

ABSTRACT
The concept of a holobiont, a host organism and its associated microbial communities, encapsulates the vital role the microbiome plays in the normal functioning of its host. Parasitic infections can disrupt this relationship, leading to dysbiosis. However, it is increasingly recognized that multicellular parasites are themselves holobionts. Intestinal parasites share space with the host gut microbiome, creating a system of nested microbiomes within the primary host. However, how the parasite, as a holobiont, interacts with the host holobiont remains unclear, as do the consequences of these interactions for host health. Here, we used 16S amplicon and shotgun metagenomics sequencing to characterize the microbiome of the intestinal cestode Eubothrium and its effect on the gut microbiome of its primary host, Atlantic salmon. Our results indicate that cestode infection is associated with salmon gut dysbiosis by acting as a selective force benefiting putative pathogens and potentially introducing novel bacterial species to the host. Our results suggest that parasitic cestodes may themselves be holobionts nested within the microbial community of their holobiont host, emphasizing the importance of also considering microbes associated with parasites when studying intestinal parasitic infections.
KEYWORDS: Atlantic salmon, Cestodes, gut dysbiosis, holobionts, host-parasite-microbiome interactions, intestinal microbiomes
INTRODUCTION
Microorganisms have been found to colonize the vast majority of multicellular life (1). It is now well established that the microbiome can modulate host phenotype (2, 3). The host immune system constantly interacts with the microbiome (4, 5), and there is mounting evidence that the underlying host genotype can influence microbiome composition (6, 7). Thus, emerging hologenomic theory posits that the genomes of the host and all its associated microorganisms are subject to coevolutionary forces and should be viewed as a “holobiont” rather than as independently evolving organisms (8). The dynamic interactions between a host and its microbiome have been most extensively studied in the gut, where the microbiome has been found to influence host adaptation (9–11), gene expression and regulation (12, 13), immune system maturation and modulation (14), maintenance of the gut mucosal barrier (15), and protection from pathogens (16, 17). Dysbiosis of the gut, defined as any change to the resident commensal gut microbiome relative to the community found in healthy individuals (18), is frequently associated with intestinal parasitic infections (19). However, it is not always clear whether dysbiosis increases susceptibility to parasite infection (20), the parasite infection triggers dysbiosis (21, 22), or a combination of both occurs (23).
There is growing recognition that multicellular parasites are themselves holobionts (24, 25). There are several well-documented cases of parasites hosting bacterial endosymbionts that are critical for parasite survival, like the dependence of filarial nematodes on their Wolbachia endosymbionts (26). Parasites thus represent an interesting case of one holobiont (the parasite) colonizing another holobiont (the parasite’s vertebrate host). This nested scenario is particularly true of intestinal parasites, which share physical space with the gut microbiome of the vertebrate host. In this case, the parasite-associated and host-associated microbiomes can be thought of as a continuum, transitioning from host-associated microbes residing in the host gut mucosa into host resident and transient microbes in the gut content, through to a combination of parasite and host microbes on or near the parasite body surface, and ending with internal parasite-specific microbes. While interactions between intestinal parasites, the host gut microbiome, and the host immune system have been characterized in previous studies (reviewed in Reynolds et al. [27]), few studies have taken into account how parasite-associated microbes might interact with the host and its associated microbes (24).
We exploited a unique, semicontrolled system to characterize microbial communities in an intestinal parasite, a cestode (or tapeworm) of the genus Eubothrium, and its definitive vertebrate host, Atlantic salmon (Salmo salar). Like most Platyhelminthes, members of Eubothrium have complex life cycles with intermediate hosts that are food sources of salmon (copepods and small pelagic fish), where the larvae develop in the body cavity, before reaching maturity in the salmon intestine after ingestion (28). Cestodes are responsible for significant health and economic burden in clinical, agriculture, and aquaculture settings (29–31). Eubothrium specifically impacts the salmon aquaculture industry, as intestinal infection of adult fish decreases growth and, in extreme cases, can be fatal (32, 33). We studied farmed adult salmon that were naturally infected with Eubothrium while housed in open seawater pens off the coast of southwestern Norway. This experimental setup allowed us to control many abiotic aspects that can affect salmon microbiome composition (e.g., feed, aquatic environment, genetic background) while still exposing the salmon to natural sources of microbes and pathogens. Cestodes lack a digestive tract and mouth, instead absorbing nutrients through their skin surface, the tegument; thus, microbes can only be associated with this tegument, or the internal body cavity. We therefore characterized the microbial communities sampled from the salmon-cestode interaction space, including the resident host microbiota of the salmon gut mucosa (“salmon gut”), the microbes loosely attached to the cestode tegument (disrupted with a phosphate-buffered saline [PBS] wash; “cestode wash”), and those associated with the cestode (whole body post-wash, including body cavity and tegument; “cestode body”). We also investigated dysbiosis in the salmon gut microbiome, comparing uninfected salmon to salmon with various levels of cestode infection. Finally, we shotgun sequenced, assembled, and functionally characterized the cestode body microbiome, generating genomes of two novel cestode-associated Mycoplasma bacteria. Our results suggest that parasitic cestodes should be considered holobionts with an associated microbiome when studying effects of intestinal parasitic infections.
RESULTS AND DISCUSSION
Cestode infection is associated with salmon gut dysbiosis.
Cestode infection was observed in 378 (81.6%) of 463 adult harvest-ready salmon (Fig. S1 in the supplemental material). The degree of cestode infection was scored using an in-house semiquantitative veterinary index ranging from 0 (no observed cestode) to 3 (excessive [>3] numbers of cestodes impeding the passage of feed along the gastrointestinal tract) (Fig. S1). There was strong evidence that salmon size (gutted weight) decreased as the cestode index increased (Fig. S2), supporting results from earlier studies demonstrating negative growth effects in salmon associated with cestode infection (32, 33). Thirty salmon were selected for the cestode-related microbiome investigations, nine each with a cestode index of 0, 1, and 2 and three with a cestode index of 3. From these 30 salmon, we generated 1,172 amplicon sequence variants (ASVs) of the 16S rRNA gene (ASVs labeled seq1 to seq1172) from 30 host gut mucosal scrapings, 17 cestode wash samples, and 21 cestode body samples (Fig. 1A), of which 116 ASVs were retained after quality control.
FIG 1.
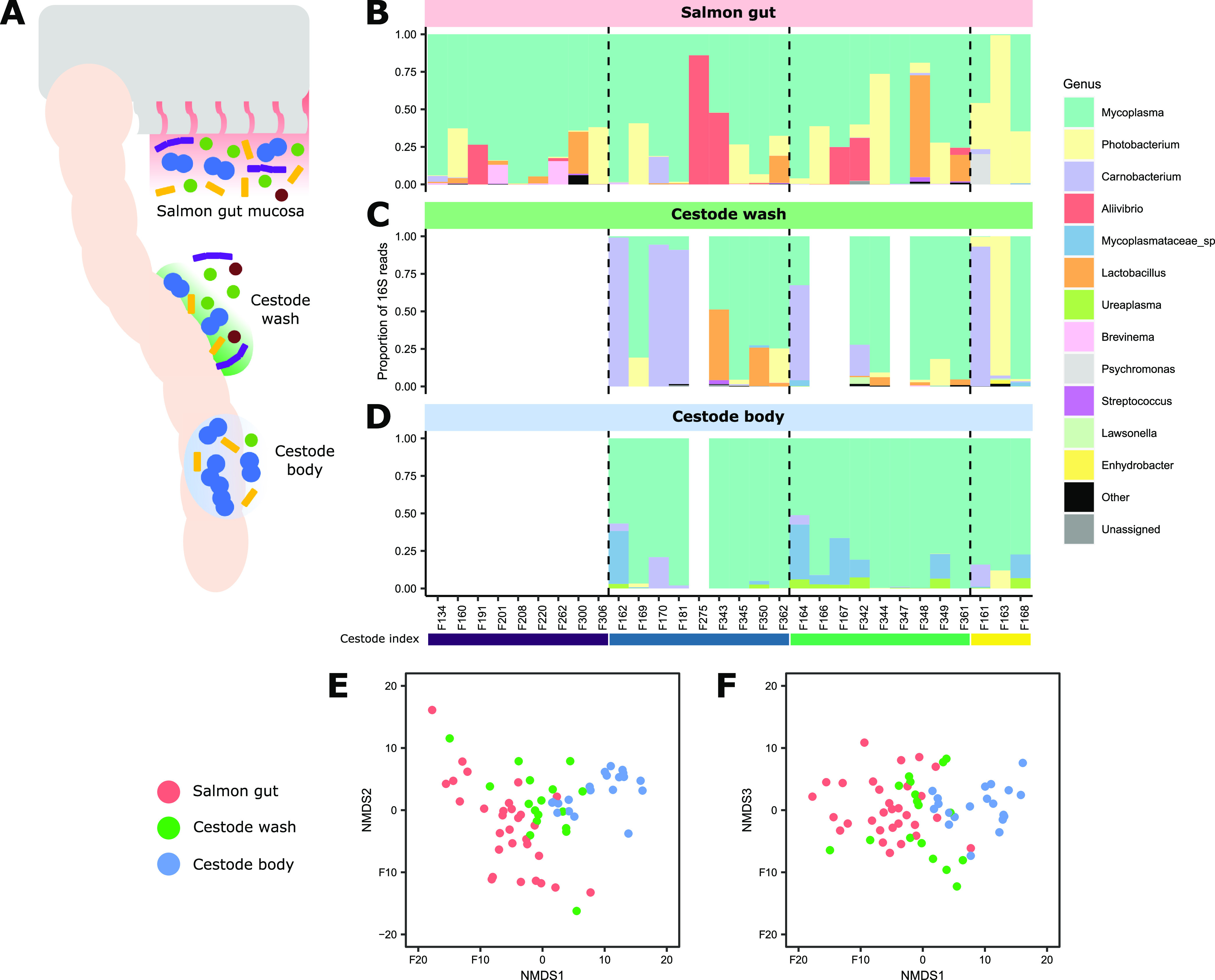
(A) Schematic of the three sampling sites, including a scrape of the salmon gut mucosa (red background), a PBS wash of the cestode tegument containing loosely attached microbes (green background), and the cestode body containing strongly attached or internal microbes (blue background). (B to D) Genus-level microbial composition of the salmon gut mucosa (B), cestode wash (C), and cestode body (D). The top 12 most abundant bacterial genera are colored, with the remainder grouped into “other.” ASVs that could not be assigned taxonomy at the genus level are grouped into “unassigned.” Individuals are ordered by cestode index (purple, 0; blue, 1; green, 2; yellow, 3), separated by the dashed vertical lines. (E and F) NMDS (k = 3) of Euclidean distances of ASV CLR-normalized abundances in samples from the salmon gut mucosa, cestode wash, and cestode body. Each point represents one sample from an individual, colored by sample type. NMDS stress, 0.117.
