

Abstract
Papain like protease (PLpro) is a cysteine protease from the coronaviridae family of viruses. Coronaviruses possess a positive sense, single-strand RNA, leading to the translation of two viral polypeptides containing viral structural, non-structural and accessory proteins. PLpro is responsible for the cleavage of nsp1–3 from the viral polypeptide. PLpro also possesses deubiquitinating and deISGlyating activity, which sequesters the virus from the host's immune system. This indispensable attribute of PLpro makes it a protein of interest as a drug target. The present study aims to analyze the structural influences of ligand binding on PLpro. First, PLpro was screened against the ZINC-in-trials library, from which four lead compounds were identified based on estimated binding affinity and interaction patterns. Next, based on molecular docking results, ZINC000000596945, ZINC000064033452 and VIR251 (control molecule) were subjected to molecular dynamics simulation. The study evaluated global and essential dynamics analyses utilising principal component analyses, dynamic cross-correlation matrix, free energy landscape and time-dependant essential dynamics to predict the structural changes observed in PLpro upon ligand binding in a simulated environment. The MM/PBSA-based binding free energy calculations of the two selected molecules, ZINC000000596945 (−41.23 ± 3.70 kcal/mol) and ZINC000064033452 (−25.10 ± 2.65 kcal/mol), displayed significant values which delineate them as potential inhibitors of PLpro from SARS-CoV-2.
Keywords: Papain like protease, Molecular dynamics simulations, Principal component analysis, Essential dynamics
Abbreviations: PLpro, Papain like protease; MD, Molecular Dynamics; RMSD, Root Mean Square Deviation; RMSF, Root Mean Square Fluctuation; SASA, Solvent Accessible Surface Area; Rg, Radius of Gyration; ED, Essential Dynamics; PCA, Principal Component Analysis; SUD, SARS Unique Domain; NAB, Nucleic acid Binding Domain
Graphical Abstract

1. Introduction
Papain like protease or PLpro is a cysteine protease specific to coronaviruses which comprises one part of the viral protease duo (the other part being the Main protease) responsible for the proteolysis of the viral polypeptide housing a myriad of proteins such as the structural, non-structural and the accessory proteins (Baez-Santos et al., 20152021 May 282022 Apr 202021 Feb, Padhi and Tripathi, 2021, Padhi and Tripathi, 2022, Mishra and Tripathi, 2021). Coronaviruses from the coronaviridae family host a large genomic organisation of 26–32 kb, which hosts a distinctive replication-transcription complex component (Lu et al., 2020, Singh, 2021a). The structural elements of coronaviruses include an enveloped virion with spikes protruding from the surface, resembling a solar corona from where the name coronavirus is derived (Fehr and Perlman, 2015). There are 39 known species of coronaviruses from which seven are known to infect humans, which include the severe acute respiratory syndrome coronavirus (SARS-CoV), middle east respiratory syndrome coronavirus (MERS-CoV) and the recent severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) responsible for the COVID-19 pandemic (Coronaviridae Study Group of the International Committee on Taxonomy of Viruses, 2020, Singh, 2021b). The mentioned coronaviruses have been associated with epidemics leading to loss of life; hence it becomes crucial to study this family of coronaviruses to find novel therapeutic agents against such pathogens (Amin et al., 2021).
Coronaviruses deploy their positive sense, single-stranded RNA into the host cell, where it undergoes immediate translation to release two polyproteins, i.e., polyprotein 1a and 1ab (Chen et al., 2020, Ziebuhr et al., 2000). These viral polyproteins host the non-structural proteins of coronaviruses (nsp1–16) which are responsible for the virulence and survival of the virus; hence they must be proteolytically cleaved to perform their function (Knoops et al., 2008, Maiti, 2020). This proteolysis is performed by two viral proteases, 3CLpro and PLpro. PLpro is a sub-component of the nsp3 protein, a multidomain protein that plays a crucial role in viral replication (Baez-Santos et al., 2014). In addition, Nsp3 hosts Ubl proteins, a domain responsible for synthesising viral subgenomic RNAs, a domain that binds G-quadruplexes, a SARS unique domain (SUD), nucleic acid binding domain (NAB) with a chaperone function, marker domain and predicted transmembrane domains. This multifunctional attribute of nsp3 and its domains make it an attractive drug target for anti-viral therapeutics (Baez-Santos et al., 20152021 May 282022 Apr 202021 Feb, Padhi and Tripathi, 2021, Padhi and Tripathi, 2022, Mishra and Tripathi, 2021).
PLpro is a multidomain protein present between the SUD and NAD domain of nsp3. While the nsp3 is localised to the ER membrane, the PLpro domain is flanked towards the cytosol (Hagemeijer et al., 2010). In the cytosol, the PLpro domain proteolytically cleaves the interdomain sites between nsp1/2, nsp2/3 and nsp3/4, leading to the release of nsp1–3 from the viral polypeptide (Han et al., 2005). Studies have shown that PLpro recognises an LXGG motif in the interdomain sites of the nsp1–4 for cleavage (Rut et al., 2020). The structure of PLpro consists of four domains with a Ubiquitin-like (Ubl) domain at the N terminus followed by a right thumb-fingers-palm domain (Gao et al., 2021, Shan et al., 2021). PLpro is a protease enzyme but also possesses deubiquitinating and deISGlyating activity (Deng et al., 2020). Both Ubl and ISG15 (interferon-induced gene 15) possess an LXGG motif, leading to recognition and subsequent hydrolyzation by PLpro (Lindner et al., 2005, Mahmoudvand and Shokri, 2021). This activity conferred by PLpro to coronaviruses plays a critical role in the host's innate immune response as Ubl and ISG15 are signalling factors for the innate immune system, which upon hydrolyzation by PLpro sequesters the virus from the host's immune system (Ratia et al., 2014, Shin et al., 2020, Liu et al., 2021) ( Fig. 1).
Fig. 1.

