

Abstract
Background
Heart failure with preserved ejection fraction (HFpEF) accounts for 50% of patients with heart failure. Clinically, HFpEF prevalence shows age and gender biases. Although the majority of patients with HFpEF are elderly, there is an emergence of young patients with HFpEF. A better understanding of the underlying pathogenic mechanism is urgently needed. Here, we aimed to determine the role of aging in the pathogenesis of HFpEF.
Methods and Results
HFpEF dietary regimen (high‐fat diet + Nω‐Nitro‐L‐arginine methyl ester hydrochloride) was used to induce HFpEF in wild type and telomerase RNA knockout mice (second‐generation and third‐generation telomerase RNA component knockout), an aging murine model. First, both male and female animals develop HFpEF equally. Second, cardiac wall thickening preceded diastolic dysfunction in all HFpEF animals. Third, accelerated HFpEF onset was observed in second‐generation telomerase RNA component knockout (at 6 weeks) and third‐generation telomerase RNA component knockout (at 4 weeks) compared with wild type (8 weeks). Fourth, we demonstrate that mitochondrial respiration transitioned from compensatory state (normal basal yet loss of maximal respiratory capacity) to dysfunction (loss of both basal and maximal respiratory capacity) in a p53 dosage dependent manner. Last, using myocardial‐specific p53 knockout animals, we demonstrate that loss of p53 activation delays the development of HFpEF.
Conclusions
Here we demonstrate that p53 activation plays a role in the pathogenesis of HFpEF. We show that short telomere animals exhibit a basal level of p53 activation, mitochondria upregulate mtDNA encoded genes as a mean to compensate for blocked mitochondrial biogenesis, and loss of myocardial p53 delays HFpEF onset in high fat diet + Nω‐Nitro‐L‐arginine methyl ester hydrochloride challenged murine model.
Keywords: aging, HFpEF, mitochondrial homeostasis, p53 activation
Subject Categories: Heart Failure, Animal Models of Human Disease, Basic Science Research, Metabolism
Nonstandard Abbreviations and Acronyms
- AMVM
adult mouse ventricular cardiomyocyte
- HFD
high‐fat diet
- HFpEF
heart failure with preserved ejection fraction
- L‐NAME
Nω‐Nitro‐L‐arginine methyl ester hydrochloride
- mTRG2
second‐generation telomerase RNA component knockout
- mTRG3
third‐generation telomerase RNA component knockout
- NMVM
neonatal mouse ventricular cardiomyocytes
- OCR
oxygen consumption rate
- p53CKO
cardiomyocyte‐specific p53 knockout
- PGC1‐α
proliferator‐activated receptor gamma, coactivator 1 alpha
- PGC1‐β
proliferator‐activated receptor gamma, coactivator 1 beta
- TERC
telomerase RNA component
- WT
wild type
Clinical Perspective
What Is New?
Onset of heart failure with preserved ejection fraction is telomere length dependent.
Mild p53 activation results in mitochondrial compensation whereas high p53 activation results in mitochondrial dysfunction.
Myocardial p53 activation is a required step for heart failure with preserved ejection fraction pathogenesis.
What Are the Clinical Implications?
p53 may act as a novel therapeutic target for heart failure with preserved ejection fraction.
Heart failure with preserved ejection fraction (HFpEF), which accounts for 50% of patients with heart failure, 1 is associated with poor quality of life, substantial health care resource use, and premature mortality. 2 The prevalence of heart failure has increased over the past 15 years due to the aging population. 3 Patients with HFpEF exhibit cardiac function impairment but with left ventricular (LV) ejection fraction ≥50% 4 , 5 ; currently there are no effective treatments for HFpEF. 6 , 7 Anatomically, HFpEF is accompanied with an increase in LV wall thickness and/or an increased in left atria resulting in impaired LV filling or suction capacity classified as diastolic dysfunction. 4 , 5 There are 2 unresolved clinical observations with HFpEF: higher prevalence in elderly 8 but not exclusive 9 and a different distribution between men and women. Clinically, male patients with HFpEF tend to be younger and obese whereas female patients with HFpEF tend to be older and often have a plethora of comorbidities such as hypertension and atrial fibrillation. 10 The underlying mechanism remains unknown.
Mitochondria produce adenosine triphosphate that fuels cardiac contraction and mitochondrial respiration is a good measure of mitochondria function. Recent studies have demonstrated the possibility of targeting mitochondria in treating HFpEF. 11 , 12 Notably, patients with HFpEF exhibit exercise intolerance 13 with a reduced peak exercise oxygen consumption. 14 , 15 In the heart, we and others have demonstrated that aging (short telomeres) can drive the onset of mitochondrial dysfunction through p53 activation. 16 , 17 Mitochondrial dysfunction is a classic hallmark of aging. 18 However, whether aging and mitochondrial dysfunction play a role in the pathogenesis of HFpEF remains to be tested. 8 , 19 , 20
Using the recently established HFpEF murine model, 21 we asked if aging, mimicked by short telomeres, can accelerate the development of HFpEF. Following the HFpEF dietary regimen, C57/B6J wild type (WT) animals developed diastolic dysfunction around 8 weeks of age in accord with previous reports. Interestingly, onset of diastolic dysfunction in second‐ (mTRG2) and third (mTRG3) generation telomerase RNA component (TERC) knockout mice was observed at 6 and 4 weeks, respectively. Adult mouse ventricular cardiomyocytes (AMVMs) isolated from HFpEF animals showed p53 activation and decreased mitochondrial biogenesis; however, transcriptome profiling showed a compensatory upregulation of mitochondrial electron transport chain genes. This compensation was reflected in mitochondrial respiration where HFpEF AMVMs exhibit a decrease only in maximal respiration but not basal respiration compared with chow AMVMs. Using neonatal mouse cardiomyocyte as model, we demonstrate that increasing p53 activation drives mitochondria from compensatory state into dysfunction in a dose‐dependent manner. Cardiomyocyte‐specific p53 knockout (MYH6‐Cre x p53f/f; p53CKO) animals did not develop HFpEF when subjected to the dietary regimen, thus confirming p53 activation is required for the development of HFpEF. Together, our data highlight that aging lowers the threshold for p53 activation and p53‐dependent repression of proliferator‐activated receptor gamma coactivator 1 alpha and beta (Pgc‐1α and Pgc‐1β, also known as Ppargc1a and Ppargc1b, respectively) drives the pathogenesis of HFpEF, which offer new insights for future therapeutic interventions.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Experimental Animals
The study was conducted in strict accordance with the recommendations of the Guidelines for the Care and Use of Laboratory Animals. All experiments were approved by the Animal Experiment Ethics Committee of Shanghai Ninth People’s Hospital (SH9H‐2019‐A416‐1). For adult mice, animals were anesthetized by deep isoflurane (5%) anesthesia and euthanized by cervical dislocation. For neonatal mice (within 36 hours) that are resistant to CO2, animals were euthanized by decapitation. WT C57BL/6J adult mice were acquired from GemPharmatech Co., Ltd. The C57BL/6J TERC knockout (mTRKO ) animals were a kind gift from Prof Ming Lei. C57BL/6J Myh6‐Cre and C57BL/6J p53f/f were acquired from Cyagen China and used to generate p53CKO. All experimental mice (8 to 12 weeks old) were maintained on a 12‐hour light/dark cycle from 06:00 to 18:00 and had unrestricted access to regular chow diet (1010001, Jiangsu Xietong Pharmaceutical Co., Ltd, for chow groups) or 60% high‐fat diet (HFD) (XTHF60, Jiangsu Xietong Pharmaceutical Co., Ltd, for the HFpEF groups). Nω‐Nitro‐L‐arginine methyl ester hydrochloride (L‐NAME) (0.5 g l−1, Sigma‐Aldrich, Germany) was administered in the drinking water for the indicated periods of time, after adjusting the pH to 7.4 as previously described. 21
Echocardiography and Doppler Imaging
For cardiac function, echocardiography was performed once every 2 weeks. Animals were anesthetized and imaged using a VisualSonics Vevo 3100 system equipped with MS400 transducer (Visual Sonics, FUJIFILM, Japan). To induce anesthesia 2% isoflurane was used, 0.5% to 1.0% isoflurane was used to maintain a heart rate in the range of 425 to 475 beats per minute to collect systolic function, and 0.5% to 1.0% isoflurane was used to maintain it. Systolic functions were measured at the midventricular long‐axis using M‐mode scanning while maintaining the heart rate at the range of 425 to 475 beats per minute. Diastolic functions were measured in the apical 4‐chamber view using pulsed‐wave tissue Doppler of the mitral valve while maintaining the heart rate at the range of 325 to 375 beats per minute. The following cardiac function parameters were assayed and calculated: heart rate, LV ejection fraction, LV end‐diastolic anterior wall, LV end‐diastolic posterior wall, LV global longitudinal strain, peak Doppler blood inflow velocity across the mitral valve during early diastole (E), peak Doppler blood inflow velocity across the mitral valve during late diastole (A), peak tissue Doppler of myocardial relaxation velocity at the mitral valve annulus during early diastole (E’), and early filling deceleration time (A’). Speckle‐tracking echocardiography was analyzed using VevoStrain software (Visual Sonics) by calculating global strain in longitudinal dimensions (B‐mode) acquired from the parasternal long‐axis view. Technical triplicates were averaged per animal and the means were used for subsequent statistical analyses.
Tail‐Cuff Blood Pressure Recordings
Blood pressure was measured noninvasively in conscious mice using a CODA instrument (BP‐2000, Visitech system, USA). Before testing, all mice were preconditioned to short‐term restraint. The blood pressure was recorded for at least 3 consecutive days, and each reading was averaged from at least 5 measurements.
Histology
For cardiomyocyte hypertrophy and fibrosis measurements, murine hearts were surgically excised and fixed in 4% paraformaldehyde in PBS overnight at 4 ℃ and processed for routine paraffin embedding 5‐μm sections were used for Masson’s trichrome and wheat germ agglutinin staining. Masson’s trichrome staining was performed by manufacturer’s instructions (G1343, Solarbio, China) and degree of fibrosis (%) was quantified using ImageJ software version 2.0 (5 microscopic fields per heart). Wheat germ agglutinin staining was conjugated to Alexa Fluor 488 (50 mg mL−1, 1 hour, room temperature) after antigen retrieval, visualized with a Leica SP8 STED 3X upright photomicroscope, and quantified using ImageJ software version 2.0 (5 microscopic fields per heart).
For telomere quantitative fluorescence in situ hybridization study, 4‐μm sections were used and quantitative fluorescence in situ hybridization staining was carried out as previously described. 17 Next, the sections were blocked with blocking buffer. Slides were incubated with antimouse α‐cardiac troponin T (1:100, Abcam, Britain) and conjugated with Alexa Fluor 488 (antimouse IgG fraction, Molecular Probes, Invitrogen, America). DAPI was stained with 1 µg mL‐1 DAPI solution in PBS. Slices were mounted with antifade reagent and images were obtained with Zeiss LSM 880 upright confocal fluorescence microscope and quantified with Imaris (Oxford Instruments, Switzerland).
