

Abstract
Approximately 12% of patients with acute myocardial infarction (AMI) experience a recurrent major adverse cardiovascular event within 1 year of their primary event, with most occurring within the first 90 days. Thus, there is a need for new therapeutic approaches that address this 90‐day post‐AMI high‐risk period. The formation and eventual rupture of atherosclerotic plaque that leads to AMI is elicited by the accumulation of cholesterol within the arterial intima. Cholesterol efflux, a mechanism by which cholesterol is removed from plaque, is predominantly mediated by apolipoprotein A‐I, which is rapidly lipidated to form high‐density lipoprotein in the circulation and has atheroprotective properties. In this review, we outline how cholesterol efflux dysfunction leads to atherosclerosis and vulnerable plaque formation, including inflammatory cell recruitment, foam cell formation, the development of a lipid/necrotic core, and degradation of the fibrous cap. CSL112, a human plasma‐derived apolipoprotein A‐I, is in phase 3 of clinical development and aims to reduce the risk of recurrent cardiovascular events in patients with AMI in the first 90 days after the index event by increasing cholesterol efflux. We summarize evidence from preclinical and clinical studies suggesting that restoration of cholesterol efflux by CSL112 can stabilize plaque by several anti‐inflammatory/immune‐regulatory processes. These effects occur rapidly and could stabilize vulnerable plaques in patients who have recently experienced an AMI, thereby reducing the risk of recurrent major adverse cardiovascular events in the high‐risk early post‐AMI period.
Keywords: acute myocardial infarction, apolipoprotein A‐I, atherosclerotic plaque, cholesterol efflux
Subject Categories: Acute Coronary Syndromes, Vascular Disease, Myocardial Infarction, Inflammation, Lipids and Cholesterol
Nonstandard Abbreviations and Acronyms
- ABCA1
ATP‐binding cassette transporter‐1
- CEC
cholesterol efflux capacity
- ICAM‐1
intercellular adhesion molecule 1
- LCAT
lecithin‐cholesterol acyltransferase
- MACE
major adverse cardiovascular event
- RCT
reverse cholesterol transport
- VCAM‐1
vascular cell adhesion molecule 1
Within a year of experiencing an acute myocardial infarction (AMI), ≈12% of patients will experience major adverse cardiovascular events (MACEs), with over half of these occurring in the 90‐day high‐risk period following the index event. 1 , 2 There is, therefore, an unmet need for effective therapies that reduce the risk of secondary MACEs in this high‐risk period post‐AMI.
The rupture or erosion of atherosclerotic plaque, caused by cholesterol retention in the arterial wall, is the primary pathophysiological step in AMI. 3 , 4 Reverse cholesterol transport (RCT) is the mechanism by which cholesterol is removed from atherosclerotic plaque and is transported to the liver for removal from the body. RCT is often impaired among patients with AMI, 5 leading to further cholesterol accumulation in the arterial wall, and recurrent cardiovascular events. The first step in RCT is cholesterol efflux, the transfer of cholesterol from plaque macrophages to apolipoprotein A‐I (apoA‐I), which is an essential component of high‐density lipoprotein (HDL). 6 ApoA‐I promotes cholesterol efflux primarily via the ATP‐binding cassette transporter‐1 (ABCA1). 6 CSL112 is human plasma‐derived apoA‐I formulated with phosphatidylcholine to form disc‐shaped particles suitable for intravenous infusion, and was designed to maximize cholesterol efflux from cells and exhibit favorable pharmacologic properties. 7 CSL112 fuses with native HDL, resulting in HDL remodeling and a rapid dose‐dependent increase in lipid‐poor pre‐β1 HDL concentration, which increases primarily ABCA1‐driven cholesterol efflux. 8 The completed phase 2b Apo‐I Event Reduction in Ischemic Syndromes I (AEGIS‐I) clinical trial demonstrated that CSL112 robustly and rapidly enhances cholesterol efflux capacity (CEC) in patients with AMI. 9 AEGIS‐II is a phase 3 randomized, placebo‐controlled trial evaluating the efficacy of CSL112 to reduce the risk of MACEs during the first 90 days after AMI. 10
In this review, we summarize the role of cholesterol efflux in protecting against the development of atherosclerosis and how enhancement of CEC by CSL112 can reduce vascular inflammation and stabilize vulnerable plaques, potentially reducing the risk of secondary MACEs.
THE CAUSE OF ATHEROSCLEROSIS
The Role of Low‐Density Lipoprotein, HDL, and Cholesterol Efflux
Atherosclerosis is a progressive inflammatory condition that develops over a patient’s lifetime and predisposes to AMI. 11 The formation of atherosclerotic plaque is elicited by low‐density lipoprotein (LDL)–mediated cholesterol deposition in the arterial wall with subsequent inflammatory cell recruitment, vessel remodeling, and ultimately plaque rupture or erosion, which may lead to thrombosis and AMI. 4 Elevated serum LDL cholesterol (LDL‐C) is a causative risk factor for atherosclerosis and readily modifiable with a range of established therapeutic approaches, including statins, which have efficacy in reducing plaque size after years of treatment. 12 HDL and apoA‐I counteract the pathogenic events leading to the formation and development of atheroma by promoting the removal of cholesterol from the artery wall. 13 HDL and apoA‐I have anti‐inflammatory effects via decreasing plasma membrane‐free cholesterol and lipid raft content in monocytes and neutrophils, causing a dose‐dependent reduction in their activation. 14 ApoA‐I is able to regulate cholesterol levels, and, in contrast to current LDL‐C–lowering agents, can rapidly remove cholesterol over several days. 15
LDL‐C causes atherosclerosis by accumulating in the arterial intima, where it can be modified by oxidation and aggregation. 16 , 17 Binding of LDL‐C to intimal proteoglycans is one of the first steps in disease initiation. 17 LDL‐C enters the intimal space where the endothelium is damaged and attracts macrophages to the site. Macrophages engulf LDL‐C and become activated, triggering a cascade of events resulting in macrophages and dendritic cells developing into foam cells as a result of excess cholesterol endocytosis. 4 , 18 , 19 , 20 As foam cells undergo apoptosis, lipid‐rich material is released to form a lipid core, also referred to as the necrotic core, as it contains cell debris resulting from the removal of apoptotic cells by efferocytosis. 4 Smooth muscle cell proliferation and increased collagen production below the endothelial cells result in the formation of a fibrotic cap (Figure 1). 3 , 4 , 18 , 19 , 21 , 22 , 23 , 24 Furthermore, oxidized lipids can damage the endothelium, leading to dysfunction and promoting a vicious cycle of vessel vulnerability to atherosclerotic progression. 25 , 26
Figure 1. Cholesterol efflux opposes the development of vulnerable plaque. 3 , 4 , 18 , 19 , 21 , 22 , 23 , 24 .