Distribution of cestode indexes across fish housed in two sea pens, each fed a different commercial diet (n = 253 for feed 1 and n = 210 for feed 2). The semiquantitative cestode index is illustrated on the right, where 0 is no observed cestode, 1 is 1 to 3 visible cestodes, 2 is >3 cestodes but normal functioning of the gastrointestinal tract, and 3 is excessive numbers of cestodes that are likely impeding the passage of feed along the gastrointestinal tract. Download FIG S1, PDF file, 0.1 MB (129.9KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Weight of individuals decreases as cestode index increases. (a) Violin plot of gutted weight of salmon (kg) by cestode index in the entire cohort (n = 463). Horizontal lines represent the median. There was strong evidence that gutted weight decreased as cestode infection increased (F5,456 = 19.5, P < 0.001, R2 = 0.17). (b) Box plot of gutted weight of salmon (kg) by cestode index in the 30 individuals selected for sequencing of cestode-related microbiomes in this study. There was some evidence that more heavily parasitized fish were smaller in this subset of individuals (F5,24 = 1.31, P = 0.29, R2 = 0.051). Download FIG S2, PDF file, 0.05 MB (53.9KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Overall, the salmon gut mucosa was dominated by the genus Mycoplasma (Fig. 1B), a known commensal of the salmon intestinal microbiome (9, 34–38). However, there was evidence that the gut microbiota differed between uninfected and infected individuals. The cestode index accounted for 15.1% of the variation among individuals (Fig. S3), and there was weak evidence that alpha diversity in the gut mucosa increased as the cestode index increased (richness, P = 0.11; Shannon, P = 0.18) (Fig. S4). There was also weak evidence (false-discovery rate [FDR] < 0.2) that the relative abundance of Mycoplasma and the probable commensal Brevinema (37) decreased with increasing cestode infection, while the relative abundance of the putative pathobionts Photobacterium (39, 40), Aliivibrio (38, 40, 41), and Carnobacterium (42) increased (Fig. S5; Table S1). Photobacterium and Aliivibrio taxa in particular have been implicated in skin infections and septicemia of fish (38–41) and have been found at increased abundances in the guts of farmed Atlantic salmon during bacterial infections (38, 43). Our results therefore connect cestode infection to salmon gut dysbiosis, consistent with studies in other systems demonstrating altered host gut microbial communities during cestode infection (21, 44, 45).
Associations between ASV or genus abundance and cestode index or sample type, as produced by MaAsLin2. Results with FDRs of <0.25 are included and those with FDRs of <0.05 are highlighted in gray. Download Table S1, PDF file, 0.2 MB (171.7KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
NMDS (k = 2) of Euclidean distances of ASV CLR-normalized abundances in the salmon fish gut mucosa. Each point represents one individual, colored by cestode index. NMDS stress, 0.150. Cestode index accounted for 15.1% of the variation among individuals (PERMANOVA, F3,26 = 1.54, P = 0.035). Download FIG S3, PDF file, 0.01 MB (8.3KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Alpha diversity of each sample type from each individual, faceted by cestode index. (a) There was weak evidence that species richness (number of ASVs) increased as cestode index increased (F4,25 = 1.53, Plinear = 0.11, and R2 = 0.07 after controlling for sequencing effort). When considering all individuals irrespective of cestode index, there was strong evidence that alpha diversity decreased in the cestode body compared to the salmon gut mucosa (F3,63 = 6.04, P = 0.0021, and R2 = 0.17 after controlling for sequencing effort). (b) There was also weak evidence that Shannon diversity (calculated using a Hill numbers framework) increased with cestode index (F4,25 = 0.93, Plinear = 0.18, R2 = −0.009 after controlling for sequencing effort) and decreased in the cestode body compared to the salmon gut (F3,63 = 1.49, P = 0.061, and R2 = 0.022 after controlling for sequencing effort). (c) Venn diagram of ASVs detected in each sample type. The salmon gut microbiota has been separated into ASVs detected in nonparasitized salmon (cestode index = 0) and those in parasitized salmon (cestode index > 0). A total of 45 ASVs were unique to the salmon gut mucosa, irrespective of cestode detection; 31 were shared between salmon gut mucosa and cestode wash, 3 between salmon gut mucosa and cestode body, and 15 between all three sample types. Download FIG S4, PDF file, 0.3 MB (312.5KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Relative abundance (proportion of 16S reads) of taxa in the salmon gut mucosa that differed with cestode infection. (a) Genera that differed with cestode index. There was weak evidence that the relative abundance of Mycoplasma decreased with increasing cestode infection (effect size, −0.349 ± 0.118; FDR, 0.173) and the relative abundance of Photobacterium increased (effect size, 0.375 ± 0.112; FDR, 0.144). (b) Genera that differed by cestode presence. There was weak evidence that Carnobacterium and Aliivibrio had higher abundances in the gut mucosa of salmon infected with cestode (Carnobacterium, effect size, 0.018 ± 0.014; FDR, 0.506; and Aliivibrio, effect size, 0.078 ± 0.92; FDR, 0.779), and the likely commensal Brevinema had higher abundance in the gut mucosa of uninfected salmon (effect size, −0.041 ± 0.014; FDR, 0.248). (c) Relative abundance of ASVs that differed with cestode index (FDR, <0.25; statistics provided in Table S1). Note that relative abundance is shown on different scales within each plot in panel c. Download FIG S5, PDF file, 0.02 MB (25.2KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Nested microbial communities within the host holobiont.
The salmon gut mucosa and cestode body microbiota were clearly differentiated, with the microbiota associated with the cestode wash being intermediate (Fig. 1E and F). Sample type explained 21.4% of the variation among samples (permutational multivariate analysis of variance [PERMANOVA], F2,64 = 8.71, P = 0.001), although the variation within the cestode body was less than within the other two sample types (F2,64 = 6.57, P = 0.005). Alpha diversity decreased in the cestode body compared to the salmon gut mucosa after controlling for sequencing effort (richness, P = 0.0021; Shannon, P = 0.061) (Fig. S4). Twenty-two ASVs were only detected in cestode-derived samples (wash or body) (Fig. S4). The salmon gut mucosa included 45 unique ASVs (10 of which were only detected in uninfected fish), 31 ASVs also present in the cestode wash, and 18 ASVs shared with the cestode body or all three sample types (Fig. S4). The mycoplasmas (Mycoplasma, Ureaplasma, and uncharacterized Mycoplasmataceae species) dominated the cestode body, while the cestode wash predominantly included Mycoplasma, Carnobacterium, or Photobacterium (Fig. 1B; Fig. S6; Table S1). Twenty-three ASVs had increased abundance in one of the three sample types, particularly mycoplasma ASVs in the cestode body, Mycoplasma and Carnobacterium ASVs in the cestode wash, and ASVs related to Photobacterium and Brevinema in the fish gut mucosa (Fig. S6; Table S1).
Relative abundance (proportion of 16S reads) of genera (a) and ASVs (b) that differed with a particular sample type (FDR < 0.2; statistics provided in Table S1). ASVs are ordered by genus (alphabetically). Note that relative abundance is shown on different scales within each plot in panel b. Download FIG S6, PDF file, 0.1 MB (118.6KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We identified one set of mycoplasma ASVs that were associated with the salmon gut mucosa and another set that were associated with the cestode body (Fig. 2; Table S1). The salmon-associated mycoplasma ASVs were closely related to the Mycoplasma 16S sequence(s) described in previous studies of the gut microbiota in Atlantic salmon (9, 38, 46) and other marine fish (47–49) (Fig. 2A). The cestode-associated mycoplasma ASVs clustered into two clades, one sister clade to salmon-associated mycoplasma and another, more divergent clade clustering close to the known fish pathogen Mycoplasma mobile (22, 50, 51) (Fig. 2A, cestode clades 1 and 2, respectively). Within an individual, the salmon mycoplasma clade was frequently at the highest abundance in the gut mucosa and at the lowest abundance in the cestode body, while the opposite pattern was observed for the two cestode-associated clades (Fig. 3).
FIG 2.

(A) Phylogeny of Mycoplasma-related ASVs (Mycoplasma, Ureaplasma, and uncharacterized Mycoplasmataceae species, labeled with “seq” prefix) with reference Mycoplasma species. The maximum-likelihood tree was constructed using RAxML with rapid bootstrapping (1,000 bootstraps). The tree was rooted using the outgroup Bacillus pumilus. The nodes are labeled with the bootstrap values. The Haemoplasma clade of Mycoplasma has been collapsed and truncated for visualization. (B) Presence and relative abundance of Mycoplasma-related ASVs in each sample. Samples are colored by sample type. Within each sample type, samples are ordered by cestode index. Circle area scales with the relative abundance of each ASV. The labels of ASVs in each of the three Mycoplasma clades are highlighted.
FIG 3.
