Partial response | ||||
| Confirmed | 0 | 0 | 0 | 0 |
| Unconfirmed | 1 | 0 | 0 | 1 |
| Total PR | — | — | — | 1 (4%) |
| Stable disease | ||||
| Confirmed | 1 | 0 | 3 | 4 |
| Unconfirmed | 2 | 1 | 1 | 4 |
| Total SD | — | — | — | 8 (31%) |
| Progressive disease | ||||
| Confirmed | 4 | 0 | 4 | 8 |
| Unconfirmed | 3 | 0 | 5 | 8 |
| Total PD | — | — | — | 16 (62%) |
| Unevaluable | 1 | 0 | 0 | 1 (4%) |
Patients received a median of 4.5 cycles of study drugs (2-21). Median time on treatment was 2.3 months. Eleven (42%) patients received treatment for ≤2 months, 10 (38%) patients received treatment for >2 and up to 6 months, and 5 (19%) patients received treatment for a more prolonged period of >6 months (range, 7–14.8 months). Of the 5 patients who were on treatment the longest, 3 had melanoma and 2 had RCC; 4 of the 5 were treated with triplet therapy (2 in cohort 2, 2 in cohort 6). The 3 patients with melanoma who were on therapy the longest had received only 1–2 prior lines of therapy. The 2 patients with RCC on treatment the longest were heavily pretreated (3 and 5 prior lines of therapy). One of these patients with melanoma metastatic to the brain who progressed on prior dabrafenib+trametinib and ipilimumab+nivolumab had a mixed response (PD by RECIST) and was treated beyond progression, achieving disease control for a total of 10.1 months. The patient with RCC with five prior lines of therapy remained on treatment for 8.9 months. The patient with melanoma with an unconfirmed PR remained on therapy for 14.8 months.
T-cell and macrophage density in pre- and on-treatment tumors
Tumor was available from 10 patients with melanoma (7 pre-treatment, 7 on-treatment, 5 paired) and 9 patients with RCC (9 pre-treatment, 2 on-treatment, 2 paired). Overall, pre-treatment tumors were highly infiltrated with immune cells. Median percent area was 6.45%, 5.22%, and 3.81% for CD4+, CD8+, and CD68+ cells, respectively. No statistically significant changes in immune cell density were detected in on-treatment samples, likely due to small sample size. For example, Fig. 1C shows an increase in tumor T-cell infiltration in a right axillary lymph node melanoma metastasis after 8 weeks on triplet therapy in cohort 6 compared with pre-treatment (Fig. 1B). This lesion measured 6.8 × 3.6 cm2 pre-treatment and mildly decreased to 6 × 3.3 cm2 after four cycles (8 weeks) on the study drugs. Figure 1E shows decreased tumor T-cell infiltration in a right-flank RCC metastasis after 8 weeks on doublet therapy in cohort 1 compared with pre-treatment (Fig. 1D). This lesion measured 10.2 × 8.2 × 9.9 cm3 pre-treatment and slightly increased to 10.9 × 8.9 cm2 after four cycles (8 weeks) on the study drugs.
Higher pre-treatment CD4+ T-cell density correlated with longer time on treatment (P = 0.013), whereas higher baseline CD8+ T-cell and macrophage density did not (P = 0.08; P = 0.42; Supplementary Fig. S2A and S2B).
Treatment-related changes in peripheral blood cytokine/chemokines
Significantly upregulated cytokines/chemokines at any on-treatment timepoint with doublet or triplet compared with baseline (P < 0.001) included pro-inflammatory cytokines which increased 4 and 24 hours after APX005M, like TNFα and CXCL10 (Fig. 2A). These changes returned to baseline prior to cycle 2, consistent with the known pharmacodynamic effects of CD40 agonists.
Figure 2.
