

Abstract
Background
Epigenetic modulators have been proposed as promising new drug targets to treat adverse remodeling in heart failure. Here, we evaluated the potential of 4 epigenetic drugs, including the recently developed histone deacetylase 6 (HDAC6) inhibitor JS28, to prevent endothelin‐1 induced pathological gene expression in cardiac myocytes and analyzed the chromatin binding profile of the respective inhibitor targets.
Methods and Results
Cardiac myocytes were differentiated and puromycin‐selected from mouse embryonic stem cells and treated with endothelin‐1 to induce pathological gene expression (938 differentially expressed genes, q<0.05). Dysregulation of gene expression was at least in part prevented by epigenetic inhibitors, including the pan‐BRD (bromodomain‐containing protein) inhibitor bromosporine (290/938 genes), the BET (bromodomain and extraterminal) inhibitor JQ1 (288/938), the broad‐spectrum HDAC inhibitor suberoylanilide hydroxamic acid (227/938), and the HDAC6 inhibitor JS28 (210/938). Although the 4 compounds were similarly effective toward pathological gene expression, JS28 demonstrated the least adverse effects on physiological gene expression. Genome‐wide chromatin binding profiles revealed that HDAC6 binding sites were preferentially associated with promoters of genes involved in RNA processing. In contrast, BRD4 binding was associated with genes involved in core cardiac myocyte functions, for example, myocyte contractility, and showed enrichment at enhancers and intronic regions. These distinct chromatin binding profiles of HDAC6 and BRD4 might explain the different effects of their inhibitors on pathological versus physiological gene expression.
Conclusions
In summary, we demonstrated, that the HDAC6 inhibitor JS28 effectively prevented the adverse effects of endothelin‐1 on gene expression with minor impact on physiological gene expression in cardiac myocytes. Selective HDAC6 inhibition by JS28 appears to be a promising strategy for future evaluation in vivo and potential translation into clinical application.
Keywords: bromodomain‐containing protein, cardiac myocyte, epigenetics, heart, heart failure, histone deacetylase
Subject Categories: Heart Failure, Remodeling, Basic Science Research
Nonstandard Abbreviations and Acronyms
- BRD
bromodomain‐containing protein
- ES‐CM
embryonic stem cell‐derived cardiac myocyte
- ET‐1
endothelin‐1
- HDAC
histone deacetylase
Clinical Perspective.
What Is New?
The pan‐BRD (bromodomain‐containing protein) inhibitor bromosporine, the BET (bromodomain and extraterminal) inhibitor JQ1, the broad‐spectrum histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid, and the HDAC6 inhibitor JS28 were able to attenuate pathological gene expression in cardiac myocytes.
The HDAC6 inhibitor JS28 showed the most desirable profile as it effectively restored gene expression with minor impact on physiological gene expression.
Chromatin immunoprecipitation enabled genome‐wide profiling of HDAC6 and BRD4 binding in cardiac myocytes.
What Are the Clinical Implications?
Pharmacological inhibition of HDAC6 by JS28 may be an effective strategy to prevent progressive organ injury in heart failure.
In chronic heart failure, impaired cardiac function leads to increased neurohormonal activity. While this serves as a compensatory mechanism to maintain blood pressure and organ perfusion in the short term, sustained activation accelerates deleterious cardiac remodeling, including cardiomyocyte (CM) hypertrophy and fibrosis. Accordingly, many established pharmacotherapies for chronic heart failure aim to mitigate disease progression by inhibiting neurohormonal activity. 1 , 2 Emerging evidence suggests that epigenetic regulation of pathological gene expression plays an important role as mediator of organ damage, thus representing a potential therapeutic target. 2 , 3
Many genes that modulate the progression of cardiac remodeling have been identified, and there is growing knowledge about the transcriptional and epigenetic mechanisms controlling their expression. In particular, reversible histone acetylation, affecting chromatin state and gene expression, is known to play a regulatory role in pathological gene expression of heart failure. 4 Histone acetyltransferases and histone deacetylases (HDACs) antagonistically control the acetylation levels at histone tails by transferring or removing acetyl groups, respectively, to lysine residues. 5 BRDs (bromodomain‐containing proteins) function as reader proteins of acetylated histone tails, translating the acetylation signals into downstream signaling cascades. 5 In addition, histone acetyltransferases and HDACs may acetylate and deacetylate numerous nonhistone proteins in both the nucleus and cytoplasm, thereby affecting multiple cellular functions. 6 , 7 Inhibitors of HDAC activity have been shown to have beneficial effects in preclinical models of heart failure. 8 , 9 , 10 The epigenetic reader protein BRD4 takes a pivotal role as transcriptional coactivator of pathological gene expression that drives CM hypertrophy and heart failure progression. 11 , 12 The importance of BRD4 in the context of heart failure has been demonstrated in experimental studies, primarily using the BET (bromodomain and extraterminal) inhibitor JQ1. 11 , 12 , 13 , 14 Treatment with JQ1 prevented stress‐induced gene expression and CM hypertrophy in primary culture models and in mouse models of heart failure. JQ1‐treated mice showed improved cardiac function and attenuation of cardiac histopathological features of heart failure. 11 , 12 , 13
Subsequently, small molecule compounds targeting BRD4 or HDACs but also numerous other epigenetic modulators have been developed. 15 In a screening experiment the recently developed HDAC6 inhibitor JS28 (previously compound 4g 16 ) showed promise to prevent adverse gene expression in CM. Here, we systematically assess the impact of JS28 on physiological and ET‐1 induced gene expression in CM in comparison to other epigenetic drugs.
METHODS
Detailed information on RNA‐seq, chromatin immunoprecipitation (ChIP)‐seq, and assay for transposase‐accessible chromatin (ATAC)‐seq experiments is listed in Table S5. Raw data are available at the National Center for Biotechnology Information Sequence Read Archive (BioProject ID PRJNA788870).
Cell Culture
Male transgenic mouse embryonic stem cell (ESC) line D3/αPIG44 expressing puromycin resistance and enhanced green fluorescent protein cassettes under control of the cardiac myocyte‐specific αMHC (alpha myosin heavy chain) promoter were generated as previously described. 17 , 18 ESC were maintained on mouse embryonic fibroblasts that were mitotically inactivated with mitomycin C, in Dulbecco's modified Eagle's medium (Gibco, #11960–085) supplemented with 15% (v/v) fetal bovine serum (Gibco, #10270–106), 2 mmol/L l‐glutamine (Gibco), 100 U/μg per mL penicillin/streptomycin (Gibco), 1x non‐essential amino acids (Gibco) and 100 μmol/L β‐mercaptoethanol and 1000 U/mL LIF (leukemia inhibitory factor; ESGRO, Merck Millipore). Cells were passaged regularly every second day.
ESC Differentiation
ESCs were differentiated by mass culture method to obtain cardiac myocytes. 19 Therefore, 106 ESCs were suspended in differentiation medium (day 0), consisting of IMDM (Gibco, #21980065) supplemented with 20% (v/v) fetal bovine serum, 2 mmol/L l‐glutamine (Gibco), 100 U/μg per mL penicillin/streptomycin (Gibco), 1x nonessential amino acids (Gibco), and 100 μmol/L β‐mercaptoethanol. Cell suspension was cultured in a sterile 10 cm bacterial petri dish on a horizontal shaker (GFL 3006, Greiner) at 75 rpm. After 2 days of incubation formed embryoid bodies (EBs) were diluted to 2000 EBs per petri dish and further cultured in differentiation medium. Medium was changed every 2 to 3 days. After detection of first GFP+ (green fluorescent protein transgenic) clusters followed by spontaneous beating inside EBs, 9 μg/mL puromycin was added on days 9 to 11 of differentiation for a total of 3 days. Following 3 days of puromycin selection, embryonic stem cell‐derived cardiac myocytes (ES‐CM) were dissociated enzymatically from EB with 0.25% trypsin–EDTA. Resulting ES‐CM were seeded in puromycin‐supplemented differentiation medium supplemented on fibronectin‐coated plates or dishes for further experiments (Figure S1A). All experiments were conducted in accordance with institutional guidelines.
