

ABSTRACT
Salmonella enterica serovar Typhimurium (S. Typhimurium) elicited strong innate immune responses in macrophages. To activate innate immunity, pattern recognition receptors (PRRs) in host cells can recognize highly conserved pathogen-associated molecular patterns (PAMPs). Here, we showed that S. Typhimurium induced a robust type I interferon (IFN) response in murine macrophages. Exposure of macrophages to S. Typhimurium activated a Toll-like receptor 4 (TLR4)-dependent type I IFN response. Next, we showed that type I IFN and IFN-stimulated genes (ISGs) were elicited in a TBK1-IFN-dependent manner. Furthermore, cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS) and immune adaptor protein stimulator of interferon genes (STING) were also required for the induction of type I IFN response during infection. Intriguingly, S. Typhimurium infection triggered mitochondrial DNA (mtDNA) release into the cytosol to activate the type I IFN response. In addition, we also showed that bacterial DNA was enriched in cGAS during infection, which may contribute to cGAS activation. Finally, we showed that cGAS and STING deficient mice and cells were more susceptible to S. Typhimurium infection, signifying the critical role of the cGAS-STING pathway in host defense against S. Typhimurium infection. In conclusion, in addition to TLR4-dependent innate immune response, we demonstrated that S. Typhimurium induced the type I IFN response in a cGAS-STING-dependent manner and the S. Typhimurium-induced mtDNA release was important for the induction of type I IFN. This study elucidated a new mechanism by which bacterial pathogen activated the cGAS-STING pathway and also characterized the important role of cGAS-STING during S. Typhimurium infection.
KEYWORDS: Salmonella, cGAS, STING, type I interferon, mtDNA, interferon
INTRODUCTION
Salmonella enterica serovar Typhimurium (S. Typhimurium) is a Gram-negative, intracellular bacterial pathogen. It is recognized as one of the most identified bacterial causes of foodborne illness worldwide (1). S. Typhimurium is also an important model organism that is widely employed to investigate the molecular mechanism in bacterial pathogenesis and pathogen-host interactions. S. Typhimurium can infect different types of cells, including epithelial cells and macrophages, and can survive within them (2). In macrophages, S. Typhimurium infection triggers strong innate immune responses that govern the outcome of infection.
Host innate immune responses to S. Typhimurium have drawn great attention. The activation of the innate immune system is featured with proinflammatory transcriptional responses, which are mediated by different pattern recognition receptors (PRRs) that sense a variety of pathogen-associated molecular patterns (PAMPs), like bacterial nucleic acid, cell wall components, or flagellin. Host cells can recognize S. Typhimurium lipopolysaccharide (LPS) by a surface innate immune receptor Toll-like receptor 4 (TLR4), which leads to the production of proinflammatory cytokines (2, 3). Accordingly, the mice that are defected in this pathway are more susceptible to S. Typhimurium infection, suggesting the sensing of essential PAMPs is vital for protection against S. Typhimurium infection (4). Additionally, TLR5 recognizes S. Typhimurium flagellin to induce cytokine and chemokine productions (4). The NOD-like receptors (NLRs) that monitor the host cell cytosol are also involved during S. Typhimurium infection (4). Two NLRs, NLRP3 (LRR and pyrin domain-containing 3) and NLRC4 (NOD-, LRR- and CARD-containing 4) can sense the infection of S. Typhimurium, which cooperatively orchestrates the protective innate immune response (5, 6). The NLRC4 is activated by S. Typhimurium flagellin (7) and type III secretion apparatus (T3SS) which is encoded on Salmonella pathogenicity island 1 (SPI-1) (8, 9), while S. Typhimurium mRNA is the possible ligand of NLRP3 (10). In addition, S. Typhimurium-secreted effectors can act as “danger-associated molecular patterns” that activate Rho-family GTPases through innate immune receptor NOD1, which leads to NF-κB activation (11).
Bacterial-derived cyclic dinucleotides (CDNs) like cyclic di-guanylate monophosphate (c-di-GMP) or cyclic di-AMP (c-di-AMP) can directly bind to STING to induce the production of type I interferons (IFNs) and transcription of IFN-stimulated genes (ISGs) (12–14). In addition, the stimulator of interferon genes (STING) can also be activated by cytosolic DNA sensor cyclic-GMP-AMP synthase (cGAS) (15, 16) which could detect the cytosolic DNA oriented from DNA viruses, bacteria, and mitochondrial DNA (17–20). Although the role of the cGAS-STING pathway in favoring antiviral innate immune response has been extensively investigated (21), its role during bacterial infection is not fully understood. Accumulating studies demonstrated that the cGAS-STING pathway, which is the vital cytosolic surveillance pathway (CSP), is essential for the induction of type I IFN response during the infection of a variety of intracellular bacterial pathogens (13, 19, 22–31). For instance, Mycobacterium tuberculosis, Francisella novicida, and Chlamydia trachomatis engage cGAS via bacterial dsDNA (19, 25, 26), whereas Listeria monocytogenes secreted cyclic di-AMP directly activates STING (13). One important feature of the innate immune response against S. Typhimurium infection is the induction of type I interferons (IFNs), including IFN-α and IFN-β (32). Although it has been reported that S. Typhimurium induces the type I IFN response through TLRs in host cells (33), whether other CSPs like the cGAS-STING pathway contribute to the induction of type I IFN response is largely unknown.
In this study, by using transcriptomic analysis, we found that type I IFN and many ISGs were potently induced during S. Typhimurium infection in murine macrophages. Consistent with a previous study (2), we also demonstrated that S. Typhimurium activated a TLR4-dependent pathway. However, we further showed that such type I IFN response could also be induced in a cGAS-STING-dependent fashion. Interestingly, the cGAS-STING-dependent type I IFN response was triggered by the release of mtDNA into the cytosol. Released mtDNA and S. Typhimurium DNA in the cytosol activated the cytosolic DNA sensor cGAS. In addition, the lack of cGAS or STING led to significantly increased susceptibility to S. Typhimurium infection in the in vitro and in vivo model, suggesting the critical role of cGAS-STING in supporting host defense against S. Typhimurium infection. In conclusion, we demonstrated that S. Typhimurium induced the type I IFN response in a cGAS-STING-dependent manner by triggering mtDNA release.
RESULTS
S. Typhimurium induces proinflammatory cytokines expression during macrophage infection.
Microbial infections elicited strong host innate immune responses through the sensing of conserved microbial products. Proinflammatory cytokines and chemokines have been shown to play essential roles in controlling bacterial infections. To explore the nature of the macrophage innate immune response during S. Typhimurium infection, mouse peritoneal macrophages (PMs) were infected with S. Typhimurium at different time points. Total RNA from infected cells or uninfected (mock) was harvested after 2 h, 4 h, or 8 h infection since these time points are used as key induction time points of innate immune activation during different intracellular bacterial infections (18–20). The samples were subjected to RT-qPCR to detect the gene expression of diverse proinflammatory cytokines and chemokines. First, cell viability was slightly decreased at 8 h postinfection (8 h.p.i.) (Fig. S1A). Next, we revealed that at the early time point after infection (2 h.p.i.), robust induction of the cytokines, including Ifnb1, Tnfa, Il12b, and inflammasome genes Nlrp3 was obtained (Fig. 1A), which is consistent with a previous study (34). In contrast, gene expression of two antimicrobial genes Nos2 and Gbp2b (35–37), one S. Typhimurium fimbrial protein FimH induced gene Il1b (38), and one IFN-activated gene Ifi205 were peaked at 8 h.p.i (Fig. 1A). Similar gene expression patterns were also confirmed in S. Typhimurium-infected murine macrophage Raw264.7 cells (Fig. S1B). To further explore the S. Typhimurium-induced proinflammatory cytokine expression, PMs were infected with S. Typhimurium at different multiplicity of infections (MOIs). While clear cell death was observed in PMs infected with S. Typhimurium at an MOI of 100 (Fig. S1C), all those above-mentioned genes were induced at an MOI of 1 and peaked at an MOI of 10 and 20, with no difference was observed between cells infected at an MOI of 10 or 20 (Fig. 1B). Consistently, significant induction of these genes was also detected in Raw264.7 cells infected at an MOI of 10 (Fig. S1D). Together, these results demonstrated that the S. Typhimurium infection triggered proinflammatory cytokine expression in murine macrophages.
FIG 1.

S. Typhimurium infection triggers proinflammatory cytokine expression in murine macrophages. (A) qRT-PCR analysis of gene expression in mouse peritoneal macrophages (PMs) uninfected (mock) or infected with S. Typhimurium for 2, 4, or 8 h at an MOI of 10 (n = 3). (B) qRT-PCR analysis of gene expression in mouse PMs uninfected (mock) or infected with S. Typhimurium for 8 h at different MOIs (n = 3). Data in (A) and (B) were normalized to uninfected control (mock, set as 1), Actin was used as the housekeeping gene. Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
S. Typhimurium infection triggers proinflammatory cytokine expression in murine macrophages. (A) CCK8 analysis of cell viability in mouse peritoneal macrophages (PMs) uninfected (mock) or infected with S. Typhimurium for 2, 4, or 8 h at an MOI of 10 (n = 3). (B) qRT-PCR analysis of gene expression in Raw264.7 cells uninfected (mock) or infected with S. Typhimurium for 2, 4, or 8 h at an MOI of 100 (n = 3). (C) CCK8 analysis of cell viability in mouse peritoneal macrophages (PMs) uninfected (mock) or infected with S. Typhimurium for 8 h at different MOIs (n = 3). (D) qRT-PCR analysis of gene expression in Raw264.7 cells uninfected (mock) or infected with S. Typhimurium for 8 h at different MOIs (n = 3). (E) KEGG pathway analysis results for comparison of upregulated genes in PMs uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 10. (F) Immunoblot analysis of protein expression in PMs uninfected (mock) or infected with S. Typhimurium for 2, 4, or 8 h at an MOI of 10. (G) Immunoblot analysis of protein expression in Raw264.7 cells uninfected (mock) or infected with S. Typhimurium for 8 h at different MOIs. Data in (A to D) were normalized to uninfected control (mock, set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. Download FIG S1, TIF file, 1.9 MB (2MB, tif) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
S. Typhimurium infection triggers type I IFN response in murine macrophages.
To comprehensively examine the macrophage innate immune responses to S. Typhimurium infection, we performed a transcriptomic analysis to assess global gene expression changes upon infection. Mouse PMs were infected with S. Typhimurium at an MOI of 10. At 8 h.p.i, total RNA from 3 biological replicates was collected and subjected to high-throughput RNA sequencing. Compared to uninfected control cells, over 3500 genes were found to be differentially expressed (≥2-fold) with a false discovery rate (FDR) cutoff of 0.05, with 1789 upregulated genes and 1745 downregulated genes (Fig. 2A and Table S1). To validate RNA-seq results, the expression of representative downregulated (Hgf, Pdgfa, and Plcb2) and upregulated (Nos2, Nlrp3, and Ccl2) genes was confirmed by RT-qPCR (Fig. 2B and C). In particular, a large subset of canonical proinflammatory cytokines, chemokines, inflammasome genes, and prostaglandins was potently upregulated in S. Typhimurium-infected macrophages (Fig. 2D). Of note, many ISGs controlled by IFNs through different IRFs (interferon regulatory factors) were also profoundly activated (Fig. 2D).
