

Abstract
Background
The small GTPase RhoA (Ras homolog gene family, member A) regulates a variety of cellular processes, including cell motility, proliferation, survival, and permeability. In addition, there are reports indicating that RhoA‐ROCK (rho associated coiled‐coil containing protein kinase) activation is essential for VEGF (vascular endothelial growth factor)‐mediated angiogenesis, whereas other work suggests VEGF‐antagonistic effects of the RhoA‐ROCK axis.
Methods and Results
To elucidate this issue, we examined human umbilical vein endothelial cells and human coronary artery endothelial cells after stable overexpression (lentiviral transduction) of constitutively active (G14V/Q63L), dominant‐negative (T19N), or wild‐type RhoA using a series of in vitro angiogenesis assays (proliferation, migration, tube formation, angiogenic sprouting, endothelial cell viability) and a human umbilical vein endothelial cells xenograft assay in immune‐incompetent NOD scid gamma mice in vivo. Here, we report that expression of active and wild‐type RhoA but not dominant‐negative RhoA significantly inhibited endothelial cell proliferation, migration, tube formation, and angiogenic sprouting in vitro. Moreover, active RhoA increased endothelial cell death in vitro and decreased human umbilical vein endothelial cell‐related angiogenesis in vivo. Inhibition of RhoA by C3 transferase antagonized the inhibitory effects of RhoA and strongly enhanced VEGF‐induced angiogenic sprouting in control‐treated cells. In contrast, inhibition of RhoA effectors ROCK1/2 and LIMK1/2 (LIM domain kinase 1/2) did not significantly affect RhoA‐related effects, but increased angiogenic sprouting and migration of control‐treated cells. In agreement with these data, VEGF did not activate RhoA in human umbilical vein endothelial cells as measured by a Förster resonance energy transfer–based biosensor. Furthermore, global transcriptome and subsequent bioinformatic gene ontology enrichment analyses revealed that constitutively active RhoA induced a differentially expressed gene pattern that was enriched for gene ontology biological process terms associated with mitotic nuclear division, cell proliferation, cell motility, and cell adhesion, which included a significant decrease in VEGFR‐2 (vascular endothelial growth factor receptor 2) and NOS3 (nitric oxide synthase 3) expression.
Conclusions
Our data demonstrate that increased RhoA activity has the potential to trigger endothelial dysfunction and antiangiogenic effects independently of its well‐characterized downstream effectors ROCK and LIMK.
Keywords: endothelial cells, endothelial dysfunction, LIM kinase 2, Rho‐associated protein kinase
Subject Categories: Angiogenesis, Endothelium/Vascular Type/Nitric Oxide, Cell Signalling/Signal Transduction
Nonstandard Abbreviations and Acronyms
- AQP1
aquaporin 1
- BrdU
5‐bromo‐2′‐deoxyuridine
- CD31/34
cluster of differentiation 31/34
- Cdc42
cell division control protein 42 homolog
- CXCL8
C‐X‐C motif chemokine 8=interleukin 8
- DIAPH1/2
diaphanous‐related formin ½
- eNOS
endothelial NO synthase
- FELASA
Federation of European Laboratory Animal Science Associations
- FRET
Förster resonance energy transfer
- GAPs
GTPase‐activating proteins
- GDI
guanosine diphosphate–dissociation inhibitors
- GEFs
guanine nucleotide exchange factors
- GO
gene ontology
- HCAEC
human coronary artery endothelial cells
- HDAC9
histone deacetylase 9
- HMG CoA
3‐hydroxy‐3‐methylglutaryl coenzyme A
- HUVEC
human umbilical vein endothelial cells
- ITGB4
integrin subunit β 4
- Ki‐67
Kiel‐67
- LIMK
LIM domain kinase
- LX7101
LIMK2 inhibitor
- NOS3
nitric oxide synthase 3
- PECAM1
platelet endothelial cell adhesion molecule 1
- PI
propidium iodide
- PKN1/2
protein kinase N1/N2
- Rac1
Ras‐related C3 botulinum toxin substrate 1
- RhoA,B,C
Ras homolog gene family, member A,B,C
- RNA‐seq
RNA sequencing
- ROCK
rho associated coiled‐coil containing protein kinase
- VEGF
vascular endothelial growth factor
- VEGFR‐2
vascular endothelial growth factor receptor 2
- VPF
vascular permeability factor
- VSV‐G
vesicular stomatitis virus glycoprotein
Clinical Perspective
What Is New?
This study demonstrates that increased RhoA (Ras homolog gene family, member A) activity has the potential to trigger endothelial dysfunction and antiangiogenic effects independently of its well‐characterized downstream effectors ROCK (rho associated coiled‐coil containing protein kinase) and LIMK (LIM domain kinase).
This way, increased RhoA activity could contribute to the pathogenesis of cardiovascular disease.
What Are the Clinical Implications?
The targeted use of specific RhoA inhibitors may represent a novel strategy for the prevention and treatment of atherosclerotic vascular disease and its complications.
The family of small Rho GTPases comprises ≈20 members in mammals and was first described >3 decades ago. 1 Rho family members, such as Cdc42 (cell division control protein 42 homolog), Rac1 (Ras‐related C3 botulinum toxin substrate 1), and RhoA (Ras homolog gene family, member A), regulate diverse signaling effectors as well as cellular and (patho)physiological functions (eg, cell division, cell migration, wound healing, or immune surveillance). By switching between 2 conformational states, a guanosine triphosphate (GTP)‐bound active state and a guanosine diphosphate–bound inactive state, they act as molecular switches of signal transduction. 2 , 3 In their active state, Rho GTPases recognize effector proteins and elicit a response until GTP hydrolysis returns them to their inactive state. This cycle is mainly controlled by GEFs (guanine nucleotide exchange factors) and GAPs (GTPase‐activating proteins). Interaction of Rho GTPases and GEFs induces exchange of guanosine diphosphate for GTP, whereas GAPs promote rapid GTP hydrolysis. To maintain Rho GTPases in their inactive cytosolic conformation, Rho GDIs (Rho guanosine diphosphate–dissociation inhibitors) interact with the GTPase and prevent nucleotide exchange and membrane association. 4 , 5
RhoA, RhoB, and RhoC form the Rho subfamily within the Rho GTPase family. 6 These 3 highly homologous proteins have an amino acid identity of about 88% and are therefore structurally similar. 7 Nevertheless, they differ on the functional level. RhoA and RhoC are mainly localized to the plasma membrane or in the cytoplasm, whereas RhoB is thought to be found mainly on the cytosolic side of early endosomes and in prelysosomal compartments. 8 , 9 All 3 isoforms are well characterized with respect to their role in cancer biology. RhoA acts as a stimulator of cell cycle progression, migration, and invasion, 10 whereas RhoC induces lamellipodia expansion through cytoskeletal reorganization, which promotes cancer cell metastasis. 11 In contrast, RhoB appears to function as a tumor suppressor, as evidenced by reduced RhoB expression in various cancer cell types. 12
Rho isoforms not only control cancer growth and metastasis, but may also regulate blood vessel formation in cancer and also play a role in the pathogenesis of endothelial dysfunction in the context of cardiovascular disease. 13 , 14 , 15 Therefore, their influence on physiological and pathophysiological blood vessel formation and endothelial homeostasis is the subject of current research efforts. 13 , 14 , 15 The best‐studied member of the Rho family in this regard is RhoA, which may play a role as a VEGF (vascular endothelial growth factor)‐induced effector of endothelial cell migration, proliferation, and cell adhesion. In this context, however, it is important to note that it remains controversial whether VEGF actually activates RhoA in human endothelial cells. 14 , 16 , 17 , 18 , 19 In addition, several studies have shown that RhoA is an important regulator of vascular leakage and transendothelial migration of leukocytes. 20 , 21 After activation of RhoA, the GTPase causes disruption of endothelial barrier function by stimulating its downstream effectors Rho‐dependent kinases ROCK1 and ROCK2 (rho associated coiled‐coil containing protein kinase 1 and 2), phosphorylation of myosin light chain, and formation of stress fibers. 22 , 23 Moreover, Rho‐dependent kinases subsequently activate LIMK1 and LIMK2 (LIM‐domain kinases 1 and 2) by phosphorylation, which thus leads to phosphorylation of the actin‐regulatory protein cofilin. By stimulating the ROCK/LIMK/cofilin axis, RhoA contributes to the reorganization of the actin cytoskeleton. 24 Nevertheless, controversial data have been published on the role of RhoA in VEGF‐mediated endothelial cell migration, 16 , 17 , 25 , 26 tubulogenesis, 27 , 28 and cell survival, 29 , 30 as well as on the importance of RhoA for neovascularization processes in vivo. 31 , 32 To elucidate the role of RhoA in human endothelial cell morphogenesis, survival, and function, we used a systematic approach in which we induced stable overexpression of constitutively active or dominant‐negative RhoA variants or RhoA wild‐type in human endothelial cells and then analyzed the angiogenic capacity of these cells in vitro and in vivo. In our work, we clearly demonstrate that an increase in RhoA activity induces antiangiogenic effects in human endothelial cells of arterial and venous origin, whereas expression of the dominant‐negative variant has no or rather a stimulatory effect on VEGF‐dependent endothelial cell functions. Consistent with these findings, the Rho inhibitor C3 transferase strongly stimulated endothelial angiogenic sprouting. Moreover, pharmacological inhibition of ROCK (Y‐27632) as well as LIMK2 (LX7101) increased angiogenic sprouting and the migration speed of human endothelial cells.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. RNA sequencing (RNA‐seq) data that support the findings of this study have been deposited in the US National Library of Medicine, National Center for Biotechnology Information Sequence Read Archive under the accession code GSE182806.
All chemicals and reagents were purchased from Sigma‐Aldrich (St. Louis, MO), unless stated otherwise. Recombinant human VEGF‐A was obtained from PeproTech (Rocky Hill, NJ). We bought the C3 transferase from Cytoskeleton (number CT04‐A; Denver, CO), the LIMK2 inhibitor LX7101 from Lexicon Pharmaceuticals (The Woodlands, TX), the LIMK1 inhibitor BMS4 from Axon Medchem (Reston, VA), and the ROCK inhibitor Y‐27632 from Santa Cruz Biotechnology (Dallas, TX). The validated RhoA biosensor was kindly provided by Jaap van Buul (Sanquin Research, Amsterdam, the Netherlands) and Yi Wu (Uconn Health, Farmington, CT). 14
Cell Culture and Lentiviral Transduction of Human Endothelial Cells
Human umbilical vein endothelial cells (HUVEC) and human coronary artery endothelial cells (HCAEC) were purchased from PromoCell and passaged in endothelial cell growth medium (PromoCell) on gelatin‐coated multiwell plates or in cell culture flasks according to the manufacturer’s specifications at 37 °C in humidified air with 5% CO2. Transduction of HUVEC and HCAEC was performed with VSV‐G (vesicular stomatitis virus glycoprotein)–pseudotyped lentiviral particles derived from the transfer plasmid pHIV‐SFiG‐1335 (a kind gift from Dr Boris Fehse, Department of Stem Cell Transplantation, University Medical Center Hamburg‐Eppendorf, Hamburg, Germany). To enable overexpression of dominant‐negative (T19N‐mutated), wild‐type, or constitutively active (G14V/Q63L) RhoA (NP_001655.1), 3×‐hemagglutinin‐tagged and codon‐optimized transcripts of the respective RhoA variants were cloned into the pHIV‐SFiG‐R135 backbone. For overexpression of the different RhoA mutants or the reporter eGFP (enhanced green fluorescent protein) alone as an adequate transduction control (pHIV‐SFiG‐1335), HUVEC or HCAEC (passages 2–5) were infected with a multiplicity of infection of 500, unless stated otherwise. Infected cells were cultured for at least 48 hours before functional or gene expression analyses. In all experiments, RhoA overexpression was determined by Western blot analyses. Transduction efficiency was additionally determined flow cytometrically by detection of the coexpressed reporter protein eGFP in endothelial cells. Transduction efficiency was generally around 80%.
