

SUMMARY
Mental experiences can become long-term memories if the hippocampal activity patterns that encode them are broadcast during network oscillations. The activity of inhibitory neurons is essential for generating these neural oscillations, but molecular control of this dynamic process during learning remains unknown. Here, we show that hippocampal oscillatory strength positively correlates with excitatory monosynaptic drive onto inhibitory neurons (E→I) in freely behaving mice. To establish a causal relationship between them, we identified γCaMKII as the long-sought mediator of long-term potentiation for E→I synapses (LTPE→I), which enabled the genetic manipulation of experience-dependent E→I synaptic input/plasticity. Deleting γCaMKII in parvalbumin interneurons selectively eliminated LTPE→I and disrupted experience-driven strengthening in theta and gamma rhythmicity. Behaviorally, this manipulation impaired long-term memory, for which the kinase activity of γCaMKII was required. Taken together, our data suggest that E→I synaptic plasticity, exemplified by LTPE→I, plays a gatekeeping role in tuning experience-dependent brain rhythms and mnemonic function.
In Brief
Activity-dependent changes traversing levels of synapses and networks are essential for memory. He et al. uncover γCaMKII as a mediator of LTPE→I and molecular control of experience-dependent brain rhythms, suggesting that E→I synaptic plasticity plays a gatekeeping role in tuning experience-driven E→I spike transmission, network activity, and therefore mnemonic function.
INTRODUCTION
Neuronal activity triggered by experience modifies the strength of excitatory synapses onto inhibitory interneurons (E→I), which in turn exerts powerful control over brain networks by regulating oscillatory neural organization (Allen and Monyer, 2015; Bartos et al., 2007; Buzsáki and Wang, 2012). Such experience-dependent network changes are essential for learning and memory (Buzsáki, 2010; Frankland and Bontempi, 2005). However, major questions remain about the molecular mechanisms through which these E→I synapses are shaped by experience, and how this process contributes to neural computation.
Synaptic plasticity, exemplified by long-term potentiation (LTP), is a fundamental process thought to underlie the long-term storage of information in neural circuits (Bliss and Collingridge, 1993). Although LTP has been studied extensively, the majority of knowledge centers on the potentiation of excitatory synapses onto excitatory neurons (LTPE→E) (Malenka and Nicoll, 1999; Malinow et al., 2000; Silva et al., 1992), with far less known about excitatory synaptic potentiation onto inhibitory neurons (LTPE→I) (Buzsáki and Eidelberg, 1982; Kullmann et al., 2012; Pelkey et al., 2017; Pelletier and Lacaille, 2008). In the canonical LTPE→E induction pathway, synchronous postsynaptic depolarization and glutamate binding allows calcium influx through N-methyl-D-aspartate receptors (NMDARs) (Morris et al., 1986), which in turn activates calcium/calmodulin-dependent protein kinase II (CaMKII) and regulates AMPAR function (Malenka and Nicoll, 1999; Malinow et al., 2000; Silva et al., 1992). As the key mediator in this process, αCaMKII is fundamentally necessary for LTPE→E (Hell, 2014; Malenka and Nicoll, 1999; Silva et al., 1992). Identification the role of αCaMKII in LTPE→E was critical in establishing a causal link between synaptic plasticity and memory without disrupting baseline function of synapses (Silva et al., 1992). Indeed, through the manipulation of αCaMKII (Silva et al., 1992), the first genetic evidence was provided to address a long-standing challenge to studies of LTPE→E: finding key molecular underpinnings, as functionally critical to synaptic plasticity as a door hinge is to an opening door (Lisman et al., 2003), and linking these processes to brain function.
Some parts of the canonical LTPE→E signaling pathway have also been observed in LTPE→I. In inhibitory interneuron dendrites, NMDARs and AMPARs are critical for mediating postsynaptic calcium signaling and driving excitatory input (Akgül and McBain, 2016; Allen and Monyer, 2015; Bartos et al., 2007). However, αCaMKII is absent from inhibitory interneurons (Liu and Jones, 1996; McBain et al., 1999; Sík et al., 1998) and is not required for LTPE→I (Lamsa et al., 2007). This apparent paradox, and the lack of the knowledge about how LTPE→I is generated at the molecular level, has hampered understanding of its function at the network and behavioral levels in vivo.
Here, we fill this gap by showing that γCaMKII is a critical mediator for LTPE→I and that deletion of this protein kinase in parvalbumin-positive (PV+) interneurons disrupts memory-related network plasticity. By uncovering molecular control of LTPE→I and experience-dependent brain rhythms, our data link synaptic plasticity in inhibitory networks to learning-related changes in network activity and in the performance of memory-guided behaviors.
RESULTS
E→I monosynaptic drive is positively correlated with LFP power in vivo
As a common measure of brain network activity, the extracellular local field potential (LFP) reflects, in part, the ongoing synaptic communication thought to underlie neural computation (Buzsáki, 2010). To test whether fluctuations in the E→I synaptic drive may produce changes in LFP power within the network, we obtained recordings of CA1 pyramidal cells and interneurons (Figure 1A) in the hippocampus from freely behaving mice. Both pyramidal cells and interneurons showed strong phase locking to theta and gamma (Figure 1B), as expected (Csicsvari et al., 2003; Mizuseki et al., 2009), with a significantly different gamma phase preference for pyramidal cells and interneurons (Figure 1C) (circular Kolmogorov-Smirnov test, p = 1.53−7). The phase lag is thought to be due to synaptic lags between pyramidal cells and interneurons (Csicsvari et al., 2003). To test the relationship between synaptic interactions and LFP properties, E→I monosynaptic coupling strength was inferred through the reliability in which interneurons (predominantly fast-spiking) fire in the milliseconds after a pyramidal spike (Csicsvari et al., 1998; English et al., 2017; Galarreta and Hestrin, 2001; Jouhanneau et al., 2018). Specifically, the spike transmission probability was calculated as the excess probability of observing a post-synaptic interneuron spike at synaptic lags after a pre-synaptic spike, accounting for the baseline rate of coincident activity (Figure 1D), a metric validated with single-cell juxtacellular stimulation (English et al., 2017). To measure the relationship between synaptic coupling strength and oscillatory network oscillatory states, presynaptic spikes were subsampled according to the instantaneous power of the band-pass-filtered LFP. Consistent with the notion that synaptic currents drive both LFP and action potential likelihood, spike transmission probability systematically related to the LFP power across multiple frequency bands(Figure 1E),with local maxima in correlation around theta (7–10 Hz) and gamma (30–80 Hz) waves (Figure 1F).
Figure 1. LFP power is positively associated with E→I monosynaptic drive.

(A) Top, the waveform of each pyramidal cell (black, n = 1,129) and inhibitory (red, n = 240) interneuron for which putative synaptic interactions were detected. Bottom, spike half-width and mean firing rate differentiate cell type.
(B) The spike phase modulation of pyramidal cells (top) and interneurons (bottom) as a function of instantaneous LFP frequency. Color bar shows Z-scored firing rate. Plots to the right show the median Raleigh Z statistic for each population as a function of LFP frequency.
(C) The distribution of the gamma phase preference of pyramidal cells and interneurons
(D) Method for estimating spike transmission probability. Putative synaptic pairs showed peaks in the CCG in the forward direction (causal test) and above a baseline defined by lower frequency co-modulation (fast test). The area of the CCG (green) above this slow baseline (red) defines the spike transmission probability.
(E) The LFP was filtered using wavelet kernels (100 logarithmically spaced Gabor wavelets from 1 to 250 Hz), and, for each frequency band, the entire recording was binned into deciles of the logarithm of the power spectrum. Each pyramidal spike was assigned to a decile bin for each frequency band, and cross-correlations CCGs (pyramidal cells → inhibitory interneurons) were conditioned upon wavelet power. The heat plot shows the spike transmission probability for each decile of each frequency band.
(F) The spike transition probability was correlated (Spearman R) with the decile for each frequency band and averaged across all pairs (gray) and separately for pairs in which the rate of the post-synaptic INT positively correlated with theta power (theta mod., blue). Inset, the distribution of correlation coefficients (Spearman R) of the interneuron firing rate versus theta power, as calculated per theta power decile.
(G) Example CCGs between a pyramidal cell and interneuron showing the probability of observing an interneuron spike immediately following a pyramidal spike as a function of theta and gamma power. Synaptic connectivity is inferred by sharp peaks at monosynaptic lags (1–4 ms) above the expected degree of synchrony given slower-timescale (15 ms) rate fluctuations (red line). CCGs were conditioned upon theta/gamma power and split into deciles.
(H) The percentage of increase in spike transition probability relative to the lowest decile bin for all pairs (gray) and for theta mod. pairs (blue) for both theta and gamma. N = 6 mice.
We first focused on the correlation between spike transmission probability and the theta rhythm, given the role of local CA1 interneurons in the generation of these currents (Freund and Antal, 1988; Korotkova et al., 2010). This correlation was strongly bimodal and depended upon whether the post-synaptic interneuron increased or decreased firing rate with theta (8–12 Hz) power (Figure 1F), in line with the presence of functionally independent subnetworks of interneurons that are modulated by locomotion (Garcia-Junco-Clemente et al., 2019). At the theta rhythm, spike transmission probability increased as a function of power (Figure 1G), beyond the baseline coincident activity observed at slower scales (the red line in Figure 1G). The largest gains in spike transmission probability were seen in those interneurons whose firing rate increased monotonically with theta power (Figure 1H) (n = 1,543 pairs; mean increase in spike transmission at max decile = 10.4% ± 0.006%, Wilcoxon signed-rank test, p = 5.4−66), though increases were observed across the whole population (Figure 1H) (n = 2,284 pairs; mean increase in spike transmission = 3.3% ± 0.005%, Wilcoxon signed-rank test, p = 5.6−7). Next, we tested the relationship between spike transmission and gamma power (Figure 1G) (30–80 Hz). Consistent with the essential role that E→I synapses play in driving the gamma rhythm (Buzsáki and Wang, 2012; Carlén et al., 2012; Fuchs et al., 2007; Pelkey et al., 2015; Polepalli et al., 2017), spike transmission probability was always higher during epochs of strong gamma power (Figure 1H) (n = 2,284 pairs; mean increase in spike transmission at max decile = 18.1% ± 0.004%, Wilcoxon signed-rank test, p = 2.7−207). Taken together, our data suggest that E→I monosynaptic drive is positively correlated with LFP power, particularly at theta and gamma rhythms.
γCaMKII is enriched in GABAergic interneurons
Given that E→I synaptic input can reliably excite its postsynaptic interneurons (Csicsvari et al., 1998; English et al., 2017; Galarreta and Hestrin, 2001; Jouhanneau et al., 2018; Miles, 1990) to generate changes in the LFP (Cardin et al., 2009; Sohal et al., 2009), one likely explanation for the positive correlation between E→I monosynaptic drive and LFP is that changes in E→I synaptic input may modulate LFP power. As E→I synaptic input can be dynamically modified (Pelkey et al., 2017), we hypothesize that E→I synaptic plasticity may regulate network oscillations in an activity-dependent manner to enable experience-driven changes in neuronal assemblies and computation (Csicsvari et al., 1998; Ognjanovski et al., 2017; Xia et al., 2017).
To manipulate experience-driven E→I synaptic input, we first sought to identify the key mediator of LTPE→I. Although αCaMKII is absent in inhibitory interneurons, LTPE→I is nevertheless prevented by broad-spectrum CaMKII inhibitors (Lamsa et al., 2007; Wang and Kelly, 2001), indicating that the “αCaMKII-like molecule” might exist in these neurons. We leveraged recent technical advances, including sensitive RNAscope in situ hybridization (ISH) and GAD2-Cre::Ai14 (tdT) mice, to investigate whether any CaMKII isoform(s) (α, β, γ, and/or δ) (Hudmon and Schulman, 2002) are expressed in GABAergic interneurons. Our RNAscope system (see STAR Methods and Figure 2E) can detect mRNAs at the level of a single transcript, while the transgenic mice, which express the fluorescent protein tdTomato under the control of the GAD2 promoter (to label an enzyme that synthesizes the inhibitory neurotransmitter GABA), allowed us to unambiguously identify inhibitory interneurons. Consistent with the classic view (Liu and Jones, 1996; McBain et al., 1999; Sík et al., 1998), we found that αCaMKII was not expressed in GAD2+ interneurons (Figures 2A and S1A). A similar lack of expression was observed for βCaMKII and δCaMKII (Figures S1B–S1E). However, γCaMKII, which is associated with impaired memory and mental retardation in humans (de Ligt et al., 2012; de Quervain and Papassotiropoulos, 2006), is robustly expressed in GAD2+ interneurons (74.8% ± 5.4%) (Figures 2A and S1A). Moreover, 72.4% ± 7.5% γCaMKII-enriched (γCaMKII+) neurons also expressed GAD2-tdTomato (Figures 2A and S1A).
Figure 2. γCaMKII is enriched in GABAergic interneurons in the mouse hippocampus and cortex.