Immunofluorescence Staining
For immunocytochemical staining, 5×104 ES‐CM were seeded after EB dissociation on fibronectin‐coated glass coverslips in 24‐well plate. Cells were rinsed with PBS and fixed with 4% paraformaldehyde for 15 minutes. After a 3‐fold washing step with PBST (0.1% Tween20 in PBS) cells were permeabilized with 0.2% Triton X‐100 in PBS for 20 minutes. Blocking of unspecific bindings was performed with 5% bovine serum albumin in PBST for 2 hours, followed by staining with primary antibody against α‐actinin (1:500, A7811, Sigma) in blocking buffer overnight at 4 °C. Cells were washed before incubation with secondary antibody AlexaFluor488 (1:500; A‐11029, Invitrogen) in PBST at room temperature for 1 hour in the dark. Subsequently, DNA was stained with 4′,6‐diamidine‐2‐phenylindole (DAPI, 50 ng/mL in PBS). Samples were washed 3 times with PBS and mounted. Confocal fluorescence microscopy was performed using an inverted Axiovert 200M microscope (Carl Zeiss, Jena, Germany) with a CSU‐X1 spinning‐disk (Yokogawa, Tokyo, Japan) equipped with an emission filter wheel and solid‐state laser lines (405, 488, and 561 nm). Image processing was performed using the ImageJ software.
Stimulation Experiment
ES‐CM were deprived of fetal bovine serum for 24 hours by changing medium to starvation medium consisting of M199 (SigmaAldrich, #M7528) supplemented with 0.1% fetal bovine serum, 2 mmol/L l‐glutamine (Gibco) and 100 U/μg per mL penicillin/streptomycin (Gibco). The following day, the medium was changed again, supplemented with 100 nmol/L endothelin‐1 (ET‐1), and incubated for additional 24 hours before RNA extraction. For inhibitor testing, ES‐CM were preincubated with epigenetic inhibitors (final concentrations, see Table S1) starting 6 hours before ET‐1 was added. Final dimethyl sulfoxide concentration was 0.5% in all experimental groups including controls. For stimulation experiments, ES‐CM were distributed to culture wells, randomly assigned to treatment groups, and treatments independently administered to each well, with n values representing the number of wells. ES‐CM differentiation and stimulation were replicated in 2 independent experiments with at least 1 well per treatment group per experiment.
Gene Expression Analysis
Total RNA was isolated from cultured ES‐CM using Quick‐RNA Micropep (Zymo Reseach) and quantified by NanoDrop (Thermo Fisher). The isolated RNA was transcribed into cDNA using QuantiTect Reverse Transcription Kit (Qiagen) according to the manufacturer's protocol and SYBR green reverse transcription quantitative polymerase chain reaction (PCR) was performed with custom primers (Table S2). Ribosomal protein S29 (Rps29) was used as reference gene.
RNA‐Sequencing
For RNA sequencing of ES‐CM, an amplified cDNA library was prepared from 10 ng of isolated RNA using the Ovation SoLo RNA‐Seq Library Preparation Kit (NuGEN) according to the manufacturer's instructions. Before amplification, the number of amplification cycles for each sample was determined individually via reverse transcription PCR. Agencourt AMPure XP Beads (Beckman Coulter) were used for purification and size selection. DNA concentration was determined using the Qubit dsDNA HS (High Sensitivity) Assay (Life Technologies). The distribution of DNA fragment sizes was determined using the High Sensitivity DNA Kit in the 2100 Bioanalyzer (Agilent). Sequencing of all samples was performed on either the HiSeq 3000 or NextSeq 500 sequencers (75 base pair [bp] paired end, Illumina) at the Max Planck Institute for Immunobiology and Epigenetics in Freiburg, Germany, or the EMBL genomics core facility, Heidelberg, Germany.
ChIP‐Sequencing
To perform ChIP of histone modifications, chromatin was isolated from 1 to 2×106 ES‐CM using the ChIP‐IT High Sensitivity Kit from Active Motif according to the manufacturer's instructions. Cell fixation was performed with 1% formaldehyde for 10 minutes and cell lysis was carried out using Bioruptor (Diagenode) with 7 to 9 cycles (30 seconds on/30 seconds off, high intensity). Chromatin shearing was performed with the same settings in the Bioruptor for 3×10 cycles. For each ChIP experiment, 0.2 to 1 μg chromatin (measured as DNA) was used.
To perform ChIP from HDAC6, BRD4, and RNA polymerase II, chromatin was isolated from 1 to 4×106 ES‐CM using the iDeal ChIP‐seq kit for Transcription Factors from Diagenode according to the manufacturer's instructions. Cells were fixed with 1% formaldehyde for 10 minutes. Chromatin was sheared using Bioruptor (Diagenode) for 3×10 cycles (30 seconds on/30 seconds off, high intensity). For each ChIP experiment, 1.5 to 30 μg chromatin (measured as DNA) was used. Immunocomplexes were purified using either protein A‐ or protein G‐coated magnetic beads, depending on the antibody. All antibodies are listed in Table S3. Library preparation was performed using the NEBNext Ultra DNA Library Prep Lot (NEB) according to the manufacturer's manual. Purification of the amplified cDNA library was performed using Agencourt AMPure XP beads (Beckman Coulter). Quantification and sequencing of the final DNA library was performed as described before.
ATAC‐Sequencing
Following the protocol of Buenrostro et al, 20 50 000 ES‐CM were lysed and processed. Tagmented and amplified DNA were purified using the MinElute Reaction Cleanup Kit from Qiagen according to the manufacturer’s instructions and eluted in 10 μL EB‐buffer (Qiagen). Size selection of libraries was performed using PAGE. The samples were loaded onto a polyacrylamide gel (6%) and run in TBE buffer (89 mmol/L Tris HCl, 89 mmol/L boric acid, 2 mmol/L Na2EDTA, pH 8.0). Subsequent DNA staining was performed with SYBR Gold Nucleic Acid Stain (Invitrogen, 1:10 000 in TBE buffer) for ≈10 minutes in the dark. Under UV light, the DNA was visualized and bands in the range of 100 to 800 bp were excised. For each 100 mg of gel, 100 μL of diffusion buffer (1 mmol/L Na2EDTA, 10 mmol/L Mg(CH3COO)2, 0.5 mol/L CH3COONH4, 0.1% SDS) was added and incubated overnight at 37 °C and 500 rpm. Remaining gel fragments were removed via a 30 μm strainer (CellTrics) and the resulting eluate was purified with the MinElute Kit (Qiagen) according to the manufacturer’s manual. Quantification and sequencing of the final DNA library was performed as described before.