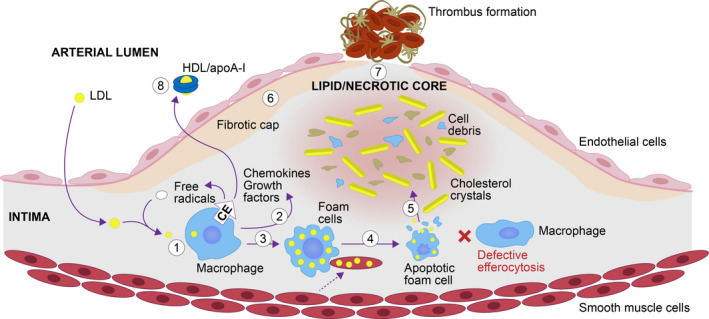
(1) Macrophages, attracted by low‐density lipoprotein (LDL), engulf LDL and become activated. (2) Activated macrophages release free radicals, chemokines, and growth factors; LDL is readily oxidized by free radicals, which further amplifies vascular inflammation. (3) Chemokines recruit macrophages and smooth muscle cells, which continue to endocytose the excess cholesterol and become foam cells. (4) Foam cells undergo apoptosis. (5) As efferocytosis is defective, apoptotic cells undergo necrosis and lipid‐rich material is released; together with cell debris, they form a lipid/necrotic core. (6) Smooth muscle cells continue to proliferate and produce collagen, forming a fibrotic cap. (7) Lipids, cytokines, and proteases from the lipid/necrotic core erode the fibrous cap, which becomes thin and susceptible to fissure and thrombosis. (8) Cholesterol efflux, mediated by high‐density lipoprotein (HDL)/apolipoprotein A‐I (apoA‐I), opposes the development of a vulnerable plaque by removing cholesterol from macrophages and preventing/reducing the formation of foam cells, reducing foam cell apoptosis, enhancing efferocytosis and lowering lipid content to prevent the formation of the lipid/necrotic core, reducing inflammation, and increasing collagen to stabilize the fibrotic cap. CE indicates cholesterol efflux.
Many atherosclerotic plaques remain stable throughout a patient’s life; however, a subset of plaques is vulnerable to rupture or erosion. Plaque disruption subsequently leads to thrombus formation and myocardial infarction (MI). 4 , 21 The characteristics of a vulnerable plaque include a thin fibrotic cap and a large lipid core. 4 , 21 Impaired CEC contributes to the development of a vulnerable plaque through plaque lipid accumulation, acute inflammation, and endothelial dysfunction. 26 , 27 On the other hand, cholesterol efflux reduces plaque cholesterol, inflammation, and apoptosis and increases efferocytosis, resulting in reduction of the lipid core. However, as atherosclerosis progresses, lipid influx outweighs efflux, 28 and consequently, apoptosis increases, efferocytosis is impeded, and apoptotic cells undergo necrosis (Figure 1). The core contains lipids, cytokines, and proteases that contribute to degradation of the plaque fibrous cap, which can then become vulnerable to rupture. When a plaque ruptures, it triggers the development of a thrombus that can block vessels and lead to an MI. 21
Cholesterol Efflux Is Atheroprotective
Cholesterol deposition incites a progressive macrophage‐dominated inflammatory response that could potentially be reversed by the activation of RCT. Cholesterol efflux is one of the counterregulatory mechanisms that oppose cholesterol accumulation and inflammation. Multiple studies have shown that a moderate cholesterol depletion leads to attenuation of the proinflammatory immune responses in various cell types that contribute to atherogenesis. Depletion of cellular cholesterol reduces the response to toll‐like receptor ligands in leukocytes, 29 decreases antigen presentation in dendritic cells, 30 reduces blood leukocyte counts, 31 and lowers platelet activation, 32 whereas elevated blood LDL levels increase adhesion molecule expression (ICAM‐1 [intercellular adhesion molecule 1] and VCAM‐1 [vascular cell adhesion molecule 1]) in endothelial cells. 33
Cholesterol efflux is considered one of the most clinically relevant atheroprotective properties of HDL as the inverse relationship between CEC and incident cardiovascular disease has been demonstrated by large prospective studies; the Dallas Heart and the European Prospective Investigation into Cancer (EPIC)‐Norfolk studies have shown an association between a high CEC and a reduced risk of cardiovascular events, including incident events. 34 , 35 , 36 , 37 A study by Khera et al showed that CEC is a stronger predictor for coronary artery disease than diabetes, hypertension, smoking, and HDL cholesterol levels. 35 CEC is also a predictor of mortality in patients with AMI, independent of lipid levels and traditional cardiovascular risk factors. 5 Furthermore, inflammation/acute‐phase response impairs CEC. 38 , 39 , 40 In line with this, CEC is reduced immediately after an AMI for at least 30 days. 41 , 42 Therefore, the reduction in CEC after AMI may contribute to the high risk of recurrent events, including death during this period. An apoA‐I–based treatment approach that increases CEC could, therefore, be highly beneficial to patients post‐AMI in reducing the risk of secondary cardiovascular events.
ATHEROPROTECTIVE EFFECTS OF CSL112
Enhanced CEC
CSL112 is designed to enhance CEC during the early post‐AMI period, when patients have particularly low CEC and, as such, it is provided as 4 weekly infusions, starting within 5 days of admission for AMI. The enhancement of CEC is hypothesized to reduce the risk of secondary MACEs in the first 90 days post‐AMI. 10
The 7 clinical trials completed to date have shown that CSL112 is well tolerated, with a renal and hepatic safety profile similar to placebo. 9 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 Infusion of CSL112 immediately increases apoA‐I levels and causes a rapid and marked increase in the capacity of serum to efflux cholesterol via ABCA1, a key transporter that mediates cholesterol efflux from macrophage foam cells (Table). 9 , 49 , 51 A 6‐g dose was selected for further development because of favorable pharmacokinetic/pharmacodynamic and safety profiles and a strong 4.3‐fold elevation in ABCA1‐dependent CEC observed in patients after AMI in a phase 2b trial. 9
Table 1.
Fold Change in CEC With Infusion of CSL112 in Patients With Stable Atherosclerotic Disease, AMI, and AMI With Renal Impairment 9 , 44 , 46
| Study name | Study type | Fold change from baseline | |
|---|---|---|---|
| Total CEC | ABCA1‐dependent CEC | ||
|
CSLCT‐HDL‐10‐70 |
Phase 2a SAD study in patients with stable atherosclerotic disease | 3.1 | … |
|
CSL112_2001 |
Phase 2 multiple‐dose study in subjects with moderate RI and AMI | 2.33 | 3.17 |
|
AEGIS‐I |
Phase 2b multiple‐dose study in patients with AMI | 2.45 | 4.3 |
AEGIS indicates Apo‐I Event Reduction in Ischemic Syndromes; ABCA1, ATP‐binding cassette transporter‐1; AMI, acute myocardial infarction; CEC, cholesterol efflux capacity; RI, renal impairment; and SAD, single‐ascending dose.
