

Abstract
Background
Elevated plasma levels of alpha‐aminoadipic acid (2‐AAA) have been associated with the development of type 2 diabetes and atherosclerosis. However, the nature of the association remains unknown.
Methods and Results
We identified genetic determinants of plasma 2‐AAA through meta‐analysis of genome‐wide association study data in 5456 individuals of European, African, and Asian ancestry from the Framingham Heart Study, Diabetes Prevention Program, Jackson Heart Study, and Shanghai Women’s and Men’s Health Studies. No single nucleotide polymorphisms reached genome‐wide significance across all samples. However, the top associations from the meta‐analysis included single‐nucleotide polymorphisms in the known 2‐AAA pathway gene DHTKD1, and single‐nucleotide polymorphisms in genes involved in mitochondrial respiration (NDUFS4) and macrophage function (MSR1). We used a Mendelian randomization instrumental variable approach to evaluate relationships between 2‐AAA and cardiometabolic phenotypes in large disease genome‐wide association studies. Mendelian randomization identified a suggestive inverse association between increased 2‐AAA and lower high‐density lipoprotein cholesterol (P=0.005). We further characterized the genetically predicted relationship through measurement of plasma 2‐AAA and high‐density lipoprotein cholesterol in 2 separate samples of individuals with and without cardiometabolic disease (N=98), and confirmed a significant negative correlation between 2‐AAA and high‐density lipoprotein (r s=−0.53, P<0.0001).
Conclusions
2‐AAA levels in plasma may be regulated, in part, by common variants in genes involved in mitochondrial and macrophage function. Elevated plasma 2‐AAA associates with reduced levels of high‐density lipoprotein cholesterol. Further mechanistic studies are required to probe this as a possible mechanism linking 2‐AAA to future cardiometabolic risk.
Keywords: 2‐aminoadipic acid, genome‐wide association study, HDL cholesterol, Mendelian randomization analysis
Subject Categories: Genetics; Diabetes, Type 2; Risk Factors; Biomarkers; Lipids and Cholesterol
Nonstandard Abbreviations and Acronyms
- 2‐AAA
alpha‐aminoadipic acid
- DPPOS
Diabetes Prevention Program Outcomes Study
- FHS
Framingham Heart Study
- JHS
Jackson Heart Study
- MR
Mendelian randomization
- PhEWAS
phenome‐wide association study
- PS
polygenic score
- SMHS
Shanghai Men’s Health Study
- SWHS
Shanghai Women’s Health Study
Clinical Perspective
What Is New?
Alpha‐aminoadipic acid levels in plasma may be partly regulated by common variants in genes involved in mitochondrial and macrophage function, including DHTKD1 and MSR1.
Elevated plasma alpha‐aminoadipic acid associates with reduced levels of high‐density lipoprotein cholesterol.
What Are the Clinical Implications?
Elevated alpha‐aminoadipic acid may be an early marker of lipoprotein dysregulation.
Further mechanistic studies may establish alpha‐aminoadipic acid as a potential target in cardiometabolic disease.
Cardiometabolic disease, including cardiovascular disease and type 2 diabetes (T2D) is a major global health concern, associated with a high incidence of comorbidities and significantly increased mortality. 1 These complex chronic diseases are polygenic and multifactorial; the underlying causes of disease are only partially delineated in most cases. Metabolites are emerging as useful biomarkers for both disease prediction and understanding disease etiology. Higher levels of the novel metabolite biomarker, alpha‐aminoadipic acid (2‐AAA) were found to increase the risk of incident diabetes in the FHS (Framingham Heart Study) participants, 2 and development of coronary artery calcification in participants in the Veterans Affairs Diabetes Trial and Follow‐Up Study. 3 Associations were independent of known risk markers (eg, age, sex, body mass index, glycemic control, family history, diet), 2 suggesting this metabolite may represent novel disease‐related biology.
2‐AAA is a mitochondrial metabolite, generated from the catabolism of the essential amino acid lysine. Little is known about the function of 2‐AAA or the determinants of variability between individuals. We hypothesized that 2‐AAA levels are, in part, genetically determined and that genetic interrogation of this metabolite would reveal novel underlying biology of cardiometabolic disease. We examined the genetic contribution to plasma 2‐AAA variation in a GWAS (genome‐wide association study) and meta‐analysis among multiple ancestries, using data from the FHS) (N=1452), the DPPOS (Diabetes Prevention Program Outcomes Study, N=1612), the JHS (Jackson Heart Study, N=1884), and the Shanghai Women’s and Men’s Health Studies (N=508). We examined the association between genetic predictors of 2‐AAA and cardiometabolic disease phenotypes through Mendelian randomization (MR) and phenome‐wide association study (PHEWAS) and validated a genetically predicted relationship with high‐density lipoprotein (HDL) cholesterol by direct measurement.
METHODS
Data and Materials Availability Statement
Several of the data sets used in the article are publicly available; the details for accessing these are indicated in the relevant sections. Other data that support the findings of this study are available from the corresponding author upon reasonable request.
Study Populations
The FHS is a prospective, observational, community‐based cohort of cardiovascular disease. Genome‐wide analysis of plasma metabolites in European ancestry participants of the FHS Offspring Cohort who attended the fifth examination (1991–1995) and underwent metabolic profiling and genome‐wide genotyping has been published previously. 4 We obtained summary statistics for the GWAS of 2‐AAA in 1452 participants.
The JHS is a longitudinal population‐based observational study designed to prospectively investigate determinants of cardiovascular disease in Black individuals. 5 A total of 5306 men and women (63.4% female, age range 21–94 years, mean age 55) in the Jackson, MS metropolitan area were recruited between September 2000 and March 2004. Medical history and physical examination, blood and urine samples, and information on diet, physical activity, socioeconomic factors, and health care access were collected for all participants. We obtained data for 1884 individuals with genome‐wide single nucleotide polymorphism (SNP) genotyping data and plasma 2‐AAA measurement. 6
The SWHS (Shanghai Women’s Health Study) and the SMHS (Shanghai Men’s Health Study) are ongoing prospective cohort studies that recruited 75 000 women and 61 500 men of Chinese ancestry, aged 35 to 75 years, from Shanghai, China. 7 , 8 Women were recruited from 1996 to 2000 and men from 2002 to 2006. At baseline, detailed information was collected on dietary intake, personal lifestyle habits, and medical history, and blood samples were obtained from 75% of study participants. Plasma 2‐AAA was measured as part of an untargeted metabolomics panel by Metabolon, Inc. (Raleigh, NC) 9 and was available for 820 participants. After quality control (QC), we included data from 508 individuals with plasma 2‐AAA measurement and genotypes for subsequent GWAS analysis.
The DPP (Diabetes Prevention Program) was a randomized clinical trial that aimed to investigate whether lifestyle changes or metformin could effectively delay diabetes in overweight and obese adults at high risk of diabetes. 10 The DPPOS measured plasma 2‐AAA as part of metabolite analyses, 11 and completed metabolomic GWAS analyses. We obtained summary statistics for the GWAS for 2‐AAA in 1612 participants of self‐identified European (70%), African (19%), Asian (4%), and other includes Native American and mixed ancestry (7%).