Production, Purification, and Titer Determination of Lentiviral Vectors
Production, purification, and titration of lentiviral vectors were performed as described previously. 33 Shortly, human embryonic kidney cells, 293T cell line T cells were seeded in 150 cm² dishes at a density of 8*106 cells per dish in DMEM high‐glucose medium supplemented with 10% FCS, 1% penicillin‐streptomycin mix, and 1% GlutaMAX. After 24 hours, the cells were used for transfection at ≈40% confluence. Six milliliters of transfection mix per dish contained the transfer vector (60 µg), the VSV‐G envelope‐expressing plasmid pMD2.G (21 µg), the second‐generation lentiviral packaging plasmid psPAX2 (39 µg), 2 M CaCl2, water, and 2× HEPES‐buffered saline at a pH 7.07. Chloroquine at a final concentration of 25 µmol/mL was added to the medium right before transfection. The medium was changed once 20 hours after transfection. Two days after transfection, the cell supernatant was collected, centrifuged (500g, 10 minutes), and filtered (pore size 0.45 µm). The filtered supernatant was mixed in centrifuge beakers with 50% polyethylene glycol 6,000, 4 M NaCl, and 1× PBS. The mixture was stored at 4 °C for 90 minutes, and the beakers were shaken every 30 minutes. For concentration of lentiviral particles, the suspension was centrifuged (7000g, 10 minutes, 4 °C), and the pellet was resuspended in 50 mM Tris‐HCl, pH 7.4. Lentiviral preparations were stored at −80 °C.
Lentiviral vector titers were determined by flow cytometry (Attune Acoustic Focusing flow cytometer; ThermoFisher Scientific, Waltham, MA). For this purpose, 3×104 HUVEC per well were plated in gelatin‐coated 12‐well plates (Greiner) and transduced with certain amounts of virus suspension. Forty‐eight hours after transduction, cells were harvested and washed with 1× PBS/10% FCS. The number of eGFP‐positive cells was measured by flow cytometry. The virus titer was calculated according to the equation: transducing units mL‐1=(F*D*N)/V, where F is the percentage of eGFP‐positive cells, D is the dilution of virus used for transduction, N is the number of cells at the time of transduction, and V is the volume of diluted virus added per well during transduction.
Live Cell Detection of Endothelial RhoA Activity Using a Förster Resonance Energy Transfer‐Based Biosensor
HUVEC were plated in a 96‐well glass bottom plate (Greiner) coated with 50 µg/mL fibronectin (Santa Cruz Biotechnology) at a density of 10, 000 cells per well and transfected with the fluorescent RhoA Förster resonance energy transfer (FRET) biosensor 14 using the TurboFect reagent (ThermoFisher Scientific) according to the manufacturer`s recommendations. As described previously, time‐lapse or normal live‐cell FRET imaging was performed at 37 °C using a Nikon A1R confocal microscope equipped with a 60× oil immersion objective (plan apo lambda, Nikon, numerical aperture=1.4), an argon laser (Melles Griot, Germany), a PMT/GaAsP detector unit (Nikon), and an O2/CO2 cage incubator (Okolab, Ottaviano, Italy). 34 Images were acquired and processed using the NIS‐Elements (Nikon) FRET module . The FRET donor mCerulean3 was exited using the 457 nm laser line (argon laser), and fluorescence emission was detected in the spectral range of the donor (465–500 nm, DD image) and the acceptor (525–555 nm, DA image). In addition, the FRET acceptor mVenus was exited using the 514 nm laser line (argon laser) and detected in the spectral range of the acceptor (525–555 nm, AA image). Laser power and detector gain were set in such a way as to obtain the best signal intensities while avoiding oversaturation of the images. Image settings were kept constant for each series of measurements and for each image measured during subsequent time‐lapse recordings. Calculation of FRET index was calibrated using donor‐ and acceptor‐only samples (mCerulean3, mVenus) to determine the correction factors for donor crosstalk (α) and the acceptor’s direct excitation (β) in the DA image. Images showing the color‐coded FRET index were calculated as the intensity of the corrected FRET image normalized to the intensity of the donor image according to the following formula (FRET index=100% * (DA–αDD–βAA)/DD).
Image Processing
NIS‐Elements and Photoshop CS2 (Adobe, San José, CA) were used for image processing according to the Nature Research Image Integrity and Standards guidelines. To improve the visibility of fluorescent structures, brightness and contrast were uniformly increased over the entire image, and the settings made were then applied identically to the image material of all experimental groups in one experiment. For time‐lapse recordings, image settings were maintained for each subsequent picture. For multicolor merged images, the contrast and brightness of the individual color channels were adjusted to optimize their visibility.
Immunofluorescence Staining of Endothelial Cells
HUVEC were grown in endothelial cell growth medium and transduced on gelatin‐coated glass coverslips in 24‐well plates. Seventy‐two hours after transduction, cells were washed with 1× PBS and fixed in 4% formaldehyde/1× PBS for 15 minutes at room temperature. Cover slips were washed again in 1× PBS, and HUVEC were permeabilized using 0.1% Triton X‐100/PBS for 15 minutes at room temperature. Visualization of F‐actin in stress fibers was performed by subsequent incubation with 1.65 µmol/L 0.1% Phalloidin Alexa Fluor 633 for 1 hour at room temperature. Again, washing was performed with 1× PBS, and subsequently, cover slips were mounted in mounting medium. For immunostaining of paxillin, an antibody (clone E228) obtained from Abcam (ab32115; lot number: GR3239000‐4, 1:100) was used. For immunostaining of hemagglutinin‐tagged RhoA variants, an antibody directed against hemagglutinin (number 3724T, lot number 5; 1:1000; Cell Signaling Technology) as well as an Alexa Fluor 594‐conjugated secondary antibody (Dianova, 1:1000) were used.
Angiogenic Sprouting (Endothelial Cell Spheroid) Assay
The potential of angiogenic sprouting from endothelial spheroids was analyzed as described by Korff and Augustin. 35 In brief, HUVEC or HCAEC were harvested and suspended in endothelial growth medium with 20% methocel (methyl cellulose in endothelial growth medium). Endothelial aggregates (spheroids) containing 400 cells each, were formed in hanging drops overnight in nonadhesive culture dishes at 37 °C in humidified air with 5% CO2. Afterward, 50 spheroids per well were embedded in 0.5 mL of embedding matrix consisting of equal amounts of rat collagen and methocel with 20% FCS in nonadhesive 48‐well plates (Greiner). Spheroids were kept in basal endothelial medium (PromoCell) with VEGF (20 ng/mL) in the presence or absence of pharmacological blockers (Y‐27632=10 µmol/L, LX7101=3 µmol/L, BMS4=0.5 µmol/L, C3 transferase=1 µg/mL) for 24 hours at 37 °C in humidified air with 5% CO2. Angiogenic sprouting was quantified by measuring the cumulative sprout length of each spheroid with the NIS‐Elements digital software.
Endothelial Tube Formation Assay
The organization of HUVEC into capillary‐like networks (tube formation) was assessed as described previously 26 , 36 by using growth factor–reduced Matrigel (Corning). In brief, Matrigel was polymerized at 37 °C for 30 minutes in 96‐well angiogenesis microplates (10 µL/well; Ibidi). Then, 1*104 HUVEC were seeded per well and incubated in basal endothelial medium (PromoCell) supplemented with 2% FCS for 24 hours at 37 °C humidified air and 5% CO2. Tube length and morphological changes were visualized using an epifluorescence microscope (Nikon). Total tube length was measured with 10× magnification and NIS‐Elements software.
Migration Assay
Directional migration of either control‐transduced endothelial cells or endothelial cells expressing the different RhoA variants was analyzed using the scratch‐wound assay. 37 Briefly, 5000 cells per well were seeded in a gelatin‐coated 96‐well plate, transduced with lentiviral vectors, and grown to confluence for 72 hours. Then, a wound was set to create a gap into which the cells could migrate. During the experiment, cells were incubated in basal endothelial growth medium supplemented with 2.5% FCS in the presence or absence of pharmacological inhibitors (Y‐27632=10 µmol/L, LX7101=3 µmol/L, BMS4=0.5 µmol/L, C3 transferase=1 µg/mL). Cell movement was imaged at 20‐minute intervals for up to 24 hours using the CFI Plan Apochromat (Nikon) 10× objective. All analyses of time‐lapse image series, including migration distance and speed, were performed using the NIS‐Elements software package.
Endothelial Cell Proliferation Assay
The proliferation rate of control‐transduced endothelial cells and endothelial cells expressing the different RhoA variants was analyzed using the 5‐bromo‐2′‐deoxyuridine (BrdU) incorporation assay and immunofluorescence staining of the nuclear proliferation marker Ki‐67 (Kiel‐67). Incorporation of BrdU in de novo–synthesized DNA was investigated using a commercially available cell proliferation ELISA (Roche) according to the manufacturer’s instructions. In brief, 2500 endothelial cells per well were plated in a gelatin‐coated 96‐well plate (Greiner) and transduced with the appropriate lentiviral vector. Seventy‐two hours after infection, cell growth medium was supplemented with BrdU (final concentration 100 µmol/L) for 3 hours at 37 °C in humidified air with 5% CO2. Afterward, cells were fixed, denatured, and incubated for 90 minutes with a BrdU antibody, following the manufacturer’s instructions. Finally, stained cells were washed once with 1× PBS, and peroxidase substrate solution was added. After stopping the peroxidase reaction with 1 M H2SO4, readout was performed at 450 nm using a Tecan infinite F200pro plate reader.
For Ki‐67 staining, endothelial cells were grown on glass coverslips in 24‐well plates (Greiner), transduced, and fixed in 4% formaldehyde/PBS 72 hours after transduction. After fixation, coverslips were washed once with PBS, blocked, and permeabilized (3% BSA, 0.2% Tween 20, 2% Triton X‐100 in 1× TBS), and incubated with mouse monoclonal Ki‐67 antibody (number 9449T, lot number 4, 1:400 in Signal stain (R) Ab diluent; Cell Signaling Technology) in a humidified chamber overnight at 4 °C. Finally, cells were incubated with an Alexa Fluor 594‐conjugated goat anti‐mouse antibody (1:1000; Dianova) for 1 hour at room temperature. Visualization of nuclei was performed with Hoechst 33342 (1 mg/mL). After final washing with 1× PBS, coverslips were mounted in Dako Fluorescence Mounting Medium (Agilent). Pictures were taken with a Nikon A1R confocal microscope.
Western Blotting
Western blot analyses were performed as previously described 38 with endothelial cells lysed using commercial lysis buffer (Cell Signaling Technology) supplemented with a premade protease and phosphatase cocktail (ThermoFisher Scientific). Cell debris were removed by centrifugation for 5 minutes at 15 000g, 4 °C. SDS‐PAGE was performed with equal amounts of protein per lane, followed by transfer to a nitrocellulose membrane. Then, membranes were incubated with primary antibody solutions directed against the hemagglutinin tag (number 3724T, lot number 5; Cell Signaling Technology; 1:1000), RhoA (number 2117, lot number 3; Cell Signaling Technology; 1:1000), or β‐Actin (clone AC‐15, lot number 046M4873V; Sigma‐ Aldrich) according to the manufacturer’s instructions. Protein detection was achieved with peroxidase‐conjugated secondary antibodies and the enhanced chemiluminescence system (Amersham Bioscience).