Bioinformatic Analysis
Sequencing data were analyzed using the tools integrated in the Galaxy platform. 21 For RNA‐seq data, low quality reads and adapters were removed from sequenced reads using Trim Galore!. 22 Reads were mapped to the mouse reference genome mm9 using RNA STAR 23 and annotated with genes. 24 PCR duplicates were removed from the mapped reads using RmDup 25 and their number was subsequently quantified using htseq‐count. 26 DESeq2 27 was used to determine differentially regulated genes between 2 samples with q<0.05 after correction for multiple testing considered statistically significant (Table S4). Functional enrichment analysis of genes (gene ontology: biological process) was performed using the Database for Annotation, Visualization and Integrated Discovery with P<0.05 considered statistically significant. 28
Similar to the RNA‐Seq data sets, Trim Galore! 22 was applied to the data sets from ChIP‐ and ATAC‐Seq. assignment of reads to the mouse genome mm9 was done with bowtie2 under default settings. 29 PCR duplicates were also removed using RmDup. 25 Subsequently, the MACS2 tool 30 was used to identify all enriched regions (peaks) of an ATAC or ChIP experiment, such as regions of higher accessibility or regions with enriched histone modifications and transcription factors.
The assignment of peaks from ChIP and ATAC experiments to nearby gene was performed using the Genomic Regions Enrichment of Annotations Tool. 31 For the analysis of enriched transcription factor binding motifs within specific regions, Hypergeometric Optimization of Motif Enrichment 32 was used with default settings.
Visualization of Data Sets
Further processing and visualization of sequencing data sets was done mainly with deeptools. 33 For genome‐wide visualization and normalization to reads per kilobase per million mapped bamcoverage was run. The bigwig files obtained were visualized in the Integrative Genomic Viewer program. 34 Heat maps for the representation of log2‐transformed RNA‐seq data were created using R software 35 using the lattice package. 36 The UpSetR Shiny App 37 was used to visualize intersecting data sets. To calculate an enriched distribution of values for specific genomic regions from ChIP‐seq data, computeMatrix was used. These values were then visualized using plotHeatmap or plotProfile.
Statistical Analysis
Statistical data analysis was performed using GraphPad Prism (version 9.0.2 for Windows, San Diego, CA). According to the data, the t test for independent samples and single factor analysis of variance followed by Dunett's T3 multiple comparison test were applied assuming normal distribution. For data sets without evidence of a normal distribution, the Mann–Whitney U test or the Kruskal‐Wallis test followed by Dunn's multiple comparison were performed. All values are presented as mean±SEM.
RESULTS
In Vitro Differentiation of ESC‐Derived CMs
CMs were differentiated from mouse ESCs, which contained a puromycin resistance cassette and eGFP (enhanced GFP) under control of the murine cardiac myocyte‐specific α‐MHC (Myh6) promoter 17 (Figure 1A, Figure S1A). After 3 weeks of culture and selection with puromycin, ES‐CM showed the typical CM sarcomere structure (Figure 1A) and stable expression of adult isoforms of troponin I (Tnni3) and β‐MHC 6 (Myh6) (Figure S1B).
Figure 1. Pathological gene expression induced by endothelin‐1 in embryonic stem cell‐derived cardiac myocytes.
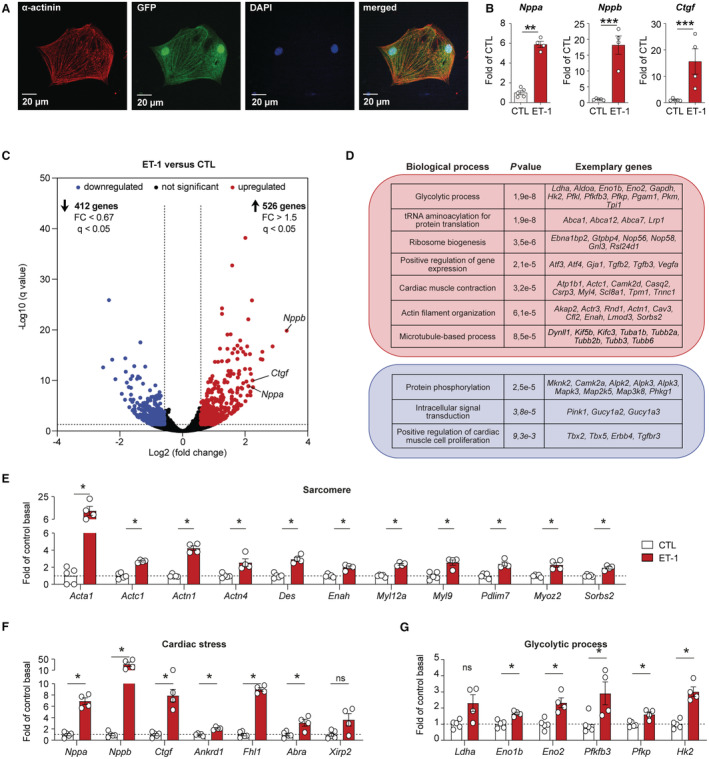
A, Representative immunofluorescence staining of cardiac α‐actinin (1:500, red) in GFP (green fluorescent protein)‐positive embryonic stem cell‐derived cardiac myocytes (ES‐CM, green). Nuclei are counterstained with DAPI (1:1000, blue). B, Gene expression of natriurectic peptide precursor A (Nppa) and B (Nppb) or connective tissue growth factor (Ctgf) in control (CTL) ES‐CM and in ES‐CM treated with endothelin‐1 (ET‐1, 100 nmol/L) were determined by quantitative reverse transcription polymerase chain reaction (n=4–8 per group, mean±SEM, Student t test, **P<0.01, ***P<0.001). C, Volcano plot showing RNA‐seq data of significantly upregulated (red) and downregulated genes (blue) in ES‐CM treated with ET‐1 compared with control (CTL) ES‐CM (n=4–5, normalized counts >1, fold change >1.5 or <0.67, q<0.05). D, Gene ontology analysis using the Database for Annotation, Visualization and Integrated Discovery for significantly upregulated genes (red box) and downregulated genes (blue box) that are shown in (C). E through G, Gene expression of differentially regulated genes associated with sarcomere, cardiac stress, or glycolytic process in control and ET‐1 treated ES‐CM as derived from RNA‐seq (n=4–5 per group, mean±SEM, *q<0.05).
Induction of Pathological Gene Expression in ES‐CM In Vitro
To induce pathological gene expression in vitro, differentiated ES‐CM were treated with ET‐1 (Figure 1B). ET‐1 induced the expression of typical cardiac stress marker genes (Nppa, Nppb, Ctgf) as determined by reverse transcription quantitative PCR (Figure 1B). In order to get in‐depth insights into the transcriptional changes induced by ET‐1 in ES‐CM, we performed RNA‐seq (Figure 1C through 1H). After ET‐1 stimulation, we observed an upregulation of 526 genes (q<0.05, 1.5‐fold up) and downregulation of 412 genes (q<0.05, 1.5‐fold down) (Figure 1D, Table S4). Gene ontology analysis revealed a strong signature of sarcomeric genes, cardiac stress‐associated genes, and metabolism‐related genes among the genes differentially regulated by ET‐1 (Figure 1D through 1G). Similar to in vivo observations that have shown a metabolic shift from fatty acid oxidation to increased glucose use, 38 ET‐1 induced upregulation of genes involved in glycolytic processes (Figure 1E and 1G). Thus, genome‐wide gene expression patterns of ES‐CM in response to ET‐1 resembled the pathological gene program of heart failure.