By significantly enhancing cellular cholesterol removal, CSL112 can potentially modulate multiple factors and processes that contribute to unstable plaque formation, such as plaque lipid content/necrotic core, inflammation and macrophage recruitment, and apoptosis of foam cells. In addition, increased cholesterol efflux by CSL112 may enhance foam cell efferocytosis and collagen expression, and improve overall plaque stability, including plaque growth and remodeling (Figure 1). 52 , 53 , 54 , 55 , 56 , 57 , 58 Overall, it may lead to a reduction in plaque vulnerability, and, consequently, rates of recurrent cardiovascular events (Figure 2). 9 , 52 , 53 , 54 , 55 , 56 , 57 , 58 , 59
Figure 2. Summary of pathways modulated by CSL112 and its precursor formulation (CSL111). 9 , 52 , 53 , 54 , 55 , 56 , 57 , 58 , 59 .
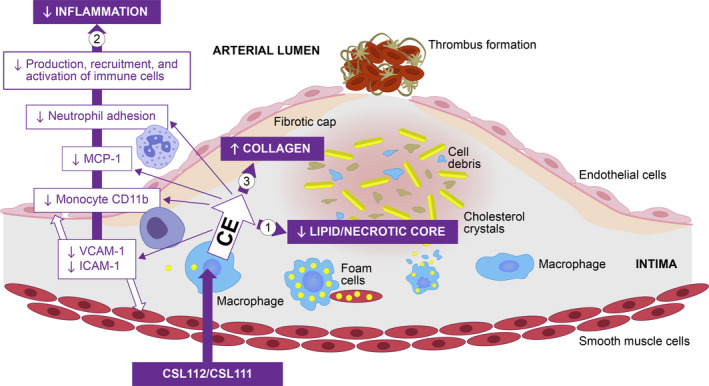
CSL112 and its precursor formulation CSL111 promote cholesterol efflux, which (1) reduces plaque lipid content and necrotic core; (2) inhibits inflammation; and (3) increases the collagen content of the plaque fibrous cap. CD11b indicates cluster of differentiation molecule 11b; CE, cholesterol efflux; ICAM‐1, intercellular adhesion molecule‐1; MCP, monocyte chemoattractant protein; and VCAM‐1, vascular cell adhesion protein 1.
Plaque Stabilization
ApoA‐I–based therapies offer the potential for stabilization/remodeling/regression of atherosclerotic plaques in animal models and humans. CSL111 is a predecessor compound formulated with higher phosphatidylcholine/apoA‐I molar ratio than CSL112. 43 , 60 A study by Murphy et al showed that when CSL111 was infused into hypercholesterolemic apolipoprotein E (apoE)−/− mice, the reduction in inflammation was accompanied by reduction in plaque lipid content, a significantly reduced necrotic core size compared with controls, and a significant increase in collagen levels. 53 Similarly, Hewing et al have reported that treating apoE−/− mice with native human apoA‐I significantly decreased lipid content and macrophage numbers in advanced aortic root atherosclerotic plaques. 61 This was associated with a reduction in markers of inflammatory M1 macrophages and an increase in anti‐inflammatory M2 macrophages within the plaque. 61 These results were similar to a randomized controlled trial that assessed coronary plaque burden using intravascular ultrasound in patients administered 4 weekly infusions of CSL111, 40 mg/kg (n=111), 80 mg/kg (n=12), or saline (n=60). 52 Following CSL111 administration, a statistically significant reduction was seen in plaque characterization index and coronary score. 52 These results were consistent with a preclinical study in mice, which showed beneficial changes in the composition of plaque. 61 The effect seen on the necrotic core was likely attributable to the reduction in both immune cell recruitment and macrophage cell death triggered by enhanced CEC. It has been shown that the removal of certain sterols by cholesterol efflux can protect macrophages from oxidized LDL‐mediated cell death. 62 Increasing CEC may, therefore, also restore efferocytosis. In another study, patients with claudication received placebo or one infusion of 80 mg/kg CSL111 5 to 7 days before percutaneous superficial femoral artery revascularization. After CSL111 treatment, both circulating monocyte activation and invasion were lower when assessed by cluster of differentiation molecule 11b (CD11b) expression, and there was evidence of a trend (P=0.05) toward decreased tumor necrosis factor‐α. Lipid content, average macrophage size, and VCAM‐I expression in the plaques were significantly lower in the CSL111 group versus placebo, demonstrating the potential for CSL111 to induce significant acute changes in plaque morphology consistent with plaque stabilization. 54
Overall, the improvement in CEC via human plasma‐derived apoA‐I in both preclinical and clinical studies is associated with plaque stabilization through plausible immune modulatory mechanisms.
Inflammatory Response and Biomarkers of Plaque Instability
Infusions of CSL111 were shown to enhance CEC and reduce systemic and plaque inflammation. 53 , 55 , 56 , 59 In hypercholesterolemic apoE−/− mice, CSL111 infusion reduced the presence of macrophages in plaques, and reduced proliferation of hematopoietic stem and progenitor cells, common myeloid progenitor cells, and granulocyte‐monocyte progenitor cells. 53 Moreover, in a mouse model of MI, an infusion of CSL111 suppressed systemic and cardiac inflammation. Cardiac chemokine levels were reduced by 60% to 80%, circulating leukocyte numbers by 30%, and monocyte expression of CD11b by up to 25%. 59
The endotoxin‐neutralizing properties of CSL111 have been demonstrated both in vitro and in animal models. 63 , 64 Furthermore, in human endotoxemia, CSL111 suppressed the endotoxin‐mediated inflammatory response, as measured by reduced inflammatory cytokines, cell activation, and clinical symptoms, such as headache, chills, nausea, vomiting, myalgia, and backache. 65 In patients with type 2 diabetes, a randomized crossover trial showed that compared with placebo, CSL111 infusion led to significant decreases in soluble VCAM‐1 levels in patient plasma, reduced CD11b expression on circulating monocytes, and reduced adhesion of patient neutrophils to fibrinogen, indicating an anti‐inflammatory effect. 55 In cultured human coronary endothelial cells, HDL isolated from patients treated with CSL111 reduced expression of VCAM‐1 and ICAM‐1. 55 Richart et al studied blood samples from this trial and found that circulating leukocyte levels were reduced by 12% after CSL111 infusion compared with placebo. 59 Similarly, CSL112 caused an inhibition of phytohemagglutinin‐M–induced upregulation of ICAM‐1 on both monocytes and neutrophils. 7
When CSL111 was infused into humans with symptomatic carotid disease (within 1 month of presentation) and the plaques were studied histologically, it was observed that systemic levels of the plasma biomarkers of inflammation associated with plaque instability, matrix metalloproteinase 9, and monocyte chemotactic factor‐1 were significantly reduced versus placebo, suggesting a reduction in monocyte activation. 56 Similarly, CSL112 was shown to inhibit the secretion of proinflammatory cytokines, including tumor necrosis factor‐α, interleukin‐1β, and interleukin‐6, and chemokines in phytohemagglutinin‐stimulated human blood, with ≈2‐fold lower concentrations of CSL112 than native HDL required to achieve a similar inhibition of tumor necrosis factor‐α and interleukin‐1β. 7 , 8 These anti‐inflammatory actions may contribute to the efficacy of CSL112 given that inhibition of interleukin‐1β (and downstream interleukin‐6) has been shown to improve cardiovascular outcomes in the CANTOS (Canakinumab Anti‐Inflammatory Thrombosis Outcome Study) trial, where the interleukin‐1β antagonist canakinumab reduced the primary composite end point of nonfatal MI, nonfatal stroke, and cardiovascular death by 15% in patients with a previous MI and baseline CRP (C‐reactive protein) ≥2 mg/L. 66
Several studies show a close relation between cholesterol efflux and inflammatory processes. It has been shown that cholesterol homeostasis contributes to hematopoietic stem and progenitor cell quiescence and, in a rheumatoid arthritis mouse model, defective CEC in hematopoietic stem and progenitor cells led to cholesterol accumulation, which, in turn, increased myelopoiesis. 67 , 68 In mouse models of autoimmunity, cholesterol accumulation in dendritic cells via defective CEC activated the inflammasome and increased inflammatory cytokine secretion. 69 Finally, a study in rats demonstrated that apoA‐I and reconstituted HDL attenuated experimentally induced arthritis and associated inflammation by inhibition of toll‐like receptor 2 expression, which can be modulated by cell membrane cholesterol content. 70 In addition to cholesterol efflux, ABCA1 receptors also mediate efflux of sphingomyelin and phosphatidylcholine. 71 , 72 Thus, it is possible that CSL112 infusion, which increases ABCA1‐dependent cholesterol efflux, 7 may alter the phospholipid composition of lipid rafts by increasing cellular efflux of other lipid species. This may also contribute to the observed anti‐inflammatory effects of CSL112, especially given that sphingomyelin depletion has been shown to reduce recruitment of toll‐like receptor 4 to the cell surface of macrophages and attenuate lipopolysaccharide‐induced inflammatory responses. 73 , 74 Overall, these findings suggest that the immunomodulatory effects of apoA‐I infusions are at least in part attributable to enhanced cholesterol efflux reducing the lipid content of immune cells.