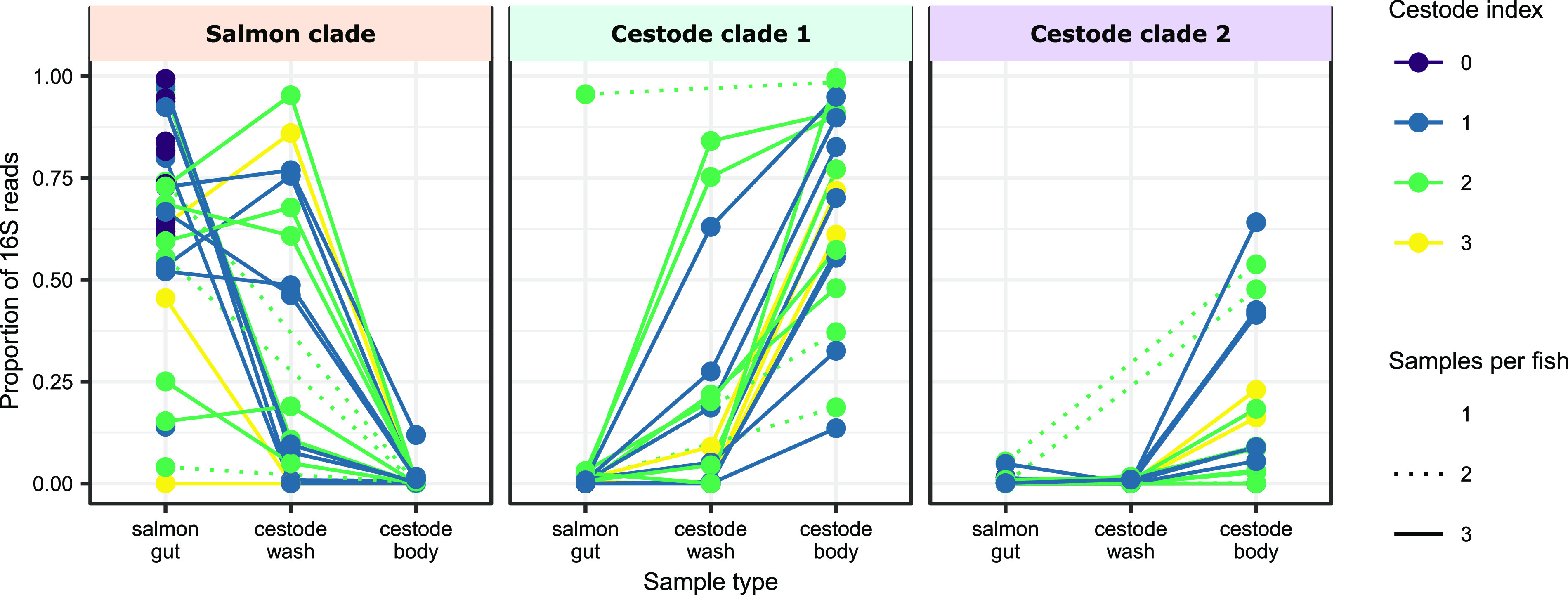
Change in relative abundance of the three Mycoplasma clades (as indicated on the phylogenetic tree in Fig. 2) between each sample type. Solid lines connect each of the 3 samples taken from each individual. Dashed lines indicate individuals where one of the two cestode samples was missing. Individuals are colored by cestode index.
Separation of host- and cestode-specific microbes is difficult due to the possibility of contamination of cestode samples by host microbiota during sampling (52). We rinsed gut content from both the salmon mucosa and the cestode before sampling. The cestode tegument was sampled by vigorous shaking in PBS, forming the cestode wash samples and likely containing a mixture of loosely attached resident and transient microbes from the salmon gut content, as well as potentially cestode-associated microbes, as suggested by the “intermediate” state of the cestode wash microbiota composition compared to the salmon mucosa and cestode body microbiota. The washed cestode body was then sampled to identify microbes either strongly attached to the surface or internal to the cestode. In future studies, a serial washing procedure could be used to sample the cestode tegument and body cavity microbiota to improve the likelihood of identifying cestode-specific microbes after the final wash (52, 53).
Our results suggest that cestodes harbor specific microbes that are not at high abundances in the host gut mucosa, although with our sampling procedure, we cannot determine whether they are external (i.e., on the tegument) or internal (i.e., inside the body cavity). These findings are consistent with studies of other cestodes and related helminths that parasitize fish, which show that a unique cestode microbiota can be observed at each life stage, including during colonization of the body cavity of intermediate hosts (21, 54). However, as adults in the intestine of the definitive host, the microbiota of these helminths tend to be more similar to the microbiota of the host gut environment (52, 54). Our study demonstrated that the microbial communities associated with adult cestodes in our system are dominated by bacteria that are of the same family (the mycoplasmas), but of distinct clades, as those residing in the host gut mucosa. These results suggest that while the three mycoplasma clades can be detected in any of the three sample types investigated, divergent selection seems to occur at the cestode and the salmon mucosa for specific mycoplasma phylotypes, reflecting a nested system of microbial communities.
Identification of different Mycoplasma phylotypes in host and parasite.
To further investigate this mycoplasma strain variation, we generated metagenomes from eight cestode body samples and de novo assembled three Mycoplasma metagenome-assembled genomes (MAGs) (Table S2). Each of these MAGs included a 16S sequence that exactly matched one of the three Mycoplasma ASVs dominating the cestode body amplicon sequencing data set (i.e., seq1 from cestode clade 1, here referred to as “CE_seq1,” and seq7 and seq10 from cestode clade 2, here “CE_seq7” and “CE_seq10,” respectively). Based on the presence of universal marker genes, the CE_seq1 and CE_seq7 MAGs were estimated to be 77.7% and 78.0% complete, respectively, whereas CE_seq10 was 38.0% complete (Table S2). We compared these novel cestode-related MAGs to Mycoplasma MAGs previously recovered from salmonids (9) (“Candidatus Mycoplasma salmoninae salar” from Atlantic salmon, “Candidatus Mycoplasma salmoninae mykiss” from rainbow trout, and “Candidatus Mycoplasma lavaretus” from European whitefish), as well as five closely related Mycoplasma and Ureaplasma references (M. iowae, M. mobile, M. penetrans, U. diversum, and U. urealyticum).
Statistics and ENA accessions of the three Mycoplasma MAGs generated from the cestode body shotgun metagenome. Download Table S2, PDF file, 0.1 MB (131.1KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenomics using concatenated bacterial single-copy genes present in all genomes generally recapitulated the relationships observed in the 16S tree (Fig. 4). CE_seq1 was related to, but distinct from, the salmon-associated “Ca. Mycoplasma salmoninae” MAGs (70.8% average nucleotide identity [ANI] compared to “Ca. Mycoplasma salmoninae salar” and 70.9% to “Ca. Mycoplasma salmoninae mykiss”) and their relatives M. penetrans and M. iowae. Instead, CE_seq1 clustered with the Ureaplasma references. CE_seq7 and CE_seq10 were closely related to “Ca. Mycoplasma lavaretus” (CE_seq7, 81.0% ANI; CE_seq10, 83.9% ANI), with some similarity to the fish pathogen M. mobile. Based on these results, we suggest that CE_seq1 is a new species of Mycoplasma, while CE_seq7 and CE_seq10 may be subspecies of “Ca. Mycoplasma lavaretus.”
FIG 4.

Phylogenomic tree of cestode Mycoplasma MAGs (CE_seq1, CE_seq7, and CE_seq10) and salmonid “Ca. Mycoplasma salmoninae” and “Ca. Mycoplasma lavaretus” MAGs (9) with reference Mycoplasma and Ureaplasma genomes. The maximum-likelihood tree was constructed based on concatenated single-copy core bacterial genes using RAxML with rapid bootstrapping (100 bootstraps). The tree was rooted using the outgroup Bacillus pumilus. The nodes are labeled with the bootstrap values. Genomes included in the functional comparison are in bold. As in Fig. 2, the salmonid Mycoplasma clade is highlighted in orange, cestode-associated clade 1 in cyan, and cestode-associated clade 2 in light purple.
The presence of these novel Mycoplasma species at high abundance in the cestode body microbiota and low abundance in the salmon gut mucosa supports the 16S results (Fig. 3), suggesting that divergent selection is occurring at the cestode and the salmon mucosa for specific Mycoplasma. The phylogenetic position of the cestode MAGs relative to “Ca. Mycoplasma salmoninae” demonstrates the establishment of distinct, yet closely related Mycoplasma phylotypes within their respective hosts, suggesting a specific interaction between salmonid hosts, cestodes, and Mycoplasma species and, thus, potential for coevolutionary relationships.
Novel cestode Mycoplasma genomes suggest holobiont-specific adaptations.
To gain insights into the adaptation of the cestode Mycoplasma to their environment, we functionally characterized the two nearly complete cestode-associated Mycoplasma MAGs and compared them to the three salmonid-associated Mycoplasma MAGs. The mycoplasmas have highly specific adaptations for survival in their respective hosts (55). The cestode has limited metabolic capacity and relies on the host for obtaining nutrients (56); thus, microbial survival inside the cestode body or on the cestode tegument may require specialized adaptations compared to microbes adapted to the salmon gut. Most gene clusters in CE_seq1 were unique (586 of 741, 79.1%), while only 152 (20.5%) were shared with at least one of the salmonid-associated MAGs (Fig. S7), suggesting that CE_seq1 may be more adapted for colonization of cestodes than survival in the salmon gut. In contrast, fewer gene clusters were unique to CE_seq7 (192 of 642, 29.9%); instead, 447 (69.6%) were shared with at least one of the salmonid-associated MAGs, mostly “Ca. Mycoplasma lavaretus” (Fig. S7).
Functional annotation of cestode-associated Mycoplasma MAGs. (a) Venn diagram of gene clusters detected in cestode-associated and salmonid-associated Mycoplasma MAGs. Gene cluster counts unique to CE_seq1 are highlighted in cyan, unique to “Ca. Mycoplasma salmoninae” (“Ca. Mycoplasma salmoninae salar” and/or “Ca. Mycoplasma salmoninae mykiss”) in orange, and those unique or shared between CE_seq7 and “Ca. Mycoplasma lavaretus” in shades of lilac. Consistent with phylogenetic patterns observed in Fig. 4, CE_seq7 and “Ca. Mycoplasma lavaretus” share many gene clusters (361), while CE_seq1 has few gene clusters in common with the “Ca. Mycoplasma salmoninae” MAGs (3 + 71 + 3 = 77), instead containing the largest number of unique gene clusters (586). (b) Probable energy production pathways detected in cestode (CE_seq1 and CE_seq7) and three previously described salmonid (“Ca. Mycoplasma salmoninae salar,” “Ca. Mycoplasma salmoninae mykiss,” and “Ca. Mycoplasma lavaretus”) Mycoplasma MAGs. Glycolysis via the EMP pathway, glycerol metabolism, and the arginine deiminase pathway of arginine degradation is shown. The major steps generating important by-products (ATP or NH3) are shown with dashed gray arrows. For each gene in the pathway, colored boxes indicate that the gene was successfully annotated in the respective MAG (white boxes indicate that the gene was not detected in the respective MAG). Dotted lines represent unknown steps in the pathway. Download FIG S7, PDF file, 0.6 MB (597.8KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Following functional annotation of these gene clusters, we identified differences in key metabolic pathways (Fig. S7). While the three salmonid MAGs and CE_seq7 had a complete glycolysis pathway, which generates ATP through carbohydrate degradation, CE_seq1 lacked many of the core components of glycolysis (although we cannot exclude the possibility that we do not observe these components due to the genome incompleteness [77.7%] of CE_seq1). However, CE_seq1 and the salmonid MAGs had a complete arginine deiminase pathway, which was missing in CE_seq7. This reversible pathway can either use ATP and ammonium to synthesize arginine or produce energy through arginine degradation, and it likely reflects an adaptive trait of salmonid Mycoplasma species to survive in the ammonia-rich salmon gut (9).
