

Abstract
The replication machinery of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is closely associated with the endoplasmic reticulum (ER) in host cells. Activation of the unfolded protein response (UPR) is a strategy hijacked by coronavirus to facilitate its replication and suppress host innate immunity. Here, we have found that SARS-CoV-2 ORF8 protein accumulates in the ER and escapes the degradation system by forming mixed disulfide complexes with ER oxidoreductases. ORF8 induces the activation of three UPR pathways through targeting key UPR components, remodels ER morphology and accelerates protein trafficking. Moreover, small molecule reducing agents release ORF8 from the mixed disulfide complexes and facilitate its degradation, therefore mitigate ER stress. Our study reveals a unique mechanism by which SARS-CoV-2 ORF8 escapes degradation by host cells and regulates ER reshaping. Targeting ORF8-involved mixed disulfide complexes could be a new strategy to alleviate SARS-CoV-2 induced ER stress and related diseases.
Keywords: Endoplasmic reticulum, ORF8, Redox, SARS-CoV-2, Unfolded protein response
Graphical abstract
Highlights
-
•
SARS-CoV-2 ORF8 protein accumulates in the ER through forming mixed disulfide complexes.
-
•
Two key protein disulfide isomerases, PDI and ERp44, are main targets of ORF8.
-
•
ORF8 induces ER stress, remodels the ER and accelerates protein trafficking.
-
•
Small molecule reducing agents facilitates the degradation of ORF8 and mitigates ER stress.
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of the ongoing coronavirus disease 2019 (COVID-19) pandemic. SARS-CoV-2 is a beta-coronavirus with a positive-sense, single-stranded RNA genome [1,2]. Among the encoding 29 proteins, the nonstructural proteins (NSP1-16) and four structural proteins: spike (S), nucleocapsid (N), membrane (M), envelope (E) proteins show high sequence similarities to the corresponding proteins in SARS-CoV-1. The conservation of nonstructural and structural proteins indicates that SARS-CoV-2 and SARS-CoV-1 may share a similar life cycle and strategies to ensure their replication, access and exit of host cells. The genome also encodes nine accessory proteins which show less sequence homology to those of other coronaviruses. Generally, coronavirus accessory proteins have been considered to be dispensable for virus replication. However, emerging evidences indicated that several accessory proteins of SARS-CoV-2 modulate host cellular processes and contribute to viral virulence and pathogenesis [3].
One accessory protein, SARS-CoV-2 ORF8, is poorly conserved among coronavirus with only 31% sequence similarity to SARS-CoV-1 ORF8ab (Fig. S1A). The ORF8 coding region is one of the most variable regions in SARS-CoV-2 genome, and several mutations in ORF8 gene have been listed in current World Health Organization “variants of concern”. Deletion of ORF8 caused less severe symptoms, milder hypoxic conditions and decreased inflammatory cytokine levels for the infected patients [4]. Recent studies suggested that ORF8 could mediate immune evasion by downregulating the cell surface expression of MHC-I molecules and inhibiting type I interferon signaling pathway [5,6]. ORF8 gene encodes a 121-amino acid residues protein with an N-terminal signal peptide for the endoplasmic reticulum (ER) import, but lacks a conventional C-terminal KDEL motif for ER retention. Therefore, whether ORF8 is retained in the ER or secreted remains unclear. An interaction landscape study revealed that ORF8 was at the top of SARS-CoV-2 proteins interacting with the early secretory pathway in host cells [7]. And the ORF8 interacting proteins were enriched in the ER protein quality control system including protein folding, glycosylation and ER-associated degradation (ERAD) [8]. However, the biological consequences of the interactions between ORF8 and clients remain largely unknown.
The ER is the site for folding of newly synthesized secretory and membrane proteins, and is also important for virion assembly and maturation. During viral infection, viruses need to hijack the translational machinery of the host cell for the expression of their own proteins, which may rapidly overwhelm the ER folding capacity and trigger the unfolded protein response (UPR). As shown by a study that SARS-CoV-2 infection indeed caused ER stress and activated the UPR [9]. The UPR pathways in mammals consist of three main signaling branches initiated by the ER transmembrane sensors: IRE1α, PERK and ATF6. Once activated under ER stress, the UPR evokes a series downstream signaling to increase the expression of folding factors to enhance protein output from the ER, and meanwhile limiting the input of newly synthesized proteins into the ER. Although ORF8 was reported to be able to induce ER stress [10], the underlying molecular mechanism is unclear at all.
In this study, we report that ORF8 forms mixed disulfide complexes with ER proteins, therefore escapes ER degradation and accumulates in the ER lumen. We show that ORF8 induces the three UPR pathways, remodels ER morphology and accelerates protein trafficking. Moreover, small molecule reducing agents, such as N-acetyl-l-cysteine (NAC) and dithiothreitol (DTT), reduce the ORF8-involved mixed disulfide complexes, enhance ORF8 degradation and mitigate ER stress. Taken together, our findings shed light on the biological significance of SARS-CoV-2 ORF8, and suggest that the usage of reducing agents may be a new strategy for alleviation of SARS-CoV-2 induced ER stress and related diseases.
2. Materials and methods
2.1. Antibodies, chemicals, oligonucleotides and recombinant DNA
The sources and identifiers of antibodies, chemicals, oligonucleotides and recombinant DNA used in this paper can be found in Table S1.
2.2. Cell lines
HEK 293T and Vero E6 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Hyclone) supplemented with 10% fetal bovine serum (Gibco). HeLa cells were cultured in DMEM supplemented with 5% fetal bovine serum. HepG2 cells were cultured in RPMI medium (Gibco) supplemented with 10% fetal bovine serum. All medium was supplemented with 100 μg/ml streptomycin and 100 units/ml penicillin (Invitrogen), and the cells were cultured at 37 °C with 5% CO2. Transfections were performed using Lipofectamine 3000 (Invitrogen) or viafect (Promega) according to the manufacture's protocol.
2.3. Lentivirus production
HEK 293T cells were co-transfected with lentiviral vectors and packaging plasmids psPAX2 (Addgene) and pMD.2G (Addgene) using Lipofectamine 3000. Lentivirus particles were collected within 24–72 h, concentrated by ultracentrifugation at 25,000 rpm at 4 °C for 2 h, and used for transduction in the presence of 10 μg/ml polybrene (YEASEN).
2.4. Lentiviral CRISPR/Cas9 and shRNA-mediated gene knockdown
Lentiviruses packaged with Lenti-CRISPRv2 PDI or Lenti-PLVTHM-ERP44 were transduced into HEK 293T cells. Lentiviruses containing Lenti-CRISPRv2-NTC (non-targeting control) or Lenti-PLVTHM-control were used as negative control. For lentiviral CRISPR/Cas9 mediated gene knockdown, HEK 293T cells were transduced with lentiviruses for 48 h, and then selected with puromycin (InvivoGen) for 7 days.
