

Abstract
Pro-inflammatory immune system development, metabolomic defects, and deregulation of autophagy play interconnected roles in driving the pathogenesis of systemic lupus erythematosus (SLE). Lupus nephritis (LN) is a leading cause of morbidity and mortality in SLE. While the causes of SLE have not been clearly delineated, skewing of T and B cell differentiation, activation of antigen-presenting cells, production of antinuclear autoantibodies and pro-inflammatory cytokines are known to contribute to disease development. Underlying this process are defects in autophagy and mitophagy that cause the accumulation of oxidative stress-generating mitochondria which promote necrotic cell death. Autophagy is generally inhibited by the activation of the mammalian target of rapamycin (mTOR), a large protein kinase that underlies abnormal immune cell lineage specification in SLE. Importantly, several autophagy-regulating genes, including ATG5 and ATG7, as well as mitophagy-regulating HRES-1/Rab4A have been linked to lupus susceptibility and molecular pathogenesis. Moreover, genetically-driven mTOR activation has been associated with fulminant lupus nephritis. mTOR activation and diminished autophagy promote the expansion of pro-inflammatory Th17, Tfh and CD3+CD4−CD8− double-negative (DN) T cells at the expense of CD8+ effector memory T (EMT) cells and CD4+ regulatory T cells (Tregs). mTOR activation and aberrant autophagy also involve renal podocytes, mesangial cells, endothelial cells, and tubular epithelial cells that may compromise end-organ resistance in LN. Activation of mTOR complexes 1 (mTORC1) and 2 (mTORC2) has been identified as biomarkers of disease activation and predictors of disease flares and prognosis in SLE patients with and without LN. This review highlights recent advances in molecular pathogenesis of LN with a focus on immuno-metabolic checkpoints of autophagy and their roles in pathogenesis, prognosis and selection of targets for treatment in SLE.
Keywords: autophagy, mitophagy, mechanistic target of rapamycin, mTOR, systemic lupus erythematosus, CD4+ T cells, CD8+ T cells, CD3+CD4−CD8− double-negative T cells, Treg, B cells, plasma cells, dendritic cells, macrophages, interferon, netosis, Rab4A, endosome traffic, lysosome, metabolomics, antinuclear antibodies, glomerulonephritis, lupus nephritis, mitochondrial dysfunction, rapamycin, sirolimus, biomarkers
Introduction to lupus nephritis
Systemic lupus erythematosus (SLE) is an autoimmune disease with wide spectrum diverse of clinical manifestations, organ involvement, and disease severity. It is driven by defects in both the innate and adaptive immune system. It disproportionately affects women, particularly of minority populations including African Americans, Hispanics, Asians, Native Americans, Alaska Natives, Native Hawaiians, and Pacific Islanders. This disparity is independent of socioeconomic status and other biopsychosocial factors, with Black women more likely to show multi-system involvement of SLE and higher disease activity (1).
Lupus nephritis (LN) is a severe manifestation of SLE and is a predictor of poor outcomes. Approximately 30–60% of SLE patients develop nephritis some time during their disease course, with nephritis not uncommonly being the first clinical manifestation (2–4). Frequency of nephritis is even higher in children with SLE, reaching 80% (5). Patients with LN often have an earlier disease onset, show increased levels of circulating inflammatory cytokines, and have a higher SLE disease activity index (SLEDAI) at the time of presentation(6, 7). Early renal involvement is associated with worse outcomes (5). The disease burden is high, both personal and economic. In a recent retrospective cohort study of 325 patients, patients with LN have more than two-fold higher mortality than SLE patients without nephritis(8). Due to the severity of disease with kidney involvement, SLE classification was recently changed from requiring 4 of 11 clinical criteria as established by the American College of Rheumatology (ACR) in 1982 (and revised in 1997) to diagnostic criteria that increase sensitivity for earlier diagnosis (previously taking 6 years on average from the first clinical manifestations)(9, 10). Under the new clinical and laboratory criteria established by the Systemic Lupus International Collaborating Clinics, a diagnosis of SLE can be made, based on the presence of a proliferative immune complex glomerulonephritis and positive antinuclear antibodies (ANA)(11). Although this reduces specificity for increased sensitivity, 85% of surveyed rheumatologists agree that a diagnosis of SLE can be made, based on a positive ANA and proliferative glomerulonephritis alone. Therefore, a high index of suspicion for LN is required in patients with a positive ANA who develop nephritic and/or nephrotic syndrome. Alternatively, patients with proliferative immune complex glomerulonephritis need to be routinely evaluated for ANA.
Pathology of lupus nephritis and current biomarkers of disease activity
A kidney biopsy is required for diagnosis of LN. Kidney pathology evaluation of LN differentiates the disease into six separate classes, based on the location of immune complexes, the presence or absence of proliferative glomerular lesions, and the degree of global glomerulosclerosis. LN classes were established by the International Society of Nephrology/Renal Pathology Society, and include minimal mesangial LN (class I), mesangial proliferative LN (class II), focal lupus nephritis (class III), diffuse LN (class IV), membranous LN (class V), and advanced sclerosing LN (class VI)(12). Focal and diffuse LN (class III or IV) contain destructive glomerular lesions, which can manifest with endocapillary hypercellularity, presence of intracapillary neutrophils and/or karyorrhexis, fibrinoid necrosis, cellular/fibrocellular crescents, wire loops, or intracapillary hyaline thrombi. Approximately 30% of patients with focal or diffuse LN and 10% of membranous lupus nephritis (MLN) develop ESRD, often from repeat disease flares (13).
Kidney biopsies are costly and invasive with potential risks, making it inadequate for effective frequent monitoring of disease activity that is critical to each patient. Current clinical practice is primarily focused on clinical manifestations included in the SLEDAI and laboratory parameters such as serum creatinine, urinary protein, complement levels, ANA, and anti-double stranded DNA (anti ds-DNA antibodies) (14–16). The use of serum and urine biomarkers present intriguing alternatives compared to serial kidney biopsies and include different effectors such as chemokines, cytokines, lymphocyte subsets, complement, and autoantibodies (16). In the kidney, cytokines and chemokines are produced by infiltrating inflammatory cells and are detected in the serum and urine (17, 18). Only a few biomarkers have been validated satisfactorily in cross-sectional longitudinal studies to be applicable in clinical practice.
In SLE, autoantibodies are formed against nuclear, cytoplasm and cell surface antigens with immune complex formation and subsequent complement deposition. Monitoring of autoantibodies is helpful, however are non-specific and present in several other conditions (19, 20). DsDNA and anti-C1q antibody titers correlate with LN flares and fall following cessation of disease activity (21, 22). However, absence of these antibodies does not rule out active LN and many SLE patients with high anti-dsDNA never develop LN (19, 20). In some patients, anti-dsDNA levels were lower in active nephritis, suggesting sequestration in immune complexes within the kidney (23, 24). Anti-Smith, anti-ribonucleoprotein, anti-nucleosome, anti-Sjogren-syndrome-related antigen A(20, 25) (26), anti RNA polymerase I, and anti-Sjogren-syndrome-related antigen B (25, 27, 28) are also increased in LN and may correlate with disease activity (7) (26), but have low sensitivity and are seen in other autoimmune diseases.
Compared to many other autoimmune diseases, the causative autoantigens in lupus nephritis are largely unknown and cannot be effectively utilized to monitor disease activity. Recently, three autoantigens were described in membranous LN with or without concurrent focal or diffuse LN. These include the exostosin 1/2 complex (EXT1/2)(29), neural cell adhesion molecule 1 (NCAM1)(30), and transforming growth factor receptor-beta 3 (TGFBR3)(31). EXT1/2-associated MLN comprises 17–38.4% of cases, and has a good prognosis with a lack of disease progression at 5–10 years (32, 33). NCAM1-associated MLN comprises 6.6% of cases and may be associated with neuropsychiatric disease in addition to nephritis (30). There are no known unique disease characteristics of TGFBR3-associated MLN, but affects 5.5% of patients (31). To date, there are no available serologic assays for non-invasive monitoring of these subtypes of disease and less than half of cases have a known inciting autoantigen.
Additionally, C3 and C4 levels can be monitored, as they are depressed in active disease (34). Depletion of serum complements are related to immune complex formation and precipitation in glomeruli (35, 36),but do not always correlate with disease activity (34, 37–40). Development of improved biomarkers and treatment targets requires further understanding of underlying disease pathogenesis.
Overview of SLE disease pathogenesis
The core pathology underlying lupus pathogenesis is the abnormal reaction to self-antigens, induced by cellular and inflammatory mediators of the innate and adaptive immune system, resulting in deregulated T-cell and B-cell activation from necrotic cell death. Lymphocyte activation increases cell energy requirements and promotes mitochondrial dysfunction. This subsequently results in oxidative stress leading to reduced generation of ATP, increasing propensity for necrosis rather than apoptotic cell death upon sustained T cell activation (41). Antigen-presenting cells take up DNA and other necrotic material and become activated through toll-like receptors. Toll-like receptor (TLR) activation (in addition to T-cell activation) and recognition of damage-associated molecular patterns (DAMPs) result in activation of autoreactive B-cells, which differentiate into plasma cells and secrete autoantibodies (42). Interestingly, there is increased expression of TLRs in target organs that correlates with disease activity, including overexpression of TLR3, TLR7, and TLR9 in the kidney in the setting of LN (43). In addition to DAMPs, pathogen-associated molecular patterns (PAMPs) can activate T-cells through molecular mimicry, epitope spreading, or superantigens that induce non-specific T cell activation and further promote inflammatory responses (44). Defective clearance of autoreactive cells is due to disturbance in both central and peripheral tolerance. Autoreactive T-cells in the thymus (impaired central tolerance) and reduced numbers and impaired function of regulatory T-cells (45, 46) (Tregs, critical for peripheral tolerance) further exacerbate disease.
T cell subsets in SLE
T-cell subset distribution is altered in patients with SLE, which lead to B-cell help, promote plasmablast differentiation, and lead to production of pro-inflammatory cytokines and pathogenic antibodies. Mechanistic studies in NZB/NZW F1 lupus-prone mice revealed that T-cells are essential for autoantibody production, as T-cell deficient athymic mice cannot produce autoantibodies (47). Additionally, adoptive transfer of CD4+ T cells from lupus-prone MRL/lpr mice induce nephritis (48). Lupus T-cells have exacerbated responses to both low and high affinity peptide antigens, of which are MHC class-II dependent primarily presented by dendritic cells, which results in increased T-cell activation (48, 49). ‘Nephrogenic’ T cells are expanded within lymph nodes in lupus-prone NSM2328 mice and are recruited to infiltrate the kidneys in active LN, primarily by the activation of CD11c+ dendritic cells in response to immune complex deposition within the kidney (50).
Patients with SLE have a higher frequency of CD4−CD8− double-negative (DN) T cells than healthy individuals without autoimmune disease, in part due to defective apoptosis and escape from the central tolerance in thymic selection. Expansion of DN T cells has been also linked to mTOR-dependent downregulation of CD8 (51). Expansion of DN T cells, both circulating and infiltrating the kidney, is associated with development of LN and correlate with disease flares (52–54). mTORC1 is activated >10-fold in DN T cell in comparison to CD4 or CD8 T cells both in healthy and SLE donors (53), although mTORC1 activity is considerably elevated in SLE T cells (55) (Figure 1). DN T cells express the costimulatory molecule CD1, which interacts with CD1c on circulating B cells, promoting B-cell help that leads to production of IgG and the pro-inflammatory cytokines IL-4, IL-17, and IFN-γ (56). SLE DN− T cells additionally can induce class-switching of IgM-producing B lymphocytes into those that produce and secrete IgG autoantibodies (56). DN T cells can infiltrate the kidneys in SLE patients (54). Activation of the mechanistic target of rapamycin (mTOR) can induce IL-4 production in DN T cells and promotes necrotic cell death in SLE patients through inhibition of organelle turnover by autophagy (57). mTOR is a serine/threonine kinase that senses metabolic cues to regulate cell growth, proliferation, and survival, which integrate the cellular response to stimulation by cytokines, hormones, growth factors, and changes in cellular metabolism and is an important regulator of T-cell specification, expansion, and survival (58).
Figure 1. Coordinate skewing of mTOR activation and autophagy in cells of the adaptive and innate immune systems underlie pro-inflammatory lineage specification in SLE.
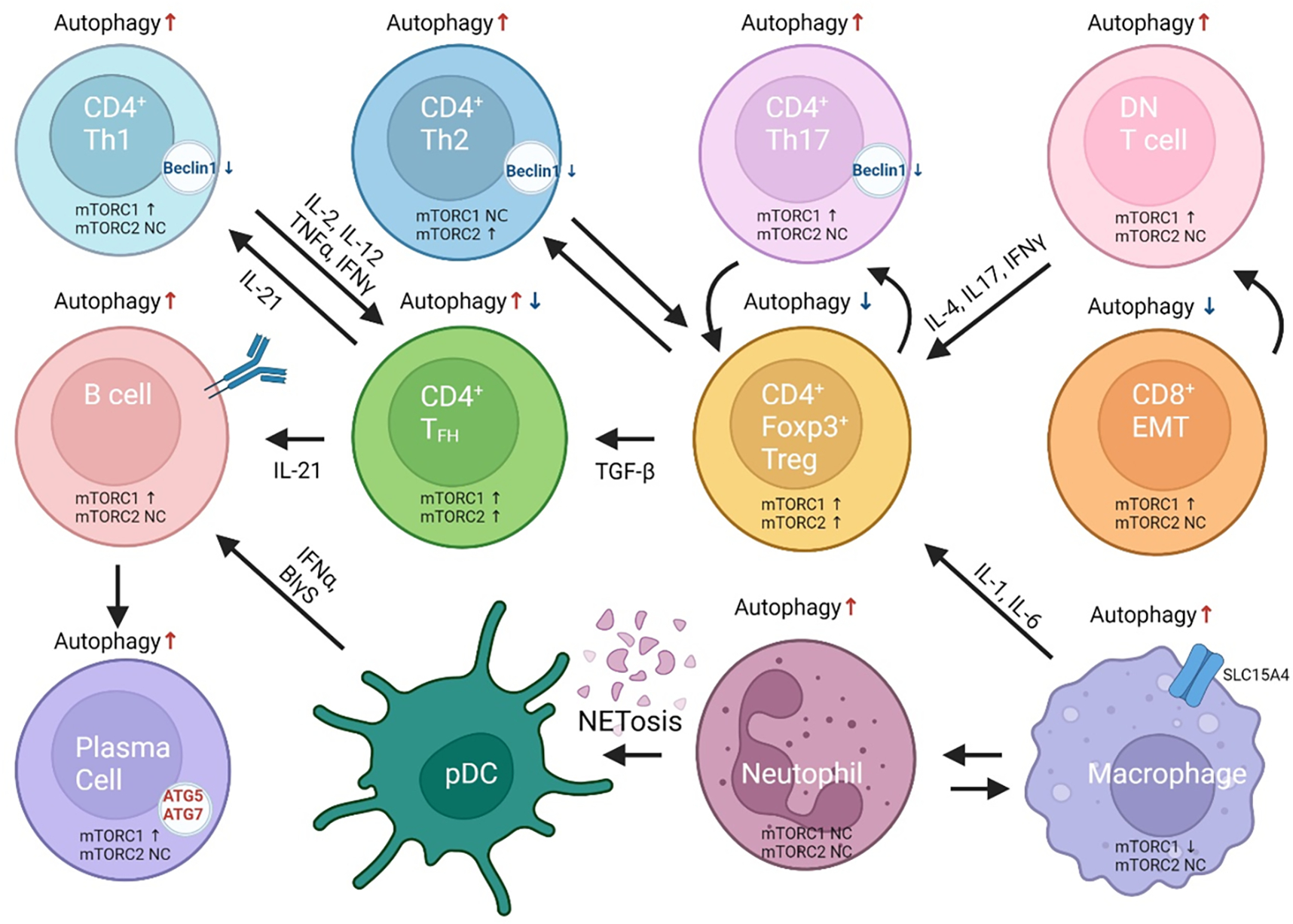
mTOR activation is considered as a central mediator of disease pathogenesis. There is a direct and well established relationship between mTOR activity and autophagy, with increased activity resulting in diminished autophagy. Different T-cell lineages, however, have discordant skewing in autophagy. Naive CD4+ T cells have increased autophagy. CD4+ Th17 cells have decreased autophagy and increased mTORc1 activity. CD4+FoxP3+Treg cells have diminished autophagy with increased activation of both complexes, mTORC1 and mTORC2. CD4+Tfh cells have mixed results with increased or decreased autophagy but mTOR complexes are both increased. CD8+ cytotoxic T cells have increased autophagy resulting in increased cell death. Activity of their mTOR complexes is not fully known. CD8 effector memory T (EMT) cells have reduced autophagy and activation of mTORC1 which are responsive to rapamycin treatment in vivo and in vitro. B cells and plasma cells have increased autophagy required for B cell differentiation, with spurious elevation of MTORC1.
Regulatory T cells (Tregs) that function to suppress inflammatory responses are deficient in SLE patients. Activation of mTORC1 reduces suppressor function of Tregs with a decrease in expression of the immunosuppressive molecules CTLA-4 and GATA3 that are restored upon mTOR blockade with rapamycin (59). Tregs are critical for the negative selection of autoreactive B-cells (60). Expansion of Tregs and restoration of Treg function serve as targets for treatment of LN (61). DN, Th17, and Tfh cells are expanded in SLE, correlate with disease activity (62), and secrete inflammatory cytokines, including IL-4, IL-17, IL-21, and IL-22. Calcium/calmodulin-dependent protein kinase IV (CAMK4) induces Th17 specification by increased activation of the mechanistic target of rapamycin (mTOR) due to binding of the cAMP response element modulator alpha (CREM-alpha)(63). Tregs can be generated through inhibition of calcium/calmodulin-dependent protein kinase IV (CAMK4), which is critical to Th17 cell differentiation. Increased IL-6 production results in Th17 cell expansion and is a target for therapy. IL-6 levels in the serum and urine were found to be higher in patients with LN and decrease in response to treatment (64, 65).