CSL112 COMPARED WITH PREVIOUS APOA‐I INFUSION THERAPIES
Two other apoA‐I infusion therapies, MDCO‐216 (recombinant dimeric mutant apoA‐I Milano) and CER‐001 (recombinant wild‐type apoA‐I), have been investigated in phase 2 clinical trials. 75 , 76 However, the development of MDCO‐216 has since been discontinued because of a lack of efficacy on plaque regression in an imaging study, and similar results were observed in relation to CER‐001. 76 , 77 In comparison to these products, CSL112 stimulates a far more substantial increase in ABCA1‐dependent CEC (as seen in the AEGIS‐I trial) than that achieved in phase 2 studies of MDCO‐216 and CER‐001 (330% versus 80%–90% and 6%, respectively). 9 , 75 , 76 Dosing of these 3 agents in clinical studies differed substantially, which may also contribute to the disparate effects on CEC. CSL112 is administered at a total dose of 6 g, corresponding to ≈80 mg/kg based on its pharmacodynamic response, whereas MDCO‐216 and CER‐001 were dosed at 20 and 3 mg/kg, respectively. 75 , 76 In addition, CSL112 is the only apoA‐I–based product that is capable of activating lecithin‐cholesterol acyltransferase (LCAT), 7 with animal model studies showing CSL112 infusion results in an immediate 1.7‐fold increase in plasma LCAT activity 7 ; in contrast, MDCO‐216 and CER‐001 have no or inhibitory effects on LCAT, respectively. 7 , 78 , 79 , 80 , 81 LCAT converts free cholesterol into cholesteryl ester, leading to the formation of larger, mature HDL particles, which are then transported to the liver for clearance. 6 The differences observed between CSL112 and these other apoA‐I–based infusion therapies could be attributed to differences in particle composition determined by apoA‐I and lipid source, and apoA‐I/lipid molar ratio. CSL112 contains human plasma‐derived apoA‐I and soy bean phosphatidylcholine, whereas CER‐001 and MDCO‐216 are both based on recombinant apoA‐I preparations and contain sphingomyelin/dipalmitoylphosphatidylglycerol and palmitoyl oleoyl phosphatidylcholine, respectively. 7 , 80 , 82 Although CSL112 has been shown to promote LCAT activation, 7 the other 2 formulations fail to activate LCAT. 7 MDCO‐216 contains a disulfide linked homodimer form of apoA‐I (apoA‐I‐Milano), which has a reduced ability to activate LCAT. Lack of LCAT stimulation by CER‐001 is attributed to inhibitory effects of sphingomyelin. 7 , 78 , 80 , 82 The potential of CSL112 to reduce future cardiovascular events by enhancing cholesterol efflux and thereby reducing cholesterol content and/or instability of atherosclerotic plaque is being evaluated in the phase 3 AEGIS‐II study. 83
CONCLUSIONS
Cholesterol efflux and RCT are important biological processes that can alter plaque characteristics and strongly modulate immune cell function. Dysfunctional cholesterol efflux underpins atherosclerosis, the development of vulnerable plaque, and cardiovascular events. Enhancing cholesterol efflux holds promise as a therapeutic approach to treating atherosclerosis, particularly in patients who have already experienced an AMI and are at high risk of an early recurrent cardiovascular event in the 90‐day high‐risk period immediately after an AMI. Human plasma‐derived apoA‐I infusion enhances plaque cholesterol efflux as well as reduces immune cell recruitment and activation in both preclinical and clinical studies. These effects occur rapidly and could stabilize vulnerable plaques in patients who have recently experienced an AMI. The AEGIS‐II clinical outcomes trial with CSL112 apoA‐I infusions will definitively evaluate the hypothesis that enhancing CEC during this high‐risk period will lead to a reduction in the risk of recurrent MACEs.
Sources of Funding
Development of this article was funded by CSL Behring.
Disclosures
Dr Kingwell is an employee of CSL Limited and a recipient of an Honorary Senior Principal Research Fellowship from the National Health and Medical Research (NHMRC) Council of Australia. Drs Velkoska, Didichenko, and Duffy are also employees of CSL Behring. Dr Nicholls is a recipient of a Principal Research Fellowship from the NHMRC of Australia; has received research support from AstraZeneca, Amgen, Anthera, CSL Behring, Cerenis, Eli Lilly, Esperion, Resverlogix, Novartis, InfraReDx, and Sanofi‐Regeneron; and is a consultant for Amgen, Akcea, AstraZeneca, Boehringer Ingelheim, CSL Behring, Eli Lilly, Esperion, Kowa, Merck, Takeda, Pfizer, Sanofi‐Regeneron, and Novo Nordisk. Dr Gibson has received research grant support from Angel Medical Corporation, Bayer Corp, CSL Behring, Janssen Pharmaceuticals, Johnson & Johnson Corporation, and Portola Pharmaceuticals; and has received modest consulting monies from Amarin Pharma, Amgen, Arena Pharmaceuticals, Bayer Corporation, Boehringer Ingelheim, Boston Clinical Research Institute, Cardiovascular Research Foundation, Chiesi, CSL Behring, Eli Lilly, Gilead Sciences, Inc, Janssen Pharmaceuticals, Johnson & Johnson Corporation, The Medicines Company, Merk & Co, Inc, Novo Nordisk, Pfizer, Pharma Mar, Portola Pharmaceuticals, Sanofi, Somahlution, St Francis Hospital, Verson Corporation, and Web MD. Dr Korjian has no disclosures to report.
