

Abstract
Objectives:
To review the current state of knowledge about the influence of specific genetic mutations that cause sensorineural hearing loss (SNHL) on cochlear implant (CI) functional outcomes, and how this knowledge may be integrated into clinical practice. A multistep and sequential population-based genetic algorithm suitable for the identification of congenital SNHL mutations before CI placement is also examined.
Data Sources, Study Selection:
A review was performed of the English literature from 2000 to 2019 using PubMed regarding the influence of specific mutations on CI outcomes and the use of next-generation sequencing for genetic screening of CI patients.
Conclusion:
CI is an effective habilitation option for patients with severe-profound congenital SNHL. However, it is well known that CI outcomes show substantial inter-patient variation. Recent advances in genetic studies have improved our understanding of genotype–phenotype relationships for many of the mutations underlying congenital SNHL, and have explored how these relationships may account for some of the variance seen in CI performance outcomes. A sequential genetic screening strategy utilizing next-generation sequencing-based population-specific gene panels may allow for more efficient mutation identification before CI placement. Understanding the relationships between specific mutations and CI outcomes along with integrating routine comprehensive genetic testing into pre-CI evaluations will allow for more effective patient counseling and open the door for the development of mutation-specific treatment strategies.
Keywords: CI, Cochlear implant, Genetic screening, Genetic testing, Genetics, Next-generation sequencing, Sensorineural hearing loss, SNHL
Congenital hearing loss (HL) is the most common sensory disorder worldwide. Significant hearing loss is present in at least 1-in-500 infants at birth, and its prevalence continues to increase through adolescence and teenage years. The majority of congenital sensorineural HL (SNHL) cases are genetic in origin, occurring either as isolated nonsyndromic HL or as part of a larger genetic syndrome (1).
Cochlear implants (CI) are a widely accepted treatment option for patients with significant SNHL who no longer receive benefit from traditional hearing aids. CI recipients typically have improved access to sound and, in many cases, open-set word understanding. Despite the success that a majority of patients experience with cochlear implants, there remains a wide variability in outcomes, specifically, speech understanding. Several patient-specific factors, such as duration of deafness and age at SNHL onset, have been associated with CI outcomes; however, these factors alone cannot successfully nor definitively predict postoperative outcomes. Patients who are considering CI must rely on preoperative evaluation and counseling to conceptualize postoperative performance with the CI.
Genetic factors have been implicated as a possible cause of variance in CI performance. Over 200 SNHL genes have been identified, involving cellular structures and mechanisms at all sites along the auditory pathway. Recent studies demonstrated differences in CI outcomes for patients with specific genetic HL disorders, raising the possibility of improved, patient-specific preoperative counseling and postoperative programming based on genotype–phenotype relationships and how these phenotypes in turn interact with CI. However, standard of care genetic screening for these patients only targets a few of the most common SNHL genes, with most patients having an “unknown” mutation at the time of CI placement. This both constrains current preoperative counseling and makes it difficult to design and execute the studies necessary to bring genetic-based precision medicine for CI into widespread clinical practice. We descriptively review the English language literature from the years 2000–2019 pertaining to the genetics of congenital SNHL as it relates to CI performance, and to the use of next-generation sequencing for genetic screening of CI patients. We also assess the current and future role of genetics in the clinical practice of cochlear implantation, and present our institution’s novel clinical genetic screening algorithm for congenital SNHL patients before CI placement.
CI PREOPERATIVE COUNSELING AND PERFORMANCE OUTCOMES
Current Practices
Preoperative cochlear implant counseling is necessarily limited as there are only a few factors that have been convincingly correlated to postoperative performance. These factors include duration of deafness, patient age, patient anatomy, and previous hearing aid use. It has been well documented that longer durations of deafness negatively impacts postoperative speech understanding, most likely due to extended auditory deprivation (2). Conversely, success with a CI is more likely if hearing loss was treated through hearing aids. Additionally, age at implantation has also been associated with worse outcomes, with older recipients performing poorly on a variety of speech understanding tasks (2,3). Etiologies of deafness associated with anatomical malformations or dysfunction of more central areas of the auditory pathway also show more variable outcomes (4,5). Despite the documented impact of these known variables on CI outcomes, we are still limited in our ability to reliably predict postoperative performance, making preoperative counseling a challenging task. Two recipients with similar hearing profiles can achieve very different postimplantation outcomes, suggesting that there are other unknown factors influencing hearing results, which are not being reflected in preoperative evaluations. One possible cause of performance variability in patients with a genetic etiology of deafness may be the specific causative mutation. As we will discuss below, there is a growing body of literature showing that CI outcomes vary between different deafness-causing mutations (2). However, the use of gene panels or other comprehensive genetic testing approaches is not standard of care in most CI programs, meaning many if not most nonsyndromic genetic hearing loss patients are implanted without their underlying mutation having been identified. As such, genetic data is not currently incorporated into the preoperative counseling process for these patients.
GENETICS OF CONGENITAL SNHL AND CI
Anatomy of Congenital SNHL
SNHL is defined as hearing loss resulting from dysfunction of structures of the inner ear and/or vestibulocochlear nerve (1). However, to understand how the phenotypes of specific mutations lead to congenital SNHL and how they intersect with CI physiology, it is useful to further subdivide this section of the peripheral auditory system into three conceptual groups: the sensory partition, the synaptic partition, and the neural partition (6,7). The sensory partition consists of the mechanoelectrical transduction mechanism of the inner ear: the organ of Corti and its outer (OHC) and inner (IHC) hair cells (6,7). The synaptic partition consists of the bases of the IHC, the synaptic interface between IHC and spiral ganglion neuron (SGN), and the terminal dendrites of the SGN (6,7). Finally, the neural partition consists of the somata of the SGN, and their axons which come together to form the auditory component of the vestibulocochlear nerve and then branch distally to synapse with the cochlear nucleus of the midbrain (6,7).