Flow Cytometry
Endothelial cell viability was analyzed with propidium iodide (PI) using flow cytometry. DNA intercalated PI was detected at an emission maximum of 617 nm. Viability was assessed by PI exclusion. For this purpose, HUVEC were plated in gelatin‐coated 24‐well plates (Greiner) at a density of 1.5*105 cells per well and transduced with the appropriate RhoA variant or control vector. Seventy‐two hours after transduction, cells were detached, washed once in 1× PBS, and pelleted (400×g, 4 °C, 3 minutes). PI staining was performed by resuspending endothelial cell pellets in basal endothelial medium supplemented with 20 µg/mL final PI (ThermoFisher Scientific) concentration and incubation of the cells at room temperature for 5 minutes in the dark. Following the staining procedure, cells were kept on ice until measurement. Cells were measured using an Attune Acoustic Focusing flow cytometer (ThermoFisher Scientific). The number of RhoA‐overexpressing green fluorescent protein‐ and PI‐double positive cells was then compared with the number of green fluorescent protein‐ and PI‐double positive control endothelial cells. Flow cytometry data were analyzed using the FlowJo software package (Tree Star) as previously described. 39
GTP Pull‐Down Assay
Levels of activated, GTP‐bound RhoA were determined using the RhoA Activation Assay Biochem Kit (Cytoskeleton) according to the manufacturer´s instructions. Briefly, endothelial cells expressing different RhoA mutants and control cells were lysed 72 hours after transduction using commercial lysis buffer (Cell Signaling Technology) supplemented with a premade protease and phosphatase cocktail (ThermoFisher Scientific). Cell lysates were cleared by centrifugation (5 minutes, 15 000g, 4 °C) and stored at −80 °C until pull down. For Rho activation pull down, 300 µg total protein was mixed with 25 µg rhotekin‐sepharose beads and incubated for 1 hour at 4 °C in an overhead shaker. Afterward, beads were pelleted at 5000g for 1 minute at 4 °C and washed with washing buffer and pelleted again. Protein elution was performed by heating the protein‐loaded beads with 2× Laemmli buffer (125 mmol/L Tris pH 6.8, 20% glycerol, 4% SDS, 0.005% Bromophenol blue, 5% β‐mercaptoethanol), followed by Western blot analysis.
Real‐Time Reverse Transcriptase–Polymerase Chain Reaction
Total RNA from HUVEC overexpressing the different RhoA mutants and control cells was isolated as outlined previously, 40 reverse transcribed, and analyzed as described previously. 41 We analyzed mRNA expression with an ABI 7500 Real‐Time Polymerase Chain Reaction System (ThermoFisher Scientific). Quantification was done according to the manufacturer’s instructions using premade probes for ITGB4 (integrin subunit β 4) (Hs00236216_m1), CXCL8 (C‐X‐C motif chemokine 8=interleukin 8) (Hs00174103_m1), and HDAC9 (histone deacetylase 9) (Hs01081558_m1). HPRT1 (hypoxanthine‐guanine phosphoribosyltransferase 1; Hs02800695_m1) was used as a housekeeping gene. We performed relative quantification of gene expression using the delta‐delta (treshold cycle) Ct method. 42
RNA‐Seq and Differential Gene Expression Analyses
HUVEC overexpressing the different RhoA mutants and control cells were lysed with TRIZOL reagent according to the manufacturer´s recommendations to extract total RNA. RNA integrity and size distribution were analyzed with a Bioanalyzer 2100 (Agilent Technologies) and by agarose gel electrophoresis. Libraries were prepared with 1‐µg input RNA per sample. All samples were analyzed as triplicates. NEBNex Ultra RNA Library Prep Kit for Illumina (New England BioLabs) was used to generate RNA‐seq libraries. Sequencing was done by the Novogene Bioinformatics Institute (Bejing) as 150‐bp paired‐end reads on an Illumina Novaseq 6000 platform. Reads were aligned to the human genome (GRCh38) with HiSat2 (version 2.0.5), and ensembl (GRCh38.p13) was used for annotations. Read numbers mapped to each gene were determined with HTSeq (version 0.6.1), and subsequently, fragments per kilobase of transcript per million mapped reads of each gene were calculated based on length and read counts mapped to the gene. DESeq2 software package (version 1.20.0) was used to perform differential gene expression analyses. Genes with a P<0.05 were considered as differentially expressed. P values were calculated using the Benjamini‐Hochberg approach. GOseq R software (version 1.34.1) was used to analyze gene ontology pathway enrichments. Gene ontology (GO) terms with a P<0.05 were considered significantly enriched.
In Vivo Endothelial Spheroid Grafting Assay
For HUVEC xenograft analyses, male and female NOD scid gamma mice (The Jackson Laboratory; stock number 005557) were used in accordance with the directive 2010/63/EU and German law (Tierschutzgesetz). All procedures followed were in accordance with institutional guidelines. Moreover, all studies were performed in accordance with the local animal welfare commission (approval number 42502‐2‐1419 MLU). Mice were housed in groups of up to 5 animals in a specific pathogen‐free environment on a 12‐hour dark/12‐hour light cycle at 22±2 °C in accordance with the Federation of European Laboratory Animal Science Associations (FELASA) guidelines.
The in vivo endothelial spheroid grafting assay was performed as described previously. 43 , 44 HUVEC were transduced with RhoA Q63L, RhoA T19N, and control vectors and spheroids were generated using the hanging drop method. Afterward, endothelial spheroids were embedded into a matrix containing endothelial growth medium, methocel, fibrinogen, growth factors, and high concentration Matrigel (Corning). These suspensions, which contained either control‐treated HUVEC or HUVEC overexpressing constitutively active RhoA or dominant‐negative RhoA, were injected subcutaneously into the left and right ventral regions of 10‐ to 12‐week‐old female and male NOD scid gamma mice according to a prespecified regimen that included similar numbers of female and male NOD scid gamma mice. Randomization of mice was therefore not performed. For explantation of plugs, mice were euthanized 21 days after injection. Plugs were immediately fixed in 4% formaldehyde and embedded in paraffin following standard protocols. Then, 8‐µm‐thick sections were sliced, and humanoid neovessels were stained using fluorescently labelled Ulex europaeus agglutinin I (Vector Laboratories). In addition, as a surrogate for the connection of blood vessels within the plug to the functional blood circulation of the murine host, murine hemoglobin associated with humanoid neovessel structures was determined by histomorphometric immunofluorescence analysis after antibody labeling of mouse hemoglobin subunit α (Abcam; ab92492, lot number GR3276791‐1, 1:250) followed by Alexa Fluor 594‐labeled secondary antibody (Dianova; 1:200). All areas of the plug sections were subsequently systematically photographed and then evaluated by computer‐aided histomorphometric analyses using the NIS elements software package (Nikon). Due to the systematic approach of the evaluation, blinding was not performed. As a primary end point, blood vessel density was analyzed by quantifying UEA I‐positive blood vessel area per total plug area. Secondary end points analyzed included the number of UEA I‐positive blood vessels per plug area and the average size of UEA I‐positive blood vessels, as well as the hemoglobin‐positive blood vessel area per total plug area, the number and the average size of hemoglobin‐positive blood vessels per high‐power field. For the calculation of the sample size, the program G*Power 3.1.9 was used. We estimated a relative within group SD for the primary end point of 0.25 and assumed relative differences of the means of at least 0.3 to be biologically significant. For a power of 80% and an alpha of 5% this corresponded to a minimal sample size of 9 plugs per group. In addition, phenotypic characterization of the vascular network derived from HUVEC was performed by immunohistochemistry using antibody labeling of human endothelial cell markers CD31 (cluster of differentiation 31) (Cell Signaling Technology; clone 89C2; 1:250) and CD34 (cluster of differentiation 34 (ThermoFisher Scientific; clone QBEnd/10; 1:250). HRP‐conjugated secondary antibodies were used as described by the manufacturer (Cell Signaling Technology; HRP‐mouse, number 8125). For staining, we used the DAB substrate kit according the manufacturer instructions (Cell Signaling Technology; number 8059) as described previously. 45
Statistical Analysis
Statistical analyses were performed using 1‐way ANOVA followed by the Bonferroni multiple comparison test (multiple group analyses) or by the unpaired 2‐tailed Student t test (2‐group analyses) as appropriate. For analysis of potential sex‐dependent differences in HUVEC‐associated vessel growth in vivo, the 2‐way ANOVA followed by the Bonferroni multiple comparisons test was used. For statistical analyses, the GraphPad Prism 6 software package was used (GraphPad Software, La Jolla, CA). Data were expressed as mean±SD or as indicated otherwise. Probability values were considered significant at a P<0.05.
Results
Activity Determination of RhoA Variants and Impact of VEGF on RhoA Activity in Human Endothelial Cells
Our initial studies focused on characterizing the different RhoA mutants in HUVEC as an endothelial cell model system. Two constitutively active RhoA variants (G14V or Q63L), a dominant‐negative (T19N) mutant, or the wild‐type protein were overexpressed in HUVEC, resulting in an increase in total RhoA levels of 1.5‐fold (G14V) to 3.1‐fold (wild‐type) compared with endogenous RhoA levels of control cells (Figure S1A and S1B). To analyze the activity state of the RhoA variants studied, protein lysates were incubated with rhotekin‐sepharose beads (Figure 1A). This method allows the pull down of GTP‐bound (active) RhoA. 46 All overexpressed RhoA variants were hemagglutinin tagged and could be detected by using anti‐hemagglutinin antibody in subsequent Western blot analyses. This allowed us to confirm activity of the G14V and Q63L RhoA mutants (Figure 1A). Because the concentration of GTP in the cytosolic environment of living endothelial cells is higher than that of guanosine diphosphate, 47 a substantial amount of the overexpressed wild‐type RhoA protein was also present in the active, GTP‐bound state (Figure 1A). In addition, we examined the local distribution of hemagglutinin‐tagged RhoA variants in HUVEC by immunofluorescence analysis using an antibody directed against the hemagglutinin tag (Figure S2). In HUVEC, which exhibit a flat adherent phenotype in vitro, both constitutively active (G14V, Q63L) and wild‐type RhoA were localized mainly at the plasma membrane, whereas inactive dominant‐negative RhoA (T19N) largely accumulated in perinuclear regions. Because it has been postulated that the proangiogenic effects of VEGF (VEGF‐A/VPF [vascular permeability factor]) depend on RhoA activity in human endothelial cells, we also analyzed the impact of VEGF (20 ng/mL) on RhoA activity in live HUVEC using a FRET‐based biosensor (for validation analyses of the sensor, please see Figure 1B and 1C). We included thrombin in these studies as a positive control. Surprisingly, we did not observe a VEGF‐induced increase in RhoA activity in our experiments, whereas thrombin (1 U/mL) significantly increased RhoA activity in HUVEC, as expected (Figure 1D through 1G). In this context, we also verified the inhibitory effect of the Rho inhibitor C3 transferase on RhoA wild‐type activity. As anticipated, the inhibitor (1 µg/mL) significantly reduced RhoA activity in HUVEC (Figure 1H).
Figure 1. Activity determination of RhoA (Ras homolog gene family, member A) variants and impact of VEGF (vascular endothelial growth factor) on RhoA activity in human endothelial cells.
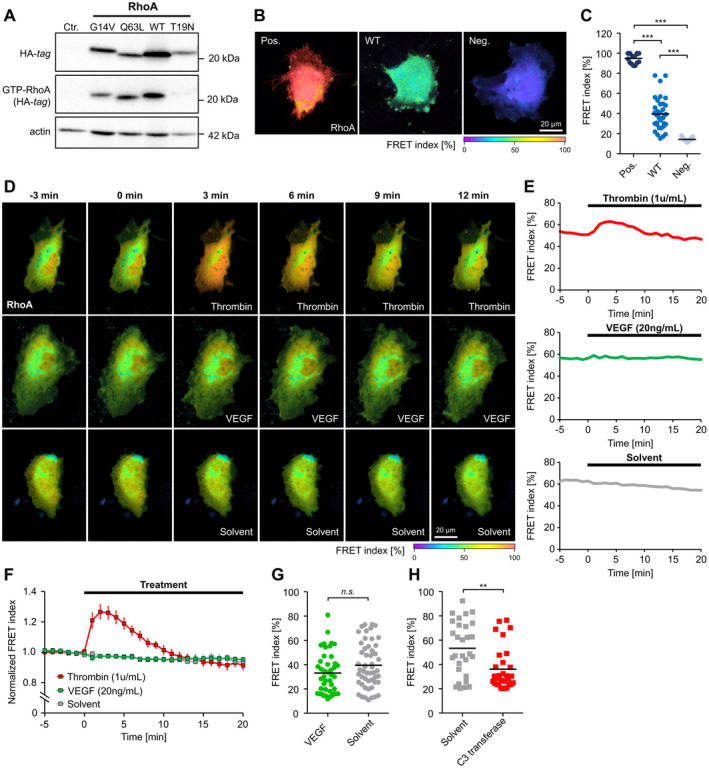
A, Pull down of GTP‐bound RhoA (GTP‐RhoA) derived from constitutively active RhoA (G14V, Q63L), wild‐type (WT) RhoA and dominant‐negative RhoA (T19N) using rhotekin‐coated beads. B and C, Validation of a RhoA Förster resonance energy transfer (FRET) biosensor after transfection of constitutively active (Pos.), WT, and dominant‐negative (Neg.) variants of the sensor in human umbilical vein endothelial cells (HUVEC). B, Representative images of the color‐coded FRET index in HUVEC transfected with the different RhoA variants. C, Statistical analysis shows that the activity of WT‐RhoA is intermediate between the RhoA activity of constitutively active and dominant‐negative controls. ***P<0.001. D, Time‐lapse recordings of HUVEC transfected with the WT RhoA biosensor. The color‐coded FRET index indicates a dynamic change in the RhoA activation state after application of thrombin (1 U/mL), but not after application of human vascular endothelial growth factor (VEGF; 20 ng/mL) or solvent. E, Changes in FRET index over time of single cells shown in (D). F, Average baseline‐normalized FRET index after stimulation of HUVEC with thrombin (n=9), VEGF (n=12), or solvent (n=6). Thrombin induces a transient RhoA activation, whereas no RhoA activation was observed after application of VEGF. G, FRET index of HUVEC measured after long‐term stimulation (18 hours) with VEGF (n=43) or solvent (n=52) indicates no VEGF‐related change in RhoA activity. H, Long‐term application (18 hours) of the RhoA inhibitor C3 transferase (n=30) reduces RhoA activity compared with solvent control (n=29). **P<0.01. Ctr. indicates control‐transduced; HA indicates hemagglutinin; and n.s., not significant.