Neonatal Cardiomyocyte Isolation and Measurements
Neonatal mice (within 36 hours) that are resistant to CO2 were euthanized by decapitation. In brief, hearts were surgically isolated and immediately digested in Trypsin‐EDTA Solution (C0201, Beyotime Biotechnology, China). After neutralization with DMEM (12430054, Gibco, USA) supplemented with 10% fetal bovine serum (10099‐141, Gibco, USA), neonatal mouse ventricular cardiomyocytes (NMVMs) were centrifuged at 200g for 5 minutes and resuspended in DMEM supplemented with 10% fetal bovine serum. The NMVMs were seeded onto a 10 cm dish for 1 hour at 5% CO2 incubator at 37 °C to remove endothelial and fibroblast cells before being transferred to a new tissue culture dish. To measure contractility, videos of isolated NMVMs were acquired using a fluorescence microscope (IX83, OLYMPUS, Japan) and contraction speed and frequencies were determined as previously described using MATLAB (MathWorks, USA). 22 For mitochondrial membrane potential measurement, 1:1000 Mitotracker (M7514, Invitrogen, USA) was added into medium and NMVMs were incubated for 30 minutes at 37 °C. Signal was acquired and quantified using a High Content Analysis Operetta CLSTM (PerkinElmer, USA).
Langendorff Isolation
For AMVM Langendorff isolation, mice were induced to a brief anesthesia by 5% isoflurane and were euthanized by cervical dislocation. Thoracic cavity was surgically exposed and the heart was surgically removed and immediately placed in ice‐cold cardiomyocyte isolation buffer (120 mmol/L NaCl, 5.4 mmol/L KCl, 0.5 mmol/L MgSO4, 0.33 mmol/L NaH2PO4, 25 mmol/L NaHCO3, and 22 mmol/L glucose, 25 mmol/L HEPES, 10 mmol/L BDM, 30 mmol/L taurine). Hearts were cannulated via the ascending aorta and hooked onto a Langendorff perfusion system. The hearts were perfused with cardiomyocyte isolation buffer at 37 ℃ for 2 to 3 minutes. AMVMs were digested in an enzyme cardiomyocyte isolation buffer containing 1 mg/mL type II collagenase (17104015, Gibco, USA) and 0.6 mg/mL type IV collagenase (17101019, Gibco, USA) for 15 minutes at 37 ℃. The digested hearts were removed and mechanically dissociated using tweezers, transferred into a 50 mL tube, and gently mixed at 37 ℃ to further release single AMVMs. AMVMs were purified with 100 mm filter and collected in 500 rpm.
For function measurement, isolated AMVMs were seeded onto laminin (1:100 in dH2O, L2020, Sigma, German) precoated confocal glass bottom dishes in DMEM supplemented with 10% fetal bovine serum. Cells were incubated for 30 minutes at 37 °C and cardiomyocyte contractions were measured using an Ionoptix HTC with 1 Hz pacing. For fluorescence live cell imaging, isolated AMVMs were seeded onto black 96‐well plates in DMEM supplemented with 10% fetal bovine serum. Medium was changed after 1 hour, and AMVMs were incubated for 30 minutes at 37 °C with Mitotracker Green FM (1:1000, M7514, Invitrogen, USA). Mitochondrial amount was measured and quantified using a High Content Analysis Operetta CLSTM (PerkinElmer, USA).
RNA‐seq
For sample collection and preparation, 3‐4×105 AMVMs from single hearts were isolated by Langendorff and lysed in Trizol (15596‐026, Invitrogen, USA) for extract RNA. After RNA quantification and quality check (1% agarose gels for RNA degradation and contamination; the NanoPhotometer® spectrophotometer [IMPLEN, CA, USA] for RNA purity; the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system [Agilent Technologies, CA, USA] for RNA integrity), a total amount of 1 µg RNA per sample was used to generate sequencing libraries using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA). Before data analysis, the clustering of the index‐coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3‐cBot‐HS (Illumia) and the library preparations were sequenced on an Illumina Novaseq platform and 150 bp paired‐end reads were generated.
Raw data (raw reads) of fastq format were first processed to obtain clean data (clean reads) by removing reads containing adapter and Q20, Q30, and GC content. Reference genome and gene model annotation files were downloaded from the genome website directly and index of the reference genome was built using Hisat2 v2.0.5 and paired‐end clean reads were aligned to the reference genome using Hisat2 v2.0.5. For novel transcripts prediction, the mapped reads of each sample were assembled by StringTie (v1.3.3b) in a reference‐based approach. 23 featureCounts v1.5.0‐p3 was used to count the reads numbers mapped to each gene, and then fragments per kilobase of transcript per million of each gene were calculated based on the length of the gene and reads count mapped to this gene. Additionally, for each sequenced library, the read counts were adjusted by edgeR program package through 1 scaling normalized factor, and then differential expression analysis of 2 conditions/groups (3 biological replicates per condition) was performed using the DESeq2 R package (1.16.1). Significance was determined as P<0.05 using multiple hypothesis test followed by Benjamini‐Hochberg correction.
We used the clusterProfiler R package to implement Gene ontology enrichment analysis of differentially expressed genes and used clusterProfiler R package to test the statistical enrichment of differential expression genes in KEGG pathways. On the other hand, GATK2 (v3.7) software was used to perform SNP calling; rMATS (v3.2.5) software was used to analyze the alternative splicing event and protein‐protein interaction analysis of differentially expressed genes was based on the STRING database. Finally, weighted correlation network analysis was used to describe the gene association modes among different samples. All data are now deposited at the GEO database (GEO accession: GSE195482).
Mitochondrial Respiration Measurement
For mitochondrial respiration measurements, 500 to 800 AMVMs/well or a density of 70% to 80% NMVMs were seeded onto laminin‐coated XFe96 Microplates (Agilent, USA) for 30 minutes or 4 days, respectively. Just before experiment, medium was replaced with XFe base medium supplemented with 5 μmol/L glucose, 1 μmol/L pyruvate, and 10 μmol/L glutamine and transferred into a CO2‐free incubator at 37 °C for another 0.5 hour to allow temperature and pH equilibration. Baseline oxygen consumption rate (OCR) was measured, then followed by injections sequentially with oligomycin (3 μmol/L for AMVMs or 1 μmol/L for NMVMs) to measure the adenosine triphosphate linked OCR, oxidative phosphorylation uncoupler FCCP (1 μmol/L for both AMVMs and NMVMs) to determine maximal respiration, and rotenone and antimycin A (10 μmol/L for AMVMs or 5 μmol/L for NMVMs) to determine the nonmitochondrial respiration per manufacturer’s instructions. A minimum of 4 technical replicates of AMVMs per mouse or technical triplicates for NMVMs were measured and used for downstream analyses.
Mitochondrial Morphology Using Transmission Electron Microscopy
The samples were separated and fixed overnight at 4 ℃ in 2% glutaraldehyde with 1% tannic acid in 0.1 M sodium cacodylate (pH=7.3). The samples were rinsed 3 times in the sodium cacodylate buffer and then incubated in 2% osmium tetroxide in the same buffer for 2 hours at room temperature. The samples were then rinsed 3 times in sodium cacodylate buffer and exposed to 1% uranylacetate in water for 15 minutes at room temperature. The samples were rinsed twice in distilled water and spun down into 3% agarose at 45℃. The agarose blocks were dehydrated in graded steps of acetone and embedded in Spurr’s low viscosity media. Following polymerization overnight at 65 ℃, 80‐nm sections were cut.
The sections were observed in a Philips CM‐10 TEM (FEI Italia, 20122 Milan, Italy) and micrographs recorded on Kodak 4489 sheet film. Mitochondrial density, area, and perimeter were quantified using ImageJ software version 2.0 (5 microscopic fields per heart) (n=3, per group), and averages were presented as number of mitochondria per 100 µm2.
Real‐Time Quantitative Polymerase Chain Reaction
For gene expression analysis, 1 μg total RNA was extracted using Trizol (15596‐026, Invitrogen, USA) reagent per manufacture’s protocol. HiScript Ⅱ Reverse Transcriptase kit (R201‐01/02, Vazyme, China) was used to reverse transcribe and generate single stranded cDNA. Gene expression for Tfam mouse gene (forward: 5’‐CAAGTCAGCTGATGGGTATGG‐3’ and reverse: 5’‐TTTCCCTGAGCCGAATCATCC‐3’), Pgc1‐α mouse gene (forward: 5’‐CCCTGCCATTGTTAAGAC‐3’ and reverse: 5’‐GCTGCTGTTCCTGTTTTC‐3’) and Pgc1‐β mouse gene (forward: 5’‐GAGGGCTCCGGCACTTCC‐3’ and reverse: 5’‐CGTACTTGCTTTTCCCAGATG‐3’), Nppb mouse gene (forward: 5’‐GAGGTCACTCCTATCCTCTGG‐3’ and reverse: 5’‐GCCATTTCCTCCGACTTTTCTC‐3’), Mhy7 mouse gene (forward: 5’‐ACTGTCAACACTAAGAGGGTCA‐3’ and reverse: 5’‐TTGGATGATTTGATCTTCCAGGG‐3’), Mhy11 mouse gene (forward: 5’‐AAGCTGCGGCTAGAGGTCA‐3’ and reverse: 5’‐CCCTCCCTTTGATGGCTGAG‐3’), Mhy7b mouse gene (forward: 5’‐CATGGGATGGTAAGAAACGGG‐3’ and reverse: 5’‐TCCTCCAGTAAGTCGAAACGG‐3’), Klf4 mouse gene (forward: 5’‐GGCGAGTCTGACATGGCTG‐3’ and reverse: 5’‐GCTGGACGCAGTGTCTTCTC‐3’), mt‐Atp mouse gene (forward: 5’‐CCTTCCACAAGGAACTCCAA‐3’ and reverse: 5’‐GGTAGCTGTTGGTGGGCTAA‐3’), mt‐Cyte mouse gene (forward: 5’‐ATTCCTTCATGTCGGACGAG‐3’ and reverse: 5’‐ACTGAGAAGCCCCCTCAAAT‐3’), mt‐Co3 mouse gene (forward: 5’‐CCTTCCACAAGGAACTCCAA‐3’ and reverse: 5’‐GGTAGCTGTTGGTGGGCTAA‐3’), and mt‐Nd4 mouse gene (forward: 5’‐TTCTTCAACCTCACCATAGCC‐3’ and reverse: 5’‐GGCTGCGAAAACTAAGATGG‐3’) were used as template. The β‐actin mouse gene (forward: 5’‐CCAGTTGGTAACAATGCCATGT‐3’ and reverse: 5’‐GAAGAGCTATGAGCTGCCTGA‐3’) was used for normalization. For mtDNA copy number assay, 100 ng total DNA was isolated using the cardiac tissue or cells Genome DNA Extract kit (D1700, Solarbio, China) according to the manufacturer’s instructions. Primers for mt‐Cytb mouse gene (forward: 5’‐GCTTTCCACTTCATCTTACCATTTA‐3’ and reverse: 5’‐TGTTGGGTTGTTTGATCCTG‐3’) and nuclear β‐actin copy number mouse gene (forward: 5’‐GGAAAAGAGCCTCAGGGCAT‐3’ and reverse: 5’‐GAAGAGCTATGAGCTGCCTGA‐3’) were used as previously described. Real‐time quantitative polymerase chain reactions were carried out on an Abi QuantStudioTM 6 Flex machine (Applied Biosystem, USA). Samples were assayed in technical duplicates and averaged for final fold enrichment calculations.