Measurement of Alpha‐Aminoadipic Acid
Measurement of plasma 2‐AAA in FHS, JHS, and DPP was carried out as part of a metabolite profiling panel at the Broad Institute as previously described. 11 , 12 , 13 Briefly, metabolites were extracted from plasma using acetonitrile and methanol and separated using a 100×2.1 mm XBridge Amide column (Waters). A high sensitivity Agilent 6490 QQQ MS (Agilent) was used to profile metabolites in the negative ion mode via multiple reaction monitoring scanning, and hydrophilic interaction liquid chromatography coupled to tandem mass spectrometry (MS) was used to analyze polar metabolites in the positive ion mode using a 4000 QTRAP triple quadrupole mass spectrometer (AB Sciex, Foster City, CA) coupled to either an 1100 Series pump (Agilent Technologies, Santa Clara, CA) or an HTS PAL autosampler (Leap Technologies, Carrboro, NC) equipped with a column heater. Raw data were processed using MassHunter Quantitative Analysis Software (Agilent). Metabolite measurements were normalized to pooled plasma samples and internal standards.
Measurement of 2‐AAA in SWHS and SMHS was carried out at Metabolon (Metabolon Inc., Morrisville, NC) as part of a global metabolomics profiling panel conducted using multiple mass spectrometry techniques. Briefly, proteins were precipitated with methanol under vigorous shaking for 2 minutes (Glen Mills GenoGrinder 2000) followed by centrifugation and removal of organic solvent (TurboVap®; Zymark). Extracts were divided into fractions for analysis: 2 for analysis by 2 separate reverse phase/ultra‐high performance liquid chromatography (UPLC)‐MS/MS methods with positive ion mode electrospray ionization (ESI), 1 for analysis by reverse phase/UPLC‐MS/MS with negative ion mode ESI, and 1 for analysis by hydrophilic interaction/UPLC‐MS/MS with negative ion mode ESI. Sample extracts were stored overnight under nitrogen before preparation for analysis. Raw data were extracted, peak‐identified, and QC processed using Metabolon’s hardware and software. All methods used a Waters ACQUITY UPLC and a Thermo Scientific Q‐Extractive high‐resolution/accurate mass spectrometer interfaced with a heated ESI source and Orbitrap mass analyzer operated at 35 000 mass resolution. Peaks were quantified using area under the curve. The raw peak intensity was rescaled to set the median across all samples equal to 1 to establish a normal distribution and values below the limit of detection were imputed with the lowest observed value in the data set.
Genome‐Wide SNP Genotyping and Genetic QC Steps
In FHS, genotyping was carried out using the Affymetrix 500K mapping array and the Affymetrix 50K gene‐focused molecular inversion probe array as described. 4 , 14 Genotypes were called using Chiamo (http://www.stats.ox.ac.uk/∼marchini/software/gwas/chiamo.html). SNPs were excluded if they had a call rate <95%, Hardy‐Weinberg equilibrium P<1×10−6, or minor allele frequency (MAF) <1%. Imputation was performed (HapMap CEU, release 22, build 36) using a hidden Markov model in MACH (v.1.0.15). Principal components were calculated using Eigenstrat. 15
In JHS, genotyping was performed as described 16 using the Affymetrix 6.0 SNP Array (Affymetrix, Santa Clara, CA). SNPs were excluded if they were genotyped successfully in <90% of samples; subjects were removed if <95% of SNPs were genotyped successfully. No SNPs were removed owing to deviation from Hardy‐Weinberg equilibrium expectations because the Black population is an admixed population, which may result in departures from Hardy‐Weinberg equilibrium expectations even under ideal conditions. Imputation to the 1000 Genomes project reference panel (Phase I, Version 3, March 2012 release) was performed using MACH 1.0 and minimac 38. Imputed data were filtered for a sample missingness rate <2%, and a SNP missingness rate <4%. Principal components (PCs) were calculated using the SNPRelate package. 17
In SWHS/SMHS genotyping was carried out using the Asian MEGA, Affymetrix 6, and Illumina Hap500/Illumina 660W panels. Imputation to the 1000 Genomes reference panel (Phase 3) was performed using Minimac on the Michigan Imputation Server. Variants with poor imputation quality (r 2<0.3) or with a MAF <1% were excluded. PCs were calculated using the SNPRelate package. 17
In DPP genotyping was performed using the Illumina Human Core Exome genotyping array (547 622 genetic markers across the genome, including 265 919 exome‐focused markers) at the Genomics Platform at the Broad Institute. Genotypes were called using Birdsuite https://www.broadinstitute.org/birdsuite). QC steps filtered for discrepant sex information, low call rates for individuals (<98%), pairs of samples with low inbreeding coefficients, SNP call rates <95%, and SNPs with Hardy‐Weinberg equilibrium P<10−8 in any ethnic group. A 2‐stage imputation procedure consisting of prephasing the genotypes into whole chromosome haplotypes followed by imputation itself was conducted. The prephasing was performed using SHAPEIT2. 18 The reference panel comprised 1000 Genomes Phase3 haplotypes, 19 and the genotype imputation was done using IMPUTE2. 20 This resulted in 9 276 901 imputed SNPs with MAF >1% and info scores >0.882 (r 2>0.8).
Genome‐Wide Association Study
GWAS of common variation (MAF >0.01) was carried out in each cohort individually and tailored to the specific characteristics of each study. All cohorts were analyzed by linear regression under an additive genetic model, with mixed‐effects models applied to cohorts with related individuals (FHS and JHS) and to SWHS/SMHS.
For FHS, GWAS was conducted as described, 4 using normalized residuals of 2‐AAA, using linear mixed‐effects models to accommodate pedigree data under an additive genetic model, adjusted for age and sex. GWASs were performed in R using the lmekin function in the kinship package. Population stratification in this well‐characterized European ancestry population was accounted for by adjusting for PC1 if P<0.0001. The final genomic control parameter lambda was 1.02. Results were filtered for MAF >5% and imputation rate of >0.80.
JHS used a mixed linear model‐based leave 1 chromosome out association analysis approach on inverse normal transformed 2‐AAA, which was implemented in GCTA 1.93.0 beta software tool. The advantages of the mixed‐linear‐model association method include the prevention of false positive associations due to population stratification and interindividual relatedness and an increase in power obtained through the application of a correction that is specific to this structure. 21 Models were adjusted for age, sex, and 10 PCs. The final genomic control parameter lambda was 1.01.
SWHS/SMHS used a linear mixed‐effects model on inverse normal transformed 2‐AAA, assuming an additive genetic model and adjusting for age, sex, and 10 PCs, which was implemented in PLINK v2.00α2LM software tool. The final genomic control parameter lambda was 1.02.
For DPP, a linear regression model was run on inverse normal transformed 2‐AAA, assuming an additive genetic model, adjusting for age, sex, and 10 PCs. Analyses were run using the GWASTools package from R Bioconductor. The final genomic control parameter lambda was 0.99.