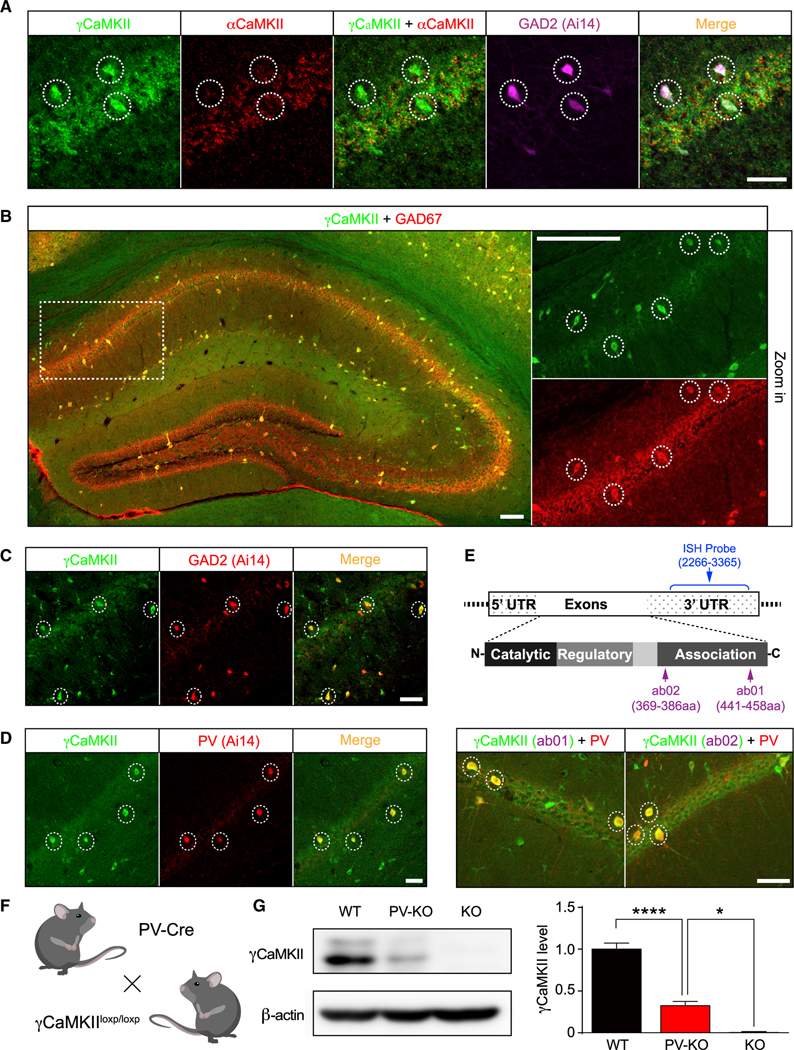
(A) γCaMKII mRNA levels are high in αCaMKII-negative/GAD2-positive neurons (dashed circles) in the hippocampus.
(B) γCaMKII protein levels are high in GAD67+ interneurons (dashed circles) in the hippocampus.
(C and D) Hippocampal sections obtained from GAD2::Ai14 (tdT) and PV::Ai14 (tdT) mice reveal high γCaMKII expression in GAD2+ and PV+ interneurons (dashed circles).
(E) Top, schematic illustration of the γCaMKII mRNA and proteins, showing the regions recognized by the ISH probe (nucleotides 2266–3365) and the antibodies ab01 (amino acids 441–458) and ab02 (amino acids 369–386). Bottom, representative images showing γCaMKII ab01 (left) and ab02 (right) immunostaining in PV+ interneurons (dashed circles) in the hippocampus. For (A)–(E), see Figures S1 and S3 for the distribution in the cortex.
(F) γCaMKII PV-KO mice were generated by crossing γCaMKIILoxP/LoxP mice with PV-Cre mice.
(G) Example western blot analysis and summary of γCaMKII in the hippocampus of wild-type (WT), PV-KO, and KO mice (n = 4–7 mice/group).
In this and subsequent figures, summary data are presented as the mean ± SEM. *p < 0.05 and ****p < 0.0001 (one-way ANOVA followed by Tukey’s test). Scale bar: 50 μm (A and C–E) and 100 μm (B). See also Figures S1–S3.
To confirm this finding, we generated an antibody against γCaMKII (ab01, which recognizes amino acids 441–458) and validated its specificity using samples from γCaMKII knockout (KO) mice (Figures 2E and S2A). Immunostaining revealed that almost all GABAergic interneurons marked by a specific GAD67 antibody (another GABA synthesis enzyme) in the hippocampus and cortex were γCaMKII-enriched neurons (96.3% ± 1.5%) (Figures 2B and S3A). Most (70.3% ± 2.3%) GABAergic interneurons identified by tdTomato in GAD2-Cre::Ai14 (tdT) mice were also labeled by our γCaMKII antibody (Figures 2C and S3B). Moreover, 85.6% ± 1.9% (Figures 2B and S3A) and 93.4% ± 0.8% (Figures 2C and S3B) γCaMKII-enriched neurons also expressed GAD67 and GAD2-tdTomato, respectively, suggesting that γCaMKII is enriched specifically in GABAergic interneurons.
γCaMKII is enriched in PV+ interneurons
Interneurons are characterized by their high structural and functional diversity (Isaacson and Scanziani, 2011; Kepecs and Fishell, 2014; Klausberger and Somogyi, 2008); here, we focused our study on PV+ interneurons, which are arguably the most thoroughly characterized GABAergic interneurons due to their fast-spiking properties and their functional importance in microcircuits and brain networks (Hu et al., 2014). We found that the vast majority of PV+ interneurons were γCaMKII-enriched (88.0% ± 3.4% in PV-Cre::Ai14 [tdT] mice [Figures 2D and S3C] and 87.7% ± 1.7% using a PV-specific antibody [Figures 2E and S3D]), as measured with our ab01 γCaMKII antibody. To confirm its specificity, we generated another specific antibody against γCaMKII (ab02, against amino acids 369–386; Figures 2E and S2A) and also found that 85.4% ± 1.5% of PV+ interneurons were γCaMKII enriched (Figures 2E and S3D). Finally, to estimate the proportion of hippocampal γCaMKII that can be attributed to PV+ cells, we selectively (Figure S2B) deleted γCaMKII in PV+ interneurons by crossing γCaMKIILoxP/LoxP mice with PV-Cre mice (hereafter referred to as γCaMKII PVKO or simply PV-KO; PV-Cre mice are termed wild type [WT]) (Figure 2F). Western blot analysis confirmed that γCaMKII expression in the hippocampus of PV-KO mice (Figure 2G) was reduced to ~1/3 of that in WT mice, consistent with the majority of hippocampal γCaMKII residing in PV+ interneurons. Taken together, our data suggest that γCaMKII is enriched specifically in GABAergic interneurons, with particular enrichment in PV+ interneurons.
γCaMKII PV-KO mice have normal synaptic transmission but impaired LTPE→I
We next asked whether the loss of γCaMKII in PV+ interneurons affects synaptic transmission and/or plasticity, measured in acute hippocampal slices. Consistent with the late onset of PV-Cre expression in development (Carlén et al., 2012; Fuchs et al., 2007; Korotkova et al., 2010) (~P14, Figure S4A), we did not find any detectable developmental, morphological or histological brain abnormalities in γCaMKII PV-KO mice (Figure S5). The expression of all the other CaMKII isoforms (αCaMKII, βCaMKII, σCaMKII), PV, and other critical proteins related to synaptic activity, including NMDA and AMPA receptor subunits, was also unchanged in the hippocampus of PV-KO mice (Figure S4B). To isolate excitatory synaptic transmission, we used the specific GABAA receptor blocker picrotoxin (100 μM) and performed whole-cell recordings of PV+ interneurons and neighboring pyramidal neurons in the CA1 stratum pyramidale (SP) of the hippocampus (Figures 3A and S7A). There was no difference between these WT and γCaMKII PV-KO neurons across a variety of intrinsic physiological properties, including the spiking frequency induced by a 2× threshold depolarizing pulse, the magnitude of afterhyperpolarization, and resting membrane potential (Figures 3B, S6B, and S7B). We also found no difference in AMPAR excitatory postsynaptic current (EPSC) rectification or NMDAR: AMPAR EPSC ratio (Figures S6C, S6D, S7D, and S7E). Moreover, we found no difference between WT and PV-KO neurons in AMPAR-mediated miniature EPSCs (mEPSCs) or spontaneous EPSCs (sEPSCs) in PV+ interneurons (Figures 3D and 3E); mEPSCs and spontaneous inhibitory postsynaptic currents (sIPSCs) also did not differ in excitatory neurons (Figures S7F and S7G). Finally, the paired-pulse ratio at various interpulse intervals, which provides a measure of presynaptic plasticity, was similar between WT and PV-KO slices in both PV+ interneurons and pyramidal neurons (Figures 3C and S7C). Taken together, these results suggest that the baseline synaptic properties and presynaptic plasticity of PV+ interneurons and excitatory neurons are not altered by the loss of γCaMKII in PV+ interneurons.
Figure 3. γCaMKII PV-KO mice have normal LTPE→E but impaired LTPE→I.

(A) Schematic illustration of recordings in PV+ interneurons. PV+ interneurons were identified by the virus-mediated expression of Cre-dependent fluorescent proteins, and pyramidal cells were identified by morphology.
(B) Left, example spike traces evoked by twice-threshold current injection in hippocampal PV+ interneurons. Right, spike frequency measured for the 1st–10th spikes (n = 5–6 cells from 2–3 mice/group).
(C) Example paired-pulse traces measured in hippocampal PV+ interneurons (left) and summary of paired-pulse ratio (right) at the indicated interstimulus intervals (n = 5–6 cells from 3–4 mice/group).
(D and E) Top, example traces of mEPSCs (D) and sEPSCs (E) measured in hippocampal PV+ interneurons of WT and PV-KO mice. Bottom, cumulative distribution plots and summary of mEPSC and sEPSC frequency and amplitude in WT and PV-KO mice (n = 5–22 cells from 2–5 mice/group).
(F) LTP in PV+ interneurons is impaired in γCaMKII PV-KO mice. Top, the stimulation pairing protocol for inducing LTP and the control protocol (no pairing) are shown schematically. Also shown are superimposed representative averaged EPSCs recorded 5 min before (dark traces) and 25 min after (light traces) LTP induction. The bottom panels show normalized amplitude before and after the TBS stimulation protocol (orange arrows) and the summary data measured 1–10, 11–20, and 21–30 min after LTP induction. There was no difference among the groups of WT (no pairing), WT (D-AP5), and γCaMKII PV-KO (n = 8–10 cells from 3–8 mice/group).
(G) LTP induced using a TBS pairing protocol in WT (black) and γCaMKII PV-KO (red) pyramidal neurons showed no difference (n = 6–8 cells from 5 mice/group). All recordings were performed with the GABAA receptor antagonist picrotoxin (100 μM) in the bath solution. PV+ interneurons were identified by the virus-mediated expression of Cre-dependent fluorescent proteins, and pyramidal cells were identified by morphology.
Data were analyzed using an unpaired Student’s t test or a two-way ANOVA followed by Tukey’s multiple comparison test, *p < 0.05 and **p < 0.01. See also Figures S4–S7.
Given that the presence of αCaMKII in excitatory neurons is critical for LTPE→E (Hell, 2014; Malenka and Nicoll, 1999; Silva et al., 1992), we next investigated whether γCaMKII might play a similar role in LTPE→I. In order to induce LTPE→I in the CA1 SP (Wang and Kelly, 2001), we paired theta-burst stimulation (TBS) of the Schaffer collateral with postsynaptic interneuron depolarization (−10 mV) (Perez et al., 2001), which results in significant LTPE→I in WT PV+ interneurons (Figure 3F). Applying TBS without the paired depolarization did not induce LTPE→I in WT slices (Figure 3F), suggesting that the LTPE→I induced by the paired paradigm was not due to propagation of LTPE→E from neighboring excitatory neuron (McBain et al., 1999). The induced LTPE→I in WT hippocampal slices is NMDAR dependent because it was prevented in the presence of the NMDAR blocker D-AP5 (Figure 3F). Remarkably, this paired stimulation protocol failed to induce LTPE→I in hippocampal slices from γCaMKII PV-KO mice (Figure 3F). In contrast, the paired stimulation paradigm induced normal LTPE→E in excitatory neurons in γCaMKII PV-KO mice (Figure 3G), suggesting that the lack of LTPE→I is not due to deficits in plasticity in feedforward excitatory networks. Taken together, these electrophysiological experiments show that γCaMKII plays a critical role in mediating LTPE→I in PV+ interneurons.
γCaMKII in hippocampal PV+ interneurons plays a critical role in long-term memory
Next, we examined the behavioral consequences of eliminating γCaMKII in PV+ interneurons. γCaMKII PV-KO mice did not show any seizure-like behaviors (Verret et al., 2012) (Figure S8A) and were normal in the three-chamber test for sociability (Figure S8D), forced-swim, and sucrose-preference tests for depression (Figures S8E and S8F). In contrast, however, γCaMKII PV-KO mice displayed impairments in classic fear learning tasks that assess long-term memory (Figures 4A–4C). In the inhibitory avoidance (IA) task, γCaMKII PV-KO mice had impaired avoidance behavior after a retention delay of 24 h (Figure 4A). Moreover, γCaMKII PV-KO mice also had significantly reduced freezing in the contextual fear conditioning (CFC) task after a retention delay of 24 h (Figure 4B). In contrast, we found no difference between WT and γCaMKII PV-KO mice in the freezing response 24 h post-training in the auditory fear memory test (Figure 4C). As contextual fear memory is sensitive to hippocampal defects, whereas auditory fear memory is not, these results suggest that hippocampus-dependent contextual memory—but not auditory memory—is impaired in γCaMKII PV-KO mice. Our behavioral results could not be explained by defects in short-term memory (short-term IA, short-term CFC, novel object recognition, and Y maze tests; Figures 4A, 4B, 4D, and 4E), anxiety (open-field and elevated plus-maze tests; Figures S8B and S8C), novelty recognition (novel object recognition test; Figure 4D), locomotor activity (open-field test; Figure S8B), or general fear responses (short-term CFC, short-term IA, and auditory fear memory tests; Figures 4A–4C).