Epigenetic Inhibitors Attenuate the Pathological Gene Expression Program
To evaluate the potential of epigenetic inhibitors to prevent pathological, ET‐1 induced gene expression, we compared the effect of the HDAC6 inhibitor JS28 16 to a the broad‐spectrum HDAC inhibitor suberoylanilide hydroxamic acid (SAHA; vorinostat), 39 the BRD inhibitor bromosporine, 40 and the BET inhibitor JQ1. 11 , 12 , 13 Epigenetic inhibitors were able to prevent differential expression of 22% to 31% of genes responsive to ET‐1 (JS28 210/938 genes, SAHA 227/938 genes, bromosporine 290/938 genes, JQ1 288/938 genes; Table S4). Gene ontology analysis revealed overlapping impact of bromosporine, JS28, and SAHA on processes related to protein translation (Figure 2A). Both JS28 and JQ1 prevented the dysregulation of genes related to myofibroblast differentiation and for JS28 we observed additional effects on inflammatory processes (Figure 2A). All epigenetic inhibitors tested modulated the upregulation of typical heart failure marker genes Nppa, Nppb, and Ctgf, although to different extent (Figure 2B). Principal component analysis revealed differential effects of the epigenetic inhibitors on ET‐1‐induced gene expression, and in particular, the effects of JQ1 appeared to be markedly different from those of the others (Figure 2C). In line with this, the 4 inhibitors affected only a small overlap of genes (49/938 genes, Figure 2D).
Figure 2. Epigenetic inhibitors prevent part of the pathological gene program.
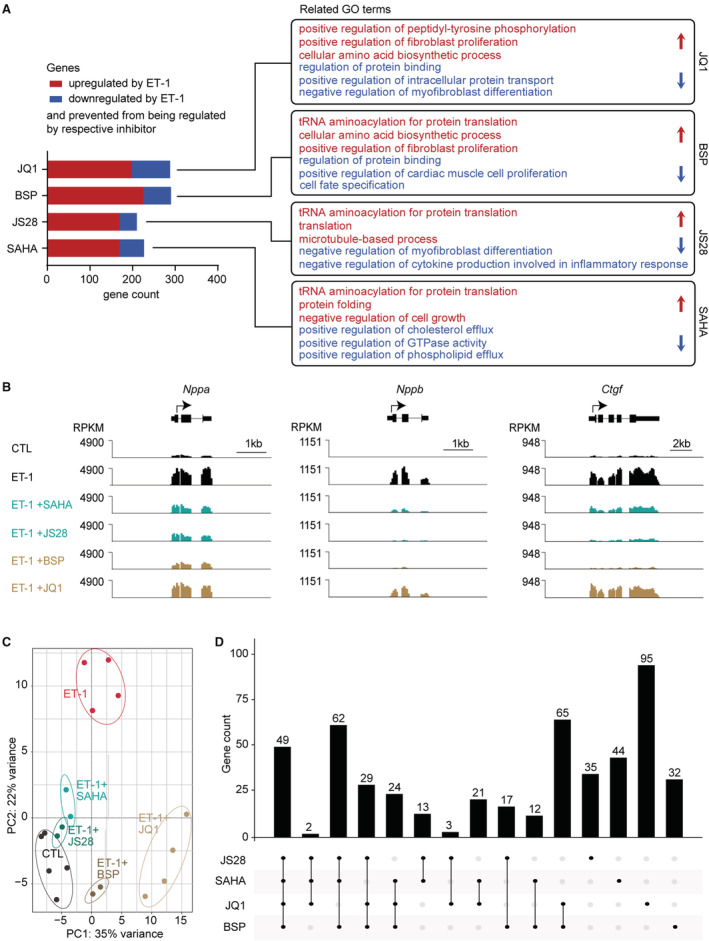
A, Gene ontology (GO) analysis of up‐ (red) or downregulated (blue) genes in embryonic stem cell‐derived cardiac myocytes (ES‐CM) by endothelin‐1 (ET‐1, normalized counts >1, 1.5‐fold down−/upregulated, q<0.05, n=2–5) that were prevented from being regulated by treatment with the indicated inhibitors. B, Representative traces showing the impact of epigenetic inhibitors on natriurectic peptide precursor A (Nppa) and B (Nppb) or connective tissue growth factor (Ctgf) gene expression. C, Principal component (PC) analysis plot from DEseq2 visualizing similarities and differences among samples from control ES‐CM, from ES‐CM treated with ET‐1 alone and ES‐CM treated with indicated inhibitors in presence of ET‐1. D, Summarizing the number of genes that were prevented from being regulated by the respective inhibitor and the overlap between different compounds. BSP indicates bromosporine; CTL, control; RPKM, reads per kilobase per million mapped; and SAHA, suberoylanilide hydroxamic acid.
Genome‐Wide Distribution of Inhibitor Targets HDAC6 and BRD4
To gain deeper insights into the underlying molecular mechanisms of the selected inhibitors, we assessed chromatin binding of 2 inhibitor targets, namely HDAC6 and BRD4, to promoter and enhancer regions in ES‐CM. We performed ChIP‐seq of HDAC6 and BRD4, as well as of epigenetic marks that define promoter (H3K27ac, H3K9ac, H3K4me1, H3K4me3, RNA Polymerase II) and enhancer (H3K27ac, H3K4me1) signatures from naїve ES‐CM and assessed chromatin accessibility by ATAC‐seq (Figures 3 and 4).
Figure 3. Genomic distribution of the JS28 inhibitor target HDAC6.
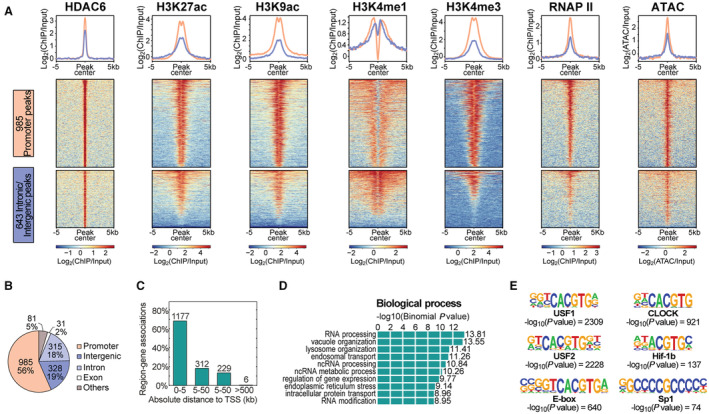
A, Heatmaps illustrating all histone deacetylase 6 (HDAC6) peaks at the promoter (pink) and at intronic/intergenic regions (blue) and their association with histone modifications, RNAP II (RNA polymerase II), and chromatin accessibility as derived from ATAC. B, Genome‐wide localization of all 1740 HDAC6 peaks and (C) the distribution of all HDAC6 peaks in distance to the transcription start site (TSS) of the nearest gene in kilobases (kb). D, Gene ontology analysis of genes associated with HDAC6 peaks. E, Significantly enriched transcription factor binding motifs within HDAC6 peaks. ATAC indicates assay for transposase‐accessible chromatin.
Figure 4. Genomic distribution of the JQ1 inhibitor target BRD4.
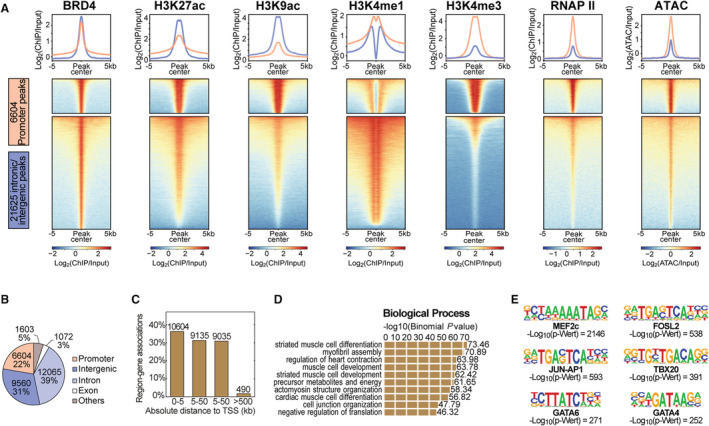
A, Heatmaps illustrating all BRD4 (bromodomain‐containing protein 4) peaks at the promoter (pink) and at intronic/intergenic regions (blue) and their association with histone modifications, RNAP II (RNA polymerase II), and chromatin accessibility as derived from ATAC. B, Genome‐wide localization of all 30 906 BRD4 peaks and (C) the distribution of all BRD4 peaks in distance to the transcription start site of the nearest gene in kilobases (kb). D, Gene ontology analysis of genes associated with BRD4 peaks. E, Significantly enriched transcription factor binding motifs within BRD4 peaks. ATAC indicates assay for transposase‐accessible chromatin.