Impact of RhoA on Stress Fiber Formation and Focal Adhesions in HUVEC
The effect of RhoA activity on stress fiber formation was analyzed by phalloidin staining of F‐actin bundles in fixed HUVEC (Figure 2A). Successful transduction of HUVEC and expression of RhoA in these cells was assumed based on coexpression of the reporter eGFP. Control‐transduced cells expressed only eGFP, but no hemagglutinin‐tagged RhoA variants. In these experiments, overexpression of inactive RhoA and RhoA wild‐type protein resulted in a cortical accumulation of F‐actin, whereas constitutively active RhoA (G14V, Q63L), as expected, caused marked stress fiber formation in transduced endothelial cells. Additionally, the size of focal adhesions in HUVEC was determined after immunostaining of the focal adhesion protein paxillin (Figure 2B through 2D). In this regard, active RhoA (Q63L) as well as wild‐type RhoA both induced a significant increase in the focal adhesion area compared with control‐transduced HUVEC (Figure 2C), as well as an increased average focal adhesion size (Figure 2D). The constitutively active RhoA G14V variant tended to exert similar effects, but without significantly increasing the focal adhesion area (Figure 2C). Nevertheless, the average size of focal adhesions was greatly increased (Figure 2D). In contrast, inactivation of RhoA (RhoA T19N) did not significantly change the focal adhesion area compared with control‐transduced cells, but also increased the average focal adhesion size.
Figure 2. Active RhoA (Ras homolog gene family, member A) induces stress fiber formation, regulates focal adhesion dynamics, and decreases endothelial cell viability.
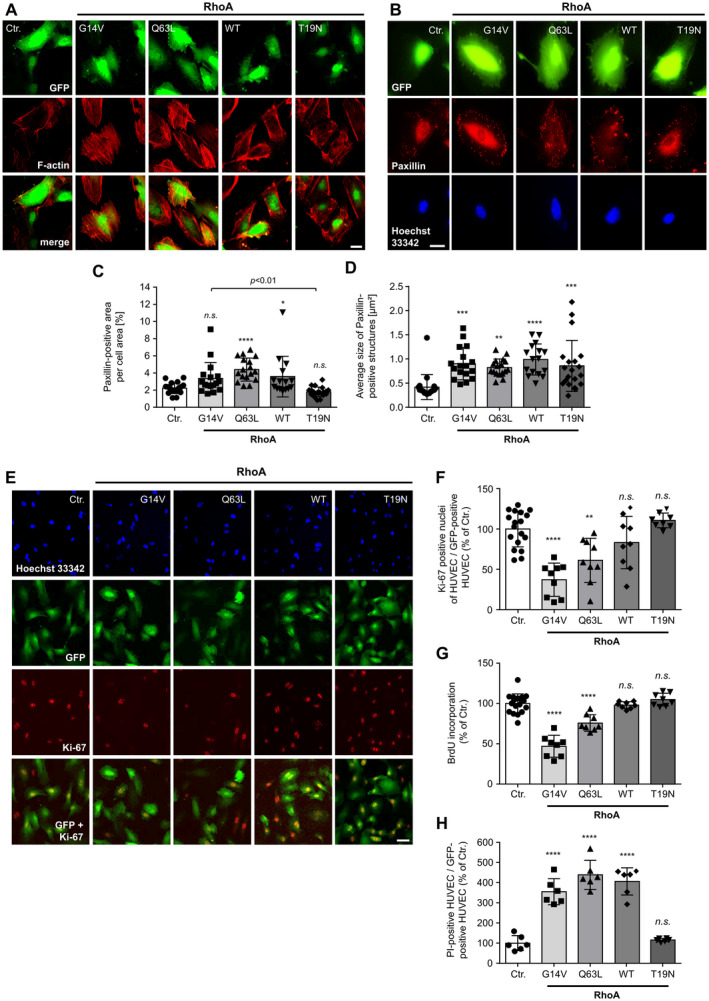
A, Stress fiber formation induced by active RhoA. Scale bar=20 µm. B, Representative microscopic images of human umbilical vein endothelial cells (HUVEC) expressing different RhoA variants and stained for selective visualization of focal adhesions (paxillin) and nuclei (Hoechst 33342). Successful transduction is indicated by coexpression of the reporter eGFP. Scale bar=20 µm. C, Quantification of paxillin‐positive area per total cell area. *P<0.05, ****P<0.0001 vs control‐transduced (Ctr.) HUVEC (n=15–20). D, Determination of the average size of paxillin‐positive focal adhesions in HUVEC. **P<0.01, ***P<0.001, ****P<0.0001 vs control‐transduced HUVEC (n=15–20). E, Expression of the cell proliferation marker Ki‐67 (Kiel‐67) in HUVEC expressing different RhoA variants. F, Quantification of Ki‐67‐positive (proliferating) cells. **P<0.01, ****P<0.0001 vs control‐transduced HUVEC (n=8–18). G, 5‐bromo‐2′‐deoxyuridine (BrdU) incorporation of HUVEC expressing different RhoA variants. ****P<0.0001 vs control‐transduced HUVEC (n=8–18). H, Flow cytometric evaluation of propidium iodide (PI) uptake of HUVEC expressing different RhoA variants. ****P<0.0001 vs control‐transduced HUVEC (n=6). eGFP indicates enhanced green fluorescent protein; GFP, green fluorescent protein; n.s., not significant; and WT, wild‐type.
Expression of Constitutively Active RhoA Reduces Endothelial Cell Viability
To clarify the influence of the small GTPase RhoA on proliferation and viability of endothelial cells in vitro, we performed 2 different proliferation assays and PI staining of RhoA‐overexpressing and control‐transduced HUVEC (Figure 2E through 2H). A reduction of BrdU incorporation during DNA synthesis in proliferating cells, as well as a reduction of nuclear Ki‐67 expression, a nuclear nonhistone protein that is universally expressed among proliferating cells and absent in quiescent cells, 48 indicated a significant decrease in HUVEC proliferation after overexpression of active RhoA (G14V and Q63L) (Figure 2E and 2F). Interestingly, overexpression of wild‐type RhoA did not negatively affect HUVEC proliferation, although a substantial fraction of wild‐type RhoA was present in its GTP‐bound active state, as demonstrated by the use of rhotekin pull down (Figure 1A). In addition, inactivation of RhoA using overexpression of the inactive T19N mutant had no effect on the proliferation of HUVEC in vitro (Figure 2E and 2F).
To investigate whether active RhoA additionally affects endothelial cell viability, HUVEC after overexpression of different RhoA variants were stained with PI. Because PI cannot penetrate intact cells, it is a suitable marker for dead or damaged cells. 49 Using flow cytometry, we detected a significant increase in the PI‐positive cell fraction of HUVEC expressing active RhoA mutants (Figure 2H). In contrast to the results of the proliferation experiments, an analysis of endothelial cell survival showed that overexpression of the RhoA wild‐type had the same detrimental effect on HUVEC survival as did overexpression of the constitutively active RhoA mutants. In contrast, inactivation of RhoA by overexpression of the T19N mutant had no effect on HUVEC viability. In summary, our data clearly demonstrate that RhoA activity has a negative effect on the proliferation and viability of human endothelial cells and thus may affect vascular endothelial homeostasis via this mechanism.
Expression of Constitutively Active RhoA Reduces Tube Formation, Angiogenic Sprouting, and Migration of Human Endothelial Cells
In addition to cell survival, tube formation, angiogenic sprouting, and endothelial cell migration represent important steps of the angiogenic process. Therefore, we next examined the influence of RhoA on these angiogenic endothelial cell functions. We first performed a 2‐dimensional Matrigel‐based tube formation assay (Figure 3A and 3B). In this assay, transduced RhoA‐overexpressing cells or control cells were plated onto a polymerized Matrigel matrix and stimulated with VEGF to form tubular structures. After expression of active RhoA mutants in HUVEC, we observed a statistically significant reduction in the total length of endothelial tubes formed in these experiments (Figure 3A and 3B). In contrast, overexpression of inactive RhoA or wild‐type RhoA apparently did not alter endothelial tube formation (Figure 3A and 3B).
Figure 3. Active RhoA (Ras homolog gene family, member A) disturbs tube formation, angiogenic sprouting, and migration of endothelial cells in vitro.
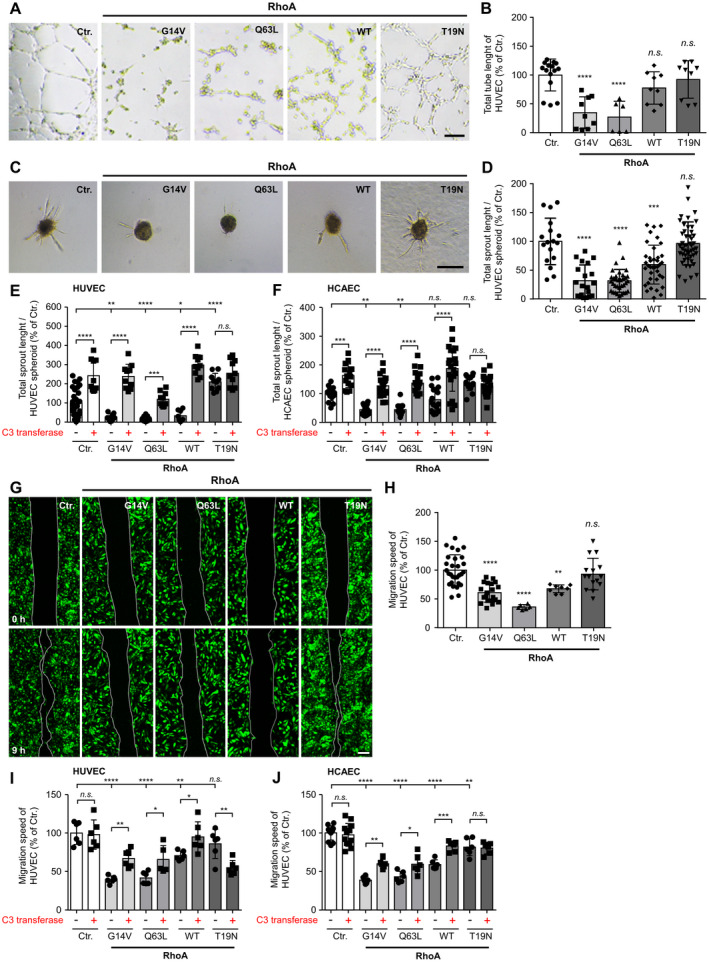
A, Representative pictures of control‐transduced (Ctr.) human umbilical vein endothelial cells (HUVEC, Ctr.) or HUVEC expressing the indicated constitutively active (G14V, Q63L), dominant‐negative (T19N), or wild‐type (WT) RhoA variants. Scale bar=250 µm. B, Statistical analysis of total tube length. ****P<0.0001 vs control‐transduced HUVEC (n=6–15 wells). C, Impact of RhoA on vascular endothelial growth factor (VEGF, 20 ng/mL)‐induced HUVEC spheroid sprouting in vitro. Scale bar=200 µm. D, Statistical analysis of total sprout length. ***P<0.001, ****P<0.0001 vs control‐transduced HUVEC (n=17–50). E, Statistical analysis of total sprout length quantified from VEGF‐induced (20 ng/mL) control‐transduced HUVEC or HUVEC expressing the different RhoA variants in presence (+) or absence (−) of the RhoA inhibitor C3 transferase (1 µg/mL). *P<0.05/***P<0.001/****P<0.0001 vs control‐transduced HUVEC (n=10–30). F, Statistical analysis of total sprout length quantified from VEGF‐induced (20 ng/mL) control‐transduced human coronary artery endothelial cells (HCAEC, Ctr.) or HCAEC expressing the different RhoA variants in presence (+) or absence (−) of the RhoA inhibitor C3 transferase (1 µg/mL). **P<0.01, ***P<0.001, ****P<0.0001 vs control‐transduced HCAEC (n=13–20). G, Scratch‐wound assay of control‐transduced HUVEC or HUVEC expressing different RhoA variants. Images depict wound size at time of wounding (0 hours) and 9 hours later. Scale bar=200 µm. H, Statistical analysis of migration speed of HUVEC. **P<0.01/****P<0.0001 vs control‐transduced HUVEC (n=6–19). I, Statistical analysis of migration speed of HUVEC kept in presence (+) or absence (−) of the RhoA inhibitor C3 transferase (1 µg/mL). *P<0.05, **P<0.01, ****P<0.0001 vs control‐transduced HUVEC. (n=10–30). J, Statistical analysis of migration speed of HCAEC kept in presence (+) or absence (−) of the RhoA inhibitor C3 transferase (1 µg/mL). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs control‐transduced HCAEC (n=6–12). n.s. indicates not significant.