Increased specification to the Th1 and Th2 lineages by expression of the transcription factors T-box expressed in T cells (Tbet) and GATA-binding protein 3 (GATA3) in naïve T cells is seen in SLE patients and increased Tbet and GATA3 expression correlates with disease activity (66). Th1-dominant immune responses are identified within peripheral blood and in kidney tissue of LN patients, with an increase in IFNγ production circulating and expressed intracellularly in T-cells infiltrating the kidney (67, 68). IFNγ production also results from effector CD8+ T lymphocytes, also known as cytotoxic T cells. While effector CD8+ T cells are expanded in SLE patients and contribute to inflammation through IFNγ production; there are decreased numbers and function of memory CD8+ T cells in lupus patients (Figure 1). The depletion of CD8+ T cells has been linked to activation of mTORC1 and deficient autophagy (59).
Interferon-α (IFNα) production results not only from skewed commitment of T cell subsets, but is also induced upstream by the innate immune system. Recognition of nucleic acids released from dying cells by plasmacytoid dendritic cells results in increased IFNα production (69). Gene expression profiling studies have identified an ‘interferon signature’ within peripheral blood mononuclear cells in greater than 50% of SLE patients (70). The IFN signature is associated with severe disease and has been demonstrated to be higher in SLE patients with nephritis (71–74). Interferon-associated genes correlate with multiple autoantibodies, including anti-dsDNA, anti-dsRNA, anti-U1-sn-RNP, anti-chromatin, anti-Ro/SSA, anti-La/SSB, anti-topoisomerase I, and anti-Scl-70. High IFN levels also can induce cross-switching of IgM to IgG autoantibodies (75). Blockade of the type I IFN receptor with anifrolumab has shown remarkable therapeutic efficacy in patients with SLE (76).
mTOR activation is responsible for skewing of T cell lineage specification in SLE
In SLE, there is activation of the interacting mTOR complexes, mTORC1 and 2. These play a critical role in lineage determination and antigen responsiveness within T-cells. mTOR controls lineage specification by transcriptional regulation in response to polarizing cytokines (77). mTORC1 promotes differentiation of Th1 and Th17 cells through activation of the transcription factors that drive their lineage commitment, T-bet and RORϒT, respectively (78, 79). In addition to driving Th1 specification, T-bet activation inhibits expression of the transcription factor eomesodermin, which prevents commitment of CD8+ T cells into memory cells, resulting in increased proportions of effector CD8+ T cells (80). In the absence of mTORC1, through genetic deletion or treatment with the mTORC1 allosteric inhibitor rapamycin, CD4+ T cells cannot polarize in response to cytokine stimulation (78, 81).
mTORC1 activation in SLE drives expansion of Th1 cells, Th17 cells, and CD4−CD8− T cells and inhibits T-cell lineage specification to Tregs by reduced expression of the key transcription factor Foxp3 (82)(Kato and Perl 2014). mTORC1 activation in CD4−CD8− T cells promotes production of the effector cytokine IL-4 (57) and reduces suppressor function of Tregs by decreased expression of the immunosuppressive molecules CTLA-4 and GATA3 (59). When mTOR is inhibited, Treg specification is also promoted by a release of inhibition on the Foxo1 and Foxo3a transcription factors, as well as promotes SMAD3 activation to drive lineage specification (81). mTORC2 activation in SLE promotes Th2 differentiation through transcriptional activation of the lineage-specific transcription factor GATA-3 (Figure 2). mTOR activity is measured through phosphorylation of the downstream targets, of which includes S6 kinase and 4E-BP1 for mTORC1 and protein kinase B (AKT) for mTORC2.
Figure 2. mTOR activation mediates pro-inflammatory T-cell lineage specification in SLE.
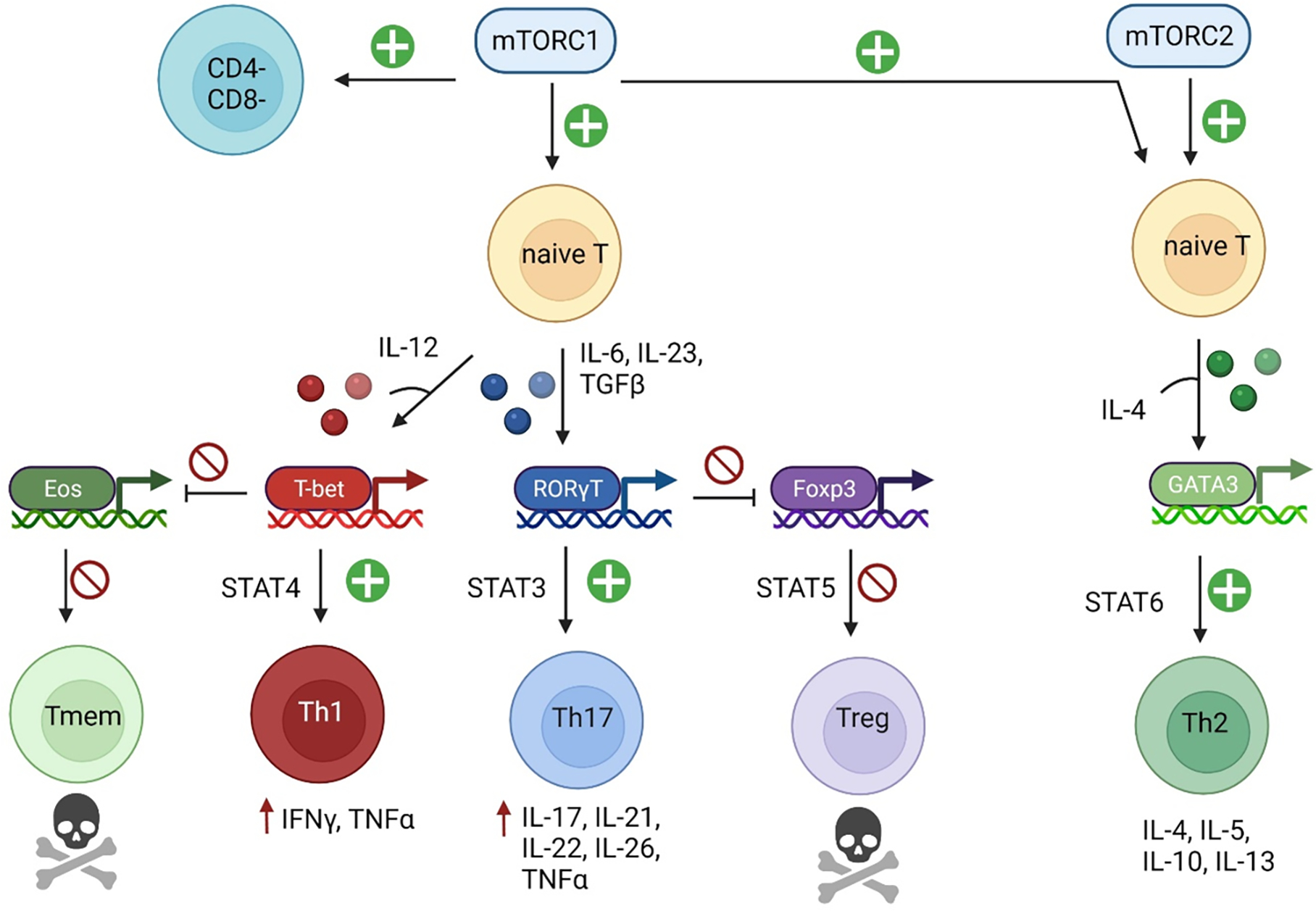
Activation of mTORC1 in naïve lupus T cells results in CD4+ T cell specification to the Th1 and Th17 lineages, through activation of lineage specific transcription factors in the presence of polarizing cytokines. This results in activation of JAK-STAT signaling and production of cytokines and other inflammatory mediators. T-bet expression during Th1 specification inhibits eomesodermin, impairing differentiation to the memory T cell lineage. Additionally, RORγT expression for Th17 specification results in reduced expression of Foxp3, inhibiting Treg development. mTORC2 activation promotes Th2 specification through the transcription factor GATA-3 in the presence of IL-4.
Metabolomic drivers of disease in SLE
Activation of the innate and adaptive immune system by the aforementioned cellular and soluble mediators ultimately results from defective metabolism within lymphocytes, resulting in a propensity of necrosis over apoptosis, which causes exposure to nucleic acids and other immune components. Within lymphocytes, there are defects in autophagy driven by activation of mTOR in lupus T cells (Figure 2). mTOR activation impacts CD4+ T cell activation through regulation of calcium fluxing and induces activation of the endocytic pathway (55, 83). Endocytic pathway components are upregulated in SLE, which include but are not limited to HRES-1/Rab4 and Rab5. HRES-1/Rab4 is an early endosomal GTPase that impacts T-cell activation through enhancing recycling of CD3 zeta chain, CD4, and CD2 adaptor protein on T-cells (84). Upregulation of these proteins increases T-cell activation and increased immunological synapse formation, leading to activation of partnering dendritic cells and/or B-lymphocytes.
Sustained T-cell activation induces exhaustion and activation-induced cell death within normal lymphocytes and is impaired in SLE. As a result, activated lymphocytes have a propensity to undergo necrotic cell death rather than apoptosis. HRES-1/Rab4 further affects this process by impairing autophagy through its direct interaction with Drp1, a protein that promotes mitochondrial fission (84). Deficient expression of this protein reduces turnover of mitochondria by autophagy (known as mitophagy). Inhibition of HRES-1/Rab4 (and other GTPases that undergo the post-translational modification by geranyl-geranylation) through treatment with a geranylgeranyl transferase inhibitor prevents nephritis in lupus-prone mice (84). Mitochondrial turnover via autophagy, specifically termed mitophagy, is critical in maintaining mitochondrial function. Impaired mitophagy results in enlarged mitochondria (or megamitochondria), with impaired oxidative phosphorylation. Impaired mitophagy is associated with overall increased autophagy in SLE. Impaired mitophagy enhances overall mitochondrial mass and also contributes to mitochondrial hyperpolarization, ATP depletion, and oxidative stress (85, 86). Mitochondrial-derived reactive oxygen species activate the innate immune system by driving formation of neutrophil extracellular traps in response to injury (87). Autoantibodies against SOD2 have been identified in glomerulonephritis, which may impair its function through being ensnared in immune complexes (88, 89). Reduction of oxidative stress with antioxidants is a therapeutic target in SLE (53). An overview of lupus pathogenesis is shown in Figure 3.
Figure 3. Overview of changes in autophagy during lupus pathogenesis.
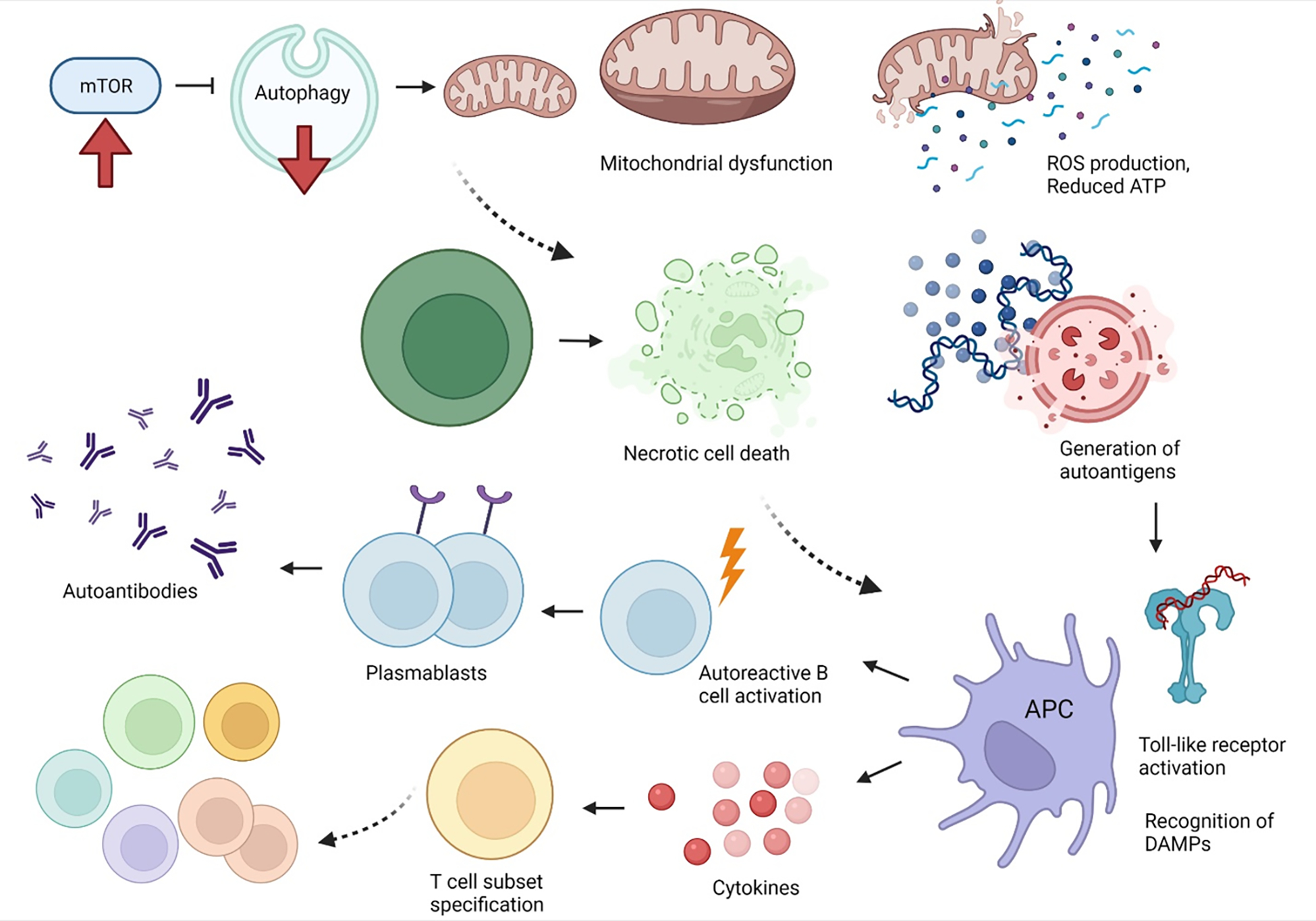
mTOR activation with subsequent inhibition of autophagy is central to lupus pathogenesis. Deficient autophagy, including autophagic turnover of mitochondria (mitophagy), results in mitochondrial dysfunction. Mitochondrial dysfunction is characterized by increased mitochondrial mass, mitochondrial hyperpolarization, increased production of reactive oxygen species, and reduced production of ATP. Upon sustained T-cell activation, mitochondrial dysfunction predisposes to necrotic cell death, rather than apoptosis. Released nucleic acids and other intracellular materials can serve as damage-associated molecular patterns (DAMPs) or autoantigens, inducing toll-like receptor activation within antigen-presenting cells. Activation of antigen presenting cells results in activation of autoreactive B cells which can differentiate into plasmablasts to produce autoantibodies. Additionally, cytokine production can preserve CD4−CD8− T cells and skew T-cell subset specification towards inflammatory Th1, Th2, Th17 cells, rather than Tregs or memory T cells.
Autophagy in SLE
Autophagy is a fundamental process of cell survival that degrades and recycles intracellular proteins and organelles, including mitochondria (Figure 3). The regular turnover of mitochondria is called mitophagy (90). Autophagy encompasses macroautophagy (responsible for degradation of organelles through the endocytic pathway), microautophagy (degradation of individual proteins and protein complexes), chaperone-mediated autophagy, and LC3-associated phagocytosis (LAP). All pathways converge on degradation of intracellular contents in the lysosome.
There are multiple mechanisms through which autophagy affects the innate and adaptive immune response and becomes dysregulated in autoimmunity. Autophagy influences the multiple components of the innate immune system, including recognition of non-selfantigens in conjunction with pattern recognition receptors, regulation of inflammasomes, direct microbial killing, and antigen processing for presentation on MHC molecules (91, 92). Autophagy impacts immune cell differentiation, as cellular differentiation requires proteolysis to remove and recycle bulk proteins for translation of components required for cellular proliferation and specification. Induction of autophagy is critical during monocyte-to-macrophage differentiation to recycle intracellular components and avoid cell death via caspase activation and apoptosis (93). Activation and proliferation of both T- and B-cells is regulated by autophagy (92). Autophagy also negatively regulates production of type I interferon (94). Immunoglobulin production by plasma cells requires autophagy to reduce endoplasmic reticulum stress that occurs during translation of immunoglobulins (93).
The exact role of autophagy in LN is not fully understood. There are pleiotropic effects of autophagy in SLE pathogenesis and changes can be cell-type specific (95, 96). Although the underlying pathways are diverse, the majority of data suggests that autophagy is overall deficient in SLE regulatory T cells and in memory CD8+ T cells, with autophagic induction being therapeutic. However, activation of autophagy can occur Th1 cells, of which produce IFN gamma. Autophagy is also activated in macrophages derived from peripheral blood mononuclear cells from SLE patients and in macrophages within the spleen and kidneys of lupus-prone mice (97). Autophagy can be activated as a normal physiologic response to injury as a salvage mechanism to regulate cellular metabolism, however hyperactivation of autophagy pathways can be pathogenic as it results in an increase in type 2 cell death (98). This can occur in effector CD4+ cells, including Th1, Th2, and Th17 cell populations, promoting production of pro-inflammatory cytokines, but also being responsible for increased activation-induced cell death.
Dysregulation of autophagy in SLE has multiple consequences and contributes to disease development. Autophagy is required for the clearance of cell debris, protein aggregates, and abnormal mitochondria, of which is impaired in SLE and serves as a source of autoantigens. Tolerance to self-antigens is impaired by reduced peripheral tolerance due to the inability of Tregs to activate autophagy due to mTOR activation (59)and increased CD4−CD8− T cells which results in further Treg depletion. Cell debris stimulate dendritic cells, of which show increased autophagy, which promotes processing and presentation of self-peptides on MHC class II molecules as well as production of type I IFN. Self-DNA from cell debris can induce neutrophil autophagy and the process of NETosis. Production of neutrophil extracellular traps (NETs) serve as a trigger for formation of immune complexes against self-DNA (ANA). Persistence of antigens occurs as degradation of NETs is impaired in SLE patients and is associated with autophagy (99, 100). These trigger toll-like receptor signaling and activation of plasmacytoid dendritic cells to further produce type I IFN (101). NETs promote activation of autoreactive B cells, of which can differentiate into plasmablasts and secrete autoantibodies. Autoreactive B cells are also activated by T-cell help, for which Th1, Th2, and Th17 cells demonstrate increased autophagy. Autophagy is activated in circulating macrophages in SLE patients, which could further enhance antigen presentation (97). In lupus-prone mice, autophagic induction is present in macrophages within the kidney. This is thought to be injurious, as adoptive transfer of autophagy-deficient Beclin-1 knockout macrophages is protective, with reduced production of anti-dsDNA antibodies, decreased proteinuria, and reduced disease severity of LN (97). While autophagy is activated in multiple innate immune cells, it is deficient in Tregs and CD8+ memory cells, largely as a consequence of mTOR activation.