Peak calling identified a total of 1740 HDAC6 peaks compared with 30 906 BRD4 peaks (Figures 3 and 4). Characterization of the HDAC6 peaks by annotation with specific genomic features (Figure 3A) revealed that 56% of the genome‐wide peaks were located at promoters (985 peaks) and <20% each in intergenic (328 peaks) or intronic regions (315 peaks), respectively (Figure 3B). Consistent with this, the majority of peaks were located 0 to 5 kb away from the transcription start site of the single nearest gene (Figure 3C). Interestingly, annotation of genes associated with the HDAC6 peaks showed an enrichment mainly for RNA‐associated processes, organelle organization, and protein transport (Figure 3D). Transcription factor binding motif analysis revealed a striking enrichment for the motif CACGTG (G‐box) (Figure 3E), a specific DNA sequence that is typically recognized and bound by general transcription factors to initiate and regulate gene transcription, 41 , 42 and SP1 (Figure 3E), known for its binding to guanine‐cytosine‐rich DNA sequences that occur in many promoters. 43
Compared with HDAC6, a far different genome‐wide pattern was found for BRD4 (Figure 4). BRD4 peaks at promoter regions and intergenic plus intronic regions were characterized by distinct promoter‐ and enhancer‐specific histone, RNAP II (RNA polymerase II), and ATAC signatures, respectively (Figure 4A). BRD4 peaks were not only localized at the promoter (6604 peaks) but also a substantial number in intergenic (9560 peaks) and intronic regions (12 065 peaks) (Figure 4B). BRD4 peaks were distributed both close to and distant from the transcription start site (Figure 4C). Biological processes associated with these genes revealed a pronounced enrichment for heart‐related processes such as striated muscle cell differentiation and development, myofibril assembly, and cardiac contraction (Figure 4D). This finding was confirmed by motif enrichment for typical transcription factors that play an essential role in regulating cardiac development and gene regulation in context of stress response (Figure 4E). For instance, motifs for GATA4, GATA6, and members of the AP‐1 (activator protein‐1) transcription factor family were overrepresented (Figure 4E).
Effects of Epigenetic Inhibitors on Physiological Gene Expression in ES‐CM
We hypothesized that epigenetic compounds, in addition to their inhibitory effects on ET‐1 signaling, may have undesired effects on basal gene expression (Figure 5). To address this question, ES‐CM were treated with selected candidate compounds (SAHA, JS28, bromosporine, JQ1) in absence of ET‐1. RNA‐seq data revealed that SAHA and JQ1 induced strong but distinct changes in physiological gene expression (Figure 5A and 5B), with 770 genes differentially expressed by SAHA and 1347 genes by JQ1, respectively (Figure 5A and 5B). Notably, genes affected by SAHA were associated with chromatin modification, histone acetylation, and in general coregulatory factors involved in the control of gene transcription (Figure 5C). In contrast, JQ1‐mediated BET inhibition affected genes involved in typical cardiac myocyte functions such as muscle contraction, fatty acid oxidation, or ATP metabolism (Figure 5C). In contrast, selective inhibition of HDAC6 by JS28 attenuated ET‐1‐induced gene expression with minimal effects on baseline gene expression, with 12 genes differentially expressed (Figure 5A and 5B). Interestingly, the broad‐spectrum BRD inhibitor bromosporine with 106 up‐ or downregulated genes also showed only a moderate impairment of the gene expression profile in ES‐CM (Figure 5A and 5B).
Figure 5. Impact of epigenetic inhibitors on physiological gene expression.
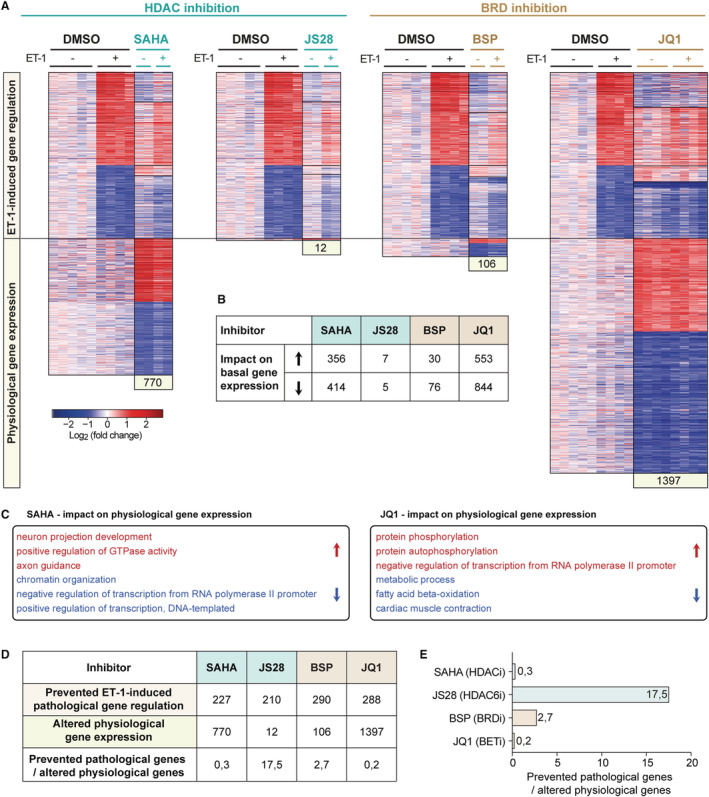
A, Heat maps showing the effects of epigenetic inhibitors on genes differentially expressed after treatment with endothelin 1 (ET‐1, upper panel) and on physiological gene expression (lower panel) (normalized counts >1, q<0.05, 1.5‐fold up−/downregulated, n=2–5). B, Numbers of genes differentially up‐ or downregulated by the respective inhibitor in absence of ET‐1. C, Gene ontology analysis of genes differentially regulated by SAHA (vorinostat) or JQ1 (normalized counts >1, 1.5× up−/downregulated, q<0.05). D and E, Numbers of genes regulated by ET‐1 and prevented from being regulated by epigenetic inhibitors compared with the impact of epigenetic inhibitors on physiological gene expression, implicating desired vs undesired effects. BRD indicates bromodomain‐containing protein; BSP, bromosporine; HDAC, histone deacetylase; and SAHA, suberoylanilide hydroxamic acid.
To estimate the respective risk–benefit ratio for each compound we compared their potential to prevent ET‐1 induced gene dysregulation (desired effect) to the number of genes altered by the inhibitor in absence of ET‐1 (undesired effect, Figure 5D and 5E). This ratio identified the HDAC6‐selective inhibitor JS28 as the most effective inhibitor to prevent pathological gene expression with minimal adverse effects in ES‐CM in vitro. JS28 was followed by the pan‐BRD inhibitor bromosporine, whereas the broad‐spectrum HDAC‐inhibitor SAHA and the BET‐inhibitor JQ1 showed a less desirable profile.