The crystal structure of PLpro. (Yellow: Ubl domain, Blue: Thumb domain, Pink: Palm domain, Green: Finger domain) (PDB Id: 6WX4). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The catalytic site of PLpro is present at the interface of the thumb and palm domain, where the catalytic triad, i.e., Cys111-His272-Asp286, performs the proteolysis of the viral polypeptide (Osipiuk et al., 2021). PLpro follows the same general catalytic mechanism as a cysteine protease, where the cysteine residue acts as a nucleophile with histidine as a general acid-base, and aspartic acid aids in stabilising the histidine residue (Klemm et al., 2020). The mechanism followed by PLpro (as proposed by Baez-Santos et al., 2015) begins with the deprotonation of the Cys111 by His273, which leads to a nucleophilic attack by the thiol group of Cys111 on the highly specific Glycine residue present in the substrate. This is followed by various intermediate states of the substrate and active site residue, which are stabilised by an oxyanion hole conferred by Trp106, which has been found to be critical for PLpro activity. The removal of cysteine residue from the intermediate states results in the formation of the N-terminus of the substrate, which is then removed from the active site of the protein.
The literature presented above makes it apparent that PLpro is an indispensable protein for coronaviruses. Hence, it must be studied structurally and dynamically to understand its activity and utilise the information to identify novel therapeutic agents against the protein and, subsequently, the virus. In the present study, we have utilised PLpro from SARS-CoV-2 to computationally investigate its protein dynamics in native and ligand-bound states through molecular docking and molecular dynamics simulation. The native state of PLpro was screened with the ZINC-In Trials library, which consisted of 9270 molecules to find suitable drug-like lead compounds upon which a global and essential dynamics approach was employed to delineate specific idiosyncrasies of the protein in the native protein and the changes observed upon ligand binding.
2. Methods
2.1. Preparation and evaluation of receptor structure
The crystal structure of PLpro from SARS-CoV-2 was downloaded from RCSB-PDB, which was resolved at 1.66 Å through X-Ray crystallography (PDB Id: 6WX4). The preparation of the receptor included the removal of all water molecules and the attached inhibitor VIR251. The resulting structure was then prepared for screening by removing all non-polar hydrogens and assigning the partial charges and atom types to the protein. Finally, this determined structure was applied as a receptor molecule for screening with a small molecule library.
2.2. Virtual screening and molecular docking studies
The virtual screening procedure was performed on the DrugDiscovery@TACC portal (drugdiscovery.tacc.utexas.edu), hosted by the Texas Advanced Computing Center, the University of Texas at Austin, which utilises AutoDock Vina (Trott and Olson, 2010) for calculating the binding affinities. These molecular docking algorithms utilise the defined coordinate values to estimate binding affinity through a force field or defined scoring functions. Compounds from the ZINC-in-trials library, which comprises 9270 molecules under clinical trials and active drug usage, were screened against the receptor molecule (PLpro). The search space for the screening studies was limited to a grid box around the active site, including the catalytic triad of PLpro. Upon completion, the portal provided a set of thousand molecules in descending order of binding affinities. The set of docked compounds was filtered based on estimated binding affinity and interaction patterns, resulting in selection of the top 4 compounds. VIR251 (PRD_002390) is a designed peptide inhibitor against the active site of PLpro and was utilised as a control molecule for the study. The top 4 ligands, along with the control molecule, were prepared for vina docking using MGLTools 1.5.7, followed by docking the ligands with the receptor through Autodock Vina for three runs utilising the same coordinates as the TACC server. These filtered top 4 ligands, along with the control molecule, were utilised for molecular docking by AutoDock 4.2.6 (Morris et al., 2009), restricting the grid box or the search space to the active site of the receptor protein (npts: 60, 60, 60; grid centre: 12.909, −19.095, −36.676). For PLpro-ligand docking studies, the Lamarckian Genetic Algorithm was employed, which was extended for 1000 GA runs while the population size was maintained at 300, the maximum number of energy evaluations was 2,500,000, and the maximum number of generations defaulted at 27,000. Other parameters included mutation and cross-over rates, which were set to 0.02 and 0.8, respectively. The docking clusters for each complex were analysed and the structure exhibiting the lowest binding affinity values (ΔG) and estimated inhibition constants (Ki) was selected for each complex. These complexes were then analysed on BIOVIA discovery studio (BIOVIA, 2019) for interaction patterns such as hydrogen bonds, electrostatic and hydrophobic interactions among others. Based on these parameters the top four compounds were filtered and the top two lead compounds which exhibited the highest binding affinity along with favourable interaction patterns were subjected to molecular dynamics (MD) simulations.
2.3. Molecular dynamics simulations
MD simulations predict the atomic movement of a protein molecule over a period of time under the influence of a force field that governs the physical laws of interatomic interactions based on the principles of classical mechanics; This attribute of MD aids in mimicking the physiological state of a protein molecule to explore the native and protein-ligand complexes to infer the dynamics of a protein molecule computationally. GROMACS 2021 was employed to execute a 100 ns molecular dynamics simulation run of the native protein and protein-ligand complexes under the influence of the selected GROMOS96 54a7 force field (Van Der Spoel et al., 2005). The pdb2gmx command was utilised to generate protein (PLpro) topology, while the ligand topologies were obtained from the PRODRG server (Schuttelkopf and van Aalten, 2004). The unit cell which hosts the native PLpro and PLpro-ligand complexes were defined utilising the gmx_editconf command in a cubic box of 10 Å with periodic boundary conditions (PBC). The solvation of the unit cell follows this to mimic the physiological environment of the protein using the gmx_solvate module with the SPC/E water model. The system was then subjected to neutralisation of charges by employing the gmx_genion module, which replaces required solvent molecules with suitable counter-ions (here Na+ for a positive charge and Cl- for a negative charge) to establish electrostatic neutrality in the system. The changes subjected to the system can introduce unnatural stresses; to avoid this, the system was minimised through the steepest descent algorithm till the maximum force < 100.0 kJ/mol/nm at a maximum of 50,000 steps. Once the system is minimised, the need arises to equilibrate the solvent and ions at the desired temperature (300 K). The equilibration was conducted in two steps; the first NVT ensemble maintains the number of particles, volume and temperature as constant, followed by the second NPT ensemble with the number of particles, pressure and temperature defined as constant. The NVT and NPT simulations were run for 500,000 steps (1 ns), where the temperature was maintained at 300 K and pressure at 1 bar via the utilisation of velocity rescaling (v-rescale) thermostat and the Parrinello-Rahman barostat, respectively. Finally, the production MD was run for 100 ns which is 50 million steps each at 2 fs using the gmx_mdrun module.
2.4. Analysis of simulated trajectories
After the production MD run, the obtained trajectory file was then corrected to remove the PBC utilising the gmx_trjconv module, which provides a new corrected trajectory file that was used for further analysis through the built-in tools present in GROMACS (Van Der Spoel et al., 2005). Parameters such as the root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), and solvent-accessible surface area (SASA) were calculated by gmx_rms, gmx_rmsf, gmx_gyrate, gmx_SASA respectfully. Next, the time-dependant variations in the secondary structure were analysed using the do_dssp module of gromacs. Next, Principal Component Analysis (PCA) using the gmx_covar and gmx_anaeig, which delineate the first two eigenvectors to plot the essential subspace for cluster distribution analysis. The gmx_anaeig module was employed again using the -extr command to calculate the two extreme projections in PDB format along the trajectory using the eigenvalues. These extreme PDB files were utilised to determine the porcupine plot of protein motion by the ModeVectors.py script in PyMOL. The xvg file obtained for the PCA analysis was then used as a template for the gmx_sham module to calculate the free energy landscape (FEL) and analyse the dynamic nature of the native and PLpro-ligand complexes. Finally, dynamics cross-correlation Matrix analysis (DCCM) was performed through the Bio3D R package (Grant et al., 2006), which delineates the protein regions with highly correlated and anti-correlated motions in the native PLpro and its complexes. Bio3D was also employed to better analyse the principal components of native PLpro and PLpro-ligand complexes.
2.5. Binding free energy calculations
Binding free energy calculations were performed for the simulated trajectories of PLpro and its ligand-bound complexes by employing the g_mmbpsa (Kumari et al., 2014) module, which utilises Molecular Mechanics with Poisson-Boltzmann and Surface Area Solvation (MM/PBSA) approach. The binding free energy in this approach is calculated by the summation of changes in Vander Waals energies (ΔE vdw), electrostatic energy (ΔE ele), polar solvation energy (ΔG pol ) and non-polar solvation energy (ΔG np ) upon complexation. The calculations can be represented as the following equation:
| ΔGbinding=ΔEvdw+ΔEele+ΔGpol+ΔGnp |
The stabilised end of the trajectory, i.e., 80–100 ns, was extracted to perform the MM/PBSA analysis where structures were observed at very 50 ps which were utilised for binding free energy calculations.
3. Results
3.1. Virtual screening and molecular docking
The results from the DrugDiscovery@TACC portal delineated a list of thousand ligands as a subset of the ZINC-in-trials library with binding affinity values ranging from − 7.2 to − 5.4 kcal/mol. First, the ligands exhibiting binding affinity less than − 6.5 kcal/mol were filtered, and the resulting 35 molecules were studied based on the highest binding affinity values, the orientation of the ligand and interactions with catalytic site residues, which resulted in the selection of four resultant compounds for further docking studies. The selected molecules, i.e., ZINC000000596945, ZINC000040165265, ZINC000040430143 and ZINC000064033452 exhibited binding affinity values of − 7.2 kcal/mol, − 6.9 kcal/mol, − 6.8 kcal/mol and − 6.8 kcal/mol respectively. These values obtained from DrugDiscovery@TACC were validated by employing AutoDock Vina, where each ligand was docked against the active site of PLpro utilising the same coordinates from the DrugDiscovery@TACC server for three runs. The results of Autodock Vina are presented in Fig. 2.
Fig. 2.