Immunoblotting
Proteins were extracted from mouse AMVMs with ice‐cold modified radioimmunoprecipitation assaybuffer (P0013C, Beyotime Biotechnology, China) containing protease and phosphatase inhibitors. Lysates were centrifugated at 14 000 rcf for 15 minutes at 4 °C and protein lysates were transferred to a new Eppendorf tube for bovine serum albumen quantification. Protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis on 7.5% to 15% gradient gels (Bio‐Rad, USA) and transferred to polyvinylidene difluoride (IPVH00010, Millipore, Germany) membranes. The membranes were blocked for 2 hours with 5% DifcoTM Skim Milk (BD10610, BD Biosciences, USA) and incubated overnight at 4 °C with primary antibodies: Pgc1‐α (4A8) (1:1000, sc‐517380, Santa Cruz, USA), Pgc1‐β (E‐9) (1:1000, sc‐373771, Santa Cruz, USA), γ‐H2A.X (1:500, 9718S, CST, USA), histone H3 (1:1000, CY6587, Abways Technology, China), p53 (1:1000, 10442‐1‐AP, Proteintech, USA), DRP1 (D6C7) (1:1000, 8570S, Cell Signaling Technology, USA), and OPA1 (D6U6N) (1:1000, 80471S, Cell Signaling Technology, USA) followed by secondary antibody for 1 hour with a 1:10 000 dilution of IgG Goat Anti‐Mouse HRP (SA00001‐1, Proteintech, USA) or IgG Goat Anti‐Rabbit HRP (SA00001‐2, Proteintech, USA). The results were visualized using an Amersham Imager 600 (General Electric Company, USA).
Statistical Analysis
All data are shown as the mean±SEM or mean±SD of multiple experiments as stated in figure legends. Statistical analyses were calculated using 1‐way ANOVA with post hoc Tukey correction, Kruskal‐Wallis test with post hoc Dunn correction, 2‐tailed Student's t test or Mann‐Whitney test and are indicated in figure legends. Statistical significance was considered at P<0.05. All statistical analyses were performed using GraphPad Prism 8.0 software (GraphPad Software, USA).
Results
mTRG2 Animals, With Marked Short Telomeres, Exhibit Accelerated HFpEF Pathogenesis
Following previously described protocol, 21 we subjected WT animals to HFD+L‐NAME dietary regimen (HFpEF group) or regular chow controls. To study the role of aging in HFpEF, we subjected second‐ and third‐generation (mTRG2 and mTRG3) telomerase RNA knockout mice (mTRKO) to HFpEF dietary regimen. mTRKO mice, which exhibit progressive telomere shortening per generation, are a model for studying aging. 24 Although it has been demonstrated that mTRG4 animals exhibit dilated cardiomyopathy, 24 we used mTRG2 and mTRG3 animals, which have relatively shorter telomeres compared with WT but are not predisposed with cardiac dysfunction (Figure S1A through S1E). Compared with WT, mTRG2 AMVMs exhibit higher levels of γ‐H2A.X, indicative of higher DNA damage response baseline (Figure S1F). Cardiac function was monitored using echocardiography every 2 weeks (Figure 1A). As shown in Table 1, both WT male and female HFpEF mice exhibited a decrease in global longitudinal strain, an increase in E/E', E/A, and as expected no change in LV ejection fraction, which is in accordance with previous study. 21 Interestingly, LV end‐diastolic anterior wall was increased in HFpEF animals at 6 weeks before the onset of diastolic dysfunction at 8 weeks (Table 1 and 2). In mTRG2 and mTRG3 animals, HFpEF dietary regimen also induced LV end‐diastolic anterior wall thickening before diastolic dysfunction in male and female mTRKO HFpEF mice; moreover, the onset of diastolic dysfunction in mTRG2 and mTRG3 HFpEF animals appeared at 6 and 4 weeks rather than at 8 weeks compared with WT‐HFpEF animals, respectively (Table 1 and 2). At 8 weeks, all HFpEF animals exhibited myocardial hypertrophy, increase in heart weight, and increase in cardiac fibrosis compared with chow animals (Figure 1B through 1E). In accordance with clinical observations, HFpEF animals exhibited elevated diastolic and systolic blood pressure (Figure 1F and 1G) and expressed higher levels of cardiac failure and cardiac hypertrophy genes compared with chow animals (Figure 1H and 1I, Figure S1G). Together, these observations suggest that short telomeres can accelerate the onset of HFpEF upon HFD+L‐NAME diet.
Figure 1. WT and mTRG2 animals develop HFpEF upon HFD+L‐NAME challenge.
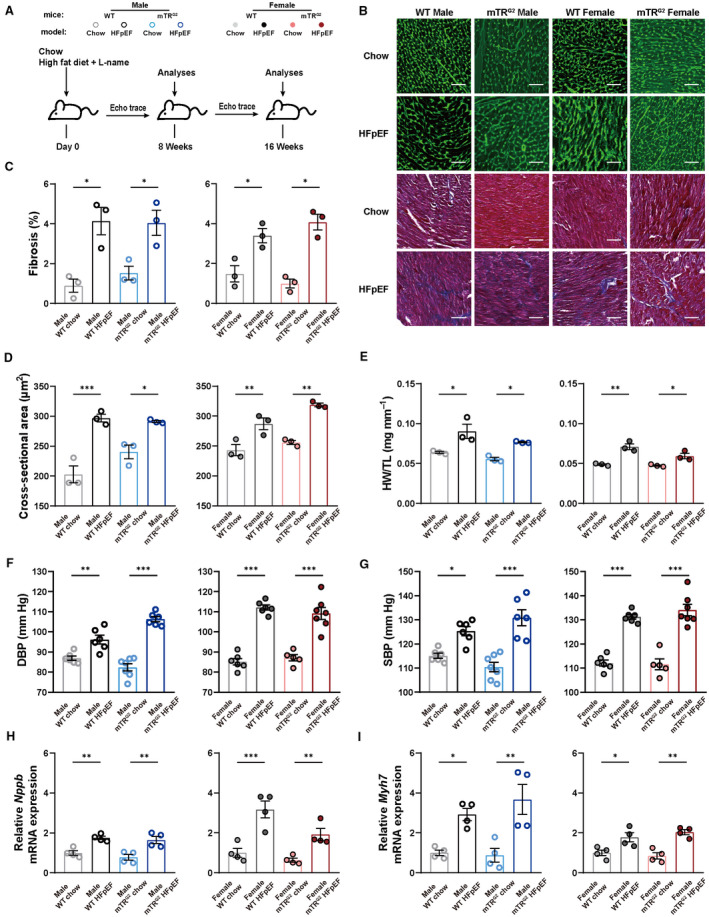
A, Experimental design. C57BL/6J and mTRG2 mice were maintained on HFpEF regimens and analyzed at 8 and 16 weeks. B, Representative images of WGA and Masson's trichrome (MT) staining in transversal sections of left ventricle from mice in different experimental groups are shown. Scale bars, 100μm. C, Percentage of fibrotic area (n=3 per group). D, WGA quantification of cardiomyocyte cross‐sectional area (n=3 per group). E, Ratio of heart weight to tibia length (HW/TL, n=3 per group). F and G, Diastolic blood pressure (DBP) and systolic blood pressure (SBP, n=6 per WT group; n=7 male chow mTRG2 group; n=6 male HFpEF mTRG2 group; n=5 female chow mTRG2 group; n=7 female HFpEF mTRG2 group). H and I, mRNA expression levels of Nppb and Myh7 in isolated AMVMs (n=4 per group). Results are shown as mean±SEM. One‐way ANOVA followed by lognormal processing and/or Tukey's multiple‐comparisons test was used. *P<0.05, **P<0.01, ***P<0.001 compared with chow group in the same genotype. AMVM indicates adult mouse ventricular cardiomyocyte; HFD, high‐fat diet; HFpEF, heart failure with preserved ejection fraction; L‐NAME, Nω‐Nitro‐L‐arginine methyl ester hydrochloride; mTRG2, second ‐generation telomerase RNA component knockout; WGA, wheat germ agglutinin staining; and WT, wild type.
Table 1.
