

Abstract
Aims
Increased resistin (Retn) levels are associated with development of cardiovascular diseases. However, the role of Retn in heart failure (HF) is still unclear. Here we probed the functional and molecular mechanism underlying the beneficial effect of Retn deletion in HF.
Methods and results
Wild-type (WT) and adipose tissue-specific Retn-knockout (RKO) mice were subjected to transverse aortic constriction (TAC)-induced HF. Cardiac function and haemodynamic changes were measured by echocardiography and left ventricular catheterization. Adipose tissue Retn deletion attenuated while Retn cardiac-selective overexpression, via a recombinant adeno-associated virus-9 vector, exacerbated TAC-induced hypertrophy, cardiac dysfunction, and myocardial fibrosis in WT and RKO mice. Mechanistically, we showed that Gadd45α was significantly increased in RKO HF mice while cardiac overexpression of Retn led to its downregulation. miR148b-3p directly targets Gadd45α and inhibits its expression. Retn overexpression upregulated miR148b-3p expression and triggered DNA damage response (DDR) in RKO-HF mice. Inhibition of miR148b-3p in vivo normalized Gadd45α expression, decreased DDR, and reversed cardiac dysfunction and fibrosis. In vitro Retn overexpression in adult mouse cardiomyocytes activated miR148b-3p and reduced Gadd45α expression. Gadd45α overexpression in H9C2-cardiomyoblasts protected against hydrogen peroxide- and Retn-induced DDR.
Conclusion
These findings reveal that diminution in circulating Retn reduced myocardial fibrosis and apoptosis, and improved heart function in a mouse model of HF, at least in part, through attenuation of miR148b-3p and DDR. The results of this study indicate that controlling Retn levels may provide a potential therapeutic approach for treating pressure overload-induced HF.
Keywords: Resistin, miR148b-3p, Gadd45α, DNA damage response, Pressure overload heart failure
Graphical Abstract
1. Introduction
Cardiac hypertrophy and ensuing heart failure (HF) are major causes of morbidity and mortality throughout the world.1 Despite substantial advances in the clinical management of HF, the diagnosis continues to carry a grave prognosis, with an overall 5-year mortality rate of ∼50%.2,3 During the last decades, enormous efforts have been focused on elucidating the signalling pathways mediating the complex response of cardiomyocytes to various hypertrophic stimuli leading to the development of cardiac hypertrophy and its progression to HF.4–6 Various types of internal and external stresses are generally known to contribute to the disease development.7 Despite the large body of research detailing the mechanisms of HF, new pathological risk factors continue to be identified, underscoring the intricacy of HF. This suggests that no single risk pathway seems to regulate the aetiology of HF alone; rather, it appears more likely that each pathway operates as a component of an orchestrated pathologic network driving the disease.
Resistin, a cysteine-rich hormone secreted by adipocytes in rodents, has been proposed to be a link between obesity, insulin resistance, and diabetes, all cardiovascular risk factors.8–10 Resistin was shown to impair glucose transport in isolated mice cardiomyocytes11 and to be upregulated by cyclic stretch and aorta-caval shunt.12 However, the pathophysiological role of resistin in humans has been questioned because the human homologue of resistin is only 59% identical to mouse resistin at the amino acid level and the source of resistin appears to differ between humans and mice.8,13 It is primarily secreted from adipose tissue in mice,8 while macrophages and monocytes, and adipocytes to a lesser degree,14 appear to be the predominant source of Retn in humans.15–17 However, recent reports, both in humans and in rodents, documented Retn expression and production by other tissues such as the heart, the liver, and the brain amongst others.12,18–21 Numerous cardiovascular effects of Retn were reported, including induction of endothelial dysfunction and the promotion of ischemia/reperfusion myocardial damage.22 However, the latter finding is debatable with at least one report showing Retn promotes a protective effect against myocardial ischemia/reperfusion injury23 while another study reported worsening outcomes.24 Clinical and epidemiological studies have shown that plasma Retn levels were elevated in patients with HF,25 were independently correlated with new onset HF,26,27 and were positively related to the severity and incidents of HF hospitalization.26,28 Higher serum levels of Retn were elevated in female patients with coronary heart disease29 and demonstrated to be a predictor of the presence and severity of coronary artery disease.30 We have previously reported that cardiac tissue from diabetic experimental animal models and human diabetic HF patients expressed high levels of Retn.18,19 We then showed that Retn overexpression in cultured rat ventricular cardiomyocytes induced cellular hypertrophy and altered cardiomyocyte mechanics and calcium handling in vitro.18,31 We have subsequently demonstrated that long-term in vivo overexpression of Retn in normal rats decreased myocardial contractility and induced a complex phenotype of ventricular remodelling.32 In addition, recombinant human resistin was shown to exacerbate cardiac ischemia/reperfusion injury and stimulate TNFα secretion and upregulate cardiac injury markers such as atrial natriuretic peptide, brain natriuretic peptide, and creatine kinase through an NF-kB signalling pathway.24 Humanized resistin mice, with macrophage-derived expression of human RETN but lacking adipocyte-derived murine Retn, developed aggravated adipose tissue inflammation, lipolysis, and insulin resistance when challenged with diet-induced obesity.33 However, a well-defined causative relationship between Retn and HF remains to be demonstrated.
MicroRNAs (miRNAs) are endogenous small noncoding RNA molecules, which belong to a class of small silencing RNAs that regulate their target gene expression post-transcriptionally.34 miRNAs that promote mRNA degradation and inhibit mRNA translation have been shown to be important in cardiac development.35 Functional miRNA studies reported that a variety of miRNAs play a role in pathogenic mechanisms leading to HF, such as remodelling, hypertrophy, apoptosis, and hypoxia.36 DNA damage and DNA damage response activation are observed in the human failing heart37 and in mouse models of cardiac hypertrophy.37–39 H2A histone family member X (H2AX) is a type of histone protein from the H2A family encoded by the H2AFX gene. γH2AX, the phosphorylated form of H2AX, is known be an early and sensitive marker of DNA damage. Excessive DNA damage recruits and activates/phosphorylates ataxia-telangiectasia-mutated (ATM) kinase, which in turn phosphorylates γH2AX and triggers downstream DDR signalling.40
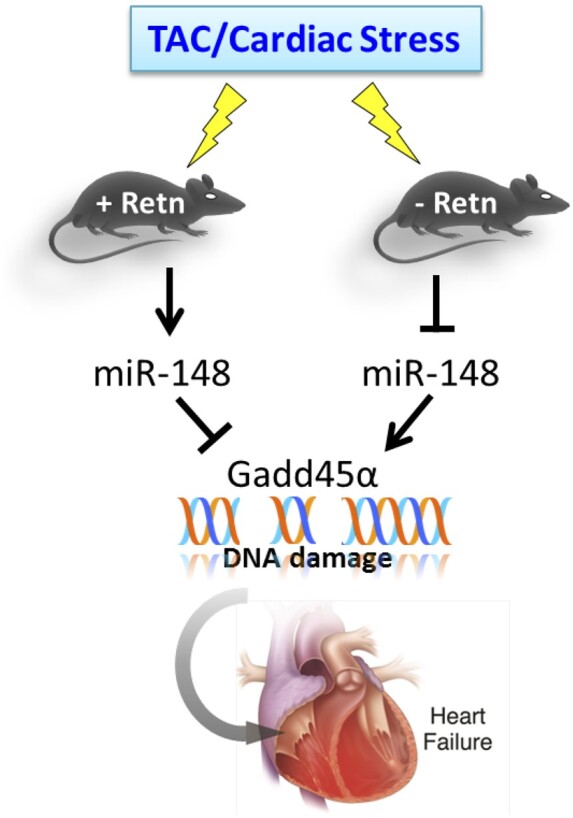
In the present study, we demonstrate that Retn plays a critical role in the development of heart disease. Loss of Retn led to downregulation of miR148b-3p and upregulation of Gadd45α activity in a pressure overload HF mouse model induced by transverse aortic constriction (TAC), resulting in protected cardiac function and decreased cardiac fibrosis and apoptosis. Retn overexpression, on the other hand, led to the development of interstitial fibrosis with defective compensation mechanisms and reverse signalling of miR148b-3p, Gadd45α activity in TAC-mice. Furthermore, the data suggest that Retn controls pathological cardiac remodelling through the regulation of Gadd45α-mitigated activation of ATM and γH2AX. Collectively, the decline in circulating Retn expression and attenuation of DDR may be involved in the cardiac protection and repair process after injury.
2. Methods
2.1 Animal studies
Wild-type (WT) and RKO mice41 were used in this study. The Retn null mice were generated by replacing the coding exons of the resistin gene with the reporter gene lacZ specifically in the white adipose tissue as described.41 WT mice are C57Bl6 background; resistin KO mice are on a mixed 129SvEv/C57Bl6 background. All experimental procedures were approved by the Mount Sinai Institutional Animal Care and Use Committee, in accordance with the ‘Principles of Laboratory Animal Care by the National Society for Medical research and the Guide for the Care and Use of Laboratory Animals’ (National Institutes of Health Publication No. 86-23, revised 1996).
2.2 Transverse aortic constriction pressure overload HF model
Male and female mice ∼8–10 weeks old (body weight of 20–25 g) were subjected to pressure overload using TAC surgery. In brief, mice were anaesthetized with 2.0% isoflurane (vol/vol) in 600 cc/min oxygen flow via inhalation, and respiration was artificially controlled during surgery. Mice were maintained under 2% isoflurane throughout surgery, and anaesthesia effects were assessed by a lack of response to toe pinching. The aortic arch was reached through a midline incision in the anterior neck, and the thoracic aorta at the arch was surgically constricted using a 27-gauge needle to generate trans-stenotic pressure.42 Sham mice underwent the same procedure without aortic banding. Animals reaching endpoints were euthanized with pentobarbital (200 mg/kg), an accepted method by the Panel on Euthanasia of the American Veterinary Medical Association. Animals were euthanized in a small area separate from the housing and research facilities.