Figure 4. Impaired hippocampus-dependent long-term memory in γCaMKII PV-KO mice.

(A) Left, schematic illustration of inhibitory avoidance test. Right, latency to enter the dark compartment before conditioning (training) and 1 or 24 h after conditioning (testing) was measured using the inhibitory avoidance test (n = 8–16 mice/group).
(B) Left, schematic illustration of contextual fear conditioning test. Right, the percentage of freezing among WT and PV-KO mice was measured 1 or 24 h after conditioning (context) in the one-trial contextual fear conditioning test and sham conditioning (walk through) (n = 7–15 mice/group).
(C) Left, schematic illustration of cued fear conditioning test. Right, the freezing response was measured 24 h after training in WT and PV-KO mice (n = 8–10 mice/group).
(D and E) Summary of the results of the novel object recognition test (D) and the Y maze test (E) (n = 8–13 mice/group).
In (A) and (B), **p < 0.01 and ****p < 0.0001 (one-way ANOVA followed by Tukey’s test). In (C), two-way ANOVA followed by Sidak’s test. In (D) and (E), **p < 0.01 and ****p < 0.0001 (unpaired or paired Student’s t test). See also Figure S8.
Impaired hippocampus-dependent long-term memory in γCaMKII PV-KO mice is reminiscent of previous findings showing that hippocampus-dependent memory function is particularly sensitive to the brain-wide manipulations of LTP-related molecules in vivo (Silva et al., 1992). To further test whether the memory defects of γCaMKII PV-KO mice are due to the loss of γCaMKII in hippocampal PV+ interneurons, we did region-specific shRNA knockdown and rescue experiments (Cohen et al., 2018; Sekeres et al., 2010). We delivered a Cre-dependent adeno-associated virus (AAV) expressing target genes specifically to the mature hippocampus through bilateral stereotactic injection (Figure 5C and S9A). Expressing γCaMKII in mature hippocampal PV+ interneurons of γCaMKII PV-KO mice reversed the hippocampal LTPE→I deficiency (Figures 5A and 5B), further confirming that impaired LTPE→I of γCaMKII PV-KO mice is due to the loss of γCaMKII in PV+ interneurons (Figure 3F). Strikingly, knocking down γCaMKII in mature hippocampal PV+ interneurons impaired memory performance in WT mice in CFC tests (Figures 5D–5F). Moreover, expressing γCaMKII—but not the kinase-dead K43R mutant—rescued the impaired memory in γCaMKII PV-KO mice in CFC tests (Figures 5G and 5H) but had no effect on anxiety or locomotor activity (Figure S9B). Taken together, these data strongly suggest that γCaMKII in hippocampal PV+ interneurons plays a critical role in hippocampus-dependent long-term memory, for which the kinase activity of γCaMKII is required. Since γCaMKII is a key mediator of LTPE→I, our results provide a piece of genetic evidence supporting that LTPE→I, in addition to well-known LTPE→E, is important for long-term memory.
Figure 5. The functional role of γCaMKII in hippocampal PV+ interneurons in LTPE→I and hippocampus-dependent long-term memory.

(A and B) LTP of PV+ interneurons was rescued by virus-mediated re-expressing γCaMKII in the hippocampal PV+ interneurons of PV-KO mice (B) and superimposed representative averaged EPSCs recorded 5 min before (dark traces) and 25 min after (light traces) LTP induction in PV-KO mice (A). Note that the WT group and PV-KO data are also shown in Figure 3F (n = 8–19 cells from 6–15 mice/group), in which PV+ interneurons were identified by the virus-mediated expression of Cre-dependent fluorescent proteins.
(C) Top, schematic illustration of AAV-mediated expression of target genes in the mature hippocampus through bilateral stereotactic injection. Bottom, the representative image showing specific delivery and expression of AAV2/9-hSyn-DIO-mCherry-P2A-HA-HsγCaMKII (red) in the hippocampus. The nuclei were counterstained with DAPI (blue).
(D and E) Representative images (D) and summary data (E) showing that γCaMKII shRNA effectively knocked down the expression of γCaMKII in hippocampal PV+ interneurons in WT (PV-Cre) mice (n = 20–83 cells from 2–3 mice/group).
(F) Memory performance measured 24 h after CFC training in mice (WT, PV-Cre) that bilaterally express AAV-DIO-scramble shRNA or -γCaMKII shRNA in hippocampal PV+ interneurons (n = 9 mice/group).
(G) To compare levels of expressed γCaMKII, we measured mCherry intensity in hippocampal PV+ interneurons of PV-KO mice injected with the AAV2/9-hSyn-DIO-mCherry-P2A-HA-HsγCaMKII WT (+WT) virus or the AAV2/9-hSyn-DIO-mCherry-P2A-HA-HsγCaMKII K43R (+K43R) virus (n = 33–34 cells from 3 mice/group).
(H) The reduced contextual fear response in PV-KO mice is rescued by overexpressing WT γCaMKII but not the kinase-dead mutant form of γCaMKII (K43R) in the hippocampus (n = 7–26 mice/group). The virus-mediated expression of Cre-dependent fluorescent protein was used as the control in the black and red groups.
The data in (B) were analyzed using a two-way ANOVA followed by Sidak’s test; the data in (E)–(G) were analyzed using an unpaired Student’s t test; the data in (H) were analyzed using a one-way ANOVA followed by Sidak’s test. *p < 0.05, **p < 0.01, and ****p < 0.0001. Scale bar: 500 μm (C) and 50 μm (D). See also Figure S9.
Experience-dependent changes in neural oscillations are impaired in γCaMKII PV-KO mice
After determining γCaMKII as the key mediator of LTPE→I and establishing its role in long-term memory, we next examined the function of E→I synaptic plasticity on network activity during learning. Given that the spiking of PV+ interneurons is critical for the generation of LFP, we first asked whether LTPE→I is required for the activity-dependent enhancement of E→I spike transmission coupling. Following TBS stimulation of the Schaffer collateral, the spiking of postsynaptic PV+ interneurons in the WT CA1 increased significantly. In contrast, this activity-dependent enhancement was prevented in γCaMKII PV-KO mice, corroborating an important role of LTPE→I in regulating the E→I spike transmission coupling (Figures 6A–6C). Next, we examined whether γCaMKII in hippocampal PV+ interneurons is required for in vivo LTPE→I at the molecular level. We measured the phosphorylation of the AMPAR subunit GluR1 at Ser831 (pGluR1) (Diering et al., 2016) following CFC training (Figures 6D and S9C), the change of which is often used to assess the expression of LTP in vivo because its critical role in AMPAR regulation (Diering et al., 2016; Whitlock et al., 2006). We found that the pGluR1 level increased in hippocampal PV+ interneurons in WT mice but not γCaMKII PV-KO mice after CFC training compared to naive mice (Figure 6D). In contrast, the basal level of pGluR1 was no different in γCaMKII PV-KO mice and WT littermates, indicating that γCaMKII acted mainly under conditions of training. Thus, and consistent with the key role of γCaMKII in LTPE→I in vitro, our data indicate that γCaMKII is critical for experience-dependent AMPAR regulation in hippocampal PV+ interneurons, a key step for the expression of in vivo LTPE→I (Malenka and Nicoll, 1999; Malinow et al., 2000; Whitlock et al., 2006) and therefore activity-dependent E→I spike transmission coupling.
Figure 6. Impaired activity-dependent spike transmission coupling and phosphorylation of GluR1 in the hippocampus of γCaMKII PV-KO mice.

(A) Example traces before (light traces) and 25 min after (dark traces) the TBS stimulation.
(B) Normalized spike transmission probability before and after the TBS stimulation in WT (black) and γCaMKII PV-KO (red) mice. Note that the spike transmission probability before the TBS stimulation between these two groups was adjusted to a similar level (see STAR Methods).
(C) The summary data measured 21–30 min after the TBS stimulation (n = 5–6 cells from 3–4 mice/group).
(D) Immunostaining of pGluR1 (S831) in the PV-positive neurons (dashed circles) in the CA1 region of WT and PV-KO mice 1.5 h after the contextual fear conditioning (CFC) training or naive conditioning.
Summary data are shown at the right (n = 118–152 cells from 5 mice/group). **p < 0.01 and ****p < 0.0001 (unpaired Student’s t test or one-way ANOVA followed by Turkey’s test). Scale bar: 50 μm. See also Figure S9.
Finally, we recorded LFP in the hippocampal CA1 region (Ognjanovski et al., 2017; Xia et al., 2017) before and after CFC training or sham (walkthrough without shock) conditioning (Figures 7A and 7B). To avoid potentially confounding influences of locomotion on LFP rhythmicity (Garcia-Junco-Clemente et al., 2019), we focused on measuring changes in network activity during sleep, a state critical for memory consolidation in both humans and rodents (Diekelmann and Born, 2010; Stickgold, 2005). We found an increase in the multi-unit activity (MUA) of fast-spiking (putative PV+) neurons during sleep after CFC training (Figures 7C and S11D). Moreover, and consistent with the notion that E→I synaptic drive is positively correlated with LFP power, during sleep—and particularly during rapid eye movement (REM) sleep (Figures S10A and S10B)—gamma wave power and theta wave power were strengthened in WT mice following CFC training (Figures 7D–7F, S10E–S10G, and S11E–S11G), whereas their delta and ripple oscillations were not affected by CFC training (Figures S11H–S11M). Interestingly, among these changes, only the increased gamma power during REM was significantly correlated with each individual animal’s context-specific freezing index (Figures 7G and S11A–S11C), indicating that this change may be most effective at predicting memory performance. Such experience-driven oscillation strengthening was significant 2 h after CFC training and lasted until the end of the recording session (Figures S10C and S10D). No increase was observed in either the MUAs of putative PV+ interneurons, or gamma and theta waves in sham-trained WT mice (Figures 7C–7F, S10C–S10G, and S11D–S11G), suggesting that these changes are experience dependent. Strikingly, although baseline (i.e., pre-training) properties and sleep conditions were similar between WT and γCaMKII PV-KO mice (Figure S12), CFC did not induce the above-mentioned increase (the MUA of putative PV+ neurons, the power of gamma and theta waves) in γCaMKII PV-KO mice (Figures 7C–7F, S10C–S10G, and S11D–S11G). Thus, by uncovering γCaMKII as a key mediator of LTP in PV+ interneurons, our data suggest that LTPE→I regulates activity-dependent E→I spike transmission coupling and therefore plays a critical role in experience-dependent rhythms and memory in vivo.
Figure 7. Impaired experience-dependent oscillations in the hippocampus of γCaMKII PV-KO mice.
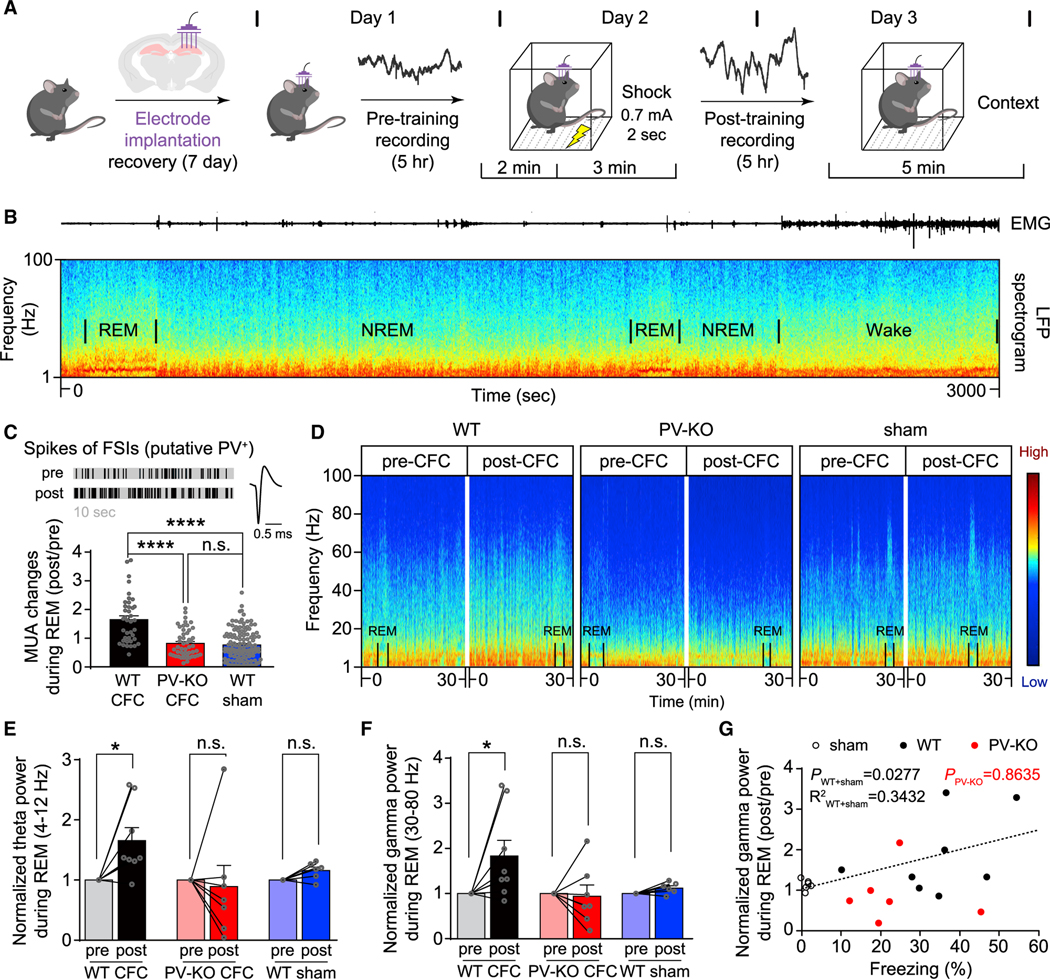
(A) The hippocampal CA1 region was recorded for 5 h pre-CFC and then recorded for 5 h post-CFC (the recordings were performed at the same time of day, see STAR Methods).