2.5. Genome editing using CRISPR/Cas9
ERP44 knockout (KO) HepG2 cells were constructed as described [11]. CRISPR targeting oligonucleotide designed for human ERP44 exon 1 (5′-GGCAGGATGCATGGTAACGCTGG-3′) was cloned into the expression vector pSpCas9 (BB)-2A-GFP (PX458) (Addgene). HepG2 cells were transfected with CRISPR construct containing the single-guide RNA (sgRNA) by ViaFect for 48 h. GFP-positive cells were then sorted as single cells into a 96-well plate using a FACS Aria-II sorter (BD BioSciences). KO efficiency was assessed by immunoblotting.
2.6. Immunofluorescence
Treated cells cultured on glass bottom dishes (NEST) were washed with PBS three times, fixed with 4% paraformaldehyde (PFA) for 15 min, and permeabilized with 0.4% Triton X-100 for 5 min. After blocking with 5% BSA for 1 h at room temperature (RT), cells were incubated with corresponding primary antibodies at 4 °C overnight and then with fluorescent-conjugated secondary antibodies for 1 h in the dark. The cells were rinsed with PBS and analyzed by Nikon Eclipse Ti2 inverted microscope.
2.7. Fluorescence protease protection (FPP)
Cells were plated on glass bottom dishes (NEST) and transfected with the indicated plasmids for 24 h. After washing with KHM buffer (100 mM potassium acetate, 20 mM HEPES, 2 mM MgCl2, pH 7.4), cells were permeabilized with 20 μM digitonin for 5 min and fixed with 4% PFA for 15 min. Then cells were treated with 50 μg/ml proteinase K in the absence or presence of 0.1% Triton X-100 for 5 min and the reaction was terminated with 1 mM phenylmethanesulfonyl fluoride (PMSF). Subsequently, cells were immunostained and analyzed by Nikon Eclipse Ti2 inverted microscope.
2.8. Immunoblotting
Cells were harvested and lysed in RIPA lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA) (Millipore) with phosphatase and protease inhibitor cocktails (Roche) on ice for 30 min. The supernatant was collected after centrifugation at 17000×g for 20 min. Protein concentration was quantified with a BCA protein assay kit (Beyotime). The same amount of proteins was loaded and analyzed by SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membrane. The membranes were blocked with 5% (w/v) nonfat milk or bovine serum albumin in TBST (50 mM Tris pH 8.0, 150 mM NaCl, 0.05% Tween-20) buffer, incubated with antibodies and visualized by a ChemiScope Mini imaging system (Clinx Science) or an Odyssey Clx infrared imager (LICOR).
2.9. Alkaline extraction assay
Collected cells were suspended in 20 mM Tricine, 250 mM sucrose, pH 7.8 with protease inhibitor cocktail, retained on ice for 20 min, and homogenized using a dounce tissue grinder. After centrifugation at 3000×g for 10 min to remove the nucleus, the post-nuclear supernatant (PNS) was further centrifuged at 100,000×g (Beckman 100.3 rotor) at 4 °C for 1 h to separate cytosol and total membrane organelles. The pellets containing total membrane organelles were then subjected to alkaline extraction (0.1 M Na2CO3, pH 11) followed by centrifugation at 100,000×g for 1 h. Both the pellets and the supernatant soluble fractions were subjected to immunoblotting for ORF8 and other protein markers.
2.10. Cycloheximide (CHX) chase assay
Transfected cells were incubated with medium containing 1 mM NAC, 1 mM VC, 200 μΜ DTT, or 100 nM Bafilomycin A1 for 15 h respectively, and then treated with 25 μg/ml CHX (Merck-Millipore) and harvested at indicated time points. The lysates were analyzed by 15% SDS-PAGE followed by immunoblotting.
2.11. Mass spectrometry (MS)
2.11.1. Sample preparation
To identify the ORF8 covalently-binding interactome, HEK 293T cells were transfected with pcDNA6B-ORF8-FLAG for 24 h, then blocked with 20 mM NEM on ice for 15 min and harvested. Cell lysates were incubated with anti-FLAG M2 affinity gel (Sigma) at 4 °C overnight. The immunoprecipitates were washed five times with ice-cold PBS. After protein separation by SDS-PAGE under nonreducing conditions, the gel was stained with Coomassie Brilliant Blue and the gel lane excised for the first dimensional MS analysis. The gel lane of same samples in another gel was excised and incubated in SDS buffer containing 50 mM DTT for 30 min, and then placed horizontally on the top of the stacking gel for electrophoresis again. The gel was visualized by silver staining and the prominent spots below the diagonal excised for the second dimensional MS analysis.
2.11.2. In-gel digestion of proteins
For ORF8 covalently-binding interactome identification, the prominent spots below the diagonal were manually excised. After destaining, reduction (10 mM DTT in 25 mM NH4HCO3 for 45 min at 56 °C) and alkylation (40 mM iodoacetamide in 25 mM NH4HCO3 for 45 min at RT in the dark), the gels plugs were washed twice with 50% acetonitrile, dried using a SpeedVac, and digested with trypsin in 25 mM NH4HCO3 overnight at 37 °C to allow complete digestion. The reaction was terminated by adding formic acid to a 1% final concentration.
2.11.3. LC-MS/MS analysis
The digested peptides were separated on a 75 μm id × 25 cm C18 column packed in-house with reversed phase silica (Reprosil-Pur C18 AQ, 1.9 μm, Dr. Maisch GmbH). A linear acetonitrile gradient was used to elute the bounded peptides at a flow rate of 300 nl/min. The eluate was electrosprayed at a 2.0 kV voltage directly into an Orbitrap Exploris 480 mass spectrometer (Thermo Fisher Scientific). In the data-dependent acquisition mode, the MS data were acquired at a high resolution of 60,000 (m/z 200) across a mass range of 350–1500 m/z. Data dependent mode was selected as cycle time mode which was set as 2 s. The precursor ions were selected from each MS full scan with isolation width of 1.6 m/z for fragmentation in the Ion Routing Multipole with normalized collision energy of 28%. Subsequently, MS/MS spectra were acquired at resolution 15,000 at m/z 200. The target value was 7.50E+04 with a maximum injection time of 22 ms. The dynamic exclusion time was 30 s.
2.11.4. Protein identification
The data were analyzed using The SEQUEST HT search engine of Thermo Proteome Discoverer (version 2.4.1.15) in the Uniprot human protein database (updated 09–2018). The search parameters were set as follows: 10 ppm mass tolerance for precursor ions; 0.02 Da mass tolerance for product ions; two missed cleavage sites allowed for trypsin digestion; the cysteine carbamidomethylation were specified as fixed modifications; the methionine oxidation was chosen as variable modifications. For ORF8 interactome identification, proteins (unique peptides ≥ 2) were selected and protein interaction was defined as significant if abundance ratio of ORF8-overexpressing group to control group is ≥ 3.
2.12. Real-time PCR, PCR and sequencing
Total RNA was isolated from transfected cells using TRIzol (Invitrogen), and then 3 μg total RNA were reverse transcribed into cDNA using GoScript Reverse Transcription System (Promega). Quantitative Real-Time PCR was performed with SYBR Select Master Mix (Applied Biosystems) by a QuantStudio 7 Flex system (Applied Biosystems). The relative levels of mRNA for the target genes were calculated using the values of comparative threshold cycle and normalized to GAPDH. The qPCR primers sequence used were described in Table S2.
cDNA of ORF8 and codon-optimized ORF8 was amplified by PCR using the primers of pcDNA6B-FLAG: T7 promoter and BGH reverse. The products were analyzed on a 2% agarose gel and visualized by SYBR Gold (Invitrogen) staining. For sequence determination, PCR products were purified using a DNA gel extraction kit (Omega) and subjected to Sanger sequencing using the above primers.