2.3 Echocardiography analysis
Echocardiography was performed under sedation with an intraperitoneal injection of ketamine (100 mg/kg). Sedation was optimized by (1) giving the lowest dose of ketamine needed to restrain the animals and to prevent motion artefact and (2) maintaining the heart rate as close as possible to 550 beats/min. Two-dimensional images and M-mode tracings were recorded on the short axis at the level of the mid-papillary muscle to determine per cent fractional shortening and ventricular dimensions (GE Vivid) as recommended by the American Society of Echocardiography.43,44
2.4 In vivo haemodynamics by pressure–volume loop analysis
One day after echocardiography, left ventricle (LV) pressure–volume (P–V) loops were obtained as previously described.44,45 Briefly, mice were anaesthetized with an intraperitoneal injection mixture of urethane (800 µg/g), etomidate (10 µg/g), and morphine (1 µg/g), and subsequently intubated and mechanically ventilated. The chest was opened through a median sternotomy. A 1.2 Fr pressure–volume (P–V) conductance catheter (Scisense, Ontario, Canada) was inserted into the LV apex through an apical stab performed with a 25GA needle. Haemodynamic recordings were performed after 5 min of stable heart rate. Pressure–volume data were analysed using IOX2 software (EMKA technologies). Linear fits were obtained for end-systolic pressure–volume relationships (ESPVRs).
2.5 Animal sacrifice, serum collection, and tissue harvesting
Upon termination of the invasive haemodynamics measurements, animals were sacrificed, and whole heart tissues were weighed. A ring of heart tissue was embedded for histological studies, and the rest of the heart tissue was frozen for RNA and protein analysis. Serum resistin content was determined using an ELISA assay kit specific for mouse resistin (Cat# ELM-Resistin) according to manufacturer’s instructions (RayBio).
2.6 RNA sequencing
Transcriptome resequencing was performed. In brief, total RNA was extracted from the hearts of WT-TAC and RKO-TAC mice at 4 weeks after TAC injury using TRIzol reagent (Ambion, Life technologies). RNA concentration and quality was determined and assessed by Agilent Bioanalyser RNA chip (Agilent 2100, Agilent). RNA samples with an RNA integrity number of >8 were used to generate sequencing libraries after depletion of the rRNA. Sequencing was performed on the BGISEQ-500 instrument using the paired-end sample preparation chemistry. Rstudio was used to generate the volcano plots and heat maps for dysregulated genes (www.rstudio.com).
2.7 Anti-microRNA administration
miR148b-3p inhibitor probe (product no.:199900, batch: 621044) and negative control (product no.: 199900, batch: 621045) were purchased from Exiqon and were each diluted with PBS to final doses of 10 mg/mL. All solutions were mixed by vortexing for 10 s and incubated for at least 15 min at 37°C prior to injection. Three weeks after TAC surgery, the mice received the inhibitor probe (10 mg/kg body weight per day) or negative control for three consecutive days by intraperitoneal injections.
2.8 AAV9-mRetn recombinant virus production and transduction
293T cells were co-transfected with pTR-Retn and pDG-9 (expressing AAV9 capsid and helper functions) under the control of CMV promoter by calcium phosphate method. Three days after transfection, cells were harvested, washed in 1× PBS, and resuspended in 150 mM NaCl, 50 mM Tris. Virus was released by three cycles of freeze/thaw, and Benzonase and MgCl2 were added to a final concentration of 150 units/mL and 2 μM, respectively. The cell lysate was incubated for 30 min at 37°C then centrifuged at 3400 g for 20 min. The supernatant was loaded onto an iodixanol gradient and centrifuged for 1 h at 69,000 rpm at 18°C. The virus was extracted from the 40%–60% interface and the bottom portion of the 40% fraction. The iodixanol fractions containing the virus were concentrated, and at the same time, buffer was exchanged to Lactated Ringer's solution, then filtered with a 0.2-μm syringe-driven filter. Stocks were stored at −80°C, and genome-containing particles were determined by real-time PCR.
2.9 Isolation of adult mouse cardiomyocytes and adenoviral infection
Retn knockout (RKO) mice were anaesthetized with ketamine (100 mg/kg ip). The heart was quickly removed from the chest and aortic perfused retrogradely at 37°C for 3 min with a Ca2+-free bicarbonate-based buffer containing (in mM) 120 NaCl, 5.4 KCl, 1.2 MgSO4, 1.2 NaH2PO4, 5.6 glucose, 20 NaHCO3, 10 2,3-butanedione monoxime (BDM; Sigma), and 5 taurine (Sigma).46 The enzymatic digestion was initiated by adding collagenase type II (0.5 mg/mL) to the perfusion solution. Cardiomyocytes were plated in laminin (5 μg/mL)-coated 6-well plates in the presence of DMEM medium supplemented with 10% bovine serum albumin and 1% of penicillin/streptomycin. Following attachment, myocytes were infected with an adenovirus-encoding Retn (Ad.Retn) or β-galactosidase (Ad-β-Gal), at a multiplicity of infection (MOI) of 100. After 24 h of infection, myocytes were harvest for molecular analyses.
2.10 Real-time quantitative PCR analysis
Total RNA was prepared from heart tissue or cardiomyocytes using TRIzol reagent (Ambion, Life technologies), and RNA was reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems) or miRCURY LNA™ Universal RT microRNA PCR (Qiagen). The cDNAs were quantified using the PerfeCTa SYBR Green FastMix (Quanta), and specific primers (Supplementary material online, Table S6) or LNA enhanced miR148b-3p-specific primers (miRCURY LNA PCR primer sets for miR148b-3p and 5S rRNA, from Qiagen). Fold changes in gene expression were determined using the 2−ΔΔCt method with normalization to endogenous 18S rRNA or 5 s rRNA controls.
2.11 Protein quantification by Western blot analysis
Protein samples were prepared from mouse heart tissue using a lysis buffer containing protease inhibitor (11852700, Roche) and phosphatase inhibitors (88667, Thermo Fisher scientific). Protein lysates were matched for protein concentration; total protein (15 ng) was separated by SDS–PAGE and transferred onto PVDF membranes (Bio-Rad) that were incubated with appropriate antibodies (Supplementary material online, Table S7). Densities of the immunoreactive bands from at least three independent experiments were evaluated using NIH Image J. Protein loading was verified against GAPDH densities.
2.12 Assessment of the histology of the heart
For histological analysis, hearts were arrested in diastole, perfusion-fixed with 4% paraformaldehyde, embedded in paraffin and cut into 5-μm sections. Paraffin sections were stained with Masson’s trichrome staining for routine histological analysis and FITC-labelled wheat-germ-agglutinin (WGA, Cat. No. L4895, Sigma) diluted 1/100 (10 µg/mL) to visualize and quantify the cell cross-sectional areas. Incubation time was 1 h protected from light. After three washing steps, the sections were cover-slipped with a water-soluble anti-fading mounting medium. Slides were visualized using a Zeiss Axiocam microscopy, Nikon Eclipse E400 microscope, or AMG EVOS FL inverted microscope. Quantification of the myocardial collagen fraction and cell surface areas was determined using NIH ImageJ software (NIH). Cell surface areas were quantified in at least 60 cells per group.
2.13 Dual-luciferase reporter assay
The Gadd45α microRNA-target pairs were predicted by miRDB, microT-CDS, Targetscan (Release 7.2), and miRSearch (V3.0). Luciferase reporters were constructed by cloning sequences from the 3’ untranslated regions (UTRs) of Gadd45α mRNAs into the psiCHECK-2 vector (Cat. No. C8021, Promega). Briefly, the WT 3’ UTRs of these genes, which contain the predicted miR148b-3p response element, as well as mutant 3’ UTRs were synthesized, and then inserted in the multiple cloning region behind the synthetic Renilla luciferase gene. HEK293T cells were co-transfected with the constructed reporter plasmid (500 ng) and mmu-miR-148b-3p mimics (100 nM) (Cat. No. YM00471310, Qiagen) or miRNA mimic negative control (Cat. No. YM00479902, Qiagen) using Lipofectamine 2000 transfection reagent (Cat. No. 11668-019, Invitrogen). Reporter assays were performed 48 h after transfection using the Dual Luciferase Reporter Assay System (Cat. No. E1910, Promega) and EnVision plate reader (PerkinElmer) according to the manufacturer’s protocol. Data are presented as a ratio of firefly luciferase activity normalized to Renilla luciferase.
2.14 Creation of Gadd45α stable cell lines and treatment
pLXSN Retroviral Vector (Cat. No. 631509, Clontech) and pCMV6-Gadd45α (Cat. No. MR201332, OriGene) were used. The 566-bp DNA fragment containing Gadd45α open-reading frame was removed from pCMV6-Gadd45α by EcoRI and XhoI digestion, and cloned into the EcoRI/XhoI sites of pLXSN to create Retroviral-Gadd45α vector. pLXSN Retroviral vector (control) and Retroviral-Gadd45α vector were transfected into two different 60-mm plates of 70% confluent 293 T cells. Retrovirus-containing conditioned medium was harvested and used to transduce H9C2 cardiomyoblasts. 48 hours post-transfection, H9C2-Gadd45α and H9C2-Control cells were selected for 10 days with complete DMEM containing 10% foetal bovine serum, 100 IU/mL penicillin, 100 μg/mL streptomycin, and 1 mg/mL G418 (Sigma Chemical, St. Louis, MO). The expression level of Gadd45α was determined by real-time PCR. DNA damage was induced by treatment with 200 μM hydrogen peroxide (H2O2) for 24 hours or 100 ng/mL recombinant Retn for 48 hours.
2.15 Immunofluorescence
Cultured cells were fixed with 4% paraformaldehyde in PBS for 15 min. Samples were blocked with 1% BSA and 1% Triton-X100 for 1 h, and subsequently incubated with anti-pATM (1:200) overnight at 4°C. After washing with PBS, secondary antibody conjugated to Alexa Fluor 488 (1:200) was added and incubated for 1 h at room temperature. Finally, the cells were counterstained with DAPI and mounted with mounting medium. Foci numbers of pATM were assessed using NIH ImageJ software (NIH, Bethesda, MD, USA).