CE_seq7 and “Ca. Mycoplasma lavaretus” also had an incomplete glycerol metabolism pathway (Fig. S7), including the presence of the glycerol uptake facilitator protein GlpF, suggesting an ability to acquire environmental glycerol to feed into glycolysis, among other pathways. Glycerol metabolism has been highlighted as an important virulence factor of pathogenic Mycoplasma species, as the oxidation of the downstream product glycerol-3-phosphate by an oxidase (GlpO) can generate reactive oxygen species like hydrogen peroxide that may be detrimental to the host (57). However, the same reaction can also be catalyzed by a dehydrogenase (e.g., gpsA) without generating hydrogen peroxide by-products. In all cestode- and salmonid-associated Mycoplasma MAGs, only the dehydrogenase was identified (Fig. S7); thus, it is unclear whether glycerol metabolism would promote virulence in these strains.
In the phylogenetic analysis, we observed that CE_seq7 shares some similarity to M. mobile, a known fish pathogen infecting the host via the gills (50). Combined with the possibility of glycerol metabolism as a virulence factor, these results raise the question of whether cestode-associated microorganisms like CE_seq7 could contribute to disease states in the salmon. There is evidence in other helminths and protozoan parasites that such obligate parasites can transfer bacterial endosymbionts to their hosts, where the bacteria can cause disease (58–60). Thus, the infectious cestode may act as a vector, introducing microbial species into the salmon gut, which can then interact with the host holobiont and affect host health. Furthermore, the vector of the cestode itself (i.e., the intermediate copepod host), may also play a role in introducing novel microbes to the salmon gut, highlighting the importance of future studies characterizing the host- and parasite-associated microbiomes at each stage of the parasite life cycle (24).
Overall, our functional comparison was limited due to the lack of functional studies of Mycoplasma that colonize nonmammalian hosts (61); thus, we were unable to further predict whether the cestode-associated Mycoplasma MAGs contain genes that aid colonization and/or pathogenesis in either the cestode or the salmon gut. It is also currently unclear whether these MAGs are symbionts with functional roles in the cestode. Thus, further studies focusing on functional interactions between parasite and microbe are needed to determine whether these microbes have a phenotypic impact on the cestode host.
Conclusions.
Parasitic helminths, like cestodes, are highly successful parasites, infecting most vertebrate taxa, including humans. They are responsible for significant health and economic burden in clinical, agriculture, and aquaculture settings (29–31). Pharmaceutical treatments are currently limited, and parasite resistance to antiparasitic drugs is increasing (29). It is therefore of paramount importance to understand host-helminth interactions that explain host susceptibility and resistance to better predict and treat parasite infections. In this study, we demonstrate that an intestinal cestode harbors its own external and/or internal microbiome. Future studies should determine the biological roles of these microbes in cestode survival and reproduction. We also establish that parasite infection is associated with host gut dysbiosis through the introduction of, and/or selection for, putatively detrimental microbes. Our study illustrates that when studying intestinal parasite infections, the gut microbial environment should be considered to have a nested, hierarchical structure, including both host-associated and parasite-associated microbes that may interact, with positive or negative consequences for the host. Beyond broad biomedical importance for both human and animal health, such an approach also has interesting implications for understanding parasite ecology and evolution (24).
MATERIALS AND METHODS
Sampling.
Samples used for this study were obtained as part of the HoloFish project (Norwegian Seafood Research Fund; project no. 901436). We sampled 463 ready-to-harvest Atlantic salmon from a commercial production site close to Bergen, Norway (60°29′58.9″N, 4°55′41.3″E) owned by Lerøy Seafood Group during April 2018. All salmon were sampled in accordance with Norwegian regulations (FOR-2015-06-18-761, The Norwegian Ministry of Agriculture and Food) (details in Text S1 in the supplemental material). All salmon were from the same commercial broodstock and production cohort and were thus reared under identical environmental conditions throughout their life cycle. Samples were obtained from two groups reared in separate sea pens and fed two different standard commercial diets (feed 1 and feed 2). These diets have been anonymized but were provided by BioMar and EWOS (produced in 2018). Inclusion of two commercial diets allowed us to replicate observations on the effect of cestode infections while controlling for specific feed effects on the infection status. Gutted weight (kg) for each fish was recorded to assess the association between overall lifetime growth of each fish and cestode infection status at the time of sampling. We also recorded the intestinal cestode infection status for each of the 463 salmon using a cestode index from 0 to 3 where 0 represents no observed cestode, 1 signifies 1 to 3 visible cestodes, 2 represents >3 cestodes but normal functioning of the gastrointestinal tract, and 3 represents excessive numbers of cestodes that are likely impeding the passage of feed along the gastrointestinal tract (Fig. S1). This index, developed by experienced Lerøy veterinarians and commonly used as part of fish health assessments in aquaculture farms (C. Kalgraff and H. Sveier, personal communication), provides a semiquantitative assessment of infection with functional consequences for fish health.
We characterized three microbial communities from the salmon intestine (salmon gut mucosa, cestode wash, and cestode body). We sampled 21 salmon infected with cestodes (cestode index from 1 to 3), as well as 9 uninfected salmon (cestode index of 0), with an equal representation of each commercial feed type. For the salmon gut sample, gut content was removed, and the intestine washed in a saline water solution before scrapes of the gut mucosal layer were taken and stored in 1× DNA/RNA Shield buffer (Zymo Research). For each infected salmon, one cestode was first rinsed in saline to remove visible gut content contamination and then placed into a phosphate-buffered saline (PBS) solution and hand shaken for at least 20 s to collect microbes loosely attached to the tegument. The cestode was then preserved separately in 96% EtOH for analysis of the cestode body microbiome. All samples were stored at −20°C until DNA extraction.
DNA extraction.
We extracted DNA from 30 salmon gut mucosa samples. For the 21 parasitized salmon, we also extracted DNA from their associated cestode wash and cestode body samples. In parallel, we extracted four mock community samples as positive controls, using ZymoBIOMICS microbial community standard (Zymo Research) as an input. This mock community comprises eight bacterial strains and two fungal strains that are not expected to be part of the salmon gut microbiome. We also extracted four extraction blanks (sterile water or extraction buffer) as negative controls. The positive and negative controls were all processed in the same manner as the rest of the samples. All extractions were performed using the DNeasy PowerLyzer PowerSoil kit (Qiagen) following the manufacturer’s protocol. Before PCR amplification, we treated each sample with the OneStep PCR inhibitor removal kit (Zymo Research).
16S library build and sequencing.
To amplify the V3-V4 region of the 16S rRNA gene, we used bacterium-specific custom primers, modified from the standard 341F and 806R primers (62), for a two-step PCR-based approach with Illumina Nextera dual indexes (Text S1). Briefly, PCR amplification with 35 or 40 cycles was performed in duplicate to control for putative bias resulting from random PCR noise. The resulting PCR products were purified using solid-phase reversible immobilization (SPRI) bead purification (63). To incorporate the Nextera dual indexes, we performed a second-stage PCR of eight cycles with a unique index combination for each sample and replicate. PCR products were purified using SPRI beads, and the DNA concentration was measured with a Qubit 2.0 fluorometer (Thermo Fisher Scientific). Libraries were pooled in equimolar ratios, including positive- and negative-control libraries for downstream quality control and contaminant filtering. Sequencing was performed at the Danish National High-throughput Sequencing Centre (University of Copenhagen, Denmark) on the Illumina MiSeq (reagent kit v3) to generate 300-bp paired-end reads. To mitigate the effects of sequencing low‐complexity libraries and to improve the clustering, 10% PhiX was added to pools prior to sequencing.
Metagenomics library build and sequencing.
Eight cestode body samples were selected for shotgun metagenomic sequencing of the cestode-associated microbiome. Metagenomic library preparation and sequencing were carried out as previously described for salmonid-related Mycoplasma (9). Briefly, fragmentation of DNA was carried out in Covaris microTube-50 AFA fiber screw-cap tubes using a Covaris M220 focused ultrasonicator. DNA mass for each sample was normalized to 200 ng prior to library preparation. Library preparation was carried out following the blunt-end single-tube method of library preparation for degraded DNA (64, 65). Prior to indexing of libraries, all libraries were analyzed with quantitative PCR (qPCR) to estimate optimal cycle settings on an Mx3005P qPCR system (Agilent Technologies). Indexed libraries were quality controlled with a Bioanalyzer 2100 (Agilent Technologies) and sequenced on an MGISEQ-2000RS at BGI Europe (Copenhagen, Denmark), generating 150-bp paired-end reads.
Data processing and taxonomy assignment for 16S profiling.
Adapter sequences were trimmed from 3′ ends of demultiplexed forward and reverse reads using AdapterRemoval v2.3.1 (66) with default parameters. The DADA2 v1.17.3 (67) package was used for the remainder of the processing steps with R v4.0.2, including quality filtering and trimming, dereplication, inference of ASVs, and chimera filtering (Text S1). Taxonomy was then assigned at the genus level using a custom database based on the SILVA nonredundant SSU v138 training set provided by DADA2 (Text S1). Species assignments at 100% sequence identity were performed where possible.