FIG 2.

RNA-seq analysis reveals the induction of type I IFN response in S. Typhimurium-infected murine macrophages. (A) Volcano plot showing gene expression analysis of PMs uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 10. The x-axis shows a fold change in gene expression by calculating log2(infected RPKM/mock RPKM) and the y-axis shows statistical significance (log10[Q-value]). Downregulated genes are plotted on the left (blue) and upregulated genes are on the right (red). Each treatment had 3 biological replicates. RPKM: reads per kilobase of exon model per Million mapped reads. Differentially expressed genes were those with a false discovery rate (FDR) cutoff of 0.05 and a fold change of ≥±2. (B and C) qRT-PCR analysis of gene expression of downregulated genes (B) and upregulated (C) genes in mouse PMs uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 10 (n = 3). (D) Heatmap of RNA-seq analysis. PMs were uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 10. Heatmap was made by calculating log2(infected RPKM/mock RPKM). Numbers 1, 2, and 3 indicate 3 biological replicates. (E) Ingenuity pathway analysis of upregulated gene expression changes in PMs infected with S. Typhimurium. (F) ELISA analysis of IFN-β production in mouse PMs uninfected (mock) or infected with S. Typhimurium for 2, 4, 6, 8, or 10 h at an MOI of 10 (n = 3). (G) qRT-PCR analysis of gene expression in C57BL/6 (WT) or TLR4−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 2, 4, and 8 h at an MOI of 10 (n = 3). (H) ELISA analysis of IFN-β production in the supernatant from C57BL/6 (WT) or TLR4−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 6 h at an MOI of 10 (n = 3). Data in (B) and (C) were normalized to uninfected control (mock, set as 1), Data in (G) were normalized to mock-infected C57BL/6 (WT) PMs, mock-infected TLR4−/− mouse-derived PMs, respectively (mock, both set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
RNA-seq analysis of uninfected and S. Typhimurium-infected macrophages. Download Table S1, XLSX file, 0.3 MB (302.9KB, xlsx) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Further KEGG pathways enrichment analysis of DEGs (differentially expressed genes) demonstrated that the immune system and signal transduction pathways were activated in S. Typhimurium-infected macrophages (Fig. S1E). Unbiased canonical pathway analysis (Ingenuity pathway analysis) showed most of the upregulated genes related to innate immune signaling, including NOD-like receptor and Toll-like receptor signaling pathways (Fig. 2E). Intriguingly, the cytosolic DNA-sensing pathway and RIG-I-like receptor signaling pathway were also enriched as the most activated pathways, implying that those pathways were involved in sensing S. Typhimurium infection (Fig. 2E). The induction of these pathways indicates type I IFN responses were activated during infection. As expected, the IFN-β production was detected in the supernatant from S. Typhimurium-infected PMs by using an enzyme-linked immunosorbent assay (ELISA) (Fig. 2F). Consistently, the hallmark of type I IFN pathway activation, the phosphorylation of IRF3 and TBK1, were observed in S. Typhimurium infected PMs and Raw264.7 cells (Fig. S1F and G). Together, we demonstrated that S. Typhimurium provoked the type I IFN response in murine macrophages.
It has been reported that S. Typhimurium infection could stimulate type I IFN response via TLRs signaling (39). In particular, S. Typhimurium lipopolysaccharides (LPS) activated the production of proinflammatory cytokines via a surface innate immune receptor Toll-like receptor 4 (TLR4) (2, 3). Consistently, we showed that S. Typhimurium infection-induced Ifnb1, proinflammatory cytokines and ISG expression (Fig. 2G), and IFN-β production (Fig. 2H) were substantially attenuated in PMs from mice that lack TLR4 (TLR4−/−). However, it is worthy to note that the expression of these genes and production of IFN-β was not completely diminished in TLR4−/− PMs in the context of S. Typhimurium infection. In particular, S. Typhimurium infection can still elicit clear Ifnb1, Tnfa, Ccl5, and Rsad2 expression at 8 h.p.i (Fig. 2G). Of note, the expression of Tnfa, which is activated by several cytosolic innate immune sensors (40), was not affected in TLR4−/− cells in response to S. Typhimurium infection (Fig. 2G). Moreover, a slight but statistically significant increased IFN-β production was detected in S. Typhimurium-infected TLR4−/− PMs (Fig. 2H), suggesting TLR4-independent pathways are involved to detect S. Typhimurium infection. Furthermore, we used primary mouse embryonic fibroblast cells (MEFs) to detect type I IFN response during S. Typhimurium infection. Consistently, the lower level of type I IFN response was induced by LPS in primary MEFs compared to those in PMs (Fig. S2A) (41). With functional DNA sensing pathways (Fig. S2A), profound induction of Ifnb1 and ISG was detected in primary MEFs during S. Typhimurium infection (Fig. S2B). Collectively, those results indicate that in addition to TLR4, there are TLR-independent pathways that allow the recognition of S. Typhimurium infection.
S. Typhimurium infection induces the type I IFN response in primary MEFs. (A) qRT-PCR analysis of gene expression in murine primary MEF cells untreated (con), treated with LPS (100 ng/mL) (LPS-MEFs), or transfected with dsDNA (1 μg/mL) (Interferon stimulatory DNA, ISD, 45 bp) (ISD-MEFs) for 6 h. PMs treated with LPS (100 ng/mL) (LPS-PMs) for 6 h were also included (n = 3). (B) qRT-PCR analysis of gene expression in murine primary MEF cells uninfected (mock) or infected with S. Typhimurium for 2, 4, 6, 8, or 10 h at an MOI of 100 (n = 3). Data in (A) were normalized to untreated MEFs and PMs, respectively (con, set as 1; con for PMs was not shown). Data in (B) were normalized to mock-infected control (mock, set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001. NS, not significant. Download FIG S2, TIF file, 2.2 MB (2.2MB, tif) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
S. Typhimurium induces type I IFN response requires TBK1-IFN axis.
To explore other signaling pathways that mediate the S. Typhimurium-induced type I IFN response, a specific inhibitor of TBK1 and an inhibitor of IKKɛ, BX795 was employed (42). BX795 pretreatment leads to a substantially decreased IRF3 phosphorylation, ISG15, and IFIT2 expression in S. Typhimurium infected macrophages (Fig. 3A). Consistently, the gene expression of Ifnb1, Il6, Cxcl10, and many ISGs was also reduced in BX795-pretreated cells during infection (Fig. 3B).
FIG 3.

TBK1-IFN axis is required for the S. Typhimurium-induced type I IFN response. (A) Immunoblot analysis of protein expression in Raw264.7 cells untreated (con) or pretreated with BX795 (2 μM) for 2 h and uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 100. (B) qRT-PCR analysis of gene expression in PMs untreated (con) or pretreated with BX795 (2 μM) for 2 h and uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 10 (n = 3). (C) qRT-PCR analysis of gene expression in C57BL/6 or IFNAR1−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 2, 4, or 8 h at an MOI of 10 (n = 3). Data in (B) were normalized to untreated, mock-infected control and BX795-treated, mock-infected control, respectively (mock, both set as 1, not shown). Data in (C) were normalized to mock-infected C57BL/6 PMs and mock-infected IFNAR1−/− PMs, respectively (mock, both set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The induction of type I IFNs will induce the expression of downstream ISGs in both autocrine and paracrine fashions. Autocrine sensing of type I IFN by IFN-α/β receptor (IFNAR1/2) induces ISGs expression. To probe the role of IFNAR1 in the induction of type I IFN response during S. Typhimurium, we examined the expression of Ifnb1, Il6, Cxcl10, Irf7, Isg15, and Ifit2 in S. Typhimurium-infected WT or IFNAR1−/− PMs by qRT-PCR at different time points postinfection. As shown in Fig. 3C, the induction of all these genes was substantially reduced in IFNAR1−/− PMs at different time points postinfection. Remarkably, significantly reduced Ifnb1 was observed in IFNAR1−/− cells, suggesting an autocrine manner in Ifnb1 induction (Fig. 3C). Together, we demonstrated that S. Typhimurium infection elicits a robust type I IFN response through the TBK1-IFN axis.
S. Typhimurium infection provokes the cGAS-STING-dependent type I IFN response.
With the observation that many cytosolic immune sensing pathways were activated during infection (Fig. 2E), we speculated cytosolic innate immune sensors may contribute to the detection of S. Typhimurium. Accumulating evidence suggests the cytosolic DNA-sensing pathway cGAS-STING plays an important role in inducing type I IFN response during intracellular bacterial pathogen infection (13, 19, 37, 43–46). We hypothesized that intracellular pathogenic S. Typhimurium could also induce the type I IFN response through this pathway. To explore this, mouse PMs derived from C57BL/6, cGAS−/− or STING−/− mice were infected with S. Typhimurium. As expected, S. Typhimurium-induced IFN-β production was significantly decreased in cGAS or STING deficient PMs, indicating the role of the cGAS-STING pathway in mediating S. Typhimurium-elicited type I IFN response (Fig. 4A and Fig. S3A). Consistently, the gene expression of Ifnb1, proinflammatory cytokines, and many ISGs was also significantly reduced in cGAS−/− or STING−/− cells (Fig. 4B), suggesting the important role of cGAS-STING in mediating S. Typhimurium-elicited type I IFN response.
FIG 4.