2.13. Flow cytometry analysis
HEK 293T cells were harvested 24 h after transfection with indicated plasmids. After centrifugation at 200×g for 5 min, cells were collected, washed with PBS twice, stained with the annexin V-FITC/propidium iodide Apoptosis Assay Kit (Beyotime) according to the manufacturer's instructions, and analyzed with a flow cytometer (BD Biosciences).
2.14. Electron microscopy analysis
Cells were transfected with pcDNA6B-ORF8 or empty vector for 24 h and then fixed with 2.5% glutaraldehyde in PB (sodium phosphate buffer) at 4 °C overnight, and washed twice for 10 min each with 0.1 M PB and ultra-pure water, separately. Then cells were post-fixed with 1% OsO4 and 1% potassium ferrocyanide at 4 °C for 2 h. After washing with ultra-pure water, cells were further treated with a graded series of ethanol solutions and 100% anhydrous acetone for dehydration. Subsequently, cells were infiltrated gradually with a mixture of acetone/Epon 812 solutions. Then samples were placed in flat embedding molds filled with fresh resin and polymerized at 45 °C for 12 h, followed by 60 °C for 24 h, and then cut by an ultramicrotome. Ultrathin sections were then stained with uranyl acetate and lead citrate. Images were examined under a 120 kV electron microscope (Tecnai Spirit, FEI) at 100 kV at RT with an CCD camera (MoradaG3, EMSIS) using RADIUS (EMSIS) software.
2.15. RUSH transport assay
HEK 293T cells were transfected with RUSH constructs using lipo3000 according to the manufacturer's instructions. Release of the RUSH reporters was initiated by addition of 100 μM biotin (Sigma) as described previously [12]. The assay was performed in the presence of 100 μg/ml CHX to inhibit nascent protein synthesis. Images were acquired by Nikon Eclipse Ti2 inverted microscope.
2.16. Quantification and statistical analysis
Graph plots and p-values were generated using GraphPad Prism 5 software. Density of immunoblot bands was quantified using Image J software (NIH Image). Data were presented as the mean ± SEM. Unpaired Student's t-test was used to compare the means of two groups. One-way ANOVA followed by the Tukey's multiple comparison test was used to compare the means of more than two groups. All data were analyzed using GraphPad Prism 5 and statistical significance is presented as *p < 0.05, **p < 0.01 and ***p < 0.001.
3. Results
3.1. SARS-CoV-2 ORF8 is not secreted but retained in the ER lumen of host cells
SARS-CoV-2 ORF8 molecule contains an N-terminal cleavable signal peptide as predicted by SignalP-5.0 Server (Fig. 1A and Fig. S1B). Since ORF8 lacks a conventional C-terminal KDEL motif for ER retention, it has been considered as a secreted protein [13]. Our experimental data showed that ORF8 protein was identified only in the cell lysate fraction but not detectable in the culture medium when ectopically expressed in HEK 293T, HepG2, HeLa or Vero E6 cells (Fig. 1B). Immunofluorescence analysis also showed that ORF8 colocalized with the ER marker protein disulfide isomerase (PDI), but not with Golgi marker GM130 (Fig. 1C and Fig. S1C). Sedimentation and alkaline extraction were further performed to investigate whether ORF8 is a soluble ER protein or an ER membrane protein. ORF8 precipitated with total membrane fractions, and was extracted into the soluble fraction upon alkaline treatment, similar to the ER soluble protein markers BiP and calreticulin, while the ER membrane protein marker calnexin was retained in the pellet fractions upon alkaline treatment (Fig. 1D). We also performed fluorescence protease K protection (FPP) assay to confirm the subcellular location of ORF8, by measuring the restricted proteolytic digestibility of ORF8. FPP assay verified that ORF8 was resistant to proteinase K after plasma membrane permeability by digitonin and was digested after Triton X-100 treatment, similar as the ER luminal protein PDI. By contrast, ORF8 Δ (1-15) lacking the signal peptide, mainly located in the cytoplasm and was digested by proteinase K upon digitonin treatment (Fig. 1E). Taken together, these data suggested that SARS-CoV-2 ORF8 is an ER luminal protein.
Fig. 1.

ORF8 is an ER luminal protein.
(A) Schematic presentation of full-length SARS-CoV-2 ORF8.
(B) Immunoblotting of ORF8 in cell extracts and FLAG immunoprecipitates from the culture medium (CM) of HEK 293T, HepG2, HeLa and Vero E6 cells overexpressing ORF8-FLAG.
(C) Immunofluorescent analysis of ORF8 localization. HEK 293T cells expressing ORF8-FLAG were immunostained using antibodies against FLAG, PDI (ER marker) and GM130 (Golgi marker). Scale bars, 10 μm.
(D) ORF8 presented in the soluble fractions. The total lysates (TL) were separated into cytosol (Cyto) and total membrane (TM) fractions. The TM fractions were extracted with Na2CO3 and separated into soluble (S) and pelleted membrane (P) fractions. FLAG, BiP, calreticulin, calnexin and α-tubulin were immunoblotted.
(E) Fluorescent photomicrographs of HepG2 cells expressing ORF8-FLAG, which were permeabilized with digitonin, and then digested with proteinase K in the absence or presence of Triton X-100. Endogenous PDI was immunostained (red) as an ER luminal protein marker. Scale bars, 10 μm. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Very recently, it has been reported that upon transfection of ORF8 expression plasmid, the mRNA expressed in cells was unexpectedly spliced at cryptic splice sites, which should result in the production of a truncated ORF8 protein lacking 42-amino acid residues in the C-terminal region [14]. By comparing original ORF8 with codon-optimized ORF8, we found that codon-optimized ORF8 encodes a protein with larger molecular size (Fig. S2A), which is due to N-glycosylation (Fig. S2B). This protein is largely secreted (Fig. S2A) as previously reported [14]. However, we did not observe mRNA splicing in the unoptimized ORF8 (Fig. S2C). More importantly, mass spectrum confirmed that ORF8 protein encoded by the original sequence was not truncated (Fig. S2D). Thus, the property of codon-optimized ORF8 is different from original ORF8 and we chose to use the original ORF8 in following study.