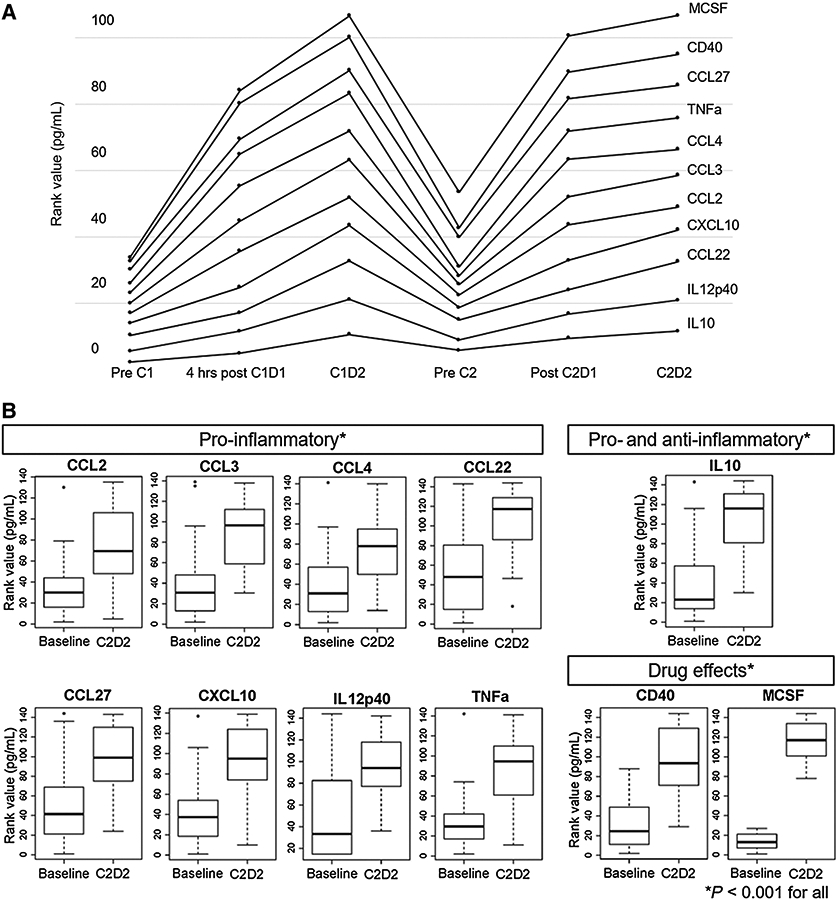
A, Rises in levels of circulating cytokines or chemokines on-treatment compared with pre-treatment samples, showing nine cytokines or chemokines with highly significant differences (P < 0.001). In addition, levels of CD40 and the ligand of the cabiralizumab target, MCSF, were also significantly higher after treatment. B, Nine cytokines/chemokines, CD40, and MCSF, the ligand of cabiralizumab, were significantly increased (P < 0.001) by rank value after two cycles of therapy (samples available from 22/26 patients) compared to pre-treatment levels (samples available from 26/26 patients).
In all patients, there were significant increases in pro-inflammatory cytokines (CCL3, CCL4, IL12p40, CXCL10, and TNFα), pro-inflammatory chemokines (CCL2, CCL22, CCL27), a dual acting cytokine (IL10; ref. 33), CD40, and MCSF, the ligand of cabiralizumab’s target (P < 0.001; Fig. 2B). There was a trend toward increase in IL34 (P = 0.004).
Some cytokines/chemokines were more significantly upregulated at C2D2 in triplet (nivolumab-containing) versus doublet (no nivolumab) cohorts, including IL10, CCL22, TNFα, CD40, and MCSF (P < 0.001), and CXCL10 (P = 0.002), CCL3 (P = 0.002), and CCL27 (P = 0.005; Fig. 3A). CD40 and MCSF increased on-treatment, irrespective of whether patients received nivolumab.
Figure 3.