(B) Representative local field potential (LFP) spectrogram (bottom) and corresponding EMG trace (top) measured during a typical 50-min sleep-wake period in a WT mouse; REM and non-REM (NREM) periods are indicated.
(C) Top left, representative MUA of the fast-spiking interneurons (FSIs, putative PV+ interneurons) during REM (10 s) in a WT mouse (pre-and post-CFC training); top right, representative trace of a spike of FSIs in WT mice. Bottom, summary of MUA changes (post/pre) for FSIs during REM (the first 30 s for each REM); compared to the groups of PV-KO and sham-treated WT mice, MUA of FSIs increased significantly in the WT group following CFC. n = 39–122 REMs from 2–4 mice/group, ****p < 0.0001 (one-way ANOVA test).
(D) Representative spectrograms waves measured in 30 min epochs pre- (left) and post-training (right) in WT, PV-KO, and sham-treated WT mice; the color represents relative power intensity (dB).
(E and F) Normalized theta wave (E) and gamma wave (F) power was measured before and after training in WT, PV-KO, and sham-treated WT mice. n = 6–8 mice/group, *p < 0.05 (Wilcoxon matched-pairs signed-rank test).
(G) Changes of gamma band power (post/pre) during REM are plotted against freezing (%) for each WT, PV-KO, and sham-treated WT mouse, note that one recorded mouse (PV-KO) was not followed by the retrieval test. Black circles: the WT sham group (n = 6 mice), black dots: the WT group (n = 8 mice), red dots: PV-KO group (n = 6 mice). PWT+sham = 0.0277, R2WT+sham = 0.3432; PPV-KO = 0.8635, and the Bonferroni-corrected p values indicate the results of a Spearman rank-order test.
See also Figures S10–S12.
DISCUSSION
Here, we identify γCaMKII as the long-sought “αCaMKII-like molecule” that is enriched in inhibitory interneurons and mediates LTPE→I in PV+ interneurons. By combining molecular, cellular, electrophysiological, and behavioral studies with a multidisciplinary analysis of genetically engineered mice, we found that γCaMKII in PV+ interneurons is critical for experience-dependent network oscillations in theta and gamma rhythmicity, and long-term memory. Taken together, our data support a model in which synaptic plasticity in PV+ interneurons plays a gatekeeping role in regulating plasticity of the neuronal network and memory.
LTP of inhibitory interneurons
Inhibitory interneurons have extensive axonal arbors distributed throughout the brain, allowing them to inhibit hundreds of pyramidal cells reliably and simultaneously (Hu et al., 2014; Isaacson and Scanziani, 2011; Kepecs and Fishell, 2014; Klausberger and Somogyi, 2008). These interneurons are highly diverse in their subtypes, morphology, distribution, and functions. Here, we focused on LTPE→I of PV+ interneurons in the hippocampus, a well-characterized type of inhibitory interneurons (Hu et al., 2014) in a brain region critical for learning and memory (Bliss and Collingridge, 1993; Morris et al., 2003). Although PV+ interneurons are critical for plasticity and learning at the network level (Carr et al., 2012; Chen et al., 2015; Donato et al., 2013; Gillespie et al., 2016; Kuhlman et al., 2013; Murray et al., 2011; Ognjanovski et al., 2017; Owen et al., 2018; Verret et al., 2012; Xia et al., 2017; Yazaki-Sugiyama et al., 2009), how this process is regulated by experience has been difficult to study experimentally. Inhibiting NMDAR or AMPAR function disrupts synaptic and network function (Allen and Monyer, 2015; Bartos et al., 2007; Carlén et al., 2012; Fuchs et al., 2007; Korotkova et al., 2010; Pelkey et al., 2015; Polepalli et al., 2017), so the observed deficits in learning and memory might have been due to such basal alterations and not to defects of LTPE→I. By identifying γCaMKII as a critical mediator of LTPE→I, we were able to follow a parallel logic in linking synaptic plasticity in inhibitory circuits to brain function (Lisman et al., 2003), as was done in the pioneering studies that linked LTPE→E and learning through the perturbation of αCaMKII (Silva et al., 1992). We disabled LTPE→I by specifically eliminating γCaMKII in PV+ interneurons, a manipulation that spared basal synaptic, network activities, and LTPE→E. Using this strategy, we found that LTPE→I is critical for experience-dependent network strengthening and memory.
The nature of LTPE→I (Kullmann et al., 2012; Pelkey et al., 2017; Pelletier and Lacaille, 2008) depends upon the precise stimulation protocol (Campanac et al., 2013; Le Roux et al., 2013; Wang and Kelly, 2001), paralleling the findings of canonical LTPE→E (Bliss and Collingridge, 1993; Malenka and Nicoll, 1999; Malinow et al., 2000). Due to the high level of heterogeneity for LTP in inhibitory interneurons, we cannot exclude the possibility that γCaMKII may affect diverse forms of NMDAR-independent LTPE→I (Kullmann et al., 2012; Pelkey et al., 2017; Pelletier and Lacaille, 2008), which in principle could participate in regulating experience-dependent neuronal spiking and network activity. Nevertheless, the activity-dependent increase of pGluR1 that regulates the expression of LTP during learning and memory (Malenka and Nicoll, 1999; Malinow et al., 2000; Whitlock et al., 2006) is impaired in the absence of γCaMKII in PV+ interneurons, supporting that γCaMKII is a key mediator for LTPE→I in vivo.
Links between LTPE→I of PV+ interneurons and brain function
Following learning, PV+ interneurons are active during sleep (Ognjanovski et al., 2017; Xia et al., 2017). Upon activation, postsynaptic γCaMKII in PV+ interneurons is driven by NMDAR-gated signals and gets activated by auto-phosphorylation to enable the early phase of LTPE→I (Hell, 2014; Malenka and Nicoll, 1999). Simultaneously, these synaptic mechanisms trigger nuclear CREB phosphorylation (Li et al., 2016; Ma et al., 2014; Wheeler et al., 2012) and control the gene expression of PV and GAD67 to produce enduring synaptic remodeling (Cohen et al., 2016). Thus, PV+ interneurons are equipped with molecular machinery for generating both the early and the late phase of LTPE→I. Through LTPE→I, the excitatory drive of PV+ interneurons is regulated by experience and therefore controls network brain synchrony, supporting the model in which LTPE→I underlies long-term network plasticity and long-term memory.
Using the CFC test, we observed that hippocampal network oscillations in the gamma and theta bands were strengthened following learning (Ognjanovski et al., 2017). Compared to regulating excitatory input onto excitatory neurons, the excitatory input onto inhibitory interneurons is more likely to drive the postsynaptic interneuron to firing threshold (Csicsvari et al., 1998; English et al., 2017; Galarreta and Hestrin, 2001; Jouhanneau et al., 2018; Miles, 1990). Our data suggest that the E→I spike transmission coupling in the hippocampus is enhanced following the LTP-inducing stimulation, which is prevented in the absence of γCaMKII in hippocampal PV+ interneurons. Given that firing of PV+ interneurons is sufficient (Cardin et al., 2009; Sohal et al., 2009) to generate brain oscillations (Buzsáki and Wang, 2012; Hu et al., 2014), LTPE→I therefore likely enjoys an advantage over LTPE→E in that small changes at the synapse can rapidly be converted into changes in spiking and network oscillations that can propagate through the network as perturbations in feedforward and feedback inhibition (Allen and Monyer, 2015; Bartos et al., 2007; Buzsáki and Wang, 2012).
Network oscillation changes after learning are thought to be critical for offline re-activation, gene expression, and enduring synaptic plasticity (Buzsáki, 2010; Frankland and Bontempi, 2005). We hypothesize that the changes in theta and gamma rhythmicity after learning were due to changes in E→I feedback loops that allow expression of the state-dependent cell assembles involved in the learning experience (Csicsvari et al., 1998). Moreover, theta and gamma oscillations are believed to be critical for synaptic and system consolidation, both of which involve multi-wave plasticity-related gene expression during memory consolidation (Abel et al., 2013; Buzsáki, 2010; Diekelmann and Born, 2010; Frankland and Bontempi, 2005). In this brain-wide crosstalk between network oscillations and synaptic plasticity, whether enhanced theta and gamma waves might facilitate the expression of multi-wave LTPE→E genes remains elusive. In this respect, it will be intriguing to check whether the activity-dependent gene expression of key LTPE→E proteins is impaired in the absence of LTPE→I, and whether this impairment is caused by the disrupted experience-driven enhancement of theta and gamma waves. Importantly, the other forms of network changes (e.g., ripples) also occur during different learning tasks (e.g., space learning), which can be detected using optimized behavioral paradigms (Ognjanovski et al., 2017; Xia et al., 2017). Although our data suggest that E→I monosynaptic drive is positively correlated with LFP power, it is not sufficient for establishing the causality between them at waves across all frequencies. For example, although gamma oscillations are thought to arise from E→I interactions, these connections are thought to be less important in causing high-frequency oscillations like ripples (Buzsáki, 2015; Donoso et al., 2018; Malerba et al., 2016), Thus, there are still open questions regarding whether and how LTPE→I of PV+ interneurons may participate in regulating these network oscillations, and related learning and memory.
γCaMKII, brain function, and disease
Over the past decades, the absence of αCaMKII in inhibitory interneurons has contributed to the debates whether and how LTPE→I exists (Buzsáki and Eidelberg, 1982; Kullmann et al., 2012; Pelkey et al., 2017; Pelletier and Lacaille, 2008). Finding that γCaMKII is the CaMKII isoform enriched in inhibitory interneurons is unexpected, as the classic view is that αCaMKII and βCaMKII are the major neuronal CaMKII isoforms (McGuinness et al., 1985; Miller and Kennedy, 1985). However, it is also not surprising because inhibitory interneurons are a small fraction of the total neurons in the brain (Kepecs and Fishell, 2014), and could be easily missed if detection methods are not sensitive or specific enough. The high expression of γCaMKII in PV+ interneurons might confer a functional advantage compared to other CaMKII isoforms, as γCaMKII has a relatively slow autophosphorylation rate (Gaertner et al., 2004), which makes it ideally suited for decoding the larger and longer-lasting calcium signals in PV+ interneurons (Cohen et al., 2016). Compared to α/βCaMKII, γCaMKII has been largely overlooked in the neuroscience field; nevertheless, γCaMKII is required for excitation-transcription coupling in excitatory neurons by synapto-nuclear shuttling, albeit in a kinase activity‒independent manner (Cohen et al., 2016; Cohen et al., 2018; Ma et al., 2014). Interestingly, many studies have found associations among γCaMKII (Cohen et al., 2018; de Ligt et al., 2012; de Quervain and Papassotiropoulos, 2006; Voineagu et al., 2011), PV+ interneurons (Gillespie et al., 2016; Hu et al., 2014; Marín, 2012; Verret et al., 2012), and brain diseases involving cognitive impairments, including Alzheimer disease, schizophrenia, epilepsy, and autism. Thus, experience-dependent network activity, tuned by LTP of PV+ interneurons and γCaMKII, might be functionally relevant in the pathogenesis of these brain disorders.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Huan Ma (mah@zju.edu.cn).
Materials availability
All unique resources generated in this study are available upon request.
Data and code availability
The datasets/code supporting the current study have not been deposited in a public repository, but are available from the corresponding author on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All mice (maintained on a C57BL/6J background) were housed at 22C under a 12 hr light/12 hr dark cycle with food and water available ad libitum. γCaMKII-KO and γCaMKIILoxP/LoxP mice were kindly provided by Dr. Eric N. Olson (UT Southwestern Medical Center). PV-Cre (Stock No. 008069), GAD2-IRES-Cre (Stock No. 010802), T29–1. CaMKIIα-Cre (Stock No. 005359), Ai14LoxP/LoxP (Stock No. 007908) and Ai32LoxP/LoxP (Stock No. 012569) mice were from The Jackson Laboratory. Male mice between 4 and 7 weeks old were used in in vitro electrophysiology experiments. Male mice between 2 and 4 months old were used in other all experiments and handled in accordance with institutional guidelines. All animal studies and experimental procedures were approved by the Animal Care and Use Committee at Zhejiang University, or conducted in accordance with the Institutional Animal Care and Use Committee of New York University Medical Center.
METHOD DETAILS
Data acquisition
All data were acquired and analyzed by experimenters who were blinded with respect to the genotype of the mice and acute slices.
Behavioral assays
All behavioral tests were performed between 8:00 am and 8:00 pm. The open field test and inhibitory avoidance test were performed as described (Cohen et al., 2018) and were analyzed using either the ANY-maze automated tracking system (Stoelting, Europe) or EthoVision tracking software (Noldus).
For the elevated plus maze, the mouse was placed at the center of the maze and allowed to explore for 10 min. The total distance traveled, entries into each arm, and time spent in each arm were videotaped and analyzed using the ANY-maze system.