3.2. ORF8 escapes ER degradation by forming mixed disulfide complexes
Crystal structure [15] revealed that ORF8 adopts an immunoglobulin (Ig)-like fold and forms an inter-chain disulfide via Cys20 and three intrachain disulfide bonds, Cys25-Cys90, Cys37-Cys102 and Cys61-Cys83 (Fig. 2A). However, the redox states and disulfide pattern of ORF8 in cells are still unknown. Immunoblotting analysis showed that ORF8 formed a lot of high molecular weight (HMW) bands on nonreducing gel, which were sensitive to the reducing agent β-mercaptoethanol, indicating that ORF8 forms disulfide-linked complexes either by itself or with other proteins in host cells (Fig. 2B). These HMW complexes significantly decreased when Cys20 of ORF8 was mutated to serine (C20S), and almost totally disappeared when all the seven cysteine residues were mutated to serine (C/S) (Fig. 2B). These data suggested that in cells ORF8 forms a more complicated disulfide pattern than that presented in the crystal. On the other side, the mixed disulfide bonds contributed to the ORF8 stability, since the protein levels of C20S and C/S mutants were much less than that of the wild-type (WT) protein (Fig. 2B, reducing gel). Indeed, the degradation of the C/S mutant in HepG2 cells was inhibited by the proteasomal inhibitor MG132 or lysosomal inhibitor Bafilomycin A1 (BafA1) (Fig. 2C), indicating that ORF8 was cleared by both proteasome and lysosome. However, in HEK 293T cells only BafA1 increased the protein level of ORF8 (Fig. 2C), which suggested that ORF8 may undergo different degradation pathways in different cell lines. Cycloheximide (CHX) is a protein synthesis inhibitor, and CHX chase assay further supported that ORF8 was stable and resistant to degradation up to 10 h, while the C/S mutant remarkably diminished within 2 h (Fig. 2D and E). Altogether, these results showed that the mixed disulfide bonds protect ORF8 against protein degradation in host cells.
Fig. 2.

ORF8 escapes ER degradation by forming mixed disulfide complexes.
(A) Crystal structure of SARS-CoV-2 ORF8 with two monomers shown in cyan and green (PDB: 7JTL). Cysteine residues were shown as spheres.
(B) Immunoblotting analysis of the redox state of ORF8 in HEK 293T cells expressing ORF8-FLAG or the mutants. Cell lysates were separated by SDS-PAGE under either reducing (R) or nonreducing (NR) conditions.
(C) Immunoblotting of ORF8 in HEK 293T and HepG2 cells expressing ORF8-FLAG or the C/S mutant treated with 100 nM Bafilomycin A1 (BafA1) or 8 μM MG132 for 12 h.
(D) Immunoblotting of ORF8 in HEK 293T cells expressing ORF8-FLAG and the C/S mutant treated with 100 nM BafA1 for 15 h and then followed by treatment with 25 μg/ml cycloheximide (CHX) for indicated time points.
(E) The relative ratios of ORF8/GAPDH normalized to that at zero time point were quantified. The band intensities ORF8 and GAPDH in (D) were analyzed using the ImageJ software. The data were shown as mean ± SEM from three independent experiments; ***p < 0.001 (two-tailed Student's t-test). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.3. ORF8 forms mixed disulfides with ER oxidoreductases
Previous study showed that ORF8 mainly interacts with proteins in the secretory pathway [7]. We identified a total of 568 proteins localized in the secretory pathway in the ORF8 interactome, by conventional co-immunoprecipitation (co-IP) and mass spectrum analysis (Fig. S3A-C). To identify the proteins forming mixed disulfides with ORF8, the interacting proteins were captured by co-IP and separated by nonreducing versus reducing 2-D gel. Nonreducing condition was used for the first dimension, and reducing condition for the second dimension to resolve the mixed disulfides. Noncovalently bound proteins migrated at the same rate under reducing and nonreducing conditions and thus appeared in a diagonal line. The proteins forming mixed disulfides migrated as covalent complexes under nonreducing conditions, then separated according to their individual sizes under reducing conditions, and therefore displayed below the diagonal. The prominent spots were excised from the gel and analyzed by mass spectrometry (Fig. 3A). Through analysis of the proteins below the diagonal we identified 375 proteins located in the ER, the Golgi and the ER-Golgi intermediate compartment (ERGIC) (Fig. 3B), which were potent clients forming mixed disulfides with ORF8. These proteins were mainly involved in protein processing in the ER, N-glycan biosynthesis, metabolic pathways etc., as analyzed by DAVID Gene Ontology (Fig. 3C).
Fig. 3.

ORF8 forms mixed disulfides with ER oxidoreductases.
(A) Identification of proteins forming mixed disulfides with ORF8 by 2-D gel electrophoresis. HEK 293T cells expressing ORF8-FLAG were treated with NEM, and FLAG immunoprecipitates were analyzed by SDS-PAGE under nonreducing conditions. Gel lanes were excised and reduced with 50 mM DTT and separated in a second dimension. Spots under the diagonal were excised from the gel and identified by mass spectrometry.
(B) Venn diagram of the 375 proteins covalently interacting with ORF8 in ER, Golgi and ERGIC based on DAVID GO term analysis.
(C) KEGG pathway mapping of the most enriched biological processes in the ORF8 covalent interactome in the ER, Golgi and ERGIC. The graph represented the top ten statistically significant enriched gene clusters ordered by FDR (false discovery rate), with the number of genes in each cluster indicated besides the bars.
(D) Co-immunoprecipitation of ORF8 and PDI/ERp44 in HEK 293T cells expressing ORF8-FLAG.
(E) Dual-color immunoblotting analysis of mixed disulfides between PDI/ERp44 and ORF8. FLAG immunoprecipitates from HEK 293T cells expressing ORF8-FLAG were analyzed by SDS-PAGE under nonreducing conditions and immunoblotted with anti-PDI/ERp44 (green) and anti-FLAG (red) antibodies. The yellow band depicted the mixed disulfide complexes.
(F) Immunoblotting of ORF8 in PDI knockout (KO) or ERP44 KO HepG2 cells.
(G, H) Immunoblotting of ORF8 in PDI KO (G) or ERP44 KO (H) HepG2 cells with ectopically expressed PDI or ERp44, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
We noticed that many protein disulfide isomerase family members are enriched in the ORF8 interactome, among which PDI and ERp44 are within the top 3 candidates (Table S3). We therefore decided to investigate whether PDI and ERp44 are targets of ORF8, since PDI family proteins are capable of catalyzing thiol-disulfide exchanges [16]. We first verified that ORF8 interacts with PDI and ERp44 by conventional co-IP experiments (Fig. 3D). Next, the formation of disulfide-linked complexes between ORF8 and PDI or ERp44 was confirmed by co-IP and nonreducing gel analysis. By using dual-fluorescent imaging, protein complexes containing both ORF8 and PDI or ERp44 were clearly observed, with molecular size larger than that of each single protein (Fig. 3E). To study whether PDI and/or ERp44 are involved in the degradation of ORF8, we employed PDI knockout (KO) [17] or ERP44 KO cells by CRISPR/Cas9 mediated gene editing. The protein level of ORF8 was much less in PDI KO and ERP44 KO HepG2 cells than that in WT cells, which was not due to increased secretion (Fig. 3F). Replenishment with PDI (Fig. 3G) or ERp44 (Fig. 3H) increased the protein level of ORF8. Similar results were observed in PDI or ERP44 knockdown HEK 293T cells (Fig. S3D). Thus, ORF8 formed mixed disulfides with multiple proteins in the secretory pathway, among which it hijacks PDI and ERp44 to enhance its stability in host cells.