CI functions by electrically stimulating the tonotopically organized SGN somata via electrode array (8). They only interact with the neural partition—bypassing the sensory and synaptic partitions—and require an intact cochlear nerve. Thus, conditions such as cochlear nerve aplasia or surgical removal of the nerve act as definitive contraindications to CI placement (9). As such, it has been hypothesized that the locations of congenital SNHL-causing mutations play a large role in determining post-CI functional outcomes, with patients with mutations expressed in the sensory and synaptic partitions predicted to have more favorable outcomes than those with neural partition mutations (6–8).
Sensory Partition Mutations
While over 200 genes involved with SNHL have been identified, only a small subset have well-delineated function and established genotype–phenotype relationships, and only a smaller subset have been examined in relation to CI outcomes (Table 1). These genes can be divided into those expressed in the sensory partition, those expressed in the synaptic partition, and those expressed in the neural partition. The most studied gene—both in the sensory partition and overall—has been GJB2 (10). GJB2 codes for connexin 26, a gap junction channel protein expressed in the cochlea. GJB2 mutations are thought to induce hearing loss via hair cell dysfunction resulting from perturbation of electrolyte transport (11–13). GJB2 mutations are the most common cause of congenital SNHL, representing 15 to 20% of cases overall, and 50% of nonsyndromic autosomal-recessive SNHL in Caucasian populations (14,15). A number of studies have demonstrated that CI patients with GJB2 mutations perform similarly or superiorly to patients with either unknown mutations or other etiologies (12–14,16–19). This has been affirmed by two systematic reviews, one of which found no difference in function compared with unknown causes of deafness (p = 0.15) and improved function compared with environmental etiologies (p = 0.03) (10,11).
TABLE 1.
Causative mutation versus CI outcome
| Gene | Partition | Function | Successful CI Outcome |
|---|---|---|---|
| GJB2 | Sensory | Connexin 26 gap junction | Yes (6–10,12,13,16) |
| SLC26A4 | Sensory | Anion transporter in stria vascularis | Yes (6,13,14,16,26) |
| MYO7A | Sensory | Myosin in hair bundle organization | Yes (17) |
| CDH23 | Sensory | Cell adhesion protein in tip linkage | Yes (6,17,22) |
| LOXHD1 | Sensory | Stereocilia cell membrane protein | Yes (23) |
| ACTG1 | Sensory | Primary actin isoform in hair cells | Yes (6,24) |
| OTOF | Synaptic | Otoferlin in synaptic vesicle release | Variable (2,6,16,33–36) |
| COCH | Synaptic | Extracellular matrix protein | Yes (42,44) |
| ROR1 | Synaptic | Tyrosine kinase-like orphan receptor | Yes (2,39) |
| OPA1 | Synaptic | GTPase in mitochondria | Yes (40,43) |
| TMPRSS3 | Neural | Type II transmembrane serine protease 3 | Variable (3,5,6,25,47–50) |
| TIMM8A | Neural | Mitochondrial translocase | No (2,51,52,55) |
| PCDH15 | Neural | Cell adhesion Protein | No (55) |
| DFNB59 | Neural | Pejvakin, protein of unknown function | No (55) |
CI outcomes have also been examined for a variety of other congenital SNHL genes expressed within the sensory partition. These include SLC26A4 (encodes an anion transporter expressed in the stria vascularis), MYO7A (encodes a myosin involved in hair bundle organization at the apex of IHC), CDH23 (encodes a cell adhesion protein involved in IHC tip linkage) LOXHD1 (encodes a protein expressed in hair cell stereocilia cell membrane), and ACTG1 (encodes a primary actin isoform in auditory hair cells) (17,20–31). Successful CI functional outcomes have been obtained in patients with all of these mutations, as would be expected with the CI bypassing the sensory partition (6,8,17,20,22,23,27–32).
Synaptic Mutations
A number of congenital SNHL-causing genes have been localized to the synaptic partition, though CI outcomes have only been investigated in a small subset. Of these, the best studied is OTOF, which codes for otoferlin, a protein involved in vesicle release at the synapse between inner hair cell and spiral ganglion neuron (33). OTOF mutations are implicated in the nonsyndromic congenital SNHL disorder DFNB9, and also are a common underlying cause of auditory neuropathy spectrum disorder (ANSD) (34). ANSD is a hearing loss disorder defined clinically by normal otoacoustic emissions coupled with abnormal auditory brainstem responses, indicating pathology located central to the IHC (6,34,35). The etiology of ANSD is heterogenous, with a variety of different genetic, congenital, and acquired conditions affecting structures spanning from the inner hair cells to the midbrain (6,34,35). CI outcomes in ANSD patients are overall positive though heterogeneous, with one study expectedly finding poorer CI function in ANSD patients with cochlear nerve insufficiency (34,36–38). However, CI outcomes in patients with ANSD secondary to OTOF mutations are consistently good, as expected given the site of lesion (22,39–42). A recent review catalogued all the genes—both synaptic and neural—associated with ANSD, though for most there have been no investigations into CI outcomes, underscoring the need for further research (6).