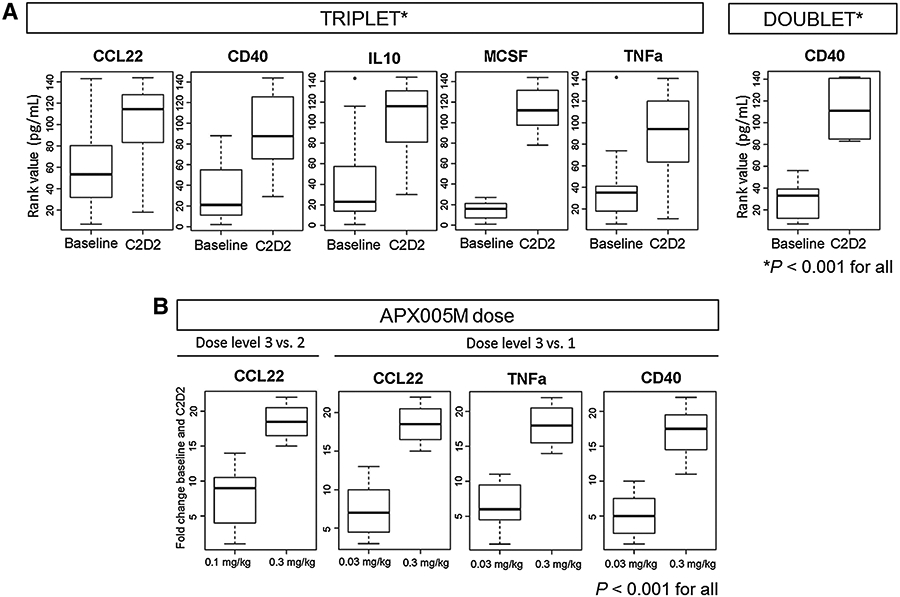
A, More cytokines/chemokines were significantly upregulated by rank value at C2D2 compared with baseline in the triplet versus the doublet cohort. Some cytokines/chemokines were more significantly upregulated at C2D2 in triplet versus doublet cohorts, including IL10, CCL22, TNFα, CD40 and MCSF (P < 0.001), and CXCL10 (P = 0.002), CCL3 (P = 0.002), and CCL27 (P = 0.005). At baseline, samples were available from 9/9 and 17/17 patients on doublet and triplet therapy, respectively. At C2D2, samples were available from 6/9 and 16/17 patients on doublet and triplet therapy, respectively. B, Comparison of the fold change of cytokine/chemokine levels at C2D2 from baseline at different dose levels of APX005M demonstrated that CCL22, TNFα and CD40 were increased in patients at the highest APX005M dose (0.3 mg/kg) compared with the lowest dose (0.03 mg/kg; P < 0.001). CCL22 was the only chemokine significantly higher at the highest APX005M dose compared with the intermediate dose (0.1 mg/kg; P < 0.001).
Comparing fold changes of cytokine/chemokine levels at C2D2 from baseline at different APX005M doses, CCL22, TNFα, and CD40 were increased in patients at the highest APX005M dose (0.3 mg/kg) compared with the lowest dose (0.03 mg/kg; P < 0.001). CCL22 was the only chemokine significantly higher at the highest APX005M dose compared with the intermediate dose (0.1 mg/kg; P < 0.001; Fig. 3B).
In assessing for an association between changes in cytokine/chemokine levels and time on treatment, the only change that correlated with time on treatment was IL3, which was higher at C2D2 for patients who remained on treatment for a prolonged time period (>200 days), P = 0.02.
Associations with clinical characteristics and outcomes
There were no significant differences in changes in cytokine/chemokine levels between patients with melanoma (n = 12) and RCC (n = 13); however, the fold change for MCSF (P = 0.008) tended to be higher in melanoma, whereas CXCL1 (P = 0.006), CD40L (P = 0.004), and VEGFA (P = 0.006) levels tended to rise more in RCC. There was no association between cytokine/chemokine changes and treatment-related fevers. Higher fold changes in IL2 (P = 0.005), TNFα (P = 0.01), MCSF (P = 0.02), and CCL15 (P = 0.03) were associated with a BOR of PR or SD compared with PD.