3.4. ORF8 induces ER stress through targeting key UPR components
Besides the above identified ORF8 interactome, we found that ORF8 interacted with multiple ER chaperones such as BiP and calnexin, as well as the ER stress sensors IRE1α, PERK and ATF6 (Fig. 4A), indicating a connection of ORF8 with UPR and ER homeostasis. Indeed, ORF8 expression in HEK 293T or HepG2 cells induced the activation of all three UPR pathways. It significantly elevated the phosphorylation of IRE1α and the level of its downstream substrate spliced XBP1 (XBP1s). ORF8 also elevated the phosphorylation of PERK and its downstream substrate eIF2α, and promoted the nuclear translocation of ATF6 cytosolic domain (ATF6c) in both HEK 293T (Fig. 4B) and HepG2 cells (Fig. S4A). By contrast, the unstable C/S mutant had little effect on the UPR activation (Fig. 4B and Fig. S4A). Consistently, the mRNA levels of GRP78 (BIP), CHOP and XBP1s were significantly upregulated in HEK 293T and HepG2 cells when ORF8 was expressed (Fig. 4C and Fig. S4B). These observations were similar to the consequence when cells were treated with ER stress inducer, thapsigargin (Tg), a sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) calcium pump inhibitor (Fig. 4D and Fig. S4C). In addition, 8-formyl-7-hydroxy-4-methylcoumarin (4μ8C), an IRE1α RNase activity inhibitor, significantly inhibited ORF8-induced XBP1s production without interfering IRE1α autophosphorylation (Fig. 4E). Interestingly, blocking the IRE1α-XBP1 pathway by 4μ8C increased the protein level of ORF8, suggesting that the activation of IRE1α-XBP1 pathway contributes to the degradation of ORF8. Subsequently, we tested whether small molecule inhibitor or chemical chaperones could relieve ORF8-induced UPR and restore ER homeostasis. 4-phenylbutyric acid (4-PBA) and tauroursodeoxycholic acid (TUDCA) are two chemical chaperones that can improve protein folding and alleviate ER stress. We found that 4-PBA and TUDCA moderately attenuated ORF8-induced activation of IRE1α-XBP1 pathway (Fig. 4F). Altogether, these results indicated that ORF8 induces the ER stress through targeting ER chaperones and key UPR components.
Fig. 4.

ORF8 induces ER stress through targeting key UPR components.
(A) Co-immunoprecipitation of ORF8 and key UPR components in HEK 293T cells expressing ORF8-FLAG.
(B) Detection of the activation of IRE1α, PERK and ATF6 by immunoblotting in HEK 293T cells expressing ORF8-FLAG or the C/S mutant.
(C) Detection of GRP78, ATF4, CHOP, XBP1u, XBP1s and BLOC1S1 mRNA levels by real-time PCR in HEK 293T cells expressing ORF8-FLAG or the C/S mutant. Data were shown as mean ± SEM of three independent experiments; **p < 0.01 (one-way ANOVA, Tukey's multiple comparison test).
(D) Detection of IRE1α and PERK activation by immunoblotting in HEK 293T cells expressing ORF8-FLAG or the C/S mutant treated without or with 0.5 μΜ Tg for 6 h.
(E) Detection of IRE1α activation by immunoblotting in HEK 293T cells expressing ORF8-FLAG or the C/S mutant treated with DMSO or 10 μΜ 4μ8C for 24 h.
(F) Detection of IRE1α activation by immunoblotting in HEK 293T cells expressing ORF8-FLAG or the C/S mutant treated with DMSO or 1 mM 4-phenylbutyric acid (4-PBA) or 150 μΜ tauroursodeoxycholic acid (TUDCA) for 24 h.
3.5. ORF8 induces ER remodeling and promotes ER export
Next, we aim to figure out the consequences of ORF8-induced ER stress. ORF8 activated UPR signaling pathways but did not induce cell apoptosis (Fig. S5A), implying that ORF8 induces the adaptive UPR rather than the terminal UPR. The IRE1α-XBP1 signaling pathway is involved in regulation of chaperone expression, ERAD, lipogenesis, and ER membrane biogenesis and expansion [[18], [19], [20]]. In accordance, we observed that the expression of many chaperones and foldases were significantly upregulated (Fig. S5B). These folding catalysts may help to cope with the high demand for the synthesis of virus proteins in the ER. Amongst, the increase of the ER sulfhydryl oxidase Ero1β may contribute to the ORF8-involved mixed disulfide formation. In addition, proteins involved in ERAD were also upregulated (Fig. S5C), which has been reported to contribute to the remodeling of the ER membrane referred to as viral replication organelles [21,22]. Although we did not find significant changes of lipogenesis-associated gene expression upon ORF8 expression (Fig. S5D), transmission electron microscopy (EM) analysis revealed that ORF8 induced a significant remodeling of ER morphology in HEK 293T cells. Unlike the tubular and bubble structures of normal ER in control cells, massive intertwined structures or convoluted membranes (CM) were observed in the EM images of HEK 293T cells expressing ORF8 (Fig. 5A). The ER membrane remodeling caused by ORF8 is in line with the observation that coronaviruses induce abnormal membrane structures, such as CM, zippered ER and double-membrane vesicles (DMVs) that are derived from the ER [23].
Fig. 5.

ORF8 induces ER remodeling and promotes ER export.
(A) EM analysis of ER morphology in control (a, b) and ORF8-expressing HEK 293T cells (c, d). Scale bars, 500 nm
(B) Schematic depicts that cargo (AAT or CTSZ) fused to streptavidin-binding peptide (SBP) and EGFP (SBP-EGFP-cargo) is retained in the ER by interacting with the ER hook, streptavidin fused with KDEL (Str-KDEL). Release is induced by addition of biotin to allow trafficking of the cargo to downstream secretory pathway.
(C, E) Representative live cell imaging of HEK 293T cells co-expressing Str-KDEL with SBP-EGFP-AAT/CTSZ and ORF8-FLAG at the indicated time points after addition of biotin. Scale bars, 10 μm.
(D, F) The plot showing fluorescence intensity in the Golgi region at each time point, normalized to the maximum value. Data were shown as mean ± SEM (n = 18 cells in AAT group; n = 22 cells in CTSZ group). And quantification of the peak time for maximum fluorescence intensity in the Golgi region was shown as mean ± SEM. **p < 0.001 (two-tailed Student's t-test).
To explore potential changes of the ER function due to ER remodeling induced by ORF8, a cargo retention system using selective hooks (RUSH) was employed to monitor protein secretion from the ER [12,24]. Cargo was fused with EGFP and streptavidin binding protein (SBP-EGFP-cargo) and initially trapped in the ER by luminal streptavidin fused with an ER retention signal KDEL. A synchronized release of cargo from the ER to downstream compartments was triggered after addition of biotin, which competes with streptavidin for SBP (Fig. 5B). In HEK 293T cells, α-1-antitrypsin (AAT), a major serum serine protease inhibitor, reached the Golgi at about 30 min after biotin addition. When co-expressed with ORF8, the majority of AAT reached the Golgi at 20 min (Fig. 5C and D). Similarly, ORF8 accelerated the ER-to-Golgi transport of another cargo protein cathepsin Z (CTSZ), a lysosomal enzyme (Fig. 5E and F). Taken together, these results suggested that ORF8 induces ER remodeling and promotes cargo protein export from the ER.