Mulitple intracellular mechanisms regulate autophagy within immune cells and show differences in SLE. mTOR activation acts as an upstream inhibitor on autophagy through phosphorylation of the autophagy-activating protein ULK1 (102). Genetic variants in genes encoding autophagy related proteins and other key components of the autophagy machinery have an increased prevalence in SLE patients. LC3-associated phagocytosis may be deficient, which impairs clearance of dying cells and perpetuate autoimmunity in murine models (103, 104). Additionally, reduced mitophagy promotes mitochondrial dysfunction, production of reactive oxygen species, and cell death. Components released from damage mitochondria activate the innate immune response and serve as DAMPs. These include mitochondrial DNA, N-formyl-peptides, and the phospholipid cardiolipin (87). Mitophagy can be controlled by both LAP-dependent and independent mechanisms. Both the processes of LAP and mitophagy (via LAP or macroautophagy) are critical to limit autoantigen production. In addition to limiting autoantigen production, autophagy protects against autoimmunity through controlling positive and negative selection of T cells in the thymus, reducing numbers of autoreactive T cells in the periphery (105). Chaperone-mediated autophagy defects may also play a role in SLE pathogenesis, as activation of chaperone-mediated autophagy increases survival in lupus-prone MRL/lpr mice.
Autophagic control of T-cell lineage specification
Energy metabolism, regulated by mTOR and autophagy, is important and a rate-limited step in T-cell differentiation, activation, and effector functions. Autophagy controls immunometabolism through affecting the shift being anaerobic respiration/glycolysis and oxidative phosphorylation (106). Th2 and Th17 cells primarily utilize glycolysis and are not as dependent on oxidative phosphorylation and fatty acid oxidation as regulatory and memory T cells (107). Tregs and memory T cells have high mitochondrial content to support their metabolism. CD4+ T cells frequently undergo lineage specification rather than true differentiation and can be metabolically reprogrammed into a separate lineage. Th17 cells can reprogram into Tregs through inhibiting glycolysis, which has been demonstrated in studies using 2-deoxyglucose to suppress glycolytic metabolism (108).
Autophagy is critical for generation and function of Tregs (109) as well as memory T cells and is deficient in SLE (Figure 4). Both Tregs and CD8+ memory T cells have high energy demands for maintenance and effector function and require oxidative metabolism. mTOR activation reduces both numbers and function of Tregs and their associated immune suppression due to inhibition of autophagy and increasing glycolysis (59). CD8+ memory T cells also require autophagy, as in its absence, there is reduced mitochondrial homeostasis and increased cell death, leading to depletion of this cell population (110). In SLE, memory T cells are depleted, have an increased propensity to undergo apoptosis, and reduced proliferative capacity (111). Type I interferon, although overall promotes autophagy, can induce increased consumption of NAD+ and result in impaired mitochondrial respiration and decrease CD8+ T cell viability (112). An ‘interferon’ signature results in reduced expression of mitochondrial derived genes and metabolism in both CD4+ and CD8+ T cells from SLE patients (112).
Figure 4.
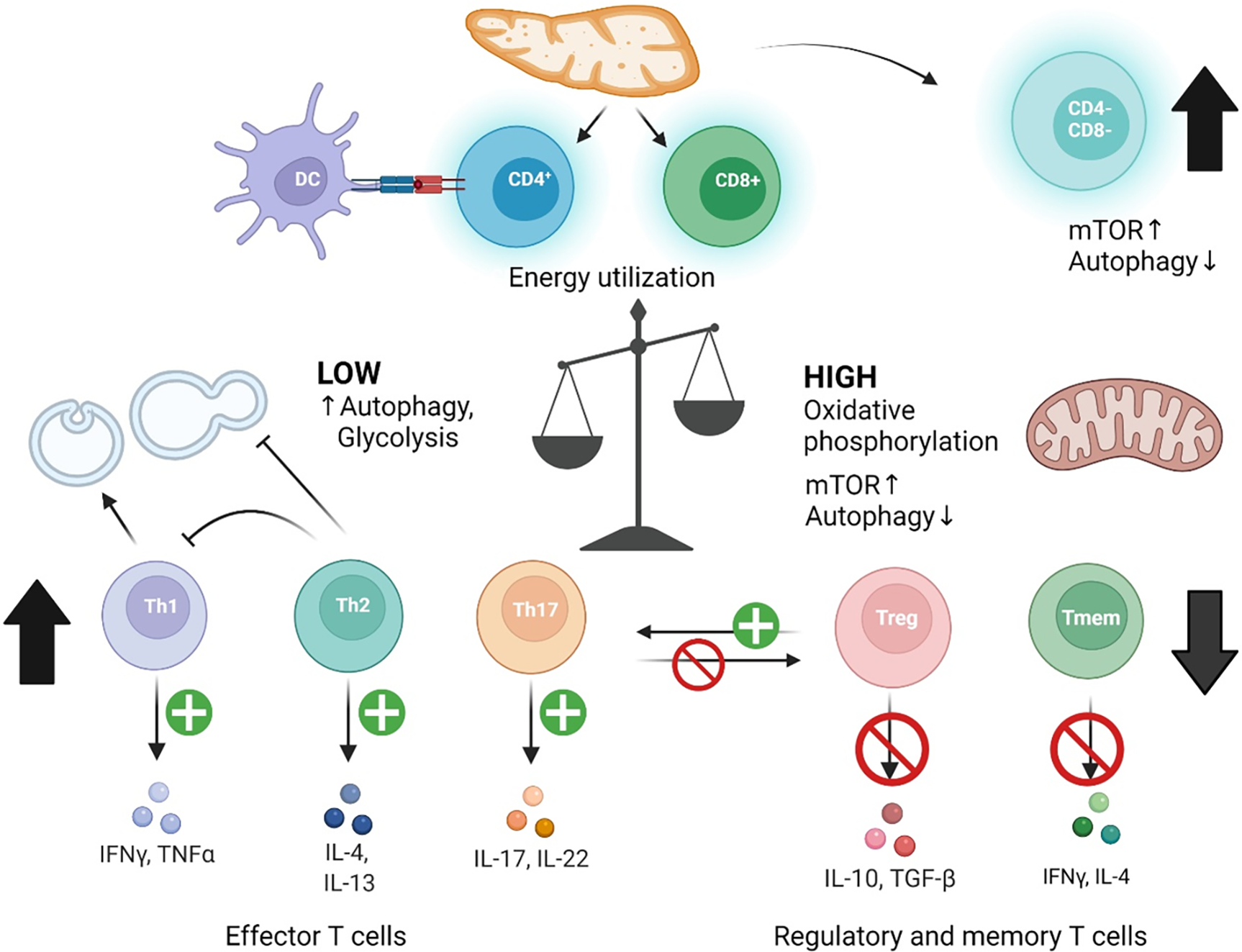
Role of autophagy in pro-inflammatory immune cell specification in SLE. Immune cell subsets have differential requirements for autophagy in activation and maintenance, of which is primarily regulated by mTOR activation and is dependent on energy metabolism. CD4−CD8− T cells of which escape thymic selection show increased mTOR activity and deficient autophagy. CD4+ Tregs and CD8+ memory T cells also have increased mTOR activity, deficient autophagy, and are depleted in SLE. Tregs are additionally depleted through increased differentiation into Th17 cells. Dendritic cells have increased autophagy that increases antigen presentation to naïve CD4+ T cells. Th subsets with low energy requirements utilize glycolysis and turnover cellular components by autophagy. Th1 and Th17 cells show activated autophagy. Th2 cells produce IL-4 and IL-13 which in turn inhibits autophagy and IFNγ production by Th1 cells.
The Th2 cytokines IL-4 and IL-13 inhibit autophagy (113), even in the presence of IFNγ. Th2 differentiation, of which leads to production of IL-4 and IL-13, is regulated by the autophagy gene ATG16L1 (114). Th1 cell polarization and production of IFN-γ promotes autophagy (113, 115). Autophagy is activated in lupus macrophages, both circulating and within the kidney (97). This may enhance antigen presentation and production of pro-inflammatory cytokines, of which include IL-6 and TNFα (97, 116). In lupus-prone mice, autophagy-related genes including ATG5, ATG12, and Beclin-1 are upregulated within splenic and renal macrophages. Similarly, circulating macrophages in SLE patients have similar signatures (97). Therefore, induction of autophagy is cell-type specific and can be dependent on differences energy utilization between immune cell subsets (Figure 4).
Mutations in autophagy-related proteins as possible lupus susceptibility genes
Autophagy-related genes (ATG) encode key components of the autophagic degradation machinery and are polymorphic in patients with SLE (117). ATG proteins initiate autophagy through nucleation of an ‘isolation membrane’ from the endoplasmic reticulum-mitochondrial junction (118) to generate a phagophore (Figures 1–3), which later develops a double-membrane autophagosome that engulfs intracellular contents to be degraded within lysosomes (119). Genome-wide association studies (GWAS) have identified polymorphisms in autophagy-related genes, which may contribute to disease pathogenesis. Polymorphisms of ATG5, a key phagophore component (120), have been implicated in both Caucasian and Asian SLE cohorts (121–123). The risk allele variants of PR domain zinc finger protein 1 (PRDM1) and ATG5 in B cells have been associated with the increased expression of ATG5, which increases IFN-α signature genes (124). In an Italian cohort ATG5, risk alleles were associated with increased anti-dsDNA antibodies and development of LN (125). Other SLE susceptibility loci include variants in further autophagy related genes: ATG7, DNA damage-regulated autophagy modulator 1 (DRAM1), Apolipoprotein L1 (APOL1), C-type lectin domain family 16 (CLECK16A), ATG16L2, immunity-related GTPase family M (IRGM) and myotubularin-related protein 3 (MTMR3) (121). Further data will be required to determine pathogenicity of these variants, as most are not found in the coding regions and are intronic, as well as have not been investigated in animal models.
Mutations of TLRs have also been identified in GWAS, which are similarly localized to endosomes and critical to antigen presentation (126). Some TLR mutations result in increased protein expression in SLE patients. For example, TLR7 is over-expressed in antigen-presenting cells, responds to nucleic acid antigens (RNA), increases differentiation of B cells to plasmablasts, and shows overexpression in plasmacytoid dendritic cells that secrete type I interferon (127, 128). Polymorphisms in APOL1 in African American patients induce a toxic gain of function due to homozygosity or compound heterozygosity of G1 and G2 risk alleles, which are triggered by increased IFN levels in SLE (129–131). The presence of APOL1 risk alleles is associated with poor renal prognosis and concurrent collapsing glomerulopathy, a severe form of focal segmental glomerulosclerosis (130). Notably, APOL1 can induce autophagic cell death in an ATG5 and ATG7-dependent manner (132). Deficiency in either of these components of the autophagy machinery can impact autophagic cell death by APOL1, which could potentially favor necrosis over apoptosis. Predisposition to cell death via necrosis is considered highly inflammatory in SLE (57, 86, 133).
Another genetic mutation in SLE that impacts autophagy is in the gene Def6, which leads to altered assembly of the protein complex raptor-p62-TRAF6, an upstream regulator of mTOR activation (134). In addition to inhibition of autophagy on mTOR activation, Def6 controls the transcription factor Bcl6 in follicular helper T cells (TFH), resulting in TFH proliferation, which provides B cell help in autoimmune responses (134).
The activation of mTOR is regulated by tuberous sclerosis proteins 1 (TSC1) and 2 (TSC2) (Figure 5)(58). Severe SLE with class IV nephritis has been documented in two case reports of patients with tuberous sclerosis (137, 138). mTOR activation also resulted in diminished FoxP3 expression, implicating Treg depletion patients with LN (139). Mutations of TSC1 and TSC2 have also been associated with lymphangioleiomyomatosis (LAM), with recent case reports of LAM in SLE (140–142). In two of these cases, a diagnosis of SLE preceded the onset of LAM by more than a decade (141, 143), thus patients with long-standing SLE should be monitored for the development of LAM and other conditions with mTOR activation.
Figure 5. Defective canonical autophagy underlies pathogenic cell-to-cell communication in SLE.
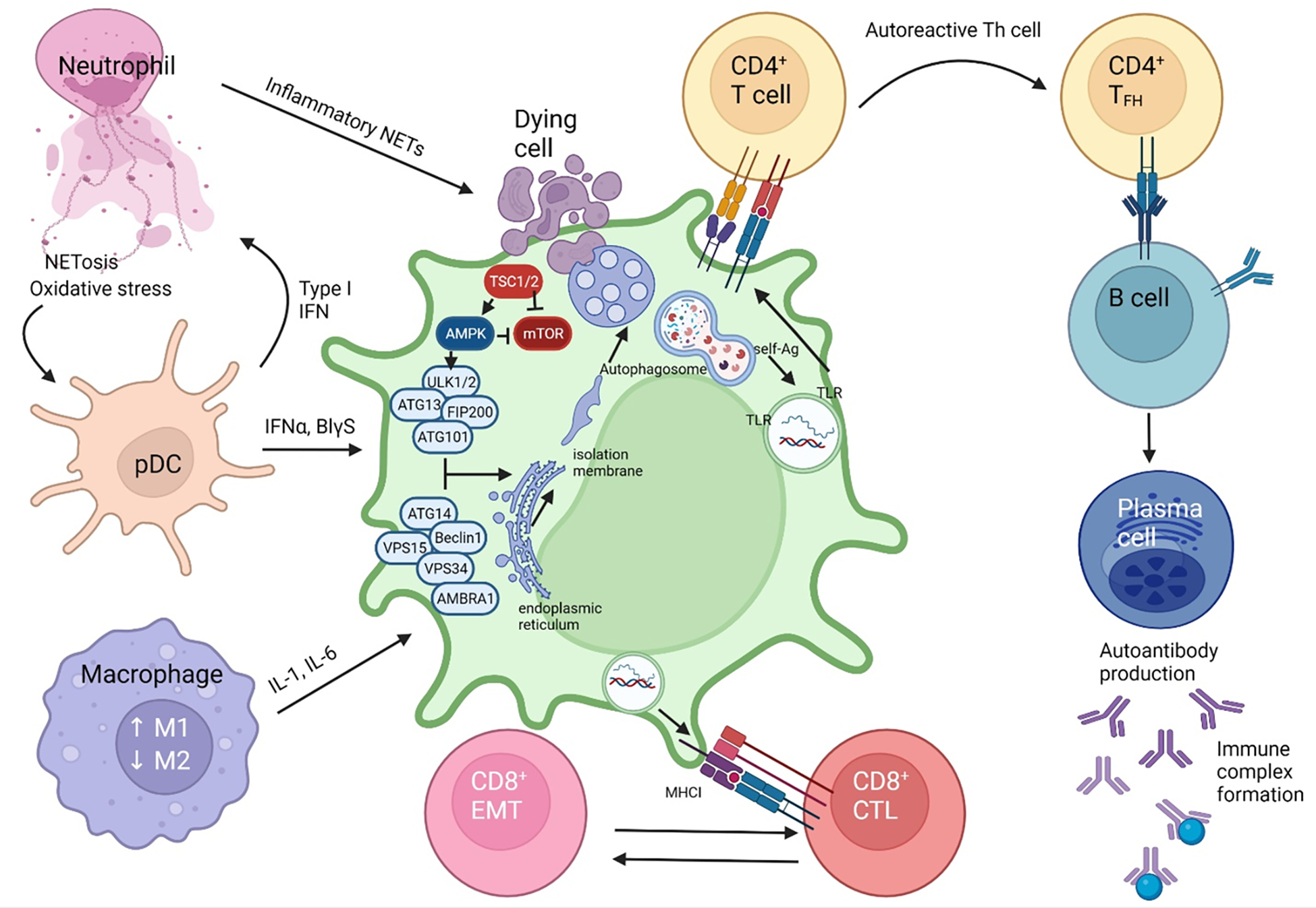
In a susceptible host, genetic material from apoptotic cells primes an autoreactive immune response. NETosis and oxidative activity increases in neutrophils. The NET DNA detection by pDC causes an increase in type 1 IFN signalling, which is a hallmark of SLE disease (135, 136).. IFNα signalling, along with the increased secretion of pro-inflammatory cytokines IL-6, IL-17, IL-18, IL-23 and type 1 IFN stimulates native dendritic cells. Homeostatic balance between macrophages shift to favor the M1 differentiation with a resultant pro-inflammatory cascade. Endocytosis of a dying cell, in the susceptible host with defective components of autophagy will result in the non-fusion of the phagosome with the lysosome. The pre-initiation complex of autophagosome formation is comprised of ULK1/2, ATG13, FIP200 and ATG101. Interaction with the complex of VPS15, ATG14, BECN1, VPS34 and AMBRA1 generates the isolation membrane from the endoplasmic reticulum or mitochondria which is required for phagosome formation (90). Defects in BECN1, CYBB/NOX2, RUBCN, ATG7, ATG5 are associated with reduced autophagosome formation and defective clearance of dying cells that trigger inflammation in SLE and lupus nephritis (90, 122, 123, 125). AMPK activates while mTOR inhibits autophagy via phosphorylation of ULK1. In turn, TSC1 and TSC2 restrain mTOR activation. The inability to complete autophagy into the final autolysosome step causes the release of self-antigens which are then presented by antigen presenting cells to CD4+ helper T cells and CD8+ cytotoxic T cells via MHC class II and MHC class I, respectively. The result is the stimulation of the adaptive immune system towards autoreactive helper T-cells and cytotoxic T-cells which lead to tissue damage. Primed CD4+ T cells form and immune synapse with and release cytokines, IL-4 and IL-21 to activate B cells which differentiate to immunoglobulin-secreting plasma cells. FN: Interferon, NET: Neutrophil extracellular traps, ER: endoplasmic reticulum, DNA: Deoxyribonucleic acid, pDC: plasmacytoid dentritic cell, CD28 & B7: costimulatory signals, TLR: toll like receptor, BCR: B-cell receptor.