Discussion
In this first-in-human study of patients with anti-PD-1/PD-L1–resistant advanced melanoma, RCC, or NSCLC treated with dual macrophage-polarizing therapy (CD40a+αCSF1R), with or without nivolumab, treatment with the doublet or triplet was safe. The RP2D for APX005M with this combination was 0.3 mg/kg and is being studied in the phase Ib trial (NCT03502330). Most grade ≥ 3 TRAEs represented asymptomatic, anticipated AST, ALT, and/or CK elevations secondary to cabiralizumab, not associated with liver, cardiac, or muscle toxicity, and gradually resolved with holding cabiralizumab. Serum LDH rose in all patients. Kuppfer cells typically clear these enzymes, but cabiralizumab’s mechanism of depleting macrophage lineage cells results in this pharmacodynamic effect (34). Periorbital edema was a unique grade 1–2 TRAE that gradually resolved when cabiralizumab was discontinued. Fatigue was common (50%).
APX005M did not cause high-grade cytokine release syndrome or infusion-related toxicity but additional premedication was sometimes required. APX005M, in contrast to other CD40a, binds to an epitope with a CD40L binding domain thought to produce more robust CD40a activity than non-CD40L–blocking antibodies (35). Higher incidences of toxicities have been reported with other CD40a. APX005M’s unique construct includes an Fc component that can cluster CD40, leading to less toxicity (26, 36). No synergistic toxicity occurred from the APX005M+cabiralizumab combination compared with the anticipated toxicity from either drug alone. No increase in irAEs occurred compared with what is expected from nivolumab monotherapy.
Most patients discontinued treatment due to PD. The response rate was low and not the primary endpoint of the phase I component; however, several heavily pretreated patients remained on study for a prolonged period and appeared to derive clinical benefit, generally in triplet cohorts, except 1 patient with RCC treated with the doublet.
Our pharmacodynamic studies included blood cytokine/chemokine analyses and measurement of tumor T-cell and macrophage infiltration. Cabiralizumab and APX005M elicited the desired inflammatory effects. CSF1R inhibition resulted in upregulation of its ligands, MCSF and IL34. CD40a increased CD40 within 4 hours. αCSF1R+CD40a increased proinflammatory cytokines/chemokines 4 hours post-APX005M, many involved in general immune cell and/or T-cell recruitment or chemoattractants for T cells and macrophages. These effects had largely reversed prior to cycle 2, consistent with the known transient effects of CD40 agonists on circulating immune cells (25, 37-39). CCL2, CCL3, and CCL4 are key for differentiation, migration, and regulation of monocytes and macrophages. TNFα is produced by macrophages in states of acute inflammation. CXCL10 is important for T-cell recruitment and was elevated 24 hours post-treatment. The CCL22 increase confirms findings in murine tumor models treated with αCSF1R+CD40a (17).
In the few paired pre-treatment and on-treatment biopsies, we were unable to detect clear changes in T-cell infiltration or macrophage density. However, both baseline and on-treatment tumor tissues were fairly well infiltrated with T cells and macrophages. Functional qualities of T cells and macrophages will be performed on samples from phase Ib. Our clinical findings of only one unconfirmed PR are consistent with a recent trial of the αCSF1R emactuzumab combined with CD40a selicrelumab in advanced solid tumors, in which BOR was SD in 40% of patients and there were no clinical responses despite pharmacodynamic activity (25). To date, selicrelumab is the most heavily studied CD40a in early-phase clinical trials with 14% PR rate as monotherapy in patients with advanced solid tumors (39), 27% ORR in combination with tremelimumab in patients with advanced melanoma (40), and PR rates of up to 20% in combination with chemotherapy in patients with various advanced solid tumors (41, 42). The CD40a APX005M, which we use here, has been studied in combination with nivolumab in patients with advanced melanoma with acquired resistance to anti-PD-1. Preliminary results from the first stage of the phase II trial demonstrated PR rate of 20% and SD rate of 20% (28). Despite this early activity documented in anti-PD-1–resistant melanoma, there are no data for its activity in patients with RCC, who comprised 50% of our study population and were heavily pre-treated. The small numbers of patients on this study and the dose-escalation schema in which only 6 of 26 patients were treated with the RP2D makes comparison to outcomes on other CD40a studies difficult, as safety was the primary outcome. We are currently evaluating the activity of this regimen in the phase Ib portion of the trial.