Other deafness-causing synaptic mutations for which CI functional outcomes have been studied include the genes COCH (encodes a extracellular matrix protein expressed throughout the mesodermal structures of inner ear including in modiolar channels surrounding SGNs and their neurites), ROR1 (encodes receptor tyrosine kinase-like orphan receptor 1 which plays a role in modulating neurite growth), and OPA1 (encodes a GTPase involved in mitochondrial fusion and stability) (43–47). Notably, mutations in all three of these genes effect the SGN themselves—causing atrophy or aberrant growth of the terminal dendrites (45,46,48,49). However, patients with these mutations have demonstrated good results with CI placement (45,46,49–51). It is hypothesized that this is due to CI stimulating the SGN central to the dendrites, thereby bypassing the lesions (45,46,49).
Neural Partition Mutations
The best studied neural partition mutations are those of the gene TMPRSS3. Encoding the type II transmembrane serine protease 3, TMPRSS3 is associated with both DFNB8 and DFNB10 nonsyndromic hearing loss depending on the location of the pathogenic mutation (52,53). Though its function is not well characterized, it is expressed in both SGN and the organ of Corti. CI outcomes in TMPRSS3 patients are inconsistent, with some studies showing poor functional outcomes and decreased auditory nerve function on electrocochleography, while others had good functional outcomes (7,8,31,54–57). Given these variable results and evidence of gene expression throughout the peripheral auditory system, more work is needed to analyze genotype–phenotype relationships for specific TMPRSS3 mutations and how they relate to CI outcomes.
CI outcomes have also been investigated in a small number of patients with TIMM8A (encodes mitochondrial translocase associated with SGN degeneration in the context of deafness-dystonia-optic neuropathy syndrome), PCDH15 (encodes a cell adhesion protein expressed in SGN, OHC, and IHC) or DFNB59 (encodes pejvakin, a protein of unknown function expression in SGN somata) mutations (58–61). CI outcomes were poor for all nine patients, as would be expected with SGN lesions (58,59,62).
GENETIC SCREENING FOR CI PLACEMENT
Current Practices
In current clinical practice, comprehensive genetic testing is not routinely performed for all congenital SNHL patients before CI placement, due in part to several limitations existing in the clinical setting that have curtailed wider spread adoption. Genetic testing—particularly whole exome (WES) or whole genome sequencing—is often expensive, and costs can vary widely between institutions (63). Though the International Pediatric Otolaryngology Group and American College of Medical Genetics and Genomics both support stepwise comprehensive genetic testing for bilateral SNHL, insurance coverage for these services is often limited due to a perceived lack of medical necessity and patients interested in genetic testing may be left with significant out-of-pocket costs (63,64). Additionally, genetic testing may require weeks to months to be completed leading to delays in treatment. This is particularly true if patients require multiple rounds of testing, or if an institution has to send testing to an outside entity. Given the strong correlation between age at implantation and hearing outcomes with CI in children, there is a sense of urgency for both parents and clinicians to implant children as early as possible. While comprehensive genetic testing may allow for more accurate preoperative counseling, parents may choose to pursue CI without obtaining genetic results. In order to overcome these obstacles and move the cost-benefit analysis further in favor of preimplantation genetic testing, more CI outcomes research and more efficient and cost-effective testing algorithms are necessary.
Sequential Screening Strategy to Identify the Genetic Cause of SNHL
Comprehensive genetic testing utilizing next-generation sequencing (NGS) has been shown in a variety of studies to increase mutation diagnosis rate for congenital SNHL compared with single gene testing alone, and can now be considered part of the standard of care for evaluation of these patients (63–67). In NGS, a DNA sequence of interest is first broken into fragments of a desired length dictated by the sequencing application. The fragments are then tagged, amplified, sequenced, and mapped to a reference sequence, allowing variants to be identified. Compared to traditional Sanger sequencing, in which only one sequencing reaction occurs at a time, NGS allows millions of reactions to occur in parallel, hugely increasing efficiency. With regard to CI, NGS has been used effectively to screen CI patients with previously undiagnosed mutations to investigate mutational differences between good and poor CI functional outcomes, helping to clarify genotype–phenotype relationships (7,25,62,68,69). However, almost all of these studies have been retrospective, with patients undergoing genetic testing after they had already received CI. Little has been published on how to best implement comprehensive genetic screening as part of standard pre-CI evaluations.
We have established an ethnic-based sequential screening strategy to identify the genetic cause of SNHL using a combination of direct sequencing, population-specific mutation arrays, and NGS complemented with a hearing-centric database (Fig. 1). The whole coding exon of the GJB2 gene is first screened by Sanger sequencing. This method has been used for genetic research and clinical genetic diagnostics for almost 40 years and remains the gold standard of genetic testing (70). This technique enables the accurate identification of all mutations present in a sequence, at a cost of approximately $5 per sample. However, using Sanger sequencing to screen each disease-associated gene to identify causative variants is time-consuming, labor intensive, and costly. To help overcome these limitations, the University of Miami developed the CapitolBioMiamiOtoArray as described in Yan, et al (71). The array screens for 9 sequence variants in 5 hearing loss genes including c.35delG, p.W44C, p.L90P, c.167delT (GJB2); 309kb deletion (GJB6); p.L236P, p.T416P (SLC26A4); and m.1555A>G, m.7444G>A (mtDNA). These sequences were chosen as the causative variants most commonly found in those with European descent (71). This chip is used for first-pass screening of GJB2-negative cases before NGS screening, and costs approximately $30 per patient. Microarrays, also known as mutation chips, have typically been used in the screening and diagnosis of HL by allowing simultaneous analysis of many genetic mutations. Mutation chips are easily customizable and can be adjusted based on the mutation frequencies in a given population. The advantages of gene-chip technology include lower costs, simplicity, and availability; however, the technology can only be used to detect the known mutations included on the chip.