Acknowledgments
Editorial assistance was provided by Meridian HealthComms Ltd, Plumley, UK, in accordance with good publication practice (GPP3), funded by CSL Behring.
For Sources of Funding and Disclosures, see page 7.
REFERENCES
- 1. Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, Horrow J, Husted S, James S, Katus H, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045–1057. doi: 10.1056/NEJMoa0904327 [DOI] [PubMed] [Google Scholar]
- 2. Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, Neumann F‐J, Ardissino D, De Servi S, Murphy SA, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001–2015. doi: 10.1056/NEJMoa0706482 [DOI] [PubMed] [Google Scholar]
- 3. Linton MF, Yancey PG, Davies SS, Jerome WG, Linton EF, Song WL, Doran AC, Vickers KC. The role of lipids and lipoproteins in atherosclerosis. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, Grossman A, Hershman JM, Hofland HJ, Kaltsas G, et al., Endotext. MDText.com, Inc.; 2000:1–114. MDText.com, Inc. Copyright © 2000‐2020. https://www.endotext.org/section/lipids/ [PubMed] [Google Scholar]
- 4. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. 2014;114:1852–1866. doi: 10.1161/CIRCRESAHA.114.302721 [DOI] [PubMed] [Google Scholar]
- 5. Guerin M, Silvain J, Gall J, Darabi M, Berthet M, Frisdal E, Hauguel‐Moreau M, Zeitouni M, Kerneis M, Lattuca B, et al. Association of serum cholesterol efflux capacity with mortality in patients with ST‐segment elevation myocardial infarction. J Am Coll Cardiol. 2018;72:3259–3269. doi: 10.1016/j.jacc.2018.09.080 [DOI] [PubMed] [Google Scholar]
- 6. Kingwell BA, Chapman MJ. Future of high‐density lipoprotein infusion therapies: potential for clinical management of vascular disease. Circulation. 2013;128:1112–1121. doi: 10.1161/CIRCULATIONAHA.113.002683 [DOI] [PubMed] [Google Scholar]
- 7. Didichenko S, Gille A, Pragst I, Stadler D, Waelchli M, Hamilton R, Leis A, Wright SD. Novel formulation of a reconstituted high‐density lipoprotein (CSL112) dramatically enhances ABCA1‐dependent cholesterol efflux. Arterioscler Thromb Vasc Biol. 2013;33:2202–2211. doi: 10.1161/ATVBAHA.113.301981 [DOI] [PubMed] [Google Scholar]
- 8. Didichenko SA, Navdaev AV, Cukier AM, Gille A, Schuetz P, Spycher MO, Thérond P, Chapman MJ, Kontush A, Wright SD. Enhanced HDL functionality in small HDL species produced upon remodeling of HDL by reconstituted HDL, CSL112: effects on cholesterol efflux, anti‐inflammatory and antioxidative activity. Circ Res. 2016;119:751–763. doi: 10.1161/CIRCRESAHA.116.308685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Michael Gibson C, Korjian S, Tricoci P, Daaboul Y, Yee M, Jain P, Alexander JH, Steg PG, Lincoff AM, Kastelein JJP, et al. Safety and tolerability of CSL112, a reconstituted, infusible, plasma‐derived apolipoprotein A‐I, after acute myocardial infarction: the AEGIS‐I trial (ApoA‐I event reducing in ischemic syndromes I). Circulation. 2016;134:1918–1930. doi: 10.1161/CIRCULATIONAHA.116.025687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gibson CM, Kastelein JJP, Phillips AT, Aylward PE, Yee MK, Tendera M, Nicholls SJ, Pocock S, Goodman SG, Alexander JH, et al. Rationale and design of ApoA‐I event reducing in ischemic syndromes II (AEGIS‐II): a phase 3, multicenter, double‐blind, randomized, placebo‐controlled, parallel‐group study to investigate the efficacy and safety of CSL112 in subjects after acute myocardial infarction. Am Heart J. 2021;231:121–127. doi: 10.1016/j.ahj.2020.10.052 [DOI] [PubMed] [Google Scholar]
- 11. Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. 2019;124:315–327. doi: 10.1161/CIRCRESAHA.118.313591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Corti R, Fuster V, Fayad ZA, Worthley SG, Helft G, Smith D, Weinberger J, Wentzel J, Mizsei G, Mercuri M, et al. Lipid lowering by simvastatin induces regression of human atherosclerotic lesions: two years' follow‐up by high‐resolution noninvasive magnetic resonance imaging. Circulation. 2002;106:2884–2887. doi: 10.1161/01.CIR.0000041255.88750.F0 [DOI] [PubMed] [Google Scholar]
- 13. Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, Rayner K, Moore K, Garabedian M, Fisher EA. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte‐derived cells. Proc Natl Acad Sci USA. 2011;108:7166–7171. doi: 10.1073/pnas.1016086108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu X, Parks JS. New roles of HDL in inflammation and hematopoiesis. Annu Rev Nutr. 2012;32:161–182. doi: 10.1146/annurev-nutr-071811-150709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eriksson M, Carlson LA, Miettinen TA, Angelin B. Stimulation of fecal steroid excretion after infusion of recombinant proapolipoprotein A‐I. Potential reverse cholesterol transport in humans. Circulation. 1999;100:594–598. doi: 10.1161/01.CIR.100.6.594 [DOI] [PubMed] [Google Scholar]
- 16. Steinberg D, Witztum JL. Oxidized low‐density lipoprotein and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:2311–2316. doi: 10.1161/ATVBAHA.108.179697 [DOI] [PubMed] [Google Scholar]
- 17. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Mechanisms of foam cell formation in atherosclerosis. J Mol Med (Berl). 2017;95:1153–1165. doi: 10.1007/s00109-017-1575-8 [DOI] [PubMed] [Google Scholar]
- 19. Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO, Cassella CP, Moore KJ, Ramsey SA, Miano JM, et al. Cholesterol loading reprograms the microRNA‐143/145‐myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage‐like phenotype. Arterioscler Thromb Vasc Biol. 2015;35:535–546. doi: 10.1161/ATVBAHA.114.304029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Owsiany KM, Alencar GF, Owens GK. Revealing the origins of foam cells in atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2019;39:836–838. doi: 10.1161/ATVBAHA.119.312557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ahmadi A, Leipsic J, Blankstein R, Taylor C, Hecht H, Stone GW, Narula J. Do plaques rapidly progress prior to myocardial infarction? The interplay between plaque vulnerability and progression. Circ Res. 2015;117:99–104. doi: 10.1161/CIRCRESAHA.117.305637 [DOI] [PubMed] [Google Scholar]