APX005M in combination with cabiralizumab and nivolumab still has strong preclinical rationale. In a phase Ib/II study of APX005M plus nivolumab in anti-PD-1/PD-L1–resistant melanoma, responses were seen in approximately 20%, suggesting promising clinical activity in a difficult-to-treat population (28). Cabiralizumab has been more extensively studied in pancreatic cancer after responses were seen in a phase I trial, but the phase II study of nivolumab and cabiralizumab with or without chemotherapy did not meet its primary PFS endpoint (43). We are currently expanding the melanoma and NSCLC cohorts in the phase Ib trial and considering if dosing or sequencing modifications may better test the regimen’s actual potential (4, 17, 18). Sequential, rather than concurrent dosing of APX005M and cabiralizumab, or less frequent cabiralizumab dosing, might be more beneficial. CD40a activate TAMs rapidly, while αCSF1R transiently inhibits them. Preclinical studies suggest that starting with a CD40a or less frequent αCSF1R dosing allows for recovery and repopulation of TAMs that can be polarized with APX005M (17). With continuous cabiralizumab administration, the combination’s maximum activity may be unrealized if TAMs are unable to repolarize. Future biomarker studies are planned.
In summary, we show that the triplet was reasonably tolerated and a RP2D was established. Our pharmacodynamic studies suggest that this regimen induces a desired pro-inflammatory state. Correlative studies to detect changes in macrophage, T-cell, and/or B-cell function/phenotype are ongoing and may aid in identification of patients more likely to respond. The phase Ib portion has been initiated. Concurrently, additional regimens are being evaluated in preclinical models to determine whether alternative dosing should be considered given the incongruity between murine and human studies of this approach to date.
Supplementary Material
{kind=link}
{kind=link}
Translational Relevance.
Resistance to immune checkpoint inhibitors (ICI) represents a substantial clinical challenge across multiple tumor types. Mechanisms of resistance to ICI are incompletely understood, making development of effective next-line therapies difficult. There is strong preclinical rationale from our institution and others that dual macrophage-polarizing therapy with a CD40 agonist and CSF1R inhibitor can overcome resistance to anti-PD-1/PD-L1 in animal models. Here we present the first in-human study of patients with anti-PD-1/PD-L1–resistant tumors treated with APX005M and cabiralizumab, with or without nivolumab. The combination demonstrated safety and evidence of pharmacodynamic activity, with upregulation of pro-inflammatory cytokines and chemokines. Despite low clinical response rates, optimization of the dosing frequency and sequence of this combination is warranted to further assess its clinical potential in a difficult-to-treat patient population whose therapeutic options are limited.
Acknowledgments
This research was supported in part by the Yale Cancer Center, Bristol Myers Squibb, Apexigen, the Yale Calabresi Immuno-oncology Training Program (K12CA215110; S.A. Weiss), a Career Enhancement Program Grant from the Yale SPORE in Lung Cancer (P50 CA196530; S.A. Weiss), the Yale SPORE in Skin Cancer (P50 CA121974; M. Bosenberg and H.M. Kluger), NIH NRSA F30 GR10226 (I. Krykbaeva), a post-doctoral fellowship from the Wenner-Gren Foundations (D. Djureinovic), and R01-CA227473 and R01-CA216846 (H.M. Kluger).