The docking results for PLpro-ligand complexes. (Blue: ZINC000000596945, Light green: ZINC000040165265, Salmon: ZINC000040430143, Violet: ZINC000064033452, Dark green: VIR251). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Next, AutoDock 4.2.6 was employed to repeat docking to validate the binding affinity values determined by AutoDock Vina of the top 4 ligands along with the control VIR251 molecule. The values for binding affinity according to AutoDock Vina and AutoDock are enumerated in Table 1. The interaction patterns between PLpro and the docked ligands are listed in Table 2.
Table 1.
Molecular docking results along with structural characteristics of the control molecule and top four ligands.
| S/No. | Selected Ligands | Chemical Formula | AutoDock binding affinity (kcal/mol) | AutoDock Inhibition constant | Structure |
|---|---|---|---|---|---|
| 1 | ZINC000000596945 | C23H19NO3 | -8.4 | 626.72 nM | 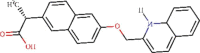 |
| 2 | ZINC000040165265 | C25H38O8 | -7.9 | 1.48 µM | 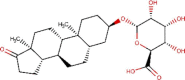 |
| 3 | ZINC000040430143 | C24H23FN4O3 | -7.7 | 2.06 µM | 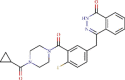 |
| 4 | ZINC000064033452 | C24H18F2N2O5 | -8.0 | 1.32 µM | 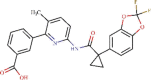 |
| 5 | VIR251 (Control) | C22H34N5O7 | -4.9 | 52.18 µM | 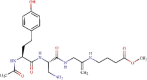 |
Table 2.
Interaction details of the selected ligands and control molecule with PLpro.
| S/No | Ligands ID | HB | D (Å) | Pi-SR | D (Å) | vdWISR |
|---|---|---|---|---|---|---|
| 1 | VIR251 | Trp106 His272 Lys274 |
1.86 2.52 2.97 |
Asn109, Thr265, Leu289 | ||
| 2 | ZINC000000596945 | His272 Tyr273 Asp286 |
2.31 2.59 2.20 |
Lys105 Cys111 Ala114 Ala288 Leu289 |
5.37 4.68 3.83 3.91 4.50 4.59 5.27 |
Ser103, Ile104, Thr115, Leu118, Lys274, His275, Ile285 |
| 3 | ZINC000040165265 | Trp106 Asn109 |
2.24 2.04 2.88 |
Trp106 His272 |
4.24 4.86 3.92 5.19 4.19 4.61 5.25 |
Lys105, Cys111, Gly271, Asp286, Gly287, Ala288, Leu289 |
| 4 | ZINC000040430143 | Trp106 Asp286 Ala288 |
2.33 1.84 2.09 2.02 |
Trp106 His272 Leu289 |
4.57 4.48 5.15 4.36 5.09 |
Lys105, Thr265, Gly266, Cys270, Gly271, Lys274, Gly287 |
| 5 | ZINC000064033452 | Trp106 Thr265 Gly271 His272 Lys274 |
2.33 2.03 2.27 2.12 1.70 |
Trp106 Cys111 Cys270 His272 |
4.67 4.80 3.37 4.95 4.62 5.47 |
Gly266 |
(HB: Hydrogen Bond interaction, D: Distance, Pi-SR: Pi-Interaction Sharing Residues, and vdWISR: van der Waals Interaction Sharing Residues)
The PLpro-ZINC000000596945 complex displayed an estimated binding affinity of − 8.4 kcal/mol and inhibition constant of 626.72 nM. The hydrogen atom associated with the Nε2 of His272 is predicted to form a hydrogen bond with the oxygen atom between the naphthalene and quinoline moieties at a distance of 2.31 Å. The carboxyl oxygen of the Tyr273 displayed a predicted hydrogen bond with the hydrogen atom associated with the nitrogen atom of the quinoline moiety of the ligand (2.59 Å). Further, the Oδ1 atom of Asp286 (2.20 Å) also forms a hydrogen bond with the same hydrogen atom as Tyr273. Leu289 displays a pi-alkyl interaction with each ring of the naphthalene moiety at distances of 4.59 Å and 5.27 Å. Ala288 (4.50 Å) and Lys105 (5.37 Å) are predicted to be in pi-alkyl interaction with the naphthalene moiety of the ligand. Cys111 (4.68 Å) and Ala114 (3.83 Å) display pi-alkyl interactions with the naphthalene moiety, and in addition, Ala114 (3.91 Å) also displayed an alkyl interaction with the pyridine ring of the naphthalene moiety of the ligand. The ligand is stabilised with several Van der Waals interactions, as listed in Table 2. The PLpro-ZINC000040165265 complex has an estimated binding affinity of − 7.9 kcal/mol and inhibition constant of 1.48 µM. The hydrogen atom of the amino group Asn109 is predicted to form a hydrogen bond with the carboxylate group associated with the tetrahydropyran moiety at a distance of 2.88 Å. The same residue also forms a hydrogen bond with a hydroxyl group of the tetrahydropyran moiety via the Oδ atom at a distance of 2.04 Å. The Hε atom of Trp106 (2.24 Å) is predicted to display another hydrogen bond with an oxygen atom of the ligand. The indole ring of Trp106 displays six pi-alkyl interactions with the ligand at distances of 4.24 Å, 4.86 Å, 3.92 Å, 5.19 Å, 4.19 Å, 4.61 Å. Further, the imidazole ring of the His272 displays another pi-alkyl interaction at a distance of 5.25 A. As listed in Table 2, various residues stabilise the ligand through Van der wals interactions.
The PLpro-ZINC000040430143 complex has an estimated binding affinity of − 7.7 kcal/mol with an inhibition constant of 2.06 µM. The hydrogen bonds of this complex were limited to the 2 H-phthalazin-1-one moiety of the ligand. First, Asp286 displays two hydrogen bonds with the 1 H-pyridazin-6-one group of the moiety via Oδ1 and carboxyl oxygen at distances of 1.84 Å and 2.09 Å, respectively. Ala288 and Trp206 form hydrogen bonds with the moiety's ketone group via the amino group's hydrogen atom of both residues at 2.02 Å and 2.33 Å. The indole ring of Trp106 displays pi-pi stacking interactions with the 2 H-phthalazin-1-one at distances of 4.57 Å, 4.48 Å, and 5.15 Å. Leu289 shows a pi-alkyl interaction with the same moiety from a distance of (5.09 Å). His272 is predicted to show another pi-alkyl interaction with the piperazine moiety of the ligand at a distance of 4.36 Å. The PLpro-ZINC000064033452 complex displayed an estimated binding affinity of −8.0 kcal/mol and a binding affinity of 1.32 μM. Lys274 and Thr265 are predicted to form individual hydrogen bonds with the carboxylate group of the ligand via Hζ1 and Hγ1 of Lys274 (1.70 Å) and Thr265 (2.03 Å), respectively. His272 (2.12 Å) and Trp106 (2.33 Å) displayed hydrogen bonds with a ketone group present in the ligand via Hδ1 and Hε1, respectively. Another hydrogen bond is observed from Gly271 to the NH group between the ketone group and pyridine ring of the ligand at a distance of 2.27 A. His272 (4.62 Å) and Trp106 (4.88 Å) also displayed pi-alkyl interaction with the cyclopropane ring of the ligand, in addition, His272 (5.47 Å) also displays another pi-alkyl interaction with the pyridine ring of the ligand. Alkyl interactions were predicted with Cys111 (3.37 Å) and Cys270 (4.95 Å) with the cyclopropane and pyridine rings, respectively. Pi-pi stacked interactions were observed from the imidazole ring Trp106 to the 1,3-benzodioxole moiety from a distance of 4.67 Å and 4.80 Å. The ligand is also stabilised with a Van der Waals interaction with Gly266.
The PLpro-VIR251 complex, which acted as the control molecule of the study, exhibited a binding affinity of − 4.9 kcal/mol along with an inhibition constant of 52.18 μM. Interestingly, the control ligand was only stabilised through hydrogen bonds with the homotyrosine moiety of VIR251. Three hydrogen bonds were observed via the Hζ1 in Lys274 (2.97 Å), Hδ1 in His272 (2.52 Å) and Hε1 of Trp106 (1.86 Å). In addition, the complex was also stabilised through Van der Waals interactions displayed by Asn109, Thr265 and Leu289.
Subsequently, the electrostatic surface potential image was rendered based on the calculations of the ABPS (Adaptive Poisson-Boltzmann Solver) plugin in PyMOL. The results suggested that the active site of PLpro is relatively neutral with a slightly acidic zone. The ligands were observed to be bound to the active site of PLpro ( Fig. 3), as seen in Fig. 4. Subsequently, the electrostatic surface potential image was rendered based on the calculations of the ABPS (Adaptive Poisson-Boltzmann Solver) plugin in PyMOL. The results suggested that the active site of PLpro is relatively neutral with a slightly acidic zone. The ligands were observed to be bound to the active site of PLpro, as seen in Fig. 4. Next, the electrostatic surface potential was plotted for the ligand molecule. The colour gradient from red to blue signifies atoms with the highest and lowest electron density, respectively ( Fig. 5). For ZINC000000596945, it can be observed that the carboxylate moiety, which is shown to have a high electron density, interacts with a positively charged Lys 105 through a salt bridge. In contrast, the quinoline moiety of the ligand displays a net positive partial charge with the NH group with the least electron density; hence, it interacts with a highly negatively charged groove in the PLpro. For ZINC000040165265, it can be observed that the hydropyran moiety shows diversity in electron density while the rest of the ligand shows neutrality. The carboxylate group associated with the hydropyran shows a high electron density. Therefore, it interacts with neutral potential on the protein surface, while the hydroxyl groups of the hydropyran display a lower electron density and hence interact with a corresponding positive potential on the protein surface. ZINC000040430143 displays an overall negative surface potential except for the nitrogen atoms of 2 H-phthalazin-1-one moiety, which show a lower electron density. Accordingly, it is observed that most of the ligand interacts with relatively neutral surface potential while the nitrogen atoms of 2 H-phthalazin-1-one moiety interact with a positive surface potential of the protein conferred by the negatively charged Asp286. Next, ZINC000064033452 displays an overall positive surface potential except for the carboxylate group on the benzoate moiety of the ligand, which displays a negative surface potential. It is observed that the ligand primarily interacts with a neutral surface potential of the protein. Finally, the control molecule VIR251 shows an overall positive surface potential and interacts with a slightly acidic neutral surface of the PLpro.
Fig. 3.

Top 4 Selected ligands bound to the active site of PLpro. Zoomed-in view - Two-dimensional (left column) and Three-dimensional (right column) representation of interacting residues with ZINC000000596945 (Cyan), ZINC000040165265 (light green), ZINC000040430143 (Salmon), ZINC000064033452 (Violet) and Vir251 (Dark Green) via van der Waals interactions (slightly green colour), hydrogen bonds (dark green colour), and pi-interactions (light pink, purple colour and blue colour). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 4.