- 22. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118:653–667. doi: 10.1161/CIRCRESAHA.115.306256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr M, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Z, O'Neill EA, Meurer RD, Gagen K, Luell S, Wang S‐P, Ichetovkin M, Frantz‐Wattley B, Eveland S, Strack AM, et al. Reconstituted HDL elicits marked changes in plasma lipids following single‐dose injection in c57bl/6 mice. J Cardiovasc Pharmacol Ther. 2012;17:315–323. doi: 10.1177/1074248411426144 [DOI] [PubMed] [Google Scholar]
- 25. Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, Hegele RA, Krauss RM, Raal FJ, Schunkert H, et al. Low‐density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38:2459–2472. doi: 10.1093/eurheartj/ehx144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Monette JS, Hutchins PM, Ronsein GE, Wimberger J, Irwin AD, Tang C, Sara JD, Shao B, Vaisar T, Lerman A, et al. Patients with coronary endothelial dysfunction have impaired cholesterol efflux capacity and reduced HDL particle concentration. Circ Res. 2016;119:83–90. doi: 10.1161/CIRCRESAHA.116.308357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vaisar T, Tang C, Babenko I, Hutchins P, Wimberger J, Suffredini AF, Heinecke JW. Inflammatory remodeling of the HDL proteome impairs cholesterol efflux capacity. J Lipid Res. 2015;56:1519–1530. doi: 10.1194/jlr.M059089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tall AR, Yvan‐Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15:104–116. doi: 10.1038/nri3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koberlin MS, Heinz LX, Superti‐Furga G. Functional crosstalk between membrane lipids and TLR biology. Curr Opin Cell Biol. 2016;39:28–36. doi: 10.1016/j.ceb.2016.01.010 [DOI] [PubMed] [Google Scholar]
- 30. Wang SH, Yuan SG, Peng DQ, Zhao SP. High‐density lipoprotein affects antigen presentation by interfering with lipid raft: a promising anti‐atherogenic strategy. Clin Exp Immunol. 2010;160:137–142. doi: 10.1111/j.1365-2249.2009.04068.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scheiermann C, Frenette PS, Hidalgo A. Regulation of leucocyte homeostasis in the circulation. Cardiovasc Res. 2015;107:340–351. doi: 10.1093/cvr/cvv099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grgurevich S, Krishnan R, White MM, Jennings LK. Role of in vitro cholesterol depletion in mediating human platelet aggregation. J Thromb Haemost. 2003;1:576–586. doi: 10.1046/j.1538-7836.2003.00087.x [DOI] [PubMed] [Google Scholar]
- 33. Verna L, Ganda C, Stemerman MB. In vivo low‐density lipoprotein exposure induces intercellular adhesion molecule‐1 and vascular cell adhesion molecule‐1 correlated with activator protein‐1 expression. Arterioscler Thromb Vasc Biol. 2006;26:1344–1349. doi: 10.1161/01.ATV.0000222152.83069.3f [DOI] [PubMed] [Google Scholar]
- 34. Qiu C, Zhao X, Zhou Q, Zhang Z. High‐density lipoprotein cholesterol efflux capacity is inversely associated with cardiovascular risk: a systematic review and meta‐analysis. Lipids Health Dis. 2017;16:212. doi: 10.1186/s12944-017-0604-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khera AV, Cuchel M, de la Llera‐Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, et al. Cholesterol efflux capacity, high‐density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–2393. doi: 10.1056/NEJMoa1409065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saleheen D, Scott R, Javad S, Zhao W, Rodrigues A, Picataggi A, Lukmanova D, Mucksavage ML, Luben R, Billheimer J, et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: a prospective case‐control study. Lancet Diabetes Endocrinol. 2015;3:507–513. doi: 10.1016/S2213-8587(15)00126-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Feingold KR, Grunfeld C. The acute phase response inhibits reverse cholesterol transport. J Lipid Res. 2010;51:682–684. doi: 10.1194/jlr.E005454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han CY, Tang C, Guevara ME, Wei H, Wietecha T, Shao B, Subramanian S, Omer M, Wang S, O’Brien KD, et al. Serum amyloid A impairs the antiinflammatory properties of HDL. J Clin Invest. 2016;126:266–281. doi: 10.1172/JCI83475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McGillicuddy FC, de la Llera MM, Hinkle CC, Joshi MR, Chiquoine EH, Billheimer JT, Rothblat GH, Reilly MP. Inflammation impairs reverse cholesterol transport in vivo. Circulation. 2009;119:1135–1145. doi: 10.1161/CIRCULATIONAHA.108.810721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Soares AAS, Tavoni TM, de Faria EC, Remalay AT, Maranhão RC, Sposito AC. HDL acceptor capacities for cholesterol efflux from macrophages and lipid transfer are both acutely reduced after myocardial infarction. Clin Chim Acta. 2018;478:51–56. doi: 10.1016/j.cca.2017.12.031 [DOI] [PubMed] [Google Scholar]
- 42. Rached F, Lhomme M, Camont L, Gomes F, Dauteuille C, Robillard P, Santos RD, Lesnik P, Serrano CV Jr, John Chapman M, et al. Defective functionality of small, dense HDL3 subpopulations in ST segment elevation myocardial infarction: relevance of enrichment in lysophosphatidylcholine, phosphatidic acid and serum amyloid A. Biochim Biophys Acta. 2015;1851:1254–1261. doi: 10.1016/j.bbalip.2015.05.007 [DOI] [PubMed] [Google Scholar]
- 43. Easton R, Gille A, D'Andrea D, Davis R, Wright SD, Shear C. A multiple ascending dose study of CSL112, an infused formulation of ApoA‐I. J Clin Pharmacol. 2014;54:301–310. doi: 10.1002/jcph.194 [DOI] [PubMed] [Google Scholar]
- 44. Gibson CM, Kerneis M, Yee MK, Daaboul Y, Korjian S, Mehr AP, Tricoci P, Alexander JH, Kastelein JJP, Mehran R, et al. The CSL112‐2001 trial: safety and tolerability of multiple doses of CSL112 (apolipoprotein A‐I [human]), an intravenous formulation of plasma‐derived apolipoprotein A‐I, among subjects with moderate renal impairment after acute myocardial infarction. Am Heart J. 2019;208:81–90. doi: 10.1016/j.ahj.2018.11.008 [DOI] [PubMed] [Google Scholar]