FIG. 1.
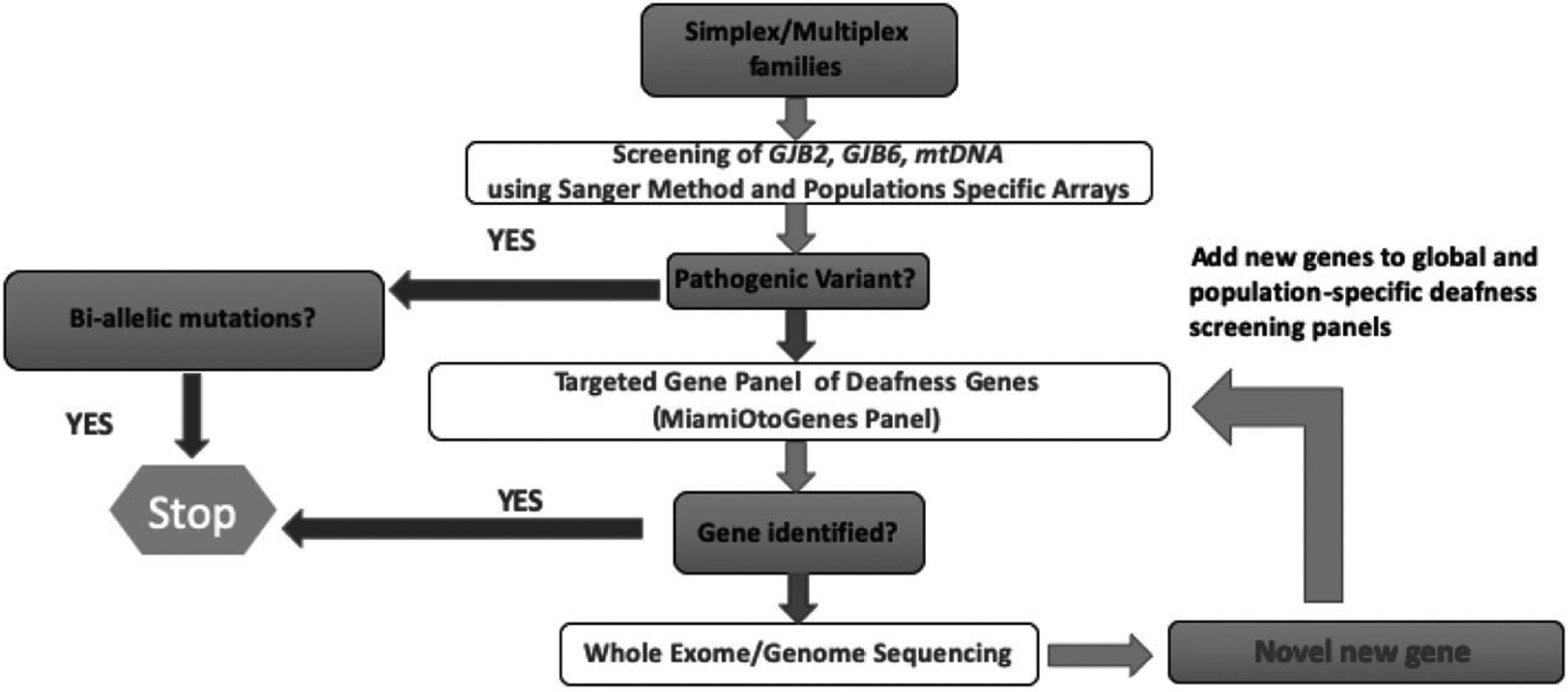
Population-based genetic workflow for mutational analysis for clinical nonsyndromic SNHL profiling. To detect nonsyndromic SNHL mutations, patients are initially screened for population-specific pathogenic variants in GJB2, GJB6, and mtDNA. If biallelic pathogenic variants are not identified, patients are screened with a global or population-specific gene panel. If a causative mutation cannot be determined, then whole exome sequencing (WES) or whole genome sequencing (WGS) is performed, and if a novel gene is discovered it is added to the appropriate panel for future screening. SNHL indicates sensorineural hearing loss.
NGS approaches allow testing a large number of genes simultaneously in a cost-effective manner (72,73). Two options are currently available: targeted gene panels or whole-exome sequencing (WES). In our screening approach, subjects with negative microarray results are further analyzed by a custom targeted capture/NGS gene panel (MiamiOtoGenes) which includes 235 known deafness genes. Using this panel, we have undertaken analysis of a multiethnic cohort of 342 GJB2 mutation-negative deaf probands (74). Overall, causative DNA variants were detected in 25% of multiplex and 7% of simplex families (74). However, the detection rate varied significantly depending on the ethnic group of interest, ranging from 0% to 57% (74). A targeted sequencing approach such as this offers good coverage overall (mean 300X, depending on platforms and number of analyzed samples), and areas of the genes with poor coverage can be supplemented with Sanger sequencing, which can also be used to validate critical areas of the NGS data (73,75,76). The cost varies based on the size of the panel but is generally less than $500, compared to around $1,000 for WES. While targeted sequencing allows for faster, more cost-effective analysis with fewer chance findings, it is not without limitations. Inherently, targeted sequencing only allows for the study of the selected number of genes, meaning all genes of interest must be previous identified and included in the panel. As genetic hearing loss is a rapidly evolving field, new clinically relevant genes are frequently discovered, meaning that panels may need to be expanded and revalidated frequently in order to keep them current with the cutting edge of research (73).