The electrostatic potential molecular surfaces of PLpro- (a) ZINC000000596945, (b) ZINC000040165265, (c) ZINC000040430143, (d) ZINC000064033452 and (e) VIR251 complexes.
Fig. 5.

The electrostatic potential molecular surfaces of (a) ZINC000000596945, (b) ZINC000040165265, (c) ZINC000040430143, (d) ZINC000064033452 and (e) VIR251. The gradient from red, yellow, green, light blue and dark blue represent a negative to positive surface potential. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2. Molecular dynamics simulations
Molecular Dynamics simulations were conducted for a period of 100 ns, with each frame being generated at a time interval of 0.01 ns. The best two compounds from molecular docking studies (PLpro-ZINC000000596945, PLpro-ZINC000064033452) along with native PLpro and control PLpro-VIR251, were subjected to MD simulations. The output of subsequent analyses was analysed through GROMACS and Bio3d R studio, which were plotted with Gnuplot (http://www.gnuplot.info/) and Bio3D. The results of the MD analysis are as follows:
3.2.1. Global dynamics analysis
The RMSD is a critical factor utilised in the analysis for the structural deviation and compactness of the protein structure, which delineates stability attributes of the protein throughout the desired time frame. The native PLpro is observed to be stabilised at around 10 ns which was maintained till 40 ns, around which a minor perturbation is observed. This is followed by a converged trajectory of the native PLpro till the end of the 100 ns frame point. The RMSD values of the trajectory for native PLpro ranged primarily between 0.2 nm and 0.4 nm, wherein the most frequent values for RMSD were 0.34 nm and 0.23 nm, as observed in Fig. 6. The PLpro-ZINC000000596945 complex shows a period of stability from 10 to 20 ns followed by a sustained period of irregularity followed by convergence attained at 40 ns maintained throughout the trajectory with minor perturbations at 60 ns and 80 ns. The RMSD values from this complex also range from 0.2 nm to 0.4 nm, wherein 0.23 nm and 0.31 nm is the most frequent value. The PLpro-ZINC000064033452 complex attains stability at around 10 ns and remains in convergence throughout the trajectory, excluding minor fluctuations around the 20–50 ns time period. The RMSD values for the complex remain between 0.2 and 0.4 nm, with the most frequent value being 0.25 nm. Finally, the control molecule, i.e., PLpro-VIR251 complex, displays stabilisation at 20 ns which is followed by an increasing trend in the RMSD of the complex and a dip around 60 ns timepoint and an eventual convergence at 80 ns. The RMSD values for this complex were within 0.2–0.5 nm, with the most frequent values being 0.25 nm and 0.36 nm.
Fig. 6.

Graphical representation for RMSD for (A) native PLpro (black), (B) PLpro-ZINC000000596945 complex (cyan), (C) PLpro-ZINC000064033452 complex (violet) and (D) PLpro-VIR251 complex (dark green). (1) represents individual RMSD graphs and (2) represents histography describing the distribution of RMSD values. The red line represents the Locally Weighted Scatterplot Smoothing. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
RMSF is a descriptive evaluation of the local fluctuations at a residue level. The majority of the fluctuations in PLpro and its complexes were observed in the N and the C terminus. In the analysis for the Ubl domain (1–60 residues), while the RMSF remains fairly low, a decrease in flexibilities is observed for the domain in ligand-bound complexes. Next, the thumb domain (61–120 residues) had significantly low RMSF values with random fluctuations, which can be credited to α-helices in the region. The α3–4 loop of the thumb region shows random fluctuations, which all remain under 0.4 nm. The Trp106 residue remains highly flexible in native PLpro, whereas a significant reduction in its flexibility is observed upon ligand binding. The fingers domain shows the most random flexibility in the protein, which can be traced to the loops in the region, which are found to be increased upon ligand binding. The palm domain also displays random fluctuations, all of which remain under 0.3 nm with the exception of the ß11–12 loop (BL2 loop), which shows the most flexibility in the native state (0.83 nm) while the flexibility is greatly reduced in ligand-bound complexes. The average RMSF values for native PLpro, PLpro-ZINC000000596945, PLpro-ZINC000064033452, PLpro-VIR251 complexes are 0.20 nm, 0.19 nm, 0.17 nm and 0.18 nm ( Fig. 7, Fig. 8, Fig. 9).
Fig. 7.

Analysis of RMSF and Rg of native black (PLpro), cyan (PLpro-ZINC000000596945), violet (PLpro-ZINC000064033452), and Dark green (PLpro-VIR251). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 8.

Analysis of SASA of native PLpro (black), cyan (PLpro-ZINC000000596945), violet (PLpro-ZINC000064033452), and Dark green (PLpro-VIR251) (A) Total SASA of all complexes; (B1,2,3,4) individual graphs for native PLpro and complexes; (C) Superimposed graph of burSASA of PLpro-ligand complexes: (C1,2,3) individual graphs for PLpro-Ligand complexes; (D1,2,3,4) individual graphs for resSASA native PLpro and complexes. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 9.

Solvent accessible surface representation of protein in white and (a) ZINC000000596945, (b) ZINC000064033452, and (c) VIR251 in a blue mesh. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Rg is indicative of a protein's compactness and folding nature; hence, it was employed to study the overall structural stability of the native PLpro and its ligands-associated complexes. The results suggested that the Rg values of the complexes were similar to the native protein. The average Rg values for native PLpro, PLpro-ZINC000000596945, PLpro-ZINC000064033452, PLpro-VIR251 complexes are 2.41 nm, 2.39 nm, 2.40 nm and 2.42 nm. The trajectory of Rg for the native protein shows an expected trend of decrease until the first 7 ns, followed by a stable trajectory until an absorbing perturbation from 34 to 35 ns, where it reaches a maximum peak of 2.48 nm at 35.06 ns. A stable decrease follows this in the trajectory where the protein reaches a minimum Rg value of 2.33 nm at 68.01 ns. The PLpro-ZINC000000596945 complex shows a similar trajectory of Rg as that of the native protein till the 14 ns mark, upon which the trajectory displays a decrease reaching a minimum of 2.32 nm followed by a similarly sharp increase in Rg from 35 to 36 ns and a subsequent reduction after the 37 ns mark. After 61 ns, the PLpro-ZINC000000596945 complex follows a relatively similar trajectory as the native protein with an increased value perturbation around 79–81 ns. The PLpro-ZINC000064033452 Rg trajectory displays a relatively stable trajectory with cycles of increase followed by decrease till 47 ns, upon which it remains stable till 85 ns, after which it reaches a peak of 2.49 nm at 86.74 ns, followed by a progressive reduction till the end of the trajectory. A significantly stable trajectory was observed for the PLpro-VIR251 complex throughout 100 ns with a notable increase during 22–28 ns, where it displays a peak value of 2.50 nm at 27.63 ns. A repetitive cycle of increase followed by decrease can be observed through the 70–85 ns time points.
Along with Rg analysis, SASA analysis is employed to analyse the interaction of the protein and its complexes with the solvent, which ultimately is a factor that delineates information regarding the protein's three-dimensional structure, i.e., the protein's conformation. First, the total SASA for the native PLpro and its complexes were explored. With an initial decrease until the 5 ns time point, the trajectory for native PLpro remains reasonably stable with minor perturbations during the 40–45 ns period observing an average SASA value of 165.201 nm2. The PLpro-ZINC000000596945 complex displays an average SASA value of 166.533 nm2, while the trajectory follows similar trends as the native PLpro, periods of 0–25 ns and 85–100 ns display higher SASA values with respect to the native PLpro trajectory. The trajectory observed for the PLpro-ZINC000064033452 complex shows higher values of SASA with respect to the native PLpro; while the similarities in trend are fewer, the complex displays an average SASA value of 168.657 nm2. Finally, the PLpro-VIR251 control complex has a significantly stable trajectory in which higher values of SASA are observed throughout. Until the 45 ns mark, the trajectory follows a similar pattern as that of native PLpro, upon which it remains stable, observing higher values. In contrast, the native PLpro trajectory decreases significantly with respect to the PLpro-VIR251 complex. While total SASA provides us with the solvent accessibility of the complex as a whole, buried SASA (burSASA) is employed to analyse the stability of the interface, i.e., the binding surface between the protein and the ligand. Buried Surface Area ( bur SASA = [SASA complex - (SASA protein + SASA ligand )]) (Hollingsworth et al., 2016) is the area of interface which is formed by the binding of the ligand to the protein, the overall Surface Accessible Area of the protein, which gets buried upon ligand binding is termed the buried Surface Area. The burial of a protein surface is directly indicative of the interaction of the ligand molecule with the protein molecule. The average burSASA value for PLpro-ZINC000000596945 complex, and PLpro-ZINC000064033452 complex is 7.135 nm2 and 7.368 nm2, respectively. Both complexes show a stable trajectory in the same trend and values, although minor perturbations are observed in the PLpro-ZINC000064033452 trajectory. A significantly lower burSASA value is observed for PLpro-VIR251 with an average value of 3.930 nm2; the trajectory for this complex remains stable throughout the simulation period with periods of slight increase at 10 and 84 ns.
Next, residue-wise SASA (resSASA) values were calculated to study the changes in the surface accessibility of the active site residues. The resSASA values of the active site residues are provided in Table 3, while the resSASA values of Trp106 and Asp286 remain relatively similar for all complexes with respect to the native protein. The values of Cys111 display a significant reduction from the native protein to PLpro-ZINC000064033452 and PLpro-VIR251 complex. The values of resSASA for His272 show a slight increase in PLpro-ZINC000000596945 complex while a significant reduction is observed for PLpro-ZINC000064033452 and PLpro-VIR251 ( Table 4).
Table 3.
SASA values for PLpro and its complexes.
| System | Protein SASA (nm2) | Ligand SASA (nm2) | Complex SASA (nm2) | Buried SASA (nm2) |
|---|---|---|---|---|
| Native PLpro | 165.201 | – | 165.201 | – |
| PLpro-ZINC000000596945 | 167.394 | 6.274 | 166.533 | 7.135 |
| PLpro-ZINC000064033452 | 168.657 | 7.100 | 168.393 | 7.368 |
| PLpro-VIR251 | 166.671 | 8.220 | 170.972 | 3.930 |
Table 4.
Residue-wise SASA values of active site residues of PLpro and its complexes.
| Residue | Native PLpro (nm2) | PLpro-ZINC000000596945 (nm2) | PLpro-ZINC000064033452 (nm2) | PLpro-VIR251 (nm2) |
|---|---|---|---|---|
| Trp106 | 1.886 | 1.454 | 1.109 | 1.121 |
| Cys111 | 0.276 | 0.222 | 0.069 | 0.093 |
| His272 | 0.530 | 0.616 | 0.175 | 0.108 |
| Asp286 | 0.462 | 0.537 | 0.503 | 0.484 |
Finally, a 3D representation of ligand SASA is used to visualise the contributions of ligand SASA in the complex. It can be observed that PLpro-ZINC000000596945 and PLpro-ZINC000064033452 bury significantly in the protein surface while the VIR251 remains relatively on the surface of the protein, which can be attributed to its large structure. The burSASA values can be visually corroborated with this surface representation.
Next, we observed the changes in secondary structures throughout the trajectory by employing the DSSP (Define Secondary Structure of Proteins) (Kabsch and Sander, 1983) module, which defines various secondary structure elements, including α-helix, β-sheet, β-bridge, turn, coil, bend, 5-helix and 3₁₀ helix. The observed changes are depicted in Fig. 10, and the obtained percentage changes in secondary structure are recorded in Table 5. While an overall 4% decrease in the structure (α-helix + β-sheet + β-bridge + turn) can be seen in the PLpro-VIR251 complex, and a minimal 2% change is seen in PLpro ligand-bound complexes. It is found that the α-helix remains constant on average throughout the trajectory, and minimal changes are observed in β-sheets and coil. Few perturbations are observed in the turn elements of all PLpro ligand-bound complexes with respect to the native structure.
Fig. 10.