To analyze the formation of vessel‐like structures in a 3‐dimensional format, HUVEC cell aggregates (spheroids) were embedded in a collagen matrix, and angiogenic sprouting from these spheroids was induced by addition of VEGF (Figure 3C through 3F). Consistent with the findings from the 2‐dimensional tube formation assays, active RhoA also inhibited sprout formation and significantly reduced the total sprout length formed by HUVEC and HCAEC (Figure 3C through 3F). Interestingly, similar to overexpression of the constitutively active RhoA mutants G14V and Q63L, overexpression of wild‐type RhoA in these experiments resulted in strong inhibition of angiogenic sprouting of HUVEC and HCAEC (Figure 3C and 3D). In contrast, dominant‐negative RhoA (T19N) again had no effect on angiogenic sprouting of HUVEC and HCAEC spheroids. The effects of active RhoA variants on sprouting were partially (RhoA Q63L) or completely (RhoA G14V, wild‐type RhoA) abolished by the addition of the exoenzyme C3 transferase from Clostridium botulinum (Figure 3E through 3F). This enzyme specifically inhibits RhoA by ADP‐ribosylation at asparagine 41 in the effector binding domain of the GTPase. 50 Moreover, C3 transferase massively induced VEGF‐induced sprouting of control‐transduced HUVEC and HCAEC, thereby indicating that Rho GTPases are negative regulators of angiogenic sprouting of human endothelial cells in vitro (Figure 3E through 3F).
Endothelial cell motility, another important aspect of the angiogenic process, was analyzed using the scratch‐wound assay (Figure 3G through 3J). HUVEC and HCAEC were transduced to express the different RhoA variants and grown to confluence. The confluent cell layer was injured (scratched) and then time‐lapse photographed at several time points to determine the movement of the cell front in the period from immediately after injury to 9 hours after wounding. The mean migration velocity was calculated after measuring the migration distance after 9 hours. With this experimental setup, an inhibitory effect of active RhoA on endothelial cell migration was detected, with RhoA Q63L tending to have the strongest effects. Again, wild‐type RhoA inhibited endothelial cell migration in a manner comparable to that observed for the constitutively active RhoA mutants (Figure 3G through 3J). Expression of the inactive RhoA T19N mutant did not have a profound effect on HUVEC migration but slightly decreased the migration speed of HCAEC (Figure 3J). Treatment of HUVEC or HCAEC with the Rho inhibitor C3 transferase, partially antagonized the inhibitory effect of the active RhoA mutants on the migration speed of these cells (Figure 3I through 3J). Moreover, inhibition of HUVEC and HCAEC migration induced by wild‐type RhoA was almost completely abolished by this intervention (Figure 3I through 3J). Interestingly, inhibition of C3 transferase decreased the migration speed of HUVEC overexpressing dominant‐negative RhoA.
Pharmacological Inhibition of ROCK or LIMK Does Not Antagonize RhoA‐Related Antiangiogenic Effects
To analyze potential downstream effectors of RhoA, we focused on the well‐characterized RhoA/ROCK/LIMK pathway and examined the effects of pharmacological inhibition of ROCK and LIMK with Y‐27632 (ROCK1/2), LX7101 (LIMK2), and BMS4 (LIMK1; Figure 4). For this purpose, we investigated the 3‐dimensional spheroid‐based angiogenic sprouting (Figure 4A) and migration (Figure 4B) of HUVEC. Inhibition of ROCK by Y‐27632 significantly promoted VEGF‐induced angiogenic sprouting in control‐transduced HUVEC. However, ROCK inhibition was not able to block the inhibitory effect of active RhoA on angiogenic sprout formation. The same observations were made using the LIMK2 inhibitor LX7101, which stimulated angiogenic sprouting of control‐transduced HUVEC but did not antagonize the inhibitory effects of active RhoA mutants. Moreover, selective inhibition of LIMK1 with BMS4 had no effect on basal or RhoA‐suppressed angiogenic sprouting (Figure 4A). Taken together, inhibition of RhoA downstream targets ROCK1/2 and LIMK2 under control conditions resulted in an increase in HUVEC migration and angiogenic sprouting (Figure 4A, 4B). In contrast, neither Rho kinases nor LIM kinases appear to mediate the inhibitory effects of active RhoA on HUVEC migration or angiogenic sprouting.
Figure 4. Impact of the RhoA (Ras homolog gene family, member A)/ROCK (rho associated coiled‐coil containing protein kinase) pathway on VEGF (vascular endothelial growth factor)‐induced angiogenic endothelial cell sprouting and basal migration.
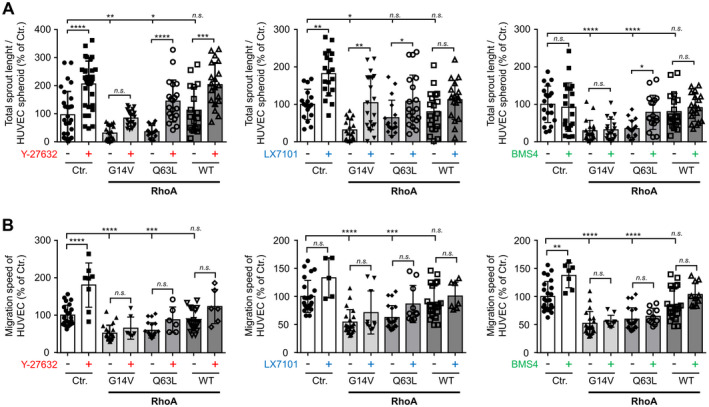
A, VEGF‐stimulated (20 ng/mL) human umbilical vein endothelial cells (HUVEC) spheroid‐based sprouting assay using control‐transduced (Ctr.) HUVEC or HUVEC expressing constitutively active RhoA (G14V, Q63L) or RhoA wild‐type (WT) in the presence (+) or absence (−) of the ROCK inhibitor Y‐27632 (10 µmol/L), the LIMK2 (LIM domain kinase 2) inhibitor LX7101 (3 µmol/L), or the LIMK1 (LIM domain kinase 1) inhibitor BMS4 (0.5 µmol/L), respectively. Statistical analysis of total sprout length. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs control‐transduced HUVEC (n=16–34). B, Statistical analysis of migration speed of HUVEC expressing different RhoA variants and kept in presence (+) or absence (−) of the ROCK inhibitor Y‐27632 (10 µmol/L), the LIMK2 inhibitor LX7101 (3 µmol/L), or the LIMK1 inhibitor BMS4 (0.5 µmol/L). ***P<0.001, ****P<0.0001 vs control‐transduced HUVEC (n=5–24). n.s. indicates not significant.
RhoA Inhibits the Angiogenic Capacity of Vascular Endothelial cells In Vivo
To elucidate the impact of RhoA activity on the angiogenic capacity of human endothelial cells in vivo, we used the HUVEC spheroid‐based grafting assay in immunocompromised NOD scid gamma mice and Ulex europaeus agglutinin I–positive blood vessels as a readout (Figure 5A). As expected, we observed frequent colocalization of Ulex europaeus agglutinin I– and hemoglobin‐positive blood vessels, showing that implanted HUVEC actively took part in the formation of functional neovessels in the murine host (Figure 5B). First, further phenotypic analyses confirmed that the humanoid intraplug blood vessel network also expressed typical human endothelial markers, such as CD31 (PECAM1 [platelet endothelial cell adhesion molecule 1]) or CD34 (Figure 5C and 5D). Stable overexpression of active RhoA (Q63L mutant) greatly reduced the angiogenic capacity of implanted HUVEC, as evidenced by a reduction in the total area of blood vessels (Figure 5E and 5G) and the number of blood vessels (Figure 5H) per high‐power field, whereas the average size of Ulex europaeus agglutinin I–positive structures was not affected (Figure 5I). In contrast, inactive RhoA (T19N mutant) did not significantly affect the angiogenic capacity of HUVEC in vivo. The functional connection of humanoid blood vessels within the plug to the vasculature and circulation of the murine host was evaluated in histomorphometric immunofluorescence analyses by quantifying hemoglobin‐positive blood vessels. Again, under the influence of active RhoA (Q63L), we observed a reduction in hemoglobin‐positive blood vessel area (Figure 5F and 5J) and a reduced number of hemoglobin‐positive blood vessels (Figure 5K), whereas the average size of hemoglobin‐positive blood vessels was not affected (Figure 5L). Moreover, inactive RhoA (T19N) did not significantly affect (but tended to reduce) the hemoglobin‐positive blood vessel area and the number of hemoglobin‐positive blood vessels in our experiments (Figure 5F, 5J and 5K). Sex‐specific evaluation of Ulex europaeus agglutinin I– or hemoglobin‐positive blood vessels yielded comparable results for plugs harvested from female and male animals (Figure S3), such that we performed a pooled evaluation of results from female and male mice.
Figure 5. In vivo blood vessel formation is affected by RhoA (Ras homolog gene family, member A) activity.