AMPK inhibition and mTOR activation impact autophagy and autoimmunity in lupus-prone mice
The master regulators of autophagy include 5’ adenosine monophosphate activated protein kinase (AMPK), of which activates autophagy and mTOR, a negative regulator (Figure 5). mTOR activity is increased in lymphocytes from SLE patients, which is partly activated through an increase in intracellular nitric oxide levels and impaired mitochondrial homeostasis (144). mTOR activation results in phosphorylation of S6 kinase and 4E-BP1 (145) with both proteins showing increased phosphorylation in SLE (84). mTOR acts as a key negative regulator of autophagy through phosphorylation of the autophagy-initiating proteins Unc-51 like autophagy activating kinases 1/2 (ULK1/ULK2), which inhibits their function (102). This results in reduced transcription of multiple genes expressed within autophagosomes, primarily driven by inhibition of transcription factor EB, a critical component of lysosomal biogenesis (146). mTOR activation impairs degradation and turnover of long-lived proteins through both autophagy and through inhibition of ubiquitin ligases impairing proteolysis via the ubiquitin-proteasome pathway (147). However, inhibition of ULK1 alone is insufficient to induce autoimmunity, as ULK1-deficient mice do not develop a lupus-like disease (103), despite a defect in a key component initiating macroautophagy. mTOR activation induces B-cell class switching (148). mTOR also promotes B-cell maturation, as treatment with rapamycin or genetic absence of the upstream activator regulatory-associated protein of mTOR (Raptor) results in a lack of B cells in the periphery due to a developmental block at the pre-B cell phase (149).
Conversely, AMPK phosphorylates ULK1 at a separate site to promote autophagy through activation of the autophagy-promoting phosphatidylinositol vacuolar protein sorting 34 (VPS34) complex. AMPK senses low ATP states and serves to promote oxidative phosphorylation, acting at mitochondrial complex I of the electron transport chain (150). Mitochondrial complex I activity is abnormal in lupus (151), although AMPK levels have not been shown to be depleted. Activation of AMPK is protective against inflammation in MRL/lpr lupus prone mice, reducing production of the pro-inflammatory mediators IL-6, inducible nitric oxide synthase (iNOS), cyclooxygenase 2 (COX2), and IFN-γ by treatment with an AMPK agonist 4-imidazole carboxamide riboside (AICAR)(152). Metformin is another agonist of AMPK and inhibits mTOR. Metformin treatment within Roquin san/san mice reduced production of dsDNA antibodies, prevented development of hepatitis and nephritis, and resulted in a favorable skewing of CD4+ T cell differentiation with increased Tregs, reduced Th17 cells, and reduction of follicular helper T cells that participate in germinal center formation to stimulate antibody production by B cells (153). Activation of AMPK resulted in inhibition of mTOR and STAT3 signaling that impaired differentiation of B cells into plasma cells (153). Metformin should clinical efficacy in clinical trials of SLE patients (154).
LC3-associated phagocytosis in autoimmunity
LC3-associated phagocytosis (LAP) is a form of autophagy that is initiated with phagocytosis and lipidation of the protein LC3 on the autophagosomal membrane (155). Lipidation of LC3, from the non-conjugated form LC3-I to the lipid conjugated form LC3-II results in elongation of autophagosomal membranes. Within this pathway, phagosomes engulf particles and dying cells and then recruit components of the autophagic machinery resulting in maturation of the phagosome and digestion of its contents. Activation of this pathway can be measured by lipidation of LC3 by a LC3-II/LC3-I ratio, rather than total LC3. LC3 levels are increased in T-cells of SLE patients, however the LC3-II/LC3-I ratio is depressed. In addition to LC3, critical components of this pathway include ATG5, ATG7, and Beclin-1 (BECN1). BECN1 levels are decreased in SLE T cells (Figure 6) (84).
Figure 6. Molecular checkpoints of distorted autophagy pathways in lupus nephritis.
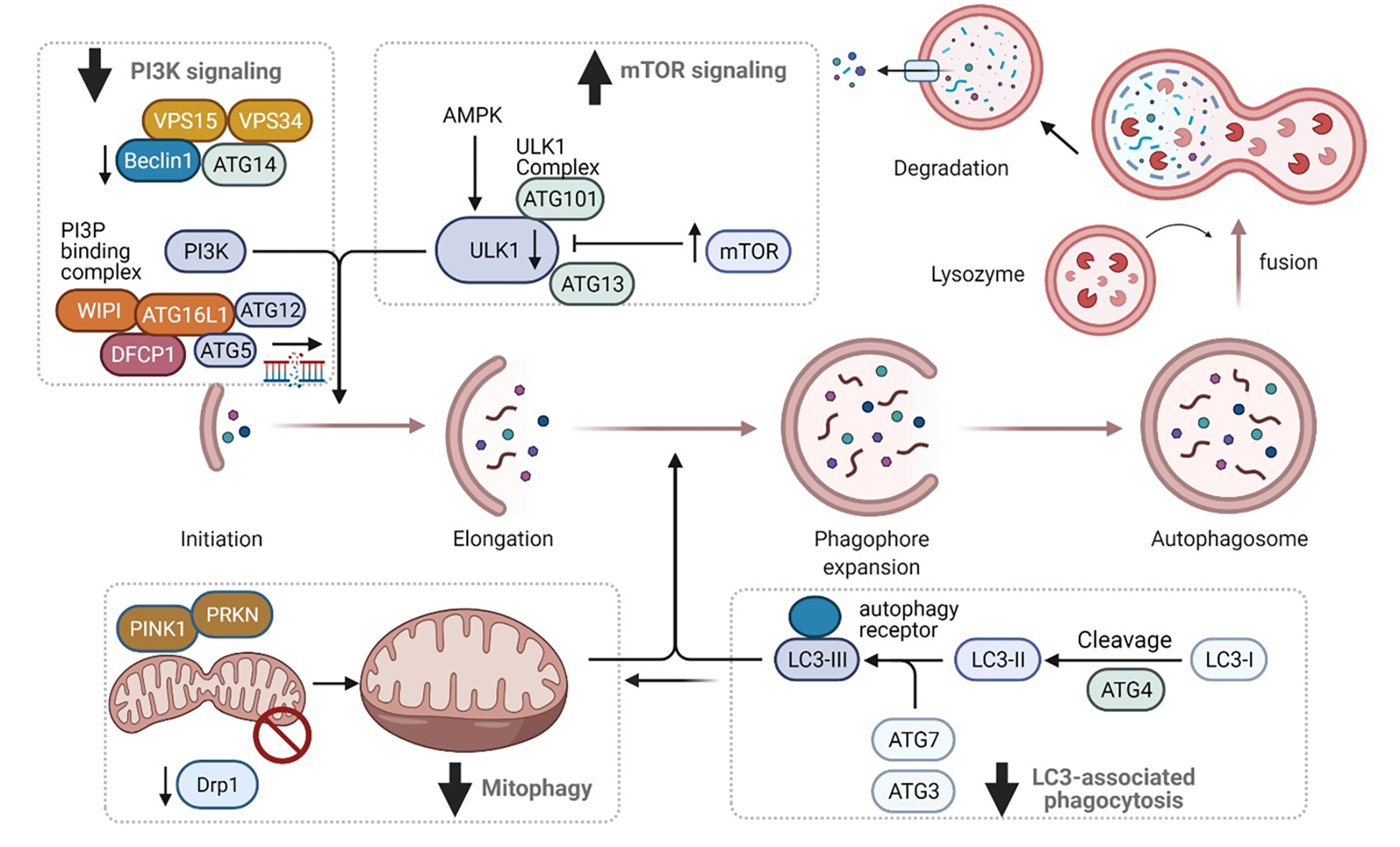
The process and main regulatory mechanisms of autophagy. The process of autophagy starts with cytoplasmic material being engulfed by double membranes with the formation of cup-like structure called the phagophore to the conversion into double membrane vesicles called the autophagosomes. The mTOR pathway and the AMP- activated Kinase (AMPK) are key determinants of autophagy activation. The process if finely regulated by autophagy-related proteins (ATG) that assemble into several complexes : Unc-51-Like Kinase (ULK1), PI3K nucleation complex and the phosphatidinol 3 phosphate binding complex. Together, they direct the distribution of the autophagy machinery allowing the formation of the autophagosome. LC3 is cleaved into LC3-I by ATG4 which is then conjugated to phosphatidylethanolamine to form LC3-II. This complex gets incorporated into autophagosome and binds with receptors harboring LC3 containing motifs. Both macroautophagy and LC3-associated autophagy can impact mitophagy, of which is largely deficient in SLE, but dependent on the energy requirements of effector cell types.
LAP is a critical pathway for the uptake of dead cells from apoptotic, necrotic, or inflammasome-mediated receptor-interacting serine/threonine protein kinase 3 (RIPK3-directed) cell death (103). This is triggered through activation of the T-cell immunoglobulin and mucin domain containing 4 (TIM4) receptor by phosphatidylserine residues displayed on the membranes of dying cells. A defect in the clearance of dying cells increases exposure to self-antigens has been widely proposed as an underlying driver of SLE pathogenesis, which could be due to deficient LAP. Reduced LAP results in increased production of pro-inflammatory cytokines, including IL-1β (which activates inflammasome signaling), IL-6, IL-12, and interferon gamma-induced protein 10 (IP-10). Additionally, inhibition of LAP results in deficient IL-10 production, an important regulatory cytokine (104). IL-10 deficiency promotes autoimmune responses, as IL-10 deficient mice develop an accelerated lupus-like disease. Similarly, IL-10 production by B cells suppresses inflammation (156). Importantly, the production of IL-2 (157), IFN-γ (57, 158), and TNF-α are reduced in SLE (159–161).
In mouse models, deficiency of LAP specific and/or core autophagy genes that are implicated in formation of the isolation membrane and phagophore (BECN1, NADPH oxidase 2 (CYBB/NOX2), rubicon autophagy regulator (RUBCN), ATG7, ATG5, ATG3, ATG12, or ATG16) results in diminished clearance of dying cells and development of an auto-inflammatory lupus-like disease (104). Mice deficient in these core autophagy genes have low body weight, accumulate activate CD8+ T-cells, develop auto-antibodies including ANA and anti-dsDNA, show an increase in IFN-α signature gene expression, and develop nephritis. Plasmablast differentiation is aberrant in ATG7-knockout mice, which could be a potential mechanism into auto-antibody production (162, 163). Similarly, autophagy inhibition in human B cells prevents differentiation into plasmablasts (131). Hyper-activation of ATG5 and ATG7 and activation of mTORC1 appear to be drivers of plasma cell development in SLE (164) (Figure 6).
Prior to being studied in the setting of autoimmunity, LAP was found to be a critical component of clearance of dying proximal tubular epithelial cells during acute tubular injury. Apoptotic cell phagocytosis is initiated by the kidney injury molecule 1 and T- cell IgG and mucin containing 1 proteins (KIM-1/TIM-1). In a mouse model that is kidney-injury molecule 1 (KIM-1) deficient, apoptotic cell clearance by proximal tubules is reduced, which results in increased pro-inflammatory cytokine secretion, increased tissue macrophages, and worsening of acute kidney injury (165).
Mitophagy is in part regulated by LAP. The LC3 autophagosomal membrane is derived from mitochondria. Co-localization of LC3 with the early endosomal component HRES-1/Rab4 increases upon mTOR inhibition and results in increased formation of autophagosomes derived from the mitochondrial membranes through activation of the endocytic machinery (166). High interferon levels in SLE prevent degradation of mitochondrial DNA within lupus monocytes (167), of which occurs due to activation of the stimulation of interferon-genes protein-mediated DNA sensing pathway (STING). Another key regulator of mitophagy is PTEN-induced kinase 1 (PINK1), which interacts with the mitophagy-inducing protein parkin (PRKN). PINK1 responds to mitochondrial hyperpolarization through recruiting PRKN to stimulate mitophagy. Genetic ablation of PINK1 or PRKN in mice results in release of mitochondrial DNA and other mitochondrial-derived self-antigens (Figure 6), resulting in activation of the STING and increased production of type I interferon (168).
LC3-associated phagocytosis may be regulated upstream through small non-coding RNAs regulating transcription. microRNA (miR)-20a is reduced in SLE patients compared to healthy controls (169). miR-20a levels are further depressed in patients with LN compared to SLE patients without kidney involvement (170). Transplantation of adipose-derived stem cells that over-express miR-20a in MRL/lpr lupus prone mice protects against development of LN (171). Overexpression of miR-20a results in inhibition of mTOR and Akt, releasing these ‘negative breaks on autophagy’. As a result, there is increased LC3 lipidation (increased LC3-II/LC3-I ratio) and consumption of p62, consistent with LAP activation (171). These data suggest that LAP has a protective mechanism against the development of SLE. Activation and deregulation of autophagy are seen in SLE patients, which is evident from autophagy-resistant T-cells from SLE patients that preferentially express negative regulators of autophagy and increased apoptosis (172). An overview of autophagic pathways in SLE are shown in Figure 6.
Autophagy is protective within podocytes and required for recovery from autoantibody-induced podocyte injury
Autoantibodies from sera from lupus patients, type I interferon, and reactive oxygen species (ROS) all result in destruction of the podocyte membrane by redistribution of the slit diaphragm protein podocin (Figure 7). As a result, an induction of autophagy occurs as a natural cellular response. Similarly, podocyte exposure to anti-DNA antibodies increases TLR9 on endosomes for immune complex clearance (173). Impaired autophagy induction results in podocyte injury (124). Inhibition of autophagy in vitro by treatment with bafilomycin A or MG132 in podocytes exposed to anti-dsDNA antibodies results in accumulation of immunoglobulin aggregates and reduced podocyte survival (172).
Figure 7. mTOR-dependent changes in autophagy involve renal and infiltrating-inflammatory cells in patients with lupus nephritis.
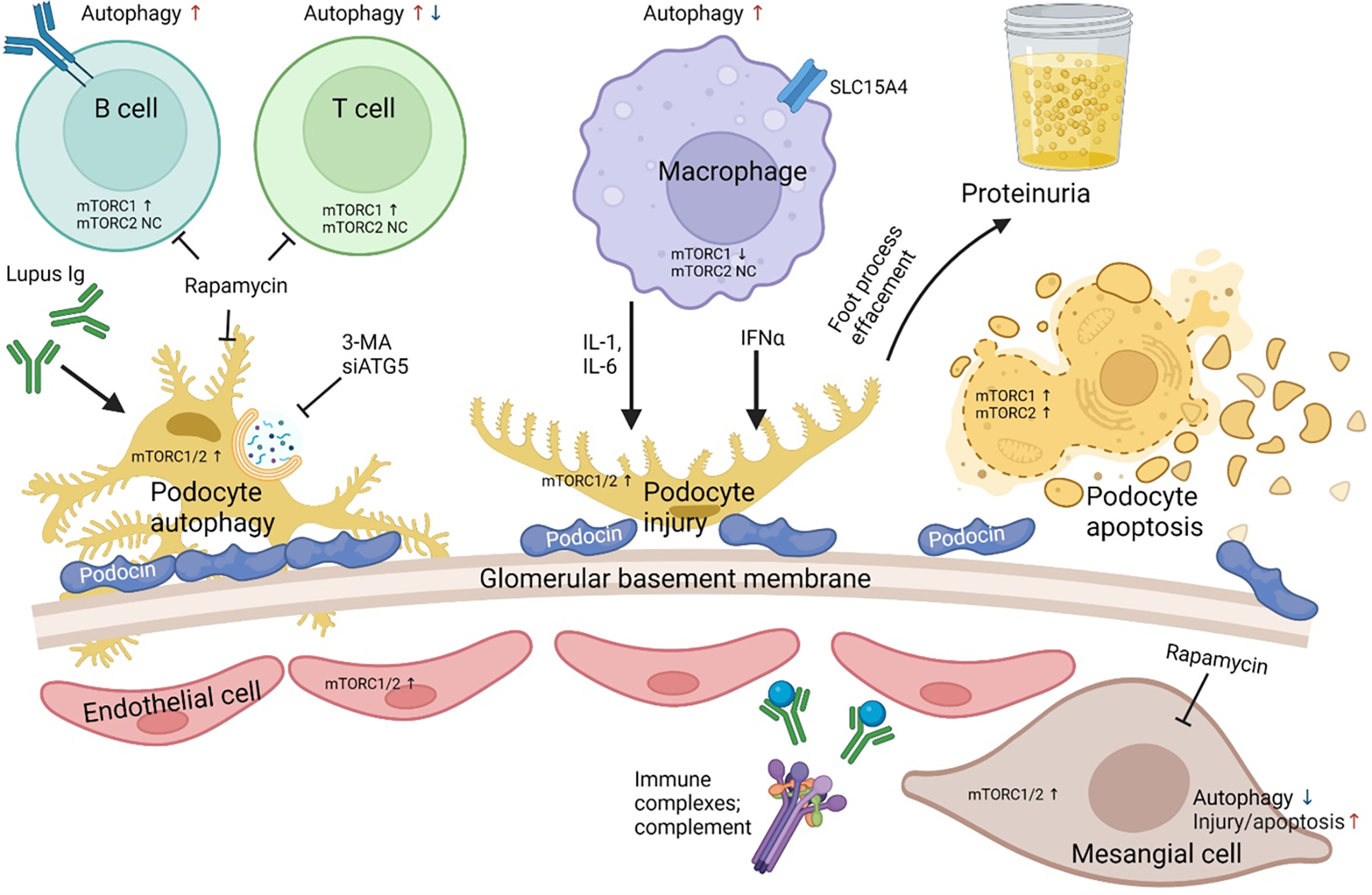
Inhibition of ATG5 by small interfering RNA and 3-MA inhibits autophagy in podocytes resulting in an aggravation of podocyte apoptosis and injury. Sera obtained from lupus nephritis patients demonstrated an increase in podocin by IFNα and an increase in podocyte injury and apoptosis by IgG. The combined cellular injury and podocyte foot process effacement leads to increase in proteinuria. Rapamycin which is a potent inhibitor of mTOR induces autophagy in such patients and reconstitute the podocyte foot processes and diminishes podocyte apoptosis leading to a decrease in albumin flux and proteinuria. mTORC1, mTORC2 and autophagy is skewed in a cell type-specific manner within the innate and adaptive immune system of SLE patients (58, 59, 178, 179).