GWAS Meta‐Analysis
The FHS, DPP, JHS, and SWHS/SMHS cohort‐specific genome‐wide association results were meta‐analyzed using the sample size–weighted Z score approach in METAL software. 22 This approach allows SNP associations to be combined across studies when the β‐coefficients and standard errors from the individual studies are in different units, as was the case in these analyses because of the differences in metabolite profiling platforms. The approach combines Z scores for each allele across studies in a weighted sum, with weights proportional to the square‐root of the study’s sample size for each. As this approach does not provide combined effect size estimates, effect size estimates used for MR and polygenic score (PS) analyses were based on SNP weights from FHS for European ancestry, and JHS for African ancestry.
Phenome‐Wide and Cardiometabolic Disease Data Sets and Analyses
BioVU: The Vanderbilt BioVU resource is a deidentified DNA biobank linked to the Synthetic Derivative, a deidentified version of the Vanderbilt electronic health record. 23 Genotyping in stored DNA samples was performed by the Vanderbilt Technologies for Advanced Genomics according to standard protocols on the MEGAEX array. QC steps for the BioVU population have been previously described. 24 Genotype data were imputed with IMPUTE4, 20 version 2.3.0 (University of Oxford), using the October 2014 release of the 1000 Genomes cosmopolitan reference haplotypes; variants imputation quality scores <0.3 were excluded. One participant from each related pair (pi‐hat >0.2) was randomly excluded. PCs were calculated using the SNPRelate package. 17 For this study, analyses were restricted to European ancestry individuals (N=74 760) and African ancestry individuals (N=16 182) who were defined by principal components analyses in conjunction with HAPMAP reference populations. The use of BioVU and other deidentified data presented in these analyses was approved by the Vanderbilt University Medical Center Institutional Review Board.
The UKBB (UKBiobank) is a British population‐based self‐reported study that is composed of ≈0.5 million participants aged 37 to 73 at recruitment. 25 GWAS summary statistics for 2173 UKBB phenotypes 26 were downloaded from the study by Bycroft et al. 27
Other data sets: Summary statistics for cardiometabolic phenotypes were obtained from existing large‐scale GWAS in European Ancestry, including GIANT (Genetic Investigation of Anthropometric Traits) 28 , 29 (body mass index, waist circumference, height; https://portals.broadinstitute.org/collaboration/giant/index.php/GIANT_consortium_data_files); Global lipids consortium phenotypes 30 (HDL, low‐density lipoprotein [LDL], total cholesterol, triglycerides); MAGIC (Meta‐Analyses of Glucose and Insulin‐Related Traits Consortium) 31 (fasting glucose, fasting insulin; www.magicinvestigators.org); DIAGRAM (Diabetes Genetics Replication and Meta‐Analysis) 2 , 32 (T2D; http://diagram‐consortium.org/downloads.html); CARDIOGRAMplusC4D 33 (coronary artery disease) and CRP (C‐reactive protein). 34
Polygenic Score
A genetic predictor for 2‐AAA was calculated using weighted genetic risk scores according to the following formula:
where the allele dosage is a value ranging from 0 and 2 and wi is the change in 2‐AAA levels (ß coefficient) for each copy of the effect allele. Only independent SNPs (r 2<0.05) that passed QC and reached significance of P<5×10−4 in the meta‐analysis, with the effect in the same direction in at least 3 of the 4 studies were included in the PS.
Heritability Analysis
We applied linkage disequilibrium (LD) score regression 35 using 2‐AAA summary statistics from the meta‐analysis of FHS and DPP GWAS (majority European ancestry, n=3064) to probe the SNP heritability of the trait (the proportion of phenotypic variance explained by all SNPs). The slope obtained from linkage disequilibrium score regression provides an estimate of 2‐AAA heritability.
PHEWAS of BioVU
A multivariable logistic regression adjusting for 5 PCs, the median age in the electronic health record, and sex was performed to test the associations between the polygenic risk score and each PHEWAS phenotype (pheCode) as the main predictor. Analyses were performed using the PheWAS R package. 36 A false discovery rate P<0.1 was considered statistically significant.
MR Analysis
We ran instrumental variable analysis to ascertain for associations between genetically determined 2‐AAA levels and selected phenotypes using an MR approach. When certain assumptions are met, MR can ascertain associations between an exposure and an outcome. 37 In this case, our exposure was plasma 2‐AAA, whereas the outcomes included 11 cardiometabolic traits (body mass index, waist circumference, HDL‐C, LDL‐C, total cholesterol, triglycerides, fasting glucose, fasting insulin, T2D, coronary artery disease, CRP). To infer relationships between exposures and outcomes, MR analyses assume (1) the genetic instrument associated with 2‐AAA; (2) the association between the genetic instrument and 2‐AAA was independent of potential confounders; (3) the genetic instrument was not pleiotropic, and could only affect the outcome through 2‐AAA; and (4) the effect was homogeneous. To construct our 2‐AAA genetic instrument, we selected independent SNPs associated with plasma 2‐AAA with P<5×10−4 and the same effect direction from at least 3 of the 4 data sets included in the meta‐analysis and tested the association with cardiometabolic outcomes using the inverse‐variance weighted average meta‐analysis (IVWA) method. Because the 2‐AAA metabolite was measured using different technologies, it was not possible to harmonize the effect sizes across data sets, even after applying a common transformation to each data set. Thus, SNP weightings for the PS were based on the largest European ancestry (FHS) and largest African ancestry (JHS) cohorts. The pleiotropy‐robust MR‐Egger and Weighted Median methods were used as sensitivity analyses and to confirm the magnitude and direction of associations identified by IVWA. To assess effects of our outcomes on 2‐AAA, we also ran the reverse MR, using cardiometabolic traits as the exposure and 2‐AAA as the outcome. The genetic instrument for each trait was constructed using SNPs P<5×10−8 in their respective GWAS, except for fasting insulin, where no SNPs passed the GWAS significance threshold, so we applied a nominal threshold of P<5×10−4. All MR methods were calculated using the Mendelian Randomization R package. 38 Bonferroni‐adjusted P<0.0046 (0.05/11) by IVWA was considered significant.
Chronic Kidney Disease Validation Study
We obtained plasma samples from 62 individuals with or without chronic kidney disease (CKD), from an existing study. 39 We measured 2‐AAA by liquid chromatography MS at the Vanderbilt Mass Spectrometry Core. Samples were spiked with internal standard (Arginine‐15N4, Sigma Aldrich), extracted with methanol, and derivatized with dansyl chloride (Sigma Aldrich) before analysis. The dansyl derivative of 2‐AAA ([M+H]+ 395.1271) was measured by targeted selected ion monitoring using a Vanquish UPLC system interfaced to a QExactive HF quadrupole/orbitrap mass spectrometer (Thermo Fisher Scientific). Data acquisition and quantitative spectral analysis were conducted using Thermo‐Finnigan Xcaliber version 4.1 and Thermo‐Finnigan LCQuan version 2.7, respectively. Calibration curves were constructed by plotting peak area ratios (2‐AAA/Arg‐15N4) against analyte concentrations for a series of 2‐AAA standards. ESI source parameters were tuned and optimized using an authentic 2‐AAA reference standard (Sigma Aldrich) derivatized with dansyl chloride and desalted by solid phase extraction before direct liquid infusion. Fasting insulin was measured by radioimmunoassay (Millipore, St. Charles, MO). The study was approved by the Vanderbilt institutional review board, and all participants provided informed consent.