Temporal Monitoring of Cardiac Function in WT, mTRG2 , and mTRG3 Male Mice Subjected to HFpEF Diet
| Male | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | mTRG2 | mTRG3 | |||||||||||||
| Group | 4 weeks | 6 weeks | 8 weeks | 16 weeks | Group | 4 weeks | 6 weeks | 8 weeks | 16 weeks | Group | 4 weeks | 6 weeks | 8 weeks | 16 weeks | |
| LV ejection fraction (%) | Chow | 82.00±3.28 | 84.35±1.37 | 85.21±1.84 | 85.91±2.11 | Chow | 80.95±1.95 | 84.56±2.74 | 85.35±1.61 | 87.76±3.92 | Chow | 87.55±1.62 | 89.58±3.00 | 85.42±2.11 | 90.70±3.34 |
| HFpEF | 88.39±3.68 | 86.88±3.72 | 86.15±3.27 | 84.12±2.72 | HFpEF | 87.17±2.92 | 82.22±1.58 | 84.41±2.92 | 80.28±1.00 | HFpEF | 84.94±1.85 | 90.52±1.49 | 84.12±2.72 | 87.70±2.95 | |
| MV E/A | Chow | 1.20±0.05 | 1.32±0.06 | 1.31±0.08 | 1.38±0.10 | Chow | 1.39±0.10 | 1.32±0.125 | 1.45±0.11 | 1.48±0.24 | Chow | 1.32±0.10 | 1.13±0.04 | 1.54±0.05 | 1.36±0.08 |
| HFpEF | 1.36±0.06 | 1.32±0.08 | 2.33±0.13‡ | 3.24±0.51† | HFpEF | 1.52±0.05 | 2.10±0.10‡ | 1.99±0.05† | 3.06±0.51† | HFpEF | 1.26±0.08 | 1.37±0.14 | 2.24±0.31* | 2.07±0.04 | |
| MV E/E' | Chow | 27.68±2.38 | 25.83±1.55 | 25.51±1.79 | 28.59±1.41 | Chow | 24.65±1.60 | 24.28±2.88 | 24.24±1.87 | 29.66±2.00 | Chow | 23.79±1.14 | 21.21±0.97 | 23.47±2.39 | 24.77±3.10 |
| HFpEF | 28.24±1.72 | 27.95±1.31 | 38.54±3.51† | 46.31±2.96† | HFpEF | 29.68±2.50 | 44.06±1.61‡ | 42.02±2.45‡ | 46.10±3.59† | HFpEF | 35.01±1.57* | 43.09±2.52‡ | 40.04±1.88† | 46.94±4.05† | |
| Global longitudinal strain (%) | Chow | −38.81±3.11 | −38.21±2.87 | −38.31±3.01 | −36.35±2.04 | Chow | −38.07±2.41 | −35.27±1.90 | −38.19±2.30 | −33.87±2.26 | Chow | −33.40±3.74 | −37.52±4.21 | −31.04±2.65 | −31.30±2.93 |
| HFpEF | −36.46±2.80 | −33.93±2.92 | −17.32±3.37‡ | −17.35±1.81‡ | HFpEF | −35.47±2.35 | −22.36±1.72† | −16.13±2.20‡ | −15.61±2.27‡ | HFpEF | −17.29±1.48* | −19.42±0.79† | −19.62±2.34 | −13.05±3.94† | |
|
LV diastolic anterior wall (mm) |
Chow | 0.93±0.04 | 0.83±0.01 | 0.93±0.03 | 0.89±0.04 | Chow | 0.91±0.05 | 0.89±0.02 | 0.90±0.02 | 0.96±0.03 | Chow | 1.01±0.01 | 0.90±0.05 | 1.16±0.05 | 1.027±0.04 |
| HFpEF | 1.01±0.05 | 1.13±0.09† | 1.11±0.02* | 1.21±0.07‡ | HFpEF | 1.09±0.05* | 1.05±0.04 | 1.14±0.05† | 1.15±0.03* | HFpEF | 1.08±0.01 | 1.16±0.01* | 1.18±0.05 | 1.22±0.02 | |
|
LV diastolic posterior wall; (mm) |
Chow | 1.18±0.10 | 0.96±0.07 | 0.86±0.04 | 0.77±0.05 | Chow | 0.88±0.03 | 0.83±0.05 | 0.85±0.07 | 0.90±0.05 | Chow | 0.86±0.13 | 0.98±0.05 | 0.84±0.06 | 0.94±0.08 |
| HFpEF | 1.09±0.17 | 1.17±0.12 | 1.08±0.17 | 1.07±0.15 | HFpEF | 1.05±0.09 | 0.98±0.07 | 0.87±0.06 | 0.91±0.07 | HFpEF | 0.86±0.08 | 0.91±0.07 | 0.89±0.10 | 0.83±0.07 | |
HFpEF indicates heart failure with preserved ejection fraction; LV, left ventricular; mTRG2, second‐generation telomerase RNA component knockout; mTRG3, third‐generation telomerase RNA component knockout; and WT, wild type. MV E/A, Ratio between mitral E wave and A wave; and MV E/E, Ratio between mitral E wave and E′ wave; n=6 male chow WT group; n=6 male HFpEF WT group; n=6 male chow mTRG2 group; n=8 at 4, 6, 8 weeks and n=7 at 16 weeks male HFpEF mTRG2 group; n=4 male chow mTRG3 group; n=5 at 4, 6, 8 weeks and n=4 at 16 weeks male HFpEF mTRG3 group. Results are shown as mean±SEM. One‐way ANOVA followed by lognormal processing and/or Tukey's multiple‐comparisons test was used. *P<0.05, † P<0.01, ‡ P<0.001 compared with chow group in the same genotype are shown.
Table 2.
Temporal Monitoring of Cardiac Function in WT, mTRG2 , and mTRG3 Female Mice Subjected to HFpEF Diet
| Female | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | mTRG2 | mTRG3 | |||||||||||||
| Group | 4 weeks | 6 weeks | 8 weeks | 16 weeks | Group | 4 weeks | 6 weeks | 8 weeks | 16 weeks | Group | 4 weeks | 6 weeks | 8 weeks | 16 weeks | |
| LV ejection fraction (%) | Chow | 84.08±1.92 | 83.74±2.98 | 88.26±1.27 | 91.14±1.86 | Chow | 82.43±0.92 | 88.09±3.02 | 81.52±1.47 | 88.23±2.27 | Chow | 82.79±1.96 | 91.13±2.79 | 82.64±1.42 | 93.62±1.92 |
| HFpEF | 92.86±1.58 | 91.44±2.90 | 89.74±2.31 | 88.26±3.53 | HFpEF | 86.73±2.83 | 83.06±3.36 | 84.98±3.18 | 86.44±2.20 | HFpEF | 84.40±1.68 | 85.34±3.03 | 80.66±2.54 | 90.01±0.13 | |
| MV E/A | Chow | 1.36±0.14 | 1.27±0.16 | 1.34±0.09 | 1.51±0.05 | Chow | 1.30±0.09 | 1.41±0.09 | 1.31±0.06 | 1.54±0.11 | Chow | 1.44±0.12 | 1.44±0.10 | 1.28±0.08 | 1.43±0.06 |
| HFpEF | 1.37±0.08 | 1.33±0.16 | 2.05±0.16† | 2.39±0.30† | HFpEF | 1.39±0.05 | 2.28±0.18† | 2.28±0.18‡ | 2.33±0.20† | HFpEF | 2.45±0.20‡ | 2.16±0.19* | 2.22±0.09‡ | 2.24±0.15† | |
| MV E/E' | Chow | 24.58±1.45 | 23.77±1.22 | 21.43±2.60 | 23.57±0.80 | Chow | 24.91±1.49 | 23.90±1.71 | 22.95±1.89 | 20.14±1.02 | Chow | 22.22±2.18 | 23.36±1.49 | 22.30±1.79 | 26.71±1.47 |
| HFpEF | 24.73±3.16 | 25.57±1.19 | 37.70±2.51† | 43.88±3.08‡ | HFpEF | 24.33±2.70 | 39.96±3.49‡ | 38.34±2.80‡ | 38.70±1.14‡ | HFpEF | 35.65±1.28† | 37.15±1.60† | 38.39±2.85† | 43.02±2.14* | |
| Global longitudinal strain (%) | Chow | −38.35±2.34 | −36.58±2.95 | −42.91±2.87 | −39.14±3.03 | Chow | −31.31±1.10 | −34.27±2.36 | −37.84±4.84 | −39.80±4.16 | Chow | −32.02±2.36 | −37.40±2.97 | −38.94±2.96 | −39.94±4.31 |
| HFpEF | −38.37±3.57 | −28.23±3.67 | −20.44±2.37‡ | −16.93±2.42‡ | HFpEF | −36.67±2.90 | −23.81±0.75* | −17.90±2.24‡ | −18.72±1.60‡ | HFpEF | −18.21±1.89* | −19.85±3.02† | −19.09±1.16† | −16.37±0.74‡ | |
|
LV diastolic anterior wall (mm) |
Chow | 0.93±0.03 | 0.91±0.05 | 0.91±0.02 | 0.87±0.05 | Chow | 0.86±0.04 | 0.87±0.03 | 0.83±0.04 | 0.98±0.03 | Chow | 0.92±0.02 | 0.94±0.03 | 0.89±0.02 | 0.90±0.01 |
| HFpEF | 1.02±0.01 | 1.15±0.05† | 1.06±0.04* | 1.13±0.06† | HFpEF | 0.96±0.05 | 0.98±0.05 | 1.07±0.02‡ | 1.18±0.04† | HFpEF | 1.02±0.03 | 1.12±0.04 | 1.20±0.05‡ | 1.10±0.02* | |
|
LV diastolic posterior wall (mm) |
Chow | 1.07±0.16 | 0.89±0.11 | 1.04±0.11 | 0.84±0.12 | Chow | 1.04±0.10 | 0.84±0.06 | 0.83±0.08 | 0.71±0.06 | Chow | 0.98±0.11 | 0.84±0.11 | 0.85±0.09 | 1.00±0.11 |
| HFpEF | 1.09±0.10 | 1.02±0.09 | 1.06±0.13 | 1.00±0.15 | HFpEF | 1.06±0.10 | 0.94±0.06 | 1.16±0.12 | 1.02±0.10 | HFpEF | 0.86±0.07 | 0.96±0.09 | 0.92±0.11 | 0.92±0.12 | |
HFpEF indicates heart failure with preserved ejection fraction; LV, left ventricular; mTRG2, second‐generation telomerase RNA component knockout; mTRG3, third‐generation telomerase RNA component knockout; and WT, wild type. MV E/A, Ratio between mitral E wave and A wave; MV E/E, Ratio between mitral E wave and E′ wave. n=6 female chow WT group; n=6 at 4, 6, 8 weeks and n=5 at 16 weeks female HFpEF WT group; n=7 at 4, 6, 8 weeks and n=6 at 16 weeks female chow mTRG2 group; n=7 female HFpEF mTRG2 group; n=5 female chow mTRG3 group; n=5 HFpEF mTRG3 group. Results are shown as mean±SEM. One‐way ANOVA followed by lognormal processing and/or Tukey's multiple‐comparisons test was used. *P<0.05, † P<0.01, ‡ P<0.001 compared with chow group in the same genotype are shown.
Next, single AMVMs from 8‐ and 16‐week HFpEF and chow animals were isolated using Langendorff perfusion and assayed using an Ionoptix HTS system paced at 1 Hz (Figure S2). There was no consistent trend in contractile dysfunction between HFpEF and chow AMVMs isolated from WT or mTRG2 animals at 8 weeks (Figure S2A through S2G). Given that the observed diastolic dysfunction stabilizes at 16 weeks, contractile functions in isolated AMVMs were also examined (Figure S2H through S2N). We observed a significant decrease in maximal diastolic velocity (Figure S2K), increase in maximal systolic velocity (Figure S2L), increase in time to maximal diastolic velocity (Figure S2M), and increase in time to maximal systolic velocity (Figure S2N) in HFpEF AMVMs compared with AMVMs from chow animals. Similar findings were also observed in mTRG3‐HFpEF AMVMs (Figure S3). Together, these data support a model where short telomeres drive the age of onset of HFpEF. As disease progresses, the contractile defects stabilize in both male and female WT HFpEF animals at 16 weeks. In mTRG2‐and mTRG3‐HFpEF animals, systolic and diastolic dysfunction appeared much earlier in female mice, but with time, cardiac dysfunction stabilizes for both sexes.
Activation of p53–Pgc1α/β Axis Reduces Mitochondrial Capacity in HFpEF Cardiomyocytes
To capture the transcriptomic changes underlying HFpEF, we performed RNA‐seq on isolated AMVMs from 8‐week animals and performed the following comparisons: male WT‐chow versus male WT‐HFpEF, male mTRG2 ‐chow versus male mTRG2‐HFpEF, female WT‐chow versus female WT‐HFpEF, and female mTRG2‐chow versus mTRG2‐HFpEF female. Gene ontology analyses of differentially expressed genes show a significant enrichment of genes in mitochondria function and muscle related pathways in all HFpEF‐chow comparisons (Figure S4). We and others have shown that myocardial p53 activation can repress Pgc1α/β and result in mitochondrial dysfunction. 16 , 17 In accordance, we observed a large number of p53 downstream target genes being differentially expressed between chow versus HFpEF in both WT and mTRG2 AMVMs (Figure S5A). To test if p53 is activated and represses Pgc1α/β in HFpEF, we isolated AMVMs and performed immunoblotting for p53, Pgc1α, and Pgc1β (Figure 2A). As expected, we observed a significant increase in p53 protein in HFpEF AMVMs compared with chow AMVMs; furthermore, p53 basal levels were higher in mTRG2 animals compared with WT animals (Figure 2B). p53 activation resulted in a decrease in Pgc1α and Pgc1β proteins (Figure 2C and 2D, Figure S4B through S4D) and with the basal p53 activation in mTRG2 AMVMs, Pgc1α, and Pgc1β proteins were further downregulated in HFpEF AMVMs (Figure 2B through 2D).