In murine lupus nephritis, treatment with the proliferation inhibitor Tris DBA reduced kidney inflammation through an increase in autophagy, suppression of inflammasome signaling, and through activation of Tregs (174). This reduced further T-cell activation through reduced priming of dendritic cells and Treg-mediated immune suppression (174). Rapamycin reduced albumin flux in podocytes through increasing nephrin and podocin expression, supporting renoprotection (175). Corticosteroids also are renoprotective through promoting autophagy, of which inhibits calcium signaling and resulting activation or proliferation of T-lymphocytes (176).
mTORC1 and mTORC2 are markedly activated that may underlie diminished autophagy in podocytes (Figure 7), mesangial cells, endothelial cells and tubular epithelial cells of LN patients as compared with those with minimal change disease (MCD) or normal controls (NC)(177). Glomerular mTORC1 activation is higher in LN patients compared with diabetic nephropathy (DN) patients. mTORC1, but not mTORC2, activation strongly correlates with serum albumin, complement C3, proteinuria, and the following pathological histological biomarkers of LN: crescent formation, interstitial inflammation and fibrosis. Moreover, mTORC1 activation is a prognostic marker in LN patients (177).
Novel targets for treatment of LN based on the disease pathophysiology
Traditional treatments of lupus nephritis are non-specific and include agents such as hydroxychloroquine, cyclophosphamide, mycophenolate mofetil and steroids with overall immune suppression and off-target effects (180). High dose steroids in conjunction with the cyclic alkylating agent cyclophosphamide are often used in the treatment of proliferative lupus nephritis and reduces risk of progression to end-stage kidney disease (181). Following this induction therapy, long-term maintenance immunosuppression is often required to avoid relapse, typicaly withmycophenolate mofetil (MMF), azathioprine, and prednisone (182).
Calcineurin inhibitors (CNI), such as tacrolimus and cyclosporine, may also be used adjunctively in treatment of LN. CNIs are therapeutic in LN through two main mechanisms: through inhibition of calcineurin-related cytokine activation (reducing levels of IL-2, TNF-alpha, and IFN-gamma) and through stabilizing the podocyte cytoskeleton to decrease proteinuria (183). Tacrolimus treatment in conjunction to MMF shows similar efficacy, but reduced toxicities, than IV cyclophosphamide (184). Cyclosporine and other CNI have adverse renal effects by inhibiting prostacyclin-dependent blood flow (185, 186). New CNIs are also under investigation. In a recent double-blind placebo-controlled clinical trial of 357 patients, there was a 17% increase in renal response in patients receiving voclosporin, compared to placebo (AURORA trial, clinicaltrials.gov identifier: NCT03021499)(187).
The aforementioned cellular and metabolic pathways can be exploited for development of novel therapies in LN (Figure 8). Antimalarial drugs, such as hydroxychloroquine, target the endocytic/autophagic machinery through altering endosome acidification. SLE patients treated with hydroxychloroquine had a reduced frequency diffuse lupus nephritis, required lower steroid doses, and had lower disease activity (188). For SLE patients with MLN, hydroxychloroquine improved rates of complete remission at one year (189). Reduction of oxidative stress through treatment of the antioxidant N-acetyl-L-cysteine (NAC), which restores intracellular glutathione levels, is therapeutic in SLE and reduces disease activity by blocking mTOR activation in T cells (53). This was demonstrated in a double-blind, placebo controlled clinical trial of 36 lupus patients (clinicaltrials.gov NCT00775476) (53). Supplementation with nicotinamide adenine dinucleotide (NAD+) also reduces oxidative stress, as it is a co-enzyme required in redox reactions. NAD+ supports T-cell metabolism and is associated with increased mitochondrial capacity and reduced cell death of SLE T-lymphocytes (112). NAD inhibits mTOR and extracellular signal-regulated kinases 1 + 2 (ERK1/2) and induces autophagy (190). Inhibition of chaperone-mediated autophagy may serve as another therapeutic target (191). Treatment with a spliceosome-derived phosphopeptide, P140, suppressed chaperone-mediated autophagy through reducing the expression levels of HSC70/Hsp73 and the refolding properties of chaperone HSC70 (192). Treatment of MRL/lpr lupus-prone mice with P140 increased survival, which was associated with the accumulation of p62 in B cells (191). Increased expression of HRES-1/Rab4 in T cells of SLE patients and T and B cells of several lupus-prone mouse strains, which preceded onset of ANA and nephritis, was associated with restriction of mitophagy and enhanced autophagy. In turn, blockade of HRES-1/Rab4 and other Rab GTPases through treatment with a geranylgeranyl transferase inhibitor (3-PEHPC) prevented nephritis in lupus-prone MRL/lpr mice (84).
Figure 8. Therapeutic targeting of autophagy in lupus nephritis.
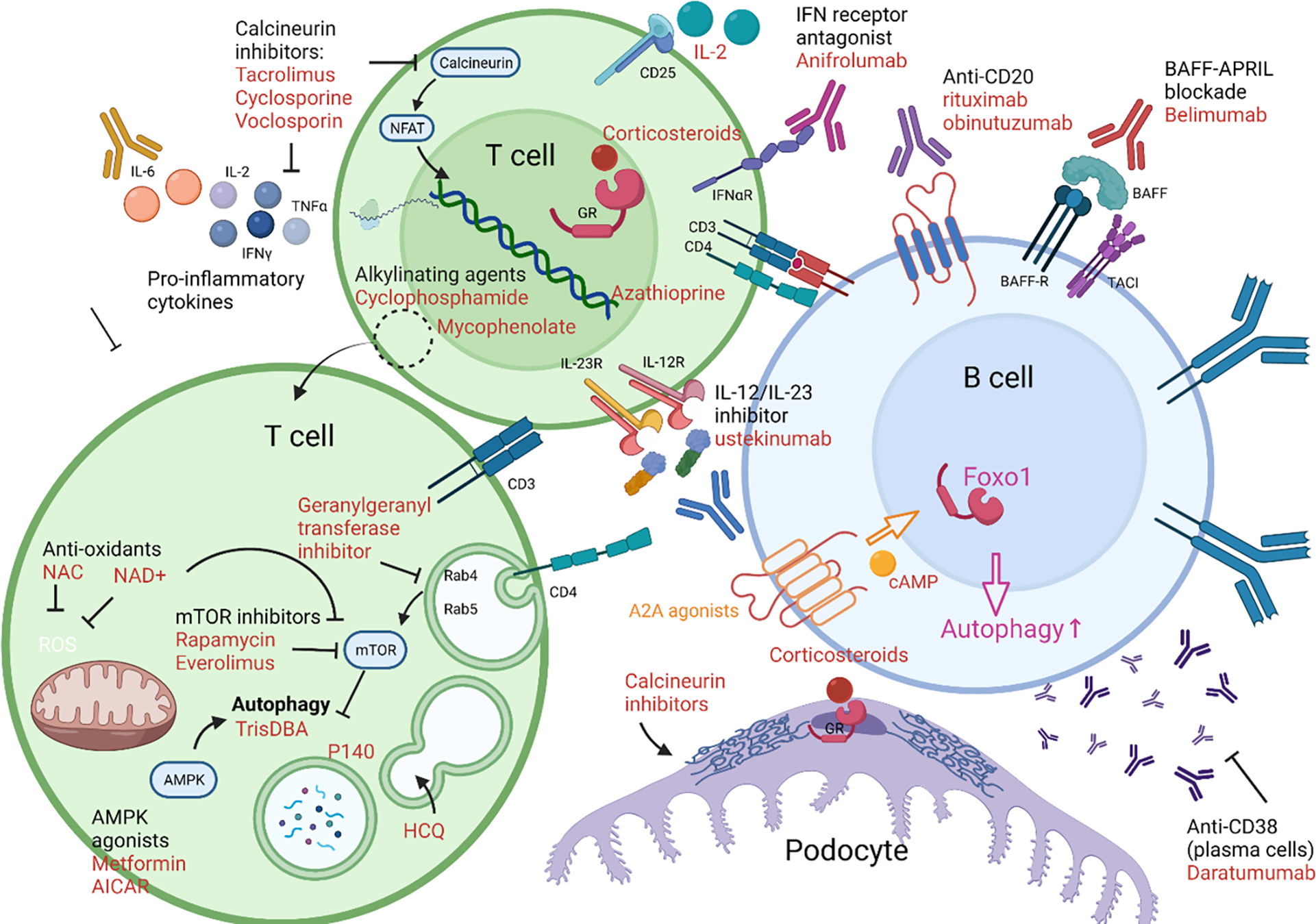
Targeting metabolism expands our repertoire of potential therapeutics in SLE. Traditional therapies have targeted DNA replication (cyclophosphamide, mycophenolate mofetil, azathioprine) or inhibiting T-cell activation through inhibiting calcineurin which prevents production of IL-2 and other cytokines (tacrolimus, cyclosporine, voclosporin), or through activating glucocorticoid receptors (corticosteroids, such as prednisone or methylprednisolone). Calcineurin inhibitors and corticosteroids can also function to stabilize the podocyte cytoskeleton. Aberrations in T-cell metabolism can be targeted through mTOR inhibition (everolimus, sirolimus/rapamycin), which restores autophagy. Anti-oxidants can inhibit mTOR, including NAC or NAD+. Autophagy can be activated through treatment with TrisDBA or agonists of AMPK (metformin, AICAR). It is also affected through altering endosomal acidification with anti-malarial drugs (hydroxychloroquine). Endosomal function can also be targeted by inhibiting RabGTPases through geranylgeranyl transferase inhibitors. Cytokines and cytokine receptors can be targeted for therapy, including the p40 domain of IL-2 and IL-23 (ustekinumab), interferon receptor (anifrolumab), and low-dose IL-2 therapy. The JAK inhibitor, baricitinib, also reduces production of pro-inflammatory cytokines through blockade of JAK-STAT signaling. Autoantibody production can be reduced through depletion of B cells by monoclonal antibodies directed against CD20 (rituximab, obinutuzumab) or depletion of plasma cells/plasmablasts with anti-CD38 monoclonal antibodies (daratumumab). Alternatively, B cell activation can be inhibited through the BAFF-APRIL axis (belimumab).
mTOR inhibition has been shown to be therapeutic in both SLE and lupus-prone mouse models (193). Rapamycin treatment of mice (84, 194) and patients with SLE has been shown to reduce disease activity, restore serum complements, reduce dsDNA antibody titers, and improve fatigue and arthritis (83, 178, 195). In MRL/lpr and NZB/NZW F1 lupus-prone mice, rapamycin treatment prevents LN and/or halts progression of established nephritis (84, 194, 196, 197). In a small study of seven lupus patients, mTOR inhibition with everolimus reduced proteinuria and improved kidney function (197). This has been shown for both selective mTORC1 inhibitors, as well as with a dual mTORC1 and mTORC2 inhibitor, INK128 (198). Inhibition of mTOR by rapamycin restores autophagy, reduces necrosis within T cells, and reduces IL-4 producing CD4−CD8− ‘double negative’ T cells. Rapamycin treatment has been shown to expand Treg populations, even in healthy individuals (199). Rapamycin was found to be effective for treatment of LN in patients with SLE (177, 195, 200). Apparently, the mechanism of action may also involve mTOR blockade in podocytes, mesangial cells, tubular epithelial cells, and vascular endothelial cells (177). Along these lines, rapamycin also blocked mTOR activation in the kidney of patients with antiphospholipid nephropathy, with the majority of these subjects also having SLE (201).
Tregs express the IL-2 receptor CD25, and low-dose IL-2 may be effective to activate and expand Tregs, as well as NK cells, to improve peripheral tolerance (61), and, overall, IL-2 production by T-cells is deficient in SLE (202). In a randomized, placebo-controlled trial of 60 SLE patients, low dose IL-2 treatment was safe and some patients showed complete remission of LN (clinicaltrials.gov identifiers: NCT02465580 and NCT02932137) (203). However, in addition to promoting Treg differentiation, IL-2 therapy promotes expansion of IFNγ-producing CD8+ T cells through activation of the transcription factors STAT6 and GATA3, of which may promote inflammation (82).
Expansion and restoration of Treg function comprise a target for treatment of LN (61). Suppression of CAMK4 by inhibitor KN-93 increased Tregs, reduced Th17 cells, and was therapeutic in MRL/lpr lupus-prone mice as well as in experimental autoimmune encephalomyelitis mouse models (63, 204). Additionally, Tregs express the IL-2 receptor CD25, and low-dose IL-2 may be effective to activate and expand Tregs, as well as NK cells, to improve peripheral tolerance (61), and, overall, IL-2 production by T-cells is deficient in SLE (202). In a randomized, placebo-controlled trial of 60 SLE patients, low dose IL-2 treatment was safe and some patients showed complete remission of LN (clinicaltrials.gov identifiers: NCT02465580 and NCT02932137) (203). However, in addition to promoting Treg differentiation, IL-2 therapy promotes expansion of IFNγ-producing CD8+ T cells through activation of the transcription factors STAT6 and GATA3, of which may promote inflammation (82).
The Treg-Th17 axis is also a treatment target in SLE (205). One method of inhibition is through targeting the effector cytokine IL-6. Anti-IL-6 monoclonal antibodies prevent autoimmunity in lupus-prone NZB/NZW F1 mice, preventing formation of anti-dsDNA antibodies (206). In a phase II clinical trial, IL-6 inhibition reduced severe disease flares (207) (clinicaltrials.gov identifier: NCT01405196). Interestingly, a dose-response relationship did not exist, as significantly reduced IL-6 levels impaired Treg function (208). Th17 cell inhibition can also be induced through blockade of IL-23. IL-23 increases transcription of RORγT that induces naïve T cells to differentiate into Th17 cells. Th17 and DN T cells infiltrate the kidney in patients and mice with lupus nephritis (52–54). IL-23 promotes the development of Th17 cells, and IL-23-treated lymph node cells from lupus-prone mice transfer disease to Rag1-deficient mice (52–54). In turn, IL-23 receptor deficiency in lupus prone mice prevents development of LN (209). These studies suggest that blockade of IL-23 and IL-17 may have therapeutic efficacy in lupus nephritis. In a phase 2 clinical trial, IL-12/IL-23 inhibition with the monoclonal antibody ustekinumab was safe and reduced IFN-gamma levels, showing promise for further investigation (clinicaltrials.gov identifier: NCT02349061)(210). Direct IL-17 inhibition did not show efficacy in murine models, as genetic absence of IL-17 in lupus-prone MRL/lpr mice did not prevent nephritis and likewise, inhibition of IL-17 with a monoclonal antibody did not ameliorate disease in NZB × NZW F1 mice (211). This is likely because IL-17 is not the only pathogenic cytokine as a cause or consequence of Th17 specification.
Type I interferon activation can also be directly targeted in SLE. In a phase 2b clinical trial, anifrolumab, a monoclonal antibody against the IFN-alpha receptor, reversed the ‘interferon signature’ in SLE patients (212) (clinicaltrials.gov identifier: NCT01438489). In a phase I trial, anti-IFN monoclonal antibody therapy reduced IFN-α and β, but there also are many other inflammatory effector molecules in SLE, including BAFF, TNF-α, IL-10, IL-1β, and GM-CSF (clinicaltrials.gov identifier: NCT00299819)(116). Further data will be required to establish clinical efficacy. IFN blockade can also be induced upstream through inhibition of the JAK-STAT pathway with the selective Janus Kinase (JAK) inhibitor baricitinib (213). Baricitinib therapy in MRL/lpr lupus-prone mice prevented systemic autoimmunity and development of nephritis, with reduced renal inflammation and stabilized the podocyte cytoskeleton (213).
Prevention of T-cell homing into target organs is another therapeutic strategy. Agonists of sphingosine 1 phosphate, including FTY720 and KRP203, have shown efficacy in preventing nephritis in lupus-prone MRL/lpr mice (214, 215). Activating sphingosine 1 phosphate signaling reduced proteinuria and increased survival in this model. This was accompanied by reduced production of anti-dsDNA antibodies and decreased IgG deposition within glomeruli (215), reduced lymphadenopathy, and decreased survival of pathogenic lymphocytes (214). . Safety for sphingosine 1 phosphate agonism was established in humans, with use of fingolimod in multiple sclerosis (216). Sphingosine 1 phosphate accumulation in cells can induce autophagy in response to endoplasmic reticulum stress (217).
On the other side of the immunological synapse, inhibiting B-cell activation or B cell depletion represent other therapeutic targets, resulting in reduction in autoantibody production. B-cell activation can be inhibited in SLE through blockade of the interaction between the B-cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL). BAFF is a key protein required for B-cell maturation and survival. Inhibition of the BAFF-APRIL axis by the monoclonal antibody belimumab was shown to have efficacy in LN, with an 11 percent increase in renal response in a recent randomized, placebo-controlled phase 3 clinical trial of 448 SLE patients with nephritis (BLISS-LN trial, clinicaltrials.gov identifier: NCT01639339) (218). Inhibiting T-B cell co-stimulation by expression of CTLA4 using a CTLA4-IgG1Fc fusion protein abatacept was an attempted therapeutic strategy in focal or diffuse lupus nephritis. Treatment reduced dsDNA titers, restored serum complements, and decreased proteinuria. However, it did not meet the primary endpoint of a complete renal response in a phase II/III clinical trial (NCT00430677(219)). Rituximab, an anti-CD20 monoclonal antibody, depletes B-cells and has been shown to be effective in maintenance therapy in an observational study of 147 patients with refractory lupus (220). A new anti-CD20 monoclonal antibody, obinutuzumab, shows increased and sustained B-cell depletion when compared to rituximab, with 22% increased efficacy in treatment of LN in a recent phase II trial (NOBILITY study, clinicaltrials.gov identifier: NCT02550652) (218). Plasma cell depletion with anti-CD38 monoclonal antibody therapy with daratumumab is another therapeutic strategy, depleting plasmablasts and long-lived plasma cells for reduced autoantibody production, although there is currently only data from case reports (221).
Adenosine A2A receptor signaling promotes autophagy (222) and mitigates mitochondrial ROS production (223). As recently uncovered, adenosine receptor 2a (A2A) agonists depleted CD11c+Tbet+ B cells and CD138+ plasma cells and reduced the production of anti-nuclear antibodies and the development of glomerulonephritis and interstitial nephritis in lupus-prone mice (224). An overview of therapeutic targets in current use and under investigation is shown in Figure 8.