DISCUSSION
Pathological gene expression is a key component of adverse tissue remodeling induced by circulating hormones in hypertension in heart failure. In this study, we evaluated the potential of epigenetic drugs to reverse the pathological gene expression response to ET‐1 in vitro. As expected, exposure of ES‐CM to ET‐1 resulted in a pronounced dysregulation of well‐known marker genes of heart failure such as Nppa, Nppb, or Ctgf. 44 , 45 In addition, we observed an upregulation of genes associated with microtubules or actin filaments, which have been reported to represent a compensatory response to contractile stress. 46 , 47 Although established heart failure drugs indirectly influence gene expression via inhibition of upstream signaling pathways, epigenetic drugs have been suggested as a new therapeutic approach to directly restore aberrant gene expression. 48 We show here that the novel HDAC6 inhibitor JS28 effectively prevented pathological gene expression with minor impact on physiological gene expression.
Among the established HDAC inhibitors, SAHA is one of the best studied compounds and has demonstrated cardioprotective effects in several experimental models of heart failure. 9 , 39 , 49 , 50 However, SAHA is a broad‐spectrum inhibitor and as our data suggest, unselective inhibition of more than 1 HDAC could have deleterious effects. Accordingly, more recently isoform‐specific inhibitors have been developed. 51 , 52 There is emerging evidence for the role of HDAC6 in cardiovascular pathophysiology. HDAC6 catalytic activity is increased in several models of cardiac stress. 53 , 54 After angiotensin II infusion, chronic pressure overload, or doxorubicin treatment HDAC6‐deficient mice and mice treated with the HDAC6‐selective inhibitor tubastatin A showed improved cardiac function compared with control mice. 55 , 56 Importantly, in this study the HDAC6‐selective compound JS28 showed similar efficacy in abrogating pathological gene expression with minimum undesired effects when compared with the broad‐spectrum inhibitor SAHA.
The BET inhibitor JQ1 had achieved significant therapeutic effects in various animal models of heart failure. 11 , 12 , 13 , 14 Here, we compared the effects of JS28 to JQ1 and bromosporine, another BET inhibitor that has not yet been tested in cardiovascular disease. Both compounds showed comparable efficacy to restore gene expression and a clear transcriptional overlap after ET‐1 treatment. Compared with HDAC inhibitors, JQ1 and bromosporine seem to address a different pathological gene subset. Importantly, in particular JQ1 also had a strong negative impact on genes involved in cardiac contraction and energy metabolism in absence of ET‐1. This has not yet been investigated but may have important implications for potential translation into clinical use, as it suggests that JQ1 may have potential cardiotoxic side effects that could become relevant with long‐term use.
To gain further insight into the molecular mechanisms involved in the different response to BET and HDAC inhibitors, we assessed the chromatin binding profile of the inhibitor targets BRD4 and HDAC6 in ES‐CM. BRD4‐bound chromatin regions were associated with genes related to CM structure and metabolism and harbored binding sites for typical myocyte transcription factors such as mouse embryonic fibroblast or GATA. Indeed, BRD4 has previously been shown to play a central role in CM energetic homeostasis. 57 , 58 On the one hand, this corresponds to the cardioprotective potential of the BET inhibitor JQ1, but it may also explain its undesired effects on cardiac myocyte gene expression. In contrast, HDAC6 binding sites were less frequent and mainly located at promoter regions. This is in line with the earlier finding that HDAC6 is recruited to active genes, interacting with RNAP II 59 , 60 and may explain why JS28 attenuated active transcription but had less impact on basal cardiac gene expression.
In this project we focused on the impact of epigenetic inhibitors on gene expression. However, selective HDAC inhibition restored the expression of genes associated with microtubule‐based processes in ET‐1 treated ES‐CM, which is of particular interest when considering the broad nongenomic actions of HDAC6. HDAC6‐dependent deacetylation of nonhistone proteins regulates protein stability and degradation, enzymatic activity, or cytoskeleton organization. 7 Microtubules, together with actin filaments, form the major components of the CM cytoskeleton. 61 In the heart, microtubules and microtubule‐based transport assist in the organization and maintenance of key structures within the cardiac myocyte, including the sarcomere and protein clearance machinery. 61 Tubulin acetylation improves its flexibility and resistance to mechanical stress, which could be beneficial for CM mechanics in heart failure. 61 In fact, a previous study showed that pharmacological deacetylation of tubulin by nocodazole reversed the beneficial effect of HDAC6 deletion, indicating a causal relation between tubulin acetylation and heart function. 56 In addition, CM mechanics and the cytoskeleton influence nuclear organization, 61 , 62 indicating that HDAC6 inhibition via nongenomic actions on tubulin could indirectly alter gene expression.
CONCLUSIONS
In summary, the HDAC6 inhibitor JS28 effectively restored pathological gene expression with minor impact on physiological gene expression in vitro and thus appears a promising candidate for future evaluation in vivo. Combining different epigenetic drugs has been proposed as a strategy for cancer therapy to reduce adverse effects. 63 , 64 Preclinical studies indicate that BRD4 and HDAC inhibitors have good synergy at low dosages, perhaps avoiding the overlapping toxicities associated with these 2 drugs. 64 For example, concomitant use of the clinically used HDAC6i ricolinostat significantly increased the antimyelomic activity of JQ1 in a panel of multiple myeloma cell lines and primary CD138+ cells obtained from patients with multiple myeloma. 65 The synergy of this combination was also associated with a reduced expression of c‐MYC and increased apoptosis. 65 As HDAC inhibitors and BRD inhibitors had distinct effects on cardiac myocyte gene expression, synergistic effects of the 2 inhibitors seem conceivable. Based on these results, further experiments with JS28 and bromosporine at different concentrations could be used to find combinations that allow enhancement of the desired effects with tolerable side effects.
Sources of Funding
Fleischmann, Hein, and Lother are members of SFB 1425, funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project ID 422681845. Jung, Hein and Ngo were funded by the Deutsche Forschungsgemeinschaft—Project ID 192904750—SFB 992 Medical Epigenetics.
Disclosures
Lother received fees for lectures and/or serving on advisory boards from Bayer and AstraZeneca not related to this work. The remaining authors have no disclosures to report.
Supporting information
Tables S1–S5
Figure S1
Acknowledgments
We thank the Freiburg Galaxy Team, Björn Grüning, Anika Erxleben, and Rolf Backofen, Bioinformatics, University of Freiburg, Germany, funded by the Deutsche Forschungsgemeinschaft (SFB 992 and SFB 1425, project S3) and German Federal Ministry of Education and Research (BMBF grant 031 A538A RBC [de.NBI]). We thank the European Molecular Biology Laboratory GeneCore (Heidelberg, Germany) and the Max Planck Institute of Immunobiology and Epigenetics Deep Sequencing Facility (Freiburg, Germany) for providing sequencing services.
Author contributions: All authors contributed to the study conception and design. Ngo performed experiments and analyzed the data. All authors interpreted the data. The article was written by Ngo, Lother, and Hein. Fleischmann and Jung critically revised the draft. All authors read and approved the final article. Lother and Hein contributed equally to this work.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.025857
For Sources of Funding and Disclosures, see page 12.