Figure 2. p53‐activation in HFpEF cardiomyocytes result in blocked mitochondrial biogenesis.
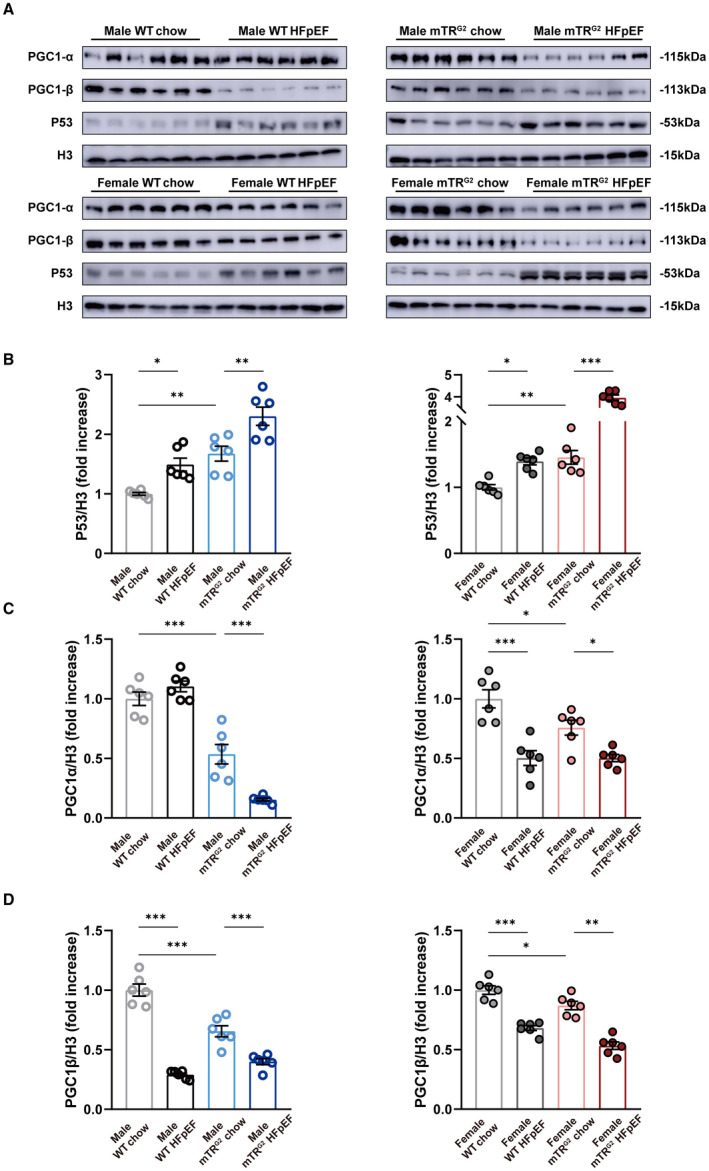
A, Representative images of p53, Pgc1‐α, and Pgc1‐β expression in AMVMs assayed by immunoblotting. Histone H3 used as loading control. (B through D) Densitometric quantifications of p53, PGC1‐α, and PGC1‐β protein expression. Results are shown as mean±SEM (n=6 per group). One‐way ANOVA followed by Tukey's multiple‐comparisons test was used. *P<0.05, **P<0.01, ***P<0.001 compared with chow group in the same genotype are shown. AMVM indicates adult mouse ventricular cardiomyocyte; HFpEF, heart failure with preserved ejection fraction; mTRG2, second ‐generation telomerase RNA component knockout; PGC1‐α, proliferator‐activated receptor gamma, coactivator 1 alpha; PGC1‐β, proliferator‐activated receptor gamma, coactivator 1 beta; and WT, wild type.
Next, we evaluated if loss of Pgc1α and Pgc1β would result in blocked mitochondrial biogenesis. To determine mitochondrial content, we used real‐time quantitative polymerase chain reaction to measure mtDNA copy number. 25 In both sexes, 8‐week HFpEF AMVMs from WT and mTRG2 animals showed a significant decrease in mitochondrial DNA copy number compared with chow AMVMs (Figure 3A). Interestingly, using Mitotracker staining to quantify mitochondrial amount, we observed a significant decrease in mitochondrial amount in HFpEF AMVMs regardless of sex or genetic background compared with chow AMVMs (Figure 3B) but magnitude of decrease is lesser compared with that of mtDNA copy number. Functionally, OCR of Langendorff isolated AMVMs were measured using a Seahorse XFe96 bioanalyzer. Compared with chow AMVMs, WT and mTRG2‐HFpEF AMVMs showed reduced maximal OCR capacity but no difference of basal OCR at 8‐weeks (Figure 3C through 3E) and 16‐weeks (Figure S6A through S6C). Notably, there was a significant increase in mtDNA transcription in HFpEF AMVMs compared with WT AMVMs suggestive of compensatory cardioprotection (Figure S7A through S7D). To evaluate mitochondrial homeostasis, we examined level of mitochondrial fission (Drp1) and mitochondrial fusion (Opa1) by immunoblotting. Among the 4 groups examined, a significant increase in Opa1 protein was observed in WT male and female HFpEF AMVMs compared with WT chow controls; this upregulation of Opa1 was lost in mTRG2 HFpEF AMVMs (Figure S7E, S7D). Ultrastructurally, mitochondria from 8‐week HFpEF hearts exhibited loss of cristae (Figure 3F), decrease in mitochondrial area (Figure 3G), and decrease in mitochondrial density (Figure 3H) compared with WT‐chow and mTRG2‐chow controls, but no difference in mitochondrial perimeter (Figure 3I). Together, these data demonstrate that myocardial p53 activation results in the loss of mitochondrial maximal respiratory capacity through downregulation of Pgc1α/β during HFpEF pathogenesis.
Figure 3. Mitochondrial compensation in HFpEF cardiomyocytes.
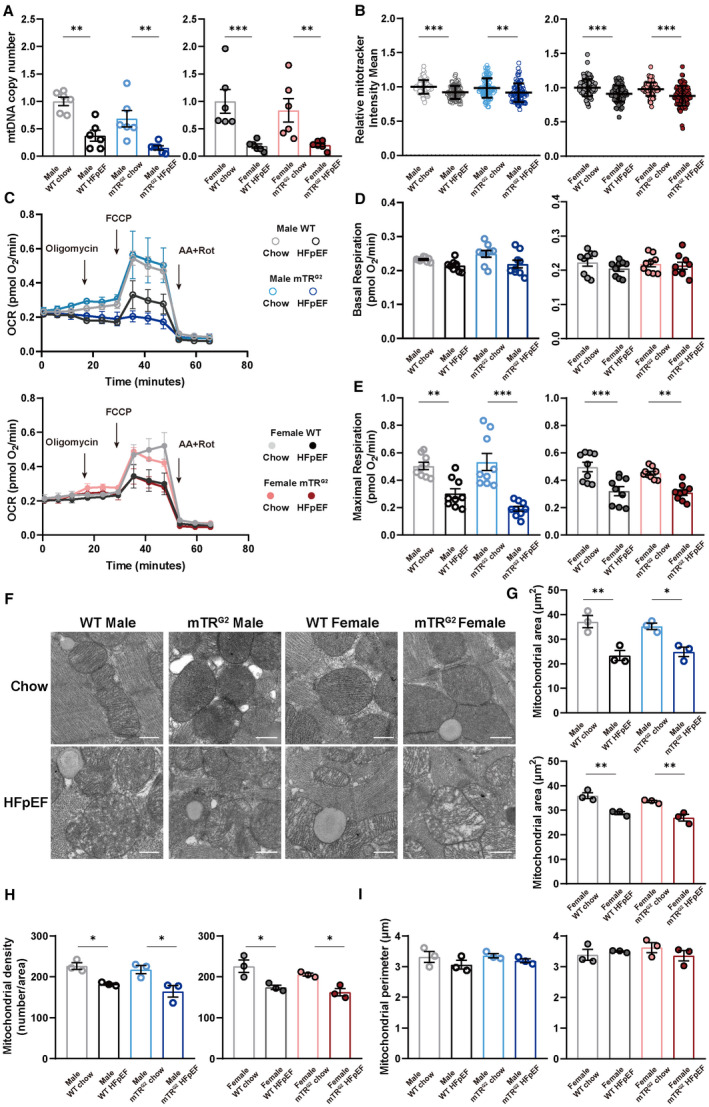
A, mtDNA copy number was determined by real‐time quantitative polymerase chain reaction and is shown as mean±SEM (n=6 per group). B, Mitotracker intensity in AMVMs was determined by Mitotracker staining and is shown as mean±SD (n=6 per group; 15 AMVMs per mouse). C, Real‐time mitochondrial respiration of isolated AMVMs (n=3 per group). D and E, Basal and maximal OCR of AMVMs were quantified and are shown as mean±SEM (n=3 per group; 3 measurements per mouse). F, Representative micrographs of heart tissue sections examined by transmission electron micrographs. Scale bar is 0.5 µm. (G through I) Mitochondrial area, mitochondria number per area and mitochondria perimeter quantifications are shown as mean±SEM (n=3 per group). Kruskal‐Wallis test followed by Dunn's multiple‐comparisons test was used for panel B; 1‐way ANOVA followed by lognormal processing and/or Tukey's multiple‐comparisons test was used for remaining analyses. *P<0.05, **P<0.01, ***P<0.001 compared with chow group in the same genotype. AMVM indicates adult mouse ventricular cardiomyocyte; HFpEF, heart failure with preserved ejection fraction; mTRG2, second ‐generation telomerase RNA component knockout; OCR, oxygen consumption rate; and WT, wild type.
p53 Activation Results in Contractile and Mitochondrial Dysfunction in Cardiomyocytes
To further verify if p53 activation is the cause of contractile and mitochondrial dysfunction seen in HFpEF cardiomyocytes, we treated NMVMs with p53 activator nutlin‐3, a murine double minute 2 antagonist, at 2.5 and 10 μmol/L concentration. Upon p53 activation, we observed a significant decline in contractile speed in NMVMs in a nutlin‐3 dose‐dependent fashion (Figure 4A). At the protein level, p53 activation blocked Pgc1α and Pgc1β protein expression (Figure 4B through 4E) and loss of mitochondrial amount (Figure 4F) in a dosage‐dependent manner similar to isolated HFpEF AMVMs. At 2.5 μmol/L nutlin‐3 stimulation, mild increase in p53 activation did not affect basal OCR but resulted in a decrease in maximal OCR (Figure 4G through 4I) mimicking what we observed in HFpEF AMVMs; however, when stimulated with 10 μmol/L nutlin‐3, NMVMs exhibited a significant decrease in both basal OCR as well as maximal OCR (Figure 4G through 4I), which is in accord with our previous observation in AMVMs isolated from dilated cardiomyopathy mice. 17 Further, loss of mitochondrial cristae was observed in a nutlin‐3 dose‐dependent manner (Figure 4J). Together, these data demonstrate that p53 activation drives contractile dysfunction in cardiomyocytes through inhibition of mitochondrial biogenesis and function.