Diabetes and Ischemia Validation Study
We obtained data for plasma 2‐AAA and HDL cholesterol in 38 individuals with or without T2D from a previous study of experimental ischemia. 40 2‐AAA was measured in plasma as part of the metabolite profiling panel at the Broad Institute as described here and previously. 40 During analysis we removed data for 2 individuals who were outliers for 2‐AAA (>2 SD from mean), and present data for the relationship between 2‐AAA and HDL in 36 (n=19 healthy controls, n=17 T2D). The study was approved by the Human Research Committees of the Brigham and Women’s Hospital, and all participants provided informed consent.
RESULTS
We conducted GWAS of plasma 2‐AAA levels using available data in 4 separate studies, including the FHS (N=1452, results previously published 4 ), the DPP (N=1612), the JHS (N=1884), and the SWHS/SMHS (N=508). Characteristics of the study participants are presented in Table S1. Several loci reached genome‐wide significance in individual studies (FHS: rs11802990 and rs10158605 located within the SPATA6 gene, P<5×10−8; JHS: intergenic rs13403315 and rs12918656 in CNTNAP4, P<5×10−8), but these associations were not replicated in the other studies (Figure S1).
We hypothesized that the absence of robust genome‐wide significant signals might be due to high polygenicity and a small sample size. To test this hypothesis, we estimated the additive genetic heritability linkage disequilibrium score regression. We found that the heritability of 2‐AAA in predominantly European ancestry participants was ≈28% (95% CI, 2–55%, N=3064), confirming a polygenic contribution to 2‐AAA variability.
We hypothesized that SNPs with modest, but biologically relevant effects on 2‐AAA would have effects that were consistent across different ancestry groups and ran a meta‐analysis across all 4 studies. Although there were no loci reaching genome‐wide significance, there were suggestive signals with consistent directions of effect across multiple studies. Of the top associations, 38 SNPs were associated with 2‐AAA at P<5×10−6 (Table S2). Notably, one of these top loci was near the DHTKD1 gene, which encodes a protein downstream of 2‐AAA in the lysine catabolic pathway and has been previously linked to 2‐AAA levels in both humans and animals. 41 , 42 We also identified SNPs in the region of other genes with known mitochondrial or cardiometabolic disease biology, including NADH:Ubiquinone Oxidoreductase Core Subunit S1 (NDUFS4) and Macrophage Scavenger Receptor 1 (MSR1).
We constructed a PS using the SNPs identified in the transethnic meta‐analysis at P<5×10−4, and a requirement of effects in the same direction in at least 3 studies (Table S2) and probed its association with disease phenotypes using the Vanderbilt BioVU electronic health record resource (European and African ancestries) and UKBB. However, there were no phenotypes significantly associated after correction for multiple testing (Tables S3 and S4). We further probed associations between the 2‐AAA PS and cardiometabolic disease phenotypes using well‐characterized GWAS including Global Lipids Genetics Consortium (HDL, LDL, triglycerides), and CARDIoGRAM (MI, coronary artery disease) (Table S5). MR analysis using the IVWA method suggested a nominal association between elevated plasma 2‐AAA and reduced HDL cholesterol (P=0.005, Figure 1). This finding was consistent when examined using the weighted median method. The direction was inconsistent with the MR‐Egger method, but the intercept P value was nonsignificant, suggesting the IVWA estimate was not biased. There was no significant association between 2‐AAA and LDL cholesterol and a nominal positive association between 2‐AAA and triglycerides (P=0.04). There was no significant association when applying the reverse MR, to assess the effect of HDL PS on 2‐AAA (Table S6), implying no effect of HDL cholesterol on 2‐AAA levels. There was also a significant association between 2‐AAA and fasting insulin (P=0.001). We conducted multivariable MR with 2‐AAA and fasting insulin and found that both remained significantly associated with HDL (Table S7). Reverse MR highlighted a potential association between the insulin genetic instrument and 2‐AAA (Table S6), suggestive of reciprocal regulation between 2‐AAA and insulin that may be independent of a 2‐AAA–HDL relationship.
Figure 1. Mendelian randomization supports causal role between elevated 2‐AAA and low HDL cholesterol.

Predicted negative association between alpha‐aminoadipic acid (2‐AAA) and HDL cholesterol, P=0.005. Single nucleotide polymorphisms (SNPs) from transethnic meta‐analysis of 2‐AAA with P<5×10−4 (n=272 SNPs) and an effect in the same direction in 3 data sets were selected as the exposure. SNPs from Global Lipids Consortium Genome‐Wide Association Study for HDL were used as the outcome. HDL indicates high‐density lipoprotein.
The significance of the HDL association was just below the multiple testing threshold. We used orthogonal methodologies to assess the validity and reproducibility of this association. To confirm whether the genetically‐predicted association between 2‐AAA and HDL cholesterol could be validated using measured values, we analyzed the relationship between plasma 2‐AAA and plasma HDL cholesterol in 2 independent samples, comprising 98 individuals in total. Of these, 62 were recruited to a study of CKD, and comprised 21 healthy controls, 20 individuals with CKD stage 3 to 5 and not yet on hemodialysis and 21 individuals with CKD stage 5 on hemodialysis. A separate study included 36 individuals in a study of T2D, including 19 healthy controls and 17 individuals with T2D. Because 2‐AAA was measured using 2 different platforms, we analyzed each study separately. In both samples, there was a significant linear correlation between higher 2‐AAA and lower HDL (r s=−0.53, P<0.0001 CKD sample; r s=−0.36, P=0.03 T2D sample; Figure 2). This was apparent both within the healthy individuals and in individuals with established disease, suggesting that this relationship is not disease dependent. In the CKD sample (n=62), we investigated whether the 2‐AAA–HDL relationship was modulated by fasting insulin. Consistent with the results from the genetic approach, the associations between both insulin and 2‐AAA with HDL remained significant in a multiple linear regression model (insulin P=0.002, 2‐AAA P=0.03).
Figure 2. Measured plasma 2‐AAA inversely correlates with HDL cholesterol in 2 independent data sets.