For the novel object recognition test, following a 10-min habituation period in the empty chamber, the mouse was placed in the chamber with two identical objects (A and B) for 10 min. After a retention period of 1 hr, the mouse was placed back in the chamber, with a familiar object (A) replaced by a novel object (C). The mouse was then allowed to explore the objects for 10 min. For the Y maze test, the mouse was placed at the end of the arm and was free to explore the arena for 10 min. The order of entries was recorded, and three consecutive choices of all three arms were regarded as a correct alternation. The percentage of alternations was then calculated by dividing the number of correct alternations by the maximum number of possible alternations (i.e., the total number of entries minus 2), and then multiplying by 100.
For the cued fear conditioning test, after 2 min of free exploration, the mouse was given 3 tone-shock pairs (90 s apart, 30 s tone [80 dB, 4 kHz] co-terminating with a 2 s foot shock delivered at 0.5 mA). Two minutes after the final tone-shock pair, the mouse was returned to its home cage. A test of the extent of fear learning was done 24 hours after the completion of the training. To assay the response to cue alone, the shape and texture of the floors in the test chamber were changed. The mouse was placed in the changed chamber and allowed 2 minutes to freely explore the chamber. The tone was then delivered 3 times at 1.5 minutes interval without delivering foot shock. All mice were allowed to freely explore the chamber for 2 minutes after the termination of the final tone. The test of the fear memory was assayed as the percent time freezing to a single tone alone, identical to the tone used for training.
For the contextual fear conditioning (CFC) test, the mouse was first habituated to the conditioning chamber for 2 min. A 2 s, 0.7-mA foot shock was delivered, and the mouse remained in the chamber for an additional 2 min after the shock. One hour or twenty-four hours after conditioning, the mouse was placed back in the chamber, and its freezing behavior was analyzed for the first 5 min. Sham (walk through) mice were subjected to the same procedure without receiving foot shock. To detect molecular changes following CFC training, the mouse was first habituated to the conditioning chamber for 2 min. A 2 s, 0.7-mA foot shock was delivered, and the mouse remained in the chamber for an additional 2 min after the shock. Mouse was returned to their home cage for 1.5 hr after the training session before perfusion.
For the social discrimination test, a three-chamber apparatus (60 cm × 40 cm × 25 cm) with two pencil holders (radius: 7.5 cm, height: 15 cm) in the two side chambers was used. After a 10-min habituation period in the apparatus, the test mouse was guided to the center chamber and allowed to explore for 10 min, during which a stranger mouse (stranger 1) was placed in one side pencil holder (the social ability). Next, another stranger mouse (stranger 2) was placed in the other pencil holder, and the test mouse was allowed to explore for an additional 10 min (social novelty). The sniffing time was defined as each instance in which the test mouse’s nose came within 2 cm of a pencil holder.
For the forced swim test, the mouse was placed for 6 min in a cylindrical tank (12 cm diameter, 25 cm in height) containing water. The depth of the water was set to prevent the mouse from touching the bottom during the test, and the mouse’s behavior was videotaped. Immobility was defined as no movement other than movement required to balance the body and keep the head above water, and only immobility time in the last 4 minutes of the 6-min test was analyzed.
For the sucrose preference test, a two-bottle paradigm for habituation (2 days with water followed by 2 days with 1% (w/v) sucrose solution drinking) was used. During the test phase, the mouse was deprived of water for 24 hr, and then given free access to two bottles (one bottle contained 1% sucrose solution, and the other contained water) for 2 hr (acute test) or 48 hr (chronic test). The two bottles’ positions were switched after 1 hr for the 2-hr test and after 24 hr for the 48-hr test. Sucrose preference (sucrose consumption, expressed as a percentage) was calculated as the ratio between the consumption of sucrose solution and the consumption of water plus sucrose solution during the test.
Stereotaxic virus injection surgery
Mice were deeply anesthetized with isoflurane (3% isoflurane for induction and 1.5% isoflurane for maintenance); deep anesthesia was defined as being unresponsive to noxious stimuli. Under anesthesia, the animal was secured in a stereotaxic frame with ear bars. The head was shaved with a razor, and a midline 5-mm incision was made with a sterile scalpel. The subcutaneous tissue was removed from the skull, and 0.5-mm burr holes were drilled. Virus particles were then injected bilaterally at the following coordinates relative to Bregma: for shRNA knockdown and CFC rescue experiments, injections were targeted to the dorsal CA1 region (AP: −2.20 mm, ML: ± 1.30 mm, and DV: −1.70 mm) and the ventral dentate gyrus (AP: −3.00 mm, ML: ± 2.60 mm, and DV: −3.20 mm); for electrophysiological experiments, the injections were targeted to AP: −1.80 mm, ML: ± 1.50 mm, and DV: −1.35 mm. A glass micropipette attached to a 10 μL Hamilton micro-syringe filled with liquid paraffin was used to inject the virus, using a controller to control the speed and volume of the injection. To prevent leakage of the virus prior to reaching the target site, the tip of the glass pipette was filled with ~1 nL virus-free saline immediately before injection. After the injections, the incision was closed with silk sutures, and medical-grade iodophor was applied to the wound to prevent infection. Behavioral experiments were performed ≥ 14 days (4 weeks for shRNA knockdown experiments) after injection. After testing, the fluorescent reporter was used to verify proper targeting of the virus. Only mice with expression of the virus in both hemispheres of the target structure in the hippocampus were included in our analysis. AAV2/9-hSyn-DIO-mCherry-P2A-HA-HsγCaMKII (WT), AAV2/9-hSyn-DIO-mCherry-P2A-HA-HsγCaMKII (K43R) and AAV2/9-hSyn-DIO-mCherry were generated by Taitool Bioscience (Shanghai, China). AAV2/9-EF1α-pDIO-DSE-mCherry-PSE-γCaMKII shRNA was generated by Sunbio Medical Biotechnology (Shanghai, China).
Plasmid generation
For the pAAV-hSyn-DIO-mCherry-P2A-HA-HsγCaMKII-WT plasmid, HA-HsγCaMKII (WT) amplified from pCDH-EF1α-HA-HsγCaMKII (WT) (Cohen et al., 2018). The pAAV-hSyn-DIO-mCherry-P2A-HA-HsγCaMKII (K43R) plasmid were generated using PCR-based mutagenesis (Fwd: 5′-TACGCAGCAAgAATCATCAATACCAAG 3′; Rev: 5′-CTCCTGCGTGGGGGTTTT 3′) (E0554S, NEB). Rat GluR1 (WT) was synthesized by Generay Biotech (Shanghai, China). S831A mutation was introduced using PCR-based mutagenesis (Fwd: 5′-CCCACAGCAAgCCATCAATGA 3′; Rev: 5′-ATCAAACAGAAACCCTTCATC 3′). To knockdown γCaMKII, an shRNA sequence (5′-GCAGCTTGCATCGCCTATATC-3′) was used.
Immunohistochemistry
Mice were deeply anesthetized and perfused with 0.01 M PBS (pH 7.4) followed by 4% paraformaldehyde in 0.1 M PB (pH 7.4). After removal, the brains were post-fixed in 4% paraformaldehyde in 0.1 M PB for 3 hr and then dehydrated for 24 hr at 4°C in 0.1 M PB containing 30% sucrose. Coronal sections (30-μm thickness) were cut in a freezing microtome (Leica CM1800, Heidelberg, Germany) at −20°C and collected in 0.01 M PBS. For staining, the sections were washed in PBS three times, and then blocked in PBS containing 10% normal donkey serum for 1 hr at room temperature. The sections were incubated with the following primary antibodies overnight at 4°C: rabbit anti-γCaMKII (1:1000) raised against γCaMKII amino acids 441–458 (ab01) and 369–386 (ab02); mouse anti-PV (1:5000, 235, Swant, Marly, Switzerland); rabbit anti-pGluR1 S831 (1:300, AB5847, Millipore); mouse anti-NeuN (1:1000, ab104224, Abcam) or mouse anti-GAD67 (1:1000, MAB5406, Millipore). After washing three times, the sections were incubated with the appropriate secondary antibodies conjugated with Alexa Fluor secondary antibodies (1:1000, A21202, A21206, A31570–31573, Invitrogen/Life Technologies) for 3 hr at room temperature. Finally, the sections were mounted on gelatin-coated glass slides and coverslipped with DAPI Fluoromount-G (36308ES20, Yeasen Biotechnology, Shanghai, China). Coronal sections of dorsal hippocampus and cortex were imaged at −1.46 mm to −2.30 mm from bregma. 3–5 sections per mouse were acquired and analyzed. Samples for each experiment were imaged in parallel with the same settings on a confocal laser scanning microscope using an 10×/0.45 or 20x/0.75 objectives (Nikon A1).
Blinded analysis was applied to all the images collected using a custom-written macro in ImageJ (NIH) to analyze the pixel intensity of immunostaining. The nuclear marker DAPI and an antibody against a molecular marker for a specific cell type were used to delineate the nucleus and cell body, respectively. To achieve blinded image collection, neurons were chosen for imaging using only the marker fluorescence or the nuclear stain without examination of the channel of interest. A region of interest (ROI) adjacent to each neuron, but not in the neuron, was selected and used as an ‘off-cell’ background. To adjust for variations in different sets of experiments, normalization across different experiments was achieved by using internal control mice (home cage), which were included in each experiment. For GAD67+, GAD2-Ai14+, PV-Ai14+ and PV+ neurons, a cell was empirically considered “positive” (confirmed by independent observers to resolve ambiguities) if the cell’s fluorescent intensity was above “off-cell” background and if the shortest diameter of ROI was greater than 5 μm (Cohen et al., 2016). Scoring pGluR1 and γCaMKII positivity was performed largely as described (Cohen et al., 2016; Cohen et al., 2018; Ma et al., 2014). For pGluR1 staining, the average pixel intensity of ROI encompassing the cytoplasm was recorded. A background ROI was chosen and the average pixel intensity was subtracted from the cytoplasmic value. For γCaMKII staining, the average pixel intensity of ROI encompassing the soma was recorded. A background ROI was chosen and the average pixel intensity was subtracted from the somatic value. Before counting γCaMKII-enriched (γCaMKII+) cells, the images were thresholded using the mean+2.5SD of average γCaMKII channel intensity of all imaged DAPI+cells in dCA1 for each set of data (sections processed and analyzed in parallel). This threshold setting was empirically determined (confirmed by independent observers to resolve ambiguities), which allows detection of γCaMKII stained with moderate to high intensity, with suppression of lightly stained γCaMKII (Cohen et al., 2016).
Transfection, drug treatment, and immunocytochemistry
HEK293 cells were maintained and transfected using Lipofectamine 3000 transfection reagent (L3000008, Thermo Fisher). HEK293 cells expressing GluR1 WT or GluR1 S831A were stimulated with 200 nM phorbol 12-myristate 13-acetate (PMA, HY-18739, Med ChemExpress) for 15 min at room temperature. The cells were then fixed immediately in 4% paraformaldehyde in phosphate-buffered saline (PBS) containing 20 mM EGTA and 4% (w/v) sucrose. The fixed cells were permeabilized with 0.1% Triton X-100, blocked with 7.5% normal donkey serum, and incubated overnight at 4°C with the following primary antibodies: rabbit anti-pGluR1 S831 (1:1000, AB5847, Millipore) and mouse anti-GluR1 (1:500, MAB2263, Millipore). The cells then were washed with PBS for 10 min, and then incubated at room temperature for 1 hr with the appropriate Alexa Fluor secondary antibodies (1:2000, A21202, A21206, A31570–31573, Invitrogen/Life Technologies), followed by a 30-min wash in PBS. The cells were imaged using a Nikon A1 confocal microscope with a 40X objective.
Nissl staining
To remove the lipids, coronal hippocampal sections mounted on gelatin-coated glass slides were treated in 75% ethanol at 37°C overnight. The sections were then stained for 10 min in a 0.1% solution of cresyl violet (C5042, Sigma) at room temperature. After rinsing with water, the sections were incubated for 3 s each in the following solutions: 70% EtOH, 80% EtOH, 90% EtOH, 95% EtOH, absolute ethanol I, and absolute ethanol II, followed by 10 min in xylene I and 30 min in xylene II. The sections were then mounted using resinene mounting agent, and the CA1, CA3, and dentate gyrus regions of the hippocampus were imaged using an optical microscope.
Golgi staining and morphology analysis
Mice were deeply anesthetized and perfused with 0.01 M PBS (pH 7.4). The whole brain was removed and incubated in Golgi-Cox solution (consisting of a mixture of 5% potassium dichromate in dH20,5% mercuric chloride in dH2O, and 5% potassium chromate in dH2O) at room temperature for 4 days. Coronal hippocampal sections (200-μm thickness) were cut using a vibratome and processed as follows: the sections were rinsed twice (5 min each) in dH2O, dehydrated in 50% alcohol for 5 min, incubated in ammonia solution containing 3 parts ammonia to 1 part dH2O for 8 min, rinsed twice (5 min each) in dH2O, and incubated in 5% sodium thiosulfate for 10 min in the dark. After rinsing, dehydrating, and clearing, hippocampal sections were imaged using a Nikon BX51 upright microscope. Relatively isolated pyramidal neurons or interneurons captured from hippocampal CA1 were selected for morphology analysis. Pyramidal neurons were identified by their characteristic structural features, including conic shaped soma, a large apical dendrite, multiple basal dendrites, and the presence of dendritic spines. Interneurons were identified by multiple and nonpolar dendrites with few spines. The apical and basal dendrites of individual pyramidal neuron, and all dendrites of individual interneuron were traced manually. Branch length and numbers of each neuron were analyzed using the Neuron J plugin in ImageJ, while Sholl analysis was carried out with sholl analysis plugin in ImageJ.