3.6. Small molecule reducing agents promotes ORF8 degradation and mitigates ER stress
Since ORF8 obtains its protein stability by forming mixed disulfides and affects ER homeostasis and induces ER stress, we reasoned that reduction of the mixed disulfides should be able to alleviate ORF8-induced ER stress. We first tested the effect of depletion of PDI or ERp44, which forms mixed disulfides with ORF8 in cells (Fig. 3). Indeed, PDI or ERP44 KO decreased ORF8 stability and alleviated ORF8-induced ER stress with less upregulated p-IRE1α, p-eIF2α and BiP (Fig. 6A and B). Next, we studied the effects of small molecule reducing agents, including N-acetyl-l-cysteine (NAC, a mucolytic agent approved by FDA), l-ascorbic acid (Vitamin C, VC, a mild reducing agent and antioxidant) and dithiothreitol (DTT, a commonly used agent for disulfide reduction). The results showed that in HEK 293T cells expressing ORF8, the amount of HMW complexes in nonreducing gel was reduced after treatment with NAC or DTT. DTT also significantly reduced the total protein level of ORF8, with NAC to a lesser extent, as demonstrated by reducing gel (Fig. 6C). Similar results have been observed in kidney epithelial cell line Vero E6 (Fig. S6A), which is commonly used for studying SARS-CoV-2 infection. CHX chase experiments showed that the degradation rate of ORF8 was significantly accelerated with the addition of NAC or DTT, while VC had little effect on the protein stability of ORF8 (Fig. 6D and E). Moreover, treatment with reducing agents alleviated ORF8-induced ER stress to some degrees (Fig. 6F and Fig. S6B). Thus, small molecule reducing agents could be used to reduce ORF8-involved mixed disulfides, promote its degradation and mitigate ORF8-induced ER stress.
Fig. 6.

Small molecule reducing agents promotes ORF8 degradation and mitigates ER stress.
(A, B) Immunoblotting of the UPR markers in PDI KO (A) or ERP44 KO (B) HepG2 cells expressing ORF8-FLAG.
(C) Immunoblotting analysis of the redox state of ORF8 in HEK 293T cells expressing ORF8-FLAG treated with 1 mM NAC or VC for 24 h, or 200 μM DTT for 6 h. Cell lysates were separated by SDS-PAGE under either reducing (R) or nonreducing (NR) conditions.
(D) Immunoblotting of ORF8 in HEK 293T cells expressing ORF8-FLAG treated with 1 mM NAC, 1 mM VC or 200 μM DTT for 15 h and then followed by treatment with 25 μg/ml CHX for indicated time points.
(E) The relative ratios of ORF8/GAPDH normalized to that at zero time point were quantified. The band intensities of ORF8 and GAPDH in (D) were analyzed using the ImageJ software. The data were shown as mean ± SEM from three independent experiments; *p < 0.05 and ***p < 0.001 (two-tailed Student's t-test).
(F) Detection of the UPR markers by immunoblotting in HEK 293T cells expressing ORF8-FLAG treated with 1 mM NAC or VC for 24 h, or 200 μM DTT for 6 h.
4. Discussion
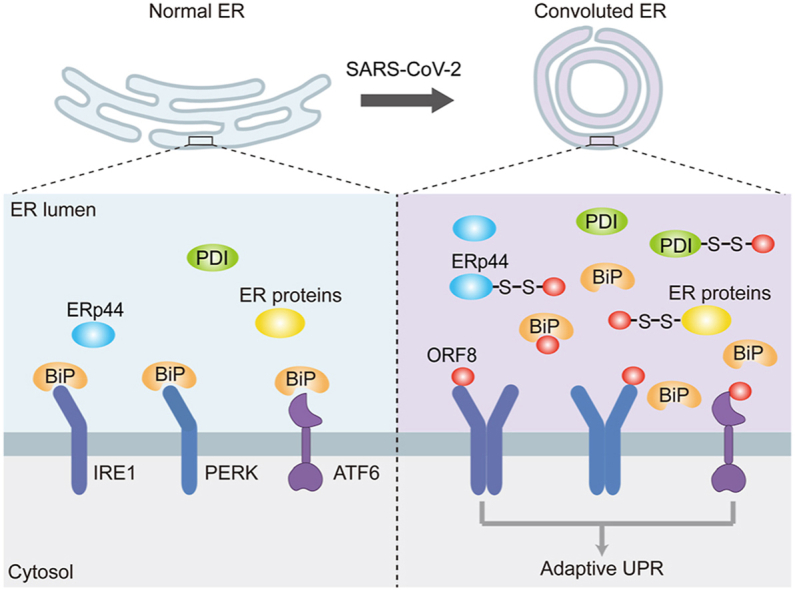
The nonstructural and structural proteins of SARS-CoV-2, have drawn worldwide attention due to their essential roles in the viral replication, access and exit of host cells, and also as promising targets for vaccines and therapeutic strategies. Different from these indispensable proteins, the accessory proteins are less conserved among the coronaviruses, but endow SARS-CoV-2 with novel features. For example, ORF3a from SARS-CoV-2 but not SARS-CoV-1 was reported to impair autophagocytic activity and induce lysosomal exocytosis [25,26]. Among the nine accessory proteins from SARS-CoV-2, ORF8 shares the least sequence similarity to the counterpart from SARS-CoV-1, however its function is still largely unknown. In this study, we characterized the interplay between ORF8 and host cells, revealing that ORF8 forms mixed disulfide complexes with several key ER oxidoreductases, therefore escapes ER degradation and accumulates in the ER lumen. Furthermore, ORF8 targets the UPR pathways and regulates the protein folding and transport machineries as well as ER homeostasis (Fig. 7).
Fig. 7.

Proposed model for ORF8-induced ER stress and ER remodeling.
SARS-CoV-2 ORF8 protein forms mixed disulfide complexes with ER oxidoreductases, such as PDI and ERp44, therefore escapes ER degradation and accumulates in the ER lumen. Furthermore, through direct binding to UPR sensors or competing for BiP chaperone binding, ORF8 induces the activation of adaptive UPR. Activation of the UPR is accompanied with changes of ER proteostasis, ER morphology and cargo proteins export, which can accommodate the requirement for virus replication and maturation.
Though ORF8 was reported to be secreted when overexpressed in A549 and 293FT cells [13], in our hands ORF8 is not secreted but retained in the ER lumen at least in four different mammalian cell lines. It should be noted that in this study we use the original ORF8 rather than the codon-optimized ORF8, as the latter displays abnormal N-glycosylation and secretion probably due to its faster folding rate (Fig. S2). ORF8 does not harbor an ER retention motif; instead, it forms mixed disulfide bonds with several ER-resident proteins, e.g. ERp44 and PDI. ERp44 and PDI are able to cycle between the ER and the Golgi to retrieve select clients lacking KDEL-like motifs back to the ER [[27], [28], [29]]. The mixed-disulfide bonds between ORF8 and PDI/ERp44 ensures its ER location as well as its stability in host cells. Either mutation of its cysteine residues or genetic depletion of PDI/ERp44, accelerates the degradation of ORF8. Although ORF8 obtained by in vitro refolding adopts an Ig-like fold as presented in the solved crystal structure [15], it forms disulfide-crosslinked oligomers and interacts with many components in the ER quality control system, suggesting that in situ it may not be a well folded protein. Nevertheless, the specific role of each cysteine involved in the folding of ORF8, complexes formation and ER stress needs to be determined in the future.