- 45. Gille A, Duffy D, Tortorici MA, Wright SD, Deckelbaum LI, D'Andrea DM. Moderate renal impairment does not impact the ability of CSL112 (apolipoprotein A‐I [human]) to enhance cholesterol efflux capacity. J Clin Pharmacol. 2019;59:427–436. doi: 10.1002/jcph.1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gille A, D'Andrea D, Tortorici MA, Hartel G, Wright SD. CSL112 (apolipoprotein A‐I [human]) enhances cholesterol efflux similarly in healthy individuals and stable atherosclerotic disease patients. Arterioscler Thromb Vasc Biol. 2018;38:953–963. doi: 10.1161/ATVBAHA.118.310538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gille A, Easton R, D'Andrea D, Wright SD, Shear CL. CSL112 enhances biomarkers of reverse cholesterol transport after single and multiple infusions in healthy subjects. Arterioscler Thromb Vasc Biol. 2014;34:2106–2114. doi: 10.1161/ATVBAHA.114.303720 [DOI] [PubMed] [Google Scholar]
- 48. Tortorici MA, Duffy D, Evans R, Feaster J, Gille A, Mant TGK, Wright SD, D'Andrea D. Pharmacokinetics and safety of CSL112 (apolipoprotein A‐I [human]) in adults with moderate renal impairment and normal renal function. Clin Pharmacol Drug Dev. 2019;8:628–636. doi: 10.1002/cpdd.618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tricoci P, D'Andrea DM, Gurbel PA, Yao Z, Cuchel M, Winston B, Schott R, Weiss R, Blazing MA, Cannon L, et al. Infusion of reconstituted high‐density lipoprotein, CSL112, in patients with atherosclerosis: safety and pharmacokinetic results from a phase 2a randomized clinical trial. J Am Heart Assoc. 2015;4:e002171. doi: 10.1161/JAHA.115.002171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tortorici M, Clementi R, Feaster J, Zheng B, Duffy D, Dalitz P, Airey J, Roberts J. CSL112 (apolipoprotein A‐I [human]) similarly enhances cholesterol efflux capacity in Japanese and caucasian subjects: findings from a phase 1 study. J Am Coll Cardiol. 2021;77:131–131. doi: 10.1016/S0735-1097(21)01490-X [DOI] [Google Scholar]
- 51. Rader DJ. Molecular regulation of HDL metabolism and function: implications for novel therapies. J Clin Invest. 2006;116:3090–3100. doi: 10.1172/JCI30163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tardif JC, Grégoire J, L'Allier PL, Ibrahim R, Lespérance J, Heinonen TM, Kouz S, Berry C, Basser R, Lavoie MA, et al. Effects of reconstituted high‐density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA. 2007;297:1675–1682. doi: 10.1001/jama.297.15.jpc70004 [DOI] [PubMed] [Google Scholar]
- 53. Murphy AJ, Funt S, Gorman D, Tall AR, Wang N. Pegylation of high‐density lipoprotein decreases plasma clearance and enhances antiatherogenic activity. Circ Res. 2013;113:e1–e9. doi: 10.1161/CIRCRESAHA.113.301112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shaw JA, Bobik A, Murphy A, Kanellakis P, Blombery P, Mukhamedova N, Woollard K, Lyon S, Sviridov D, Dart AM. Infusion of reconstituted high‐density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ Res. 2008;103:1084–1091. doi: 10.1161/CIRCRESAHA.108.182063 [DOI] [PubMed] [Google Scholar]
- 55. Patel S, Drew BG, Nakhla S, Duffy SJ, Murphy AJ, Barter PJ, Rye K‐A, Chin‐Dusting J, Hoang A, Sviridov D, et al. Reconstituted high‐density lipoprotein increases plasma high‐density lipoprotein anti‐inflammatory properties and cholesterol efflux capacity in patients with type 2 diabetes. J Am Coll Cardiol. 2009;53:962–971. doi: 10.1016/j.jacc.2008.12.008 [DOI] [PubMed] [Google Scholar]
- 56. Nasr H, Torsney E, Poston RN, Hayes L, Gaze DC, Basser R, Thompson MM, Loftus IM, Cockerill GW. Investigating the effect of a single infusion of reconstituted high‐density lipoprotein in patients with symptomatic carotid plaques. Ann Vasc Surg. 2015;29:1380–1391. doi: 10.1016/j.avsg.2015.04.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Spieker LE, Sudano I, Hürlimann D, Lerch PG, Lang MG, Binggeli C, Corti R, Ruschitzka F, Lüscher TF, Noll G. High‐density lipoprotein restores endothelial function in hypercholesterolemic men. Circulation. 2002;105:1399–1402. doi: 10.1161/01.CIR.0000013424.28206.8F [DOI] [PubMed] [Google Scholar]
- 58. Tanaka S, Genève C, Zappella N, Yong‐Sang J, Planesse C, Louedec L, Viranaïcken W, Bringart M, Montravers P, Denamur E, et al. Reconstituted high‐density lipoprotein therapy improves survival in mouse models of sepsis. Anesthesiology. 2020;132:825–838. doi: 10.1097/ALN.0000000000003155 [DOI] [PubMed] [Google Scholar]
- 59. Richart AL, Reddy M, Khalaji M, Natoli AL, Heywood SE, Siebel AL, Lancaster GL, Murphy AJ, Carey AL, Drew BG, et al. Apo AI nanoparticles delivered post myocardial infarction moderate inflammation. Circ Res. 2020;127:1422–1436. doi: 10.1161/CIRCRESAHA.120.316848 [DOI] [PubMed] [Google Scholar]
- 60. Herzog E, Pragst I, Waelchli M, Gille A, Schenk S, Mueller‐Cohrs J, Diditchenko S, Zanoni P, Cuchel M, Seubert A, et al. Reconstituted high‐density lipoprotein can elevate plasma alanine aminotransferase by transient depletion of hepatic cholesterol: role of the phospholipid component. J Appl Toxicol. 2016;36:1038–1047. doi: 10.1002/jat.3264 [DOI] [PubMed] [Google Scholar]
- 61. Hewing B, Parathath S, Barrett T, Chung WKK, Astudillo YM, Hamada T, Ramkhelawon B, Tallant TC, Yusufishaq MSS, DiDonato JA, et al. Effects of native and myeloperoxidase‐modified apolipoprotein A‐I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2014;34:779–789. doi: 10.1161/ATVBAHA.113.303044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Terasaka N, Wang N, Yvan‐Charvet L, Tall AR. High‐density lipoprotein protects macrophages from oxidized low‐density lipoprotein‐induced apoptosis by promoting efflux of 7‐ketocholesterol via ABCG1. Proc Natl Acad Sci USA. 2007;104:15093–15098. doi: 10.1073/pnas.0704602104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Parker TS, Levine DM, Chang JC, Laxer J, Coffin CC, Rubin AL. Reconstituted high‐density lipoprotein neutralizes gram‐negative bacterial lipopolysaccharides in human whole blood. Infect Immun. 1995;63:253–258. doi: 10.1128/iai.63.1.253-258.