2.16 Terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL) assay
A TUNEL kit (Cat. No. S7165, EMD Millipore) was used to assess myocardial apoptosis following the manufacturer’s instructions. Briefly, tissue sections were deparaffinized and rehydrated, then pretreated with proteinase K (20 µg/mL) for 15 min at room temperature. The TdT reaction was performed for 1 h at 37°C in a humidified chamber. Finally, sections were stained with DAPI for 5 min and mounted with mounting medium. Slides were visualized using AMG EVOS FL inverted microscope with 40× magnification. Quantification of the apoptotic cells was determined using NIH ImageJ software (NIH, Bethesda, MD, USA). The apoptosis rate refers to the number of TUNEL positive nuclei divided by the total number of DAPI-stained cells ×100%.
2.17 Human left ventricle samples
The current investigation conforms to the principles outlined in the Declaration of Helsinki. Human heart tissue specimens were obtained from the National Disease Research Interchange through the Human Tissues and Organs for Research Resource program. In accordance with the Institutional Review Board of the Icahn School of Medicine at Mount Sinai, the authors had no access to any identifying patient information (e.g. name, social security number, medical record number, pathology accession number, or any other code) that would permit specimens used in this study to be linked to individually identifiable living individuals. These specimens were completely de-identified and were treated as any commercially available cell line. Failing heart samples were obtained from explanted hearts at the time of cardiac transplantation from patients with ischaemic heart disease: 1) Caucasian, female, 66 years old, died on 10/11/2000; 2) Afro-American, female, 62 years old, died on 10/12/2001; or dilated cardiomyopathy 3) Caucasian, male, 53 years old, died on 9/23/1997. The average EF of the patients was 20 ± 3%. Control non-failing heart samples were obtained from donors who had normal cardiac function and died from neurological diseases or road traffic accidents: 1) Caucasian, male, 54 years old, died on 4/19/1994; 2) Caucasian, male, 58 years old, died on 9/24/2000; and 3) Caucasian, female, 68 years old, died on 9/1/1993.
2.18 Statistical analysis
The results are presented as mean ± standard error of the mean (S.E.M.). Unpaired Student’s t-test or one-way ANOVA with Tukey’s post-hoc test was performed as indicated to determine statistical differences. Probability values of P < 0.05 were considered significant. Statistical analyses were conducted using GraphPad Prism 5 software (Graph Pad, CA, USA).
3. Results
3.1 Inhibition of Retn prevents and partially reverses cardiac remodelling and dysfunction in pressure overload-induced HF
We first sought to examine the effect of direct manipulation of Retn levels on the pathogenesis of cardiac hypertrophy and HF. To this end, WT and adipose tissue RKO mice41 were used; the RKO mice showed complete deletion of resistin in adipose tissue and significant reduction in circulating serum levels compared to WT (Supplementary material online, Figure S1A and B), in agreement with previous findings.41 However, WT and RKO mice expressed comparable levels of resistin in heart samples (Supplementary material online, Figure S1C and D). WT and RKO mice were subjected to either sham operation or TAC pressure overload for 4 or 10 weeks (Figure 1A). The heart weight/body weight (HW/BW) and HW/tibia length (HW/TL) ratios were significantly increased 4 weeks (Supplementary material online, Figure S2A) and 10 weeks after TAC injury in both WT-TAC and RKO-TAC mice compared to sham mice (Figure 1B). Consistent with these data, histological analysis confirmed the cardiac chamber remodelling and increased cardiomyocytes cross-sectional areas in both groups (Figure 1C; Supplementary material online, Figure S2B for 4 weeks group). Interestingly, RKO-TAC mice developed a concentric hypertrophic phenotype characterized by marked increase in left ventricular (LV) thickness with increased cell size (Figure 1C; Supplementary material online, Figure S2B for 4 weeks group) and increased foetal genes expression (i.e. albeit less apparent compared to WT-TAC; Figure 1D; Supplementary material online, Figure S2C for 4 weeks group). Physiological and functional assessment was performed to confirm the apparent gross hearts abnormalities. Transthoracic echocardiographic analysis showed a significant increase in contractility, as assessed by the fractional shortening at 4 weeks (RKO-TAC 50.52 ± 0.49% vs. WT-TAC 35.17 ± 0.73%, P < 0.001; Supplementary material online, Figure S1D and Table S1) and at 10 weeks RKO-TAC 52.21 ± 0.54% vs. WT-TAC 30.91 ± 1.81%; Figure 1E and Supplementary material online, Table S2, P < 0.0001) and the ejection fraction (RKO-TAC 68.14 ± 4.59% vs. WT-TAC 49.66 ± 6.82%, P < 0.05; Supplementary material online, Table S3), accompanied by a marked decrease in ventricular geometry, characterized by significantly decreased LV internal dimension at end-systole (LVIDs) in RKO-TAC mice compared to WT-TAC mice (Figure 1E; Supplementary material online, Figure S2D and Table S1 for 4 weeks group). In addition, LV catheterization and haemodynamic analysis revealed decreased LV chamber dimensions and improved contractility, assessed by end-systolic pressure–volume relationship (ESPVR), in RKO-TAC mice compared to WT-TAC mice (Figure 1F and Supplementary material online, Table S3), which is in agreement with the echocardiographic measurements. Moreover, diastolic parameters were also preserved in the RKO-TAC group as evidenced by the decrease in the time constant of LV pressure decay during the isovolumic relaxation phase tau at 10 weeks post-TAC (17.6 ± 1.46 vs. 33.36 ± 7.5; P < 0.05; Supplementary material online, Table S3). These data suggest that Retn loss of function reverses pressure overload-induced myocardial dysfunction and remodelling and blunted the progression of HF, eventually leading to a longer survival rate in RKO-TAC mice compared with WT-TAC (Figure 1G).
Figure 1.

Resistin inhibition attenuates TAC-induced hypertrophy, cardiac dysfunction, and fibrosis in RKO mice. (A) Study protocol for TAC induction in WT or RKO mice. (B) Gravimetric analysis of heart weight/body weight (left panel; n = 7–8), heart weight/tibia length (middle panel; n = 7–8), and lung weight/tibia length (right panel; n = 5) in WT and RKO mice subjected to sham or TAC for 10 weeks. (C) Representative images of Masson’s trichrome (top panels, scale bar, 2 mm) and WGA (lower panels, scale bar, 100 µm, n = 60) stained hearts sections; quantification of cardiomyocytes cross-sectional area. (D) Real-time PCR analysis of the expression levels of Anf, Myh7, Myh6, and Acta1 (n = 4). (E) Representative echocardiographic images; quantification of fractional shortening and LVIDs of WT or RKO mice subjected to sham or TAC for 10 weeks (n = 7–8). (F) Assessment of the load-independent parameter ESPVR by pressure–volume conductance catheters (n = 4). (G) Survival curve of WT or RKO mice subjected to sham or pressure overloaded 10 weeks post-TAC (n = 7–14). (H) Representative images of Masson’s trichrome-stained hearts sections and cardiac fibrosis quantification as percentage of blue stained area vs. control (scale bar, 100 µm) in the indicated groups of mice (n = 4). Real-time PCR analysis of the expression levels of Col1a1, FN, CCN2, and LOX (n = 4). N, each animal is shown as an individual point whereas horizontal lines represent median values. Values are shown as mean ± S.E.M. with each experiment performed in biological and technical replicates as indicated. Significance was evaluated by Student’s t-test or one-way ANOVA with Tukey’s post-hoc test. P values <0.05 were considered significant.
3.2 Loss of resistin inhibits cardiac fibrosis
Since cardiac fibrosis and ventricular stiffness are prominent features in the transition from compensatory hypertrophy to HF, we sought to examine the potential involvement of Retn deletion in the regulation of cardiac extracellular matrix (ECM) remodelling. Fibrosis is a pathological feature of cardiac adaptation to stress, where the proliferation of fibroblasts and increased deposition of ECM components results in myocardial stiffness and diastolic dysfunction. Histological examination of stained LV sections and subsequent quantification of the fibrotic area revealed that TAC induced, as expected, a profound increase in interstitial fibrosis in the WT-TAC hearts compared to WT sham-operated hearts both at 4 weeks (Supplementary material online, Figure S2E) and at 10 weeks groups (Figure 1H). In contrast, RKO-TAC hearts showed a significant decrease in fibrosis (Figure 1H; Supplementary material online, Figure S2E for 4 weeks group), paralleled by reductions in the mRNA expression of the pro-fibrotic genes, collagen type I alpha 1 (Col1a1), fibronectin (FN), cellular communication network factor 2 (CCN2), and lysyl hydroxylase (LOX) in the RKO-TAC hearts when compared to WT-TAC hearts (Figure 1H; Supplementary material online, Figure S2F for 4 weeks group). These findings are in agreement with the observed preservation of diastolic parameters in the RKO-TAC group (Supplementary material online, Table S3).
3.3 Resistin overexpression exacerbates pressure overload-induced cardiac dysfunction and myocardial fibrosis in Retn KO mice
To directly validate the action of Retn on cardiac performance, we overexpressed Retn using a recombinant adeno-associated virus 9 vector that confers high tropism to cardiac tissue and then performed physiological and functional assessment of RKO failing hearts after TAC induction. RKO mice received gene transfer of AAV9-empty or AAV9-mRetn for 6 weeks, and then subjected to TAC for an additional 7 weeks (Figure 2A). Post-mortem analysis revealed that the HW/BW and HW/TL ratios weren’t different between AAV9-empty and AAV9-mRetn-treated mice after pressure overload setting (Figure 2B). AAV9-mediated Retn gene transfer in vivo showed increased Retn protein and mRNA expression in the heart (Figure 2C). Molecular analysis of the re-induction of the hypertrophic foetal cardiac genes showed a significant increase in atrial natriuretic factor (Anf) and a decrease in alpha myosin heavy chain (Myh6) but no change in beta-myosin heavy chain (Myh7) or skeletal muscle alpha-actin (Acta1) (Figure 2D).
Figure 2.