Western blot analysis
Mice were deeply anesthetized and perfused with 0.01 M PBS. The hippocampal tissues were removed and lysed in RIPA buffer containing 10 mM Tris, 150 mM NaCl, 1% Triton X-100, 0.5% NP-40, and 1 mM EDTA (pH 7.4), as well as a 1:100 (v/v) ratio of protease inhibitor cocktail (Roche). The lysates were sonicated, and the protein concentration was measured using a BCA protein assay (Thermo Fisher Scientific). A total of 30 μg total protein per lane was resolved by SDS-PAGE and transferred to a PVDF membrane (Millipore). The membrane was blocked in 5% (w/v) non-fat milk in TBST for 1 hr at room temperature, and then incubated overnight at 4°C with the following primary antibodies: rabbit anti-γCaMKII (1:1000, ab01); mouse anti-αCaMKII (1:1000, MA1–048, Thermo Fisher Scientific); mouse anti-pCaMKII (1:1000, 139800, Thermo Fisher Scientific); rabbit anti-δCaMKII (1:10000, PA5–22168, Invitrogen); rabbit anti-PV (1:1000, ab11427, Abcam); mouse anti-GluR1 (1:1000, MAB2263, Millipore); rabbit anti-NR2A (1:1000, ab133265, Abcam); or rabbit anti-β-actin (1:1500, 4970, Cell Signaling Technology). The next day, the membrane was washed 3 times with TBST, and then incubated with the appropriate peroxidase-conjugated secondary antibodies (Jackson). Proteins were detected using an enhanced chemiluminescence (ECL) detection system (Yeasen Biotechnology, Shanghai, China) and an Amersham Imager 600 imager (GE Healthcare). The protein bands were analyzed using ImageJ.
RNAscope in situ hybridization
The mRNA levels of CaMKII isoforms were detected using in situ hybridization (ISH) with an RNAscope assay (Advanced Cell Diagnostics, Hayward, CA). In briefly, brain slices were dehydrated by sequential incubation in 50%, 70%, and 100% ethanol at room temperature. The slides were then dried at room temperature for 5 min, and a hydrophobic barrier was created around the brain slices using a hydrophobic pen. The slides were treated with hydrogen peroxide for 10 min at room temperature, followed by a protease solution at room temperature for 30 min. After washing, the slides were hybridized with the following probes at 40°C for 2 hr in a HybEZ oven (all from Advanced Cell Diagnostics): Mm-αCaMKII-C3 (445231-C3); Mm-βCaMKII (453601); Mm-γCaMKII-C2 (522071-C2); and Mm-δCaMKII (508941). After washing off the excess probes, the slides were then incubated with preamplifier and amplifier probes as follows: AMP1 (40°C for 30 min), AMP2 (40°C for 30 min), and AMP3 (40°C for 15 min). Three fluorescently labeled probes (fluorescein-Cy3, Cy5, and FITC) were selected for each channel, and the slides were incubated in the labeled probes for 15 min at 40° C (with washes in washing buffer twice between incubations). After washing, the slides were coverslipped using DAPI Fluoromount-G (Yeasen Biotechnology) and imaged using a Nikon A1 confocal laser scanning microscope. Blinded analysis was applied to all the images collected, as stated in the “Immunohistochemistry” section. Before counting γCaMKII+ cells, the images were thresholded using the mean+2SD (empirically determined and confirmed by independent observers to resolve ambiguities) of average γCaMKII channel intensity of all imaged DAPI+ cells in dCA1 for each set of data (sections processed and analyzed in parallel).
Electrophysiology
Acute hippocampal slice preparation
Mice (4–7 weeks old) were anesthetized with diethyl ether and the brain was rapidly removed and placed in ice-cold, high-sucrose artificial cerebrospinal fiuid (ACSF) consisting of (in mM): 206 sucrose, 26 NaHCO3,11 glucose, 2.5 KCl, 1 NaH2PO4,10 MgCl2, and 0.5 CaCl2. Slices were cut on a Leica vibratome in high-sucrose ACSF and immediately transferred to an incubation chamber containing normal ACSF consisting of (in mM): 122 NaCl, 26 NaHCO3, 11 glucose, 3 KCl, 1.25 NaH2PO4, 1.3 MgCl2, and 2 CaCl2. The slices were allowed to recover at 32°C for 30 min, followed by another 60 min at room temperature. The solutions used to prepare the slices and the recording solutions were continuously bubbled with carbogen gas containing 95% O2 and 5% CO2.
Electrophysiological recordings
Recordings were performed in a submerged chamber at room temperature with constant bath perfusion of normal ACSF at a rate of ~4 ml/min. Slices were visualized using infrared differential interference contrast (DIC) imaging on an upright microscope. PV+ interneurons were identified by the expression of Cre-dependent fluorescent proteins, and pyramidal cells were identified by morphology. All recordings were performed with the GABAa receptor antagonist picrotoxin (100 μM) in the bath solution unless otherwise indicated. Whole-cell current-clamp recordings were performed using an internal solution consisting of (in mM): 130 K-gluconate, 1 MgCl2, 10 HEPES, 0.3 EGTA, 4 Mg2ATP, 0.3 Na3GTP, and 10 phosphocreatine. Resting potential was measured just after the patched membrane was ruptured by suction. To analyze action potential properties, PV+ interneurons were recorded at rest and depolarized with 500-ms (or 250-ms for pyramidal cells) current injection pulses at 10-pA increments. The spike frequency of PV+ interneurons was measured for the first 10 spikes evoked by a 2X-threshold current. For all voltage-clamp recordings (except the sIPSC and LTP experiments), glass pipettes (3–6 MΩ tip resistance) were filled with an internal solution consisting of (in mM): 130 CsMeSO4,6 CsCl, 1 MgCl2,10 HEPES, 0.3 EGTA, 4 Mg2ATP, 0.3 Na3GTP, 0.1 spermine, 10 phosphocreatine, and 5 QX-314 (pH 7.25–7.3; 290–299 mOsm). mEPSCs were recorded at −65 mV in the presence of 100 μM picrotoxin, 50 μM D-AP5, and 0.1 μM TTX; sEPSCs were recorded in the conditions except that TTX was omitted. For evoked EPSCs, either a bipolar stimulating electrode or a glass pipette filled with normal ACSF was placed in the stratum radiatum, and responses were evoked at 0.1 Hz. To measure the NMDAR to AMPAR EPSC ratio in PV+ interneurons, EPSCs were recorded at a holding potential of +40 mV and −65 mV, respectively, and 20–40 consecutive events were averaged. The EPSC magnitude at +40 mV, 50 ms post stimulus was taken as the NMDAR EPSC, and the AMPAR EPSC was measured as the amplitude at the holding potential of −65 mV. For pyramidal cells, AMPAR EPSCs were recorded at a holding potential of −70 mV. To measure AMPAR rectification, 70 μM D-AP5 was added in order to completely block NMDARs. The AMPAR rectification ratio was calculated as the peak amplitude of the AMPAR EPSC at +40 mV divided by the amplitude at −65 mV for PV+ interneurons. The rectification index for pyramidal cells was calculated as the ratio of the slopes of the two lines connecting average EPSC values at −70 and 0 mV, and, 0 and +40 mV, respectively. The paired-pulse ratio was calculated by dividing the peak amplitude of the second EPSC by the peak amplitude of first EPSC, with an interstimulus interval ranging from 50 ms to 450 ms. sIPSCs were recorded using a high Cl− internal solution containing (in mM): 136 CsCl, 1MgCl2, 10 HEPES, 0.3 EGTA, 4 Mg2ATP, 0.3 Na3GTP, 10 phosphocreatine and 5 QX-314 (pH 7.25–7.3; 290–299 mOsm). 10 mM NBQX and 50 μM D-AP5 were added in the bath solution to isolate sIPSC.
For LTP recordings, glass pipettes (3–6 MΩ tip resistance) were filled with an internal solution consisting of (in mM): 130 K-gluconate, 1 MgCl2, 10 HEPES, 0.3 EGTA, 4 Mg2ATP, 0.3 Na3GTP, 10 phosphocreatine. 5 QX-314 was applied in internal solution in voltage clamp. When indicated, 70 μM AP5 was added to block the activity of NMDARs. An incision was made in the slice between CA1 and CA3 in order to prevent the propagation of current excitation via the Schaffer collaterals. Responses were evoked at 0.1 Hz. The cell was held at −70 mV to record AMPAR EPSCs, and the stimulus intensity in PV+ interneurons was set to evoke a mixture of successes and failures of synaptic transmission for a 5-min stable baseline. LTP was induced by three trains of theta-burst stimulation (each train consisted four bursts of 5 pulses at 100 Hz delivered at 200-ms intervals) separated by 30 s, paired with four 60-ms depolarization steps to −10 mV. This induction protocol was applied within 12 min of achieving the whole-cell configuration in order to avoid “wash-out” of LTP components. In vitro spike transmission data were recorded in current clamp without picrotoxin, TBS was applied after a 3-min stable baseline. The stimulus intensity was set to evoke a baseline spike probability lower than 33%. Series resistance was monitored online, and recordings in which series resistance increased to > 30 MΩ or changed >20% during an experiment were discarded. Data were collected using a MultiClamp 700B amplifier (Molecular Devices), filtered at 2 kHz, and digitized at 10 kHz using a Digidata 1550B data acquisition system (Molecular Devices).
In vivo electrophysiology
Electrode implantation and signal acquisition
For data related to spike transmission probability, T29–1. CaMKIIα:: AI32 mice were anesthetized with 1.5%−2% isoflurane and provided with a local anesthetic to the incision site (bupivicaine at 0.05 mg/kg, 2.5 mg/ml, S.C.). The skull was cleaned with saline and hydrogen peroxide and ground wires (bare stainless steel) were positioned intracranially over the cerebellum. The skull was then coated with Optibond (Kerr Dental, Brea, CA) and a craniotomy (~1.5×0.5 mm) was performed at AP: −2.2, ML: −2.0 (left hemisphere), 45° angle from the midline. The dura was removed and the probe was implanted ~0.5 mm into the cortex. The probe and custom driver were cemented to the skull with C & B Metabond Quick Adhesive Cement (Parkell) and Unifast Trad acrylic (GC America). The craniotomy was capped with a mixture of mineral oil (one part) and dental wax (three parts), and a Faraday cage was constructed using copper mesh and connected to the cerebellar ground wire. Following surgery, an opioid analgesic was injected (Buprenexat 0.06 mg/kg, 0.015 mg/ml, IM) and given as needed for the next 1–3 days. After implantation, animals were housed individually on a reversed 12/12 hr day/night schedule. Following one week of recovery, mice were recorded 5–7 days/week for two months before being euthanized with pentobarbital cocktail (Euthasol®, transcardial 300 mg/kg) and perfused with formalin (10%). Neural data was acquired using 32 site, 4-shank μLED probes (Neuralight, MI). Data were amplified and digitized at 30 kHz with Intan amplifier boards (RHD2132/RHD2000 Evaluation System, Intan). For all the other data, male mice (~2 months old) were anesthetized with isoflurane. A small craniotomy was performed on the right hemisphere 2.0 mm posterior to Bregma and 0.9 mm lateral to the midline. The microelectrode array (3×5, inter-wire interval, 300 μm; wire diameter, 33 μm) was implanted in the CA1 region at a depth of 1.4 ± 0.2 mm from the skull surface). Two miniature steel screws (one for reference and one for ground) were anchored at the skull over the cerebellum, and wires were inserted into the neck muscle for EMG recording. After recovery for 7 days, data were acquired for two consecutive days over a period of 5 hr (the time window critical for memory consolidation (Abel et al., 2013; Ognjanovski et al., 2017)) each day; on the second day, the mice received an electric shock (0.7 mA, 2 s) prior to the start of recording; recordings were performed at the same time of the day, and always starting at 12:30 PM during the light period, that is, when mice spend most of the time sleeping. After the experiments, the hippocampus was post-fixed to verify the proper placement of the electrodes in the target region. Data were sampled at 30 kHz using a brain signal acquisition system (NeuroStudio, Jiangsu Brain medical technology company, China) and down-sampled to 1 kHz to generate a local field potential (LFP).
Data analysis related to spike transmission probability
Data analysis in this section was done largely based on (English et al., 2017). Unit isolation. Spikes were extracted and classified using Kilosort. Global principal components were calculated (three perchannel,8 channels/shank) and spikes were extracted from the high pass filtered wideband signal (3rd order butterworth filter, passband: 0.5–15kHz). Manual unit curation was done using Klusters. Spike sorting quality was assessed with L-Ratio, Isolation distance, inter-spike interval violation, and visual inspection of cross-correlations suggestive of erroneous splitting of single units.
Cell type classification
Spike waveform (width and asymmetry), autocorrelation properties, and mean firing rate (mean inter-spike interval) were used to classify neurons into excitatory cells and interneurons. The autocorrelation was parameterized with a double exponential model:
for x ≤ 0 otherwise ACGBASE = 0
forx >0 otherwise ACGBURST = 0. Where.
Units were defined by: rate, autocorrelation peak above baseline β1, spike width, and spike asymmetry. Then k-means clustering (k = 2) was performed on the z-score normalized feature matrix which separated excitatory cells from putative interneurons (including fast spiking and regular spiking interneurons). The validity of the cluster labels was confirmed through the cross-correlation (CCG) analysis, revealing increased synchrony at synaptic time-scales.