cGAS-STING contributes to S. Typhimurium-induced type I IFN response. (A) ELISA analysis of IFN-β production in the supernatant from C57BL/6 (WT), cGAS−/− or STING−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 6 h at an MOI of 10 (n = 3). (B) and (C) qRT-PCR analysis of gene expression in C57BL/6 (WT), cGAS−/− or STING−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 10 (n = 3). Data in (B) and (C) were normalized to mock-infected C57BL/6 (WT) PMs, mock-infected cGAS−/− PMs, and mock-infected STING−/− PMs, respectively (mock, all set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
cGAS-STING is required for the type I IFN response during S. Typhimurium infection. (A) ELISA analysis of IFN-β production in the supernatant from C57BL/6 (WT), cGAS−/− or STING−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 2, 4, 6, or 8 h at an MOI of 10 (n = 3). (B) qRT-PCR analysis of gene expression in C57BL/6 (WT) or cGAS−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 2 and 4 h at an MOI of 10 (n = 3). (C) Immunoblot analysis of protein expression in C57BL/6 or STING−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 10, or transfected with dsDNA (Interferon stimulatory DNA, ISD) for 6 h. (D) qRT-PCR analysis of gene expression in C57BL/6 (WT) or STING−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 2 and 4 h at an MOI of 10 (n = 3). (E) qRT-PCR analysis of gene expression in cecum excised from S. Typhimurium-infected C57BL/6 (WT) or STING−/− mice at 24 or 72 h postinfection (n = 3). Data in (B) were normalized to mock-infected C57BL/6 (WT) PMs or mock-infected cGAS−/− PMs, respectively (mock, both set as 1). Data in (D) were normalized to mock-infected C57BL/6 (WT) PMs or mock-infected STING−/− PMs, respectively (mock, both set as 1). Data in (E) were normalized to mock-infected C57BL/6 (WT) cecum samples and mock-infected STING−/− cecum samples, respectively (mock, both set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Download FIG S3, TIF file, 2.0 MB (2.1MB, tif) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To further profile the type I IFN response elicited by S. Typhimurium in cGAS−/− cells, the expression pattern of many ISG early postinfection was detected. As expected, the induction of Ifnb1 and many ISGs was significantly attenuated in cGAS−/− PMs at 2 h.p.i and 4 h.p.i (Fig. S3B). Similarly, S. Typhimurium infection-induced expression of IRF3-responsive genes ISG15 and IFIT2 and phosphorylation of TBK1 was diminished in STING−/− cells (Fig. S3C). In addition, STING−/− mice derived PMs also exhibited significant lower type I IFN response during S. Typhimurium infection at 2 h.p.i and 4 h.p.i (Fig. S3D), suggesting the cGAS-STING pathway was also involved in the initial activation of type I IFN response during infection (Fig. S3B). Next, cecum samples were excised from PBS-treated or S. Typhimurium-infected WT and STING−/− mice at 24 or 72 h postinfection. A marked elevated ISG induction was observed at 72 h.p.i, whereas all ISGs were induced to a lower extent in S. Typhimurium-infected STING−/− mice (Fig. S3E). Together, these results demonstrated cGAS-STING pathway is a bona fide contributor to the host type I IFN response during S. Typhimurium infection.
Activation of the STING potentiates S. Typhimurium-induced type I IFN response.
Further experiments were performed to validate the role of STING in mediating type I IFN during S. Typhimurium infection. Mouse PMs were treated with 5,6-dimethylxanthenone-4-acetic acid (DMXAA, Vadimezan), a small molecule of mouse STING agonist, to activate the STING pathway (47). As shown in Fig. 5A, DMXAA treatment profoundly potentiated the S. Typhimurium-induced IRF3 and TBK1 phosphorylation. Furthermore, the gene expression of Ifnb1, proinflammatory cytokines, and ISGs was also significantly upregulated in DMXAA-treated S. Typhimurium-infected PMs compared with that in the untreated condition (Fig. 5A). Furthermore, cells were stimulated with 2′3′-cGAMP, the natural ligand of STING and the S. Typhimurium-induced expression of Ifnb1, proinflammatory cytokines and ISGs was also significantly activated in PMs and MEFs (Fig. 5B and Fig. S4A). Notably, the S. Typhimurium-induced type I IFN response was further enhanced by 2′3′-cGAMP-transfection in TLR4−/− cells (Fig. S4B). Collectively, these results demonstrated that the activation of the cGAS-STING cascade potentiated the S. Typhimurium-induced type I IFN response.
FIG 5.

Activation of STING potentiates S. Typhimurium-induced type I IFN response. (A) Immunoblot analysis of protein expression in Raw264.7 untreated (con) or pretreated with DMXAA (100 μg/mL) for 12 h and uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 100. (B) qRT-PCR analysis of gene expression in PMs untreated (con) or pretreated with DMXAA (100 μg/mL) for 12 h and uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 10 (n = 3). (C) PMs were untreated or lipofectamine-transfected with 5 μM 2′3′-cGAMP (cGAMP) for 4 h and then uninfected (mock) or infected with S. Typhimurium at an MOI of 10. At 6 h postinfection, qRT-PCR analysis of gene expression (n = 3). Data in (B) were normalized to untreated, mock-infected control (mock, set as 1, not shown). Data in (C) were normalized to untreated, mock-infected control (mock, set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Activation of the STING potentiates S. Typhimurium-induced type I IFN response. (A) Primary MEFs were untreated (S. T.) or lipofectamine-transfected with 5 μM 2′3′-cGAMP (S. T. + cGAMP) for 4 h and then infected with S. Typhimurium at an MOI of 100. At 6 h postinfection, qRT-PCR analysis of gene expression (n = 3). (B) TLR4−/− PMs were untreated or lipofectamine-transfected with 5 μM 2′3′-cGAMP (cGAMP) for 4 h and then uninfected (mock) or infected with S. Typhimurium at an MOI of 10. At 6 h postinfection, qRT-PCR analysis of gene expression (n = 3). Data in (A) were normalized to untreated, mock-infected control and 2′3′-cGAMP-transfected, mock-infected control, respectively (mock, both set as 1, not shown). Data in (B) were normalized to untreated, mock-infected control (mock, set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Download FIG S4, TIF file, 0.8 MB (858.2KB, tif) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
S. Typhimurium triggers mtDNA release to induce type I IFN response.
Emerging studies suggest the self-DNA like mitochondrial DNA (mtDNA) abnormally present within the cytosol also stimulates the cGAS-STING pathway (17, 48). In addition, Salmonella can target mitochondria, possibly using its type III secretion system (T3SS) effectors (49–51). Therefore, we hypothesized that S. Typhimurium infection can lead to the release of mtDNA into cytosol that engages cGAS in macrophages. To explore this, the mitochondrial membrane potential (MMP) in S. Typhimurium-infected cells was measured, which acts as an indicator of mitochondrial injury. The MMP was significantly decreased during S. Typhimurium infection by using the TMRE (tetramethylrhodamine ethyl ester) (Fig. 6A and B), a positively charged dye that fails to retain in mitochondria has decreased membrane potential (52). Of note, although in Fig. 6A, fewer cells were captured in S. Typhimurium-infected condition, we calculated the average Integrated density of TMRE fluorescence from 20 different cells, respectively, with 3 independent experiments. We found that the TMRE-associated signal was significantly lower in infected cells. In addition, JC-1 dye, which forms polymers in mitochondria with normal MMP and is released from the mitochondria to form monomers with reduced MMP, was also employed (53). The formation of JC-1 monomer in S. Typhimurium-infected cells further confirmed the reduction of MMP (Fig. S5A).
FIG 6.

S. Typhimurium triggers mtDNA release to induce the type I IFN response. (A) Fluorescence microscopy analysis of mitochondrial membrane potential by assessing TMRE in Raw264.7 cells uninfected (mock) or infected with S. Typhimurium for 2 h at an MOI of 100. Scale bar: 20 μm. (B) The average integrated density of TMRE fluorescence. The average density was obtained from 20 individual cells for different conditions. 3 independent experiments were performed. (C) Immunoblot analysis of protein expression in different fractions in Raw264.7 cells uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 10 or 100. Cells were treated with Tunicamycin (4 μg/mL) for 8 h as a positive-control for mtDNA release. (D) qPCR analysis of mtDNA in the cytosolic fraction of Raw264.7 cells uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 10 or 100 (n = 3). (E) qRT-PCR analysis of gene expression in EB-treated and untreated (con) Raw264.7 cells uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 100 (n = 3). (F) qRT-PCR analysis of gene expression in PMs pretreated with Mito Tempo (100 μM) or NecroX-5 (40 μM) for 2 h and uninfected (mock) or infected with S. Typhimurium for 6 h at an MOI of 10 (n = 3). Data in (D) were normalized to mock-infected control (con, set as 1). Data in (E) and (F) were normalized to untreated, mock-infected control and pretreated, mock-infected control, respectively (mock, all set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
S. Typhimurium-induced mtDNA release is involved in the induction of type I IFN. (A) Fluorescence microscopy analysis of mitochondrial membrane potential by assessing JC-1 aggregates and JC-1 monomer in HeLa uninfected (mock) or infected with S. Typhimurium for 2 h at an MOI of 10. Green: JC-1 monomer; Red: JC-1 aggregates. Scale bar: 75 μm. Right: fluorescence spectrophotometer analysis of the ratio of JC-1 aggregates/JC-1 monomer in HeLa cells uninfected (mock) or infected with S. Typhimurium for 2 h at an MOI of 10 or 100 (n = 3). (B) Fluorescence microscopy analysis of the localization of mitochondria and S. Typhimurium in Raw264.7 cells infected with S. Typhimurium for 2 h at an MOI of 100. Hoechst33342: nuclei; GFP, Mito-tracker green to indicate mitochondria; mCherry, S. Typhimurium carries a mCherry expressing plasmid. Scale Bar: 20 μm. (C) qPCR analysis of mtDNA in the cytosolic fraction of C57BL/6 (WT) or TLR4−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 100 (n = 3). (D) qPCR analysis of the total mtDNA in EB-treated and untreated (con) Raw264.7 cells (n = 3). (E) qPCR analysis of mtDNA in the cytosolic fraction of PMs pretreated with Mito Tempo (100 μM) or NecroX-5 (40 μM) for 2 h and uninfected (mock) or infected with S. Typhimurium for 6 h at an MOI of 100 (n = 3). (F) qRT-PCR analysis of gene expression in TLR4−/− PMs pretreated with Mito Tempo (100 μM) for 2 h and uninfected (mock) or infected with S. Typhimurium for 6 h at an MOI of 10 (n = 3). Data in (C) were normalized to uninfected C57BL/6 (WT) and TLR4−/− PMs, respectively (mock, both set as 1). Data in (D) were normalized to untreated control (con, set as 1). Data in (E) were normalized to untreated mock-infected control (mock, set as 1). Data in (F) were normalized to untreated, mock-infected control and Mito Tempo pretreated, mock-infected control, respectively (mock, both set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. NS, not significant. Download FIG S5, TIF file, 2.4 MB (2.4MB, tif) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Next, to directly probe the release of mtDNA into the cytosol during S. Typhimurium infection. Different fractions of S. Typhimurium infected cells were separated and the immunoblot analysis confirmed the isolation of the cytoplasm fraction, with no cross-contamination of mitochondria-associated proteins in the cytosolic fractions (Fig. 6C). S. Typhimurium infection results in the release of mtDNA into the cytosol, as manifested by a higher level of mitochondrial genes in cytosolic fractions (Fig. 6D), although no colocalization between S. Typhimurium and mitochondria was detected (Fig. S5B). The Tunicamycin that inhibits GlcNAc phosphotransferase is used as a positive-control for mtDNA release (54). Interestingly, the release of mtDNA into the cytosol was also detected in TLR4−/− PMs (Fig. S5C).