Footnotes
Authors’ Disclosures
S.A. Weiss reports grants from NIH/NCI Yale Calabresi Immuno-oncology Training Program and Yale SPORE in Lung Cancer Career Enhancement Program, as well as other support from Bristol Myers Squibb and Apexigen during the conduct of the study; S.A. Weiss also reports personal fees from Array BioPharma and MagellanRx outside the submitted work. D. Djureinovic reports grants from Wenner-Gren Foundations during the conduct of the study. I. Krykbaeva reports non-financial support from Apexigen and Bristol Myers Squibb during the conduct of the study. O. Trifan reports other support from Apexigen and BMS during the conduct of the study. M. Bosenberg reports other support from Apexigen and Bristol Myers Squibb during the conduct of the study, as well as personal fees from Eli Lilly and Company outside the submitted work. S.M. Kaech reports personal fees from EvolveImmune and GigaGen outside the submitted work. S. Gettinger reports other support from Apexigen and Bristol Myers Squibb during the conduct of the study. M. Sznol reports other support from BMS and Apexigen during the conduct of the study. M. Sznol also reports other support from Adaptive Biotechnologies, Amphivena, Intensity, Actym, Johnson and Johnson, and GSK; personal fees and other support from Evolveimmune, Nextcure, Repertoire, and Oncohost; and personal fees from Asher, Jazz Pharma, Targovax, Gilead, Sapience, iTEOS, Incyte, Pfizer, Tessa, Oncosec, Biontech, Trillium, STcube, Regeneron, Simcha, Numab, AstraZeneca, Molecular Partners, Idera, Apexigen, Alligator, Verastem, Agenus, Rubius, BMS, Genentech, Boston Pharma, Servier, Adaptimmune, Immunocore, Dragonfly, Pierre-Fabre, Boehringer Ingelheim, Innate Pharma, Nektar, Pieris, AbbVie, Seattle Genetics, Genocea, and Lilly outside the submitted work. M. Hurwitz reports advisory board membership with Bristol Myers Squibb, CRISPR Therapeutics, Exelixis, Nektar Therapeutics, and Janssen; research support from Achilles Therapeutics, Apexigen, Arrowhead, Astellas, AstraZeneca, Bayer, Bristol Myers Squibb, CRISPR Therapeutics, Corvus, Eli Lilly, Endocyte, Genentech, Genmab, GSK, Innocrin, Iovance, KSQ, Merck, Nektar Therapeutics, Novartis, Pfizer, Progenics, Sanofi Aventis, Seattle Genetics, Tmunity, Torque, and Unum; and other support from Gamida Cell and Arvinas. H.M. Kluger reports grants from Bristol Myers Squibb and Apexigen during the conduct of the study, as well as grants from Apexigen and Bristol Myers Squibb outside the submitted work; H.M. Kluger also reports consulting fees from Nektar, Iovance, Immunocore, Celldex, Array Biopharma, Merck, Elevate Bio, Instil Bio, Bristol Myers Squibb, Clinigen, Shionogi, Chemocentryx, Calithera, and Signatero. No disclosures were reported by the other authors.