Secondary structure changes observed in (a) PLpro, (b) PLpro-ZINC000000596945, (c) PLpro-ZINC000064033452, and PLpro-VIR251.
Table 5.
Percentages of secondary structural elements of native PLpro and its complexes.
| System | Structure | Coil | β-sheet | β-bridge | Bend | Turn | α-helix | Other |
|---|---|---|---|---|---|---|---|---|
| Native PLpro | 68 | 20 | 31 | 1 | 11 | 10 | 26 | 1 |
| PLpro-ZINC000000596945 | 66 | 21 | 30 | 1 | 12 | 8 | 26 | 1 |
| PLpro-ZINC000064033452 | 66 | 20 | 31 | 2 | 13 | 7 | 26 | 2 |
| PLpro-VIR251 | 64 | 22 | 28 | 2 | 13 | 9 | 26 | 1 |
(Structure = α-helix + β-sheet + β-bridge + turn) (Other = π-helix + 3₁₀-helix).
3.2.2. Essential dynamics analysis
Molecular dynamics simulations provide the global dynamics trajectory, which delineates all the motions presented by the protein and its complexes under the selected force field, which include all large and small amplitude motions. Due to the vast degree of freedom presented by the protein in global dynamics, it becomes difficult to understand the relevant motions essential for the activity of the protein. In MD simulations, various techniques are applied for data dimensionality reduction to obtain the collective motions of the protein, which are large amplitude and small frequency motions which are said to be relevant to the activity of the protein. It has been observed that a relatively smaller subset of the global motions which possess a collective degree of motion can be associated with protein activity; hence they are collectively termed essential motions. PCA is a multivariate data dimensionality reduction technique that formulates a covariance matrix based on the fluctuations of the Cα atoms. The diagonalization of the covariance matrix presents eigenvectors and eigenvalues, which determine the atomic motion and atomic contribution in motion, respectively. The principal components (PCs) obtained from PCA are plotted onto a conformational subspace which depicts the variances in atomic positions, i.e., protein conformation; hence it is termed the essential subspace. It has been found that PC1–3 accounts for the majority of the variance observed in the collective motion of protein and its complexes. In Fig. 11, the proportion of variance vs eigenvalue rank graph, it can be observed that the first three PCs of the native PLpro account for 58.7% of the variance, while the PLpro-ZINC000000596945, PLpro-ZINC000064033452, and Plpro-VIR251 complexes account for 53.7%, 50.8% and 55.7% of the total variance in Cα atom motion. It should be noted that for native PLpro, PC1 and PC2 were sufficient to account for about 50% of the total variance, while for PLpro complexes, PC3 contribution is required to account for the same variance. PC1 is a critical factor in analysing the dominant motions in essential dynamics; As observed in the graph, PC1 of native PLpro accounts for 40.3% of the variance, while the PLpro-ZINC000000596945, PLpro-ZINC000064033452 and Plpro-VIR251 complex account for 24%, 32% and 33.7% of the total variance. It can be inferred from the data above that upon ligand binding; a significant drop can be observed in the Cα atoms while the native PLpro displays distinct conformational changes, whereas the ligand-bound PLpro displays a reduction in such conformational changes. Next, PC1 and PC2 were plotted as a scatter plot onto the essential subspace in Fig. 11 , where each dot represents a specific conformation of the protein and the shift from blue to white, then red represents the simulation period. The blue dots represent the initial conformations in the simulation, followed by white dots, which represent the intermediate states and ultimately, the red dots represent the conformations towards the end of the simulation time period. The native PLpro plot shows clusters assimilated towards the right and left sides of the plot with distinct intermediate states between the two clusters, which is suggestive of stable conformations throughout the simulation time period. The essential subspace scatter plot for PLpro-ZINC000000596945 and PLpro-ZINC000064033452 displays dispersed confirmations in the essential subspace with reduced intermediate states, which is indicative of more flexible protein conformations.
Fig. 11.

PCA analysis for (A) native PLpro, (B) PLpro-ZINC000000596945 complex, (C) PLpro-ZINC000064033452 complex and (D) PLpro-VIR251 complex. (1) Essential subspace projection of PC1 vs PC2; (2) Proportion of variance vs eigenvalue graphs and (3) Residue-wise PCA contribution where black and blue lines show PC1 and PC2 contribution, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
PLpro-ZINC000064033452 complex displays distinct parallel blue and red conformations in the scatter plot, both convened on two distinct sides. It can be observed that the blue dots are more dispersed, followed by convergence in the intermediate states, ultimately a distinct smaller final cluster of red dots which indicates stable convergence of conformations. The flexibility contributed by PC1 (black line) and PC2 (blue line) were analysed at a residue level for native PLpro and its complexes (Fig. 11). The data suggests that while the majority of the protein exhibits limited flexibilities as the majority structure of PLpro is composed of secondary structures; hence the flexibilities are attributed to the interspersed loop regions in inter and intradomain regions. Greater flexibility is observed in the PLpro-ZINC000000596945 complex in comparison to the native PLpro and PLpro-ZINC000064033452 and PLpro-VIR251 complexes. The majority of flexibilities are observed in the Ubl domain (1–60 residues) and the region between the palm and fingers domain (200–250 residues), which can be attributed to the loops in the regions. An increase in the flexibilities of active site residues is observed in PLpro-ZINC000000596945 and PLpro-ZINC000064033452 complexes, indicating conformational changes in the active site of the protein upon ligand binding.
Next, to analyse the amplitude and directionality of fluctuations observed in the native protein and ligand-bound complexes, DCCM was constructed. Protein dynamics indicate that regions in protein display correlated (same) and anti-correlated (opposite) motion, which delineates the dynamic motion of the protein. This suggests that changes in one part of the protein can influence other motifs and thereby the activity of the protein. DCCM is performed through covariance diagonalization of the displacement vector of each Cα atom which provides a cross-correlation coefficient C(i,j). A positive value of the coefficient corresponds to a correlated motion of the same phase, period and direction, whereas a negative value implicates an anti-correlated motion of the opposite direction. In Fig. 12 presented below, the red colour and blue colours indicate correlated and anti-correlated motions, respectively, while the colour shift is indicative of the extent of correlation or anti-correlation, i.e., deeper red or blue shows more correlation or anti-correlation. For native PLpro, the DCCM plot displays heavy intradomain correlations in all protein domains. A significant correlation in the residues of the fingers and the Ubl domains can also be observed; the same is also seen for the palm and thumb domains with few dispersed anti-correlations. The Ubl and palm domains show a significant degree of anti-correlation. The PLpro-ZINC000000596945 complex shows a reduction in the interdomain correlations while maintaining the intradomain correlations. A significant decrease is observed in both correlated and anti-correlated motions between the thumb and palm domain, which host the protein's active site. A heavy increase can be observed in the low-intensity anti-correlated motion between the fingers and thumb domain of the protein. The PLpro-ZINC000064033452 complex displayed similar correlated and anti-correlated motions as that in the PLpro-ZINC000000596945 complex. However, a significant reduction in both motions is seen between the thumb-palm and Ubl-palm domains. For the control or PLpro-VIR251 complex, an increase is observed in the overall correlated motion that can be explicitly seen between the palm and thumb domain with respect to other complexes and native protein.
Fig. 12.