To explore the requirement of mtDNA for S. Typhimurium-induced type I IFN response, Raw264.7 cells were treated with Ethidium bromide (EtBr) to delete the mtDNA (54). MtDNA depletion in EtBr-treated Raw264.7 cells was confirmed by qPCR (Fig. S5D). After removing the EtBr, both untreated and EtBr-treated cells were infected by S. Typhimurium. The induction of type I IFN response was significantly attenuated in EtBr-treated cells (Fig. 6E). This result proved that mtDNA is important for the activation of type I IFN response during S. Typhimurium in murine macrophages. To further explore the release of mtDNA could active type I IFN response, Raw264.7 cells were treated with a mitochondrial-targeted antioxidant, Mito Tempo ([2-(2,2,6,6-tetramethylpiperidin-1-oxyl-4-ylamino)-2-oxoethyl] triphenylphosphonium chloride), to reduce the release of mtDNA into the cytoplasm (Fig. S5E) (55). Notably, S. Typhimurium-induced type I IFN response was significantly decreased in Mito Tempo treated WT cells, further supporting the notion that mtDNA release elicited the type I IFN response (Fig. 6F). Importantly, this Mito Tempo also inhibits S. Typhimurium-induced type I IFN response in TLR4−/− PMs (Fig. S5F). Furthermore, NecroX-5, an inhibitor that scavenges mitochondrial reactive oxygen (56), also reduced mtDNA release and type I IFN response during S. Typhimurium infection (Fig. 6F and Fig. S5E). Collectively, these results indicated that S. Typhimurium infection induces the release of mtDNA to the cytosol and mtDNA can act as the danger signal to induce type I IFN pathways.
Bacterial DNA binds to cGAS during S. Typhimurium infection.
Pathogenic cytoplasmic DNAs bind to cGAS to produce 2′3′-cGAMP that activates STING and downstream signaling pathways (48, 57). Direct transfection of S. Typhimurium genomic DNA induces type I IFN response in a STING-dependent fashion in macrophages (Fig. S6A), suggesting the bacterial genomic DNA may act as a PAMPs. Moreover, we pretreated PMs with bafilomycin A1 (BafA1, a specific inhibitor of the vacuolar H+ ATPase that interferes with endosomal acidification and changes the formation of phagolysosomes) and chymostatin (an inhibitor of lysosomal serine and cysteine proteinases) (58) and found that those pretreated cells exhibited reduced type I IFN induction during S. Typhimurium infection (Fig. 7A). This indicates the degradation of bacteria in phagolysosomes contributes to the activation of type I IFN response during infection.
FIG 7.

Bacterial DNA binds to cGAS during S. Typhimurium infection. (A) qRT-PCR analysis of gene expression in PMs untreated (con) or pretreated with BafA1 (200 nM) or chymostatin (100 μM) for 2 h and uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 10 (n = 3). (B) qPCR analysis of S. Typhimurium-derived sequences (dnaE and sciK) in the cytosolic fraction of PMs uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 100 (n = 3). ND, not detected. (C) Raw264.7 cells were uninfected or infected with S. Typhimurium for 8 h at an MOI of 100, then immunoprecipitated with IgG or cGAS antibody. qPCR of S. Typhimurium-derived sequences (sciK and dnaE) from DNA isolated from IPs (n = 3). Data in (A) were normalized to untreated, mock-infected control and BafA1-treated, mock-infected, and chymostatin-treated, mock-infected, respectively (mock, all set as 1, not shown). Quantities in (C) were normalized to inputs. Actin was used as the housekeeping gene. Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
S. Typhimurium DNA induces type I IFN response and the cGAS-STING pathway constrains S. Typhimurium infection. (A) qRT-PCR analysis of gene expression in C57BL/6 (WT) or STING−/− mouse-derived PMs transfected with S. Typhimurium genomic DNA (1 μg per well in 24-well plate) for 6 h (n = 3). (B) Immunoblotting of lysates of Raw264.7 cells was uninfected or infected with S. Typhimurium for 8 h at an MOI of 100, then immunoprecipitated with IgG or cGAS antibody. (C) qPCR of mtDNA sequences (Cox1, Cox2, and Cox3) from DNA isolated from IPs in (B) (n = 3). (D) Fluorescence microscopy analysis of C57BL/6 or cGAS−/− mouse-derived PMs infected with S. Typhimurium for 2 h at an MOI of 10. Hoechst33342: nuclei, GFP, S. Typhimurium carries a GFP expressing plasmid. Scale bar: 75 μm. The ratio of GFP fluorescence integrated density to Hoechst33342 fluorescence integrated density is on the right (n = 3). (E) qPCR analysis of S. Typhimurium 16S rRNA level in primary MEFs untreated (con) or pretreated with DMXAA (100 μg/mL) for 12 h, or pretreated lipofectamine-transfected with 5 μM 2′3′-cGAMP (cGAMP) for 4 h and then infected with S. Typhimurium for 8 h at an MOI of 100 (n = 3). (F) qPCR analysis of S. Typhimurium 16S rRNA level in TLR4−/− PMs untreated (con) or pretreated lipofectamine-transfected with 5 μM 2′3′-cGAMP (cGAMP) for 4 h and infected with S. Typhimurium for 8 h at an MOI of 100 (n = 3). Data in (A) were normalized to mock-transfected C57BL/6 (WT) PMs and mock-transfected STING−/− PMs, respectively (mock, both set as 1). Quantities in (C) were normalized to inputs. Data in (E and F) were normalized to untreated S. Typhimurium-infected control (con, set as 1). Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Download FIG S6, TIF file, 1.5 MB (1.5MB, tif) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Next, we probed the S. Typhimurium genes in the cytosolic fraction during S. Typhimurium infection. As shown in Fig. 7B, S. Typhimurium DNA was detected in the cytosolic fraction during S. Typhimurium infection. Together with the detection of mtDNA in the cytosolic fraction (Fig. 6D), these results suggest both mtDNA and S. Typhimurium DNA contribute to cGAS activation. To further explore factors that bind to cGAS during S. Typhimurium infection, a coimmunoprecipitation assay that can detect the DNA sequences enriched in cGAS was employed (19, 31). Successful immunoprecipitation of cGAS was confirmed by immunoblotting (Fig. S6B). Notably, two S. Typhimurium genes sciK and dnaE were detected in cGAS compared to IgG controls (Fig. 7C), suggesting S. Typhimurium DNA also binds to cGAS during infection. Subsequently, to distinguish the contribution of mtDNA and bacterial genomic DNA in activating cGAS, we aimed to measure mtDNA in cGAS immunoprecipitates. However, all cGAS immunoprecipitates were positive for mtDNA sequences and the abundance is similar (Fig. S6C). This may be because the strong lysis buffer causes the deconstruction of mitochondria. Together, physical interactions between cGAS and S. Typhimurium DNA have been identified in the context of infection. However, the relative contribution of bacterial DNA and mtDNA to cGAS activation needs further investigation.
cGAS-STING pathway contributes to host defenses against S. Typhimurium infection.
We further explored whether cGAS-STING contributes to the host defense against S. Typhimurium infection. To this end, WT and STING−/− mice were intragastrically infected with 1 × 108 CFU (CFU) of S. Typhimurium and the lethality was monitored over time. As shown in Fig. 8A, STING−/− mice showed an early mortality rate compared with WT mice. On day 6, more than 60% of STING−/− mice had died while the rate in WT mice was less than 20%, which is consistent with the previous study (59). Furthermore, we examined the bacterial load in different tissues of WT, cGAS−/− and STING−/− mice at 120 h postinfection. Significantly increased bacterial burden was detected from the spleen and liver of cGAS−/− and STING−/− mice, further fostering the role of cGAS-STING in defending S. Typhimurium infection (Fig. 8B).
FIG 8.

cGAS-STING contributes to host defense against S. Typhimurium infection. (A) and (B) C57BL/6, cGAS−/−, STING−/− mice were intragastrically inoculated with 1 × 108 CFU S. Typhimurium. (A) The survival rate of the mice was determined (C57BL/6, n = 10; STING−/−, n = 10). (B) Homogenates of the liver and spleen were plated to determine the bacterial CFU counts per gram in the indicated organs at 120 h postinfection (n = 6). (C) Fluorescence microscopy analysis of C57BL/6 or STING−/− mouse-derived PMs infected with S. Typhimurium for 2 h or 4 h at an MOI of 10. Hoechst33342: nuclei, GFP, S. Typhimurium carry a GFP expressing plasmid. Scale bar: 75 μm. The ratio of GFP fluorescence integrated density to Hoechst33342 fluorescence integrated density is on the right (n = 3). (D) qRT-PCR analysis of S. Typhimurium 16S rRNA level in PMs untreated (con) or pretreated with DMXAA (100 μg/mL) for 12 h, or pretreated lipofectamine-transfected with 5 μM 2′3′-cGAMP (cGAMP) for 4 h and infected with S. Typhimurium for 8 h at an MOI of 100 (n = 3). Data in (D) were normalized to untreated S. Typhimurium-infected control (S. T, set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *, P < 0.05; **, P < 0.01.