Finally, patients for whom the two panels fail to provide a meaningful result undergo WES to achieve a comprehensive interrogation of the full spectrum of variants, and to detect single-nucleotide variants (SNVs), insertion/deletions (Indels), and copy number variations (CNVs). However, it is important to note that while WES allows for testing of a greater number of genes, in practice obtaining thorough coverage of all coding exons is not feasible, with limiting factors including probes that are not tiled for particular genes, repetitive sequences, or probes with GC-rich sequences (73,77). Overall, approximately 10% of targeted bases sequenced in WES do not get to a read depth of 20, making confident interpretation challenging (73,78). We believe that this type of stepwise, population-specific approach allows for the best balance between high rates of mutation identification and cost and time efficiency. Notably, that all the prices listed here are rough estimates, and that cost and time for each step can vary greatly based on laboratory practices and workflows, labor costs, array designs, and analytical performance parameters.
FUTURE DIRECTIONS
Over the last decade, significant progress has been made in establishing CI outcomes for some of the most common genetic causes of congenital SNHL and mapping the genotype–phenotype relationships that may provide a physiologic explanation for these results. Yet outside this handful of common hearing loss genes, there is a little to no data on CI outcomes, with many mutations not even having a putative mechanism of SNHL. Furthermore, while this study focuses specifically on hearing loss genes, there are a number of other inherited genetic conditions—such as Friedrichs ataxia and other hereditary neurodegenerative diseases—that also cause SNHL, for which patients also receive CI, but about which little is known regarding geno-type-pheno-type relationships, mechanisms of deafness, or CI outcomes (79,80). To fill all of these knowledge gaps, causative mutations must be screened for and identified in a larger percentage of congenital SNHL patients, and these patients must then be seamlessly and routinely entered into research studies. Doing this requires institutions to break down barriers between clinical and research programs and establish multidisciplinary comprehensive care programs that integrate clinical geneticists, otolaryngologists, and audiologists with both basic and translational science programs. At our institution, we have established a Center for Hereditary Deafness, a multidisciplinary collaborative initiative that includes a dedicated hereditary HL clinic, clinical and molecular otogenetics research programs, a robust clinical CI program, and molecular and bioengineering CI research labs. The clinical programs provide data for the gathering of population-based cohort data, gene mapping, and whole exome sequencing, which in turn provides the rationale for new in vitro and in vivo therapeutic gene, cell, and/or drug-based studies, with an ultimate goal of bringing new therapies back to the clinic.
To power this pipeline, we are currently working to more seamlessly incorporate our previously discussed genetic testing algorithm into our clinics. A genetic testing order set is being integrated into the electronic medical record that will allow otolaryngologists to more easily schedule their patients for testing and refer them for a consult by the medical genetics service. Clinical genetic testing is being brought in house to allow for faster turnaround and the use of custom gene chips. New population-based gene chips are being developed to better tailor testing for patients from specific ethnic groups. A new trial will add prospective genetic testing to the preoperative workup of all congenital SNHL patients undergoing CI placement. All of these initiatives share the same goal: achieving a genetic diagnosis in a higher percentage of patients before receiving CI. At present, this will allow for better preoperative counseling, helping providers to give patients and their families more accurate information about expected hearing outcome. Furthermore, it will also identify new targets for basic and translational research and provide the data necessary for larger scale CI outcome studies. In turn, this sets the stage for potential CI clinical advances such as mutation-specific CI programming strategies, electrode choice, or even device design. Outside of CI, achieving higher rates of genetic diagnosis and a better understanding of genotype–phenotype relationships is also a necessary step in the development of alternative therapeutic approaches such as gene therapy and hair cell regeneration, approaches that could be particularly important for patients with mutations not amiable to CI. There is great potential for the application of personalized medicine approaches to CI and to genetic deafness in general, but the first step must be improved genetic screening.
CONCLUSIONS
Genetic testing shows promise in helping to predict functional outcomes for CI recipients. The location of the structural and functional manifestations of specific SNHL-causing mutations within the peripheral auditory system may provide a mechanistic explanation for the variable CI outcomes observed in patients with congenital SNHL. The integration of a comprehensive algorithmic genetic testing approach with a multidisciplinary CI program combining both clinical medicine and biomedical research is necessary for the development and application of patient-specific CI approaches.
Acknowledgments
Dr. Liu’s lab is supported by NIH grants of R01DC005575, R01DC012115, R01DC107624. Drs. E.N. and A.N. are supported by T32 DC015995.
Footnotes
The authors disclose no conflicts of interest.