DCCM plots for (a) native PLpro, (b) PLpro-ZINC000000596945 complex, (c) PLpro-ZINC000064033452 complex and (d) PLpro-VIR251 complex. Three-dimensional representation of correlated motions in red (above) and anticorrelated motions in blue (below) are presented. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Porcupine Analysis was performed to analyse the essential motions in native PLpro and its ligand-bound complexes. The spikes represent the direction of fluctuations presented by PC1, and their size represents the magnitude of the fluctuation observed in the Cα atom backbone of the protein. For the native PLpro protein opposing motions of large magnitudes are observed in the Ubl domain and the loops of the fingers domain presenting a vibrational up and down motion. Another dominant high magnitude motion can be observed in the loop between ß11 and ß12 made of residues 266–271 towards the loop between β13 and β14, while small motions are also observed in α4 towards α7. This indicates a closing movement of the β14–15 loop towards the β13–14loop, which brings the active site residues closer ( Fig. 13).
Fig. 13.

Porcupine analysis for (a) native PLpro, (b) PLpro-ZINC000000596945 complex, (c) PLpro-ZINC000064033452 complex and (d) PLpro-VIR251 complex. The red spikes represent the direction of fluctuations presented by PC1, and their size represents the magnitude of the fluctuation observed in the Cα atom backbone of the protein. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The PLpro-ZINC000000596945 complex shows a significant reduction in the motions observed in the protein. The Ubl and loops of the fingers domain show a downward motion, while a reduced motion in the β11–12 loop can be observed. Increased motions are introduced in α6 helix upon ligand binding opposite to the direction of β11–12 loop. In the PLpro-ZINC000064033452 complex, an even further reduction in the motion of the Ubl domain can be observed while the loops of the fingers domain show significantly high fluctuations. A concerted motion of the β11–12 and β13–14 loop can be observed in the complex, and an opposite motion of equal magnitude can be observed in the loop region between α3–4, which indicates a cleft formation for ligand binding. The control PLpro-VIR251 complex shows a significantly reduced motion in the Ubl domain while the fingers domain and β11–12 loop fluctuations persist in the complex. Random small fluctuations are observed in the α helices of the thumb domain.
3.2.2.1. Time-based essential dynamics analysis
While porcupine analysis was employed to analyse the overall localised concerted motion of the protein, a time-dependent essential dynamics analysis was employed to validate the obtained results and better elucidate the motions relevant to the protein throughout the protein trajectory. A concatenated trajectory comprising the motions contributed by the first ten principal components was extracted from the global dynamics trajectory upon which structures were extracted from the concatenated trajectory.
The native PLpro protein ( Fig. 14) from 0 ns to 20 ns maintains a to and fro motion along the reference of the 0 ns structure. From 20–40 ns, the vibrational motions in the Ubl domain, loops of fingers domain and inter helical domain persists while the β11–12 loop (His 272 is present in β12) begins to move towards the β13–14 loop (which hosts Asp286) and α3–4 loop (which hosts Trp106 and Cys111 is present at the tip of α4). From 40–60 ns, the β11–12 loop moves back to a similar position as the 0 ns structure. From 60–80 ns, a similar movement is observed as seen in the 20–40 ns time period, bringing the active sites closer to each other. Until the end of the simulation (100 ns), the loop remains in the same closer orientation while the random up and down vibrations remain persistent in the Ubl domain, loops of fingers domain and inter helical loops. These results are consistent with that of porcupine analysis.
Fig. 14.

Time-based essential dynamics analysis of native PLpro at every 20 ns with secondary structures depicted with red, cyan and magenta for beta stands, alpha helices and loops, respectively. The changes are depicted with orange arrows. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
For the PLpro-ZINC000000596945 complex from 0 to 20 ns, the fingers and Ubl domain show an upwards motion with respect to the reference structure of 0 ns, while the β11–12 loop shows a trend of moving significantly closer to the ligand and the ligand's orientation becomes closer to the β13–14 loop. From 20–40 ns, while the loop between β11–12 shifts, the β12 possessing the His272 catalytic residue remains close to the ligand, and the β13–14 loop moves slightly closer to the ligand as the ligand displaces towards the α3–4 loop. No significant changes were observed between 40 and 60 ns. From 60–80 ns, Thr265-His272 residues comprising the β11–12 loop transition to a two-residue loop of Tyr268-Gln289 while the rest assimilate into β-sheets creating a cleft for the bound ligand. Asn109-Asn110 of α3–4 loop transition into a helix extending α3 helix while the downward motion of Ubl domain and loop of fingers domain. While no significant changes in motion are observed for the 80–100 ns period, a shortening of β11 into a loop is observed ( Fig. 15, Fig. 16, Fig. 17).
Fig. 15.

Time-based essential dynamics analysis of PLpro-ZINC000000596945 complex at every 20 ns with secondary structures depicted with red, cyan and magenta for beta stands, alpha helices and loops, respectively. The changes are depicted with orange arrows. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 16.

Time-based essential dynamics analysis of PLpro-ZINC000064033452 at every 20 ns with secondary structures depicted with red, cyan and magenta for beta stands, alpha helices and loops, respectively. The changes are depicted with orange arrows. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 17.

Time-based essential dynamics analysis of PLpro-VIR251 at every 20 ns with secondary structures depicted with red, cyan and magenta for beta stands, alpha helices and loops, respectively. The changes are depicted with orange arrows. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Next, the PLpro-ZINC000064033452 complex displayed no significant changes during the 0–40 ns time period. While the motions in the Ubl domain and loops of the fingers domain remain limited, the transition of Gly266-Asn267 from β sheet to loop is observed. From 40–60 ns, the ligand shifts towards the β11–12 loop along with a shift towards the same are observed in β13–14loop and α3–4 loop. No significant changes are observed during 60–80 ns, yet the slight fluctuation in loops remains persistent in a vibrational motion. From 80–100 ns, a drastic shift in the orientation of the ß11–12 and ß13–14 loops is observed while β11 moves towards the ligand, β14 moves away from the ligand. It should be noted that the fluctuations in the remaining protein structures remained limited with respect to native PLpro as most of the relevant fluctuations were accounted for by β11–12, β12–14, and α3–4 loops.
The control or PLpro-VIR251 complex exhibits limited fluctuations overall while the vibrational trend of the Ubl domain, loops of fingers domain, β13–14loop and α3–4 loop persists significantly refused with respect to other complexes and native PLpro. The majority of fluctuations were observed in the β11–12 loop. From 0–20 ns, the β11–12 loop moves towards the ligand, upon which it fluctuates around a mean position till the end of the simulation.
3.2.2.2. Free energy landscape analysis
Free energy landscapes provide insights into the conformational variability of a protein structure. The first two principal components were used to obtain Gibbs free energy values associated with each protein conformer. The contour plots in Fig. 18 show the transition from high to low energy states, i.e., the energy minima basins, which are represented by a colour shift of red to blue where the concentrated blue spots represent the metastable states of the protein and its associated complexes. For the native PLpro complex, the energy basin is divided into two clusters of low-energy conformers, but only one displays concentrated minima, indicating that while the protein remains stable during the simulation, it possesses inherent flexibilities. Similarly, the PLpro-ZINC000000596945 divides the energy basin into three minima, but only one displays concentrated minima, suggesting the same as native PLpro. For the PLpro-ZINC000064033452 complex, the energy basin is divided into two distinct minima, which is suggestive of distinctive changes in the protein confirmations upon ligand binding. Finally, the PLpro-VIR251 complex displays six concentrated minima in the energy basin, indicating significantly unstable conformations.
Fig. 18.