The heterogenous states of ORF8 raises the possibility that ORF8 mimics an ‘unfolded’ protein substrate to induce the ER stress. UPR is a signal transduction pathway to adjust and match the protein-folding capacity of the ER when cells encounter the ER stress. Although ORF8ab and S proteins from SARS-CoV-1 could induce the UPR as reported previously [30,31], the underlying molecular mechanism is unclear. We found that ORF8 activates the UPR pathways in two different ways, either competing for BiP chaperone binding with the UPR sensors, or directly binding to IRE1 and PERK, and both mechanisms contribute to the UPR activation [32,33]. Notably, ORF8 expression induces the adaptive UPR rather than the terminal UPR, as it does not promote cell death. Activation of the UPR is accompanied with changes of ER proteostasis, ER morphology and cargo proteins export, which suggest that ORF8 reshapes the ER to accommodate the requirement for virus replication and maturation. Indeed, accumulating evidences have shown that induction of the UPR may constitute a major aspect of coronavirus-host interaction [34,35]. Interestingly, EM images show that ORF8 promotes the ER membrane remodeling, which probably provide the major membrane source for virion assembly [36]. The ER morphology change also accelerates the ER-to-Golgi transport, which could be important for the virion maturation. Future studies are required to determine whether ORF8 affects viral protein trafficking and viral replication. The process that ORF8 induces UPR is very similar to the scenario of B lymphocytes maturation, which is accomplished by a large expansion of the ER membranes and upregulation of ER quality control systems [37]. Thus, it is likely that through mimicking an unfolded or folding intermediate of Ig, ORF8 deceives the host cell to launch the UPR for viral replication.
Many recent studies show that there is direct crosstalk between the UPR and immune responses, and cytopathic effect [[38], [39], [40]]. In line with a recent study [10], we also found that ORF8 slightly, but statistically significantly, inhibits the phosphorylation of interferon regulatory factor 3 (IRF3), downregulates the expression of IRF3-dependent innate immune responses (Fig. S7). Moreover, in some immune cell types like dendritic cells and B cells, particular UPR sensors appear constitutively active and are necessary for Ig synthesis and antigen presentation. Our findings that ERp44 and PDI are targets of ORF8 are of particular interests, since ERp44 is critical for the maturation of secretory IgM [41], and PDI is essential for selecting optimal peptides by major histocompatibility complex class Ι (MHC-Ι) [42]. Currently, there are conflict results on whether ORF8 suppresses MHC-I expression [5,43]. Several MHC-I and MHC-Ⅱ molecules were detected in our ORF8 interactome study (Table S3). Nevertheless, future studies are needed to clarify the relationship between ORF8-induced UPR and immune evasion.
Finally, we show that reducing agents can reduce mixed disulfides, promotes the degradation of ORF8 and further mitigates ORF8-induced ER stress. Recently, it has been reported that SARS-CoV-2 infection interferes with the metabolism and redox of cellular thiols in the host cell, and antivirals and NAC can prevent such defect [44]. Reducing compounds can also disrupt key disulfides in S protein and decrease the binding to ACE2 and infectivity of SARS-CoV-2 [45]. Thus, future drug development based on small molecule reducing compounds could kill two birds with one stone, by targeting the disulfides of both S and ORF8 proteins. In sum, our findings of the redox sensitive stability of ORF8 may provide novel intervention strategies against SARS-CoV-2.
Author contributions
P.L., X.W., and L.W. designed the experiments and analyzed the data; P.L., X.W., Y.S., and F.C. performed the experiments. H.Z. analyzed the EM data. J.W. and F.Y. performed the MS analysis. J.H., H.Z., C.C.W., and L.W. conceived and supervised the projects. P.L., X.W., and L.W. wrote the manuscript. All authors approved the final version of the manuscript.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [46] partner repository with the dataset identifier PXD032035.
Declaration of interests
The authors declare no competing interests.
Acknowledgments
We thank Peihui Wang (Shandong University) for the cDNA of ORF8, Jianchao Zhang (Institute of Biophysics, CAS) for assisting in MS analysis, Hao Song (Beijing Institutes of Life Science, CAS) and Yan Li (Institute of Microbiology, CAS) for helpful discussion. This work was supported by the National Key R&D Program of China (2021YFA1300800, 2017YFA0504000); the National Natural Science Foundation of China (32022033, 31870761); the Strategic Priority Research Program of CAS (XDB37020303); and the Youth Innovation Promotion Association, CAS, to L.W.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102388.
Contributor Information
Xi Wang, Email: wangxi@ibp.ac.cn.