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Levine DM, Parker TS, Donnelly TM, Walsh A, Rubin AL. In vivo protection against endotoxin by plasma high density lipoprotein. Proc Natl Acad Sci USA. 1993;90:12040–12044. doi: 10.1073/pnas.90.24.12040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pajkrt D, Doran JE, Koster F, Lerch PG, Arnet B, van der Poll T, ten Cate JW, van Deventer SJ. Antiinflammatory effects of reconstituted high‐density lipoprotein during human endotoxemia. J Exp Med. 1996;184:1601–1608. doi: 10.1084/jem.184.5.1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- 67. Dragoljevic D, Kraakman MJ, Nagareddy PR, Ngo D, Shihata W, Kammoun HL, Whillas A, Lee MKS, Al‐Sharea A, Pernes G, et al. Defective cholesterol metabolism in haematopoietic stem cells promotes monocyte‐driven atherosclerosis in rheumatoid arthritis. Eur Heart J. 2018;39:2158–2167. doi: 10.1093/eurheartj/ehy119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Westerterp M, Gourion‐Arsiquaud S, Murphy AJ, Shih A, Cremers S, Levine RL, Tall AR, Yvan‐Charvet L. Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell. 2012;11:195–206. doi: 10.1016/j.stem.2012.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Westerterp M, Gautier EL, Ganda A, Molusky MM, Wang W, Fotakis P, Wang N, Randolph GJ, D'Agati VD, Yvan‐Charvet L, et al. Cholesterol accumulation in dendritic cells links the inflammasome to acquired immunity. Cell Metab. 2017;25:1294–1304.e1296. doi: 10.1016/j.cmet.2017.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wu BJ, Ong KL, Shrestha S, Chen K, Tabet F, Barter PJ, Rye KA. Inhibition of arthritis in the lewis rat by apolipoprotein A‐I and reconstituted high‐density lipoproteins. Arterioscler Thromb Vasc Biol. 2014;34:543–551. doi: 10.1161/ATVBAHA.113.302832 [DOI] [PubMed] [Google Scholar]
- 71. Hotta N, Abe‐Dohmae S, Taguchi R, Yokoyama S. Preferential incorporation of shorter and less unsaturated acyl phospholipids into high density lipoprotein‐like particles in the ABCA1‐ and ABCA7‐mediated biogenesis with apoA‐I. Chem Phys Lipids. 2015;187:1–9. doi: 10.1016/j.chemphyslip.2015.01.005 [DOI] [PubMed] [Google Scholar]
- 72. Schifferer R, Liebisch G, Bandulik S, Langmann T, Dada A, Schmitz G. ApoA‐I induces a preferential efflux of monounsaturated phosphatidylcholine and medium chain sphingomyelin species from a cellular pool distinct from HDL(3) mediated phospholipid efflux. Biochim Biophys Acta. 2007;1771:853–863. doi: 10.1016/j.bbalip.2007.04.011 [DOI] [PubMed] [Google Scholar]
- 73. Gowda S, Yeang C, Wadgaonkar S, Anjum F, Grinkina N, Cutaia M, Jiang XC, Wadgaonkar R. Sphingomyelin synthase 2 (SMS2) deficiency attenuates LPS‐induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2011;300:L430–L440. doi: 10.1152/ajplung.00208.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hailemariam TK, Huan C, Liu J, Li Z, Roman C, Kalbfeisch M, Bui HH, Peake DA, Kuo MS, Cao G, et al. Sphingomyelin synthase 2 deficiency attenuates NFkappaB activation. Arterioscler Thromb Vasc Biol. 2008;28:1519–1526. doi: 10.1161/ATVBAHA.108.168682 [DOI] [PubMed] [Google Scholar]
- 75. Nicholls SJ, Puri R, Ballantyne CM, Jukema JW, Kastelein JJP, Koenig W, Wright RS, Kallend D, Wijngaard P, Borgman M, et al. Effect of infusion of high‐density lipoprotein mimetic containing recombinant apolipoprotein A‐I Milano on coronary disease in patients with an acute coronary syndrome in the MILANO‐PILOT trial: a randomized clinical trial. JAMA Cardiol. 2018;3:806–814. doi: 10.1001/jamacardio.2018.2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nicholls SJ, Andrews J, Kastelein JJP, Merkely B, Nissen SE, Ray KK, Schwartz GG, Worthley SG, Keyserling C, Dasseux JL, et al. Effect of serial infusions of CER‐001, a pre‐beta high‐density lipoprotein mimetic, on coronary atherosclerosis in patients following acute coronary syndromes in the CER‐001 atherosclerosis regression acute coronary syndrome trial: a randomized clinical trial. JAMA Cardiol. 2018;3:815–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Business Wire . The medicines company discontinues development of MDCO‐216, its investigational cholesterol efflux promoter. 2016. Available at: https://www.businesswire.com/news/home/20161107006553/en/the‐medicines‐company‐discontinues‐development‐of‐MDCO‐216‐its‐investigational‐cholesterol‐efflux‐promoter. Accessed June 4, 2021.
- 78. Calabresi L, Franceschini G, Burkybile A, Jonas A. Activation of lecithin cholesterol acyltransferase by a disulfide‐linked apolipoprotein A‐I dimer. Biochem Biophys Res Commun. 1997;232:345–349. doi: 10.1006/bbrc.1997.6286 [DOI] [PubMed] [Google Scholar]
- 79. Kempen HJ, Gomaraschi M, Simonelli S, Calabresi L, Moerland M, Otvos J, Jeyarajah E, Kallend D, Wijngaard PLJ. Persistent changes in lipoprotein lipids after a single infusion of ascending doses of MDCO‐216 (apoA‐IMilano/POPC) in healthy volunteers and stable coronary artery disease patients. Atherosclerosis. 2016;255:17–24. doi: 10.1016/j.atherosclerosis.2016.10.042 [DOI] [PubMed] [Google Scholar]
- 80. Tardy C, Goffinet M, Boubekeur N, Ackermann R, Sy G, Bluteau A, Cholez G, Keyserling C, Lalwani N, Paolini JF, et al. CER‐001, a HDL‐mimetic, stimulates the reverse lipid transport and atherosclerosis regression in high cholesterol diet‐fed LDL‐receptor deficient mice. Atherosclerosis. 2014;232:110–118. doi: 10.1016/j.atherosclerosis.2013.10.018 [DOI] [PubMed] [Google Scholar]
- 81. Bolin DJ, Jonas A. Sphingomyelin inhibits the lecithin‐cholesterol acyltransferase reaction with reconstituted high density lipoproteins by decreasing enzyme binding. J Biol Chem. 1996;271:19152–19158. doi: 10.1074/jbc.271.32.19152 [DOI] [PubMed] [Google Scholar]
- 82. Alexander ET, Weibel GL, Joshi MR, Vedhachalam C, de la Llera‐Moya M, Rothblat GH, Phillips MC, Rader DJ. Macrophage reverse cholesterol transport in mice expressing ApoA‐I Milano. Arterioscler Thromb Vasc Biol. 2009;29:1496–1501. doi: 10.1161/ATVBAHA.109.191379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rader DJ. Apolipoprotein A‐I infusion therapies for coronary disease: two outs in the ninth inning and swinging for the fences. JAMA Cardiol. 2018;3:799–801. doi: 10.1001/jamacardio.2018.2168 [DOI] [PubMed] [Google Scholar]