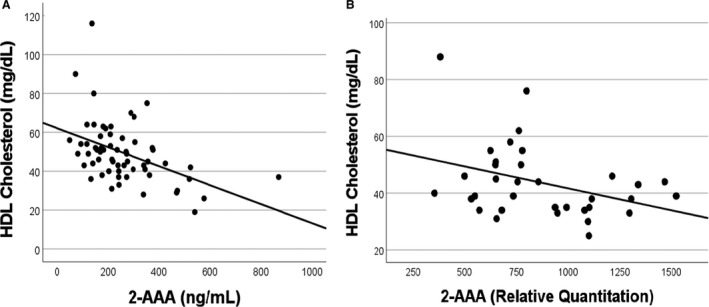
A, N=62 individuals with or without chronic kidney disease (r s=−0.53, P<0.0001). B, N=36 individuals with or without type 2 diabetes (r s=−0.36, P=0.03). 2‐AAA indicates alpha‐aminoadipic acid; and HDL, high‐density lipoprotein.
DISCUSSION
Plasma 2‐AAA has been associated with cardiometabolic disease; however, the determinants of elevated 2‐AAA are unknown. We probed the genetic determinants of 2‐AAA through GWAS and meta‐analysis of plasma 2‐AAA levels in epidemiological cohorts. Some previous genetic studies of metabolites have included 2‐AAA, but without replication and in small numbers. 4 , 43 Our sample represents the largest GWAS of 2‐AAA to date and the only one to include multiple ancestries. Although we did not detect significant signals at the genome‐wide significance threshold, we highlight several suggestive regions of interest including a known 2‐AAA pathway gene, DHTKD1. Interrogation of a 2‐AAA PS revealed a nominal genetic association between elevated 2‐AAA and reduced HDL cholesterol, which we validated experimentally.
The determinants of high 2‐AAA within the population have not yet been established; however, genetic approaches are emerging as useful tools to probe novel biomarkers, 44 with utility to identify novel disease‐related biology even for biomarkers under complex multifactorial control. Variants in the 2‐AAA pathway gene DHTKD1 have been associated with Mendelian cases of 2‐aminoaciduria, 41 whereas this same gene has been associated with elevated 2‐AAA in mouse models. 42 Heritability estimates for metabolites vary; heritability of 2‐AAA was previously estimated at ≈48%. 43 Analysis in our sample confirmed a heritable component, estimated at 28%, albeit with a wide CI of 2% to 55%. The estimate suggests there may be comparable heritability for 2‐AAA as for other circulating biomarkers such as LDL cholesterol (≈17%). 45 Thus, plasma 2‐AAA levels are influenced by heritable genetic variation.
Our analysis suggested a putative association between 2‐AAA and DHTKD1. The dehydrogenase E1 and transketolase domain containing 1 (DHTKD1) gene encodes part of a mitochondrial super complex that catalyzes the conversion of 2‐oxoadipate to glutaryl‐CoA within the 2‐AAA catabolic pathway. 41 A mouse genetic reference study 42 found that variation in Dhtkd1 was associated with hepatic expression of Dhtkd1 at the mRNA and protein level, as well as with serum 2‐AAA. Knockout of Dhtkd1 in mouse results in increased 2‐AAA in brain and liver. 46 Mendelian variation in DHTKD1 is associated with 2‐aminoadipic, 2‐ketoadipic, and 2‐oxoadipic aciduria 41 , 47 and with Charcot‐Marie‐Tooth disease. 48 Our data highlight associations between common variants in DHTKD1 and 2‐AAA, suggesting that direct modulation of the 2‐AAA pathway by DHTKD1 may be a contributor to circulating levels of 2‐AAA.
Our analysis also revealed suggestive candidate genetic associations with plasma 2‐AAA, including between SNPs in NDUFS4 and 2‐AAA. This gene, NADH:ubiquinone oxidoreductase subunit S4, encodes the complex I subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase, and as such plays a key role in mitochondrial oxidative phosphorylation. Interestingly, 2‐AAA was reported to be significantly reduced in muscle of Ndufs4 knockout mice, confirming a biological relationship 49 and further highlighting a relationship between altered levels of 2‐AAA and mitochondrial dysfunction. Other genes implicated in the analysis included TRAML1, a translocation associated membrane protein with limited functional characterization, which has been associated with obesity. 50 The mechanistic relevance of this gene to 2‐AAA remains to be determined.
We also identified candidate associations with SNPs mapping in an intergenic region near FGF20 and MSR1. FGF20 (fibroblast growth factor 20) is involved in multiple cellular processes and has been associated with Parkinson’s disease. 51 Of note, 2‐AAA has also been implicated in Parkinson’s disease. 52 Macrophage scavenger receptor 1 (MSR1), also known as class A scavenger receptor (SR‐A), plays a role in macrophage endocytosis and uptake of cholesterol esters 53 and has been implicated in several disease processes, including atherosclerosis. 54 MSR1 has been associated with both lipoprotein and glucose metabolism, 55 highlighting potential mechanistic links underlying the association between 2‐AAA and both type 2 diabetes and atherosclerosis. Given our findings linking 2‐AAA to HDL cholesterol, this is a particularly interesting avenue for future mechanistic interrogation.
Based on MR analysis using a risk score for 2‐AAA derived from the meta‐analysis, we confirmed a relationship with fasting insulin 2 and identified a suggestive association between elevated 2‐AAA and reduced HDL. There was no association between a genetic risk score for HDL and 2‐AAA, suggesting that high 2‐AAA may lead to reduced HDL cholesterol rather than the inverse. However, this remains to be probed experimentally. Further, although insulin itself associates with HDL, our data suggest that the association between 2‐AAA and HDL is partially independent of modulation of insulin levels. Although there remains uncertainty surrounding the relative importance of HDL levels compared with HDL function as a causal factor in disease development, 56 , 57 HDL cholesterol metabolism is thought to play a crucial role in both diabetes and atherosclerosis. 58 , 59 We validated the predicted relationship through direct measurement and confirmed a negative correlation between 2‐AAA and HDL. Although this does not establish causality, taken together these data suggest that elevated 2‐AAA in individuals may contribute to cardiometabolic risk through modulation of HDL cholesterol levels. Further mechanistic studies are required to probe this relationship.
Our study had a number of strengths but also some limitations. Despite being the largest GWAS meta‐analysis ever conducted of plasma 2‐AAA, we are likely underpowered to detect genetic signals at genome‐wide significance. The 2‐AAA measurements and genotyping in the different cohorts were conducted at different times and on different platforms, limiting our ability to fully harmonize analyses or directly compare measurements across cohorts. Previous metabolite GWAS often did not include measurement of 2‐AAA, limiting our ability to further meta‐analyze our results with published studies. However, review of suggestive loci confirmed an association between variation in DHTKD1 and 2‐AAA, which is supported by multiple independent lines of evidence and could be considered as a positive control. Thus, we consider that our suggestive loci, including MSR1 and NDUFS4, may be biologically relevant in mediating the relationship between 2‐AAA and disease. Future larger studies are required to confirm this. Because the GWAS did not identify strong SNP associations, we constructed 2‐AAA genetic instruments using subsignificant SNPs, which can introduce bias to an MR analysis, including false positive associations. Given the diverse representation of ancestries and platforms and small sample sizes, we could not accurately determine how much of the 2‐AAA variability was captured by the 2‐AAA genetic instruments. Thus, the MR genetic instrument was susceptible to violating the first assumption of MR (instrument validity) and weak instrument bias. Further, there remain other limitations, common to MR studies, such as potential for pleiotropy, heterogeneity, reverse causation, or presence of confounders, which can lead to inaccurate inferences and cannot be fully excluded. Our MR analysis identified a relationship between 2‐AAA and HDL cholesterol. Given the limitations of the MR genetic instrument, we validated this experimentally; however, we caution that neither the MR or the correlation analyses establish causality. Further studies are required to establish the mechanistic basis of the association between 2‐AAA and HDL. Our analysis included individuals of European, African, and Asian ancestry. Non‐White individuals have increased risk of diabetes given the same risk factors as White individuals. 60 Studying non‐White populations is important, both to address health and research disparities in understudied populations and because studying individuals with diverse genetic ancestry improves ability to detect causal genetic variation. 61
In conclusion, genetic analysis of plasma 2‐AAA revealed several loci of potential relevance to cardiometabolic disease. Further, our data predicted an association between 2‐AAA and HDL cholesterol, which we confirmed through direct measurement. Further in‐depth mechanistic studies are required; however, these data provide a link between 2‐AAA and lipoprotein metabolism and may represent a mechanism whereby elevated 2‐AAA associates with future risk of T2D and atherosclerosis.