Further processing was conducted in RStudio v1.3.959 using phyloseq v1.32.0 (68). To remove PCR false positives, we only retained ASVs detected at least twice in both PCR replicates. Putative contaminant sequences were identified and removed with decontam v1.8.0 (69) using both the frequency and prevalence functions. Several ASVs detected in the blanks originated from the mock control samples, indicating low-level cross-contamination had occurred between samples (70). We therefore filtered all ASVs present in the mock samples that matched the known (genus-level) composition of the mock community. Finally, we removed all eukaryotic, chloroplast, and mitochondrial ASVs. After these processing steps, salmon and cestode sample replicates retained, on average, 39,355 sequences (lowest sequencing effort, 1,014 sequences; highest sequencing effort, 99,151 sequences). Species accumulation curves indicated that sequencing reached saturation for most samples (see additional figures at https://github.com/jcbrealey/cestode_microbiome). The mock samples had a genus-level composition that matched the theoretical bacterial community composition (see additional figures on GitHub). There was good concordance between PCR replicates of both the mock and the salmon and cestode samples (see additional figures on GitHub). We therefore merged the ASV count data for each replicate in a sample using the phyloseq function merge_samples.
Statistical analysis.
For all analyses, cestode index was treated as an ordered factor, while sample type (salmon gut, cestode wash, or cestode body) was treated as an unordered factor. Associations between cestode index and gutted fish weight were evaluated in linear regression, including feed type and sex as covariables. For visualization of 16S data, relative abundance was calculated at the ASV level and summed at the genus level. To account for the compositional nature of microbiome sequence data (71), for statistical analyses, ASV and genus counts were transformed with a pseudocount of 1 and the centered log ratio (CLR) calculated using the R package microbiome (http://microbiome.github.io/microbiome). Nonmetric multidimensional scaling (NMDS) was performed using CLR-normalized abundance values and Euclidean distance with the ordinate function in phyloseq. The NMDS stress value, which is a measure of the degree to which the distance between samples in the reduced dimensional space corresponds with the actual distance between samples (similar to a goodness-of-fit value), has been included in the figure legend of each NMDS plot. PERMANOVAs were performed on the Euclidean distances using the adonis function in the R package vegan (https://github.com/vegandevs/vegan). Homogeneity of group dispersions was assessed using the betadisper and permutest functions. Species richness was calculated using the specnumber function in vegan and compared among groups using linear regression. Shannon diversity was calculated using a Hill numbers framework with the R package hillR (q = 1) (72). MaAsLin2 (73) was used to identify changes in ASV- and genus-level relative abundances associated with sample type (fixed effect, salmon gut as reference level), with individual ID and sampling date included as random effects. To identify changes in relative abundance associated with cestode index, MaAsLin2 was run with cestode index, feed type, sex, and size category (small category as reference level) as fixed effects and sampling date as a random effect. The same model was also run using cestode presence as a binary variable (yes/no) instead of cestode index, but no taxon was associated with feed type, sex, or size at false-discovery rate (FDR) of <0.2. When interpreting statistical tests, we have used “language of evidence” as outlined in Muff et al. (74). Thus, P values and FDRs of <0.05 are interpreted as providing moderate or strong evidence for the respective association, while values between 0.05 to 0.2 are interpreted as providing little or weak evidence.
Phylogenetic tree based on 16S profiling.
ASVs classified as Mycoplasmataceae (unknown genus), Mycoplasma, or Ureaplasma were placed in a phylogenetic tree with a reference set of mycoplasma 16S sequences (Mycoplasma, Ureaplasma) and Bacillus pumilus as an outgroup to root the tree (accessions available at https://github.com/jcbrealey/cestode_microbiome). Using QIIME v2-2020.8 (75), sequences were aligned with MAFFT (76), and uninformative positions were masked (--p-max-gap-frequency 0.2, --p-min-conservation 0.4). A bootstrapped, maximum-likelihood phylogenetic tree was constructed with RAxML rapid bootstrapping (77, 78), using the GTRGAMMA substitution model and 1,000 bootstrap replicates. The resulting tree was visualized in FigTree v1.4.3 (https://github.com/rambaut/figtree/).
Metagenomic data processing and MAG generation.
Processing steps of the shotgun metagenomics data included adapter removal (66), PCR duplicate removal (79), and filtering of reads mapping to the sequencing control phiX174, human, salmon (80), and cestode reference genomes (81–84) (Text S1). A coassembly of reads from all samples was then performed with MEGAHIT v1.1.1 (85), and reads for each sample were mapped back to the coassembly with bowtie2 v2.3.4.3 (86) (Text S1). The majority of the remaining analyses were performed through Anvi’o v6.2 and v7.0 (87) as previously described (9), including contig taxonomy assignment (88), gene calling (89), identification of single-copy genes (90), gene annotation (91–93), and manual binning and curation of three MAGs (Text S1). MAG quality statistics were generated using CheckM v1.1.3 (94), which also placed all three MAGs in the Mycoplasmataceae. More specific MAG taxonomic identification was carried out by extracting each 16S gene and matching it to ASVs from the 16S amplicon sequencing data set. To confirm the 16S gene associated with each MAG, single assembly and binning were performed as described above for select samples (162E, 166E, and 361E).
Pangenome comparisons and phylogenomics.
The three generated mycoplasma MAGs were compared to selected Mycoplasma and Ureaplasma reference genomes (accessions available at https://github.com/jcbrealey/cestode_microbiome) and related salmonid Mycoplasma genomes, including “Candidatus Mycoplasma salmoninae salar,” generated from gut content samples obtained from eight host salmon originating from the same sample cohort analyzed in this study, “Candidatus Mycoplasma salmoninae mykiss” from rainbow trout (Oncorhynchus mykiss), and “Candidatus Mycoplasma lavaretus” from European whitefish (Coregonus lavaretus) (9). The reference FASTA reads were annotated with the Anvi’o platform, as described above, before performing comparative pangenome analyses with Anvi’o (Text S1). Pairwise average nucleotide identity (ANI) was then calculated for each genome/MAG with pyANI (95). Gene cluster and functional annotation were extracted for each genome. Additional functional annotation was performed with RAST (96, 97), taking into account Mycoplasma readthrough of the stop codon (98). Exploratory analysis was then performed in R v4.0.2.
To construct a phylogenomic tree, 38 additional Mycoplasma reference genomes were processed in Anvi’o in the same manner as above using B. pumilus as an outgroup (accessions available at https://github.com/jcbrealey/cestode_microbiome). Concatenated amino acid sequences for all HMM hits for each genome were used to generate a maximum-likelihood phylogeny using RAxML v8.2.11 (77, 78) (Text S1). The resulting tree was visualized in FigTree v1.4.3.
Supplemental Methods. Download Text S1, PDF file, 0.2 MB (193.5KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data availability.
The raw amplicon and metagenomic sequencing data, as well as the binned metagenome assemblies of the three individual draft MAGs, have been deposited in the European Nucleotide Archive under BioProject accession no. PRJEB51496. Individual and sample metadata, ASV count data, MAG gene cluster data, additional quality control figures, and R scripts used for analysis are available on GitHub (https://github.com/jcbrealey/cestode_microbiome).
ACKNOWLEDGMENTS
The work was funded by the FHF-Norwegian Seafood Research Fund (“HoloFish,” grant no. 901436), the Danish National Research Foundation (CEH-DNRF143) and the European Union’s Horizon 2020 Action “HoloFood” (817729) to MTL and MDM. This study is part of the Parasite Microbiome Project.
We thank the following people for their assistance with this project: Cathrine Kalgraff, Marcus Thomas Pius Gilbert, Mikkel Sinding, Filipe Vieira, Maria Karm, Martin Nielsen, Even Fjære and Annette Bernhard for assisting with sampling in the field, Luisa dos Santos Bay Nielsen for assistance with data generation, Tom Delmont for metagenome assembly and Anvi’o advice, and Håkon Hansen for discussions on Eubothrium infections in salmon.
Harald Sveier is employed at Lerøy Seafood Group, who produces, markets, and sells farmed Atlantic salmon, including the fish used in the current investigation. Furthermore, Lerøy Seafood Group provided parts of the funding for this study. All other authors declare no competing interests.
Contributor Information
Jaelle C. Brealey, Email: jaelle.brealey@ntnu.no.
Morten T. Limborg, Email: morten.limborg@sund.ku.dk.
Avery August, Cornell University.