To probe the role of cGAS-STING in effecting intracellular growth of S. Typhimurium in murine macrophages, S. Typhimurium with a GFP plasmid was used to infect WT, cGAS−/− or STING−/− PMs and the GFP signal in S. Typhimurium-infected cells was imaged. Fluorescence microscopy illustrated that the S. Typhimurium-infected cGAS−/− and STING−/− cells contained stronger GFP fluorescence compared with those S. Typhimurium-infected WT cells (Fig. 8C and Fig. S6D). These findings demonstrated that the lack of cGAS-STING led to an improved S. Typhimurium infection in mice models and intracellular growth in murine macrophages.
To explore whether the activation of the cGAS-STING pathway could eradicate intracellular S. Typhimurium growth, STING agonist DMXAA was used to pretreat cells and the reduced S. Typhimurium 16s RNA level was obtained in DMXAA-treated PMs and MEFs (Fig. 8D). Furthermore, the 2′3′-cGAMP transfection induced STING activation also decreased intracellular S. Typhimurium growth level in PMs (Fig. 8D) and primary MEFs (Fig. S6E). Intriguingly, cGAMP transfection in TLR4−/− PMs also decreased intracellular S. Typhimurium growth (Fig. S6F). Taken together, these results proved that cGAS-STING plays an important role in the host defense against S. Typhimurium infection.
DISCUSSION
Salmonella induces a robust innate immune response in a TLR-dependent manner (3, 46). In addition to TLRs, cytosolic NLRs, including NLRP3 and NLRC4 are both activated during S. Typhimurium infection (5) and S. Typhimurium mRNA might act as a danger signal that induces the activation of NLRP3 (10). We also noticed macrophages derived from mice that lack the surface receptors TLR4 exhibited substantially attenuated type I IFN responses during S. Typhimurium infection (Fig. 2). However, whether other innate immune sensing pathways are involved during S. Typhimurium in macrophages remains elusive. In this study, we showed that the cytosolic surveillance pathway cGAS-STING was critically involved in the induction of the type I IFN response through the TBK1-IFN cascade.
To delineate the contribution of the different signaling pathways that mediate the type I IFN response during infection, the cGAS−/−, STING−/− and TLR4−/− mice derived PMs were used to infect S. Typhimurium and the type I IFN and ISG induction was measured at different time postinfection. The lack of TLR4 substantially attenuated the type I IFN and ISG expression from 2 h.p.i to 8 h.p.i (Fig. S2C). However, there is still significant induction of Ifnb1, proinflammatory cytokines, and ISGs in TLR4−/− cells, suggesting the TLR-independent recognition mechanisms during S. Typhimurium infection. The release of mtDNA in TLR4−/− PMs (Fig. S5F) and reduced type I IFN response in Mito Tempo treated TLR4−/− PMs (Fig. S6B) further underscored the TLR-independent mechanism. Intriguingly, compared to WT cells, the type I IFN response was also significantly reduced in cGAS−/− or STING−/− cells, which is consistent with a previous study that demonstrated a clear reduction of both IFN-β and IFIT1 mRNAs in S. Typhimurium-infected cGAS−/− and STING−/− BMDMs (19). In particular, the S. Typhimurium-induced Ifnb1 expression was attenuated in cGAS−/− or STING−/− PMs from 2 h.p.i, suggesting the cGAS-STING pathway was also involved in the initial activation of type I IFN response during infection.
It is not surprising that intracellular bacteria induce host innate immune response in STING-dependent manners since bacterial CDNs like c-di-GMP or c-di-AMP are efficiently recognized by STING, which leads to the production of type I IFN (12–14, 60). However, the induction of type I IFN by most bacteria is largely dependent on cGAS (19, 22–31). This may be due to the possibility that bacterial CDNs are constrained in bacterial cells, which are not easily accessed to the cytoplasm. In addition, c-di-GMP in intracellular S. Typhimurium was maintained at low levels (61), possibly to avoid the cytosolic detection of bacterial infection.
Both pathogenic DNAs and self-DNA like mitochondrial DNA (mtDNA) within the cytosol can stimulate the cGAS-STING pathway (17, 48, 57). The S. Typhimurium genomic DNA may also activate cGAS, supported by the observation that the transfection of S. Typhimurium DNA into macrophages can also provoke type I IFN response (Fig. S6A) and bafilomycin A1 and chymostatin pretreated reduced type I IFN induction during infection (Fig. 7A). Speculatively, phagocytosis of S. Typhimurium may liberate some genomic DNA as PAMPs from bacteria. Coupled with this speculation, we also detected S. Typhimurium genes in the cytosolic fraction and showed physical interactions between cGAS and S. Typhimurium DNA in the context of infection (Fig. 7B and C). This observation indicates S. Typhimurium DNA also contributes to cGAS activation.
Concomitantly, we demonstrated that mtDNA is also an important stimulus that activates the cGAS-STING pathway during S. Typhimurium infection (Fig. 6 and Fig. S5). Importantly, in EtBr-treated mtDNA depletion cells, induction of type I IFN response was significantly attenuated during S. Typhimurium infection (Fig. 6E), further fostering the notion that mtDNA is important for the activation of type I IFN response. However, the temptation to measure mtDNA in the cGAS immunoprecipitates to determine the abundance of mtDNA enriched in cGAS was not succeeded, making it difficult to distinguish the relative contribution of mtDNA and S. Typhimurium DNA in cGAS activation, and such a question needs further investigations.
Intriguingly, the release of mtDNA into cytosol can be induced by internalized bacterial endotoxin LPS, which activated the Gasdermin D to form pores in mitochondria (62). Here, we demonstrated S. Typhimurium infection results in the release of mtDNA into the cytosol (Fig. 6D) as internalized S. Typhimurium might induce mtDNA release through Gasdermin D. In addition, it has been reported that Salmonella can target mitochondria by deploying T3SS effectors (49). S. Typhimurium secreted a T3SS effector SipB with membrane fusion activity to target mitochondria (50). Another T3SS effector SopA was also proved to target mitochondria in HeLa or COS cells (51). These pieces of evidence imply that S. Typhimurium may cause mitochondria damage that led to the release of mtDNA. Similarly, Mycobacterium abscessus and Streptococcus pneumoniae infection also lead to the release of mtDNA into the cytosol of murine macrophages, which activates the cGAS-STING-dependent type I IFN production (55, 63). Building on this information, it is likely that the liberating of mtDNA into the cytosol may represent a universal mechanism to activate the cGAS-STING pathway during intracellular bacterial infection, albeit further experimental evidence was required to confirm this notion.
It has been shown that STING−/− mice are highly susceptible to S. Typhimurium infection compared with WT animals with the perspective of the protective role of STING in regulating intestinal homeostasis (59). In addition, the STING-IRF1 signaling axis is required for the control of mucosal TH17 cell responses that have protective effects against S. Typhimurium infection (64). In particular, the uptake and transport of S. Typhimurium to MLNs (mesenteric lymph nodes) were slightly enhanced in STING−/− mice compared to WT mice, albite no statistical significance (64). In this study, in vivo, and in vitro experiments showed the cGAS-STING pathway was potently implicated in the host defense against S. Typhimurium infection. We further demonstrated that the activation of the STING potently inhibits S. Typhimurium growth (Fig. 8D and Fig. S6E and F). Furthermore, we also noticed that in TLR4−/− PMs, S. Typhimurium-induced type I IFN response was further enhanced by 2′3′-cGAMP-transfection and cGAMP transfection also inhibited S. Typhimurium infection (Fig. S4B and S6F), suggesting the cGAS-STING pathway plays an important role in the host defense against S. Typhimurium infection in addition to TLR4 pathway. Type I IFN production is the main feature of cGAS-STING activation, but both protective and detrimental role of type I IFN has been found during S. Typhimurium infection (65, 66). Thus, the underlying mechanisms of cGAS-STING against S. Typhimurium infection need further investigations.
In conclusion, we demonstrated that in addition to TLR4-dependent response, S. Typhimurium could induce type I IFN response in a STING-dependent manner by triggering mtDNA release (Fig. 9). This study highlighted the important role of cGAS-STING in meditating type I IFN response and host defense in the context of S. Typhimurium infection. Furthermore, this study uncovered a mechanism by which type I IFN response is elicited through the liberating of mtDNA into the cytosol during S. Typhimurium infection. Our new paradigms deciphering the requirement of the cGAS-STING pathway largely expanded the current understanding of S. Typhimurium pathogenesis and innate immunity.
FIG 9.
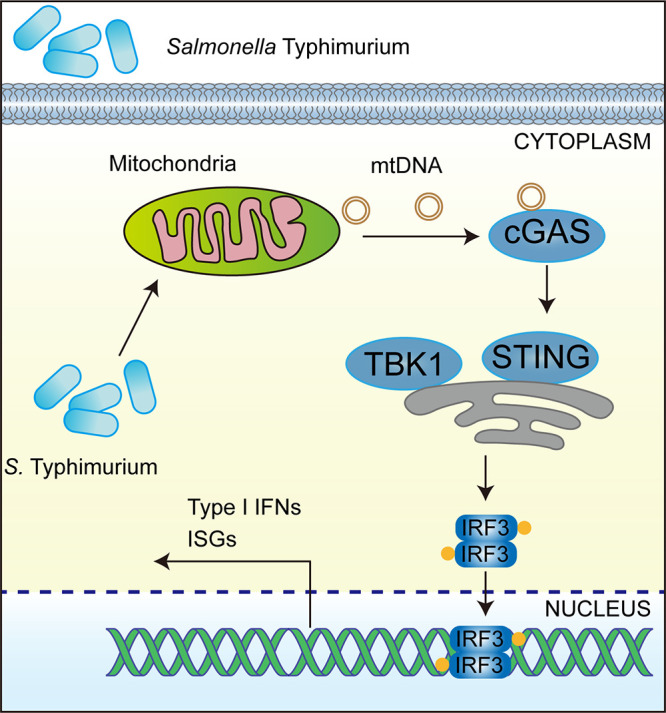
S. Typhimurium triggers the cGAS-STING-dependent type I IFN response by inducing mtDNA release. In murine macrophages, S. Typhimurium infection led to the release of mtDNA into cytosol that possibly engages cytosolic DNA sensor cGAS. The activated cGAS led to the activation of immune adaptor protein STING and phosphorylation of kinase TBK1, which activates the transcription factor IRF3. Translocation of IRF3 resulted in transcriptional induction of the type I IFN response.