Sources of Funding
This work was funded by the National Institutes of Health (NIH) (R01 DK117144 and R01 HL142856 to Ferguson). Gamboa was supported by NIH (K23 DK100533). Data sets used for the analyses described were obtained from Vanderbilt University Medical Center’s BioVU, which is supported by numerous sources: institutional funding, private agencies, and federal grants. These include the NIH funded Shared Instrumentation Grant S10OD017985 and S10RR025141; and Clinical and Translational Science Awards grants UL1TR002243, UL1TR000445, and UL1RR024975. Genomic data are also supported by investigator‐led projects that include NIH (U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, and R01HD074711); and additional funding sources listed at https://victr.vumc.org/biovu‐funding/. The Shanghai Women’s Health Study (UM1 CA182910) and Shanghai Men’s Health Study (UM1 CA173640) are supported by the National Institutes of Health. The Jackson Heart Study is supported by and conducted in collaboration with Jackson State University (HHSN268201800013I), Tougaloo College (HHSN268201800014I), the Mississippi State Department of Health (HHSN268201800015I), and the University of Mississippi Medical Center (HHSN268201800010I, HHSN268201800011I, and HHSN268201800012I) contracts from the National Heart, Lung, and Blood Institute (NHLBI) and the National Institute on Minority Health and Health Disparities (NIMHD). The views expressed in this article are those of the authors and do not necessarily represent the views of the NHLBI; the NIH; or the US Department of Health and Human Services. Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the NIH under award numbers U01 DK048489, U01 DK048339, U01 DK048377, U01 DK048349, U01 DK048381, U01 DK048468, U01 DK048434, U01 DK048485, U01 DK048375, U01 DK048514, U01 DK048437, U01 DK048413, U01 DK048411, U01 DK048406, U01 DK048380, U01 DK048397, U01 DK048412, U01 DK048404, U01 DK048387, U01 DK048407, U01 DK048443, and U01 DK048400; by providing funding during DPP (Diabetes Prevention Program) and DPPOS (Diabetes Prevention Program Outcomes Study) to the clinical centers and the Coordinating Center for the design and conduct of the study; and collection, management, analysis, and interpretation of the data. Funding was also provided by the National Institute of Child Health and Human Development, the National Institute on Aging, the National Eye Institute, the NHLBI, the National Cancer Institute, the Office of Research on Women’s Health, the NIMHD, the Centers for Disease Control and Prevention, and the American Diabetes Association. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The Southwestern American Indian Centers were supported directly by the NIDDK, including its Intramural Research Program, and the Indian Health Service. The General Clinical Research Center Program, National Center for Research Resources, and the Department of Veterans Affairs supported data collection at many of the clinical centers. Merck KGaA provided medication for DPPOS. DPP/DPPOS have also received donated materials, equipment, or medicines for concomitant conditions from Bristol‐Myers Squibb, Parke‐Davis, and LifeScan Inc., Health O Meter, Hoechst Marion Roussel, Inc., Merck‐Medco Managed Care, Inc., Merck and Co., Nike Sports Marketing, Slim Fast Foods Co., and Quaker Oats Co., McKesson BioServices Corp., Matthews Media Group, Inc., and the Henry M. Jackson Foundation provided support services under subcontract with the DPP Coordinating Center. The sponsor of this study was represented on the steering committee and played a part in study design, how the study was done, and publication. All authors in the writing group had access to all data. The opinions expressed are those of the study group and do not necessarily reflect the views of the funding agencies. A complete list of centers, investigators, and staff can be found in the Appendix.
Disclosures
None.
Supporting information
Appendix S1
Table S1, Tables S2–S7
Figure S1
Acknowledgments
The DPP Research Group gratefully acknowledges the commitment and dedication of the participants of the DPP and DPPOS. The authors also thank the staffs and participants of the JHS.
For Sources of Funding and Disclosures, see page 10.