REFERENCES
- 1.McFall-Ngai M, Hadfield MG, Bosch TCG, Carey HV, Domazet-Lošo T, Douglas AE, Dubilier N, Eberl G, Fukami T, Gilbert SF, Hentschel U, King N, Kjelleberg S, Knoll AH, Kremer N, Mazmanian SK, Metcalf JL, Nealson K, Pierce NE, Rawls JF, Reid A, Ruby EG, Rumpho M, Sanders JG, Tautz D, Wernegreen JJ. 2013. Animals in a bacterial world, a new imperative for the life sciences. Proc Natl Acad Sci USA 110:3229–3236. doi: 10.1073/pnas.1218525110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li M, Wang B, Zhang M, Rantalainen M, Wang S, Zhou H, Zhang Y, Shen J, Pang X, Zhang M, Wei H, Chen Y, Lu H, Zuo J, Su M, Qiu Y, Jia W, Xiao C, Smith LM, Yang S, Holmes E, Tang H, Zhao G, Nicholson JK, Li L, Zhao L. 2008. Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci USA 105:2117–2122. doi: 10.1073/pnas.0712038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Palma G, Blennerhassett P, Lu J, Deng Y, Park AJ, Green W, Denou E, Silva MA, Santacruz A, Sanz Y, Surette MG, Verdu EF, Collins SM, Bercik P. 2015. Microbiota and host determinants of behavioural phenotype in maternally separated mice. Nat Commun 6:7735. doi: 10.1038/ncomms8735. [DOI] [PubMed] [Google Scholar]
- 4.Wu H-J, Wu E. 2012. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 3:4–14. doi: 10.4161/gmic.19320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelly C, Salinas I. 2017. Under pressure: interactions between commensal microbiota and the teleost immune system. Front Immunol 8:559. doi: 10.3389/fimmu.2017.00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qin Y, Havulinna AS, Liu Y, Jousilahti P, Ritchie SC, Tokolyi A, Sanders JG, Valsta L, Brożyńska M, Zhu Q, Tripathi A, Vázquez-Baeza Y, Loomba R, Cheng S, Jain M, Niiranen T, Lahti L, Knight R, Salomaa V, Inouye M, Méric G. 2022. Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort. Nat Genet 54:134–142. doi: 10.1038/s41588-021-00991-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopera-Maya EA, Kurilshikov A, van der Graaf A, Hu S, Andreu-Sánchez S, Chen L, Vila AV, Gacesa R, Sinha T, Collij V, Klaassen MAY, Bolte LA, Gois MFB, Neerincx PBT, Swertz MA, Harmsen HJM, Wijmenga C, Fu J, Weersma RK, Zhernakova A, Sanna S, LifeLines Cohort Study . 2022. Effect of host genetics on the gut microbiome in 7,738 participants of the Dutch Microbiome Project. Nat Genet 54:143–151. doi: 10.1038/s41588-021-00992-y. [DOI] [PubMed] [Google Scholar]
- 8.Theis KR, Dheilly NM, Klassen JL, Brucker RM, Baines JF, Bosch TCG, Cryan JF, Gilbert SF, Goodnight CJ, Lloyd EA, Sapp J, Vandenkoornhuyse P, Zilber-Rosenberg I, Rosenberg E, Bordenstein SR. 2016. Getting the hologenome concept right: an eco-evolutionary framework for hosts and their microbiomes. mSystems 1:e00028-16. doi: 10.1128/mSystems.00028-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rasmussen JA, Villumsen KR, Duchene D, Puetz LC, Delmont TO, Sveier H, Præbel K, Jørgensen L von G, Martin MD, Gilbert MTP, Bojesen AM, Kristiansen K, Limborg MT. 2021. Genome-resolved metagenomics suggests a mutualistic relationship between Mycoplasma and salmonid hosts. Commun Biol 4:579. doi: 10.1038/s42003-021-02105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zepeda Mendoza ML, Xiong Z, Escalera-Zamudio M, Runge AK, Thézé J, Streicker D, Frank HK, Loza-Rubio E, Liu S, Ryder OA, Samaniego Castruita JA, Katzourakis A, Pacheco G, Taboada B, Löber U, Pybus OG, Li Y, Rojas-Anaya E, Bohmann K, Carmona Baez A, Arias CF, Liu S, Greenwood AD, Bertelsen MF, White NE, Bunce M, Zhang G, Sicheritz-Pontén T, Gilbert MPT. 2018. Hologenomic adaptations underlying the evolution of sanguivory in the common vampire bat. Nat Ecol Evol 2:659–668. doi: 10.1038/s41559-018-0476-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rudman SM, Greenblum S, Hughes RC, Rajpurohit S, Kiratli O, Lowder DB, Lemmon SG, Petrov DA, Chaston JM, Schmidt P. 2019. Microbiome composition shapes rapid genomic adaptation of Drosophila melanogaster. Proc Natl Acad Sci USA 116:20025–20032. doi: 10.1073/pnas.1907787116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strand MA, Jin Y, Sandve SR, Pope PB, Hvidsten TR. 2021. Transkingdom network analysis provides insight into host-microbiome interactions in Atlantic salmon. Comput Struct Biotechnol J 19:1028–1034. doi: 10.1016/j.csbj.2021.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richards AL, Muehlbauer AL, Alazizi A, Burns MB, Findley A, Messina F, Gould TJ, Cascardo C, Pique-Regi R, Blekhman R, Luca F. 2019. Gut microbiota has a widespread and modifiable effect on host gene regulation. mSystems 4:e00323-18. doi: 10.1128/mSystems.00323-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. 2005. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 15.Singh R, Chandrashekharappa S, Bodduluri SR, Baby BV, Hegde B, Kotla NG, Hiwale AA, Saiyed T, Patel P, Vijay-Kumar M, Langille MGI, Douglas GM, Cheng X, Rouchka EC, Waigel SJ, Dryden GW, Alatassi H, Zhang H-G, Haribabu B, Vemula PK, Jala VR. 2019. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat Commun 10:89. doi: 10.1038/s41467-018-07859-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Theriot CM, Koenigsknecht MJ, Carlson PE, Jr., Hatton GE, Nelson AM, Li B, Huffnagle GB, Z Li J, Young VB. 2014. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 5:3114. doi: 10.1038/ncomms4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuijt TJ, Lankelma JM, Scicluna BP, de Sousa e Melo F, Roelofs JJTH, de Boer JD, Hoogendijk AJ, de Beer R, de Vos A, Belzer C, de Vos WM, van der Poll T, Wiersinga WJ. 2016. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut 65:575–583. doi: 10.1136/gutjnl-2015-309728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nyholm L, Koziol A, Marcos S, Botnen AB, Aizpurua O, Gopalakrishnan S, Limborg MT, Gilbert MTP, Alberdi A. 2020. Holo-omics: integrated host-microbiota multi-omics for basic and applied biological research. iScience 23:101414. doi: 10.1016/j.isci.2020.101414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams AR, Myhill LJ, Stolzenbach S, Nejsum P, Mejer H, Nielsen DS, Thamsborg SM. 2021. Emerging interactions between diet, gastrointestinal helminth infection, and the gut microbiota in livestock. BMC Vet Res 17:62. doi: 10.1186/s12917-021-02752-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koch H, Schmid-Hempel P. 2011. Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proc Natl Acad Sci USA 108:19288–19292. doi: 10.1073/pnas.1110474108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hahn MA, Piecyk A, Jorge F, Cerrato R, Kalbe M, Dheilly NM. 2022. Host phenotype and microbiome vary with infection status, parasite genotype, and parasite microbiome composition. Mol Ecol 31:1577–1594. doi: 10.1111/mec.16344. [DOI] [PubMed] [Google Scholar]
- 22.Gaulke CA, Martins ML, Watral VG, Humphreys IR, Spagnoli ST, Kent ML, Sharpton TJ. 2019. A longitudinal assessment of host-microbe-parasite interactions resolves the zebrafish gut microbiome’s link to Pseudocapillaria tomentosa infection and pathology. Microbiome 7:10. doi: 10.1186/s40168-019-0622-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.White EC, Houlden A, Bancroft AJ, Hayes KS, Goldrick M, Grencis RK, Roberts IS. 2018. Manipulation of host and parasite microbiotas: survival strategies during chronic nematode infection. Sci Adv 4:eaap7399. doi: 10.1126/sciadv.aap7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dheilly NM, Martínez JM, Rosario K, Brindley PJ, Fichorova RN, Kaye JZ, Kohl KD, Knoll LJ, Lukeš J, Perkins SL, Poulin R, Schriml L, Thompson LR. 2019. Parasite microbiome project: grand challenges. PLoS Pathog 15:e1008028. doi: 10.1371/journal.ppat.1008028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jorge F, Brealey JC, Brindley PJ, Buysse M, Cantacessi C, Duron O, Fichorova R, Fitzpatrick CR, Hahn M, Hunter C, Hervé V, Knoll LJ, Kohl KD, Lalle M, Lukeš J, Martínez JM, Perkins SL, Poulin R, Rosario K, Schneider AC, Schriml LM, Thompson LR, Walls RL, Dheilly NM. 2022. MIxS-SA: a MIxS extension defining the minimum information standard for sequence data from symbiont-associated micro-organisms. Isme Commun 2:9. doi: 10.1038/s43705-022-00092-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Landmann F, Voronin D, Sullivan W, Taylor MJ. 2011. Anti-filarial activity of antibiotic therapy is due to extensive apoptosis after Wolbachia depletion from filarial nematodes. PLoS Pathog 7:e1002351. doi: 10.1371/journal.ppat.1002351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynolds LA, Finlay BB, Maizels RM. 2015. Cohabitation in the Intestine: interactions among helminth parasites, bacterial microbiota, and host immunity. J Immunol 195:4059–4066. doi: 10.4049/jimmunol.1501432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saksvik M, Nylund A, Nilsen F, Hodneland K. 2001. Experimental infection of Atlantic salmon (Salmo salar) with marine Eubothrium sp. (Cestoda: Pseudophyllidea): observations on the life cycle, aspects of development and growth of the parasite. Folia Parasitol (Praha) 48:118–126. doi: 10.14411/fp.2001.018. [DOI] [PubMed] [Google Scholar]
- 29.Idris OA, Wintola OA, Afolayan AJ. 2019. Helminthiases; prevalence, transmission, host-parasite interactions, resistance to common synthetic drugs and treatment. Heliyon 5:e01161. doi: 10.1016/j.heliyon.2019.e01161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fitzpatrick JL. 2013. Global food security: the impact of veterinary parasites and parasitologists. Vet Parasitol 195:233–248. doi: 10.1016/j.vetpar.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Shinn AP, Pratoomyot J, Bron JE, Paladini G, Brooker EE, Brooker AJ. 2015. Economic costs of protistan and metazoan parasites to global mariculture. Parasitology 142:196–270. doi: 10.1017/S0031182014001437. [DOI] [PubMed] [Google Scholar]
- 32.Bristow GA, Berland B. 1991. The effect of long term, low level Eubothrium sp. (Cestoda: Pseudophyllidea) infection on growth in farmed salmon (Salmo salar L.). Aquaculture 98:325–330. doi: 10.1016/0044-8486(91)90315-X. [DOI] [Google Scholar]
- 33.Saksvik M, Nilsen F, Nylund A, Berland B. 2001. Effect of marine Eubothrium sp. (Cestoda: Pseudophyllidea) on the growth of Atlantic salmon, Salmo salar L. J Fish Diseases 24:111–119. doi: 10.1046/j.1365-2761.2001.00276.x. [DOI] [PubMed] [Google Scholar]
- 34.Holben WE, Williams P, Gilbert MA, Saarinen M, Särkilahti LK, Apajalahti JHA. 2002. Phylogenetic analysis of intestinal microflora indicates a novel Mycoplasma phylotype in farmed and wild salmon. Microb Ecol 44:175–185. doi: 10.1007/s00248-002-1011-6. [DOI] [PubMed] [Google Scholar]