Resistin overexpression exacerbates TAC-induced hypertrophy, cardiac dysfunction, and cardiac fibrosis in RKO mice. (A) Study protocol for Retn overexpression via AAV9 in TAC-induced RKO mice. (B) Analysis of heart weights (HW)/body weight (BW) and HW/tibia length ratios in AAV9-empty or AAV9-mRetn RKO mice subjected to TAC for 7 weeks (n = 6–11). (C) Western blot quantification and mRNA heart expression of Retn in AAV9-mRetn vs. AAV9-empty controls (n = 4–5). Protein levels were normalized to GAPDH, a loading control; mRNA levels were normalized to 18S. (D) Real-time PCR analysis of the expression levels of Anf, Myh7, Myh6, and Acta1 (n = 5). (E) Representative echocardiographic images and quantification of fractional shortening (FS%) and the left ventricular internal diameters in systole (LVIDs) of AAV9-empty or AAV9-mRetn RKO mice subjected to TAC for 7 weeks (n = 5). (F) Assessment of the load-independent parameter end-systolic pressure–volume relationship (ESPVR) by pressure–volume conductance catheters (n = 5). (G) Survival curve of AAV9-empty or AAV9-mRetn RKO mice subjected to TAC for 7 weeks (n = 6–22). (H) Representative images of Masson’s trichrome-stained hearts sections and myocardial fibrosis quantification as percentage of blue-stained area (fibrotic) vs. control (scale bar, 100 µm, n = 3). (I) Real-time PCR analysis of the expression levels of fibrosis-related factors: Col1a1, FN, CCN2, and LOX (n = 5). N, each animal is shown as an individual point whereas horizontal lines represent median values. Values are shown as mean ± S.E.M. with each experiment performed in biological and technical replicates as indicated. Significance was evaluated by Student’s t-test. P values <0.05 were considered significant.
Echocardiographic analysis revealed significant changes in cardiac contractility and systolic function. A decrease in fractional shortening (50.14 ± 0.39% vs. 33.34 ± 0.37%, P < 0.0001; Figure 2E and Supplementary material online, Table S4) accompanied by a marked increase in LV internal dimension at end-systole (LVIDs) (1.61 ± 0.06 vs. 2.79 ± 0.05 mm, P < 0.0001, Figure 2E and Supplementary material online, Table S4) are documented in AAV9-mRetn mice when compared to AAV9-empty mice. In vivo haemodynamic analysis further confirmed the pronounced defect in systolic and diastolic functions caused by Retn overexpression. The end-systolic pressure volume relationship (ESPVR) was significantly decreased (6.45 ± 1.01 vs. 1.94 ± 0.1 mmHg/μl, P < 0.01; Figure 2F, Supplementary material online, Table S4) while the relaxation tau constant was increased (20 ± 2.67 vs. 30.86 ± 4.18 msec, P < 0.05; Supplementary material online, Table S4) in AAV9-mRetn mice compared to AAV9-empty mice. The Retn-induced decline in cardiac performance led to shorter survivals in the AAV9-mRetn groups (Figure 2G). These functional data suggest that Retn gain of function contributes to the deterioration of myocardial function and further exacerbates pressure overload HF.
Extending these observations, histological analysis showed advanced cardiac fibrosis in heart receiving AAV9-mRetn gene transfer after TAC induction (Figure 2H), as well as increased expression of fibrotic markers (Figure 2I) compared to AAV9-empty hearts. Collectively, these data demonstrate that Retn cardiac-specific overexpression exacerbates systolic and diastolic functions following cardiac injury.
3.4 Resistin controls cardiac remodelling through regulation of miR148b-3p and Gadd45α
To gain an insight into the molecular function of Retn in the setting of cardiac failure, we employed transcriptional and experimental approaches for the identification of biologically relevant target genes. Interrogation of whole-genome transcriptional responses by RNA sequencing of WT-TAC and RKO-TAC mouse hearts showed a total of 2,486 transcripts were dysregulated (P < 0.05) with 1,790 upregulated and 696 downregulated genes (Figure 3A). The RNA-seq data showed that inhibition of Retn was associated with a significant upregulation of the growth arrest DNA-damage-inducible protein 45 alpha (Gadd45α) (Figure 3A and B), a suggested diabetes-associated gene which may be involved in diabetic cardiomyopathy.47,48 Ingenuity pathway analysis (IPA) showed that the top enriched biological processes are associated with pathways involved in cell apoptosis (i.e. p55 and JNK signalling), cell cycle, and DNA damage [GADD45α, ATM, BRCA1 signalling (Supplementary material online, Figure S3A)]. Interestingly, Gadd45α was represented in all biological processes depicted (Supplementary material online, Figure S3B), highlighting its critical role in apoptosis and DNA damage response pathways. These observations prompted us to focus on Gadd45α and probe its expression, and its involvement in Retn’s regulation of cardiac function. Gadd45α protein and mRNA expression were significantly downregulated in WT-TAC hearts (Figure 3C; Supplementary material online, Figure S4A for 4 weeks group at the protein level). Retn inhibition upregulated (Figure 3C; Supplementary material online, Figure S4A for 4 weeks group) while Retn overexpression (via AAV9-mRetn) downregulated Gadd45α expression in the RKO-TAC hearts (Figure 3D). We next determined whether Retn-driven downregulation of Gadd45α expression is regulated by non-coding RNAs, which have recently emerged as key regulators of gene expression in cardiomyocytes.49 Using bioinformatics tools, Gadd45α was identified as a putative target gene of miR148b-3p (Supplementary material online, Figure S5). The mRNA sequence of Gadd45α is predicted to contain a conserved ‘seed’ sequence complimentary to miR148b-3p in the 3′-untranslated region (3’-UTR) (Figure 3E; Supplementary material online, Figure S5). We performed a luciferase-based expression assays in vitro to verify that Gadd45α is bona fide miR148b-3p target. A partial of the Gadd45α 3′-UTR containing miR148b-3p binding sites or a mutant fragment was cloned downstream of the stop codon of the Renilla luciferase in a dual-luciferase reporter vector. The construct was co-transfected in HEK293 cells with synthetic miR148b-3p mimics or control miR mimics. In the presence of miR148b-3p, we observed a significant decrease (P < 0.05) in luciferase activity of Gadd45α construct compared to control miR (Figure 3E). Additionally, co-transfection of miR148b-3p mimics or control miR with construct containing mutated Gadd45α 3’-UTR sequence had no effect on the luciferase activity (Figure 3E).
Figure 3.

Retn controls cardiac remodelling through regulation of miR148b-3p and Gadd45α. (A) Volcano plots showing differentially expressed genes (P < 0.05) in the hearts of the RKO-TAC vs. WT-TAC mice. Blue reflects downregulated and red upregulated genes. The dash line denotes significance level at P = 0.05. (B) Heat map of differentially expressed genes, red indicating gene-upregulated and blue indicating gene-downregulated. (C) Gadd45α mRNA and protein expression in groups of mice indicated (n = 3–4). (D) Western blot and real-time PCR analysis of the expression of Gadd45α in TAC-induced RKO mice hearts following AA9-empty or AAV9-mRetn overexpression (n = 3–4). (E) Bioinformatics analysis identified the Gadd45α gene as a putative target of miR148b-3p. Direct interaction was confirmed using dual-luciferase reporter assays (n = 3). (F) Real-time PCR analysis of the expression levels of miR148b-3p in sham and TAC-induced WT or RKO mice hearts for 10 weeks (n = 4). (G) Real-time PCR analysis of the expression levels of miR148b-3p in AAV9-empty or AAV9-mRetn overexpressing TAC-induced RKO mice hearts (n = 5). (H, I) Real-time PCR analysis of the expression levels of miR148b-3p and Gadd45α following adenovirus-encoding resistin (Ad.Retn) or Ad.β-Gal infection of cardiomyocytes (MOI 100) isolated from RKO mice (n = 3). (J) Western blot analysis and quantification (left panel), and real-time PCR verification (right panel) of Retn expression in cardiomyocytes (from H and I) following Ad.β-gal or Ad.Retn infection (MOI 100) (n = 3). Protein levels were normalized to GAPDH, a loading control; mRNA levels were normalized to 18S. N, each animal is shown as an individual point whereas horizontal lines represent median values. Values are shown as mean± S.E.M. with each experiment performed in biological and technical replicates as indicated. Significance was evaluated by Student’s t-test or one-way ANOVA with Tukey’s post-hoc test. P values <0.05 were considered significant.
We next assessed the expression of miR148b-3p and found it to be significantly increased in pressure overload WT-TAC hearts compared to sham WT both at 4 weeks (Supplementary material online, Figure S4B) and at 10 weeks (Figure 3F). Retn inhibition decreased (Figure 3F) while Retn overexpression increased miR148b-3p expression in the RKO-TAC hearts (Figure 3G). Furthermore, infection of isolated mouse adult cardiomyocytes with adenovirus overexpressing Retn (Ad.Retn) also showed an increase in miR148b-3p levels (Figure 3H) and a decrease in Gadd45α mRNA expression compared to β-gal (Figure 3I). Increased Retn expressions were confirmed at protein and mRNA levels (Figure 3J). In addition, failing human cardiac tissues showed increased expression of RETN and miR-148b-3p and decreased expression of GADD45α, further corroborating the mouse data (Supplementary material online, Figure S6). These data suggest that Retn may regulate Gadd45α expression through miR148b-3p.