Conclusions
Evidence has been mounting of the increased importance of intracellular metabolic pathways, such as mTOR activation and autophagy, in the development of SLE (225, 226). Metabolic changes can result in skewing of T cell subset specification, production of pro-inflammatory cytokines, and promoting B-cell activation. Deregulation of autophagy and defects in associated genes are pathogenic in SLE and may promote auto-antibody production. It is evident that genetically enforced defects in regulatory components of the autophagy machinery contribute to pro-inflammatory immune cell lineage development and compromise the natural response to podocyte injury. Current SLE treatments are non-specific, although multiple agents are under development for targeted therapies, such as the autophagy inducers rapamycin, with the premise for protection against end-organ damage in LN.
Acknowledgements
This work was supported in part by grants AI072648, AI122176, and AR076092 from the National Institutes of Health, the Phillips Lupus and Autoimmunity Center of Excellence, and the Central New York Community Foundation.
Abbreviations:
- 3-MA
3-methyladenine
- ACR
American College of Rheumatology
- AICAR
AMPK-agonist 4-imidazole carboxamide riboside
- AKT
AKT serine/threonine kinase 1
- AMBRA1
autophagy and beclin 1 regulator 1
- ANA
antinuclear antibodies
- APRIL
a proliferation-inducing ligand
- ATG
autophagy related
- ATG5
autophagy related 5
- ATG7
autophagy related 7
- APOL1
apolipoprotein L1
- ATG16L2
autophagy related 16 like 2
- ATP
adenosine triphosphate
- BAFF
B-cell activating factor
- BCR
B-cell receptor
- BECN1
beclin 1
- CD
cluster of differentiation
- CNI
calcineurin inhibitor
- COX2
cyclooxygenase 2
- CTLA4
cytotoxic T-lymphocyte associated protein 4
- CYBB
cytochrome b-245 beta chain
- DAMPs
damage-associated molecular patterns
- DC
dendritic cell
- Def6
DEF guanine nucleotide exchange factor 6
- DN
CD4−CD8− T cell
- ERK1/2
extracellular signal related kinases 1/2
- DRAM1
DNA damage regulated autophagy modulator 1
- DRP1
dynamin-related protein 1
- dsDNA
double stranded DNA
- GATA3
GATA binding protein 3
- GWAS
genome-wide association study
- ESKD
end-stage kidney disease
- EXT1/2
exostosin 1/exostosin 2 complex
- Foxp3
forkhead box protein P3
- GATA3
GATA-binding protein 3
- GWAS
genome-wide association study
- HRES-1/Rab4
Human Retroviral Endogenous Sequence-1/Rab GTPase 4
- HSP90
heat shock protein-90
- IFN
interferon
- IFN-α
interferon alpha
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- IP-10
interferon gamma-inducible protein 10
- IRGM
immunity related GTPase M
- JAK
Janus Kinase
- KIM-1
kidney injury molecule-1
- LAP
LC3-associated phagocytosis
- LC3
microtubule-associated protein 1 light chain 3 alpha
- LN
lupus nephritis
- MHC
major histocompatibility complex
- MLN
membranous lupus nephritis
- MMF
mycophenolate mofetil
- MTMR3
myotubularin related protein 3
- mTOR
mechanistic target of rapamycin
- NAC
N-acetyl-L-cysteine
- NAD+
nicotinamide adenine dinucleotide
- NCAM1
neural cell adhesion molecule 1
- NET
neutrophil extracellular traps
- NETosis
neutrophil cell death with extrusion of neutrophil extracellular traps
- NOX2
NADPH oxidase 2
- NZB/NZW
New Zealand Black/New Zealand white F1 lupus-prone mice
- NZM2328
New Zealand mixed lupus-prone mice
- p62
sequestosome 1
- PAMPs
pathogen-associated molecular patterns
- pDC
plasmacytoid dendritic cell
- PI3K
phosphoinositide 3-kinase
- PINK-1
PTEN-induced kinase 1
- Raptor
regulatory-associated protein of mTOR
- PRDM1
PR domain zinc finger protein 1
- PRKN
Parkin RBR E3 ubiquitin protein ligase
- Rag1
recombination activating 1
- RIPK3
receptor-interacting serine/threonine protein kinase 3
- RNA
ribonucleic acid
- RNP
ribonucleoprotein
- RORγT
retinoic orphan receptor gamma
- SLC15A4
Solute carrier family 15 member 4
- SLE
systemic lupus erythematosus
- STING
stimulation of interferon genes protein-mediated DNA sensing pathway
- SOD2
superoxide dismutase 2
- STAT
signal transducer and activator of transcription
- Tbet
T-box expressed in T-cells
- TCR
T-cell receptor
- TGFBR3
transforming growth factor beta receptor 3
- TLR
toll-like receptor
- TIM4
T-cell immunoglobulin and mucin domain containing 4
- TNF
tumor necrosis factor
- Treg
regulatory T cell
- TSC
tuberous sclerosis complex
- ULK 1/2
UNC-51-like autophagy activating kinases 1/2
- Vps34
vacuolar protein sorting 34
Footnotes
The authors have read the journal’s policy on disclosure of potential conflicts of interest and declare no conflicts of interest. All authors have read the journal’s authorship agreement and that the manuscript has been reviewed by and approved by all named authors.
References
- 1.Alarcón GS, Roseman J, Bartolucci AA, Friedman AW, Moulds JM, Goel N, et al. Systemic lupus erythematosus in three ethnic groups: II. Features predictive of disease activity early in its course. LUMINA Study Group. Lupus in minority populations, nature versus nurture. Arthritis Rheum. 1998;41(7):1173–80. Epub 1998/07/15. doi: . [DOI] [PubMed] [Google Scholar]
- 2.Madhok R Systemic lupus erythematosus: lupus nephritis. BMJ Clin Evid. 2015;2015. Epub 2015/12/20. [PMC free article] [PubMed] [Google Scholar]
- 3.Mok CC. Biomarkers for lupus nephritis: a critical appraisal. J Biomed Biotechnol. 2010;2010:638413. Epub 2010/04/24. doi: 10.1155/2010/638413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aragón CC, Tafúr RA, Suárez-Avellaneda A, Martínez MT, Salas AL, Tobón GJ. Urinary biomarkers in lupus nephritis. J Transl Autoimmun. 2020;3:100042. Epub 2020/08/04. doi: 10.1016/j.jtauto.2020.100042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahmoud SS, Bazaraa HM, Lotfy HM, Abd-El-Aziz DM. Renal involvement in childhood-onset systemic lupus erythematosus in Egypt. Rheumatol Int. 2012;32(1):47–51. Epub 2010/07/27. doi: 10.1007/s00296-010-1554-7. [DOI] [PubMed] [Google Scholar]
- 6.Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum. 1992;35(6):630–40. Epub 1992/06/01. doi: 10.1002/art.1780350606. [DOI] [PubMed] [Google Scholar]
- 7.Diaz-Gallo LM, Oke V, Lundström E, Elvin K, Ling Wu Y, Eketjäll S, et al. Four Systemic Lupus Erythematosus Subgroups, Defined by Autoantibodies Status, Differ Regarding HLA-DRB1 Genotype Associations and Immunological and Clinical Manifestations. ACR Open Rheumatol. 2021. Epub 2021/10/19. doi: 10.1002/acr2.11343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thompson JC, Mahajan A, Scott DA, Gairy K. The Economic Burden of Lupus Nephritis: A Systematic Literature Review. Rheumatol Ther. 2021. Epub 2021/11/04. doi: 10.1007/s40744-021-00368-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. Epub 1997/10/27. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 10.Al Sawah S, Daly RP, Foster S, Naegeli A, Benjamin K, Doll H, et al. Understanding Delay in Diagnosis, Access to Care and Satisfaction with Care in Lupus: Findings from a Cross-Sectional Online Survey in the United States. Annals of Rheumatic Disease. 2015;74(2). [Google Scholar]
- 11.Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677–86. doi: DOI: 10.1002/art.34473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bajema IM, Wilhelmus S, Alpers CE, Bruijn JA, Colvin RB, Cook HT, et al. Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int. 2018;93(4):789–96. doi: 10.1016/j.kint.2017.11.023. [DOI] [PubMed] [Google Scholar]
- 13.Dasari S, Chakraborty A, Truong L, Mohan C. A Systematic Review of Interpathologist Agreement in Histologic Classification of Lupus Nephritis. Kidney Int Rep. 2019;4(10):1420–5. Epub 2019/11/09. doi: 10.1016/j.ekir.2019.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chedid A, Rossi GM, Peyronel F, Menez S, Atta MG, Bagnasco SM, et al. Low-Level Proteinuria in Systemic Lupus Erythematosus. Kidney Int Rep. 2020;5(12):2333–40. Epub 2020/12/12. doi: 10.1016/j.ekir.2020.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishizaki J, Saito K, Nawata M, Mizuno Y, Tokunaga M, Sawamukai N, et al. Low complements and high titre of anti-Sm antibody as predictors of histopathologically proven silent lupus nephritis without abnormal urinalysis in patients with systemic lupus erythematosus. Rheumatology (Oxford). 2015;54(3):405–12. Epub 2014/09/04. doi: 10.1093/rheumatology/keu343. [DOI] [PubMed] [Google Scholar]
- 16.Lech M, Anders HJ. The pathogenesis of lupus nephritis. J Am Soc Nephrol. 2013;24(9):1357–66. Epub 2013/08/10. doi: DOI: 10.1681/asn.2013010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaur S, Bansal Y, Kumar R, Bansal G. A panoramic review of IL-6: Structure, pathophysiological roles and inhibitors. Bioorg Med Chem. 2020;28(5):115327. Epub 2020/01/30. doi: 10.1016/j.bmc.2020.115327. [DOI] [PubMed] [Google Scholar]
- 18.Linker-Israeli M, Deans RJ, Wallace DJ, Prehn J, Ozeri-Chen T, Klinenberg JR. Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis. J Immunol. 1991;147(1):117–23. Epub 1991/07/01. [PubMed] [Google Scholar]
- 19.Dema B, Charles N. Autoantibodies in SLE: Specificities, Isotypes and Receptors. Antibodies (Basel). 2016;5(1). Epub 2016/01/04. doi: 10.3390/antib5010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caster DJ, Powell DW. Utilization of Biomarkers in Lupus Nephritis. Adv Chronic Kidney Dis. 2019;26(5):351–9. Epub 2019/11/18. doi: 10.1053/j.ackd.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kianmehr N, Khoshmirsafa M, Shekarabi M, Falak R, Haghighi A, Masoodian M, et al. High frequency of concurrent anti-C1q and anti-dsDNA but not anti-C3b antibodies in patients with Lupus Nephritis. J Immunoassay Immunochem. 2021;42(4):406–23. Epub 2021/04/01. doi: 10.1080/15321819.2021.1895215. [DOI] [PubMed] [Google Scholar]
- 22.Linnik MD, Hu JZ, Heilbrunn KR, Strand V, Hurley FL, Joh T. Relationship between anti-double-stranded DNA antibodies and exacerbation of renal disease in patients with systemic lupus erythematosus. Arthritis Rheum. 2005;52(4):1129–37. Epub 2005/04/09. doi: 10.1002/art.20980. [DOI] [PubMed] [Google Scholar]
- 23.Macanovic M, Hogarth MB, Lachmann PJ. Anti-DNA antibodies in the urine of lupus nephritis patients. Nephrol Dial Transplant. 1999;14(6):1418–24. Epub 1999/06/26. doi: 10.1093/ndt/14.6.1418. [DOI] [PubMed] [Google Scholar]
- 24.Yamada A, Miyakawa Y, Kosaka K. Entrapment of anti-DNA antibodies in the kidney of patients with systemic lupus erythematosus. Kidney Int. 1982;22(6):671–6. Epub 1982/12/01. doi: 10.1038/ki.1982.228. [DOI] [PubMed] [Google Scholar]
- 25.Sciascia SA, Dickensheets H, Picking W, Robson K, Wang D, Ye BH, et al. Autoantibodies and autoantigens in the urine of SLE patients. Autoimmunity. 2004;37(6–7):503–14. Epub 2004/12/29. doi: 10.1080/08916930410001713016. [DOI] [PubMed] [Google Scholar]
- 26.Field M, Williams DG, Charles P, Maini RN. Specificity of anti-Sm antibodies by ELISA for systemic lupus erythematosus: increased sensitivity of detection using purified peptide antigens. Ann Rheum Dis. 1988;47(10):820–5. Epub 1988/10/01. doi: 10.1136/ard.47.10.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meryhew NL, Messner RP, Tan EM. Urinary excretion of antinuclear antibodies. J Rheumatol. 1983;10(6):913–9. Epub 1983/12/01. [PubMed] [Google Scholar]
- 28.Picking WL, Smith C, Petruci R, Scheffel J, Levich JD, Stetler DA. Anti-RNA polymerase I antibodies in the urine of patients with systemic lupus erythematosus. J Rheumatol. 1990;17(10):1308–13. Epub 1990/10/01. [PubMed] [Google Scholar]
- 29.Sethi S, Madden BJ, Debiec H, Charlesworth MC, Gross L, Ravindran A, et al. Exostosin 1/Exostosin 2-Associated Membranous Nephropathy. J Am Soc Nephrol. 2019;30(6):1123–36. Epub 2019/05/08. doi: 10.1681/ASN.2018080852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caza TN, Hassen SI, Kuperman M, Sharma SG, Dvanajscak Z, Arthur J, et al. Neural cell adhesion molecule 1 is a novel autoantigen in membranous lupus nephritis. Kidney Int. 2021;100(1):171–81. Epub 2020/10/13. doi: 10.1016/j.kint.2020.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caza TN, Hassen SI, Kenan DJ, Storey A, Arthur JM, Herzog C, et al. Transforming growth factor beta receptor 3 (TGFBR3)-associated membranous nephropathy. Kidney 360. 2021;Publish Ahead of Print June 11, 2021. Epub June 11, 2021. doi: 10.34067/KID.0001492021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ravindran A, Casal Moura M, Fervenza FC, Nasr SH, Alexander MP, Fidler ME, et al. In Patients with Membranous Lupus Nephritis, Exostosin-Positivity and Exostosin-Negativity Represent Two Different Phenotypes. Journal of the American Society of Nephrology. 2021:ASN.2020081181. doi: 10.1681/asn.2020081181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saïdi M, Brochériou I, Estève E, Tuffet S, Amoura Z, Miyara M, et al. The Exostosin Immunohistochemical Status Differentiates Lupus Membranous Nephropathy Subsets With Different Outcomes. Kidney Int Rep. 2021;6(7):1977–80. Epub 2021/07/27. doi: 10.1016/j.ekir.2021.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birmingham DJ, Hebert LA. The Complement System in Lupus Nephritis. Semin Nephrol. 2015;35(5):444–54. doi: 10.1016/j.semnephrol.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 35.Kim H, Kim T, Kim M, Lee HY, Kim Y, Kang MS, et al. Activation of the alternative complement pathway predicts renal outcome in patients with lupus nephritis. Lupus. 2020;29(8):862–71. Epub 2020/05/16. doi: 10.1177/0961203320925165. [DOI] [PubMed] [Google Scholar]
- 36.Troldborg A, Thiel S, Trendelenburg M, Friebus-Kardash J, Nehring J, Steffensen R, et al. The Lectin Pathway of Complement Activation in Patients with Systemic Lupus Erythematosus. J Rheumatol. 2018;45(8):1136–44. Epub 2018/06/17. doi: 10.3899/jrheum.171033. [DOI] [PubMed] [Google Scholar]
- 37.Martin M, Trattner R, Nilsson SC, Björk A, Zickert A, Blom AM, et al. Plasma C4d Correlates With C4d Deposition in Kidneys and With Treatment Response in Lupus Nephritis Patients. Front Immunol. 2020;11:582737. Epub 2020/11/03. doi: 10.3389/fimmu.2020.582737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gou SJ, Yuan J, Wang C, Zhao MH, Chen M. Alternative Complement Pathway Activation Products in Urine and Kidneys of Patients with ANCA-Associated GN. Clin J Am Soc Nephrol. 2013;8(11):1884–91. Epub 2013/10/12. doi: DOI: 10.2215/CJN.02790313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelly RH, Carpenter AB, Sudol KS, Jagarlapudi SP, Manzi S. Complement C3 fragments in urine: detection in systemic lupus erythematosus patients by western blotting. Appl Theor Electrophor. 1993;3(6):265–9. Epub 1993/01/01. [PubMed] [Google Scholar]
- 40.Manzi S, Rairie JE, Carpenter AB, Kelly RH, Jagarlapudi SP, Sereika SM, et al. Sensitivity and specificity of plasma and urine complement split products as indicators of lupus disease activity. Arthritis Rheum. 1996;39(7):1178–88. Epub 1996/07/01. doi: 10.1002/art.1780390716. [DOI] [PubMed] [Google Scholar]
- 41.Caza TN, Talaber G, Perl A. Metabolic regulation of organelle homeostasis in lupus T cells. Clin Immunol. 2012;144(3):200–13. Epub 2012/07/28. doi: 10.1016/j.clim.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fillatreau S, Manfroi B, Dörner T. Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nat Rev Rheumatol. 2021;17(2):98–108. Epub 2020/12/20. doi: 10.1038/s41584-020-00544-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conti F, Spinelli FR, Truglia S, Miranda F, Alessandri C, Ceccarelli F, et al. Kidney Expression of Toll Like Receptors in Lupus Nephritis: Quantification and Clinicopathological Correlations. Mediators Inflamm. 2016;2016:7697592. Epub 2016/09/17. doi: 10.1155/2016/7697592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caza T, Oaks Z, Perl A. Interplay of infections, autoimmunity, and immunosuppression in systemic lupus erythematosus. Int Rev Immunol. 2014;33(4):330–63. Epub 2014/01/30. doi: 10.3109/08830185.2013.863305. [DOI] [PubMed] [Google Scholar]
- 45.Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110–21. Epub 2011/12/02. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 46.Crispin JC, Martínez A, Alcocer-Varela J. Quantification of regulatory T cells in patients with systemic lupus erythematosus. Journal of autoimmunity. 2003;21(3):273–6. Epub 2003/11/06. doi: 10.1016/s0896-8411(03)00121-5. [DOI] [PubMed] [Google Scholar]
- 47.Mihara M, Ohsugi Y, Saito K, Miyai T, Togashi M, Ono S, et al. Immunologic abnormality in NZB/NZW F1 mice. Thymus-independent occurrence of B cell abnormality and requirement for T cells in the development of autoimmune disease, as evidenced by an analysis of the athymic nude individuals. J Immunol. 1988;141(1):85–90. Epub 1988/07/01. [PubMed] [Google Scholar]
- 48.Okamoto A, Fujio K, Tsuno NH, Takahashi K, Yamamoto K. Kidney-infiltrating CD4+ T-cell clones promote nephritis in lupus-prone mice. Kidney Int. 2012;82(9):969–79. Epub 2012/07/06. doi: 10.1038/ki.2012.242. [DOI] [PubMed] [Google Scholar]