Free energy landscape in three dimensional and two dimensional representations for (a) native PLpro, (b) PLpro-ZINC000000596945 complex, (c) PLpro-ZINC000064033452 complex and (d) PLpro-VIR251 complex.
3.3. Binding free energy analysis
Binding free energy calculations were performed by employing the g_mmpbsa module, which performs an MM/PBSA-based approach to provide the binding free energies of PLpro and its associated complexes. A concatenated trajectory comprising of stable 80–100 ns of the global trajectory is extracted where a structure is analysed at every 50 ps. The obtained results are represented in Fig. 19, and the calculated values are presented in Table 6. Both PLpro-ZINC000000596945 (−41.23 ± 3.70 kcal/mol) and PLpro-ZINC000064033452 (−25.10 ± 2.65 kcal/mol) exhibit uniformity throughout the plot (Fig. 19) which is indicative of stability of both complexes. On the contrary, the plot for PLpro-VIR251 (−7.13 ± 5.69 kcal/mol) shows greater perturbations with values showing positive values at certain instances, for example, at 91.65 ns, 93.25 ns, 96.30 and 96.45 ns (3D representations provided in Fig. 19). Upon investigation, it was found that during these time points, the ligand remains greatly far from the protein, with interactions only being contributed by the ß11–12 loop with unfavourable interactions, which can be the contributors to positive values of binding free energy. It should also be noted that due to the ligand orientation, the control molecule displays a high value of polar solvation energy, which greatly decreases the binding free energy of VIR251.
Fig. 19.

Binding free energy plot for (a) cyan: PLpro-ZINC000000596945, (b) purple: PLpro-ZINC000064033452 and (c) dark green: PLpro-VIR251. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 6.
Individual component contribution in binding free energy calculations for native PLpro and its ligand-bound complexes.
| System | Van der Waals energy (kcal/mol) | Electrostatic energy (kcal/mol) | Polar solvation energy (kcal/mol) | SASA energy (kcal/mol) | Binding free energy (kcal/mol) |
|---|---|---|---|---|---|
| PLpro-ZINC000000596945 | -44.70 ± 3.39 | -9.86 ± 4.59 | 17.34 ± 3.52 | -4.00 ± 0.21 | -41.23 ± 3.70 |
| PLpro-ZINC000064033452 | -39.25 ± 2.93 | -5.78 ± 2.54 | 24.07 ± 3.43 | -4.13 ± 0.27 | -25.10 ± 2.65 |
| Plpro-VIR251 | -17.38 ± 2.48 | -22.68 ± 6.01 | 35.11 ± 7.70 | -2.17 ± 0.26 | -7.13 ± 5.69 |
Next, using the g_mmbpsa programme, binding free energy for each contributing residue was calculated ( Fig. 20). For PLpro-ZINC000000596945 complex Lys105 (−6.19 kcal/mol), Lys94 (−6.15 kcal/mol) and Lys274 (−5,52 kcal/mol) were the top contributors whereas for the PLpro-ZINC000064033452 complex Asp108 (−6.85 kcal/mol), Asp286 (−4.89 kcal/mol) and (−4.67 kcal/mol) were the residue that contributed the most to the binding free energy. The control complex (PLpro-VIR251) displayed the least values, as evident with docking, where Cys111 (−1.13 kcal/mol), Tyr106 (−1.12 kcal/mol), and Leu162 (−0.75 kcal/mol) were the top three contributors.
Fig. 20.