- 35.Dehler CE, Secombes CJ, Martin SAM. 2017. Seawater transfer alters the intestinal microbiota profiles of Atlantic salmon (Salmo salar L.). Sci Rep 7:13877. doi: 10.1038/s41598-017-13249-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Llewellyn MS, McGinnity P, Dionne M, Letourneau J, Thonier F, Carvalho GR, Creer S, Derome N. 2016. The biogeography of the Atlantic salmon (Salmo salar) gut microbiome. ISME J 10:1280–1284. doi: 10.1038/ismej.2015.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Bruni L, Jaramillo-Torres A, Gajardo K, Kortner TM, Krogdahl Å. 2021. Differential response of digesta- and mucosa-associated intestinal microbiota to dietary insect meal during the seawater phase of Atlantic salmon. Anim Microbiome 3:8. doi: 10.1186/s42523-020-00071-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bozzi D, Rasmussen JA, Carøe C, Sveier H, Nordøy K, Gilbert MTP, Limborg MT. 2021. Salmon gut microbiota correlates with disease infection status: potential for monitoring health in farmed animals. Anim Microbiome 3:30. doi: 10.1186/s42523-021-00096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Love M, Teebken-Fisher D, Hose JE, Farmer JJ, III, Hickman FW, Fanning GR. 1981. Vibrio damsela, a marine bacterium, causes skin ulcers on the damselfish Chromis punctipinnis. Science 214:1139–1140. doi: 10.1126/science.214.4525.1139. [DOI] [PubMed] [Google Scholar]
- 40.Ina-Salwany MY, Al-Saari N, Mohamad A, Mursidi FA, Mohd-Aris A, Amal MNA, Kasai H, Mino S, Sawabe T, Zamri-Saad M. 2019. Vibriosis in fish: a review on disease development and prevention. J Aquat Anim Health 31:3–22. doi: 10.1002/aah.10045. [DOI] [PubMed] [Google Scholar]
- 41.Egidius E, Wiik R, Andersen K, Hoff KA, Hjeltnes B. 1986. Vibrio salmonicida sp. nov., a new fish pathogen. Int J Syst Bacteriol 36:518–520. doi: 10.1099/00207713-36-4-518. [DOI] [Google Scholar]
- 42.Leisner JJ, Laursen BG, Prévost H, Drider D, Dalgaard P. 2007. Carnobacterium: positive and negative effects in the environment and in foods. FEMS Microbiol Rev 31:592–613. doi: 10.1111/j.1574-6976.2007.00080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang C, Sun G, Li S, Li X, Liu Y. 2018. Intestinal microbiota of healthy and unhealthy Atlantic salmon Salmo salar L. in a recirculating aquaculture system. J Ocean Limnol 36:414–426. doi: 10.1007/s00343-017-6203-5. [DOI] [Google Scholar]
- 44.McKenney EA, Williamson L, Yoder AD, Rawls JF, Bilbo SD, Parker W. 2015. Alteration of the rat cecal microbiome during colonization with the helminth Hymenolepis diminuta. Gut Microbes 6:182–193. doi: 10.1080/19490976.2015.1047128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aivelo T, Norberg A. 2018. Parasite–microbiota interactions potentially affect intestinal communities in wild mammals. J Anim Ecol 87:438–447. doi: 10.1111/1365-2656.12708. [DOI] [PubMed] [Google Scholar]
- 46.Green TJ, Smullen R, Barnes AC. 2013. Dietary soybean protein concentrate-induced intestinal disorder in marine farmed Atlantic salmon, Salmo salar is associated with alterations in gut microbiota. Vet Microbiol 166:286–292. doi: 10.1016/j.vetmic.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 47.Bano N, Smith AD, Bennett W, Vasquez L, Hollibaugh JT. 2007. Dominance of Mycoplasma in the guts of the Long-Jawed mudsucker, Gillichthys mirabilis, from five California salt marshes. Environ Microbiol 9:2636–2641. doi: 10.1111/j.1462-2920.2007.01381.x. [DOI] [PubMed] [Google Scholar]
- 48.Xing M, Hou Z, Yuan J, Liu Y, Qu Y, Liu B. 2013. Taxonomic and functional metagenomic profiling of gastrointestinal tract microbiome of the farmed adult turbot (Scophthalmus maximus). FEMS Microbiol Ecol 86:432–443. doi: 10.1111/1574-6941.12174. [DOI] [PubMed] [Google Scholar]
- 49.Kim D-H, Brunt J, Austin B. 2007. Microbial diversity of intestinal contents and mucus in rainbow trout (Oncorhynchus mykiss). J Appl Microbiol 102:1654–1664. doi: 10.1111/j.1365-2672.2006.03185.x. [DOI] [PubMed] [Google Scholar]
- 50.Stadtl Ander CT, Lotz W, Körting W, Kirchhoff H. 1995. Piscine gill epithelial cell necrosis due to Mycoplasma mobile strain 163 K: comparison of in-vivo and in-vitro infection. J Comp Pathol 112:351–359. doi: 10.1016/S0021-9975(05)80016-7. [DOI] [PubMed] [Google Scholar]
- 51.Burns AR, Watral V, Sichel S, Spagnoli S, Banse AV, Mittge E, Sharpton TJ, Guillemin K, Kent ML. 2018. Transmission of a common intestinal neoplasm in zebrafish by cohabitation. J Fish Dis 41:569–579. doi: 10.1111/jfd.12743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kashinskaya EN, Simonov EP, Izvekova GI, Parshukov AN, Andree KB, Solovyev MM. 2020. Composition of the microbial communities in the gastrointestinal tract of perch (Perca fluviatilis L. 1758) and cestodes parasitizing the perch digestive tract. J Fish Dis 43:23–38. doi: 10.1111/jfd.13096. [DOI] [PubMed] [Google Scholar]
- 53.Izvekova GI, Lapteva NA. 2004. Microflora associated with the digestive-transport surfaces of fish and their parasitic cestodes. Russian J Ecol 35:176–180. doi: 10.1023/B:RUSE.0000025968.97632.30. [DOI] [Google Scholar]
- 54.Jorge F, Dheilly NM, Poulin R. 2020. Persistence of a core microbiome through the ontogeny of a multi-host parasite. Front Microbiol 11:954. doi: 10.3389/fmicb.2020.00954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dandekar T, Snel B, Schmidt S, Lathe W, Suyama M, Huynen M, Bork P. 2002. Comparative genome analysis of the mollicutes, p 255–278. In Razin S, Herrmann R (ed), molecular biology and pathogenicity of Mycoplasmas. Springer, Boston, MA. [Google Scholar]
- 56.Persson ME, Larsson P, Stenroth P. 2007. Fractionation of δ15N and δ13C for Atlantic salmon and its intestinal cestode Eubothrium crassum. J Fish Biology 71:441–452. doi: 10.1111/j.1095-8649.2007.01500.x. [DOI] [Google Scholar]
- 57.Blötz C, Stülke J. 2017. Glycerol metabolism and its implication in virulence in Mycoplasma. FEMS Microbiol Rev 41:640–652. doi: 10.1093/femsre/fux033. [DOI] [PubMed] [Google Scholar]
- 58.Vaughan JA, Tkach VV, Greiman SE. 2012. Neorickettsial endosymbionts of the digenea: diversity, transmission and distribution. Adv Parasitol 79:253–297. doi: 10.1016/B978-0-12-398457-9.00003-2. [DOI] [PubMed] [Google Scholar]
- 59.Dessi D, Rappelli P, Diaz N, Cappuccinelli P, Fiori PL. 2006. Mycoplasma hominis and Trichomonas vaginalis: a unique case of symbiotic relationship between two obligate human parasites. Front Biosci 11:2028–2034. doi: 10.2741/1944. [DOI] [PubMed] [Google Scholar]
- 60.Deenonpoe R, Chomvarin C, Pairojkul C, Chamgramol Y, Loukas A, Brindley PJ, Sripa B. 2015. The carcinogenic liver fluke Opisthorchis viverrini is a reservoir for species of Helicobacter. Asian Pac J Cancer Prev 16:1751–1758. doi: 10.7314/apjcp.2015.16.5.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Y, Huang JM, Zhou YL, Almeida A, Finn RD, Danchin A, He LS. 2020. Phylogenomics of expanding uncultured environmental Tenericutes provides insights into their pathogenicity and evolutionary relationship with Bacilli. BMC Genomics 21:408. doi: 10.1186/s12864-020-06807-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu Y, Lee C, Kim J, Hwang S. 2005. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol Bioeng 89:670–679. doi: 10.1002/bit.20347. [DOI] [PubMed] [Google Scholar]
- 63.DeAngelis MM, Wang DG, Hawkins TL. 1995. Solid-phase reversible immobilization for the isolation of PCR products. Nucleic Acids Res 23:4742–4743. doi: 10.1093/nar/23.22.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carøe C, Gopalakrishnan S, Vinner L, Mak SST, Sinding MHS, Samaniego JA, Wales N, Sicheritz‐Pontén T, Gilbert MTP. 2018. Single‐tube library preparation for degraded DNA. Methods Ecol Evol 9:410–419. doi: 10.1111/2041-210X.12871. [DOI] [Google Scholar]
- 65.Mak SST, Gopalakrishnan S, Carøe C, Geng C, Liu S, Sinding MHS, Kuderna LFK, Zhang W, Fu S, Vieira FG, Germonpré M, Bocherens H, Fedorov S, Petersen B, Sicheritz-Pontén T, Marques-Bonet T, Zhang G, Jiang H, Gilbert MTP. 2017. Comparative performance of the BGISEQ-500 vs Illumina HiSeq2500 sequencing platforms for palaeogenomic sequencing. Gigascience 6:gix049. doi: 10.1093/gigascience/gix049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schubert M, Lindgreen S, Orlando L. 2016. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res Notes 9:88. doi: 10.1186/s13104-016-1900-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. 2018. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6:226. doi: 10.1186/s40168-018-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Minich JJ, Sanders JG, Amir A, Humphrey G, Gilbert JA, Knight R. 2019. Quantifying and understanding well-to-well contamination in microbiome research. mSystems 4:e00186-19. doi: 10.1128/mSystems.00186-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gloor GB, Wu JR, Pawlowsky-Glahn V, Egozcue JJ. 2016. It’s all relative: analyzing microbiome data as compositions. Ann Epidemiol 26:322–329. doi: 10.1016/j.annepidem.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 72.Alberdi A, Gilbert MTP. 2019. A guide to the application of Hill numbers to DNA-based diversity analyses. Mol Ecol Resour 19:804–817. doi: 10.1111/1755-0998.13014. [DOI] [PubMed] [Google Scholar]
- 73.Mallick H, Rahnavard A, McIver LJ, Ma S, Zhang Y, Nguyen LH, Tickle TL, Weingart G, Ren B, Schwager EH, Chatterjee S, Thompson KN, Wilkinson JE, Subramanian A, Lu Y, Waldron L, Paulson JN, Franzosa EA, Bravo HC, Huttenhower C. 2021. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput Biol 17:e1009442. doi: 10.1371/journal.pcbi.1009442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Muff S, Nilsen EB, O’Hara RB, Nater CR. 2021. Rewriting results sections in the language of evidence. Trends Ecol Evol 37:203–210. doi: 10.1016/j.tree.2021.10.009. [DOI] [PubMed] [Google Scholar]
- 75.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- 79.Shen W, Le S, Li Y, Hu F. 2016. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One 11:e0163962. doi: 10.1371/journal.pone.0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lien S, Koop BF, Sandve SR, Miller JR, Kent MP, Nome T, Hvidsten TR, Leong JS, Minkley DR, Zimin A, Grammes F, Grove H, Gjuvsland A, Walenz B, Hermansen RA, Von Schalburg K, Rondeau EB, Di Genova A, Samy JKA, Olav Vik J, Vigeland MD, Caler L, Grimholt U, Jentoft S, Inge Våge D, De Jong P, Moen T, Baranski M, Palti Y, Smith DR, Yorke JA, Nederbragt AJ, Tooming-Klunderud A, Jakobsen KS, Jiang X, Fan D, Hu Y, Liberles DA, Vidal R, Iturra P, Jones SJM, Jonassen I, Maass A, Omholt SW, Davidson WS. 2016. The Atlantic salmon genome provides insights into rediploidization. Nature 533:200–205. doi: 10.1038/nature17164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 1303.3997 [q-bio.GN]. https://arxiv.org/abs/1303.3997v2.