REFERENCES
- 1. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Bohm M, Burri H, Butler J, Celutkiene J, Chioncel O, et al. 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2021;42:3599–3726. doi: 10.1093/eurheartj/ehab368 [DOI] [PubMed] [Google Scholar]
- 2. Lother A, Hein L. Pharmacology of heart failure: from basic science to novel therapies. Pharmacol Ther. 2016;166:136–149. doi: 10.1016/j.pharmthera.2016.07.004 [DOI] [PubMed] [Google Scholar]
- 3. Alexanian M, Padmanabhan A, McKinsey TA, Haldar SM. Epigenetic therapies in heart failure. J Mol Cell Cardiol. 2019;130:197–204. doi: 10.1016/j.yjmcc.2019.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Qin J, Guo N, Tong J, Wang Z. Function of histone methylation and acetylation modifiers in cardiac hypertrophy. J Mol Cell Cardiol. 2021;159:120–129. doi: 10.1016/j.yjmcc.2021.06.011 [DOI] [PubMed] [Google Scholar]
- 5. Gillette TG, Hill JA. Readers, writers, and erasers: chromatin as the whiteboard of heart disease. Circ Res. 2015;116:1245–1253. doi: 10.1161/CIRCRESAHA.116.303630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co‐regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371 [DOI] [PubMed] [Google Scholar]
- 7. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non‐histone protein acetylation. Nat Rev Mol Cell Biol. 2019;20:156–174. doi: 10.1038/s41580-018-0081-3 [DOI] [PubMed] [Google Scholar]
- 8. Travers JG, Wennersten SA, Pena B, Bagchi RA, Smith HE, Hirsch RA, Vanderlinden LA, Lin YH, Dobrinskikh E, Demos‐Davies KM, et al. HDAC inhibition reverses preexisting diastolic dysfunction and blocks covert extracellular matrix remodeling. Circulation. 2021;143:1874–1890. doi: 10.1161/CIRCULATIONAHA.120.046462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wallner M, Eaton DM, Berretta RM, Liesinger L, Schittmayer M, Gindlhuber J, Wu J, Jeong MY, Lin YH, Borghetti G, et al. HDAC inhibition improves cardiopulmonary function in a feline model of diastolic dysfunction. Sci Transl Med. 2020;12:eaay7205. doi: 10.1126/scitranslmed.aay7205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gallo P, Latronico MV, Grimaldi S, Borgia F, Todaro M, Jones P, Gallinari P, De Francesco R, Ciliberto G, Steinkuhler C, et al. Inhibition of class I histone deacetylase with an apicidin derivative prevents cardiac hypertrophy and failure. Cardiovasc Res. 2008;80:416–424. doi: 10.1093/cvr/cvn215 [DOI] [PubMed] [Google Scholar]
- 11. Spiltoir JI, Stratton MS, Cavasin MA, Demos‐Davies K, Reid BG, Qi J, Bradner JE, McKinsey TA. BET acetyl‐lysine binding proteins control pathological cardiac hypertrophy. J Mol Cell Cardiol. 2013;63:175–179. doi: 10.1016/j.yjmcc.2013.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anand P, Brown JD, Lin CY, Qi J, Zhang R, Artero PC, Alaiti MA, Bullard J, Alazem K, Margulies KB, et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013;154:569–582. doi: 10.1016/j.cell.2013.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duan Q, McMahon S, Anand P, Shah H, Thomas S, Salunga HT, Huang Y, Zhang R, Sahadevan A, Lemieux ME, et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci Transl Med. 2017;9:eaah5084. doi: 10.1126/scitranslmed.aah5084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stratton MS, Lin CY, Anand P, Tatman PD, Ferguson BS, Wickers ST, Ambardekar AV, Sucharov CC, Bradner JE, Haldar SM, et al. Signal‐dependent recruitment of BRD4 to cardiomyocyte super‐enhancers is suppressed by a microRNA. Cell Rep. 2016;16:1366–1378. doi: 10.1016/j.celrep.2016.06.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, Han J, Wei X. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther. 2019;4:62. doi: 10.1038/s41392-019-0095-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Senger J, Melesina J, Marek M, Romier C, Oehme I, Witt O, Sippl W, Jung M. Synthesis and biological investigation of oxazole hydroxamates as highly selective histone deacetylase 6 (HDAC6) inhibitors. J Med Chem. 2016;59:1545–1555. doi: 10.1021/acs.jmedchem.5b01493 [DOI] [PubMed] [Google Scholar]
- 17. Kolossov E, Bostani T, Roell W, Breitbach M, Pillekamp F, Nygren JM, Sasse P, Rubenchik O, Fries JW, Wenzel D, et al. Engraftment of engineered ES cell‐derived cardiomyocytes but not BM cells restores contractile function to the infarcted myocardium. J Exp Med. 2006;203:2315–2327. doi: 10.1084/jem.20061469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R. The in vitro development of blastocyst‐derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. J Embryol Exp Morphol. 1985;87:27–45. [PubMed] [Google Scholar]
- 19. Fuegemann CJ, Samraj AK, Walsh S, Fleischmann BK, Jovinge S, Breitbach M. Differentiation of mouse embryonic stem cells into cardiomyocytes via the hanging‐drop and mass culture methods. Curr Protoc Stem Cell Biol. 2010;Chapter 1:Unit 1F.11. doi: 10.1002/9780470151808.sc01f11s15 [DOI] [PubMed] [Google Scholar]
- 20. Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC‐seq: a method for assaying chromatin accessibility genome‐wide. Curr Protoc Mol Biol. 2015;109:21.29.21–21.29.29. doi: 10.1002/0471142727.mb2129s109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Eberhard C, et al. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016;44:W3–W10. doi: 10.1093/nar/gkw343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. F Krueger. Trim Galore! A wrapper tool around Cutadept and FastQC to consistently apply quality and adapter trimming to FastQ files. 2015.