Phase locking
The LFP was extracted by low-pass filtering the 30 kHz raw data (sinc filter with a 450 Hz cut-off band) and then downsampled to 1250 Hz. Fluctuations in power were assessed through wavelet filtering (50 logarithmically spaced Gabor wavelets from 1–250 Hz). Phase is only meaningful with sufficient power in that frequency band. To avoid setting a cut-off threshold in units of standard deviations (which likely differ as a function of frequency), power thresholds at each spectral band were set at the level in which multi-unit activity first displayed weak phase locking (mean complex vector length = 0.05). Instantaneous spike phase and Raleigh-Z statistic were then calculated during moments of sufficient power at each frequency band.
Detection of monosynaptic pairs and calculation of spike transmission probability
Cross-correlograms (CCGs) were generated to compare the spike trains of pyramidal cells with those of each interneuron (0.8 ms binning). To detect connected pairs, two criteria were required (see English etal., 2017). First, the peak in the CCG needed to exceed that from the slowly co-modulated baseline, and second the peak in the causal direction (positive lags) needed to be significantly larger than the peak in the anti-causal direction (negative lags). To generate the lower frequency baseline, λslow, the observed CCG was convolved with a “partially hollow” Gaussian kernel with a standard deviation of 15 ms. The probability of obtaining an observed (or higher) synchrony count in the mth time lag of the observed CCG (0.8–2.4ms), given the expected, low frequency baseline rate λslow(m) in the same bin was estimated using the Poisson distribution
The probability of obtaining the observed (or higher) synchrony count in the positive mth time lag of the observed CCG (0.8–2.8ms), higher than the maximum λanticausal (-m) at a negative time lag from −2.0 to 0 ms, was similarly estimated using the Poisson distribution.
Pcausal and Pfast were calculated at the peak CCG count observed at lags between 0.8–2.8ms; synaptic peaks were not observed beyond these time points. Putative monosynaptic connectivity was established for those pairs with Pfast < 0.001 and Pcausal < 0.0026 (see English et al., 2017). The synaptic strength was estimated as the excess in causal spike transmission probability from that expected given λslow.
Therefore, the spike transmission probability was defined as:
Spike transmission probability and LFP power
The LFP was extracted by low-pass filtering the 30 kHz raw data (sinc filter with a 450 Hz cut-off band) and then downsampled to 1250 Hz. Fluctuations in power were assessed through wavelet filtering of the LFP (100 logarithmically spaced Gabor wavelets from 3–250 Hz). To assess spike transmission probability during different LFP states, the logarithm of the LFP power at each band pass was calculated (as this transform approaches a normal distribution) and the distribution was binned into deciles. Every moment in the recording was classified in terms of a 100-element vector, where each element describes the power decile of each (N = 100) frequency band. Each presynaptic spike was then classified according to the power decile of each frequency band, and for each frequency band, 10 CCGs were calculated, one per decile. Then conditional spike transmission probability for each CCG was calculated as described above.
Data analysis related to experience-dependent network changes
Sleep state classification
Sleep state was identified by low EMG activity, wake state was identified by high EMG activity. An increase in theta wave (4–12 Hz) power was considered a marker of REM sleep. Periods of an increased theta-delta power ratio (> 1) were identified as REM epochs (Weber et al., 2015).
Spectrum analysis
The power spectrum of continuous LFP was computed using multi-taper estimation in MATLAB with 256 windows to divide the signal into segments containing 250 samples providing overlap between adjoining segments. Before this process, LFP was used to calculate the discrete Fourier transform.
Wave detection
Delta (1–4 Hz), theta (4–12 Hz), and gamma (30–80 Hz) waves were filtered by band-pass filtering of LFP data. To detect ripples, the LFP obtained from the CA1 was first band-pass filtered (100–200 Hz), and the amplitude was calculated using the Hilbert transform. Signals were defined as ripples of they exceeded the peak z-score amplitude threshold (5 times the standard deviation), and the minimum and maximum duration was 20 and 100 ms, respectively.
Spike sorting
Based on three waveform features—the first principle component (PC1), the peak-valley, and the waveform nonlinear energy—we divided spikes into single units using the Offline Sorter (Plexon Inc., Dallas, TX, USA). Units were excluded when the absolute refractory period of single unit autocorrelation was < 1 ms. FSIs were identified with the half-width and peak-trough ratio as key features (Cardin et al., 2009; Owen et al., 2018).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data processing, figure generation, and statistical analyses were performed in Prism (GraphPad Software), Excel (Microsoft), Clampfit (Version 10.4; Molecular device), NeuroExplorer5 or MATALAB (MathWorks). All data are presented as the mean ± SEM. The exact sample size (e.g., the number of mice, slices, or neurons) of each experiment is indicated in the figure legends. For the comparison of parameters including only 2 conditions, Student’s t test (unpaired or paired) or Wilcoxon matched-pairs signed rank test was used. For the comparison of more than 2 conditions within the same graph, One-way or Two-way ANOVAs were performed, with post hoc comparison of all conditions to one another by Tukey’s or Sidak’s test. Cumulative frequency distributions were statistically analyzed using Kolmogorov-Smirnov test.
Statistical outcomes were based on p < 0.05 and displayed throughout figures as: ‘n.s.’ not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p< 0.0001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Rabbit anti-γCaMKII (ab01) | This paper | N/A |
| Rabbit anti-γCaMKII (ab02) | This paper | N/A |
| Mouse anti-PV | Swant | Cat#235; RRID: AB_10000343 |
| Rabbit anti-PV | Abcam | Cat#ab11427; RRID: AB_298032 |
| Rabbit anti-pGluR1 (S831) | Millipore | Cat#AB5847; RRID: AB_11211981 |
| Mouse anti-GluRI | Millipore | Cat#MAB2263; RRID: AB_11212678 |
| Mouse anti-NeuN | Abcam | Cat#ab104224; RRID: AB_10711040 |
| Mouse anti-GAD67 | Millipore | Cat#MAB5406; RRID: AB_2278725 |
| Mouse anti-αCaMKII | Thermo Fisher Scientific | Cat#MA1-048; RRID: AB_325403 |
| Mouse anti-βCaMKII | Thermo Fisher Scientific | Cat#139800; RRID: AB_2533045 |
| Rabbit anti-δCaMKII | Invitrogen | Cat#PA5-22168; RRID: AB_11153337 |
| Rabbit anti-NR2A | Abcam | Cat#ab133265; RRID: AB_11158532 |
| Rabbit anti-β-actin | Cell Signaling Technology | Cat#4970; RRID: AB_2223172 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| AAV2/9-hSyn-DIO-mCherry-P2A-HA-HsγCaMKII-WT/K43R | Taitool Bioscience | AAV2/9-WY2498 AAV2/9-WY2499 |
| AAV2/9-hEF1 α-DIO-mCherry | Taitool Bioscience | AAV2/9-S0197 |
| AAV2/9-EF1 α-pDIO-DSE-mCherry-PSE-γCaMKII shRNA | Sunbio Medical Biotechnology | pAAVE3082 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| DAPI Fluoromount-G | Yeasen Biotechnology | Cat#36308ES20 |
| Cresyl violet | Sigma-Aldrich | Cat#C5042 |
| Protease inhibitor cocktail | Roche | Cat#04693132001 |
| Picrotoxin | Tocris | Cat#1128 |
| D-AP5 | Abcam | Cat#ab120003 |
| TTX | Absin | Cat#abs44200985a |
| NBQX | Abcam | Cat#ab120046 |
| 12-myristate 13-acetate (PMA) | MedChemExpress | Cat#HY-18739 |
|
| ||
| Critical commercial assays | ||
|
| ||
| BCA protein assay | Thermo Fisher Scientific | Cat#23227 |
| Enhanced chemiluminescence (ECL) detection system | Yeasen Biotechnology | Cat#36222 |
| RNAscope Multiplex Fluorescent v2 | Advanced Cell Diagnostics | Cat#323110 |
| RNAscope H2O2 and Protease Reagents | Advanced Cell Diagnostics | Cat#322381 |
| Lipofectamine 3000 Transfection Reagent | Thermo Fisher Scientific | Cat# L3000008 |
|
| ||
| Experimental models: cell lines | ||
|
| ||
| HEK293 | ATCC | CRL-321 |
|
| ||
| Experimental models: organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664; RRID: IMSR_JAX:000664 |
| Mouse: γCaMKII-KO | Lab of Eric N. Olson | N/A |
| Mouse: γCaMKIILoxP/LoxP | Lab of Eric N. Olson | N/A |
| Mouse: PV-Cre | The Jackson Laboratory | JAX: 008069; RRID: IMSR_JAX:008069 |
| Mouse: GAD2-IRES-Cre | The Jackson Laboratory | JAX: 010802; RRID: IMSR_JAX:010802 |
| Mouse: T29-1. CaMKIIα-Cre | The Jackson Laboratory | JAX: 005359; RRID: IMSR_JAX:005359 |
| Mouse: Ai14LoxP/LoxP | The Jackson Laboratory | JAX: 007908; RRID: IMSR_JAX:007908 |
| Mouse: Ai32LoxP/LoxP | The Jackson Laboratory | JAX: 012569; RRID: IMSR_JAX:012569 |
|
| ||
| Oligonucleotides | ||
|
| ||
| shRNA targeting sequence: γCaMKII | This paper | N/A |
| 5′-GCAGCTTGCATCGCCTATATC-3′ | ||
|
| ||
| Recombinant DNA | ||
|
| ||
| Plasmid: GluR1 WT | This paper | N/A |
| Plasmid: GluR1 S831A | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ANY-maze automated tracking system | Stoelting | https://www.stoeltingco.com/any-maze-video-tracking-software-1224.html |
| EthoVision tracking software | Noldus | https://www.noldus.com/ethovision-xt |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Prism | Graphpad | https://www.graphpad-prism.cn |
| Clampfit | Molecular Devices | v.10.4 |
| NeuroExplorer 5 | Mizuseki et al., 2009 | https://www.neuroexplorer.com/ |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab/ |
| Statistical Toolbox | MathWorks | https://www.mathworks.com/products/statistics.html |
| Offline Sorter | Plexon | https://plexon.com/products/offline-sorter/ |
| Klusters | Mizuseki et al., 2009 | http://neurosuite.sourceforge.net/ |
| Kilosort | Github | https://github.com/cortex-lab/KiloSort |
| FMAToolbox | M.Z., College de France | http://fmatoolbox.sourceforge.net |
| Buzcode | Github | https://github.com/buzsakilab/buzcode |
|
| ||
| Other | ||
|
| ||
| RNAscope probe: Mm-αCaMKII-C3 | Advanced Cell Diagnostics | Cat#445231-C3 |
| RNAscope probe: Mm-βCaMKII | Advanced Cell Diagnostics | Cat#453601 |
| RNAscope probe: Mm-γCaMKII-C2 | Advanced Cell Diagnostics | Cat#522071-C2 |
| RNAscope probe: Mm-δCaMKII | Advanced Cell Diagnostics | Cat#508941 |
Highlights.
E→I monosynaptic drive is positively correlated with LFP power in vivo
γCaMKII is a mediator of long-term potentiation for E→I synapses
γCaMKII controls experience-driven E→I spike transmission and brain rhythms
Synaptic plasticity in inhibitory interneurons tunes network plasticity and memory
ACKNOWLEDGMENTS
We thank Eric Olson for providing γCaMKIILoxP/LoxP mice. We thank Chris McBain and Jean-Claude Lacaille for insightful comments on the manuscript. We thank Gyorgy Buzsáki for generous support in collecting spike transmission probability data. We thank Paul Frankland, Michael Tadross, Robert Froemke, Richard Tsien, Adrien Peyrache, Lisa Roux, and Scott Owen for their valuable suggestions. We thank Prof. Xiaoming Li for providing the GAD2-IRES-Cre and Ai14 mice. We thank Bo Hu, Yuta Senzai, and Shen Lin for consultation on in vivo recording. We thank Yunchang Zhang for assistance in data collection and preprocessing. We also thank the members of the Ma laboratory for helpful discussions. This study was supported by the National Natural Science Foundation of China (grant numbers 81930030, 31771109, and 31722023 to H.M.; 81901154 to J.Y.; 31900696 to W.L.), the National Key R&D Program of China (2019YFA0508603 to H.M.), CAMS Innovation Fund for Medical Sciences (2019-I2M-5–057 to H.M.), the Fundamental Research Funds for the Central Universities of China (2018XZZX002–02, 2019XZZX001–01-04, and 2019FZA7009 to H.M.), the 111 Project (B13026 to H.M.), and NIMH grant (K99MH118423 to S.M.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.neuron.2021.01.014.
DECLARATION OF INTERESTS
H.H. and S.D. are members of advisory boards for the journal Neuron.