REFERENCES
- 1.Korver AMH, Smith RJH, Van Camp G, et al. Congenital hearing loss. Nat Rev Dis Primers 2017;3:16094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holden LK, Firszt JB, Reeder RM, et al. Factors affecting outcomes in cochlear implant recipients implanted with a perimodiolar electrode array located in scala tympani. Otol Neurotol 2016;37:1662–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedland DR, Runge-Samuelson C, Baig H, et al. Case-control analysis of cochlear implant performance in elderly patients. Arch Otolaryngolo Head Neck Surg 2010;136:432–8. [DOI] [PubMed] [Google Scholar]
- 4.Farhood Z, Nguyen SA, Miller SC, et al. Cochlear implantation in inner ear malformations: Systematic review of speech perception outcomes and intraoperative findings. Otolaryngol Head Neck Surg 2017;156:783–93. [DOI] [PubMed] [Google Scholar]
- 5.Peng KA, Kuan EC, Hagan S, et al. Cochlear nerve aplasia and hypoplasia: predictors of cochlear implant success. Otolaryngol Head Neck Surg 2017;157:392–400. [DOI] [PubMed] [Google Scholar]
- 6.Shearer AE, Hansen MR. Auditory synaptopathy, auditory neuropathy, and cochlear implantation. Laryngoscope Investig Otolaryngol 2019;4:429–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shearer AE, Eppsteiner RW, Frees K, et al. Genetic variants in the peripheral auditory system significantly affect adult cochlear implant performance. Hear Res 2017;348:138–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eppsteiner RW, Shearer AE, Hildebrand MS, et al. Prediction of cochlear implant performance by genetic mutation: The spiral ganglion hypothesis. Hear Res 2012;292:51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deep NL, Roland JT. Auditory brainstem implantation: Candidacy evaluation, operative technique, and outcomes. Otolaryngol Clin North Am 2020;53:103–13. [DOI] [PubMed] [Google Scholar]
- 10.Nishio S-y, Usami S-i. Outcomes of cochlear implantation for the patients with specific genetic etiologies: A systematic literature review. Acta Otolaryngol 2017;137:730–42. [DOI] [PubMed] [Google Scholar]
- 11.Abdurehim Y, Lehmann A, Zeitouni AG. Predictive value of GJB2 mutation status for hearing outcomes of pediatric cochlear implantation. Otolaryngol Head Neck Surg 2017;157:16–24. [DOI] [PubMed] [Google Scholar]
- 12.Kim SH, Nepali R, Yoo MH, et al. Long term speech perception outcomes of cochlear implantation in gap junction protein beta 2 related hearing loss. J Audiol Otol 2017;21:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Angeli SI, Suarez H, Lopez A, et al. Influence of DFNB1 status on expressive language in deaf children with cochlear implants. Otol Neurotol 2011;32:1437–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green GE, Scott DA, McDonald JM, et al. Performance of cochlear implant recipients with GJB2-related deafness. Am J Med Genet 2002;109:167–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Azaiez H, Chamberlin GP, Fischer SM, et al. GJB2: The spectrum of deafness-causing allele variants and their phenotype. Hum Mutat 2004;24:305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bauer PW, Geers AE, Brenner C, et al. The effect of GJB2 allele variants on performance after cochlear implantation. Laryngoscope 2003;113:2135–40. [DOI] [PubMed] [Google Scholar]
- 17.Wu C-M, Ko H-C, Tsou Y-T, et al. Long-term cochlear implant outcomes in children with GJB2 and SLC26A4 mutations. PLoS One 2015;10:e0138575–138580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Connell SS, Angeli SI, Suarez H, et al. Performance after cochlear implantation in DFNB1 patients. Otolaryngol Head Neck Surg 2007;137:596–602. [DOI] [PubMed] [Google Scholar]
- 19.Vivero RJ, Fan K, Angeli S, et al. Cochlear implantation in common forms of genetic deafness. Int J Pediatr Otorhinolaryngol 2010;74:1107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiong CM, Reyes-Quintos MRT, Yarza TKL, et al. The SLC26A4 c.706C > G (p.Leu236Val) variant is a frequent cause of hearing impairment in filipino cochlear implantees. Otol Neurotol 2018;39:e726–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alper SL, Sharma AK. The SLC26 gene family of anion transporters and channels. Mol Aspects Med 2013;34:494–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu C-C, Liu T-C, Wang S-H, et al. Genetic characteristics in children with cochlear implants and the corresponding auditory performance. Laryngoscope 2011;121:1287–93. [DOI] [PubMed] [Google Scholar]
- 23.Pennings RJE, Damen GWJA, Snik AFM, et al. Audiologic performance and benefit of cochlear implantation in usher syndrome Type I. Laryngoscope 2006;116:717–22. [DOI] [PubMed] [Google Scholar]
- 24.Frejo L, Giegling I, Teggi R, et al. Genetics of vestibular disorders: Pathophysiological insights. J Neurol 2016;263 (suppl 1):S45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyagawa M, Nishio S-Y, Usami S. A comprehensive study on the etiology of patients receiving cochlear implantation with special emphasis on genetic epidemiology. Otol Neurotol 2016;37:e126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim BJ, Kim AR, Lee C, et al. Discovery of CDH23 as a significant contributor to progressive postlingual sensorineural hearing loss in Koreans. PLoS One 2016;11:e0165680–165690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu XZ, Angeli SI, Rajput K, et al. Cochlear implantation in individuals with Usher type 1 syndrome. Int J Pediatr Otorhinolaryngol 2008;72:841–7. [DOI] [PubMed] [Google Scholar]