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017;377:1345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med 2018;378:1277–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med 2016;375:1823–33. [DOI] [PubMed] [Google Scholar]
- 4.Wiehagen KR, Girgis NM, Yamada DH, Smith AA, Chan SR, Grewal IS, et al. Combination of CD40 agonism and CSF-1R blockade reconditions tumor-associated macrophages and drives potent antitumor immunity. Cancer Immunol Res 2017;5:1109–21. [DOI] [PubMed] [Google Scholar]
- 5.Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013;24:589–602. [DOI] [PubMed] [Google Scholar]
- 6.Singh M, Khong H, Dai Z, Huang XF, Wargo JA, Cooper ZA, et al. Effective innate and adaptive antimelanoma immunity through localized TLR7/8 activation. J Immunol 2014;193:4722–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grugan KD, McCabe FL, Kinder M, Greenplate AR, Harman BC, Ekert JE, et al. Tumor-associated macrophages promote invasion while retaining Fc-dependent anti-tumor function. J Immunol 2012;189:5457–66. [DOI] [PubMed] [Google Scholar]
- 8.Solinas G, Schiarea S, Liguori M, Fabbri M, Pesce S, Zammataro L, et al. Tumor-conditioned macrophages secrete migration-stimulating factor: a new marker for M2-polarization, influencing tumor cell motility. J Immunol 2010;185:642–52. [DOI] [PubMed] [Google Scholar]
- 9.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 2014;74:5057–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hussein MR. Tumour-associated macrophages and melanoma tumourigenesis: integrating the complexity. Int J Exp Pathol 2006;87:163–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Komohara Y, Jinushi M, Takeya M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci 2014;105:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen P, Huang Y, Bong R, Ding Y, Song N, Wang X, et al. Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin Cancer Res 2011;17:7230–9. [DOI] [PubMed] [Google Scholar]
- 13.Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014;25:846–59. [DOI] [PubMed] [Google Scholar]
- 14.Hao NB, Lu MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol 2012;2012:948098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jensen TO, Schmidt H, Moller HJ, Hoyer M, Maniecki MB, Sjoegren P, et al. Macrophage markers in serum and tumor have prognostic impact in American Joint Committee on Cancer stage I/II melanoma. J Clin Oncol 2009;27:3330–7. [DOI] [PubMed] [Google Scholar]
- 16.Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory microenvironment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol 2008;66:1–9. [DOI] [PubMed] [Google Scholar]
- 17.Hoves S, Ooi CH, Wolter C, Sade H, Bissinger S, Schmittnaegel M, et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J Exp Med 2018;215:859–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perry CJ, Munoz-Rojas AR, Meeth KM, Kellman LN, Amezquita RA, Thakral D, et al. Myeloid-targeted immunotherapies act in synergy to induce inflammation and antitumor immunity. J Exp Med 2018;215:877–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaib M, Chauhan SC, Makowski L. Friend or foe? Recent strategies to target myeloid cells in cancer. Front Cell Dev Biol 2020;8:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez-Yrigoyen M, Cassetta L, Pollard JW. Macrophage targeting in cancer. Ann N Y Acad Sci 2020. May 22 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 21.Komohara Y, Fujiwara Y, Ohnishi K, Takeya M. Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy. Adv Drug Deliv Rev 2016;99:180–5. [DOI] [PubMed] [Google Scholar]
- 22.Autio KA, Klebanoff CA, Schaer DA, Kauh JS, Slovin SF, Adamow M, et al. Immunomodulatory activity of a colony-stimulating factor-1 receptor inhibitor in patients with advanced refractory breast or prostate cancer: a phase 1 study. Clin Cancer Res 2020;26:5609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majety M, Runza V, Lehmann C, Hoves S, Ries CH. A drug development perspective on targeting tumor-associated myeloid cells. FEBS J 2018;285:763–76. [DOI] [PubMed] [Google Scholar]
- 24.Bellovin D, Wondyfraw N, Levin A, DeNardo DG, Masteller E, Brennan T. cmFPA008, an anti-mouse CSF-1R antibody, combines with multiple immunotherapies to reduce tumor growth in nonclinical models. J Immunother Cancer 2015;3:P351. [Google Scholar]