3.5 miR148b-3p silencing prevents and partially reverses myocardial dysfunction and remodelling
Anti-sense oligonucleotides that inhibit the expression of miRNAs are currently under development as therapeutic modalities for a diverse set of diseases associated with miRNAs aberrant gain-of-function. Therefore, we considered whether inhibition of miR148b-3p in the failing heart could have a beneficial effect in mitigating cardiac dysfunction associated with pressure overload. To test the therapeutic potential of silencing miR148b-3p in vivo, WT mice were subjected to pressure overload induced by TAC. After three weeks of TAC surgery, mice were randomly assigned to receive either anti-miR148b-3p or control anti-miR for 3 consecutive days (Figure 4A). At three weeks post-treatment (i.e. 6 weeks of TAC), mice treated with anti-miR148b-3p had a significant decline in miR148b-3p cardiac expression (Figure 4B) and demonstrated preserved systolic function when compared to control‐treated animals as assessed by fractional shortening (anti-miR148b-3p 47.79 ± 5.21% vs. control 29.49 ± 5.72%, P < 0.05; Figure 4C, Supplementary material online, Table S5). Anti-miR148b-3p treatment also prevented ventricular dilatation as evinced by the preservation of LV diameter at systole in anti-miR148b-3p-treated mice (Figure 4C and Supplementary material online, Table S5). Furthermore, the improvement of systolic function was accompanied by a reduction in HW/BW and HW/TL ratios (Figure 4D), and a decrease in Anf expression (Figure 4E) (at Week 6) in anti-miR148b-3p-treated TAC mice, although the expression of other hypertrophic gene markers (i.e. Acta1, MyH7, MyH6) did not show changes (Figure 4E), which might be very likely due to the relatively short treatment time with the anti-miR148p. In addition, anti-miR148b-3p treatment attenuated the interstitial myocardial fibrosis and fibrotic markers associated with chronic pressure overload (Figure 4F). As expected, TAC-operated mouse hearts treated with anti-miR148b-3p showed increased Gadd45α mRNA and protein expression (Figure 4G). These data suggest that miR148b-3p plays a role in cardiac dysfunction and mediates Retn-induced HF potentially through negative regulation of Gadd45α.
Figure 4.

Inhibition of miR148b-3p prevents and partially reverses cardiac remodelling and dysfunction. (A) Study protocol for anti-miR-148 therapy in TAC-induced WT mice HF. (B) Real-time PCR analysis of the expression levels of miR148b-3p in sham and TAC-WT mice ± anti-miR-148b-3p (n = 4). (C) Representative echocardiographic images and quantification of fractional shortening (FS%) and the left ventricular internal diameters in systole (LVIDs) (n = 4–6). (D) Analysis of heart weights (HW)/body weight (BW) and HW/tibia length ratios in sham and TAC-induced WT mice ± anti-miR-148b-3p (n = 4–6). (E) Real-time PCR analysis of the expression levels Anf, Myh7, Myh6, and Acta1(n = 4). (F) Representative images of Masson’s trichrome-stained hearts sections and myocardial fibrosis quantification as percentage of blue stained area (fibrotic) vs. control (scale bar, 50 µm, n = 4). Real-time PCR analysis of the expression levels of Col1a1, FN, CCN2, and LOX (n = 4). (G) Western blot (and densitometric quantification) and real-time PCR analysis of the expression of Gadd45α in sham and TAC-induced WT mice ± anti-miR-148b-3p (n = 4). Protein levels were normalized to GAPDH, a loading control; mRNA levels were normalized to 18S. N, each animal is shown as an individual point whereas horizontal lines represent median values as indicated. Values shown are mean± S.E.M., with each experiment performed in biological and technical replicates. Significance was evaluated by Student’s t-test or one-way ANOVA with Tukey’s post-hoc test. P values <0.05 were considered significant.
3.6 Retn overexpression promotes/triggers DNA damage response
To further delineate the molecular mechanisms driving Retn pathological effect on cardiac function, we focused on the potential involvement of DNA damage and the DNA damage response (DDR) given the role Gadd45α plays in the maintenance of genomic stability and DNA repair.50 We hypothesize that Retn promotes pathological cardiac remodelling through inhibition of Gadd45α and activation of DDR. To this end, we found that the expression levels of the essential DNA repair-related genes, Parp1 and Xrcc1, were decreased in WT-TAC-operated hearts remarkably after 10 weeks (Figure 5A) or following AAV9-mRetn overexpression (Figure 5B), but no changes after 4 weeks of TAC (Supplementary material online, Figure S7A). Retn inhibition on the other hand restored and/or further increased the levels of Parp1 and Xrcc1 in RKO-TAC or sham hearts (Figure 5A), suggesting that Retn, like TAC injury, may compromise the DNA damage repair activity. Furthermore, the expression of the phosphorylated histone variant H2AX (γH2AX), known to be an early and sensitive marker of DNA damage,51 was significantly increased in WT-TAC-operated hearts compared to sham mice (Figure 5C; Supplementary material online, Figure S7B for 4 weeks group) or following AAV9-mRetn overexpression (Figure 5D), suggesting that TAC injury or Retn upregulation induces DNA damage. Retn inhibition, in contrast, decreased γH2AX expression in the RKO-TAC hearts compared to WT-TAC mice (Figure 5C), further indicating that Retn gene deletion indeed suppresses DNA damage activation. Collectively, these data demonstrate that increased γH2AX activation in TAC and AAV9-mRetn mice was likely due to defective DNA repair represented by downregulation of Parp1 and Xrcc1 repair enzymes (Figure 5A and B).
Figure 5.

Effect of Retn and inhibition of miR148b-3p on DNA damage and DDR after TAC-induced mice HF. (A) Real-time PCR analysis of the expression levels of DNA repair-related genes (Parp1 and Xrcc1) in WT and RKO mice subjected to sham or TAC for 10 weeks (n = 4). (B) Real-time PCR analysis of the expression levels of Parp1 and Xrcc1 in AAV9-empty or AAV9-mRetn RKO mice subjected to TAC for 7 weeks (n = 5). (C) Western blot and quantification heart expression of γH2AX in WT and RKO mice subjected to sham or TAC for 10 weeks (n = 3–4). (D) Western blot and quantification heart expression of γH2AX in AAV9-mRetn vs. AAV9-empty controls (n = 5). (E) Real-time PCR analysis of the expression levels of Parp1 and Xrcc1 in sham and TAC-induced WT mice ± anti-miR-148b-3p (n = 4). (F) Western blot and quantification heart expression of γH2AX in sham and TAC-induced WT mice ± anti-miR-148b-3p (n = 4). Protein levels were normalized to GAPDH, a loading control; mRNA levels were normalized to 18S. N, each animal is shown as an individual point whereas horizontal lines represent median values. Values are shown as mean± S.E.M. with each experiment performed in biological and technical replicates as indicated. Significance was evaluated by Student’s t-test or one-way ANOVA with Tukey’s post-hoc test. P values <0.05 were considered significant.
We demonstrated earlier that Retn negatively regulates Gadd45α through activation of miR148b-3p (Figures 3 and 4). We therefore assessed the effects of miR148b-3p on DNA damage and repair activities. As expected, TAC-operated mouse hearts treated with anti-miR148b-3p showed increased Parp1 and Xrcc1 expression (Figure 5E) and decreased γH2AX expression (Figure 5F) compared to control hearts, paralleling the Retn gene deletion data above. These results clearly demonstrate that silencing Retn and miR148b-3p prevent DNA damage and DDR and potentially protect against cardiac dysfunction in response to TAC injury.
3.7 Gadd45α mitigates Retn-triggered DNA damage response
We next examined the direct involvement of Gadd45α and whether its overexpression prevents Retn-induced DNA damage and DDR. To this end, we generated H9C2-Gadd45α stable cell lines (Figure 6A) and challenged them with H2O2 or recombinant Retn to trigger DNA damage. Following exposure to H2O2 or Retn, control cells showed significant decrease in the abundance of Parp1 (Figure 6B, left panel) and Xrcc1 (Figure 6B, right panel) whereas Gadd45α-overexpressing cells showed increased expression of both enzymes (Figure 6B), reflecting that the delayed repair caused by H2O2 or Retn was partially rescued by Gadd45α expression. A similar pattern was observed with the accumulation of γH2AX. Treatment with H2O2 (Figure 6C) or Retn (Figure 6C) increased γH2AX accumulation and elevated γH2AX compared to control cells. This increase in γH2AX abundance was significantly reduced by Gadd45α (Figure 6C and D), intuitively supporting the hypothesis that Gadd45α protects against Retn-induced DNA damage.
Figure 6.

Overexpression of Gadd45α protects H9C2 rat myocytes against hydrogen peroxide (H2O2) and recombinant Retn-induced DDR. H9C2 cells were transfected with Gadd45α or empty control by Retroviral Gene Transfer and Expression system, then treated with 200 μM H2O2 for 24 h or 100 ng/mL recombinant Retn for 48 h. (A) Real-time PCR analysis of the expression levels of Gadd45α in Gadd45α overexpressing H9C2 cells vs. control (n = 3). (B) Expression levels of Parp1 and Xrcc1 were analysed by real-time PCR (n = 3). (C and D) Representative images and analysis of Western blots of γH2AX (n = 3). Protein levels were normalized to GAPDH, a loading control; mRNA levels were normalized to 18S. (E and F). Representative images and analysis of immunostaining of nuclear foci of pATM (green) and DAPI (blue) (n = 6, scale bar, 100 µm). Values are shown as mean± S.E.M. with each experiment performed in biological and technical replicates as indicated. Significance was evaluated by Student’s t-test or one-way ANOVA with Tukey’s post-hoc test. P values <0.05 were considered significant.
To verify these findings of marked genomic damage further, we examined the phosphorylation of Ataxia telangiectasia mutated (ATM) kinase, a well-established marker and inducer of DDR.40 Using immunostaining, we detected significantly higher counts of foci in cells positive for p-ATP after exposure to H2O2 (Figure 6E) or Retn (Figure 6F), reflecting increased DDR. Gadd45α overexpression, in contrast, led to significant reduction in pATM-positive foci (Figure 6E and F), underscoring the role of Gadd45α in mitigating the rates of DNA damage. Collectively, these results strengthen our conclusion that Retn predisposes cardiomyocytes to accumulation of DNA damage and defective DNA repair whereas Gadd45α has an opposite effect on these processes.