- 28.Usami S-i, Miyagawa M, Nishio S-y, et al. Patients with CDH23 mutations and the 1555A > G mitochondrial mutation are good candidates for electric acoustic stimulation (EAS). Acta Otolaryngol 2012;132:377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edvardson S, Jalas C, Shaag A, et al. A deleterious mutation in the LOXHD1 gene causes autosomal recessive hearing loss in Ashkenazi Jews. Am J Med Genet A 2011;155:1170–2. [DOI] [PubMed] [Google Scholar]
- 30.Miyagawa M, Nishio S-y, Ichinose A, et al. Mutational spectrum and clinical features of patients with ACTG1 mutations identified by massively parallel DNA sequencing. Ann Otol Rhinol Laryngol 2015;124:84S–93S. [DOI] [PubMed] [Google Scholar]
- 31.Miyagawa M, Nishio S-y, Ikeda T, et al. Massively parallel DNA sequencing successfully identifies new causative mutations in deafness genes in patients with cochlear implantation and EAS. PLoS One 2013;8:e75793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roh KJ, Park S, Jung JS, et al. Hearing preservation during cochlear implantation and electroacoustic stimulation in patients with SLC26A4 mutations. Otol Neurotol 2017;38:1262–7. [DOI] [PubMed] [Google Scholar]
- 33.Roux I, Safieddine S, Nouvian R, et al. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell 2006;127:277–89. [DOI] [PubMed] [Google Scholar]
- 34.Roush P, Frymark T, Venediktov R, et al. Audiologic management of auditory neuropathy spectrum disorder in children: A systematic review of the literature. Am J Audiol 2011;20:159–70. [DOI] [PubMed] [Google Scholar]
- 35.Moser T, Starr A. Auditory neuropathy — neural and synaptic mechanisms. Nature Rev Neurol 2016;12:135–49. [DOI] [PubMed] [Google Scholar]
- 36.Walton J, Gibson WPR Sanli H, et al. Predicting cochlear implant outcomes in children with auditory neuropathy. Otol Neurotol 2008;29:302–9. [DOI] [PubMed] [Google Scholar]
- 37.Berlin CI, Hood LJ, Morlet T, et al. Multi-site diagnosis and management of 260 patients with auditory neuropathy/dys-synchrony (auditory neuropathy spectrum disorder*). Int J Audiol 2010;49:30–43. [DOI] [PubMed] [Google Scholar]
- 38.Shallop JK, Peterson A, Facer GW, et al. Cochlear implants in five cases of auditory neuropathy: Postoperative findings and progress. Laryngoscope 2001;111:555–62. [DOI] [PubMed] [Google Scholar]
- 39.Santarelli R Information from cochlear potentials and genetic mutations helps localize the lesion site in auditory neuropathy. Genome Med 2010;2:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodríguez-Ballesteros M, del Castillo FJ, Martín Y, et al. Auditory neuropathy in patients carrying mutations in the otoferlin gene (OTOF). Hum Mutat 2003;22:451–6. [DOI] [PubMed] [Google Scholar]
- 41.Chen K, Liu M, Wu X, et al. Targeted next generation sequencing reveals OTOF mutations in auditory neuropathy spectrum disorder. Int J Pediatr Otorhinolaryngol 2018;115:19–23. [DOI] [PubMed] [Google Scholar]
- 42.Rouillon I, Marcolla A, Roux I, et al. Results of cochlear implantation in two children with mutations in the OTOF gene. Int J Pediatr Otorhinolaryngol 2006;70:689–96. [DOI] [PubMed] [Google Scholar]
- 43.Mielczarek M, Olszewski J, Pietkiewicz P. Sequencing of exons 4, 5, 12 of COCH gene in patients with postlingual sensorineural hearing loss accompanied by vestibular lesion. Arch Med Sci 2018;14:625–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robertson NG, Lu L, Heller S, et al. Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nature Genet 1998;20:299–303. [DOI] [PubMed] [Google Scholar]
- 45.Diaz-Horta O, Abad C, Sennaroglu L, et al. ROR1 is essential for proper innervation of auditory hair cells and hearing in humans and mice. Proc Natl Acad Sci U S A 2016;113:5993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santarelli R, Rossi R, Scimemi P, et al. OPA1-related auditory neuropathy: Site of lesion and outcome of cochlear implantation. Brain 2015;138:563–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robertson NG, Cremers CWRJ, Huygen PLM, et al. Cochlin immunostaining of inner ear pathologic deposits and proteomic analysis in DFNA9 deafness and vestibular dysfunction. Hum Mol Genet 2006;15:1071–85. [DOI] [PubMed] [Google Scholar]
- 48.Burgess BJ, O’Malley JT, Kamakura T, et al. Histopathology of the human inner ear in the p.L114P COCH mutation (DFNA9). Audiol Neurootol 2016;21:88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vermeire K, Brokx JPL, Wuyts FL, et al. Good speech recognition and quality-of-life scores after cochlear implantation in patients with DFNA9. Otol Neurotol 2006;27:44–9. [DOI] [PubMed] [Google Scholar]
- 50.Huang T, Santarelli R, Starr A. Mutation of OPA1 gene causes deafness by affecting function of auditory nerve terminals. Brain Res 2009;1300:97–104. [DOI] [PubMed] [Google Scholar]
- 51.Parzefall T, Frohne A, Koenighofer M, et al. Identification of a rare COCH mutation by whole-exome sequencing: Implications for personalized therapeutic rehabilitation in an Austrian family with non-syndromic autosomal dominant late-onset hearing loss. Wien Klin Wochenschr 2018;130:299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fasquelle L, Scott HS, Lenoir M, et al. Tmprss3, a transmembrane serine protease deficient in human DFNB8/10 deafness, is critical for cochlear hair cell survival at the onset of hearing. J Biol Chem 2011;286:17383–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guipponi M, Toh M-Y, Tan J, et al. An integrated genetic and functional analysis of the role of type II transmembrane serine proteases (TMPRSSs) in hearing loss. Hum Mutat 2008;29:130–41. [DOI] [PubMed] [Google Scholar]