- 49.Vratsanos GS, Jung S, Park YM, Craft J. CD4(+) T cells from lupus-prone mice are hyperresponsive to T cell receptor engagement with low and high affinity peptide antigens: a model to explain spontaneous T cell activation in lupus. J Exp Med. 2001;193(3):329–37. Epub 2001/02/07. doi: 10.1084/jem.193.3.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bagavant H, Deshmukh US, Wang H, Ly T, Fu SM. Role for nephritogenic T cells in lupus glomerulonephritis: progression to renal failure is accompanied by T cell activation and expansion in regional lymph nodes. J Immunol. 2006;177(11):8258–65. Epub 2006/11/23. doi: 10.4049/jimmunol.177.11.8258. [DOI] [PubMed] [Google Scholar]
- 51.Hedrich CM, Rauen T, Crispin JC, Koga T, Ioannidis C, Zajdel M, et al. cAMP-responsive element modulator α (CREMα) trans-represses the transmembrane glycoprotein CD8 and contributes to the generation of CD3+CD4−CD8− T cells in health and disease. J Biol Chem. 2013;288(44):31880–7. Epub 2013/09/21. doi: 10.1074/jbc.M113.508655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shivakumar S, Tsokos GC, Datta SK. T cell receptor alpha/beta expressing double-negative (CD4−/CD8−) and CD4+ T helper cells in humans augment the production of pathogenic anti-DNA autoantibodies associated with lupus nephritis. J Immunol. 1989;143(1):103–12. Epub 1989/07/01. [PubMed] [Google Scholar]
- 53.Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012;64(9):2937–46. Epub 2012/05/03. doi: 10.1002/art.34502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crispín JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. 2008;181(12):8761–6. Epub 2008/12/04. doi: 10.4049/jimmunol.181.12.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fernandez DR, Telarico T, Bonilla E, Li Q, Banerjee S, Middleton FA, et al. Activation of mammalian target of rapamycin controls the loss of TCRzeta in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. J Immunol. 2009;182(4):2063–73. Epub 2009/02/10. doi: 10.4049/jimmunol.0803600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sieling PA, Porcelli SA, Duong BT, Spada F, Bloom BR, Diamond B, et al. Human double-negative T cells in systemic lupus erythematosus provide help for IgG and are restricted by CD1c. J Immunol. 2000;165(9):5338–44. Epub 2000/10/25. doi: 10.4049/jimmunol.165.9.5338. [DOI] [PubMed] [Google Scholar]
- 57.Lai ZW, Borsuk R, Shadakshari A, Yu J, Dawood M, Garcia R, et al. Mechanistic target of rapamycin activation triggers IL-4 production and necrotic death of double-negative T cells in patients with systemic lupus erythematosus. J Immunol. 2013;191(5):2236–46. Epub 2013/08/06. doi: 10.4049/jimmunol.1301005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perl A Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat Rev Rheumatol. 2016;12(3):169–82. Epub 2015/12/25. doi: 10.1038/nrrheum.2015.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kato H, Perl A. Blockade of Treg Cell Differentiation and Function by the Interleukin-21-Mechanistic Target of Rapamycin Axis Via Suppression of Autophagy in Patients With Systemic Lupus Erythematosus. Arthritis & rheumatology (Hoboken, NJ). 2018;70(3):427–38. Epub 2017/11/22. doi: 10.1002/art.40380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen JW, Schickel JN, Tsakiris N, Sng J, Arbogast F, Bouis D, et al. Positive and negative selection shape the human naïve B cell repertoire. J Clin Invest. 2021. Epub 2021/11/24. doi: 10.1172/jci150985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mizui M, Tsokos GC. Targeting Regulatory T Cells to Treat Patients With Systemic Lupus Erythematosus. Front Immunol. 2018;9:786. Epub 2018/05/15. doi: 10.3389/fimmu.2018.00786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shah K, Lee WW, Lee SH, Kim SH, Kang SW, Craft J, et al. Dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Res Ther. 2010;12(2):R53. Epub 2010/03/26. doi: 10.1186/ar2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koga T, Hedrich CM, Mizui M, Yoshida N, Otomo K, Lieberman LA, et al. CaMK4-dependent activation of AKT/mTOR and CREM-α underlies autoimmunity-associated Th17 imbalance. J Clin Invest. 2014;124(5):2234–45. Epub 2014/03/29. doi: 10.1172/jci73411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.El-Shereef RR, Lotfi A, Abdel-Naeam EA, Tawfik H. Serum and Urinary Interleukin-6 in Assessment of Renal Activity in Egyptian Patients with Systemic Lupus Erythematosus. Clin Med Insights Arthritis Musculoskelet Disord. 2016;9:29–36. Epub 2016/03/12. doi: 10.4137/cmamd.S32269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Herrera-Esparza R, Barbosa-Cisneros O, Villalobos-Hurtado R, Avalos-Díaz E. Renal expression of IL-6 and TNFalpha genes in lupus nephritis. Lupus. 1998;7(3):154–8. Epub 1998/06/02. doi: 10.1191/096120398678919949. [DOI] [PubMed] [Google Scholar]
- 66.Lit LC, Wong CK, Li EK, Tam LS, Lam CW, Lo YM. Elevated gene expression of Th1/Th2 associated transcription factors is correlated with disease activity in patients with systemic lupus erythematosus. J Rheumatol. 2007;34(1):89–96. Epub 2006/11/23. [PubMed] [Google Scholar]
- 67.Akahoshi M, Nakashima H, Tanaka Y, Kohsaka T, Nagano S, Ohgami E, et al. Th1/Th2 balance of peripheral T helper cells in systemic lupus erythematosus. Arthritis Rheum. 1999;42(8):1644–8. Epub 1999/08/14. doi: . [DOI] [PubMed] [Google Scholar]
- 68.Masutani K, Akahoshi M, Tsuruya K, Tokumoto M, Ninomiya T, Kohsaka T, et al. Predominance of Th1 immune response in diffuse proliferative lupus nephritis. Arthritis Rheum. 2001;44(9):2097–106. Epub 2001/10/11. doi: . [DOI] [PubMed] [Google Scholar]
- 69.Biermann MH, Veissi S, Maueröder C, Chaurio R, Berens C, Herrmann M, et al. The role of dead cell clearance in the etiology and pathogenesis of systemic lupus erythematosus: dendritic cells as potential targets. Expert Rev Clin Immunol. 2014;10(9):1151–64. Epub 2014/08/02. doi: 10.1586/1744666x.2014.944162. [DOI] [PubMed] [Google Scholar]
- 70.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100(5):2610–5. Epub 2003/02/27. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morimoto AM, Flesher DT, Yang J, Wolslegel K, Wang X, Brady A, et al. Association of endogenous anti-interferon-α autoantibodies with decreased interferon-pathway and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2011;63(8):2407–15. Epub 2011/04/21. doi: 10.1002/art.30399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harris BD, Kuruganti S, Deshpande A, Goepfert PA, Chatham WW, Walter MR. Characterization of Type-I IFN subtype autoantibodies and activity in SLE serum and urine. Lupus. 2020;29(9):1095–105. Epub 2020/07/03. doi: 10.1177/0961203320935976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Becker AM, Dao KH, Han BK, Kornu R, Lakhanpal S, Mobley AB, et al. SLE peripheral blood B cell, T cell and myeloid cell transcriptomes display unique profiles and each subset contributes to the interferon signature. PloS one. 2013;8(6):e67003. Epub 2013/07/05. doi: 10.1371/journal.pone.0067003. LLC, which develops assays for autoimmune diseases and she has also obtained funding from MedImmune, Novo Nordisk, Genentech/Roche and Human Genome Sciences. Dr. David Karp has received research funding from Human Genome Sciences. None of these funds were used for the current study. This does not alter the authors’ adherence to all the PLOS ONE policies on sharing data and materials. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chiche L, Jourde-Chiche N, Whalen E, Presnell S, Gersuk V, Dang K, et al. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis & rheumatology (Hoboken, NJ). 2014;66(6):1583–95. Epub 2014/03/20. doi: 10.1002/art.38628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li QZ, Zhou J, Lian Y, Zhang B, Branch VK, Carr-Johnson F, et al. Interferon signature gene expression is correlated with autoantibody profiles in patients with incomplete lupus syndromes. Clinical and experimental immunology. 2010;159(3):281–91. Epub 2009/12/01. doi: 10.1111/j.1365-2249.2009.04057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med. 2020;382(3):211–21. Epub 2019/12/19. doi: 10.1056/NEJMoa1912196. [DOI] [PubMed] [Google Scholar]
- 77.Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol. 2012;30:39–68. Epub 2011/11/29. doi: 10.1146/annurev-immunol-020711-075024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12(4):295–303. Epub 2011/03/02. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bi Y, Liu G, Yang R. mTOR regulates T-cell differentiation and activation in immunity and autoimmunity. Crit Rev Eukaryot Gene Expr. 2011;21(4):313–22. Epub 2011/12/21. doi: 10.1615/critreveukargeneexpr.v21.i4.20. [DOI] [PubMed] [Google Scholar]
- 80.Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32(1):67–78. Epub 2010/01/12. doi: 10.1016/j.immuni.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30(6):832–44. Epub 2009/06/23. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kato H, Perl A. Double-Edged Sword: Interleukin-2 Promotes T Regulatory Cell Differentiation but Also Expands Interleukin-13- and Interferon-γ-Producing CD8(+) T Cells via STAT6-GATA-3 Axis in Systemic Lupus Erythematosus. Front Immunol. 2021;12:635531. Epub 2021/03/26. doi: 10.3389/fimmu.2021.635531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fernandez D, Bonilla E, Mirza N, Niland B, Perl A. Rapamycin reduces disease activity and normalizes T cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheum. 2006;54(9):2983–8. Epub 2006/09/02. doi: 10.1002/art.22085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Caza TN, Fernandez DR, Talaber G, Oaks Z, Haas M, Madaio MP, et al. HRES-1/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis and represents a target for treatment in SLE. Ann Rheum Dis. 2014;73(10):1888–97. Epub 2013/07/31. doi: 10.1136/annrheumdis-2013-203794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lai ZW, Marchena-Mendez I, Perl A. Oxidative stress and Treg depletion in lupus patients with anti-phospholipid syndrome. Clin Immunol. 2015;158(2):148–52. Epub 2015/04/12. doi: 10.1016/j.clim.2015.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gergely P Jr., Grossman C, Niland B, Puskas F, Neupane H, Allam F, et al. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46(1):175–90. Epub 2002/01/31. doi: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Banoth B, Cassel SL. Mitochondria in innate immune signaling. Transl Res. 2018;202:52–68. Epub 2018/08/31. doi: 10.1016/j.trsl.2018.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Prunotto M, Carnevali ML, Candiano G, Murtas C, Bruschi M, Corradini E, et al. Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. J Am Soc Nephrol. 2010;21(3):507–19. Epub 2010/02/13. doi: 10.1681/asn.2008121259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murtas C, Bruschi M, Candiano G, Moroni G, Magistroni R, Magnano A, et al. Coexistence of Different Circulating Anti-Podocyte Antibodies in Membranous Nephropathy. Clin J Am Soc Nephrol. 2012;7(9):1394–400. Epub 2012/07/10. doi: DOI:2215/CJN.02170312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ye X, Zhou X-J, Zhang H. Exploring the Role of Autophagy-Related Gene 5 (ATG5) Yields Important Insights Into Autophagy in Autoimmune/Autoinflammatory Diseases. Frontiers in Immunology. 2018;9(2334). doi: 10.3389/fimmu.2018.02334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Valečka J, Almeida CR, Su B, Pierre P, Gatti E. Autophagy and MHC-restricted antigen presentation. Molecular immunology. 2018;99:163–70. Epub 2018/05/23. doi: 10.1016/j.molimm.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 92.Merkley SD, Chock CJ, Yang XO, Harris J, Castillo EF. Modulating T Cell Responses via Autophagy: The Intrinsic Influence Controlling the Function of Both Antigen-Presenting Cells and T Cells. Front Immunol. 2018;9:2914. Epub 2019/01/09. doi: 10.3389/fimmu.2018.02914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Riffelmacher T, Richter FC, Simon AK. Autophagy dictates metabolism and differentiation of inflammatory immune cells. Autophagy. 2018;14(2):199–206. Epub 2017/08/15. doi: 10.1080/15548627.2017.1362525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci U S A. 2007;104(35):14050–5. Epub 2007/08/22. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alessandri C, Barbati C, Vacirca D, Piscopo P, Confaloni A, Sanchez M, et al. T lymphocytes from patients with systemic lupus erythematosus are resistant to induction of autophagy. Faseb j. 2012;26(11):4722–32. Epub 2012/07/28. doi: 10.1096/fj.12-206060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gros F, Arnold J, Page N, Décossas M, Korganow AS, Martin T, et al. Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy. 2012;8(7):1113–23. Epub 2012/04/24. doi: 10.4161/auto.20275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li B, Yue Y, Dong C, Shi Y, Xiong S. Blockade of macrophage autophagy ameliorates activated lymphocytes-derived DNA induced murine lupus possibly via inhibition of proinflammatory cytokine production. Clin Exp Rheumatol. 2014;32(5):705–14. Epub 2014/08/26. [PubMed] [Google Scholar]
- 98.Denton D, Kumar S. Autophagy-dependent cell death. Cell death and differentiation. 2019;26(4):605–16. Epub 2018/12/19. doi: 10.1038/s41418-018-0252-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A. 2010;107(21):9813–8. Epub 2010/05/05. doi: 10.1073/pnas.0909927107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guo Y, Gao F, Wang X, Pan Z, Wang Q, Xu S, et al. Spontaneous formation of neutrophil extracellular traps is associated with autophagy. Sci Rep. 2021;11(1):24005. Epub 2021/12/16. doi: 10.1038/s41598-021-03520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Science translational medicine. 2011;3(73):73ra20–73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41. Epub 2011/01/25. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108(42):17396–401. Epub 2011/10/05. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533(7601):115–9. Epub 2016/04/21. doi: 10.1038/nature17950. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 105.Jacquin E, Apetoh L. Cell-Intrinsic Roles for Autophagy in Modulating CD4 T Cell Functions. Front Immunol. 2018;9:1023. Epub 2018/06/06. doi: 10.3389/fimmu.2018.01023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–65. Epub 2016/07/12. doi: 10.1038/nri.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Almeida L, Lochner M, Berod L, Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol. 2016;28(5):514–24. Epub 2016/11/09. doi: 10.1016/j.smim.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 108.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208(7):1367–76. Epub 2011/06/29. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol. 2016;17(3):277–85. Epub 2016/01/26. doi: 10.1038/ni.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol. 2014;15(12):1152–61. Epub 2014/11/05. doi: 10.1038/ni.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fritsch RD, Shen X, Illei GG, Yarboro CH, Prussin C, Hathcock KS, et al. Abnormal differentiation of memory T cells in systemic lupus erythematosus. Arthritis Rheum. 2006;54(7):2184–97. Epub 2006/06/28. doi: 10.1002/art.21943. [DOI] [PubMed] [Google Scholar]
- 112.Buang N, Tapeng L, Gray V, Sardini A, Whilding C, Lightstone L, et al. Type I interferons affect the metabolic fitness of CD8(+) T cells from patients with systemic lupus erythematosus. Nat Commun. 2021;12(1):1980. Epub 2021/04/02. doi: 10.1038/s41467-021-22312-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, et al. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity. 2007;27(3):505–17. Epub 2007/09/26. doi: 10.1016/j.immuni.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 114.Kabat AM, Harrison OJ, Riffelmacher T, Moghaddam AE, Pearson CF, Laing A, et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. Elife. 2016;5:e12444. Epub 2016/02/26. doi: 10.7554/eLife.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Harris J, Master SS, De Haro SA, Delgado M, Roberts EA, Hope JC, et al. Th1-Th2 polarisation and autophagy in the control of intracellular mycobacteria by macrophages. Vet Immunol Immunopathol. 2009;128(1–3):37–43. Epub 2008/11/26. doi: 10.1016/j.vetimm.2008.10.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Crotzer VL, Blum JS. Autophagy and its role in MHC-mediated antigen presentation. J Immunol. 2009;182(6):3335–41. Epub 2009/03/07. doi: 10.4049/jimmunol.0803458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhou XJ, Lu XL, Lv JC, Yang HZ, Qin LX, Zhao MH, et al. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis. 2011;70(7):1330–7. Epub 2011/05/31. doi: 10.1136/ard.2010.140111. [DOI] [PubMed] [Google Scholar]
- 118.Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495(7441):389–93. Epub 2013/03/05. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 119.Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–64. Epub 2018/04/06. doi: 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 120.Nakatogawa H Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol. 2020;21(8):439–58. Epub 2020/05/07. doi: 10.1038/s41580-020-0241-0. [DOI] [PubMed] [Google Scholar]