Figure 4. Does‐dependent p53 activation determines transition from mitochondrial compensation to mitochondrial dysfunction.
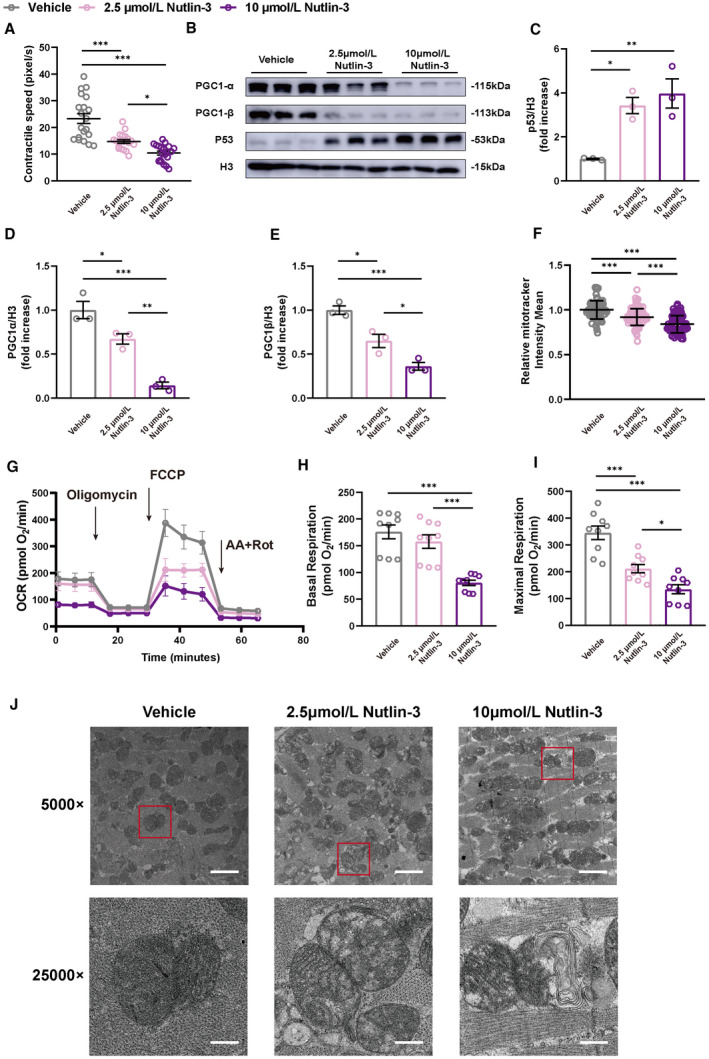
A, Contractile speed of NMVMs determined by live cell imaging are shown as mean±SEM (n=20 cells per group). B, Representative micrographs of p53, PGC1‐α, and PGC1‐β using immunoblotting are shown. Histone H3 used as loading control. (C through E) Densitometric quantifications are shown as mean±SEM (n=3 per group). F, Mitochondrial amount assayed by Mitotracker staining is shown as mean±SD (triplicates, n=30 NMVMs). G, Real‐time mitochondrial respiration of NMVMs (n=3 per group). H and I, Basal and maximal OCR of NMVMs were quantified and are shown as mean±SEM (n=3 per group; 3 measurements per group). J, Representative micrographs of NMVMs examined by transmission electron micrographs. Scale bars are 2 µm and 0.4 µm at 5000 and 25000× magnification, respectively. Kruskal‐Wallis test followed by Dunn’s multiple‐comparisons test was used for panel F; 1‐way ANOVA followed by Tukey’s multiple‐comparisons test was used for remaining analyses. *P<0.05, **P<0.01, ***P<0.001 compared with vehicle group. HFpEF, heart failure with preserved ejection fraction; NMVM, neonatal mouse ventricular cardiomyocyte; OCR, oxygen consumptio rate; p53CKO, cardiomyocyte‐specific p53 knockout; PGC1‐α, proliferator‐activated receptor gamma, coactivator 1 alpha; and PGC1‐β, proliferator‐activated receptor gamma, coactivator 1 beta.
Loss of Myocardial p53 Delays the Onset of HFpEF Upon HFD+L‐NAME Challenge
To test if p53‐Pgc1α/β inhibition is key to the pathogenesis of HFpEF in vivo, we knocked out p53 in AMVMs by crossing p53f/f with MYH6cre to generate p53CKO (Figure 5A). Unexpectedly, in the absence of myocardial p53, induction of diastolic dysfunction in p53CKO animals was delayed to 16 weeks compared with 8 weeks (Table 3). At single cardiomyocyte level, p53CKO‐HFpEF AMVMs exhibited enhanced contractile function compared with p53CKO‐chow AMVMs at 8 weeks (Figure S8) suggestive of cardioprotective compensation is at play. Notably, p53CKO‐HFpEF AMVMs showed no significant difference in Pgc1α/β protein expression (Figure 5B through 5D) and no change in mitochondrial DNA copy number (Figure 5E) and mitochondrial amount (Figure 5F) at 8 weeks compared with p53CKO‐chow AMVMs. Moreover, we observed no difference in mitochondrial respiration function between HFpEF and p53CKO‐chow AMVMs (Figure 5G through 5I). Together, these data demonstrate that p53 activation accelerates HFpEF development but is not required, and the p53–Pgc1α/β signaling axis is crucial in maintaining mitochondrial respiratory compacity.
Figure 5. p53CKO animals fail to develop HEpEF upon HFD+L‐NAME challenge.
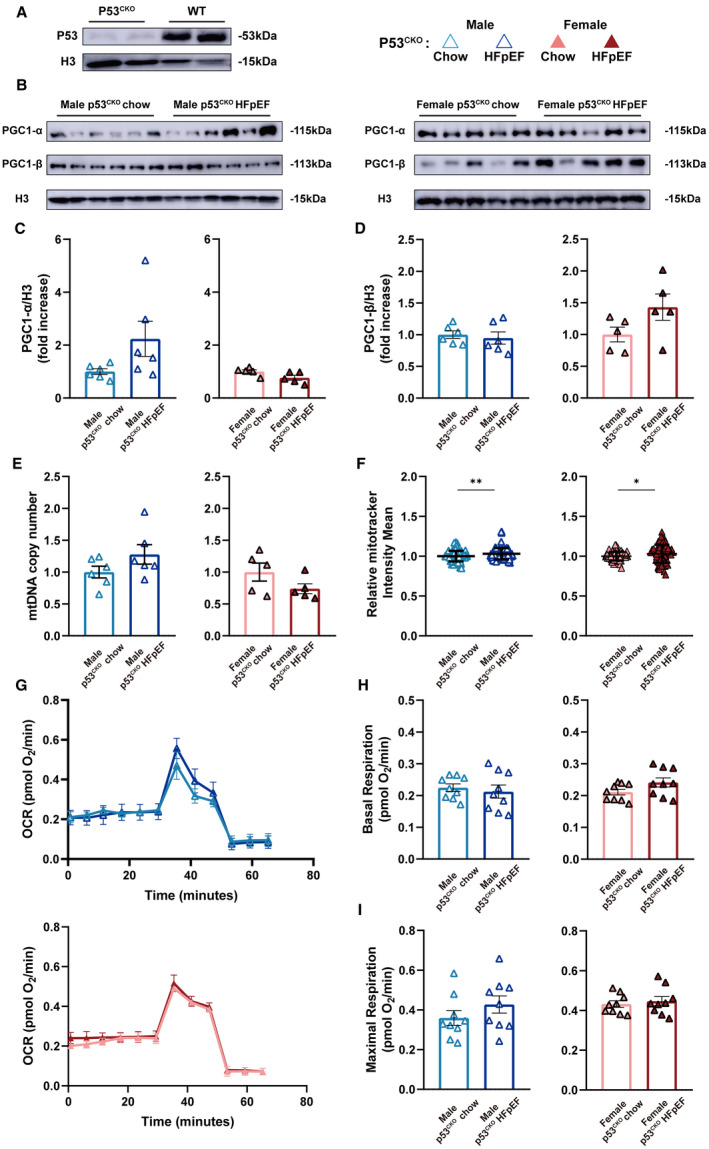
A and B, Representative immunoblots of p53, Pgc1‐α, and Pgc1‐β expression in isolated AMVMs are shown. Histone H3 used as loading control. C and D, Densitometric quantifications of Pgc1‐α and Pgc1‐β expression. Results are shown as mean±SEM (n=6 per male p53CKO group; n=5 per female p53CKO group). E, mtDNA copy number in AMVMs was determined by real‐time quantitative polymerase chain reaction and is shown as mean±SEM (n=6 per male p53CKO group; n=5 per female p53CKO group). F, Mitochondrial amount was assayed using Mitotracker staining and is shown as mean±SD (n=6 per male p53CKO group; n=5 per female p53CKO group; 15 AMVMs per mouse). G, Real‐time mitochondrial respiration of AMVMs (n=3 per group). H and I, Basal and maximal OCR of AMVMs were quantified and are shown as mean±SEM (n=3 per group; 3 measurements per mouse). Mann‐Whitney test was used for panel F; 2‐tailed unpaired Student's t test was used for remaining analyses. *P<0.05, **P<0.01, ***P<0.001 compared with chow group. AMVM indicates adult mouse ventricular cardiomyocyte; HFD, high‐fat diet; HFpEF, heart failure with preserved ejection fraction; L‐NAME, Nω‐Nitro‐L‐arginine methyl ester hydrochloride; OCR, oxygen consumption rate; p53CKO, cardiomyocyte‐specific p53 knockout; PGC1‐α, proliferator‐activated receptor gamma, coactivator 1 alpha; and PGC1‐β, proliferator‐activated receptor gamma, coactivator 1 beta.
Table 3.