REFERENCES
- 1. CDC . National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States, 2014. US Department of Health and Human Services; 2014. [Google Scholar]
- 2. Wang TJ, Ngo D, Psychogios N, Dejam A, Larson MG, Vasan RS, Ghorbani A, O’Sullivan J, Cheng S, Rhee EP, et al. 2‐Aminoadipic acid is a biomarker for diabetes risk. J Clin Invest. 2013;123:4309–4317. doi: 10.1172/JCI64801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Saremi A, Howell S, Schwenke DC, Bahn G, Beisswenger PJ, Reaven PD; Vadt Investigators . Advanced glycation end products, oxidation products, and the extent of atherosclerosis during the VA Diabetes Trial and Follow‐up Study. Diabetes Care. 2017;40:591–598. doi: 10.2337/dc16-1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rhee E, Ho J, Chen M‐H, Shen D, Cheng S, Larson M, Ghorbani A, Shi XU, Helenius I, O’Donnell C, et al. A genome‐wide association study of the human metabolome in a community‐based cohort. Cell Metab. 2013;18:130–143. doi: 10.1016/j.cmet.2013.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Taylor HA. The Jackson Heart Study: an overview. Ethn Dis. 2005;15:S6‐1‐3. [PubMed] [Google Scholar]
- 6. Wilson JG, Rotimi CN, Ekunwe L, Royal CDM, Crump ME, Wyatt SB, Steffes MW, Adeyemo A, Zhou J, Taylor HA, et al. Study design for genetic analysis in the Jackson Heart Study. Ethn Dis. 2005;15:S6‐30‐37. [PubMed] [Google Scholar]
- 7. Shu X‐O, Li H, Yang G, Gao J, Cai H, Takata Y, Zheng W, Xiang Y‐B. Cohort profile: the Shanghai Men’s Health Study. Int J Epidemiol. 2015;44:810–818. doi: 10.1093/ije/dyv013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zheng W, Chow W‐H, Yang G, Jin F, Rothman N, Blair A, Li H‐L, Wen W, Ji B‐T, Li QI, et al. The Shanghai Women’s Health Study: rationale, study design, and baseline characteristics. Am J Epidemiol. 2005;162:1123–1131. doi: 10.1093/aje/kwi322 [DOI] [PubMed] [Google Scholar]
- 9. Yu D, Moore SC, Matthews CE, Xiang Y‐B, Zhang X, Gao Y‐T, Zheng W, Shu X‐O. Plasma metabolomic profiles in association with type 2 diabetes risk and prevalence in Chinese adults. Metabolomics. 2016;12:3. doi: 10.1007/s11306-015-0890-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Knowler WC, Barrett‐Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, Nathan DM; Diabetes Prevention Program Research Group . Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Walford GA, Ma Y, Clish C, Florez JC, Wang TJ, Gerszten RE; Diabetes Prevention Program Research G . Metabolite profiles of diabetes incidence and intervention response in the diabetes prevention program. Diabetes. 2016;65:1424–1433. doi: 10.2337/db15-1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rhee EP, Cheng S, Larson MG, Walford GA, Lewis GD, McCabe E, Yang E, Farrell L, Fox CS, O’Donnell CJ, et al. Lipid profiling identifies a triacylglycerol signature of insulin resistance and improves diabetes prediction in humans. J Clin Invest. 2011;121:1402–1411. doi: 10.1172/JCI44442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, et al. Metabolite profiles and the risk of developing diabetes. Nat Med. 2011;17:448–453. doi: 10.1038/nm.2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wilk JB, Chen T‐H, Gottlieb DJ, Walter RE, Nagle MW, Brandler BJ, Myers RH, Borecki IB, Silverman EK, Weiss ST, et al. A genome‐wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009;5:e1000429. doi: 10.1371/journal.pgen.1000429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome‐wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847 [DOI] [PubMed] [Google Scholar]
- 16. Raffield LM, Ellis J, Olson NC, Duan Q, Li J, Durda P, Pankratz N, Keating BJ, Wassel CL, Cushman M, et al. Genome‐wide association study of homocysteine in African Americans from the Jackson Heart Study, the Multi‐Ethnic Study of Atherosclerosis, and the Coronary Artery Risk in Young Adults study. J Hum Genet. 2018;63:327–337. doi: 10.1038/s10038-017-0384-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS. A high‐performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28:3326–3328. doi: 10.1093/bioinformatics/bts606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Delaneau O, Marchini J, Zagury J‐F. A linear complexity phasing method for thousands of genomes. Nat Methods. 2011;9:179–181. doi: 10.1038/nmeth.1785 [DOI] [PubMed] [Google Scholar]
- 19. Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome‐wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang J, Zaitlen NA, Goddard ME, Visscher PM, Price AL. Advantages and pitfalls in the application of mixed‐model association methods. Nat Genet. 2014;46:100–106. doi: 10.1038/ng.2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roden D, Pulley J, Basford M, Bernard G, Clayton E, Balser J, Masys D. Development of a large‐scale de‐identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther. 2008;84:362–369. doi: 10.1038/clpt.2008.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ruderfer DM, Walsh CG, Aguirre MW, Tanigawa Y, Ribeiro JD, Franklin JC, Rivas MA. Significant shared heritability underlies suicide attempt and clinically predicted probability of attempting suicide. Mol Psychiatry. 2020;25:2422–2430. doi: 10.1038/s41380-018-0326-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Medicine. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang L, Zheng Z, Qi T, Kemper KE, Wray NR, Visscher PM, Yang J. A resource‐efficient tool for mixed model association analysis of large‐scale data. Nat Genet. 2019;51:1749–1755. doi: 10.1038/s41588-019-0530-8 [DOI] [PubMed] [Google Scholar]
- 27. Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Turcot V, Lu Y, Highland HM, Schurmann C, Justice AE, Fine RS, Bradfield JP, Esko T, Giri A, Graff M, et al. Protein‐altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat Genet. 2018;50:26–41. doi: 10.1038/s41588-017-0011-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shungin D, Winkler TW, Croteau‐Chonka DC, Ferreira T, Locke AE, Mägi R, Strawbridge RJ, Pers TH, Fischer K, Justice AE, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518:187–196. doi: 10.1038/nature14132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283. doi: 10.1038/ng.2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. DIAGRAM Consortium, GIANT Consortium, Global BPgen Consortium, Anders Hamsten on behalf of Procardis Consortium, the MAGIC Investigators , Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia‐Naji N, Gloyn AL, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–116. doi: 10.1038/ng.520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. Fine‐mapping type 2 diabetes loci to single‐variant resolution using high‐density imputation and islet‐specific epigenome maps. Nat Genet. 2018;50:1505–1513. doi: 10.1038/s41588-018-0241-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nikpay M, Goel A, Won H‐H, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, et al. A comprehensive 1,000 Genomes‐based genome‐wide association meta‐analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ligthart S, Vaez A, Võsa U, Stathopoulou MG, Vries PS, Prins BP, Van der Most PJ, Tanaka T, Naderi E, Rose LM, et al. Genome analyses of >200,000 individuals identify 58 loci for chronic inflammation and pathways that link inflammation and complex disorders. Am J Hum Genet. 2018;103:691–706. doi: 10.1016/j.ajhg.2018.