3.8 Loss of Retn prevents induction of myocardial apoptosis in vivo
To determine whether inhibition of Retn plays a role in the protection against cardiomyocyte apoptosis and further support the findings regarding DNA damage, we probed myocardial apoptosis in cardiac tissues following TAC. TUNEL analysis revealed that TAC induced increased apoptotic nuclei in the WT-TAC hearts compared to WT sham-operated hearts both at 4 weeks (Supplementary material online, Figure S8) and at 10 weeks groups (Figure 7). In contrast, RKO-TAC hearts showed a marked decrease in DNA fragmentation of nuclei detected by TUNEL compared to WT-TAC (Figure 7; Supplementary material online, Figure S8 for the 4-week group). These results are in line with the DNA damage findings and suggest that inhibition of Retn plays a critical protective role against DNA fragmentation following pressure overload-induced HF.
Figure 7.
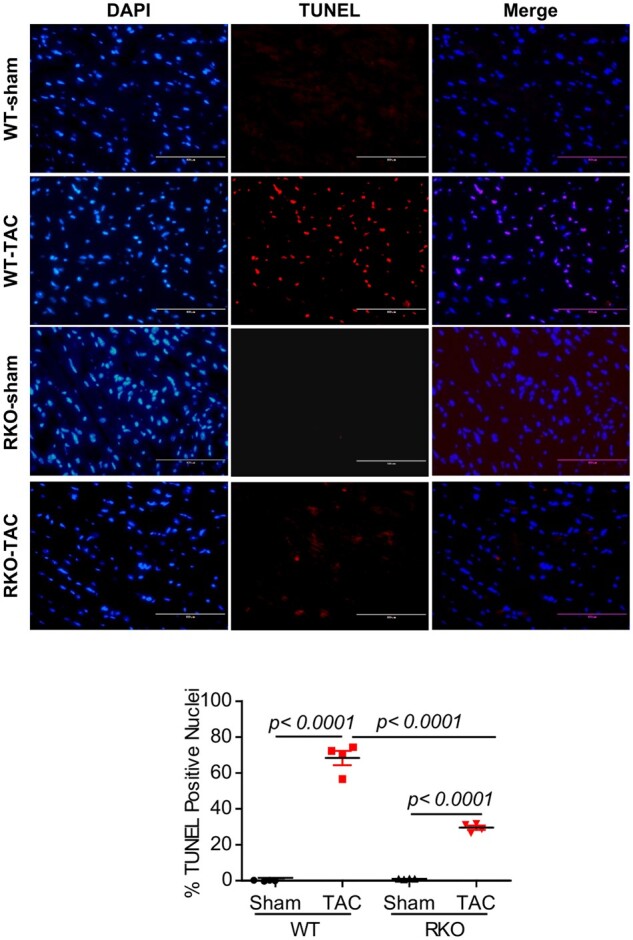
Inhibition of Retn blocks myocardial apoptosis. TUNEL staining was performed on LV tissues from the different groups at 10 weeks post-TAC as indicated. (A) TUNEL-positive apoptotic cells are shown as red-stained cells under fluorescent microscope. (B) Quantification of apoptosis shown as a percentage of apoptotic nuclei (red by TUNEL) vs. total nuclei (blue by DAPI) (scale bar, 100 µm, n = 4, 5–6 random fields/group were quantified). Values are shown as mean ± S.E.M. Significance was evaluated by Student’s t-test or one-way ANOVA with Tukey’s post-hoc test. P values <0.05 were considered significant.
4. Discussion
HF is a major and deadly health problem that is increasing in prevalence. There is an increasing need to understand the molecular aetiologies of HF in order to develop positive therapies to correct it potentially by directly affecting the molecular basis of the condition. The current study represents the first detailed investigation documenting the direct implication of Retn in the modulation of cardiac function and remodelling. Using gain/loss-of-function approaches, we demonstrated that Retn is able and sufficient to negatively drive pathways promoting cardiac dysfunction and fibrotic remodelling after pressure overload injury. Deletion of Retn in vivo blocked pressure overload-induced myocardial dysfunction and remodelling, and partially attenuated the progression to HF. This was evidenced by improvement in cardiac contractility parameters, decreased myocardial fibrosis and apoptosis, and improved overall survival following Retn deletion after pressure overload. On the other hand, cardiac-selective overexpression of Retn was sufficient to deteriorate cardiac function and induce HF and premature death in Retn-KO mice. It is worthy to note that our studies were performed on a mouse model that lacks Retn specifically in the white adipose tissue and not whole body knockout, highlighting the potential role of adipose-derived Retn in driving the observed cardiac phenotypes. Although adipocytes are considered the main source of secrete Retn, we and others have nevertheless demonstrated that it is also detectable in cardiomyocytes.11,12,18,31 Therefore, a clear limitation in our study is that the RKO mice still have some residual Retn expressed in the heart, which may conceivably exert its negative effects and therefore explain the partial reversal in cardiac function and remodelling observed in the study. However, our findings still provide a strong evidence for a critical negative role of Retn in HF and add to a growing body of evidence associating Retn with HF. Higher serum levels of Retn were found in patients with HF25 and demonstrated to be a predictor of the presence and severity of coronary artery disease30 and positively related to the severity and incidents of HF hospitalization.26,28 Furthermore, Retn has been associated with markers of insulin resistance and inflammation which have in turn been shown to predict HF incidence.30,47,52 Others, however, have observed that higher circulating Retn levels were strongly associated with increased risk of new-onset HF and the association of Retn with HF persisted after adjustment for well-known HF risk factors, such as B-type natriuretic peptide, obesity, insulin resistance, and inflammation,26,27 suggesting that Retn may promote HF independent of insulin resistance and inflammation, further supporting our findings for a direct effect.
Other lines of evidence from our laboratory and others strongly indicate that hyper-resistinemia contributes to the impairment of cardiac contractility and development of cardiac dysfunction. Cyclic stretch and aorta-caval shut upregulated Retn.12 We have previously showed that overexpression of Retn in cultured rat ventricular myocytes altered cardiomyocyte function and calcium transients in vitro and induced cellular hypertrophy.18,31 We have subsequently demonstrated that long-term overexpression of Retn in vivo in normal rats impaired left ventricular contractility and induced a complex phenotype myocardial remodelling characterized by oxidative stress, fibrosis, and apoptosis.32 Furthermore, by measuring cardiac tissue levels of Retn in several animal models of cardiac hypertrophy and failure, we demonstrated that mouse models of cardiac hypertrophy that is associated with fibrosis (diabetes, pressure overload, and HF) exhibited increased tissue levels of Retn compared to non-fibrosing hypertrophy (volume overload) where Retn is not or minimally augmented.53 These observations are corroborated by our finding in the current study that Retn silencing attenuated while Retn overexpression exacerbated cardiac fibrosis in TAC-induced RKO mice. Associating Retn with cardiac fibrosis is of particular clinical and therapeutic interest, since 1) the level of serum Retn is elevated in hypertrophy and HF conditions, diseases where cardiac fibrosis is emerging as a predictor of arrhythmias and as a potential benchmark for device therapy and 2) the accumulation of collagen fibres and their progressive organization and cross-linking into irreversible fibrosis are histological characteristics of pressure overload HF.54
The mechanisms by which Retn promotes HF remain unknown. To gain molecular mechanistic insight regarding Retn effects on HF signalling, we explored potential transcriptional regulation mediated by microRNAs, given the key roles these molecules play in cardiac function and fibrosis.55 We have observed that miR148b-3p was significantly upregulated by pressure overload, and interestingly, Retn inhibition decreased while Retn overexpression—in vitro and in vivo—increased miR148b-3p expression in the TAC-induced mice heart, suggesting that miR148b-3p may be associated with pressure overload HF. This assumption is supported by a previous report indicating that circulating miR148b-3p might function as a biomarker for HF in patients with mitral regurgitation.56 Therefore, we considered whether inhibition of miR148b-3p in the failing heart could have a beneficial effect in mitigating cardiac dysfunction associated with pressure overload. Treatment with anti-miR148b-3p partially preserved systolic function and ventricular dilatation, and attenuated cardiac hypertrophy and interstitial myocardial fibrosis associated with chronic pressure overload. The partial effects exerted by anti-miR148b-3p on cardiac dysfunction and the hypertrophic response in our study were likely due to the short treatment time (i.e. 3 weeks). Although this novel class of locked nucleic acid (LNA)-based oligonucleotides (anti-miR148b-3p) are efficient and specific silencers of endogenous miRNAs in mice, their efficiency is often limited by the LNA dose, the treatment time, the delivery route (systemic vs. local), and the cellular uptake in vivo. In our case, the inhibition of miR148b-3p caused a significant reduction in the endogenous expression of miR148b-3p following TAC injury; however, it potentially needs longer treatment protocols to provide profound effects.