Temporal Monitoring of Cardiac Function in p53CKO Male and Female Mice Subjected to HFpEF Diet
| p53CKO | ||||||||
|---|---|---|---|---|---|---|---|---|
| Male | Female | |||||||
| Group | 6 weeks | 8 weeks | 16 weeks | Group | 6 weeks | 8 weeks | 16 weeks | |
| LV ejection fraction (%) | Chow | 92.83±2.13 | 90.37±1.68 | 93.08±1.74 | Chow | 94.27±1.73 | 91.08±3.17 | 94.27±173 |
| HFpEF | 87.42±2.29 | 87.70±1.62 | 90.86±2.14 | HFpEF | 93.87±1.00 | 94.32±2.13 | 95.92±2.34 | |
| MV E/A | Chow | 1.37±0.19 | 1.38±0.09 | 1.39±0.05 | Chow | 1.39±0.12 | 1.52±0.09 | 1.26±0.08 |
| HFpEF | 1.52±0.07 | 1.47±0.10 | 1.92±0.18* | HFpEF | 1.38±0.12 | 1.35±0.10 | 1.93±0.09‡ | |
| MV E/E' | Chow | 19.35±1.78 | 25.66±1.26 | 17.41±1.24 | Chow | 19.25±1.47 | 22.49±1.42 | 18.92±1.69 |
| HFpEF | 23.28±1.29 | 28.03±1.81 | 37.27±2.32‡ | HFpEF | 20.52±1.28 | 22.28±2.48 | 46.15±4.02‡ | |
| Global longitudinal strain (%) | Chow | −34.42±2.18 | −31.39±2.55 | −43.99±2.88 | Chow | −36.37±1.97 | −30.91±1.05 | −36.71±2.58 |
| HFpEF | −32.56±2.92 | −37.70±2.84 | −20.59±3.18‡ | HFpEF | −38.30±4.85 | −31.78±2.95 | −16.97±1.84‡ | |
| LV diastolic anterior wall (mm) | Chow | 0.92±0.02 | 1.04±0.09 | 1.05±0.05 | Chow | 0.96±0.01 | 1.03±0.06 | 1.08±0.04 |
| HFpEF | 0.89±0.07 | 1.11±0.05 | 1.18±0.03* | HFpEF | 1.11±0.06 | 1.16±0.07 | 1.22±0.06 | |
| LV diastolic posterior wall (mm) | Chow | 0.76±0.03 | 1.08±0.09 | 0.89±0.09 | Chow | 0.90±0.08 | 0.81±0.02 | 0.93±0.09 |
| HFpEF | 0.73±0.05 | 0.94±0.08 | 0.96±0.04 | HFpEF | 0.87±0.03 | 0.88±0.03 | 1.08±0.12 | |
HFpEF indicates heart failure with preserved ejection fraction; LV, left ventricular; and p53CKO, cardiomyocyte‐specific p53 knockout. MV E/A, Ratio between mitral E wave and A wave; and MV E/E, Ratio between mitral E wave and E′ wave. n=6 male chow p53CKO group; n=6 at 6, 8 weeks and n=5 at 16 weeks male HFpEF p53CKO group; n=5 female chow p53CKO group; n=5 at 6, 8 weeks and n=4 at 16 weeks female chow p53CKO group. Results are shown as mean±SEM. Two‐tailed unpaired Student’s t test or 2‐tailed unpaired Student’s t test followed by lognormal processing was used. *P<0.05, † P<0.01, ‡ P<0.001 compared with chow group in the same genotype are shown.
Discussion
In this study, we asked if sex and aging play a role in the pathogenesis of HFpEF using the new model developed by the Hill laboratory. 21 Here we report no difference in diastolic dysfunction onset between male and female mice similar to previous report. 26 mTRG2 and mTRG3 animals, with progressive shorter telomeres, exhibit accelerated HFpEF development compared with WT HFpEF animals. Molecularly, we found that p53 activation in cardiomyocytes is critical to HFpEF progression and at lower dosage, p53 is capable of suppressing mitochondrial biogenesis but not sufficient to drive mitochondrial dysfunction. Using a cardiomyocyte‐specific p53 knockout mouse model (p53CKO), we demonstrate that p53 accelerates HFpEF pathogenesis upon HFD+L‐NAME challenge. The observation where at 16‐weeks, p53CKO can still develop diastolic dysfunction under HFD+L‐NAME challenge is intriguing and suggest myocardial p53 activation may not be the only pathway responsible for diastolic dysfunction. Further experiments are warranted.
Here we recapitulate all the HFpEF phenotypes at 8‐weeks (cardiac hypertrophy, upregulation of BNP, and diastolic dysfunction) compared with 5 weeks as previously reported. 21 This difference is likely due to the mouse strains used: C57/B6N by the Hill laboratory versus C57/B6J in this study. It is well known that C57/B6J exhibits a mutation in mitochondrial Nnt gene and produces less mitochondrial reactive oxygen species upon stimulation. 27 The delayed onset of HFpEF phenotype in C57/B6J suggests the possibility that mitochondrial reactive oxygen species participates in but is not essential for the pathogenesis of HFpEF. Compared with hypertensive heart failure rats (onset at 20 months of age), 28 diabetic mouse model, 29 transverse aortic constriction surgery model, 30 and subtotal nephrectomy model, 31 our work here confirms the robustness of the HFD+L‐NAME HFpEF model on C57/B6J background and is currently the most cost‐effective and complication‐free HFpEF model available.
Cardiac hypertrophy is used to diagnose patients with HFpEF. 4 , 5 A recent report showed that LV myocardial stiffness in patients with LV hypertrophy represents a transitional state from a normal healthy heart to HFpEF, 32 suggesting hypertrophy precedes HFpEF. We too also observed LV diastolic anterior wall thickening in the HFpEF treatment group before the onset of diastolic dysfunction. With this knowledge, we speculate that compensatory metabolic changes promote regional hypertrophic response, which is necessary for the onset of diastolic dysfunction the development of heart failure. 33 , 34 Impaired adaptation of energy metabolism during the hypertrophic response exacerbates pathological hypertrophy and increases cardiomyocyte death. Further perturbations in mitochondrial metabolism may directly worsen contractile function and progress into heart failure. 35 , 36
Telomere shortening has been identified as a biomarker of lifetime stress, as early as in childhood, 37 and this stress‐related telomere shortening could be responsible for accelerated biological aging. 38 Previously we have shown that telomere shortening coincides with genetic cardiomyopathies 17 , 39 and “humanizing” the telomere lengths unmasks the dilated cardiomyopathy phenotype. 17 Telomere attrition occurs passively in dividing systems because of DNA end‐replication problems during cell division 40 , 41 ; however, in healthy human cardiomyocytes telomeres do not shorten with aging. 39 Here we show that compared with WT mice, mTRG2 and mTRG3 animals developed HFpEF phenotype 2 and 4 weeks earlier, respectively. Despite different kinetics in reaching the HFpEF state, WT, mTRG2, and mTRG3 HFpEF animals all stabilized and did not progress into heart failure. It has long been known that the major risk factor for the vast majority of chronic diseases is age itself. 42 Here our findings support the notion that critically aged systems (mTRG2 and mTRG3) are more prone to activate p53 and develop HFpEF.
Molecularly, we demonstrate that it is indeed the slight increase in p53 activation that dampens mitochondrial biogenesis (Pgc1α and Pgc1β) but not to the extent of mitochondrial dysfunction is key to the formation of HFpEF. In the heart, critically short telomeres alone can drive the development of dilated cardiomyopathy in mice in a p53‐dependent manner through Pgc1α and Pgc1β repression. 16 , 43 Mitochondria is responsible for producing the majority of adenosine triphosphate consumed by the heart (≈95%) and it has been documented that patients with HFpEF exhibit metabolic dysfunction and are exercise intolerant. 44 , 45 We show that mild p53 activation coincided with mitochondrial compensation in HFpEF murine cardiomyocytes—a decrease in mitochondrial DNA copy number and mitochondrial amount, loss of mitochondrial ultrastructure, but a normal basal mitochondrial respiration coupled with an increase in mtDNA transcription. This observed mitochondrial compensation is nicely supported by the observed upregulation of mitochondrial adenosine triphosphate synthesis pathways in patients with HFpEF. 46 Thus the question becomes when do patients with HFpEF worsen and end up with mitochondrial dysfunction? Clinically, peak oxygen consumption reserve is an independent predictor of exercise intolerance in patients with HFpEF 15 and likely a good readout of mitochondrial status. Given that adult cardiomyocytes do not maintain sarcomeric structure under prolong culture, we activated p53 in neonatal murine cardiomyocytes using nutlin‐3 to address our question. Strikingly, only a decrease in maximal OCR was observed with mild p53 activation (2.5 μmol/L nutlin‐3) similar to HFpEF cardiomyocytes. When we induced higher p53 activation (10 μmol/L Nutlin‐3), a decrease in both basal and maximal oOCR similar to dilated cardiomyopathy cardiomyocytes 17 was observed. Lastly, using a myocardial‐p53 knockout animal, p53CKO, we show that HFD+L‐NAME induced HFpEF was delayed.
CONCLUSIONS
In summary, we show that HFpEF pathogenesis is multifactorial yet short telomeres can accelerate this process via p53 activation. Here we provide in vivo and in vitro functional characterization of HFpEF myocardium and reveal that mitochondria can enter a compensatory state before mitochondrial dysfunction and this phenomenon is p53 dose dependent. Together, our data suggest that interventions that limit p53 activation or restore mitochondrial biogenesis may delay the onset of HFpEF.
Sources of Funding
This research was supported by National Natural Science Foundation of China (81570037 to H.Z. and 82070248 to A.C.Y.C.); Shanghai Pujiang Program (19PJ1407000 to A.C.Y.C.); Guiding Medical Project of Science and Technology Committee of Shanghai Municipality (19411963300 to H.Z.); The Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (0900000024 to A.C.Y.C.); Innovative Research Team of High‐Level Local Universities in Shanghai (A.C.Y.C.).
Disclosures
None.
Supporting information
Figures S1–S8
Acknowledgments
We thank Prof. Joseph Hill for providing guidance in establishing the HFpEF model. We thank staff at Electron Microscopy Center, Bioimaging Facility, and Protein Facility of Shanghai Institute of Precision Medicine for their expert assistance. We also thank the staff members of the Animal Facility at the National Facility for Protein Science in Shanghai (NFPS), China for providing animal support.
Supplemental Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.024582
For Sources of Funding and Disclosures, see page 17.
Contributor Information
Alex Chia Yu Chang, Email: alexchang@shsmu.edu.cn.
Huili Zhang, Email: huilizhang815@163.com.