MATERIALS AND METHODS
Mice.
Mice used in this study were on a C57BL/6 background. WT C57BL/6 mice were purchased from the Animal Center of Xi’an Jiaotong University. cGAS−/−, STING−/− mice, and IFNAR1−/− mice based on the C57BL/6 background were kindly provided by Zhengfan Jiang (67). C57BL/10ScNJGpt (TLR4−/−) and wild-type C57BL/10JGpt mice were purchased from GemPharmatech, Jiangsu, China. All the mouse experimental procedures were performed under the Regulations for the Administration of Affairs Concerning Experimental Animals approved by the State Council of the People’s Republic of China. The protocol was approved by the Animal Welfare and Research Ethics Committee of Northwest A&F University (protocol number: NWAFUSM2018001). Male and female mice were sex-matched and used at 6 to 12 weeks of age and were kept under specific pathogen-free conditions.
S. Typhimurium infection.
S. Typhimurium was cultured in LB overnight at 37°C with 100 μg/mL streptomycin or 50 μg/mL kanamycin (for strains carried pKT100 plasmids). The following day, pelleted bacteria at 4500 rpm by centrifugation and washed bacteria 3 times with sterile PBS and resuspended in RPMI 1640 or DMEM medium. When the OD600 = 2.5, there are approximately 108 CFU S. Typhimurium in 1 mL resuspension. Different volumes of bacterial resuspension were mixed with antibiotic-free RPMI 1640 or DMEM to obtain the desired multiplicity of infection (MOI). Peritoneal macrophages (PMs) were washed 3 times with sterile PBS before infection and cultured in RPMI 1640 medium without FBS and penicillin-streptomycin before infection. The cells were infected at indicated MOI and then the plate was centrifuged at 300 × g for 5 min to synchronize infection (34). Infected cells were incubated at 37°C for 20 min and then the infection medium was removed, and the cells were washed 3 times by sterile PBS and replaced with RPMI 1640 containing FBS (10%) and gentamicin (200 μg/mL) for indicated times to kill extracellular bacteria. Raw264.7, HeLa, and primary mouse embryonic fibroblast (MEF) cells were cultured in DMEM and changed to antibiotic-free and FBS-free DMEM medium. Infected cells were incubated at 37°C for 2 h and then the infection medium was removed and replaced with DMEM containing FBS (10%) and gentamicin (200 μg/mL) for indicated times to kill extracellular bacteria.
For mice infection, food and water were deprived 12 h before infection. Mid-exponential-phase S. Typhimurium was collected and washed twice in sterile PBS. Each mouse was intragastrically infected with 1 × 108 CFU S. Typhimurium by ball-tipped feeding needles and food and water were resupplied from 12 h postinfection. For survival assays, the daily survival was observed and recorded for 2 weeks to calculate the death rate of the mice in different groups (59). For the detection of bacterial load in different organs, mice were sacrificed by carbon dioxide asphyxiation followed by cervical dislocation at 120 h postinfection. Different tissues were weighed and homogenized in 0.9% NaCl, and serial dilutions of the homogenates were plated on LB plates with 200 μg/mL streptomycin.
The rest of the Materials and Methods are described in detail in Text S1 and Table S2. The uncropped versions of immunoblotting results were provided in Fig. S7.
Unprocessed western blots results. Download FIG S7, JPG file, 1.07 MB (1.1MB, jpg) .
{kind=link}
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Bacterial strains, cells, mice, plasmids, primers, reagents, and resources used in this study. Download Table S2, XLSX file, 0.02 MB (22.2KB, xlsx) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental materials and methods. Download Text S1, DOCX file, 0.05 MB (50.9KB, docx) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
ACKNOWLEDGMENTS
We thank Zhengfan Jiang (Beijing University) for generously providing the cGAS−/−, STING−/− mice, IFNAR1−/− mice, and HeLa cells. We thank the Teaching and Research Core Facility at the College of Life Science (Min Duan, Ningjuan Fan, Xiyan Chen, and Hui Duan) and Life Science Research Core Services (LSRCS), Northwest A&F University for the technical support.
This work was supported by the grant of the National Natural Science Foundation of China (31800113 to L.X., 31725003 and 31670053 to X.S., 31970114 and 32170130 to Y.W.), the Open Project Program of the State Key Laboratory of Pathogen and Biosecurity (SKLPBS1825 to X.S.), China Postdoctoral Science Foundation (2018M631201 to L.X. and 2020T130543 to L.X.), Chinese Universities Scientific Fund (the Starting Research Fund from the Northwest A&F University, No. 2452018045 to L.X.), Shaanxi Postdoctoral Science Foundation (2018BSHTDZZ20 to L.X.) and Natural Science Basis Research Plan in Shaanxi Province of China (2020JQ-245 to L.X.).
L.X., X.S., and Y.W. conceived the ideas and designed the experiments. L.X. and M.L. performed the majority of experiments. Y.Y., C.Z., Z.X., J.T., Z.S., S.C., G.L., F.Z., Y.G., and X.W. helped in in vivo infection, cell culture, molecular biology, and biochemical experiments. Y.Y. performed the computational analyses and provided technical support. L.X. and M.L. analyzed data and wrote the paper. X.S. and Y.W. critically revised the manuscript. X.S. supervised the study. All authors discussed the results and commented on the manuscript.
We declare no conflict of interest.
Contributor Information
Xihui Shen, Email: xihuishen@nwsuaf.edu.cn.
Alejandro Aballay, School of Medicine, Oregon Health and Science University.
REFERENCES
- 1.Wain J, Hendriksen RS, Mikoleit ML, Keddy KH, Ochiai RL. 2015. Typhoid fever. Lancet 385:1136–1145. doi: 10.1016/S0140-6736(13)62708-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Royle MC, Totemeyer S, Alldridge LC, Maskell DJ, Bryant CE. 2003. Stimulation of Toll-like receptor 4 by lipopolysaccharide during cellular invasion by live Salmonella typhimurium is a critical but not exclusive event leading to macrophage responses. J Immunol 170:5445–5454. doi: 10.4049/jimmunol.170.11.5445. [DOI] [PubMed] [Google Scholar]
- 3.Miller SI, Ernst RK, Bader MW. 2005. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol 3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 4.Keestra-Gounder AM, Tsolis RM, Baumler AJ. 2015. Now you see me, now you don't: the interaction of Salmonella with innate immune receptors. Nat Rev Microbiol 13:206–216. doi: 10.1038/nrmicro3428. [DOI] [PubMed] [Google Scholar]
- 5.Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. 2010. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med 207:1745–1755. doi: 10.1084/jem.20100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, Wright JA, Cicuta P, Monie TP, Bryant CE. 2014. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci USA 111:7403–7408. doi: 10.1073/pnas.1402911111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gram AM, Wright JA, Pickering RJ, Lam NL, Booty LM, Webster SJ, Bryant CE. 2021. Salmonella flagellin activates NAIP/NLRC4 and canonical NLRP3 inflammasomes in human macrophages. J Immunol 206:631–640. doi: 10.4049/jimmunol.2000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A. 2010. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA 107:3076–3080. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 10.Sander LE, Davis MJ, Boekschoten MV, Amsen D, Dascher CC, Ryffel B, Swanson JA, Muller M, Blander JM. 2011. Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature 474:385–389. doi: 10.1038/nature10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keestra AM, Winter MG, Auburger JJ, Frassle SP, Xavier MN, Winter SE, Kim A, Poon V, Ravesloot MM, Waldenmaier JF, Tsolis RM, Eigenheer RA, Baumler AJ. 2013. Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1. Nature 496:233–237. doi: 10.1038/nature12025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. 2011. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodward JJ, Iavarone AT, Portnoy DA. 2010. C-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, Vance RE. 2011. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun 79:688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V. 2013. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature 498:380–384. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS. 2015. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. 2012. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11:469–480. doi: 10.1016/j.chom.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, Cox JS. 2015. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type i interferons and activate autophagy. Cell Host Microbe 17:811–819. doi: 10.1016/j.chom.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watson RO, Manzanillo PS, Cox JS. 2012. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150:803–815. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hopfner KP, Hornung V. 2020. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol 21:501–521. doi: 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- 22.Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, Schmid-Burgk JL, Schmidt T, Hornung V, Cole ST, Ablasser A. 2015. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe 17:799–810. doi: 10.1016/j.chom.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 23.Hansen K, Prabakaran T, Laustsen A, Jørgensen SE, Rahbæk SH, Jensen SB, Nielsen R, Leber JH, Decker T, Horan KA, Jakobsen MR, Paludan SR. 2014. Listeria monocytogenes induces IFNbeta expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J 33:1654–1666. doi: 10.15252/embj.201488029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. 2015. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18:157–168. doi: 10.1016/j.chom.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Storek KM, Gertsvolf NA, Ohlson MB, Monack DM. 2015. cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. J Immunol 194:3236–3245. doi: 10.4049/jimmunol.1402764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Yeruva L, Marinov A, Prantner D, Wyrick PB, Lupashin V, Nagarajan UM. 2014. The DNA sensor, cyclic GMP-AMP synthase, is essential for induction of IFN-beta during Chlamydia trachomatis infection. J Immunol 193:2394–2404. doi: 10.4049/jimmunol.1302718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barker JR, Koestler BJ, Carpenter VK, Burdette DL, Waters CM, Vance RE, Valdivia RH. 2013. STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. mBio 4:e00018-13–e00013. doi: 10.1128/mBio.00018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andrade WA, Agarwal S, Mo S, Shaffer SA, Dillard JP, Schmidt T, Hornung V, Fitzgerald KA, Kurt-Jones EA, Golenbock DT. 2016. Type I interferon induction by Neisseria gonorrhoeae: dual requirement of cyclic GMP-AMP synthase and Toll-like receptor 4. Cell Rep 15:2438–2448. doi: 10.1016/j.celrep.2016.