- 25.Machiels JP, Gomez-Roca C, Michot JM, Zamarin D, Mitchell T, Catala G, et al. Phase Ib study of anti-CSF-1R antibody emactuzumab in combination with CD40 agonist selicrelumab in advanced solid tumor patients. J Immunother Cancer 2020;8:e001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Filbert EL, Björck PK, Srivastava MK, Bahjat FR, Yang X. APX005M, a CD40 agonist antibody with unique epitope specificity and Fc receptor binding profile for optimal therapeutic application. Cancer Immunol Immunother 2021;70:1853–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bajor DL, Xu X, Torigian DA, Mick R, Garcia LR, Richman LP, et al. Immune activation and a 9-year ongoing complete remission following CD40 antibody therapy and metastasectomy in a patient with metastatic melanoma. Cancer Immunol Res 2014;2:1051–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kluger H, Weiss SA, Olszanski AJ, Schuchter L, Linette GP, Garland L, et al. Phase Ib/II of CD40 agonistic antibody APX005M in combination with nivolumab (nivo) in subjects with metastatic melanoma (M) or non-small cell lung cancer (NSCLC) [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2019; 2019 Mar 29–Apr 3; Atlanta, GA. Philadelphia (PA): AACR; Cancer Res; 2019;79(13 Suppl):Abstract nr CT089. [Google Scholar]
- 29.Kluger HM, Chiang V, Mahajan A, Zito CR, Sznol M, Tran T, et al. Long-term survival of patients with melanoma with active brain metastases treated with pembrolizumab on a phase II trial. J Clin Oncol 2019;37:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baine MK, Turcu G, Zito CR, Adeniran AJ, Camp RL, Chen L, et al. Characterization of tumor infiltrating lymphocytes in paired primary and metastatic renal cell carcinoma specimens. Oncotarget 2015;6:24990–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kluger H, Zito CR, Barr M, Baine M, Chiang VL, Sznol M, et al. Characterization of PD-L1 expression and associated T cell infiltrates in metastatic melanoma samples from variable anatomic sites. Clin Cancer Res 2015;21:3052–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kluger HM, Zito CR, Turcu G, Baine MK, Zhang H, Adeniran A, et al. PD-L1 studies across tumor types, its differential expression and predictive value in patients treated with immune checkpoint inhibitors. Clin Cancer Res 2017;23:4270–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Perez-Gracia JL, et al. Cytokines in clinical cancer immunotherapy. Br J Cancer 2019;120:6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Radi ZA, Koza-Taylor PH, Bell RR, Obert LA, Runnels HA, Beebe JS, et al. Increased serum enzyme levels associated with kupffer cell reduction with no signs of hepatic or skeletal muscle injury. Am J Pathol 2011;179:240–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barr TA, Heath AW. Functional activity of CD40 antibodies correlates to the position of binding relative to CD154. Immunology 2001;102:39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bjorck P, Filbert E, Zhang Y, Yang X, Trifan O The CD40 agonistic monoclonal antibody APX005M has potent immune stimulatory capabilities. J Immunother Cancer 2015;3:P198. [Google Scholar]
- 37.O’Hara MH, O’Reilly EM, Varadhachary G, Wolff RA, Wainberg ZA, Ko AH, et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: an open-label, multicentre, phase 1b study. Lancet Oncol 2021;22:118–31. [DOI] [PubMed] [Google Scholar]
- 38.Li DK, Wang W. Characteristics and clinical trial results of agonistic anti-CD40 antibodies in the treatment of malignancies. Oncol Lett 2020;20:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol 2007;25:876–83. [DOI] [PubMed] [Google Scholar]
- 40.Bajor DL, Mick R, Riese MJ, Huang AC, Sullivan B, Richman LP, et al. Long-term outcomes of a phase I study of agonist CD40 antibody and CTLA-4 blockade in patients with metastatic melanoma. Oncoimmunology 2018;7:e1468956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vonderheide RH, Burg JM, Mick R, Trosko JA, Li D, Shaik MN, et al. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology 2013;2:e23033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res 2013;19:6286–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Five Prime Therapeutics. Five Prime Therapeutics provides update on phase 2 trial of cabiralizumab combined with Opdivo in pancreatic cancer [news release]. San Francisco, CA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.