References
- 1. Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2017;14:591–602. doi: 10.1038/nrcardio.2017.65 [DOI] [PubMed] [Google Scholar]
- 2. Pfeffer MA, Shah AM, Borlaug BA. Heart failure with preserved ejection fraction in perspective. Circ Res. 2019;124:1598–1617. doi: 10.1161/CIRCRESAHA.119.313572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355:251–259. doi: 10.1056/NEJMoa052256 [DOI] [PubMed] [Google Scholar]
- 4. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Colvin MM, Drazner MH, Filippatos G, Fonarow GC, Givertz MM, et al. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2016;134:e282–e293. doi: 10.1161/CIR.0000000000000435 [DOI] [PubMed] [Google Scholar]
- 5. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, González‐Juanatey JR, Harjola V‐P, Jankowska EA, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37:2129–2200. doi: 10.1093/eurheartj/ehw128 [DOI] [PubMed] [Google Scholar]
- 6. Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA, Paulus WJ. Phenotype‐specific treatment of heart failure with preserved ejection fraction: a multiorgan roadmap. Circulation. 2016;134:73–90. doi: 10.1161/CIRCULATIONAHA.116.021884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lam CS, Xanthakis V, Sullivan LM, Lieb W, Aragam J, Redfield MM, Mitchell GF, Benjamin EJ, Vasan RS. Aortic root remodeling over the adult life course: longitudinal data from the Framingham Heart Study. Circulation. 2010;122:884–890. doi: 10.1161/CIRCULATIONAHA.110.937839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loffredo FS, Nikolova AP, Pancoast JR, Lee RT. Heart failure with preserved ejection fraction: molecular pathways of the aging myocardium. Circ Res. 2014;115:97–107. doi: 10.1161/CIRCRESAHA.115.302929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zacharias M, Joffe S, Konadu E, Meyer T, Kiernan M, Lessard D, Goldberg RJ. Clinical epidemiology of heart failure with preserved ejection fraction (HFpEF) in comparatively young hospitalized patients. Int J Cardiol. 2016;202:918–921. doi: 10.1016/j.ijcard.2015.09.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tromp J, Shen LI, Jhund PS, Anand IS, Carson PE, Desai AS, Granger CB, Komajda M, McKelvie RS, Pfeffer MA, et al. Age‐related characteristics and outcomes of patients with heart failure with preserved ejection fraction. J Am Coll Cardiol. 2019;74:601–612. doi: 10.1016/j.jacc.2019.05.052 [DOI] [PubMed] [Google Scholar]
- 11. Tong D, Schiattarella GG, Jiang N, Altamirano F, Szweda PA, Elnwasany A, Lee DI, Yoo H, Kass DA, Szweda LI, et al. NAD(+) repletion reverses heart failure with preserved ejection fraction. Circ Res. 2021;128:1629–1641. doi: 10.1161/CIRCRESAHA.120.317046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deng Y, Xie M, Li Q, Xu X, Ou W, Zhang Y, Xiao H, Yu H, Zheng Y, Liang YU, et al. Targeting mitochondria‐inflammation circuit by β‐hydroxybutyrate mitigates HFpEF. Circ Res. 2021;128:232–245. doi: 10.1161/CIRCRESAHA.120.317933 [DOI] [PubMed] [Google Scholar]
- 13. Kumar AA, Kelly DP, Chirinos JA. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation. 2019;139:1435–1450. doi: 10.1161/CIRCULATIONAHA.118.036259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Houstis NE, Eisman AS, Pappagianopoulos PP, Wooster L, Bailey CS, Wagner PD, Lewis GD. Exercise intolerance in heart failure with preserved ejection fraction: diagnosing and ranking its causes using personalized O(2) pathway analysis. Circulation. 2018;137:148–161. doi: 10.1161/CIRCULATIONAHA.117.029058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haykowsky MJ, Brubaker PH, John JM, Stewart KP, Morgan TM, Kitzman DW. Determinants of exercise intolerance in elderly heart failure patients with preserved ejection fraction. J Am Coll Cardiol. 2011;58:265–274. doi: 10.1016/j.jacc.2011.02.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chang AC, Ong SG, LaGory EL, Kraft PE, Giaccia AJ, Wu JC, Blau HM. Telomere shortening and metabolic compromise underlie dystrophic cardiomyopathy. Proc Natl Acad Sci USA. 2016;113:13120–13125. doi: 10.1073/pnas.1615340113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. López‐Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rando TA, Wyss‐Coray T. Asynchronous, contagious and digital aging. Nature Aging. 2021;1:29–35. doi: 10.1038/s43587-020-00015-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gude NA, Broughton KM, Firouzi F, Sussman MA. Cardiac ageing: extrinsic and intrinsic factors in cellular renewal and senescence. Nat Rev Cardiol. 2018;15:523–542. doi: 10.1038/s41569-018-0061-5 [DOI] [PubMed] [Google Scholar]
- 21. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019;568:351–356. doi: 10.1038/s41586-019-1100-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang ACY, Chang ACH, Nicin L, Weber GJ, Holbrook C, Davies MF, Blau HM, Bertaccini EJ. An in vitro model for identifying cardiac side effects of anesthetics. Anesth Analg. 2020;130:e1–e4. doi: 10.1213/ANE.0000000000003757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nat Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee HW, Blasco MA, Gottlieb GJ, Horner JW II, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–574. doi: 10.1038/33345 [DOI] [PubMed] [Google Scholar]
- 25. Ylikallio E, Page JL, Xu X, Lampinen M, Bepler G, Ide T, Tyynismaa H, Weiss RS, Suomalainen A. Ribonucleotide reductase is not limiting for mitochondrial DNA copy number in mice. Nucleic Acids Res. 2010;38:8208–8218. doi: 10.1093/nar/gkq735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tong D, Schiattarella GG, Jiang N, May HI, Lavandero S, Gillette TG, Hill JA. Female sex is protective in a preclinical model of heart failure with preserved ejection fraction. Circulation. 2019;140:1769–1771. doi: 10.1161/CIRCULATIONAHA.119.042267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ronchi JA, Figueira TR, Ravagnani FG, Oliveira HC, Vercesi AE, Castilho RF. A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radic Biol Med. 2013;63:446–456. doi: 10.1016/j.freeradbiomed.2013.05.049 [DOI] [PubMed] [Google Scholar]
- 28. Gómez‐Garre D, González‐Rubio ML, Muñoz‐Pacheco P, Caro‐Vadillo A, Aragoncillo P, Fernández‐Cruz A. Rosuvastatin added to standard heart failure therapy improves cardiac remodelling in heart failure rats with preserved ejection fraction. Eur J Heart Fail. 2010;12:903–912. doi: 10.1093/eurjhf/hfq101 [DOI] [PubMed] [Google Scholar]
- 29. Methawasin M, Strom JG, Slater RE, Fernandez V, Saripalli C, Granzier H. Experimentally increasing the compliance of titin through RNA Binding Motif‐20 (RBM20) inhibition improves diastolic function in a mouse model of heart failure with preserved ejection fraction. Circulation. 2016;134:1085–1099. doi: 10.1161/CIRCULATIONAHA.116.023003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reil J‐C, Hohl M, Reil G‐H, Granzier HL, Kratz MT, Kazakov A, Fries P, Müller A, Lenski M, Custodis F, et al. Heart rate reduction by If‐inhibition improves vascular stiffness and left ventricular systolic and diastolic function in a mouse model of heart failure with preserved ejection fraction. Eur Heart J. 2013;34:2839–2849. doi: 10.1093/eurheartj/ehs218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Primessnig U, Schönleitner P, Höll A, Pfeiffer S, Bracic T, Rau T, Kapl M, Stojakovic T, Glasnov T, Leineweber K, et al. Novel pathomechanisms of cardiomyocyte dysfunction in a model of heart failure with preserved ejection fraction. Eur J Heart Fail. 2016;18:987–997. doi: 10.1002/ejhf.524 [DOI] [PubMed] [Google Scholar]
- 32. Hieda M, Sarma S, Hearon CM, Dias KA, Martinez J, Samels M, Everding B, Palmer D, Livingston S, Morris M, et al. Increased myocardial stiffness in patients with high‐risk left ventricular hypertrophy: the hallmark of stage‐b heart failure with preserved ejection fraction. Circulation. 2020;141:115–123. doi: 10.1161/CIRCULATIONAHA.119.040332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang L, Jaswal JS, Ussher JR, Sankaralingam S, Wagg C, Zaugg M, Lopaschuk GD. Cardiac insulin‐resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure‐overload hypertrophy. Circ Heart Fail. 2013;6:1039–1048. doi: 10.1161/CIRCHEARTFAILURE.112.000228 [DOI] [PubMed] [Google Scholar]
- 34. Liew CW, Xu S, Wang X, McCann M, Whang Kong H, Carley AC, Pang J, Fantuzzi G, O'Donnell JM, Lewandowski ED. Multiphasic regulation of systemic and peripheral organ metabolic responses to cardiac hypertrophy. Circ Heart Fail. 2017;10. doi: 10.1161/CIRCHEARTFAILURE.117.003864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709–724. doi: 10.1161/CIRCRESAHA.113.300376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052 [DOI] [PubMed] [Google Scholar]
- 37. Mitchell C, Hobcraft J, McLanahan SS, Siegel SR, Berg A, Brooks‐Gunn J, Garfinkel I, Notterman D. Social disadvantage, genetic sensitivity, and children's telomere length. Proc Natl Acad Sci USA. 2014;111:5944–5949. doi: 10.1073/pnas.1404293111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Geronimus AT, Hicken MT, Pearson JA, Seashols SJ, Brown KL, Cruz TD. Do us black women experience stress‐related accelerated biological aging?: a novel theory and first population‐based test of black‐white differences in telomere length. Hum Nat. 2010;21:19–38. doi: 10.1007/s12110-010-9078-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chang ACY, Chang ACH, Kirillova A, Sasagawa K, Su W, Weber G, Lin J, Termglinchan V, Karakikes I, Seeger T, et al. Telomere shortening is a hallmark of genetic cardiomyopathies. Proc Natl Acad Sci USA. 2018;115:9276–9281. doi: 10.1073/pnas.1714538115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Arnoult N, Karlseder J. Complex interactions between the DNA‐damage response and mammalian telomeres. Nat Struct Mol Biol. 2015;22:859–866. doi: 10.1038/nsmb.3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sfeir A, de Lange T. Removal of shelterin reveals the telomere end‐protection problem. Science. 2012;336:593–597. doi: 10.1126/science.1218498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Niccoli T, Partridge L. Ageing as a risk factor for disease. Curr Biol. 2012;22:R741–R752. doi: 10.1016/j.cub.2012.07.024 [DOI] [PubMed] [Google Scholar]
- 43. Chang ACY, Pardon G, Chang ACH, Wu H, Ong S‐G, Eguchi A, Ancel S, Holbrook C, Ramunas J, Ribeiro AJS, et al. Increased tissue stiffness triggers contractile dysfunction and telomere shortening in dystrophic cardiomyocytes. Stem Cell Reports. 2021;16:2169–2181. doi: 10.1016/j.stemcr.2021.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lewis GA, Schelbert EB, Williams SG, Cunnington C, Ahmed F, McDonagh TA, Miller CA. Biological phenotypes of heart failure with preserved ejection fraction. J Am Coll Cardiol. 2017;70:2186–2200. doi: 10.1016/j.jacc.2017.09.006 [DOI] [PubMed] [Google Scholar]
- 45. Hunter WG, Kelly JP, McGarrah RW III, Khouri MG, Craig D, Haynes C, Ilkayeva O, Stevens RD, Bain JR, Muehlbauer MJ, et al. Metabolomic profiling identifies novel circulating biomarkers of mitochondrial dysfunction differentially elevated in heart failure with preserved versus reduced ejection fraction: evidence for shared metabolic impairments in clinical heart failure. J Am Heart Assoc. 2016;5. doi: 10.1161/JAHA.115.003190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hahn VS, Knutsdottir H, Luo X, Bedi K, Margulies KB, Haldar SM, Stolina M, Yin J, Khakoo AY, Vaishnav J, et al. Myocardial gene expression signatures in human heart failure with preserved ejection fraction. Circulation. 2021;143:120–134. doi: 10.1161/CIRCULATIONAHA.120.050498 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S8