miR148b-3p belongs to the miR148/152 family which includes miR148a, miR148b, and miR152 and are involved in various biological processes including growth, development, and tumourigenesis.57 Currently there is a paucity of information regarding the role of miR148b-3p in cardiovascular function with a single report showing miR148b-3p regulates cardiomyocyte apoptosis induced by hypoxia/reoxygenation injury in vitro.58 Computational prediction of targets of miR148b-3p identified a putative miR148b ‘seed sequence’ in the 3’-UTR of the Gadd45α gene. Notably, only miR148b-3p family member was identified in all the four databases intersection analysis as indicated in the ‘Methods’ section (Supplementary material online, Figure S5), suggesting that Gadd45α is a cognate target of miR148b-3p in the heart. Gadd45α is a member of a group of genes that are induced by DNA damaging agents and growth arrest signals. Gadd45α has been widely studied as a common expressed protein involved in cell growth, DNA repair and apoptosis59 (Supplementary material online, Figure S3). It has been reported that Gadd45α potentially acts as a linker between diabetic cardiomyopathy and baroreflex dysfunction.48 Gadd45α attenuates hepatic fibrosis by regulating the activation of hepatic stellate cells inhibition of TGF-β/Smad signalling,60 supporting its role in fibrotic response. We observed that the downregulation of Gadd45α is strongly induced by TAC injury, Retn overexpression and miR148b-3p stimulation both in cultured myocytes and in vivo in mouse hearts. Inhibition of Retn and/or miR148b-3p correlated with a significant upregulation of Gadd45α expression, strongly indicating that miR148b-3p promotes cardiac dysfunction and mediates Retn-induced HF potentially through the regulation of Gadd45α. Our study uncovers the dysregulation of miR148b-3p in the heart during cardiac remodelling and provides evidence that it is a crucial regulator recruited by Retn to promote cardiac hypertrophy and fibrosis leading to the induction of HF after pressure overload. These findings have conceivably prompted us to examine whether and how Gadd45α umpires Retn-driven cardiac remodelling. We focused on the potential involvement of DNA damage and DDR given the role Gadd45α plays in the maintenance of genomic stability and DNA repair50 (Supplementary material online, Figure S3). In addition, accumulation of unrepaired DNA damage and activation of DDR are observed in cardiomyocytes, including human failing heart37 and mouse models of cardiac hypertrophy.37–39 We therefore hypothesized that Gadd45α enhances base excision repair and mitigates Retn-evoked pathological cardiac remodelling. In the current study, we showed that pressure overload (TAC injury) and Retn overexpression resulted in (i) insufficient DNA damage repair due to downregulation of Parp1 and Xrcc1, (ii) significant increase in phosphorylated ATM and γH2AX, primary effectors of DDR pathway, in cardiomyocytes, and (iii) increased DNA fragmentation and cardiomyocyte apoptosis. However, these changes were reversed by either Retn silencing or Gadd45α enhancement, clearly supporting our hypothesis that Retn predisposes cardiomyocytes to accumulation of DNA damage and defective DNA repair, whereas Gadd45α protects against these processes. Our results are in line with recent findings demonstrating that unrepaired DNA accumulation was more pronounced in mice-deficient in Xrcc1,38 while knockdown of ATM attenuated persistent DDR activation and protected against MI and pressure overload-induced HF.38,39,61 In the light of these findings, it is conceivable to suggest that Retn accentuates cardiac dysfunction through compromising Gadd45α safeguarding of the genomic integrity.
Taken together, our present study demonstrates for the first time that Retn directly controls cardiac remodelling though the activation of the miR148b-3p/Gadd45α axis and activation of DDR. This proof-of-concept study suggests that silencing of Retn and/or miR148b-3p activity will protect the heart against DNA damage-induced pathological cardiac remodelling and improve systolic function in patients with HF. Future therapeutic approaches might benefit from targeted approaches, such as adeno-associated virus-based Retn/anti-miR148/Gadd45α gene delivery, which may restrict inhibition to the cardiac myocyte in patients. Repression of aberrant DDR may also emerge as a potential objective in the treatment of HF and in hyper-resistinemia conditions.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
D.L. conceived and supervised the project. B.Z. conducted the experiments and analysed the data. R.B. performed experiments. D.L. and B.Z. wrote the manuscript; all authors commented on the manuscript.
Supplementary Material
Acknowledgements
The authors thank Mitch Lazar for the resistin knockout mice.
Conflict of interest: none declared.
Funding
R.B. was supported in part by a T32 HL7824-19, and D.L. was supported by R01HL097357 and R01HL137220 from the National Institutes of Health.
Data availability
The data supporting the findings of this study are available within the article, its supplementary materials, and/or from the corresponding author, D.L., upon reasonable request.
Contributor Information
Baoyin Zhao, Cardiovascular Research Institute, New York, NY 10029, USA.
Rihab Bouchareb, Cardiovascular Research Institute, New York, NY 10029, USA.
Djamel Lebeche, Cardiovascular Research Institute, New York, NY 10029, USA; Department of Medicine, Diabetes, Obesity and Metabolism Institute, New York, NY 10029, USA; Graduate School of Biomedical Sciences, The Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Translational perspective.
Heart failure (HF) is a major cause of morbidity and mortality worldwide. Various types of pathological risk factors contribute to HF development. Population studies have associated the levels of the adipokine resistin with incidences of HF and worst cardiovascular outcomes; however, a direct role of resistin in the development of HF has not been demonstrated. In this regard, resistin is documented to exert a critical negative role in directly promoting cardiac dysfunction and fibrotic remodelling after pressure overload injury. Controlling resistin levels may provide a potential therapeutic approach for treating HF, particularly if combined with targeted approaches restricting resistin silencing in cardiac myocyte in patients.
References
- 1. Savarese G, Lund LH.. Global public health burden of heart failure. Card Fail Rev 2017;3:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB.. Heart disease and stroke statistics–2015 update: a report from the American Heart Association. Circulation 2015;131:e29–322. [DOI] [PubMed] [Google Scholar]
- 3. Ambrosy AP, Fonarow GC, Butler J, Chioncel O, Greene SJ, Vaduganathan M, Nodari S, Lam CSP, Sato N, Shah AN, Gheorghiade M.. The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J Am Coll Cardiol 2014;63:1123–1133. [DOI] [PubMed] [Google Scholar]
- 4. Hunter JJ, Chien KR.. Signaling pathways for cardiac hypertrophy and failure. N Engl J Med 1999;341:1276–1283. [DOI] [PubMed] [Google Scholar]
- 5. Molkentin JD, Dorn GW 2nd. Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol 2001;63:391–426. [DOI] [PubMed] [Google Scholar]
- 6. Frey N, Olson EN.. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol 2003;65:45–79. [DOI] [PubMed] [Google Scholar]
- 7. Bozkurt B, Aguilar D, Deswal A, Dunbar SB, Francis GS, Horwich T, Jessup M, Kosiborod M, Pritchett AM, Ramasubbu K, Rosendorff C, Yancy C, American Heart Association Heart Failure and Transplantation Committee of the Council on Clinical Cardiology; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular and Stroke Nursing; Council on Hypertension; and Council on Quality and Outcomes Research . Contributory risk and management of comorbidities of hypertension, obesity, diabetes mellitus, hyperlipidemia, and metabolic syndrome in chronic heart failure: a scientific statement from the American Heart Association. Circulation 2016;134:e535–e578. [DOI] [PubMed] [Google Scholar]
- 8. Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA.. The hormone resistin links obesity to diabetes. Nature 2001;409:307–312. [DOI] [PubMed] [Google Scholar]
- 9. Fontana A, Spadaro S, Copetti M, Spoto B, Salvemini L, Pizzini P, Frittitta L, Mallamaci F, Pellegrini F, Trischitta V, Menzaghi C.. Association between resistin levels and all-cause and cardiovascular mortality: a new study and a systematic review and meta-analysis. PloS One 2015;10:e0120419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Park HK, Kwak MK, Kim HJ, Ahima RS.. Linking resistin, inflammation, and cardiometabolic diseases. Korean J Intern Med 2017;32:239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Graveleau C, Zaha VG, Mohajer A, Banerjee RR, Dudley-Rucker N, Steppan CM, Rajala MW, Scherer PE, Ahima RS, Lazar MA, Abel ED.. Mouse and human resistins impair glucose transport in primary mouse cardiomyocytes, and oligomerization is required for this biological action. J Biol Chem 2005;280:31679–31685. [DOI] [PubMed] [Google Scholar]
- 12. Wang BW, Hung HF, Chang H, Kuan P, Shyu KG.. Mechanical stretch enhances the expression of resistin gene in cultured cardiomyocytes via tumor necrosis factor-alpha. Am J Physiol Heart Circ Physiol 2007;293:H2305–H2312. [DOI] [PubMed] [Google Scholar]
- 13. Ghosh S, Singh AK, Aruna B, Mukhopadhyay S, Ehtesham NZ.. The genomic organization of mouse resistin reveals major differences from the human resistin: functional implications. Gene 2003;305:27–34. [DOI] [PubMed] [Google Scholar]
- 14. McTernan CL, McTernan PG, Harte AL, Levick PL, Barnett AH, Kumar S.. Resistin, central obesity, and type 2 diabetes. Lancet (London, England) 2002;359:46–47. [DOI] [PubMed] [Google Scholar]
- 15. Yang R-Z, Huang Q, Xu A, McLenithan JC, Eisen JA, Shuldiner AR, Alkan S, Gong D-W, Eison JA.. Comparative studies of resistin expression and phylogenomics in human and mouse. Biochem Biophys Res Commun 2003;310:927–935. [DOI] [PubMed] [Google Scholar]
- 16. Kaser S, Kaser A, Sandhofer A, Ebenbichler CF, Tilg H, Patsch JR.. Resistin messenger-RNA expression is increased by proinflammatory cytokines in vitro. Biochem Biophys Res Commun 2003;309:286–290. [DOI] [PubMed] [Google Scholar]
- 17. Lu SC, Shieh WY, Chen CY, Hsu SC, Chen HL.. Lipopolysaccharide increases resistin gene expression in vivo and in vitro. FEBS Lett 2002;530:158–162. [DOI] [PubMed] [Google Scholar]