Residue wise binding free contribution energy for (a) Cyan: PLpro-ZINC000000596945, (b) Purple: PLpro-ZINC000064033452 and (c) Dark-green: PLpro-VIR251. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
In the present study, we have investigated the structural influences of ligand binding on PLpro, which is a viral protease responsible for proteolytic cleavage of viral polypeptides. After retrieval and preparation of protein structure obtained from the RCSB-PDB databank, it was utilised in molecular docking studies. Through TACC an Vina docking studies the ligand showed binding affinities in the following trend, ZINC000000596945 > ZINC000040165265 > ZINC000040430143 > ZINC000064033452. The same ligands were then utilised for Autodock where the binding affinity was observed as ZINC000000596945 > ZINC000064033452 > ZINC000040165265 > ZINC000040430143. Next, the free protein, PLpro-ZINC000000596945 complex, PLpro-ZINC000064033452 complex and the control complex (PLpro-VIR251) were subjected to a 100 ns molecular dynamics simulation. The RMSD analysis of the obtained trajectories delineated a trend of PLpro-VIR251 > PLpro > PLpro-ZINC000000596945 > PLpro-ZINC000064033452 which suggests a stable trajectory for ZINC000064033452 and ZINC000000596945 whereas VIR251 appears to make the complex unstable. The RMSD density graphs show a converged and uniformly stable trajectory for ZINC000064033452 while ZINC000000596945 remains fairly stable, attaining two peak density distributions. RMSF analysis was performed to study the protein's inherent flexibilities, which displayed a trend of PLpro > PLpro-ZINC000000596945 > PLpro-VIR251 > PLpro-ZINC000064033452, which displays a decrease in the flexibility of the protein upon ligand binding which can be especially observed in ZINC000064033452. Next, Rg analysis was employed to study the overall compactness of protein structures, which showed a pattern of PLpro-VIR251 > PLpro > PLpro-ZINC000064033452 > PLpro-ZINC000000596945. The observed values are fairly similar to each other with minor perturbations, suggesting a localised protein compactness change. Since the protein is present in a solvent for the simulation, we have studied the SASA for the protein and its complexes which showed a trend of PLpro-VIR251 > PLpro-ZINC000064033452 > PLpro-ZINC000000596945 > PLpro, which shows that upon ligand binding significant changes occur in the tertiary structure of the protein. The high SASA value of PLpro-VIR251 is credited to the larger size of the ligand, the subsequently high ligand SASA, while the ligand SASA for ZINC000000596945 and ZINC000064033452 remain similar. Next, it was observed that the surface area values buried upon ligand binding are observed as PLpro-ZINC000064033452 > PLpro-ZINC000000596945 > PLpro-VIR251, which suggests ZINC000000596945 and ZINC000064033452 effectively bury inside the protein meanwhile most of VIR251 remains outside the protein surface. This indicates that the burial of ligand is observed most in ZINC000000596945 and ZINC000064033452; hence, they can show better affinity and interactions with PLpro. On the contrary, the VIR251 molecule does not bury well within the protein surface and possesses lower binding affinity and interactions with the protein molecule. Due to ligand binding, a significant decrease in the SASA values of Cys111 and His272 residues is observed for PLpro-ZINC000064033452 and PLpro-VIR251, while the residue SASA values for Asp286 remain similar for all complexes, and a general decrease is also observed in Trp106 (present in the α3–4 loop) which suggests stabilisation and burial of the loop upon ligand binding in all complexes. In the secondary structure analysis, no significant changes are observed in α-helices, while minute differences are seen in β-sheets ( Table 7).
Table 7.
Average values for the global dynamics analysis of PLpro and its ligand-bound complexes.
| System | Average RMSD (nm) | Average RMSF (nm) | Average Rg (nm) | Average SASA (nm2) |
|---|---|---|---|---|
| Native PLpro | 0.341 | 0.200 | 2.411 | 165.201 |
| PLpro-ZINC000000596945 | 0.332 | 0.193 | 2.393 | 166.533 |
| PLpro-ZINC000064033452 | 0.308 | 0.179 | 2.409 | 168.393 |
| PLpro-VIR251 | 0.402 | 0.185 | 2.423 | 170.972 |
Next, the principal component analysis was applied to filter the essential motions from global dynamics. The changes observed in variances of principle components suggest a significant decrease in the Cα fluctuations for ligand-bound complexes, which suggests considerable changes are induced in the protein structure upon ligand binding in the trend PLpro > PLpro-ZINC000000596945 > PLpro-VIR251 > PLpro-ZINC000064033452. The essential subspace plots suggest a significant decrease in the essential mobilities of ZINC000000596945 and VIR251 bound complexes, and a greatly decreased plot PLpro-ZINC000064033452 suggests distinctive conformational changes in the structure with respect to native PLpro. The contribution of PC1 in the movement of each residue was recorded for native protein and complexes, which suggested that the BL2 loop region contributes more essential motion in ligand-bound complexes. The DCCM analysis suggested a general decrease in correlations and anti-correlations of the PLpro-ZINC000000596945 and PLpro-ZINC000064033452 complexes which corroborates with the decrease in essential mobilities observed in the PCA analysis. Next, Porcupine analysis delineates a decrease in essential motions in the overall protein structure, mostly in the Ubl and fingers domain, while an increase in the motions of the BL2 loop and β12–13 loop is observed in PLpro-ZINC000000596945 and PLpro-ZINC000064033452 which is suggestive of an induced fit model of binding which is corroborated by structural studies. This is followed by a time-dependant essential dynamics analysis, which shows the 3D representation of the protein's essential motion trajectory. For the free protein, a highly flexible BL2 loop is observed with perturbations of motion in the Ubl and fingers domain, while in a ligand-bound state, the loop remains stable and shows a concentrated closed orientation. Next, in the free energy landscape analyses, PLpro and PLpro-ZINC000000596945 attain one global minimum while PLpro-ZINC000064033452 observed two minima and the PLpro-VIR251 complex displayed six minima which suggests the highly unstable nature of the control complex. Relating the FEL and PCA results, it is observed that in PLpro, the metastable minima are achieved towards the end of the simulation, while for PLpro-ZINC000000596945, the initial states of simulation comprise the low energy basin. It should be noted that for the PLpro-ZINC000064033452 complex, the simulation's initial and final stages display a minimum, each suggestive of a stable transition from one conformation to another. Finally, binding free energy calculations corroborate the results displayed by docking, where the PLpro-ZINC000000596945 complex displayed binding free energy of − 41.23 ± 3.70 kcal/mol, PLpro-ZINC000064033452 showed a value of − 25.10 ± 2.65 kcal/mol and the control complex displays the least value of − 7.13 ± 5.69 kcal/mol.
5. Conclusion
In conclusion, the present study investigated the structural influences of ligand binding on Papain like protease from SARS-CoV-2. PLpro was screened with the Zinc-In Trials library for ligand molecules. ZINC000000596945 is an experimental drug with a known inhibitory effect against Arachidonate 5-lipoxygenase (ALXO5), a pharmaceutical target against many diseases (Funk et al., 1989). ALXO5 contributes to inflammatory responses and non-allergic reactions of the respiratory system (Haeggstrom and Funk, 2011). ZINC000064033452 is a known drug named Lumacaftor, which is used in conjugation with another drug Ivacaftor to treat patients with cystic fibrosis, which showed improvement in lung function and general symptoms of cystic fibrosis (Mayer, 2016). The global dynamics studies suggested stabilisation of protein upon binding of ZINC000000596945 and ZINC000064033452 based on RMSD, RMSF and Rg analysis, while SASA suggested sufficient binding of ZINC000000596945 and ZINC000064033452, resulting in conformational changes. The essential dynamics suggested a relative decrease in the essential mobilities of ligand-bound complexes and delineated a critical role of the BL2 loop in binding as suggested by structure studies. The FEL analyses suggested highly stable metastable states in PLpro-ZINC000064033452 at the beginning and end of the trajectory suggesting a stable transition of states. The binding free energy calculations showed the least binding energy for VIR251, followed by appreciable values for both ZINC000000596945 and ZINC000064033452, displaying them as potential inhibitors against the PLpro from SARS-CoV-2. These ligands can be further studied as lead drug molecules with appropriate experimental validation to study their applicability as drug molecules against PLpro and subsequently the disease caused by SARS-CoV-2.
CRediT authorship contribution statement
Ekampreet Singh: Data curation, Formal analysis, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. Rajat Kumar Jha: Data curation, Formal analysis, Methodology, Software, Visualization, Writing – review & editing. Rameez Jabeer Khan: Data curation, Methodology, Software, Visualization, Writing - review & editing. Ankit Kumar: Data curation, Methodology, Software, Writing – review & editing. Monika Jain: Formal analysis, Methodology, Software, Validation, Writing – review & editing. Jayaraman Muthukumaran: Conceptualization, Investigation, Supervision, Validation, Writing – review & editing. Amit Kumar Singh: Conceptualization, Investigation, Supervision, Validation, Writing – review & editing.
Acknowledgements
Dr. Amit Kumar Singh thanks Indian Council of Medical Research (ICMR) and Indian National Science Academy (INSA), New Delhi, India. The authors thank Sharda University for support.
Declaration of competing interest
None.
References
- Amin S.A., et al. Protease targeted COVID-19 drug discovery and its challenges: insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorg. Med. Chem. 2021;29 doi: 10.1016/j.bmc.2020.115860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez-Santos Y.M. X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J. Med. Chem. 2014;57(6):2393–2412. doi: 10.1021/jm401712t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez-Santos Y.M., John S.E., St, Mesecar A.D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Liu Q., Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 2020;92(4):418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020;5(4):536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIOVIA, D.S., 2019. Discovery Studio Visualization Dassault Systèmes BIOVIA.
- Deng X., et al. Structure-guided mutagenesis alters deubiquitinating activity and attenuates pathogenesis of a murine coronavirus. J. Virol. 2020;94:11. doi: 10.1128/JVI.01734-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr A.R., Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol. 2015;1282:1–23. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk C.D., et al. Characterization of the human 5-lipoxygenase gene. Proc. Natl. Acad. Sci. USA. 1989;86(8):2587–2591. doi: 10.1073/pnas.86.8.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X., et al. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B. 2021;11(1):237–245. doi: 10.1016/j.apsb.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B.J., et al. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22(21):2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- Haeggstrom J.Z., Funk C.D. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem. Rev. 2011;111(10):5866–5898. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- Hagemeijer M.C., et al. Dynamics of coronavirus replication-transcription complexes. J. Virol. 2010;84(4):2134–2149. doi: 10.1128/JVI.01716-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.S., et al. Papain-like protease 2 (PLP2) from severe acute respiratory syndrome coronavirus (SARS-CoV): expression, purification, characterization, and inhibition. Biochemistry. 2005;44(30):10349–10359. doi: 10.1021/bi0504761. [DOI] [PubMed] [Google Scholar]
- Hollingsworth S.A., et al. Conformational selectivity in cytochrome P450 redox partner interactions. Proc. Natl. Acad. Sci. USA. 2016;113(31):8723–8728. doi: 10.1073/pnas.1606474113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22(12):2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Klemm T., et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020;39(18) doi: 10.15252/embj.2020106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops K. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008;6(9) doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari R., Kumar R., Lynn A. g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Lindner H.A., et al. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 2005;79(24):15199–15208. doi: 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat. Microbiol. 2021;6(4):467–478. doi: 10.1038/s41564-021-00884-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudvand S., Shokri S. Interactions between SARS coronavirus 2 papain-like protease and immune system: a potential drug target for the treatment of COVID-19. Scand. J. Immunol. 2021;94(4) doi: 10.1111/sji.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti B.K. Can papain-like protease inhibitors halt SARS-CoV-2 replication? ACS Pharmacol. Transl. Sci. 2020;3(5):1017–1019. doi: 10.1021/acsptsci.0c00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M. Lumacaftor-ivacaftor (Orkambi) for cystic fibrosis: behind the ‘breakthrough’. Evid. Based Med. 2016;21(3):83–86. doi: 10.1136/ebmed-2015-110325. [DOI] [PubMed] [Google Scholar]
- Mishra S.K., Tripathi T. One year update on the COVID-19 pandemic: where are we now? Acta Trop. 2021;214 doi: 10.1016/j.actatropica.2020.105778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G.M. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipiuk J. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021;12(1):743. doi: 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhi A.K., Tripathi T. Targeted design of drug binding sites in the main protease of SARS-CoV-2reveals potential signatures of adaptation. Biochem. Biophys. Res. Commun. 2021;555:147–153. doi: 10.1016/j.bbrc.2021.03.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhi A.K., Tripathi T. High-throughput design of symmetrical dimeric SARS-CoV-2 main protease:structural and physical insights into hotspots for adaptation and therapeutics. Phys. Chem. Chem. Phys. 2022;24(16):9141–9145. doi: 10.1039/d2cp00171c. [DOI] [PubMed] [Google Scholar]
- Ratia K., et al. Structural Basis for the Ubiquitin-Linkage Specificity and deISGylating activity of SARS-CoV papain-like protease. PLoS Pathog. 2014;10(5) doi: 10.1371/journal.ppat.1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rut W. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti-COVID-19 drug design. Sci. Adv. 2020;6:42. doi: 10.1126/sciadv.abd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuttelkopf A.W., van Aalten D.M. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol. Crystallogr. 2004;60(Pt. 8):1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Shan H. Development of potent and selective inhibitors targeting the papain-like protease of SARS-CoV-2. Cell Chem. Biol. 2021;28(6):855–865. doi: 10.1016/j.chembiol.2021.04.020. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D., et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587(7835):657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh E., et al. In: In Silico Modeling of Drugs Against Coronaviruses: Computational Tools and Protocols. Roy K., editor. Springer US; New York, NY: 2021. Ligand-based approaches for the development of drugs against SARS-CoV-2; pp. 117–134. [Google Scholar]
- Singh E., et al. In: In Silico Modeling of Drugs Against Coronaviruses: Computational Tools and Protocols. Roy K., editor. Springer US; New York, NY: 2021. Computational drug repurposing for the development of drugs against coronaviruses; pp. 135–162. [Google Scholar]
- Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spoel D., et al. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81(Pt. 4):853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]