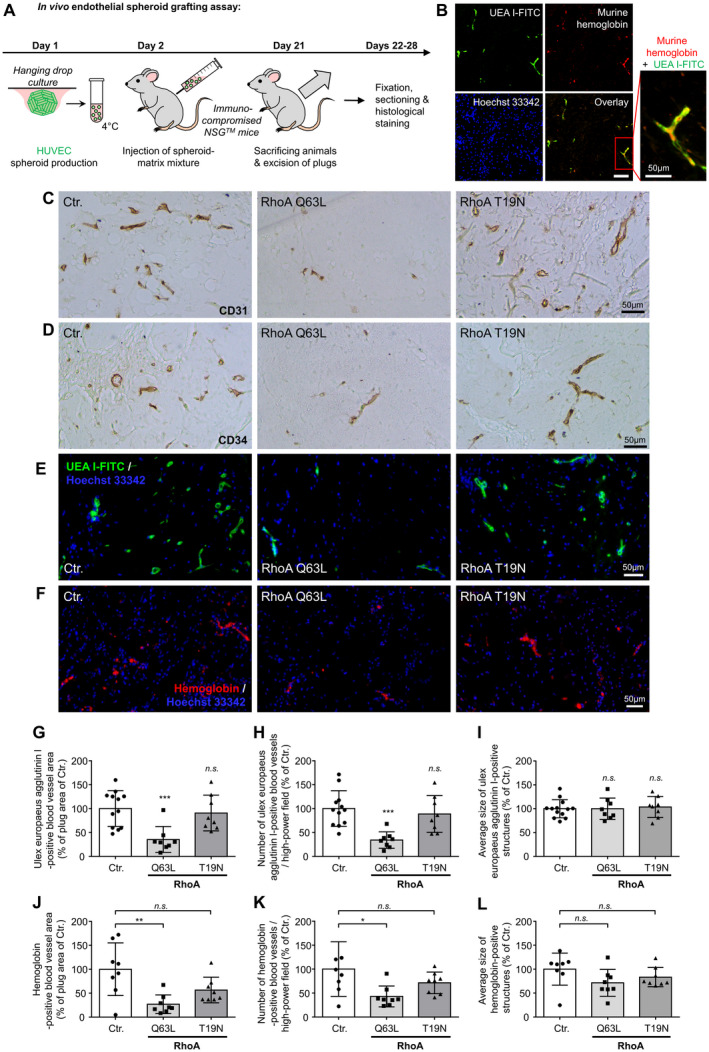
A, Schematic illustration of the experimental setup of the human umbilical vein endothelial cells (HUVEC) spheroid‐based grafting assay in immunodeficient NSG mice. B, Colocalization of fluorescein‐5‐isothiocyanat‐labeled (FITC) europaeus agglutinin I (UEA I) and murine hemoglobin‐positive blood vessels. Scale bar=100 µm. C and D, Representative microscopic pictures of xenograft plug sections stained for visualization of endothelial cell markers CD31 and CD34 (cluster of differentiation 31/34). E, Representative microscopic pictures of xenograft plug sections stained with FITC‐labeled Ulex europaeus agglutinin (UEA I‐FITC) for selective visualization of the vascular network derived from embedded HUVEC. Nuclei were stained with Hoechst 33342. F, Representative microscopic images of sections of xenograft plugs stained with an antibody directed against murine hemoglobin to selectively visualize humanoid blood vessels within the plug that are connected to the murine host circulation. Nuclei were stained with Hoechst 33342. G, Blood vessel growth was analyzed by quantification of the UEA I–positive blood vessel area per total plug area. The number of UEA I–positive blood vessels per plug area (H) and average size of UEA I–positive blood vessels (I) were also determined. ***P<0.001 vs control‐transduced (Ctr.) HUVEC (n=8–12). J, Quantification of the hemoglobin‐positive blood vessel area per total plug area. **P<0.01 vs control plugs (n=8). Determination of the number (K) and average size (L) of hemoglobin‐positive blood vessels per high‐power field. *P<0.05 vs control plugs. (n=8). n.s. indicates not significant; and NSG, NOD scid gamma.
Both Activation and Inactivation of RhoA Lead to Profound Changes in the HUVEC Transcriptome
To further elucidate the role of RhoA in HUVEC, we performed global transcriptome analyses of HUVEC overexpressing either constitutively active (Q63L) or dominant‐negative (T19N) RhoA or were subjected to control transduction (Figure 6). RNA‐seq analysis of 3 biological replicates revealed that constitutive activation of RhoA induced differential expression of 7768 genes, whereas dominant‐negative RhoA significantly affected the expression of 2172 genes (Figure 6A and 6B). Examples of significantly regulated mRNAs in RhoA Q63L‐ as well as RhoA T19N‐overexpressing HUVEC are shown in Figure 6C. Interestingly, expression of RhoA Q63L significantly downregulated the key angiogenic mediator VEGFR‐2 (vascular endothelial growth factor receptor 2) and the vascular tone regulator NOS3 (nitric oxide synthase 3)/eNOS (endothelial NO synthase; Figure 6C). In addition, it strongly induced the expression of HDAC9, a member of the class IIa histone deacetylases that catalyzes the deacetylation of histones and transcription factors and thereby generally suppresses gene transcription. 51 We observed that ≈94% of all genes differentially regulated by RhoA Q63L showed reduced expression, suggesting that HDAC9 may function as an important effector of RhoA‐mediated antiangiogenic signal transduction in human endothelial cells. Additionally, RhoA expression modifies the transcription of inflammatory molecules (CXCL8) as well as adhesion molecules (ITGB4) and AQP1 (aquaporin 1). To validate the results obtained by RNA‐seq, we used quantitative real‐time polymerase chain reaction and obtained similar results for the expression of IGTB4, CXCL8, and HDAC9 (Figure S4). Consistent with our observations, GO enrichment analyses revealed that constitutively active RhoA as compared with dominant‐negative RhoA induced a differentially expressed gene pattern that was enriched for GO terms associated with mitotic nuclear division, mitotic cell cycle, cell proliferation, cell adhesion, cell motility, locomotion, cytoskeleton organization, and cell morphogenesis (Figure 6D through 6E). Significantly regulated genes are summarized in Table S1. Taken together, the data obtained in the transcriptome and subsequent bioinformatic analyses provide first mechanistic explanations for the inhibitory effect of active RhoA on the angiogenic capacity and cell viability of human endothelial cells.
Figure 6. RhoA (Ras homolog gene family, member A) activity affects gene expression of human umbilical vein endothelial cells (HUVEC).
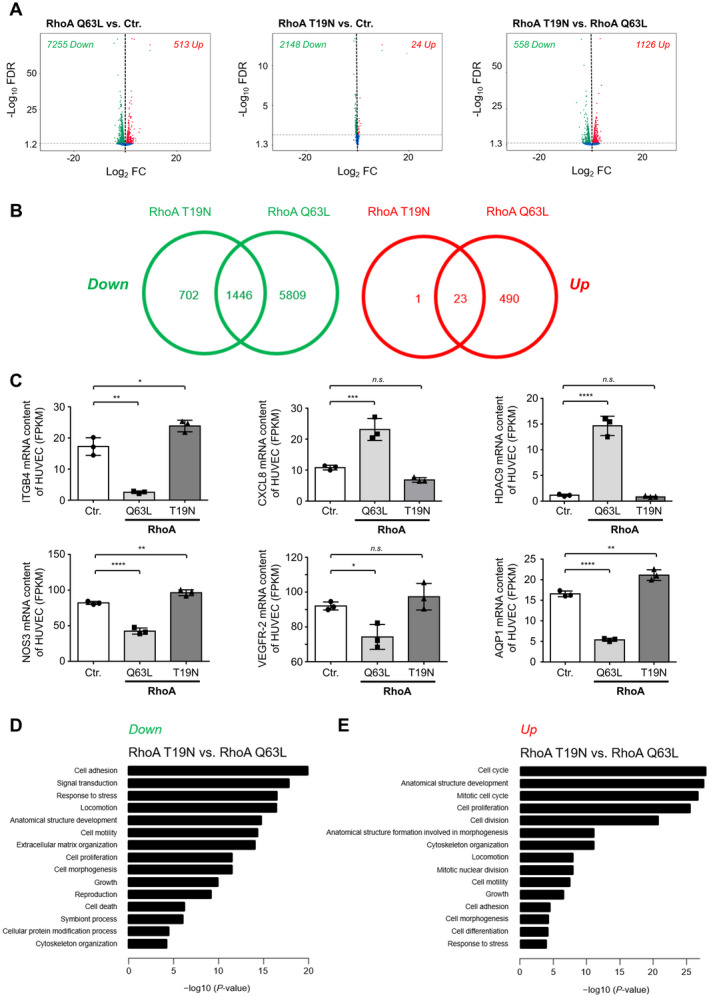
A, Volcano plots depicting differential gene expression in HUVEC expressing constitutively active (Q63L) or dominant‐negative RhoA compared with appropriate control‐transduced (Ctr.) cells as determined by RNA‐sequencing (RNA‐seq). B, Significantly upregulated (red; Up) or downregulated (green; Down) mRNA in RhoA Q63L‐ and RhoA T19N‐expressing HUVEC are depicted by Venn diagrams. Numbers indicate the number of transcripts with significantly deregulated expression in RhoA Q63L‐ and/or RhoA T19N‐expressing HUVEC. C, The abundance of ITGB4 (integrin subunit β 4), CXCL8 (C‐X‐C motif chemokine 8/interleukin 8), HDAC9 (histone deacetylase 9), NOS3 (nitric oxide synthase 3), VEGFR‐2 (vascular endothelial growth factor receptor 2), and AQP1 (aquaporin 1) mRNA were determined by RNA‐seq in control‐transduced as well as in HUVEC expressing active (Q63L) or inactive (T19N) RhoA. Results are shown as fragments per kilobase of transcript per million mapped reads (FPKM) (n=3). D and E, Top gene ontology biological process terms significantly enriched in upregulated (D) or downregulated (E) genes of RhoA T19N‐overexpressing vs RhoA Q63L‐overexpressing HUVEC. FC indicates fold change; FDR, false discovery rate; n.s., not significant.
Discussion
Numerous publications have shown that small GTPases in general and the 3 members of the Rho GTPase family, RhoA, RhoB, and RhoC, in particular regulate the biological functions of endothelial cells and act as regulators of the angiogenic process. 18 , 52 , 53 , 54 , 55 , 56 However, the complexity of the signaling pathways and the diversity of the 20 small Rho GTPases make it difficult to characterize the function of a particular GTPase in the context of living cells. This is particularly the case because cellular processes such as migration, cell polarity, actin cytoskeleton organization, and vesicle transport are organized by well‐orchestrated spatiotemporal activation and inactivation of various Rho GTPases that are difficult to manipulate and study separately. 57 , 58 , 59 However, it is not only the interplay of Rho GTPases in the organization of cellular processes that is of fundamental importance. It has also been suggested that knockdown of 1 of the 3 members of the Rho family, RhoA, RhoB, or RhoC, affects the expression of the remaining Rho GTPases, so that targeted interventions on the expression of Rho GTPases can trigger counter‐regulations in the affected cells, which in turn compensate for and thus mask the function of the defective Rho GTPase. 53 , 55 For this reason, expression of RhoA variants fixed in well‐defined activation states, together with overexpression of the wild‐type protein as an additional reference group, seems to be an advantageous experimental strategy to specifically investigate the influence of the respective activation state of the Rho GTPase on cell function and gene expression while maintaining potential protein–protein interactions.