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andrade WA, Firon A, Schmidt T, Hornung V, Fitzgerald KA, Kurt-Jones EA, Trieu-Cuot P, Golenbock DT, Kaminski PA. 2016. Group B Streptococcus degrades cyclic-di-AMP to modulate STING-dependent type I interferon production. Cell Host Microbe 20:49–59. doi: 10.1016/j.chom.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu ZZ, Yang YJ, Zhou CK, Yan SQ, Ma K, Gao Y, Chen W. 2021. STING contributes to host defense against Staphylococcus aureus pneumonia through suppressing necroptosis. Front Immunol 12:636861. doi: 10.3389/fimmu.2021.636861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou CM, Wang B, Wu Q, Lin P, Qin SG, Pu QQ, Yu XJ, Wu M. 2021. Identification of cGAS as an innate immune sensor of extracellular bacterium Pseudomonas aeruginosa. iScience 24:101928. doi: 10.1016/j.isci.2020.101928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hess CB, Niesel DW, Klimpel GR. 1989. The induction of interferon production in fibroblasts by invasive bacteria: a comparison of Salmonella and Shigella species. Microb Pathog 7:111–120. doi: 10.1016/0882-4010(89)90030-2. [DOI] [PubMed] [Google Scholar]
- 33.Kawashima T, Kosaka A, Yan H, Guo Z, Uchiyama R, Fukui R, Kaneko D, Kumagai Y, You DJ, Carreras J, Uematsu S, Jang MH, Takeuchi O, Kaisho T, Akira S, Miyake K, Tsutsui H, Saito T, Nishimura I, Tsuji NM. 2013. Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-beta. Immunity 38:1187–1197. doi: 10.1016/j.immuni.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 34.Perkins DJ, Rajaiah R, Tennant SM, Ramachandran G, Higginson EE, Dyson TN, Vogel SN. 2015. Salmonella Typhimurium co-opts the host type I IFN system to restrict macrophage innate immune transcriptional responses selectively. J Immunol 195:2461–2471. doi: 10.4049/jimmunol.1500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, Roose-Girma M, Bumann D, Kayagaki N, Takeda K, Yamamoto M, Broz P. 2014. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509:366–370. doi: 10.1038/nature13157. [DOI] [PubMed] [Google Scholar]
- 36.McLaughlin PA, Bettke JA, Tam JW, Leeds J, Bliska JB, Butler BP, van der Velden AWM. 2019. Inflammatory monocytes provide a niche for Salmonella expansion in the lumen of the inflamed intestine. PLoS Pathog 15:e1007847. doi: 10.1371/journal.ppat.1007847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoffpauir CT, Bell SL, West KO, Jing T, Wagner AR, Torres-Odio S, Cox JS, West AP, Li P, Patrick KL, Watson RO. 2020. TRIM14 is a key regulator of the type I IFN response during Mycobacterium tuberculosis infection. J Immunol 205:153–167. doi: 10.4049/jimmunol.1901511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uchiya KI, Kamimura Y, Jusakon A, Nikai T. 2019. Salmonella fimbrial protein FimH is involved in expression of proinflammatory cytokines in a Toll-like receptor 4-dependent manner. Infect Immun 87:e00881-18. doi: 10.1128/IAI.00881-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arpaia N, Godec J, Lau L, Sivick KE, McLaughlin LM, Jones MB, Dracheva T, Peterson SN, Monack DM, Barton GM. 2011. TLR signaling is required for Salmonella typhimurium virulence. Cell 144:675–688. doi: 10.1016/j.cell.2011.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.LeBlanc PM, Yeretssian G, Rutherford N, Doiron K, Nadiri A, Zhu L, Green DR, Gruenheid S, Saleh M. 2008. Caspase-12 modulates NOD signaling and regulates antimicrobial peptide production and mucosal immunity. Cell Host Microbe 3:146–157. doi: 10.1016/j.chom.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Niu S, Xi D, Zhao S, Sun J, Jiang Y, Liu J. 2019. Differences in lipopolysaccharides-induced inflammatory response between mouse embryonic fibroblasts and bone marrow-derived macrophages. J Interferon Cytokine Res 39:375–382. doi: 10.1089/jir.2018.0167. [DOI] [PubMed] [Google Scholar]
- 42.Clark K, Plater L, Peggie M, Cohen P. 2009. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J Biol Chem 284:14136–14146. doi: 10.1074/jbc.M109.000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vail KJ, da Silveira BP, Bell SL, Cohen ND, Bordin AI, Patrick KL, Watson RO. 2021. The opportunistic intracellular bacterial pathogen Rhodococcusequi elicits type I interferon by engaging cytosolic DNA sensing in macrophages. PLoS Pathog 17:e1009888. doi: 10.1371/journal.ppat.1009888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.West KO, Scott HM, Torres-Odio S, West AP, Patrick KL, Watson RO. 2019. The splicing factor hnRNP M is a critical regulator of innate immune gene expression in macrophages. Cell Rep 29:1594–1609.e5. doi: 10.1016/j.celrep.2019.09.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weindel CG, Bell SL, Vail KJ, West KO, Patrick KL, Watson RO. 2020. LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. Elife 9:e51071. doi: 10.7554/eLife.51071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wagner AR, Scott HM, West KO, Vail KJ, Fitzsimons TC, Coleman AK, Carter KE, Watson RO, Patrick KL. 2021. Global transcriptomics uncovers distinct contributions from splicing regulatory proteins to the macrophage innate immune response. Front Immunol 12:656885. doi: 10.3389/fimmu.2021.656885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu J, Dobbs N, Yang K, Yan N. 2020. Interferon-independent activities of mammalian STING mediate antiviral response and tumor immune evasion. Immunity 53:115–126.e5. doi: 10.1016/j.immuni.2020.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Motwani M, Pesiridis S, Fitzgerald KA. 2019. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet 20:657–674. doi: 10.1038/s41576-019-0151-1. [DOI] [PubMed] [Google Scholar]
- 49.Nandi I, Aroeti L, Ramachandran RP, Kassa EG, Zlotkin-Rivkin E, Aroeti B. 2021. Type III secreted effectors that target mitochondria. Cell Microbiol 23:e13352. doi: 10.1111/cmi.13352. [DOI] [PubMed] [Google Scholar]
- 50.Hernandez LD, Pypaert M, Flavell RA, Galan JE. 2003. A Salmonella protein causes macrophage cell death by inducing autophagy. J Cell Biol 163:1123–1131. doi: 10.1083/jcb.200309161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Layton AN, Brown PJ, Galyov EE. 2005. The Salmonella translocated effector SopA is targeted to the mitochondria of infected cells. J Bacteriol 187:3565–3571. doi: 10.1128/JB.187.10.3565-3571.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crowley LC, Christensen ME, Waterhouse NJ. 2016. Measuring mitochondrial transmembrane potential by TMRE staining. Cold Spring Harb Protoc 2016:pdb.prot087361. doi: 10.1101/pdb.prot087361. [DOI] [PubMed] [Google Scholar]
- 53.Zhang T, Liu CF, Zhang TN, Wen R, Song WL. 2020. Overexpression of peroxisome proliferator-activated receptor gamma coactivator 1-alpha protects cardiomyocytes from lipopolysaccharide-induced mitochondrial damage and apoptosis. Inflammation 43:1806–1820. doi: 10.1007/s10753-020-01255-4. [DOI] [PubMed] [Google Scholar]
- 54.Aarreberg LD, Esser-Nobis K, Driscoll C, Shuvarikov A, Roby JA, Gale M, Jr. 2019. Interleukin-1β induces mtDNA release to activate innate immune signaling via cGAS-STING. Mol Cell 74:801–815.e6. doi: 10.1016/j.molcel.2019.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim BR, Kim BJ, Kook YH, Kim BJ. 2020. Mycobacterium abscessus infection leads to enhanced production of type 1 interferon and NLRP3 inflammasome activation in murine macrophages via mitochondrial oxidative stress. PLoS Pathog 16:e1008294. doi: 10.1371/journal.ppat.1008294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim HJ, Koo SY, Ahn BH, Park O, Park DH, Seo DO, Won JH, Yim HJ, Kwak HS, Park HS, Chung CW, Oh YL, Kim SH. 2010. NecroX as a novel class of mitochondrial reactive oxygen species and ONOO(-) scavenger. Arch Pharm Res 33:1813–1823. doi: 10.1007/s12272-010-1114-4. [DOI] [PubMed] [Google Scholar]
- 57.Xiao TS, Fitzgerald KA. 2013. The cGAS-STING pathway for DNA sensing. Mol Cell 51:135–139. doi: 10.1016/j.molcel.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Charrel-Dennis M, Latz E, Halmen KA, Trieu-Cuot P, Fitzgerald KA, Kasper DL, Golenbock DT. 2008. TLR-independent type I interferon induction in response to an extracellular bacterial pathogen via intracellular recognition of its DNA. Cell Host Microbe 4:543–554. doi: 10.1016/j.chom.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Canesso MCC, Lemos L, Neves TC, Marim FM, Castro TBR, Veloso ES, Queiroz CP, Ahn J, Santiago HC, Martins FS, Alves-Silva J, Ferreira E, Cara DC, Vieira AT, Barber GN, Oliveira SC, Faria AMC. 2018. The cytosolic sensor STING is required for intestinal homeostasis and control of inflammation. Mucosal Immunol 11:820–834. doi: 10.1038/mi.2017.88. [DOI] [PubMed] [Google Scholar]
- 60.Marinho FV, Benmerzoug S, Oliveira SC, Ryffel B, Quesniaux VFJ. 2017. The emerging roles of STING in bacterial infections. Trends Microbiol 25:906–918. doi: 10.1016/j.tim.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Petersen E, Mills E, Miller SI. 2019. Cyclic-di-GMP regulation promotes survival of a slow-replicating subpopulation of intracellular Salmonella Typhimurium. Proc Natl Acad Sci USA 116:6335–6340. doi: 10.1073/pnas.1901051116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, Rehman J, Malik AB. 2020. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity 52:475–486.e5. doi: 10.1016/j.immuni.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu X, Peng X, Lu C, Zhang X, Gan L, Gao Y, Yang S, Xu W, Wang J, Yin Y, Wang H. 2019. Type I IFN expression is stimulated by cytosolic MtDNA released from pneumolysin-damaged mitochondria via the STING signaling pathway in macrophages. FEBS J 286:4754–4768. doi: 10.1111/febs.15001. [DOI] [PubMed] [Google Scholar]