REFERENCES
- Abel T, Havekes R, Saletin JM, and Walker MP (2013). Sleep, plasticity and memory from molecules to whole-brain networks. Curr. Biol. 23, R774–R788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akgül G, and McBain CJ (2016). Diverse roles for ionotropic glutamate receptors on inhibitory interneurons in developing and adult brain. J. Physiol. 594, 5471–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen K, and Monyer H. (2015). Interneuron control of hippocampal oscillations. Curr. Opin. Neurobiol. 31, 81–87. [DOI] [PubMed] [Google Scholar]
- Bartos M, Vida I, and Jonas P. (2007). Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat. Rev. Neurosci. 8, 45–56. [DOI] [PubMed] [Google Scholar]
- Bliss TV, and Collingridge GL (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. (2010). Neural syntax: cell assemblies, synapsembles, and readers. Neuron 68, 362–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G. (2015). Hippocampal sharp wave-ripple: A cognitive biomarker for episodic memory and planning. Hippocampus 25, 1073–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, and Eidelberg E. (1982). Direct afferent excitation and long-term potentiation of hippocampal interneurons. J. Neurophysiol. 48, 597–607. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, and Wang XJ (2012). Mechanisms of gamma oscillations. Annu. Rev. Neurosci. 35, 203–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanac E, Gasselin C, Baude A, Rama S, Ankri N, and Debanne D. (2013). Enhanced intrinsic excitability in basket cells maintains excitatory-inhibitory balance in hippocampal circuits. Neuron 77, 712–722. [DOI] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, and Moore CI (2009). Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 459, 663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlén M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, Rühlmann C, Jones SR, Deisseroth K, Sheng M, et al. (2012). A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol. Psychiatry 17, 537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr MF, Karlsson MP, and Frank LM (2012). Transient slow gamma synchrony underlies hippocampal memory replay. Neuron 75, 700–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SX, Kim AN, Peters AJ, and Komiyama T. (2015). Subtype-specific plasticity of inhibitory circuits in motor cortex during motor learning. Nat. Neurosci. 18, 1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SM, Ma H, Kuchibhotla KV, Watson BO, Buzsáki G, Froemke RC, and Tsien RW (2016). Excitation-Transcription Coupling in Parvalbumin-Positive Interneurons Employs a Novel CaM Kinase-Dependent Pathway Distinct from Excitatory Neurons. Neuron 90, 292–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SM, Suutari B, He X, Wang Y, Sanchez S, Tirko NN, Mandelberg NJ, Mullins C, Zhou G, Wang S, et al. (2018). Calmodulin shuttling mediates cytonuclear signaling to trigger experience-dependent transcription and memory. Nat. Commun. 9, 2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Czurko A, and Buzsáki G. (1998). Reliability and state dependence of pyramidal cell-interneuron synapses in the hippocampus: an ensemble approach in the behaving rat. Neuron 21, 179–189. [DOI] [PubMed] [Google Scholar]
- Csicsvari J, Jamieson B, Wise KD, and Buzsáki G. (2003). Mechanisms of gamma oscillations in the hippocampus of the behaving rat. Neuron 37, 311–322. [DOI] [PubMed] [Google Scholar]
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, et al. (2012). Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929. [DOI] [PubMed] [Google Scholar]
- de Quervain DJ, and Papassotiropoulos A. (2006). Identification of a genetic cluster influencing memory performance and hippocampal activity in humans. Proc. Natl. Acad. Sci. USA 103, 4270–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekelmann S, and Born J. (2010). The memory function of sleep. Nat. Rev. Neurosci. 11, 114–126. [DOI] [PubMed] [Google Scholar]
- Diering GH, Heo S, Hussain NK, Liu B, and Huganir RL (2016). Extensive phosphorylation of AMPA receptors in neurons. Proc. Natl. Acad. Sci. USA 113, E4920–E4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato F, Rompani SB, and Caroni P. (2013). Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature 504, 272–276. [DOI] [PubMed] [Google Scholar]
- Donoso JR, Schmitz D, Maier N, and Kempter R. (2018). Hippocampal Ripple Oscillations and Inhibition-First Network Models: Frequency Dynamics and Response to GABA Modulators. J. Neurosci. 38, 3124–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English DF, McKenzie S, Evans T, Kim K, Yoon E, and Buzsáki G. (2017). Pyramidal Cell-Interneuron Circuit Architecture and Dynamics in Hippocampal Networks. Neuron 96, 505–520.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankland PW, and Bontempi B. (2005). The organization of recent and remote memories. Nat. Rev. Neurosci. 6, 119–130. [DOI] [PubMed] [Google Scholar]
- Freund TF, and Antal M. (1988). GABA-containing neurons in the septum control inhibitory interneurons in the hippocampus. Nature 336, 170–173. [DOI] [PubMed] [Google Scholar]
- Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, Rozov A, Whittington MA, Traub RD, Rawlins JN, and Monyer H. (2007). Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron 53, 591–604. [DOI] [PubMed] [Google Scholar]
- Gaertner TR, Kolodziej SJ, Wang D, Kobayashi R, Koomen JM, Stoops JK, and Waxham MN (2004). Comparative analyses of the three-dimensional structures and enzymatic properties of alpha, beta, gamma and delta isoforms of Ca2+-calmodulin-dependent protein kinase II. J. Biol. Chem. 279, 12484–12494. [DOI] [PubMed] [Google Scholar]
- Galarreta M, and Hestrin S. (2001). Spike transmission and synchrony detection in networks of GABAergic interneurons. Science 292, 2295–2299. [DOI] [PubMed] [Google Scholar]
- Garcia-Junco-Clemente P, Tring E, Ringach DL, and Trachtenberg JT (2019). State-Dependent Subnetworks of Parvalbumin-Expressing Interneurons in Neocortex. Cell Rep. 26, 2282–2288.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie AK, Jones EA, Lin YH, Karlsson MP, Kay K, Yoon SY, Tong LM, Nova P, Carr JS, Frank LM, and Huang Y. (2016). Apolipoprotein E4 Causes Age-Dependent Disruption of Slow Gamma Oscillations during Hippocampal Sharp-Wave Ripples. Neuron 90, 740–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW (2014). CaMKII: claiming center stage in postsynaptic function and organization. Neuron 81, 249–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Gan J, and Jonas P. (2014). Interneurons. Fast-spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function. Science 345, 1255263. [DOI] [PubMed] [Google Scholar]
- Hudmon A, and Schulman H. (2002). Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu. Rev. Biochem. 71, 473–510. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, and Scanziani M. (2011). How inhibition shapes cortical activity. Neuron 72, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouhanneau JS, Kremkow J, and Poulet JFA (2018). Single synaptic inputs drive high-precision action potentials in parvalbumin expressing GABAergic cortical neurons in vivo. Nat. Commun. 9, 1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepecs A, and Fishell G. (2014). Interneuron cell types are fit to function. Nature 505, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, and Somogyi P. (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, and Monyer H. (2010). NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron 68, 557–569. [DOI] [PubMed] [Google Scholar]
- Kuhlman SJ, Olivas ND, Tring E, Ikrar T, Xu X, and Trachtenberg JT (2013). A disinhibitory microcircuit initiates critical-period plasticity in the visual cortex. Nature 501, 543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Moreau AW, Bakiri Y, and Nicholson E. (2012). Plasticity of inhibition. Neuron 75, 951–962. [DOI] [PubMed] [Google Scholar]
- Lamsa K, Irvine EE, Giese KP, and Kullmann DM (2007). NMDA receptor-dependent long-term potentiation in mouse hippocampal interneurons shows a unique dependence on Ca(2+)/calmodulin-dependent kinases. J. Physiol. 584, 885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roux N, Cabezas C, Böhm UL, and Poncer JC (2013). Input-specific learning rules at excitatory synapses onto hippocampal parvalbumin-expressing interneurons. J. Physiol. 591, 1809–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Tadross MR, and Tsien RW (2016). Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science 351, 863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Lichtman JW, and Sanes JR (2003). LTP: perils and progress. Nat. Rev. Neurosci. 4, 926–929. [DOI] [PubMed] [Google Scholar]
- Liu XB, and Jones EG (1996). Localization of alpha type II calcium calmodulin-dependent protein kinase at glutamatergic but not gamma-aminobutyric acid (GABAergic) synapses in thalamus and cerebral cortex. Proc. Natl. Acad. Sci. USA 93, 7332–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Groth RD, Cohen SM, Emery JF, Li B, Hoedt E, Zhang G, Neubert TA, and Tsien RW (2014). γCaMKII shuttles Ca2+/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell 159, 281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, and Nicoll RA (1999). Long-term potentiation–a decade of progress? Science 285, 1870–1874. [DOI] [PubMed] [Google Scholar]
- Malerba P, Krishnan GP, Fellous JM, and Bazhenov M. (2016). Hippocampal CA1 Ripples as Inhibitory Transients. PLoS Comput. Biol. 12, e1004880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Mainen ZF, and Hayashi Y. (2000). LTP mechanisms: from silence to four-lane traffic. Curr. Opin. Neurobiol. 10, 352–357. [DOI] [PubMed] [Google Scholar]
- Marín O. (2012). Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 13, 107–120. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Freund TF, and Mody I. (1999). Glutamatergic synapses onto hippocampal interneurons: precision timing without lasting plasticity. Trends Neurosci. 22, 228–235. [DOI] [PubMed] [Google Scholar]
- McGuinness TL, Lai Y, and Greengard P. (1985). Ca2+/calmodulin-dependent protein kinase II. Isozymic forms from rat forebrain and cerebellum. J. Biol. Chem. 260, 1696–1704. [PubMed] [Google Scholar]
- Miles R. (1990). Synaptic excitation of inhibitory cells by single CA3 hippocampal pyramidal cells of the guinea-pig in vitro. J. Physiol. 428, 61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SG, and Kennedy MB (1985). Distinct forebrain and cerebellar isozymes of type II Ca2+/calmodulin-dependent protein kinase associate differently with the postsynaptic density fraction. J. Biol. Chem. 260, 9039–9046. [PubMed] [Google Scholar]
- Mizuseki K, Sirota A, Pastalkova E, and Buzsáki G. (2009). Theta oscillations provide temporal windows for local circuit computation in the entorhinal-hippocampal loop. Neuron 64, 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, and Baudry M. (1986). Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature 319, 774–776. [DOI] [PubMed] [Google Scholar]
- Morris RG, Moser EI, Riedel G, Martin SJ, Sandin J, Day M, and O’Carroll C. (2003). Elements of a neurobiological theory of the hippocampus: the role of activity-dependent synaptic plasticity in memory. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 773–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ, Sauer JF, Riedel G, McClure C, Ansel L, Cheyne L, Bartos M, Wisden W, and Wulff P. (2011). Parvalbumin-positive CA1 interneurons are required for spatial working but not for reference memory. Nat. Neurosci. 14, 297–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ognjanovski N, Schaeffer S, Wu J, Mofakham S, Maruyama D, Zochowski M, and Aton SJ (2017). Parvalbumin-expressing interneurons coordinate hippocampal network dynamics required for memory consolidation. Nat. Commun. 8, 15039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen SF, Berke JD, and Kreitzer AC (2018). Fast-Spiking Interneurons Supply Feedforward Control of Bursting, Calcium, and Plasticity for Efficient Learning. Cell 172, 683–695.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Barksdale E, Craig MT, Yuan X, Sukumaran M, Vargish GA, Mitchell RM, Wyeth MS, Petralia RS, Chittajallu R, et al. (2015).Pentraxins coordinate excitatory synapse maturation and circuit integration of parvalbumin interneurons. Neuron 85, 1257–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, and McBain CJ (2017). Hippocampal GABAergic Inhibitory Interneurons. Physiol. Rev. 97, 1619–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier JG, and Lacaille JC (2008). Long-term synaptic plasticity in hippocampal feedback inhibitory networks. Prog. Brain Res. 169, 241–250. [DOI] [PubMed] [Google Scholar]
- Perez Y, Morin F, and Lacaille JC (2001). A hebbian form of long-term potentiation dependent on mGluR1a in hippocampal inhibitory interneurons. Proc. Natl. Acad. Sci. USA 98, 9401–9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polepalli JS, Wu H, Goswami D, Halpern CH, Südhof TC, and Malenka RC (2017). Modulation of excitation on parvalbumin interneurons by neuroligin-3 regulates the hippocampal network. Nat. Neurosci. 20, 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekeres MJ, Neve RL, Frankland PW, and Josselyn SA (2010). Dorsal hippocampal CREB is both necessary and sufficient for spatial memory. Learn. Mem. 17, 280–283. [DOI] [PubMed] [Google Scholar]
- Sík A, Hájos N, Gulácsi A, Mody I, and Freund TF (1998). The absence of a major Ca2+ signaling pathway in GABAergic neurons of the hippocampus. Proc. Natl. Acad. Sci. USA 95, 3245–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, and Tonegawa S. (1992). Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science 257, 206–211. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, and Deisseroth K. (2009). Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickgold R. (2005). Sleep-dependent memory consolidation. Nature 437, 1272–1278. [DOI] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, and Geschwind DH (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, and Kelly P. (2001). Calcium-calmodulin signalling pathway upregulates glutamatergic synaptic function in non-pyramidal, fast spiking rat hippocampal CA1 neurons. J. Physiol. 533, 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F, Chung S, Beier KT, Xu M, Luo L, and Dan Y. (2015). Control of REM sleep by ventral medulla GABAergic neurons. Nature 526, 435–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, and Tsien RW (2012). Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell 149, 1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, and Bear MF (2006). Learning induces long-term potentiation in the hippocampus. Science 313, 1093–1097. [DOI] [PubMed] [Google Scholar]
- Xia F, Richards BA, Tran MM, Josselyn SA, Takehara-Nishiuchi K, and Frankland PW (2017). Parvalbumin-positive interneurons mediate neocortical-hippocampal interactions that are necessary for memory consolidation. eLife 6, e27868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazaki-Sugiyama Y, Kang S, Caˆ teau H, Fukai T, and Hensch TK (2009). Bidirectional plasticity in fast-spiking GABA circuits by visual experience. Nature 462, 218–221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets/code supporting the current study have not been deposited in a public repository, but are available from the corresponding author on request.