The results of the present study clearly demonstrate the inhibitory effects of constitutively active RhoA on the angiogenic capacity of HUVEC and HCAEC. We found a significantly reduced proliferation rate of HUVEC expressing the constitutively active RhoA mutants G14V or Q63L and a significantly reduced cell viability after expression of active RhoA variants, but also after increasing the cellular levels of wild‐type RhoA. In addition, HUVEC and HCAEC overexpressing constitutively active RhoA showed severely inhibited 2‐dimensional tube formation and significantly impaired angiogenic sprouting in vitro. The inhibitory effect of active RhoA was also demonstrated in the analysis of cell migration of endothelial cells expressing RhoA G14V, RhoA Q63L, or increased levels of wild‐type RhoA protein. Treatment of RhoA‐transduced cells with C3 transferase as a potent RhoA inhibitor was able to partially or completely reverse these inhibitory effects. Consistent with these findings, C3 transferase treatment also strongly stimulated angiogenic sprouting of control‐transduced HUVEC and HCAEC, demonstrating that endogenously expressed Rho proteins have an important role in limiting VEGF‐induced sprout formation of human endothelial cells. It has to be mentioned that these findings are in contrast to other publications that suggest an important role of RhoA activity both in VEGF‐induced angiogenesis in vitro 16 , 17 , 18 and in vivo. 25 For instance, van Nieuw Amerongen and colleagues showed that RhoA and ROCK are important mediators of VEGF‐induced angiogenesis in vitro. 17 Nonetheless, our analyses using a validated FRET‐based biosensor in HUVEC instead indicate that VEGF does not induce RhoA activity, at least under 2‐dimensional culture conditions in vitro, whereas typical RhoA stimuli, such as thrombin, in our hands clearly do. This lack of RhoA activation after VEGF stimulation of human endothelial cells has also been observed by Reinhard and colleagues. 14 Overexpression of dominant‐negative RhoA in endothelial cells does not suppress VEGF‐ and basic fibroblast growth factor‐related angiogenesis in vitro and in vivo, which is in line with in vivo data from Zahra and colleagues who show that vascular endothelial–specific knockout of RhoA in mice does not affect embryonic development and retinal angiogenesis. 32 In contrast to the findings of other groups but in good agreement with our observations made in the experiments using C3 transferase, pharmacological inhibition of the RhoA effector ROCK strongly enhanced VEGF‐induced angiogenic sprouting and basal migration speed of human endothelial cells. These observations are consistent with the findings of Kroll and colleagues, 31 as well as those of other groups 60 , 61 , 62 who have demonstrated that RNA interference‐mediated knockdown or pharmacological inhibition of ROCK activates VEGF‐driven retinal neovascularization in vivo and sprouting angiogenesis in vitro and stabilizes newly formed blood vessels in vivo. In addition, the results of Breyer and colleagues show an acceleration of the migration velocity of endothelial cells after inhibition of ROCK. 63 Interestingly, preliminary results from another group also suggest that inhibition of RhoA/ROCK plays an important role in mediating Protein kinase A‐induced angiogenesis, suggesting suppression of RhoA/ROCK as an important mechanistic principle for angiogenesis induction. 64 Together with the findings of other research groups, our results therefore suggest that the RhoA/ROCK axis exerts an inhibitory influence on VEGF‐induced angiogenesis and endothelial homeostasis, and that further studies are needed to investigate the context‐specificity of these effects, for example, in the pathogenesis of cardiovascular diseases. Therefore, especially with regard to ROCK independence of RhoA‐mediated effects on vascular endothelial homeostasis in our study, we believe it would be relevant to investigate specific RhoA inhibitors in disease models of atherosclerosis and myocardial infarction. In this context, for example, pleiotropic beneficial effects of HMG CoA (3‐hydroxy‐3‐methylglutaryl coenzyme A) reductase inhibitors (statins) in the treatment of cardiovascular disease have also been attributed to inhibition of posttranslational modification and thus inactivation of RhoA, 65 effects that could potentially be enhanced by targeted use of specific RhoA inhibitors.
In contrast and a little surprisingly, ROCK inhibition did not antagonize the inhibitory effect of active RhoA on migration and 3‐dimensional sprouting of human endothelial cells. In addition, pharmacological inhibition of F‐actin stabilizing LIMK1/2 downstream of ROCK could not restore angiogenetic sprouting or migration of HUVEC expressing active RhoA variants. Thus, the mechanisms by which active RhoA suppresses the angiogenic capacity of endothelial cells and reduces endothelial cell viability in our study remain largely unclear. Therefore, it is likely that other RhoA effectors, such as DIAPH1 or DIAPH2 (diaphanous‐related formin 1 or 2), citron kinase, or PKN1 or PKN2 (protein kinase N1 or N2), play a role in mediating these inhibitory RhoA effects. 66 In addition, microtubule depolymerization may represent a mechanistic link between RhoA activation and the collapse of endothelial capillary tubes in vitro. 67 Nonetheless, further studies are necessary to unravel antiangiogenic signal transduction pathways initiated by active RhoA.
Moreover, our functional observations are supported by global transcriptome analyses, revealing that active RhoA affects the expression of numerous important mediators of endothelial homeostasis, such as HDAC9, VEGFR‐2, AQP1, CXCL8, ITGB4, and NOS3.
GO enrichment analyses performed separately for down‐ and upregulated genes revealed that constitutively active RhoA (compared with RhoA T19N‐induced gene regulation) induced a differentially expressed gene pattern that was enriched for GO terms associated with mitotic nuclear division, mitotic cell cycle, cell proliferation, cell cycle, cell adhesion, cell motility, locomotion, cell adhesion, cytoskeleton organization, and cell morphogenesis. In these analyses, a strongly increased expression of HDAC9, a member of the class IIa histone deacetylases that catalyzes the deacetylation of histones and transcription factors and thereby generally suppresses gene transcription, 51 was observed on the single‐gene level. This is an interesting finding that agrees well with the rather surprising observation that ≈94% of all genes differentially regulated by RhoA Q63L showed reduced expression. This may indicate that HDAC9 could function as an important effector of RhoA‐mediated antiangiogenic signal transduction in human endothelial cells and warrants further research to elucidate the role of pharmacological HDAC9 inhibition in the treatment of endothelial dysfunction and cardiovascular disease.
By using constitutively active and dominant‐negative variants of RhoA, we clearly demonstrated that active RhoA has the potential to induce endothelial dysfunction and inhibit the angiogenic capacity of human endothelial cells in vitro and in vivo. Furthermore, we found that inhibition of RhoA activity had no clear effects on VEGF‐induced angiogenesis, suggesting that other Rho GTPases may compensate for the loss of RhoA activity. Our results and the fact that active RhoA induces the loss of endothelial barrier function 53 , 68 , 69 reinforce the view that active RhoA is a potential disturbing factor for endothelial cell homeostasis and function.
Sources of Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG INST 271/342‐1 and BE 3246/6‐1) and by the European Regional Development Fund of the European Commission (W21029490) to R.A.B.
Disclosures
None.
Supporting information
Figures S1–S4
Table S1
Acknowledgments
The authors gratefully acknowledge the expert technical assistance of D. Frenzel.
Supplemental Materials for this article are available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.025119
For Sources of Funding and Disclosures, see page 18.
References
- 1. Madaule P, Axel R. A novel ras‐related gene family. Cell. 1985;41:31–40. doi: 10.1016/0092-8674(85)90058-3 [DOI] [PubMed] [Google Scholar]
- 2. Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci. 2005;118:843–846. doi: 10.1242/jcs.01660 [DOI] [PubMed] [Google Scholar]
- 3. Wittinghofer A, Vetter IR. Structure‐function relationships of the G domain, a canonical switch motif. Annu Rev Biochem. 2011;80:943–971. doi: 10.1146/annurev-biochem-062708-134043 [DOI] [PubMed] [Google Scholar]
- 4. Cherfils J. Gefs and gaps: Mechanisms and structures. In: Wittinghofer A, ed. Ras Superfamily Small G Proteins: Biology and Mechanisms 1. Vienna: Springer; 2014. [Google Scholar]
- 5. Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013;93:269–309. doi: 10.1152/physrev.00003.2012 [DOI] [PubMed] [Google Scholar]
- 6. Boureux A, Vignal E, Faure S, Fort P. Evolution of the Rho family of ras‐like GTPases in eukaryotes. Mol Biol Evol. 2007;24:203–216. doi: 10.1093/molbev/msl145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ridley AJ. RhoA, RhoB and RhoC have different roles in cancer cell migration. J Microsc. 2013;251:242–249. doi: 10.1111/jmi.12025 [DOI] [PubMed] [Google Scholar]
- 8. Adamson P, Marshall CJ, Hall A, Tilbrook PA. Post‐translational modifications of p21rho proteins. J Biol Chem. 1992;267:20033–20038. doi: 10.1016/S0021-9258(19)88661-1 [DOI] [PubMed] [Google Scholar]
- 9. Adamson P, Paterson HF, Hall A. Intracellular localization of the p21rho proteins. J Cell Biol. 1992;119:617–627. doi: 10.1083/jcb.119.3.617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haga RB, Ridley AJ. Rho GTPases: regulation and roles in cancer cell biology. Small GTPases. 2016;7:207–221. doi: 10.1080/21541248.2016.1232583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thomas P, Pranatharthi A, Ross C, Srivastava S. RhoC: a fascinating journey from a cytoskeletal organizer to a cancer stem cell therapeutic target. J Exp Clin Cancer Res. 2019;38:328. doi: 10.1186/s13046-019-1327-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen W, Niu S, Ma X, Zhang P, Gao YU, Fan Y, Pang H, Gong H, Shen D, Gu L, et al. RhoB acts as a tumor suppressor that inhibits malignancy of clear cell renal cell carcinoma. PLoS One. 2016;11:e0157599. doi: 10.1371/journal.pone.0157599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shimokawa H, Sunamura S, Satoh K. RhoA/Rho‐kinase in the cardiovascular system. Circ Res. 2016;118:352–366. doi: 10.1161/CIRCRESAHA.115.306532 [DOI] [PubMed] [Google Scholar]
- 14. Reinhard NR, van Helden SF, Anthony EC, Yin T, Wu YI, Goedhart J, Gadella TW, Hordijk PL. Spatiotemporal analysis of RhoA/B/C activation in primary human endothelial cells. Sci Rep. 2016;6:25502. doi: 10.1038/srep25502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bryan BA, D'Amore PA. What tangled webs they weave: Rho‐GTPase control of angiogenesis. Cell Mol Life Sci. 2007;64:2053–2065. doi: 10.1007/s00018-007-7008-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zeng H, Zhao D, Mukhopadhyay D. KDR stimulates endothelial cell migration through heterotrimeric g protein Gq/11‐mediated activation of a small GTPase RhoA. J Biol Chem. 2002;277:46791–46798. doi: 10.1074/jbc.M206133200 [DOI] [PubMed] [Google Scholar]
- 17. van Nieuw Amerongen GP, Koolwijk P, Versteilen A, van Hinsbergh VW. Involvement of RhoA/Rho kinase signaling in VEGF‐induced endothelial cell migration and angiogenesis in vitro. Arterioscler Thromb Vasc Biol. 2003;23:211–217. doi: 10.1161/01.ATV.0000054198.68894.88 [DOI] [PubMed] [Google Scholar]
- 18. Bryan BA, Dennstedt E, Mitchell DC, Walshe TE, Noma K, Loureiro R, Saint‐Geniez M, Campaigniac JP, Liao JK, D'Amore PA. RhoA/ROCK signaling is essential for multiple aspects of VEGF‐mediated angiogenesis. FASEB J. 2010;24:3186–3195. doi: 10.1096/fj.09-145102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen W, Mao K, Liu Z, Dinh‐Xuan AT. The role of the Rhoa/Rho kinase pathway in angiogenesis and its potential value in prostate cancer (review). Oncol Lett. 2014;8:1907–1911. doi: 10.3892/ol.2014.2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heemskerk N, Schimmel L, Oort C, van Rijssel J, Yin T, Ma B, van Unen J, Pitter B, Huveneers S, Goedhart J, et al. F‐actin‐rich contractile endothelial pores prevent vascular leakage during leukocyte diapedesis through local RhoA signalling. Nat Commun. 2016;7:10493. doi: 10.1038/ncomms10493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Szulcek R, Beckers CM, Hodzic J, de Wit J, Chen Z, Grob T, Musters RJ, Minshall RD, van Hinsbergh VW, van Nieuw Amerongen GP. Localized RhoA GTPase activity regulates dynamics of endothelial monolayer integrity. Cardiovasc Res. 2013;99:471–482. doi: 10.1093/cvr/cvt075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carbajal JM, Schaeffer RC Jr. RhoA inactivation enhances endothelial barrier function. Am J Physiol. 1999;277:C955–C964. doi: 10.1152/ajpcell.1999.277.5.C955 [DOI] [PubMed] [Google Scholar]