Lei Wang, Email: wanglei@ibp.ac.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Wu F., Zhao S., Yu B., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou P., Yang X.L., Wang X.G., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Redondo N., Zaldívar-López S., Garrido J.J., et al. SARS-CoV-2 accessory proteins in viral pathogenesis: knowns and unknowns. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.708264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Young B.E., Fong S.W., Chan Y.H., et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: an observational cohort study. Lancet. 2020;396(10251):603–611. doi: 10.1016/s0140-6736(20)31757-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y., Chen Y., Li Y., et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-Ι. Proc. Natl. Acad. Sci. U. S. A. 2021;118(23) doi: 10.1073/pnas.2024202118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J.Y., Liao C.H., Wang Q., et al. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020;286 doi: 10.1016/j.virusres.2020.198074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon D.E., Jang G.M., Bouhaddou M., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sicari D., Chatziioannou A., Koutsandreas T., et al. Role of the early secretory pathway in SARS-CoV-2 infection. J. Cell Biol. 2020;219(9) doi: 10.1083/jcb.202006005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosa-Fernandes L., Lazari L.C., da Silva J.M., et al. SARS-CoV-2 activates ER stress and Unfolded protein response. bioRxiv. 2021;2021 doi: 10.1101/2021.06.21.449284. 06.21.449284. [DOI] [Google Scholar]
- 10.Rashid F., Dzakah E.E., Wang H., et al. The ORF8 protein of SARS-CoV-2 induced endoplasmic reticulum stress and mediated immune evasion by antagonizing production of interferon beta. Virus Res. 2021;296 doi: 10.1016/j.virusres.2021.198350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ran F.A., Hsu P.D., Wright J., et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8(11):2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boncompain G., Divoux S., Gareil N., et al. Synchronization of secretory protein traffic in populations of cells. Nat. Methods. 2012;9(5):493–498. doi: 10.1038/nmeth.1928. [DOI] [PubMed] [Google Scholar]
- 13.Wang X., Lam J.Y., Wong W.M., et al. Accurate diagnosis of COVID-19 by a novel immunogenic secreted SARS-CoV-2 orf8 protein. mBio. 2020;11(5) doi: 10.1128/mBio.02431-20. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuoka K., Imahashi N., Ohno M., et al. SARS-CoV-2 accessory protein ORF8 is secreted extracellularly as a glycoprotein homodimer. J. Biol. Chem. 2022;298(3) doi: 10.1016/j.jbc.2022.101724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flower T.G., Buffalo C.Z., Hooy R.M., et al. Structure of SARS-CoV-2 ORF8, a rapidly evolving immune evasion protein. Proc. Natl. Acad. Sci. U. S. A. 2021;118(2) doi: 10.1073/pnas.2021785118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L., Wang X., Wang C.C. Protein disulfide-isomerase, a folding catalyst and a redox-regulated chaperone. Free Radic. Biol. Med. 2015;83:305–313. doi: 10.1016/j.freeradbiomed.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 17.Yu J., Li T., Liu Y., et al. Phosphorylation switches protein disulfide isomerase activity to maintain proteostasis and attenuate ER stress. EMBO J. 2020;39(10) doi: 10.15252/embj.2019103841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee A.H., Iwakoshi N.N., Glimcher L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 2003;23(21):7448–7459. doi: 10.1128/mcb.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sriburi R., Jackowski S., Mori K., et al. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004;167(1):35–41. doi: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sha H., He Y., Chen H., et al. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metabol. 2009;9(6):556–564. doi: 10.1016/j.cmet.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reggiori F., Monastyrska I., Verheije M.H., et al. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe. 2010;7(6):500–508. doi: 10.1016/j.chom.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Byun H., Gou Y., Zook A., et al. ERAD and how viruses exploit it. Front. Microbiol. 2014;5:330. doi: 10.3389/fmicb.2014.00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du Toit A. Coronavirus replication factories. Nat. Rev. Microbiol. 2020;18(8):411. doi: 10.1038/s41579-020-0406-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niu L., Ma T., Yang F., et al. Atlastin-mediated membrane tethering is critical for cargo mobility and exit from the endoplasmic reticulum. Proc. Natl. Acad. Sci. U. S. A. 2019;116(28):14029–14038. doi: 10.1073/pnas.1908409116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miao G., Zhao H., Li Y., et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell. 2021;56(4):427–442. doi: 10.1016/j.devcel.2020.12.010. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen D., Zheng Q., Sun L., et al. ORF3a of SARS-CoV-2 promotes lysosomal exocytosis-mediated viral egress. Dev. Cell. 2021;56(23):3250–3263. doi: 10.1016/j.devcel.2021.10.006. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otsu M., Bertoli G., Fagioli C., et al. Dynamic retention of Ero1alpha and Ero1beta in the endoplasmic reticulum by interactions with PDI and ERp44. Antioxidants Redox Signal. 2006;8(3–4):274–282. doi: 10.1089/ars.2006.8.274. [DOI] [PubMed] [Google Scholar]
- 28.Anelli T., Alessio M., Mezghrani A., et al. ERp44, a novel endoplasmic reticulum folding assistant of the thioredoxin family. EMBO J. 2002;21(4):835–844. doi: 10.1093/emboj/21.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang K., Li D.F., Wang X., et al. Crystal structure of the ERp44-peroxiredoxin 4 complex reveals the molecular mechanisms of thiol-mediated protein retention. Structure. 2016;24(10):1755–1765. doi: 10.1016/j.str.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Sung S.C., Chao C.Y., Jeng K.S., et al. The 8ab protein of SARS-CoV is a luminal ER membrane-associated protein and induces the activation of ATF6. Virology. 2009;387(2):402–413. doi: 10.1016/j.virol.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan C.P., Siu K.L., Chin K.T., et al. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006;80(18):9279–9287. doi: 10.1128/jvi.00659-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertolotti A., Zhang Y., Hendershot L.M., et al. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000;2(6):326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 33.Gardner B.M., Walter P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. 2011;333(6051):1891–1894. doi: 10.1126/science.1209126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fung T.S., Liu D.X. Human coronavirus: host-pathogen interaction. Annu. Rev. Microbiol. 2019;73:529–557. doi: 10.1146/annurev-micro-020518-115759. [DOI] [PubMed] [Google Scholar]
- 35.Xue M., Feng L. The role of unfolded protein response in coronavirus infection and its implications for drug design. Front. Microbiol. 2021;12 doi: 10.3389/fmicb.2021.808593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cortese M., Lee J.Y., Cerikan B., et al. Integrative imaging reveals SARS-CoV-2-induced reshaping of subcellular morphologies. Cell Host Microbe. 2020;28(6):853–866. doi: 10.1016/j.chom.2020.11.003. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Anken E., Romijn E.P., Maggioni C., et al. Sequential waves of functionally related proteins are expressed when B cells prepare for antibody secretion. Immunity. 2003;18(2):243–253. doi: 10.1016/s1074-7613(03)00024-4. [DOI] [PubMed] [Google Scholar]
- 38.Bettigole S.E., Glimcher L.H. Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 2015;33:107–138. doi: 10.1146/annurev-immunol-032414-112116. [DOI] [PubMed] [Google Scholar]
- 39.Glimcher L.H., Lee A.H., Iwakoshi N.N. XBP-1 and the unfolded protein response (UPR) Nat. Immunol. 2020;21(9):963–965. doi: 10.1038/s41590-020-0708-3. [DOI] [PubMed] [Google Scholar]
- 40.Bartolini D., Stabile A.M., Vacca C., et al. Endoplasmic reticulum stress and NF-kB activation in SARS-CoV-2 infected cells and their response to antiviral therapy. IUBMB Life. 2022;74(1):93–100. doi: 10.1002/iub.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giannone C., Chelazzi M.R., Orsi A., et al. Biogenesis of secretory immunoglobulin M requires intermediate non-native disulfide bonds and engagement of the protein disulfide isomerase ERp44. EMBO J. 2022;41(3) doi: 10.15252/embj.2021108518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park B., Lee S., Kim E., et al. Redox regulation facilitates optimal peptide selection by MHC class I during antigen processing. Cell. 2006;127(2):369–382. doi: 10.1016/j.cell.2006.08.041. [DOI] [PubMed] [Google Scholar]
- 43.Yoo J.S., Sasaki M., Cho S.X., et al. SARS-CoV-2 inhibits induction of the MHC class I pathway by targeting the STAT1-IRF1-NLRC5 axis. Nat. Commun. 2021;12(1):6602. doi: 10.1038/s41467-021-26910-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bartolini D., Stabile A.M., Bastianelli S., et al. SARS-CoV2 infection impairs the metabolism and redox function of cellular glutathione. Redox Biol. 2021;45 doi: 10.1016/j.redox.2021.102041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi Y., Zeida A., Edwards C.E., et al. Thiol-based chemical probes exhibit antiviral activity against SARS-CoV-2 via allosteric disulfide disruption in the spike glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 2022;119(6) doi: 10.1073/pnas.2120419119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perez-Riverol Y., Bai J., Bandla C., et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50(D1):D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [46] partner repository with the dataset identifier PXD032035.