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bulik‐Sullivan BK, Loh P‐R, Finucane HK, Ripke S, Yang J; Schizophrenia Working Group of the Psychiatric Genomics Consortium , Patterson N, Daly MJ, Price AL, Neale BM. LD Score regression distinguishes confounding from polygenicity in genome‐wide association studies. Nat Genet. 2015;47:291–295. doi: 10.1038/ng.3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Carroll RJ, Bastarache L, Denny JC. R PheWAS: data analysis and plotting tools for phenome‐wide association studies in the R environment. Bioinformatics. 2014;30:2375–2376. doi: 10.1093/bioinformatics/btu197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Skrivankova VW, Richmond RC, Woolf BAR, Davies NM, Swanson SA, VanderWeele TJ, Timpson NJ, Higgins JPT, Dimou N, Langenberg C, et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomisation (STROBE‐MR): explanation and elaboration. BMJ. 2021;375:n2233. doi: 10.1136/bmj.n2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yavorska OO, Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol. 2017;46:1734–1739. doi: 10.1093/ije/dyx034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gamboa JL, Roshanravan B, Towse T, Keller CA, Falck AM, Yu C, Frontera WR, Brown NJ, Ikizler TA. Skeletal muscle mitochondrial dysfunction is present in patients with CKD before initiation of maintenance hemodialysis. Clin J Am Soc Nephrol. 2020;15:926–936. doi: 10.2215/CJN.10320819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Beckman JA, Hu J‐R, Huang S, Farber‐Eger E, Wells QS, Wang TJ, Gerszten RE, Ferguson JF. Metabolomics reveals the impact of Type 2 diabetes on local muscle and vascular responses to ischemic stress. Clin Sci (Lond). 2020;134:2369–2379. doi: 10.1042/CS20191227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Danhauser K, Sauer S, Haack T, Wieland T, Staufner C, Graf E, Zschocke J, Strom T, Traub T, Okun Jürgen G, et al. DHTKD1 mutations cause 2‐aminoadipic and 2‐oxoadipic aciduria. Am J Hum Genet. 2012;91:1082–1087. doi: 10.1016/j.ajhg.2012.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu Y, Williams E, Dubuis S, Mottis A, Jovaisaite V, Houten S, Argmann C, Faridi P, Wolski W, Kutalik Z, et al. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell. 2014;158:1415–1430. doi: 10.1016/j.cell.2014.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Long T, Hicks M, Yu H‐C, Biggs WH, Kirkness EF, Menni C, Zierer J, Small KS, Mangino M, Messier H, et al. Whole‐genome sequencing identifies common‐to‐rare variants associated with human blood metabolites. Nat Genet. 2017;49:568–578. doi: 10.1038/ng.3809 [DOI] [PubMed] [Google Scholar]
- 44. Mosley JD, Benson MD, Smith JG, Melander O, Ngo D, Shaffer CM, Ferguson JF, Herzig MS, McCarty CA, Chute CG, et al. Probing the virtual proteome to identify novel disease biomarkers. Circulation. 2018;138:2469–2481. doi: 10.1161/CIRCULATIONAHA.118.036063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hoffmann TJ, Theusch E, Haldar T, Ranatunga DK, Jorgenson E, Medina MW, Kvale MN, Kwok P‐Y, Schaefer C, Krauss RM, et al. A large electronic‐health‐record‐based genome‐wide study of serum lipids. Nat Genet. 2018;50:401–413. doi: 10.1038/s41588-018-0064-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Biagosch C, Ediga R, Hensler SV, Faerberboeck M, Kuehn R, Wurst W, Meitinger T, Kolker S, Sauer S, Prokisch H. Elevated glutaric acid levels in Dhtkd1‐/Gcdh‐ double knockout mice challenge our current understanding of lysine metabolism. Biochim Biophys Acta Mol Basis Dis. 2017;1863:2220–2228. doi: 10.1016/j.bbadis.2017.05.018 [DOI] [PubMed] [Google Scholar]
- 47. Stiles AR, Venturoni L, Mucci G, Elbalalesy N, Woontner M, Goodman S, Abdenur JE. New cases of DHTKD1 mutations in patients with 2‐ketoadipic aciduria. JIMD Rep. 2016;25:15–19. doi: 10.1007/8904_2015_462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu W‐Y, Gu M‐M, Sun L‐H, Guo W‐T, Zhu H‐B, Ma J‐F, Yuan W‐T, Kuang Y, Ji B‐J, Wu X‐L, et al. A nonsense mutation in DHTKD1 causes Charcot‐Marie‐Tooth disease type 2 in a large Chinese pedigree. Am J Hum Genet. 2012;91:1088–1094. doi: 10.1016/j.ajhg.2012.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Terburgh K, Lindeque Z, Mason S, van der Westhuizen F, Louw R. Metabolomics of Ndufs4‐/‐ skeletal muscle: adaptive mechanisms converge at the ubiquinone‐cycle. Biochim Biophys Acta Mol Basis Dis. 2019;1865:98–106. doi: 10.1016/j.bbadis.2018.10.034 [DOI] [PubMed] [Google Scholar]
- 50. Cotsapas C, Speliotes EK, Hatoum IJ, Greenawalt DM, Dobrin R, Lum PY, Suver C, Chudin E, Kemp D, Reitman M, et al. Common body mass index‐associated variants confer risk of extreme obesity. Hum Mol Genet. 2009;18:3502–3507. doi: 10.1093/hmg/ddp292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang X, Sun X, Zhang X, Li H, Xie A. Quantitative assessment of the effect of FGF20 rs12720208 variant on the risk of Parkinson’s disease: a meta‐analysis. Neurol Res. 2017;39:374–380. doi: 10.1080/01616412.2017.1286542 [DOI] [PubMed] [Google Scholar]
- 52. O’Neill E, Chiara Goisis R, Haverty R, Harkin A. L‐alpha‐aminoadipic acid restricts dopaminergic neurodegeneration and motor deficits in an inflammatory model of Parkinson’s disease in male rats. J Neurosci Res. 2019;97:804–816. doi: 10.1002/jnr.24420 [DOI] [PubMed] [Google Scholar]
- 53. Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, Rhee JS, Silverstein R, Hoff HF, Freeman MW. Scavenger receptors class A‐I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. 2002;277:49982–49988. doi: 10.1074/jbc.M209649200 [DOI] [PubMed] [Google Scholar]
- 54. Durst R, Neumark Y, Meiner V, Friedlander Y, Sharon N, Polak A, Beeri R, Danenberg H, Erez G, Spitzen S, et al. Increased risk for atherosclerosis of various macrophage scavenger receptor 1 alleles. Genet Test Mol Biomarkers. 2009;13:583–587. doi: 10.1089/gtmb.2009.0048 [DOI] [PubMed] [Google Scholar]
- 55. Fukuhara‐Takaki K, Sakai M, Sakamoto Y, Takeya M, Horiuchi S. Expression of class A scavenger receptor is enhanced by high glucose in vitro and under diabetic conditions in vivo: one mechanism for an increased rate of atherosclerosis in diabetes. J Biol Chem. 2005;280:3355–3364. doi: 10.1074/jbc.M408715200 [DOI] [PubMed] [Google Scholar]
- 56. Tuteja S, Rader DJ. High‐density lipoproteins in the prevention of cardiovascular disease: changing the paradigm. Clin Pharmacol Ther. 2014;96:48–56. doi: 10.1038/clpt.2014.79 [DOI] [PubMed] [Google Scholar]
- 57. Ouimet M, Barrett TJ, Fisher EA. HDL and reverse cholesterol transport. Circ Res. 2019;124:1505–1518. doi: 10.1161/CIRCRESAHA.119.312617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Goldberg IJ. Diabetic dyslipidemia: causes and consequences. J Clin Endocrinol Metab. 2001;86:965–971. doi: 10.1210/jcem.86.3.7304 [DOI] [PubMed] [Google Scholar]
- 59. Wu L, Parhofer KG. Diabetic dyslipidemia. Metabolism. 2014;63:1469–1479. doi: 10.1016/j.metabol.2014.08.010 [DOI] [PubMed] [Google Scholar]
- 60. Gujral UP, Vittinghoff E, Mongraw‐Chaffin M, Vaidya D, Kandula NR, Allison M, Carr J, Liu K, Narayan KM, Kanaya AM. Cardiometabolic abnormalities among normal‐weight persons from five racial/ethnic groups in the United States: a cross‐sectional analysis of two cohort studies. Ann Intern Med. 2017. 166:628. doi: 10.7326/M16-1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li YR, Keating BJ. Trans‐ethnic genome‐wide association studies: advantages and challenges of mapping in diverse populations. Genome Med. 2014;6:91. doi: 10.1186/s13073-014-0091-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Table S1, Tables S2–S7
Figure S1