- 18. Kim M, Oh JK, Sakata S, Liang I, Park W, Hajjar RJ, Lebeche D.. Role of resistin in cardiac contractility and hypertrophy. J Mol Cell Cardiol 2008;45:270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karakikes I, Kim M, Hadri L, Sakata S, Sun Y, Zhang W, Chemaly ER, Hajjar RJ, Lebeche D.. Gene remodeling in type 2 diabetic cardiomyopathy and its phenotypic rescue with SERCA2a. PloS One 2009;4:e6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Szalowska E, Elferink MG, Hoek A, Groothuis GM, Vonk RJ.. Resistin is more abundant in liver than adipose tissue and is not up-regulated by lipopolysaccharide. J Clin Endocrinol Metab 2009;94:3051–3057. [DOI] [PubMed] [Google Scholar]
- 21. Morash BA, Willkinson D, Ur E, Wilkinson M.. Resistin expression and regulation in mouse pituitary. FEBS Lett 2002;526:26–30. [DOI] [PubMed] [Google Scholar]
- 22. Gualillo O, González-Juanatey JR, Lago F.. The emerging role of adipokines as mediators of cardiovascular function: physiologic and clinical perspectives. Trends Cardiovasc Med 2007;17:275–283. [DOI] [PubMed] [Google Scholar]
- 23. Gao J, Chang Chua C, Chen Z, Wang H, Xu X, C Hamdy R, McMullen JR, Shioi T, Izumo S, Chua BHL.. Resistin, an adipocytokine, offers protection against acute myocardial infarction. J Mol Cell Cardiol 2007;43:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rothwell SE, Richards AM, Pemberton CJ.. Resistin worsens cardiac ischaemia-reperfusion injury. Biochem Biophys Res Commun 2006;349:400–407. [DOI] [PubMed] [Google Scholar]
- 25. Takeishi Y, Niizeki T, Arimoto T, Nozaki N, Hirono O, Nitobe J, Watanabe T, Takabatake N, Kubota I.. Serum resistin is associated with high risk in patients with congestive heart failure–a novel link between metabolic signals and heart failure. Circ J 2007;71:460–464. [DOI] [PubMed] [Google Scholar]
- 26. Frankel DS, Vasan RS, D'Agostino RB Sr., Benjamin EJ, Levy D, Wang TJ, Meigs JB.. Resistin, adiponectin, and risk of heart failure the Framingham offspring study. J Am Coll Cardiol 2009;53:754–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang MH, Na B, Schiller NB, Whooley MA.. Association of resistin with heart failure and mortality in patients with stable coronary heart disease: data from the heart and soul study. J Card Fail 2011;17:24–30. [DOI] [PubMed] [Google Scholar]
- 28. Burnett MS, Devaney JM, Adenika RJ, Lindsay R, Howard BV.. Cross-sectional associations of resistin, coronary heart disease, and insulin resistance. J Clin Endocrin Metab 2006;91:64–68. [DOI] [PubMed] [Google Scholar]
- 29. Pischon T, Bamberger CM, Kratzsch J, Zyriax BC, Algenstaedt P, Boeing H, Windler E.. Association of plasma resistin levels with coronary heart disease in women. Obes Res 2005;13:1764–1771. [DOI] [PubMed] [Google Scholar]
- 30. Reilly MP, Lehrke M, Wolfe ML, Rohatgi A, Lazar MA, Rader DJ.. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation 2005;111:932–939. [DOI] [PubMed] [Google Scholar]
- 31. Kang S, Chemaly ER, Hajjar RJ, Lebeche D.. Resistin promotes cardiac hypertrophy via the AMP-activated protein kinase/mammalian target of rapamycin (AMPK/mTOR) and c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathways. J Biol Chem 2011;286:18465–18473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chemaly ER, Hadri L, Zhang S, Kim M, Kohlbrenner E, Sheng J, Liang L, Chen J, K-Raman P, Hajjar RJ, Lebeche D.. Long-term in vivo resistin overexpression induces myocardial dysfunction and remodeling in rats. J Mol Cell Cardiol 2011;51:144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qatanani M, Szwergold NR, Greaves DR, Ahima RS, Lazar MA.. Macrophage-derived human resistin exacerbates adipose tissue inflammation and insulin resistance in mice. J Clin Invest 2009;119:531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, Dangwal S, Kumarswamy R, Bang C, Holzmann A, Remke J, Caprio M, Jentzsch C, Engelhardt S, Geisendorf S, Glas C, Hofmann TG, Nessling M, Richter K, Schiffer M, Carrier L, Napp LC, Bauersachs J, Chowdhury K, Thum T.. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun 2012;3:1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hodgkinson CP, Kang MH, Dal-Pra S, Mirotsou M, Dzau VJ.. MicroRNAs and Cardiac Regeneration. Circ Res 2015;116:1700–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vegter EL, van der Meer P, de Windt LJ, Pinto YM, Voors AA.. MicroRNAs in heart failure: from biomarker to target for therapy. Eur J Heart Fail 2016;18:457–468. [DOI] [PubMed] [Google Scholar]
- 37. Takahashi T, Shishido T, Kinoshita D, Watanabe K, Toshima T, Sugai T, Narumi T, Otaki Y, Tamura H, Nishiyama S, Arimoto T, Takahashi H, Miyamoto T, Watanabe T, Woo CH, Abe JI, Takeishi Y, Kubota I, Watanabe M.. Cardiac nuclear high-mobility group box 1 ameliorates pathological cardiac hypertrophy by inhibiting DNA damage response. JACC Basic Transl Sci 2019;4:234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Higo T, Naito AT, Sumida T, Shibamoto M, Okada K, Nomura S, Nakagawa A, Yamaguchi T, Sakai T, Hashimoto A, Kuramoto Y, Ito M, Hikoso S, Akazawa H, Lee JK, Shiojima I, McKinnon PJ, Sakata Y, Komuro I.. DNA single-strand break-induced DNA damage response causes heart failure. Nat Commun 2017;8:15104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakada Y, Nhi Nguyen NU, Xiao F, Savla JJ, Lam NT, Abdisalaam S, Bhattacharya S, Mukherjee S, Asaithamby A, Gillette TG, Hill JA, Sadek HA.. DNA damage response mediates pressure overload-induced cardiomyocyte hypertrophy. Circulation 2019;139:1237–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shiloh Y, Ziv Y.. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013;14:197–210. [PubMed] [Google Scholar]
- 41. Banerjee RR, Rangwala SM, Shapiro JS, Rich AS, Rhoades B, Qi Y, Wang J, Rajala MW, Pocai A, Scherer PE, Steppan CM, Ahima RS, Obici S, Rossetti L, Lazar MA.. Regulation of fasted blood glucose by resistin. Science 2004;303:1195–1198. [DOI] [PubMed] [Google Scholar]
- 42. Zaw AM, Williams CM, Law HK, Chow BK.. Minimally invasive transverse aortic constriction in mice. JoVE 2017;303:e55293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ, European Association of Echocardiography . Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 2005;18:1440–1463. [DOI] [PubMed] [Google Scholar]
- 44. LaRocca TJ, Fabris F, Chen J, Benhayon D, Zhang S, McCollum L, Schecter AD, Cheung JY, Sobie EA, Hajjar RJ, Lebeche D.. Na+/Ca2+ exchanger-1 protects against systolic failure in the Akitains2 model of diabetic cardiomyopathy via a CXCR4/NF-kappaB pathway. Am J Physiol Heart Circ Physiol 2012;303:H353–H367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pacher P, Nagayama T, Mukhopadhyay P, Batkai S, Kass DA.. Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat Protoc 2008;3:1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B, Wang S, Lakatta EG, Cheng H, Xiao RP.. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am J Physiol Heart Circ Physiol 2000;279:H429–H436. [DOI] [PubMed] [Google Scholar]
- 47. Ohmori R, Momiyama Y, Kato R, Taniguchi H, Ogura M, Ayaori M, Nakamura H, Ohsuzu F.. Associations between serum resistin levels and insulin resistance, inflammation, and coronary artery disease. J Am Coll Cardiol 2005;46:379–380. [DOI] [PubMed] [Google Scholar]
- 48. Wang N, Yang C, Xie F, Sun L, Su X, Wang Y, Wei R, Zhang R, Li X, Yang B, Ai J.. Gadd45alpha: a novel diabetes-associated gene potentially linking diabetic cardiomyopathy and baroreflex dysfunction. PloS One 2012;7:e49077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Divakaran V, Mann DL.. The emerging role of microRNAs in cardiac remodeling and heart failure. Circ Res 2008;103:1072–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Siafakas AR, Richardson DR.. Growth arrest and DNA damage-45 alpha (GADD45alpha). Int J Biochem Cell Biol 2009;41:986–989. [DOI] [PubMed] [Google Scholar]
- 51. Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y.. GammaH2AX and cancer. Nat Rev Cancer 2008;8:957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vasan RS, Sullivan LM, Roubenoff R, Dinarello CA, Harris T, Benjamin EJ, Sawyer DB, Levy D, Wilson PW, D'Agostino RB, Framingham Heart Study . Inflammatory markers and risk of heart failure in elderly subjects without prior myocardial infarction: the Framingham Heart Study. Circulation 2003;107:1486–1491. [DOI] [PubMed] [Google Scholar]
- 53. Chemaly ER, Kang S, Zhang S, McCollum L, Chen J, Benard L, Purushothaman KR, Hajjar RJ, Lebeche D.. Differential patterns of replacement and reactive fibrosis in pressure and volume overload are related to the propensity for ischaemia and involve resistin. J Physiol 2013;591:5337–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Creemers EE, Pinto YM.. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc Res 2011;89:265–272. [DOI] [PubMed] [Google Scholar]
- 55. Gurha P. MicroRNAs in cardiovascular disease. Curr Opin Cardiol 2016;31:249–254. [DOI] [PubMed] [Google Scholar]
- 56. Chen MC, Chang TH, Chang JP, Huang HD, Ho WC, Lin YS, Pan KL, Liu WH, Huang YK.. Circulating miR-148b-3p and miR-409-3p as biomarkers for heart failure in patients with mitral regurgitation. Int J Cardiol 2016;222:148–154. [DOI] [PubMed] [Google Scholar]
- 57. Chen Y, Song YX, Wang ZN.. The microRNA-148/152 family: multi-faceted players. Mol Cancer 2013;12:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sun M, Zhai M, Zhang N, Wang R, Liang H, Han Q, Jia Y, Jiao L.. MicroRNA-148b-3p is involved in regulating hypoxia/reoxygenation-induced injury of cardiomyocytes in vitro through modulating SIRT7/p53 signaling. Chem Biol Interact 2018;296:211–219. [DOI] [PubMed] [Google Scholar]
- 59. Salvador JM, Brown-Clay JD, Fornace AJ Jr. Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv Exp Med Biol 2013;793:1–19. [DOI] [PubMed] [Google Scholar]
- 60. Hong L, Sun QF, Xu TY, Wu YH, Zhang H, Fu RQ, Cai FJ, Zhou QQ, Zhou K, Du QW, Zhang D, Xu S, Ding JG.. New role and molecular mechanism of Gadd45a in hepatic fibrosis. World J Gastroenterol 2016;22:2779–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Foster CR, Daniel LL, Daniels CR, Dalal S, Singh M, Singh K.. Deficiency of ataxia telangiectasia mutated kinase modulates cardiac remodeling following myocardial infarction: involvement in fibrosis and apoptosis. PloS One 2013;8:e83513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the article, its supplementary materials, and/or from the corresponding author, D.L., upon reasonable request.