- 23. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karolchik D, Barber GP, Casper J, Clawson H, Cline MS, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, et al. The UCSC genome browser database: 2014 update. Nucleic Acids Res. 2014;42:D764–D770. doi: 10.1093/nar/gkt1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; Genome Project Data Processing S . The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3 [PubMed] [Google Scholar]
- 29. Langmead B, Salzberg SL. Fast gapped‐read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model‐based analysis of ChIP‐seq (MACS). Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis‐regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T. deepTools2: a next generation web server for deep‐sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. R Development Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2010. [Google Scholar]
- 36. D Sarkar. Lattice: multivariate data visualization with R. Use R. 2008:1–265. doi: 10.1007/978-0-387-75969-2 [DOI]
- 37. Lex A, Gehlenborg N, Strobelt H, Vuillemot R, Pfister H. UpSet: visualization of intersecting sets. IEEE Trans Vis Comput Graph. 2014;20:1983–1992. doi: 10.1109/TVCG.2014.2346248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tran DH, Wang ZV. Glucose metabolism in cardiac hypertrophy and heart failure. J Am Heart Assoc. 2019;8:e012673. doi: 10.1161/JAHA.119.012673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nagata S, Marunouchi T, Tanonaka K. Histone deacetylase inhibitor SAHA treatment prevents the development of heart failure after myocardial infarction via an induction of heat‐shock proteins in rats. Biol Pharm Bull. 2019;42:453–461. doi: 10.1248/bpb.b18-00785 [DOI] [PubMed] [Google Scholar]
- 40. Picaud S, Leonards K, Lambert JP, Dovey O, Wells C, Fedorov O, Monteiro O, Fujisawa T, Wang CY, Lingard H, et al. Promiscuous targeting of bromodomains by bromosporine identifies BET proteins as master regulators of primary transcription response in leukemia. Sci Adv. 2016;2:e1600760. doi: 10.1126/sciadv.1600760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kawagoe Y, Campbell BR, Murai N. Synergism between CACGTG (G‐box) and CACCTG cis‐elements is required for activation of the bean seed storage protein beta‐phaseolin gene. Plant J. 1994;5:885–890. doi: 10.1046/j.1365-313x.1994.5060885.x [DOI] [PubMed] [Google Scholar]
- 42. Liu L, Xu W, Hu X, Liu H, Lin Y. W‐box and G‐box elements play important roles in early senescence of rice flag leaf. Sci Rep. 2016;6:20881. doi: 10.1038/srep20881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Briggs MR, Kadonaga JT, Bell SP, Tjian R. Purification and biochemical characterization of the promoter‐specific transcription factor, Sp1. Science. 1986;234:47–52. doi: 10.1126/science.3529394 [DOI] [PubMed] [Google Scholar]
- 44. Dirkx E, da Costa Martins PA, De Windt LJ. Regulation of fetal gene expression in heart failure. Biochim Biophys Acta. 2013;1832:2414–2424. doi: 10.1016/j.bbadis.2013.07.023 [DOI] [PubMed] [Google Scholar]
- 45. Harada M, Itoh H, Nakagawa O, Ogawa Y, Miyamoto Y, Kuwahara K, Ogawa E, Igaki T, Yamashita J, Masuda I, et al. Significance of ventricular myocytes and nonmyocytes interaction during cardiocyte hypertrophy: evidence for endothelin‐1 as a paracrine hypertrophic factor from cardiac nonmyocytes. Circulation. 1997;96:3737–3744. doi: 10.1161/01.cir.96.10.3737 [DOI] [PubMed] [Google Scholar]
- 46. Sequeira V, Nijenkamp LL, Regan JA, van der Velden J. The physiological role of cardiac cytoskeleton and its alterations in heart failure. Biochim Biophys Acta. 2014;1838:700–722. doi: 10.1016/j.bbamem.2013.07.011 [DOI] [PubMed] [Google Scholar]
- 47. Hein S, Kostin S, Heling A, Maeno Y, Schaper J. The role of the cytoskeleton in heart failure. Cardiovasc Res. 2000;45:273–278. doi: 10.1016/s0008-6363(99)00268-0 [DOI] [PubMed] [Google Scholar]
- 48. Papait R, Greco C, Kunderfranco P, Latronico MV, Condorelli G. Epigenetics: a new mechanism of regulation of heart failure? Basic Res Cardiol. 2013;108:361. doi: 10.1007/s00395-013-0361-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, Wang ZV, Morales C, Luo X, Cho G, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129:1139–1151. doi: 10.1161/CIRCULATIONAHA.113.002416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Iyer A, Fenning A, Lim J, Le GT, Reid RC, Halili MA, Fairlie DP, Brown L. Antifibrotic activity of an inhibitor of histone deacetylases in DOCA‐salt hypertensive rats. Br J Pharmacol. 2010;159:1408–1417. doi: 10.1111/j.1476-5381.2010.00637.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Batchu SN, Brijmohan AS, Advani A. The therapeutic hope for HDAC6 inhibitors in malignancy and chronic disease. Clin Sci (Lond). 2016;130:987–1003. doi: 10.1042/CS20160084 [DOI] [PubMed] [Google Scholar]
- 52. Romanick SS, Ferguson BS. The nonepigenetic role for small molecule histone deacetylase inhibitors in the regulation of cardiac function. Future Med Chem. 2019;11:1345–1356. doi: 10.4155/fmc-2018-0311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lemon DD, Horn TR, Cavasin MA, Jeong MY, Haubold KW, Long CS, Irwin DC, McCune SA, Chung E, Leinwand LA, et al. Cardiac HDAC6 catalytic activity is induced in response to chronic hypertension. J Mol Cell Cardiol. 2011;51:41–50. doi: 10.1016/j.yjmcc.2011.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kee HJ, Bae EH, Park S, Lee KE, Suh SH, Kim SW, Jeong MH. HDAC inhibition suppresses cardiac hypertrophy and fibrosis in DOCA‐salt hypertensive rats via regulation of HDAC6/HDAC8 enzyme activity. Kidney Blood Press Res. 2013;37:229–239. doi: 10.1159/000350148 [DOI] [PubMed] [Google Scholar]
- 55. Demos‐Davies KM, Ferguson BS, Cavasin MA, Mahaffey JH, Williams SM, Spiltoir JI, Schuetze KB, Horn TR, Chen B, Ferrara C, et al. HDAC6 contributes to pathological responses of heart and skeletal muscle to chronic angiotensin‐II signaling. Am J Physiol Heart Circ Physiol. 2014;307:H252–H258. doi: 10.1152/ajpheart.00149.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Song R, Yang Y, Lei H, Wang G, Huang Y, Xue W, Wang Y, Yao L, Zhu Y. HDAC6 inhibition protects cardiomyocytes against doxorubicin‐induced acute damage by improving alpha‐tubulin acetylation. J Mol Cell Cardiol. 2018;124:58–69. doi: 10.1016/j.yjmcc.2018.10.007 [DOI] [PubMed] [Google Scholar]
- 57. Kim SY, Zhang X, Schiattarella GG, Altamirano F, Ramos TAR, French KM, Jiang N, Szweda PA, Evers BM, May HI, et al. Epigenetic reader BRD4 (bromodomain‐containing protein 4) governs nucleus‐encoded mitochondrial transcriptome to regulate cardiac function. Circulation. 2020;142:2356–2370. doi: 10.1161/CIRCULATIONAHA.120.047239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Padmanabhan A, Alexanian M, Linares‐Saldana R, Gonzalez‐Teran B, Andreoletti G, Huang Y, Connolly AJ, Kim W, Hsu A, Duan Q, et al. BRD4 (bromodomain‐containing protein 4) interacts with GATA4 (GATA binding protein 4) to govern mitochondrial homeostasis in adult cardiomyocytes. Circulation. 2020;142:2338–2355. doi: 10.1161/CIRCULATIONAHA.120.047753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome‐wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen PB, Hung JH, Hickman TL, Coles AH, Carey JF, Weng Z, Chu F, Fazzio TG. Hdac6 regulates Tip60‐p400 function in stem cells. Elife. 2013;2:e01557. doi: 10.7554/eLife.01557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Uchida K, Scarborough EA, Prosser BL. Cardiomyocyte microtubules: control of mechanics, transport, and remodeling. Annu Rev Physiol. 2022;84:257–283. doi: 10.1146/annurev-physiol-062421-040656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Heffler J, Shah PP, Robison P, Phyo S, Veliz K, Uchida K, Bogush A, Rhoades J, Jain R, Prosser BL. A balance between intermediate filaments and microtubules maintains nuclear architecture in the cardiomyocyte. Circ Res. 2020;126:e10–e26. doi: 10.1161/CIRCRESAHA.119.315582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ramadoss M, Mahadevan V. Targeting the cancer epigenome: synergistic therapy with bromodomain inhibitors. Drug Discov Today. 2018;23:76–89. doi: 10.1016/j.drudis.2017.09.011 [DOI] [PubMed] [Google Scholar]
- 64. Chen J, Li Y, Zhang J, Zhang M, Wei A, Liu H, Xie Z, Ren W, Duan W, Zhang Z, et al. Discovery of selective HDAC/BRD4 dual inhibitors as epigenetic probes. Eur J Med Chem. 2021;209:112868. doi: 10.1016/j.ejmech.2020.112868 [DOI] [PubMed] [Google Scholar]
- 65. Carew JS, Espitia CM, Zhao W, Visconte V, Anwer F, Kelly KR, Nawrocki ST. Rational cotargeting of HDAC6 and BET proteins yields synergistic antimyeloma activity. Blood Adv. 2019;3:1318–1329. doi: 10.1182/bloodadvances.2018026484 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S5
Figure S1