- 64.Park SM, Omatsu T, Zhao Y, Yoshida N, Shah P, Zagani R, Reinecker HC. 2019. T cell fate following Salmonella infection is determined by a STING-IRF1 signaling axis in mice. Commun Biol 2:464. doi: 10.1038/s42003-019-0701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. 2012. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol 13:954–962. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Owen KA, Anderson CJ, Casanova JE. 2016. Salmonella suppresses the TRIF-dependent. mBio 7:e02051-15–e02015. doi: 10.1128/mBio.02051-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang C, Guan Y, Lv M, Zhang R, Guo Z, Wei X, Du X, Yang J, Li T, Wan Y, Su X, Huang X, Jiang Z. 2018. Manganese increases the sensitivity of the cGAS-STING pathway for double-stranded DNA and is required for the host defense against DNA viruses. Immunity 48:675–687.e7. doi: 10.1016/j.immuni.2018.03.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S. Typhimurium infection triggers proinflammatory cytokine expression in murine macrophages. (A) CCK8 analysis of cell viability in mouse peritoneal macrophages (PMs) uninfected (mock) or infected with S. Typhimurium for 2, 4, or 8 h at an MOI of 10 (n = 3). (B) qRT-PCR analysis of gene expression in Raw264.7 cells uninfected (mock) or infected with S. Typhimurium for 2, 4, or 8 h at an MOI of 100 (n = 3). (C) CCK8 analysis of cell viability in mouse peritoneal macrophages (PMs) uninfected (mock) or infected with S. Typhimurium for 8 h at different MOIs (n = 3). (D) qRT-PCR analysis of gene expression in Raw264.7 cells uninfected (mock) or infected with S. Typhimurium for 8 h at different MOIs (n = 3). (E) KEGG pathway analysis results for comparison of upregulated genes in PMs uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 10. (F) Immunoblot analysis of protein expression in PMs uninfected (mock) or infected with S. Typhimurium for 2, 4, or 8 h at an MOI of 10. (G) Immunoblot analysis of protein expression in Raw264.7 cells uninfected (mock) or infected with S. Typhimurium for 8 h at different MOIs. Data in (A to D) were normalized to uninfected control (mock, set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. Download FIG S1, TIF file, 1.9 MB (2MB, tif) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
RNA-seq analysis of uninfected and S. Typhimurium-infected macrophages. Download Table S1, XLSX file, 0.3 MB (302.9KB, xlsx) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
S. Typhimurium infection induces the type I IFN response in primary MEFs. (A) qRT-PCR analysis of gene expression in murine primary MEF cells untreated (con), treated with LPS (100 ng/mL) (LPS-MEFs), or transfected with dsDNA (1 μg/mL) (Interferon stimulatory DNA, ISD, 45 bp) (ISD-MEFs) for 6 h. PMs treated with LPS (100 ng/mL) (LPS-PMs) for 6 h were also included (n = 3). (B) qRT-PCR analysis of gene expression in murine primary MEF cells uninfected (mock) or infected with S. Typhimurium for 2, 4, 6, 8, or 10 h at an MOI of 100 (n = 3). Data in (A) were normalized to untreated MEFs and PMs, respectively (con, set as 1; con for PMs was not shown). Data in (B) were normalized to mock-infected control (mock, set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001. NS, not significant. Download FIG S2, TIF file, 2.2 MB (2.2MB, tif) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
cGAS-STING is required for the type I IFN response during S. Typhimurium infection. (A) ELISA analysis of IFN-β production in the supernatant from C57BL/6 (WT), cGAS−/− or STING−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 2, 4, 6, or 8 h at an MOI of 10 (n = 3). (B) qRT-PCR analysis of gene expression in C57BL/6 (WT) or cGAS−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 2 and 4 h at an MOI of 10 (n = 3). (C) Immunoblot analysis of protein expression in C57BL/6 or STING−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 10, or transfected with dsDNA (Interferon stimulatory DNA, ISD) for 6 h. (D) qRT-PCR analysis of gene expression in C57BL/6 (WT) or STING−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 2 and 4 h at an MOI of 10 (n = 3). (E) qRT-PCR analysis of gene expression in cecum excised from S. Typhimurium-infected C57BL/6 (WT) or STING−/− mice at 24 or 72 h postinfection (n = 3). Data in (B) were normalized to mock-infected C57BL/6 (WT) PMs or mock-infected cGAS−/− PMs, respectively (mock, both set as 1). Data in (D) were normalized to mock-infected C57BL/6 (WT) PMs or mock-infected STING−/− PMs, respectively (mock, both set as 1). Data in (E) were normalized to mock-infected C57BL/6 (WT) cecum samples and mock-infected STING−/− cecum samples, respectively (mock, both set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Download FIG S3, TIF file, 2.0 MB (2.1MB, tif) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Activation of the STING potentiates S. Typhimurium-induced type I IFN response. (A) Primary MEFs were untreated (S. T.) or lipofectamine-transfected with 5 μM 2′3′-cGAMP (S. T. + cGAMP) for 4 h and then infected with S. Typhimurium at an MOI of 100. At 6 h postinfection, qRT-PCR analysis of gene expression (n = 3). (B) TLR4−/− PMs were untreated or lipofectamine-transfected with 5 μM 2′3′-cGAMP (cGAMP) for 4 h and then uninfected (mock) or infected with S. Typhimurium at an MOI of 10. At 6 h postinfection, qRT-PCR analysis of gene expression (n = 3). Data in (A) were normalized to untreated, mock-infected control and 2′3′-cGAMP-transfected, mock-infected control, respectively (mock, both set as 1, not shown). Data in (B) were normalized to untreated, mock-infected control (mock, set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Download FIG S4, TIF file, 0.8 MB (858.2KB, tif) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
S. Typhimurium-induced mtDNA release is involved in the induction of type I IFN. (A) Fluorescence microscopy analysis of mitochondrial membrane potential by assessing JC-1 aggregates and JC-1 monomer in HeLa uninfected (mock) or infected with S. Typhimurium for 2 h at an MOI of 10. Green: JC-1 monomer; Red: JC-1 aggregates. Scale bar: 75 μm. Right: fluorescence spectrophotometer analysis of the ratio of JC-1 aggregates/JC-1 monomer in HeLa cells uninfected (mock) or infected with S. Typhimurium for 2 h at an MOI of 10 or 100 (n = 3). (B) Fluorescence microscopy analysis of the localization of mitochondria and S. Typhimurium in Raw264.7 cells infected with S. Typhimurium for 2 h at an MOI of 100. Hoechst33342: nuclei; GFP, Mito-tracker green to indicate mitochondria; mCherry, S. Typhimurium carries a mCherry expressing plasmid. Scale Bar: 20 μm. (C) qPCR analysis of mtDNA in the cytosolic fraction of C57BL/6 (WT) or TLR4−/− mouse-derived PMs uninfected (mock) or infected with S. Typhimurium for 8 h at an MOI of 100 (n = 3). (D) qPCR analysis of the total mtDNA in EB-treated and untreated (con) Raw264.7 cells (n = 3). (E) qPCR analysis of mtDNA in the cytosolic fraction of PMs pretreated with Mito Tempo (100 μM) or NecroX-5 (40 μM) for 2 h and uninfected (mock) or infected with S. Typhimurium for 6 h at an MOI of 100 (n = 3). (F) qRT-PCR analysis of gene expression in TLR4−/− PMs pretreated with Mito Tempo (100 μM) for 2 h and uninfected (mock) or infected with S. Typhimurium for 6 h at an MOI of 10 (n = 3). Data in (C) were normalized to uninfected C57BL/6 (WT) and TLR4−/− PMs, respectively (mock, both set as 1). Data in (D) were normalized to untreated control (con, set as 1). Data in (E) were normalized to untreated mock-infected control (mock, set as 1). Data in (F) were normalized to untreated, mock-infected control and Mito Tempo pretreated, mock-infected control, respectively (mock, both set as 1). Actin was used as the housekeeping gene. Error bars represent ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. NS, not significant. Download FIG S5, TIF file, 2.4 MB (2.4MB, tif) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
S. Typhimurium DNA induces type I IFN response and the cGAS-STING pathway constrains S. Typhimurium infection. (A) qRT-PCR analysis of gene expression in C57BL/6 (WT) or STING−/− mouse-derived PMs transfected with S. Typhimurium genomic DNA (1 μg per well in 24-well plate) for 6 h (n = 3). (B) Immunoblotting of lysates of Raw264.7 cells was uninfected or infected with S. Typhimurium for 8 h at an MOI of 100, then immunoprecipitated with IgG or cGAS antibody. (C) qPCR of mtDNA sequences (Cox1, Cox2, and Cox3) from DNA isolated from IPs in (B) (n = 3). (D) Fluorescence microscopy analysis of C57BL/6 or cGAS−/− mouse-derived PMs infected with S. Typhimurium for 2 h at an MOI of 10. Hoechst33342: nuclei, GFP, S. Typhimurium carries a GFP expressing plasmid. Scale bar: 75 μm. The ratio of GFP fluorescence integrated density to Hoechst33342 fluorescence integrated density is on the right (n = 3). (E) qPCR analysis of S. Typhimurium 16S rRNA level in primary MEFs untreated (con) or pretreated with DMXAA (100 μg/mL) for 12 h, or pretreated lipofectamine-transfected with 5 μM 2′3′-cGAMP (cGAMP) for 4 h and then infected with S. Typhimurium for 8 h at an MOI of 100 (n = 3). (F) qPCR analysis of S. Typhimurium 16S rRNA level in TLR4−/− PMs untreated (con) or pretreated lipofectamine-transfected with 5 μM 2′3′-cGAMP (cGAMP) for 4 h and infected with S. Typhimurium for 8 h at an MOI of 100 (n = 3). Data in (A) were normalized to mock-transfected C57BL/6 (WT) PMs and mock-transfected STING−/− PMs, respectively (mock, both set as 1). Quantities in (C) were normalized to inputs. Data in (E and F) were normalized to untreated S. Typhimurium-infected control (con, set as 1). Error bars represent ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Download FIG S6, TIF file, 1.5 MB (1.5MB, tif) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Unprocessed western blots results. Download FIG S7, JPG file, 1.07 MB (1.1MB, jpg) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Bacterial strains, cells, mice, plasmids, primers, reagents, and resources used in this study. Download Table S2, XLSX file, 0.02 MB (22.2KB, xlsx) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental materials and methods. Download Text S1, DOCX file, 0.05 MB (50.9KB, docx) .
Copyright © 2022 Xu et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.