- 82.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31:1674–1676. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 86.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eren AM, Esen OC, Quince C, Vineis JH, Morrison HG, Sogin ML, Delmont TO. 2015. Anvi’o: an advanced analysis and visualization platform for ’omics data. PeerJ 3:e1319. doi: 10.7717/peerj.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wood DE, Lu J, Langmead B. 2019. Improved metagenomic analysis with Kraken 2. Genome Biol 20:257. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee MD. 2019. GToTree: a user-friendly workflow for phylogenomics. Bioinformatics 35:4162–4164. doi: 10.1093/bioinformatics/btz188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Galperin MY, Wolf YI, Makarova KS, Vera Alvarez R, Landsman D, Koonin EV. 2021. COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res 49:D274–D281. doi: 10.1093/nar/gkaa1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, Tosatto SCE, Paladin L, Raj S, Richardson LJ, Finn RD, Bateman A. 2021. Pfam: the protein families database in 2021. Nucleic Acids Res 49:D412–D419. doi: 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. 2021. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res 49:D545–D551. doi: 10.1093/nar/gkaa970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pritchard L, Glover RH, Humphris S, Elphinstone JG, Toth IK. 2016. Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal Methods 8:12–24. doi: 10.1039/C5AY02550H. [DOI] [Google Scholar]
- 96.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R. 2014. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Inamine JM, Ho KC, Loechel S, Hu PC. 1990. Evidence that UGA is read as a tryptophan codon rather than as a stop codon by Mycoplasma pneumoniae, Mycoplasma genitalium, and Mycoplasma gallisepticum. J Bacteriol 172:504–506. doi: 10.1128/jb.172.1.504-506.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Distribution of cestode indexes across fish housed in two sea pens, each fed a different commercial diet (n = 253 for feed 1 and n = 210 for feed 2). The semiquantitative cestode index is illustrated on the right, where 0 is no observed cestode, 1 is 1 to 3 visible cestodes, 2 is >3 cestodes but normal functioning of the gastrointestinal tract, and 3 is excessive numbers of cestodes that are likely impeding the passage of feed along the gastrointestinal tract. Download FIG S1, PDF file, 0.1 MB (129.9KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Weight of individuals decreases as cestode index increases. (a) Violin plot of gutted weight of salmon (kg) by cestode index in the entire cohort (n = 463). Horizontal lines represent the median. There was strong evidence that gutted weight decreased as cestode infection increased (F5,456 = 19.5, P < 0.001, R2 = 0.17). (b) Box plot of gutted weight of salmon (kg) by cestode index in the 30 individuals selected for sequencing of cestode-related microbiomes in this study. There was some evidence that more heavily parasitized fish were smaller in this subset of individuals (F5,24 = 1.31, P = 0.29, R2 = 0.051). Download FIG S2, PDF file, 0.05 MB (53.9KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Associations between ASV or genus abundance and cestode index or sample type, as produced by MaAsLin2. Results with FDRs of <0.25 are included and those with FDRs of <0.05 are highlighted in gray. Download Table S1, PDF file, 0.2 MB (171.7KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
NMDS (k = 2) of Euclidean distances of ASV CLR-normalized abundances in the salmon fish gut mucosa. Each point represents one individual, colored by cestode index. NMDS stress, 0.150. Cestode index accounted for 15.1% of the variation among individuals (PERMANOVA, F3,26 = 1.54, P = 0.035). Download FIG S3, PDF file, 0.01 MB (8.3KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Alpha diversity of each sample type from each individual, faceted by cestode index. (a) There was weak evidence that species richness (number of ASVs) increased as cestode index increased (F4,25 = 1.53, Plinear = 0.11, and R2 = 0.07 after controlling for sequencing effort). When considering all individuals irrespective of cestode index, there was strong evidence that alpha diversity decreased in the cestode body compared to the salmon gut mucosa (F3,63 = 6.04, P = 0.0021, and R2 = 0.17 after controlling for sequencing effort). (b) There was also weak evidence that Shannon diversity (calculated using a Hill numbers framework) increased with cestode index (F4,25 = 0.93, Plinear = 0.18, R2 = −0.009 after controlling for sequencing effort) and decreased in the cestode body compared to the salmon gut (F3,63 = 1.49, P = 0.061, and R2 = 0.022 after controlling for sequencing effort). (c) Venn diagram of ASVs detected in each sample type. The salmon gut microbiota has been separated into ASVs detected in nonparasitized salmon (cestode index = 0) and those in parasitized salmon (cestode index > 0). A total of 45 ASVs were unique to the salmon gut mucosa, irrespective of cestode detection; 31 were shared between salmon gut mucosa and cestode wash, 3 between salmon gut mucosa and cestode body, and 15 between all three sample types. Download FIG S4, PDF file, 0.3 MB (312.5KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Relative abundance (proportion of 16S reads) of taxa in the salmon gut mucosa that differed with cestode infection. (a) Genera that differed with cestode index. There was weak evidence that the relative abundance of Mycoplasma decreased with increasing cestode infection (effect size, −0.349 ± 0.118; FDR, 0.173) and the relative abundance of Photobacterium increased (effect size, 0.375 ± 0.112; FDR, 0.144). (b) Genera that differed by cestode presence. There was weak evidence that Carnobacterium and Aliivibrio had higher abundances in the gut mucosa of salmon infected with cestode (Carnobacterium, effect size, 0.018 ± 0.014; FDR, 0.506; and Aliivibrio, effect size, 0.078 ± 0.92; FDR, 0.779), and the likely commensal Brevinema had higher abundance in the gut mucosa of uninfected salmon (effect size, −0.041 ± 0.014; FDR, 0.248). (c) Relative abundance of ASVs that differed with cestode index (FDR, <0.25; statistics provided in Table S1). Note that relative abundance is shown on different scales within each plot in panel c. Download FIG S5, PDF file, 0.02 MB (25.2KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Relative abundance (proportion of 16S reads) of genera (a) and ASVs (b) that differed with a particular sample type (FDR < 0.2; statistics provided in Table S1). ASVs are ordered by genus (alphabetically). Note that relative abundance is shown on different scales within each plot in panel b. Download FIG S6, PDF file, 0.1 MB (118.6KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Statistics and ENA accessions of the three Mycoplasma MAGs generated from the cestode body shotgun metagenome. Download Table S2, PDF file, 0.1 MB (131.1KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Functional annotation of cestode-associated Mycoplasma MAGs. (a) Venn diagram of gene clusters detected in cestode-associated and salmonid-associated Mycoplasma MAGs. Gene cluster counts unique to CE_seq1 are highlighted in cyan, unique to “Ca. Mycoplasma salmoninae” (“Ca. Mycoplasma salmoninae salar” and/or “Ca. Mycoplasma salmoninae mykiss”) in orange, and those unique or shared between CE_seq7 and “Ca. Mycoplasma lavaretus” in shades of lilac. Consistent with phylogenetic patterns observed in Fig. 4, CE_seq7 and “Ca. Mycoplasma lavaretus” share many gene clusters (361), while CE_seq1 has few gene clusters in common with the “Ca. Mycoplasma salmoninae” MAGs (3 + 71 + 3 = 77), instead containing the largest number of unique gene clusters (586). (b) Probable energy production pathways detected in cestode (CE_seq1 and CE_seq7) and three previously described salmonid (“Ca. Mycoplasma salmoninae salar,” “Ca. Mycoplasma salmoninae mykiss,” and “Ca. Mycoplasma lavaretus”) Mycoplasma MAGs. Glycolysis via the EMP pathway, glycerol metabolism, and the arginine deiminase pathway of arginine degradation is shown. The major steps generating important by-products (ATP or NH3) are shown with dashed gray arrows. For each gene in the pathway, colored boxes indicate that the gene was successfully annotated in the respective MAG (white boxes indicate that the gene was not detected in the respective MAG). Dotted lines represent unknown steps in the pathway. Download FIG S7, PDF file, 0.6 MB (597.8KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental Methods. Download Text S1, PDF file, 0.2 MB (193.5KB, pdf) .
Copyright © 2022 Brealey et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
The raw amplicon and metagenomic sequencing data, as well as the binned metagenome assemblies of the three individual draft MAGs, have been deposited in the European Nucleotide Archive under BioProject accession no. PRJEB51496. Individual and sample metadata, ASV count data, MAG gene cluster data, additional quality control figures, and R scripts used for analysis are available on GitHub (https://github.com/jcbrealey/cestode_microbiome).