- 54.Elbracht M, Senderek J, Eggermann T, et al. Autosomal recessive postlingual hearing loss (DFNB8): Compound heterozygosity for two novel TMPRSS3 mutations in German siblings. J Med Genet 2007;44:e81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shearer AE, Tejani VD, Brown CJ, et al. In vivo electrocochleography in hybrid cochlear implant users implicates TMPRSS3 in spiral ganglion function. Sci Rep 2018;8:14165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weegerink NJD, Schraders M, Oostrik J, et al. Genotype-phenotype correlation in DFNB8/10 families with TMPRSS3 mutations. J Assoc Res Otolaryngol 2011;12:753–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miyagawa M, Nishio S-y, Sakurai Y, et al. The patients associated with TMPRSS3 mutations are good candidates for electric acoustic stimulation. Ann Otol Rhinol Laryngol 2015;124:193S–204S. [DOI] [PubMed] [Google Scholar]
- 58.Brookes JT, Kanis AB, Tan LY, et al. Cochlear implantation in deafness-dystonia-optic neuronopathy (DDON) syndrome. Int J Pediatr Otorhinolaryngol 2008;72:121–6. [DOI] [PubMed] [Google Scholar]
- 59.Cif L, Gonzalez V, Garcia-Ptacek S, et al. Progressive dystonia in Mohr-Tranebjaerg syndrome with cochlear implant and deep brain stimulation. Movement Disord 2013;28:737–8. [DOI] [PubMed] [Google Scholar]
- 60.Zallocchi M, Meehan DT, Delimont D, et al. Role for a novel Usher protein complex in hair cell synaptic maturation. PLoS One 2012;7:e30573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Delmaghani S, del Castillo FJ, Michel V, et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nature Genet 2006;38:770–8. [DOI] [PubMed] [Google Scholar]
- 62.Wu C-C, Lin Y-H, Liu T-C, et al. Identifying children with poor cochlear implantation outcomes using massively parallel sequencing. Medicine (Baltimore) 2015;94:e1073–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liming BJ, Carter J, Cheng A, et al. International Pediatric Otolaryngology Group (IPOG) consensus recommendations: Hearing loss in the pediatric patient. Int J Pediatr Otorhinolaryngol 2016;90:251–8. [DOI] [PubMed] [Google Scholar]
- 64.Alford RL, Arnos KS, Fox M, et al. American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet Med 2014;16:347–55. [DOI] [PubMed] [Google Scholar]
- 65.Shearer AE, Smith RJH. Massively parallel sequencing for genetic diagnosis of hearing loss: The new standard of care. Otolaryngol Head Neck Surg 2015;153:175–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boudewyns A, van den Ende J, Sommen M, et al. Role of targeted next generation sequencing in the etiological work-up of congenitally deaf children. Otol Neurotol 2018;39:732–8. [DOI] [PubMed] [Google Scholar]
- 67.Downie L, Halliday J, Burt R, et al. Exome sequencing in infants with congenital hearing impairment: A population-based cohort study. Eur J Hum Genet 2020;28:587–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu W-H, Chang P-Y, Chang S-C, et al. Mutation screening in non-syndromic hearing loss patients with cochlear implantation by massive parallel sequencing in Taiwan. PLoS One 2019; 14: e0211261–211270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park JH, Kim AR, Han JH, et al. Outcome of cochlear implantation in prelingually deafened children according to molecular genetic etiology. Ear Hear 2017;38:e316–24. [DOI] [PubMed] [Google Scholar]
- 70.Gardner P, Oitmaa E, Messner A, et al. Simultaneous multigene mutation detection in patients with sensorineural hearing loss through a novel diagnostic microarray: A new approach for newborn screening follow-up. Pediatrics 2006; 118:985. [DOI] [PubMed] [Google Scholar]
- 71.Yan D, Xiang G, Chai X, et al. Screening of deafness-causing DNA variants that are common in patients of European ancestry using a microarray-based approach. PLoS One 2017;12:e0169219–169220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Williams ES, Hegde M. Implementing genomic medicine in pathology. Adv Anatomic Pathol 2013;20. [DOI] [PubMed] [Google Scholar]
- 73.Di Resta C, Galbiati S, Carrera P, et al. Next-generation sequencing approach for the diagnosis of human diseases: open challenges and new opportunities. EJIFCC 2018;29:4–14. [PMC free article] [PubMed] [Google Scholar]
- 74.Yan D, Tekin D, Bademci G, et al. Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents. Hum Genet 2016;135:953–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aziz N, Zhao Q, Bry L, et al. College of American Pathologists’ Laboratory standards for next-generation sequencing clinical tests. Arch Pathol Laboratory Med 2014;139:481–93. [DOI] [PubMed] [Google Scholar]
- 76.Johnston JJ, Rubinstein WS, Facio FM, et al. Secondary variants in individuals undergoing exome sequencing: Screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am J Hum Genet 2012;91:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yao R, Zhang C, Yu T, et al. Evaluation of three read-depth based CNV detection tools using whole-exome sequencing data. Mol Cytogenet 2017;10:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rehm HL. Disease-targeted sequencing: A cornerstone in the clinic. Nat Rev Genet 2013;14:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barsottini OG, Pedroso JL, Martins CR, et al. Deafness and vestibulopathy in cerebellar diseases: A practical approach. Cerebellum 2019;18:1011–6. [DOI] [PubMed] [Google Scholar]
- 80.Frewin B, Chung M, Donnelly N. Bilateral cochlear implantation in Friedreich’s ataxia: A case study. Cochlear Implants Int 2013;14:287–90. [DOI] [PubMed] [Google Scholar]