- 23. Mikelis CM, Simaan M, Ando K, Fukuhara S, Sakurai A, Amornphimoltham P, Masedunskas A, Weigert R, Chavakis T, Adams RH, et al. RhoA and ROCK mediate histamine‐induced vascular leakage and anaphylactic shock. Nat Commun. 2015;6:6725. doi: 10.1038/ncomms7725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao ZS, Manser E. Pak and other Rho‐associated kinases–effectors with surprisingly diverse mechanisms of regulation. Biochem J. 2005;386:201–214. doi: 10.1042/BJ20041638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoang MV, Whelan MC, Senger DR. Rho activity critically and selectively regulates endothelial cell organization during angiogenesis. Proc Natl Acad Sci USA. 2004;101:1874–1879. doi: 10.1073/pnas.0308525100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Benndorf RA, Schwedhelm E, Gnann A, Taheri R, Kom G, Didié M, Steenpass A, Ergün S, Böger RH. Isoprostanes inhibit vascular endothelial growth factor‐induced endothelial cell migration, tube formation, and cardiac vessel sprouting in vitro, as well as angiogenesis in vivo via activation of the thromboxane a(2) receptor: a potential link between oxidative stress and impaired angiogenesis. Circ Res. 2008;103:1037–1046. [DOI] [PubMed] [Google Scholar]
- 27. Shih YP, Yuan SY, Lo SH. Down‐regulation of DLC1 in endothelial cells compromises the angiogenesis process. Cancer Lett. 2017;398:46–51. doi: 10.1016/j.canlet.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. El Atat O, Fakih A, El‐Sibai M. RHOG activates RAC1 through CDC42 leading to tube formation in vascular endothelial cells. Cells. 2019;8:171. doi: 10.3390/cells8020171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petrache I, Crow MT, Neuss M, Garcia JG. Central involvement of Rho family GTPases in TNF‐alpha‐mediated bovine pulmonary endothelial cell apoptosis. Biochem Biophys Res Commun. 2003;306:244–249. [DOI] [PubMed] [Google Scholar]
- 30. Giry M, Popoff MR, von Eichel‐Streiber C, Boquet P. Transient expression of RhoA, ‐B, and ‐C GTPases in HeLa cells potentiates resistance to clostridium difficile toxins A and B but not to clostridium sordellii lethal toxin. Infect Immun. 1995;63:4063–4071. doi: 10.1128/iai.63.10.4063-4071.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kroll J, Epting D, Kern K, Dietz CT, Feng Y, Hammes HP, Wieland T, Augustin HG. Inhibition of rho‐dependent kinases rock I/II activates VEGF‐driven retinal neovascularization and sprouting angiogenesis. Am J Physiol Heart Circ Physiol. 2009;296:H893–H899. doi: 10.1152/ajpheart.01038.2008 [DOI] [PubMed] [Google Scholar]
- 32. Zahra FT, Sajib MS, Ichiyama Y, Akwii RG, Tullar PE, Cobos C, Minchew SA, Doçi CL, Zheng YI, Kubota Y, et al. Endothelial RhoA GTPase is essential for in vitro endothelial functions but dispensable for physiological in vivo angiogenesis. Sci Rep. 2019;9:11666. doi: 10.1038/s41598-019-48053-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eckenstaler R, Ripperger A, Hauke M, Petermann M, Hemkemeyer SA, Schwedhelm E, Ergün S, Frye M, Werz O, Koeberle A, et al. A thromboxane a(2) receptor‐driven cox‐2‐dependent feedback loop that affects endothelial homeostasis and angiogenesis. Arterioscler Thromb Vasc Biol. 2022:Atvbaha121317380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eckenstaler R, Benndorf RA. A combined acceptor photobleaching and donor fluorescence lifetime imaging microscopy approach to analyze multi‐protein interactions in living cells. Front Mol Biosci. 2021;8:635548. doi: 10.3389/fmolb.2021.635548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Korff T, Augustin HG. Integration of endothelial cells in multicellular spheroids prevents apoptosis and induces differentiation. J Cell Biol. 1998;143:1341–1352. doi: 10.1083/jcb.143.5.1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Benndorf R, Boger RH, Ergun S, Steenpass A, Wieland T. Angiotensin II type 2 receptor inhibits vascular endothelial growth factor‐induced migration and in vitro tube formation of human endothelial cells. Circ Res. 2003;93:438–447. doi: 10.1161/01.RES.0000088358.99466.04 [DOI] [PubMed] [Google Scholar]
- 37. Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30 [DOI] [PubMed] [Google Scholar]
- 38. Ripperger A, Benndorf RA. The C421A (Q141K) polymorphism enhances the 3'‐untranslated region (3'‐UTR)‐dependent regulation of ATP‐binding cassette transporter ABCG2. Biochem Pharmacol. 2016;104:139–147. doi: 10.1016/j.bcp.2016.02.011 [DOI] [PubMed] [Google Scholar]
- 39. Gehling UM, Willems M, Schlagner K, Benndorf RA, Dandri M, Petersen J, Sterneck M, Pollok JM, Hossfeld DK, Rogiers X. Mobilization of hematopoietic progenitor cells in patients with liver cirrhosis. World J Gastroenterol. 2010;16:217–224. doi: 10.3748/wjg.v16.i2.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weil J, Benndorf R, Fredersdorf S, Griese DP, Eschenhagen T. Norepinephrine upregulates vascular endothelial growth factor in rat cardiac myocytes by a paracrine mechanism. Angiogenesis. 2003;6:303–309. doi: 10.1023/B:AGEN.0000029411.76494.33 [DOI] [PubMed] [Google Scholar]
- 41. Deppe S, Ripperger A, Weiss J, Ergün S, Benndorf RA. Impact of genetic variability in the ABCG2 gene on ABCG2 expression, function, and interaction with AT1 receptor antagonist telmisartan. Biochem Biophys Res Commun. 2014;443:1211–1217. doi: 10.1016/j.bbrc.2013.12.119 [DOI] [PubMed] [Google Scholar]
- 42. Weiss J, Sauer A, Herzog M, Böger RH, Haefeli WE, Benndorf RA. Interaction of thiazolidinediones (glitazones) with the ATP‐binding cassette transporters P‐glycoprotein and breast cancer resistance protein. Pharmacology. 2009;84:264–270. doi: 10.1159/000241734 [DOI] [PubMed] [Google Scholar]
- 43. Laib AM, Bartol A, Alajati A, Korff T, Weber H, Augustin HG. Spheroid‐based human endothelial cell microvessel formation in vivo. Nat Protoc. 2009;4:1202–1215. doi: 10.1038/nprot.2009.96 [DOI] [PubMed] [Google Scholar]
- 44. Braun H, Hauke M, Ripperger A, Ihling C, Fuszard M, Eckenstaler R, Benndorf RA. Impact of DICER1 and DROSHA on the angiogenic capacity of human endothelial cells. Int J Mol Sci. 2021;22. doi: 10.3390/ijms22189855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Najjar SM, Ledford KJ, Abdallah SL, Paus A, Russo L, Kaw MK, Ramakrishnan SK, Muturi HT, Raphael CK, Lester SG, et al. Ceacam1 deletion causes vascular alterations in large vessels. Am J Physiol Endocrinol Metab. 2013;305:E519–E529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP‐binding protein rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Scheele JS, Rhee JM, Boss GR. Determination of absolute amounts of GDP and GTP bound to Ras in mammalian cells: comparison of parental and Ras‐overproducing NIH 3T3 fibroblasts. Proc Natl Acad Sci USA. 1995;92:1097–1100. doi: 10.1073/pnas.92.4.1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weigel MT, Dowsett M. Current and emerging biomarkers in breast cancer: prognosis and prediction. Endocr Relat Cancer. 2010;17:R245–R262. doi: 10.1677/ERC-10-0136 [DOI] [PubMed] [Google Scholar]
- 49. Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, Traganos F. Features of apoptotic cells measured by flow cytometry. Cytometry. 1992;13:795–808. doi: 10.1002/cyto.990130802 [DOI] [PubMed] [Google Scholar]
- 50. Sehr P, Joseph G, Genth H, Just I, Pick E, Aktories K. Glucosylation and ADP ribosylation of rho proteins: effects on nucleotide binding, GTPase activity, and effector coupling. Biochemistry. 1998;37:5296–5304. [DOI] [PubMed] [Google Scholar]
- 51. Hu S, Cho EH, Lee JY. Histone deacetylase 9: its role in the pathogenesis of diabetes and other chronic diseases. Diabetes Metab J. 2020;44:234–244. doi: 10.4093/dmj.2019.0243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barlow HR, Cleaver O. Building blood vessels‐one rho GTPase at a time. Cells. 2019;8:545. doi: 10.3390/cells8060545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pronk MCA, van Bezu JSM, van Nieuw Amerongen GP, van Hinsbergh VWM, Hordijk PL. RhoA, RhoB and RhoC differentially regulate endothelial barrier function. Small GTPases. 2019;10:466–484. doi: 10.1080/21541248.2017.1339767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gerald D, Adini I, Shechter S, Perruzzi C, Varnau J, Hopkins B, Kazerounian S, Kurschat P, Blachon S, Khedkar S, et al. RhoB controls coordination of adult angiogenesis and lymphangiogenesis following injury by regulating VEZF1‐mediated transcription. Nat Commun. 2013;4:2824. doi: 10.1038/ncomms3824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Howe GA, Addison CL. RhoB controls endothelial cell morphogenesis in part via negative regulation of RhoA. Vasc Cell. 2012;4:1. doi: 10.1186/2045-824X-4-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hoeppner LH, Sinha S, Wang Y, Bhattacharya R, Dutta S, Gong X, Bedell VM, Suresh S, Chun C, Ramchandran R, et al. RhoC maintains vascular homeostasis by regulating VEGF‐induced signaling in endothelial cells. J Cell Sci. 2015;128:3556–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pertz O. Spatio‐temporal Rho GTPase signaling – where are we now? J Cell Sci. 2010;123:1841–1850. doi: 10.1242/jcs.064345 [DOI] [PubMed] [Google Scholar]
- 58. Fritz RD, Pertz O. The dynamics of spatio‐temporal rho GTPase signaling: formation of signaling patterns. F1000Res. 2016;5:749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Müller PM, Rademacher J, Bagshaw RD, Wortmann C, Barth C, van Unen J, Alp KM, Giudice G, Eccles RL, Heinrich LE, et al. Systems analysis of RhoGEF and RhoGAP regulatory proteins reveals spatially organized RAC1 signalling from integrin adhesions. Nat Cell Biol. 2020;22:498–511. doi: 10.1038/s41556-020-0488-x [DOI] [PubMed] [Google Scholar]
- 60. Borikova AL, Dibble CF, Sciaky N, Welch CM, Abell AN, Bencharit S, Johnson GL. Rho kinase inhibition rescues the endothelial cell cerebral cavernous malformation phenotype. J Biol Chem. 2010;285:11760–11764. doi: 10.1074/jbc.C109.097220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Im E, Kazlauskas A. Src family kinases promote vessel stability by antagonizing the Rho/ROCK pathway. J Biol Chem. 2007;282:29122–29129. doi: 10.1074/jbc.M702637200 [DOI] [PubMed] [Google Scholar]
- 62. Mavria G, Vercoulen Y, Yeo M, Paterson H, Karasarides M, Marais R, Bird D, Marshall CJ. ERK‐MAPK signaling opposes rho‐kinase to promote endothelial cell survival and sprouting during angiogenesis. Cancer Cell. 2006;9:33–44. doi: 10.1016/j.ccr.2005.12.021 [DOI] [PubMed] [Google Scholar]
- 63. Breyer J, Samarin J, Rehm M, Lautscham L, Fabry B, Goppelt‐Struebe M. Inhibition of rho kinases increases directional motility of microvascular endothelial cells. Biochem Pharmacol. 2012;83:616–626. doi: 10.1016/j.bcp.2011.12.012 [DOI] [PubMed] [Google Scholar]
- 64. Aslam M, Gündüz D. Poster communications: Rhoa/ROCK signalling as an important target for CAMP/PKA‐mediated angiogenesis. Proc Physiol Soc. 2016;37:PCA328. [Google Scholar]
- 65. Cai A, Zhou Y, Li L. Rho‐GTPase and atherosclerosis: pleiotropic effects of statins. J Am Heart Assoc; 2015:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348:241–255. doi: 10.1042/bj3480241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bayless KJ, Davis GE. Microtubule depolymerization rapidly collapses capillary tube networks in vitro and angiogenic vessels in vivo through the small GTPase rho. J Biol Chem. 2004;279:11686–11695. doi: 10.1074/jbc.M308373200 [DOI] [PubMed] [Google Scholar]
- 68. Birukova AA, Adyshev D, Gorshkov B, Bokoch GM, Birukov KG, Verin AD. GEF‐H1 is involved in agonist‐induced human pulmonary endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2006;290:L540–L548. doi: 10.1152/ajplung.00259.2005 [DOI] [PubMed] [Google Scholar]
- 69. Holinstat M, Mehta D, Kozasa T, Minshall RD, Malik AB. Protein kinase calpha‐induced p115rhogef phosphorylation signals endothelial cytoskeletal rearrangement. J Biol Chem. 2003;278:28793–28798. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S4
Table S1