- 121.Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41(11):1228–33. Epub 2009/10/20. doi: DOI:1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009;41(11):1234–7. Epub 2009/10/20. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 123.Ciccacci C, Perricone C, Alessandri C, Latini A, Politi C, Delunardo F, et al. Evaluation of ATG5 polymorphisms in Italian patients with systemic lupus erythematosus: contribution to disease susceptibility and clinical phenotypes. Lupus. 2018;27(9):1464–9. Epub 2018/05/16. doi: 10.1177/0961203318776108. [DOI] [PubMed] [Google Scholar]
- 124.Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018;19(9):579–93. Epub 2018/07/15. doi: 10.1038/s41580-018-0033-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhou XJ, Nath SK, Qi YY, Cheng FJ, Yang HZ, Zhang Y, et al. Brief Report: identification of MTMR3 as a novel susceptibility gene for lupus nephritis in northern Han Chinese by shared-gene analysis with IgA nephropathy. Arthritis & rheumatology (Hoboken, NJ). 2014;66(10):2842–8. Epub 2014/06/20. doi: 10.1002/art.38749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee YH, Choi SJ, Ji JD, Song GG. Association between toll-like receptor polymorphisms and systemic lupus erythematosus: a meta-analysis update. Lupus. 2016;25(6):593–601. Epub 2016/01/15. doi: 10.1177/0961203315622823. [DOI] [PubMed] [Google Scholar]
- 127.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312(5780):1669–72. Epub 2006/05/20. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 128.Bekeredjian-Ding IB, Wagner M, Hornung V, Giese T, Schnurr M, Endres S, et al. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J Immunol. 2005;174(7):4043–50. Epub 2005/03/22. doi: 10.4049/jimmunol.174.7.4043. [DOI] [PubMed] [Google Scholar]
- 129.Freedman BI, Langefeld CD, Andringa KK, Croker JA, Williams AH, Garner NE, et al. End-stage kidney disease in African Americans with lupus nephritis associates with APOL1. Arthritis & rheumatology (Hoboken, NJ). 2014;66(2):390–6. doi: 10.1002/art.38220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Larsen CP, Beggs ML, Saeed M, Walker PD. Apolipoprotein L1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. J Am Soc Nephrol. 2013;24(5):722–5. Epub 2013/03/23. doi: DOI:1681/ASN.2012121180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Clarke AJ, Ellinghaus U, Cortini A, Stranks A, Simon AK, Botto M, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis. 2015;74(5):912–20. Epub 2014/01/15. doi: 10.1136/annrheumdis-2013-204343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhaorigetu S, Wan G, Kaini R, Jiang Z, Hu CA. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy. 2008;16(4(8)):1079–82/ 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Perl A, Gergely P Jr., Nagy G, Koncz A, Banki K. Mitochondrial hyperpolarization: a checkpoint of T-cell life, death and autoimmunity. Trends Immunol. 2004;25(7):360–7. Epub 2004/06/23. doi: 10.1016/j.it.2004.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yi W, Gupta S, Ricker E, Manni M, Jessberger R, Chinenov Y, et al. The mTORC1–4E-BP-eIF4E axis controls de novo Bcl6 protein synthesis in T cells and systemic autoimmunity. Nat Commun. 2017;8(1):254. Epub 2017/08/16. doi: 10.1038/s41467-017-00348-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Crow MK. Type I interferon in the pathogenesis of lupus. J Immunol. 2014;192(12):5459–68. Epub 2014/06/08. doi: 10.4049/jimmunol.1002795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhou XJ, Klionsky DJ, Zhang H. Podocytes and autophagy: a potential therapeutic target in lupus nephritis. Autophagy. 2019;15(5):908–12. Epub 2019/02/14. doi: 10.1080/15548627.2019.1580512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Singh N, Birkenbach M, Caza T, Perl A, Cohen PL. Tuberous sclerosis and fulminant lupus in a young woman. J Clin Rheumatol. 2013;19(3):134–7. Epub 2013/03/23. doi: 10.1097/RHU.0b013e318289c033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Carrasco Cubero C, Bejarano Moguel V, Fernández Gil M, Álvarez Vega JL. Coincidence of tuberous sclerosis and systemic lupus erythematosus-a case report. Reumatol Clin. 2016;12(4):219–22. Epub 2015/11/04. doi: 10.1016/j.reuma.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 139.Zeng H, Chi H. mTOR signaling and transcriptional regulation in T lymphocytes. Transcription. 2014;5(2):e28263. Epub 2015/03/13. doi: 10.4161/trns.28263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sam R, Khalid S, Brecklin C, Schwartz M, Dunea G. A case of lymphangioleiomyomatosis with membranous nephropathy and likely systemic lupus. Clin Exp Nephrol. 2009;13(2):166–9. Epub 2008/09/30. doi: 10.1007/s10157-008-0083-0. [DOI] [PubMed] [Google Scholar]
- 141.Futami S, Arai T, Hirose M, Sugimoto C, Ikegami N, Akira M, et al. Comorbid connective tissue diseases and autoantibodies in lymphangioleiomyomatosis: a retrospective cohort study. Orphanet J Rare Dis. 2018;13(1):182. Epub 2018/10/22. doi: 10.1186/s13023-018-0933-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cui H, Cheng C, Xu W, Tian X, Yang Y, Wang Y, et al. The etiology of diffuse cystic lung diseases: an analysis of 1010 consecutive cases in a LAM clinic. Orphanet J Rare Dis. 2021;16(1):273. Epub 2021/06/14. doi: 10.1186/s13023-021-01905-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Olde Bekkink M, Ahmed-Ousenkova YM, Netea MG, van der Velden WJ, Berden JH. Coexistence of systemic lupus erythematosus, tuberous sclerosis and aggressive natural killer-cell leukaemia: coincidence or correlated? Lupus. 2016;25(7):766–71. Epub 2016/03/08. doi: 10.1177/0961203316636466. [DOI] [PubMed] [Google Scholar]
- 144.Nagy G, Koncz A, Perl A. T cell activation-induced mitochondrial hyperpolarization is mediated by Ca2+- and redox-dependent production of nitric oxide. J Immunol. 2003;171(10):5188–97. Epub 2003/11/11. doi: 10.4049/jimmunol.171.10.5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hara K, Yonezawa K, Kozlowski MT, Sugimoto T, Andrabi K, Weng QP, et al. Regulation of eIF-4E BP1 phosphorylation by mTOR. J Biol Chem. 1997;272(42):26457–63. Epub 1997/10/23. doi: 10.1074/jbc.272.42.26457. [DOI] [PubMed] [Google Scholar]
- 146.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–7. Epub 2009/06/27. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 147.Zhao J, Zhai B, Gygi SP, Goldberg AL. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc Natl Acad Sci U S A. 2015;112(52):15790–7. Epub 2015/12/17. doi: 10.1073/pnas.1521919112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chiu H, Jackson LV, Oh KI, Mai A, Ronai ZA, Ruggero D, et al. The mTORC1/4E-BP/eIF4E Axis Promotes Antibody Class Switching in B Lymphocytes. J Immunol. 2019;202(2):579–90. Epub 2018/12/12. doi: 10.4049/jimmunol.1800602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Iwata TN, Ramírez JA, Tsang M, Park H, Margineantu DH, Hockenbery DM, et al. Conditional Disruption of Raptor Reveals an Essential Role for mTORC1 in B Cell Development, Survival, and Metabolism. J Immunol. 2016;197(6):2250–60. Epub 2016/08/16. doi: 10.4049/jimmunol.1600492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ, et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016;23(4):649–62. Epub 2016/04/15. doi: 10.1016/j.cmet.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Doherty E, Oaks Z, Perl A. Increased mitochondrial electron transport chain activity at complex I is regulated by N-acetylcysteine in lymphocytes of patients with systemic lupus erythematosus. Antioxid Redox Signal. 2014;21(1):56–65. Epub 2014/03/29. doi: 10.1089/ars.2013.5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Peairs A, Radjavi A, Davis S, Li L, Ahmed A, Giri S, et al. Activation of AMPK inhibits inflammation in MRL/lpr mouse mesangial cells. Clin Exp Immunol. 2009;156(3):542–51. Epub 2009/05/15. doi: 10.1111/j.1365-2249.2009.03924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lee SY, Moon SJ, Kim EK, Seo HB, Yang EJ, Son HJ, et al. Metformin Suppresses Systemic Autoimmunity in Roquin(san/san) Mice through Inhibiting B Cell Differentiation into Plasma Cells via Regulation of AMPK/mTOR/STAT3. J Immunol. 2017;198(7):2661–70. Epub 2017/03/01. doi: 10.4049/jimmunol.1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sun F, Geng S, Wang H, Wang H, Liu Z, Wang X, et al. Effects of metformin on disease flares in patients with systemic lupus erythematosus: post hoc analyses from two randomised trials. Lupus Sci Med. 2020;7(1). Epub 2020/10/24. doi: 10.1136/lupus-2020-000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Heckmann BL, Boada-Romero E, Cunha LD, Magne J, Green DR. LC3-Associated Phagocytosis and Inflammation. Journal of molecular biology. 2017;429(23):3561–76. Epub 2017/08/25. doi: 10.1016/j.jmb.2017.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3(10):944–50. Epub 2002/09/24. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 157.Mizui M, Koga T, Lieberman LA, Beltran J, Yoshida N, Johnson MC, et al. IL-2 protects lupus-prone mice from multiple end-organ damage by limiting CD4−CD8− IL-17-producing T cells. J Immunol. 2014;193(5):2168–77. Epub 2014/07/27. doi: 10.4049/jimmunol.1400977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tsokos GC, Boumpas DT, Smith PL, Djeu JY, Balow JE, Rook AH. Deficient gamma-interferon production in patients with systemic lupus erythematosus. Arthritis Rheum. 1986;29(10):1210–5. Epub 1986/10/01. doi: 10.1002/art.1780291005. [DOI] [PubMed] [Google Scholar]
- 159.Jacob N, Jacob CO. On anti-tumor necrosis factor-induced systemic lupus erythematosus. J Rheumatol. 2010;37(1):3–5. Epub 2009/12/31. doi: 10.3899/jrheum.091071. [DOI] [PubMed] [Google Scholar]
- 160.Rabilloud T, Heller M, Gasnier F, Luche S, Rey C, Aebersold R, et al. Proteomics analysis of cellular response to oxidative stress. Evidence for in vivo overoxidation of peroxiredoxins at their active site. J Biol Chem. 2002;277(22):19396–401. Epub 2002/03/21. doi: 10.1074/jbc.M106585200. [DOI] [PubMed] [Google Scholar]
- 161.Soforo E, Baumgartner M, Francis L, Allam F, Phillips PE, Perl A. Induction of systemic lupus erythematosus with tumor necrosis factor blockers. J Rheumatol. 2010;37(1):204–5. Epub 2009/12/31. doi: 10.3899/jrheum.081312. [DOI] [PubMed] [Google Scholar]
- 162.Weindel CG, Richey LJ, Bolland S, Mehta AJ, Kearney JF, Huber BT. B cell autophagy mediates TLR7-dependent autoimmunity and inflammation. Autophagy. 2015;11(7):1010–24. Epub 2015/06/30. doi: 10.1080/15548627.2015.1052206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Weindel CG, Richey LJ, Mehta AJ, Shah M, Huber BT. Autophagy in Dendritic Cells and B Cells Is Critical for the Inflammatory State of TLR7-Mediated Autoimmunity. J Immunol. 2017;198(3):1081–92. Epub 2016/12/30. doi: 10.4049/jimmunol.1601307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jones DD, Gaudette BT, Wilmore JR, Chernova I, Bortnick A, Weiss BM, et al. mTOR has distinct functions in generating versus sustaining humoral immunity. J Clin Invest. 2016;126(11):4250–61. Epub 2016/11/02. doi: 10.1172/jci86504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yang L, Brooks CR, Xiao S, Sabbisetti V, Yeung MY, Hsiao LL, et al. KIM-1-mediated phagocytosis reduces acute injury to the kidney. J Clin Invest. 2015;125(4):1620–36. Epub 2015/03/10. doi: 10.1172/jci75417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Talaber G, Miklossy G, Oaks Z, Liu Y, Tooze SA, Chudakov DM, et al. HRES-1/Rab4 promotes the formation of LC3(+) autophagosomes and the accumulation of mitochondria during autophagy. PloS one. 2014;9(1):e84392. Epub 2014/01/10. doi: 10.1371/journal.pone.0084392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gkirtzimanaki K, Kabrani E, Nikoleri D, Polyzos A, Blanas A, Sidiropoulos P, et al. IFNα Impairs Autophagic Degradation of mtDNA Promoting Autoreactivity of SLE Monocytes in a STING-Dependent Fashion. Cell Rep. 2018;25(4):921–33.e5. Epub 2018/10/26. doi: 10.1016/j.celrep.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561(7722):258–62. Epub 2018/08/24. doi: 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhang H, Huang X, Ye L, Guo G, Li X, Chen C, et al. B Cell-Related Circulating MicroRNAs With the Potential Value of Biomarkers in the Differential Diagnosis, and Distinguishment Between the Disease Activity and Lupus Nephritis for Systemic Lupus Erythematosus. Front Immunol. 2018;9:1473. Epub 2018/07/17. doi: 10.3389/fimmu.2018.01473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Carlsen AL, Schetter AJ, Nielsen CT, Lood C, Knudsen S, Voss A, et al. Circulating microRNA expression profiles associated with systemic lupus erythematosus. Arthritis Rheum. 2013;65(5):1324–34. Epub 2013/02/13. doi: 10.1002/art.37890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wei S, Zhang Z, Yan L, Mo Y, Qiu X, Mi X, et al. miR-20a Overexpression in Adipose-Derived Mesenchymal Stem Cells Promotes Therapeutic Efficacy in Murine Lupus Nephritis by Regulating Autophagy. Stem Cells Int. 2021;2021:3746335. Epub 2021/11/02. doi: 10.1155/2021/3746335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Qi YY, Zhou XJ, Cheng FJ, Hou P, Ren YL, Wang SX, et al. Increased autophagy is cytoprotective against podocyte injury induced by antibody and interferon-α in lupus nephritis. Ann Rheum Dis. 2018;77(12):1799–809. Epub 2018/09/14. doi: 10.1136/annrheumdis-2018-213028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Machida H, Ito S, Hirose T, Takeshita F, Oshiro H, Nakamura T, et al. Expression of Toll-like receptor 9 in renal podocytes in childhood-onset active and inactive lupus nephritis. Nephrol Dial Transplant. 2010;25(8):2530–537. Epub 2010/02/26. doi: 10.1093/ndt/gfq058. [DOI] [PubMed] [Google Scholar]
- 174.Wu CY, Hua KF, Chu CL, Yang SR, Arbiser JL, Yang SS, et al. Tris DBA Ameliorates Accelerated and Severe Lupus Nephritis in Mice by Activating Regulatory T Cells and Autophagy and Inhibiting the NLRP3 Inflammasome. J Immunol. 2020;204(6):1448–61. Epub 2020/02/16. doi: 10.4049/jimmunol.1801610. [DOI] [PubMed] [Google Scholar]
- 175.Stratakis S, Stylianou K, Petrakis I, Mavroeidi V, Poulidaki R, Petra C, et al. Rapamycin ameliorates proteinuria and restores nephrin and podocin expression in experimental membranous nephropathy. Clin Dev Immunol. 2013;2013:941893. Epub 2013/09/27. doi: 10.1155/2013/941893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Harr MW, McColl KS, Zhong F, Molitoris JK, Distelhorst CW. Glucocorticoids downregulate Fyn and inhibit IP(3)-mediated calcium signaling to promote autophagy in T lymphocytes. Autophagy. 2010;6(7):912–21. Epub 2010/09/04. doi: 10.4161/auto.6.7.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mao Z, Tan Y, Tao J, Li L, Wang H, Yu F, et al. Renal mTORC1 activation is associated with disease activity and prognosis in lupus nephritis. Rheumatology (Oxford). 2022. Epub 2022/01/19. doi: 10.1093/rheumatology/keac037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lai ZW, Kelly R, Winans T, Marchena I, Shadakshari A, Yu J, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet. 2018;391(10126):1186–96. Epub 2018/03/20. doi: 10.1016/s0140-6736(18)30485-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kobayashi T, Nguyen-Tien D, Sorimachi Y, Sugiura Y, Suzuki T, Karyu H, et al. SLC15A4 mediates M1-prone metabolic shifts in macrophages and guards immune cells from metabolic stress. Proc Natl Acad Sci U S A. 2021;118(33). Epub 2021/08/14. doi: 10.1073/pnas.2100295118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Anders HJ, Saxena R, Zhao MH, Parodis I, Salmon JE, Mohan C. Lupus nephritis. Nat Rev Dis Primers. 2020;6(1):7. Epub 2020/01/25. doi: 10.1038/s41572-019-0141-9. [DOI] [PubMed] [Google Scholar]
- 181.Austin III HA, Muenz LR, Joyce KM, Antonovych TT, Balow JE. Diffuse proliferative lupus nephritis: identification of specific pathologic features affecting renal outcome. Kidney Int. 1984;25(4):689–95. doi: DOI:1038/ki.1984.75. [DOI] [PubMed] [Google Scholar]
- 182.Gourley MF, Austin HA 3rd, Scott D, Yarboro CH, Vaughan EM, Muir J, et al. Methylprednisolone and cyclophosphamide, alone or in combination, in patients with lupus nephritis. A randomized, controlled trial. Ann Intern Med. 1996;125(7):549–57. Epub 1996/10/01. doi: 10.7326/0003-4819-125-7-199610010-00003. [DOI] [PubMed] [Google Scholar]
- 183.Yasuda H, Fukusumi Y, Ivanov V, Zhang Y, Kawachi H. Tacrolimus ameliorates podocyte injury by restoring FK506 binding protein 12 (FKBP12) at actin cytoskeleton. Faseb j. 2021;35(11):e21983. Epub 2021/10/19. doi: 10.1096/fj.202101052R. [DOI] [PubMed] [Google Scholar]
- 184.Hannah J, Casian A, D’Cruz D. Tacrolimus use in lupus nephritis: A systematic review and meta-analysis. Autoimmun Rev. 2016;15(1):93–101. Epub 2015/10/03. doi: 10.1016/j.autrev.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 185.Role of a prostaglandin E1 analogue in the prevention of acute graft rejection by cyclosporine. N Engl J Med. 1990;323(12):831–3. Epub 1990/09/20. doi: 10.1056/nejm199009203231214. [DOI] [PubMed] [Google Scholar]
- 186.Sennesael JJ, Lamote JG, Violet I, Tasse S, Verbeelen DL. Divergent effects of calcium channel and angiotensin converting enzyme blockade on glomerulotubular function in cyclosporine-treated renal allograft recipients. Am J Kidney Dis. 1996;27(5):701–8. Epub 1996/05/01. doi: 10.1016/s0272-6386(96)90106-7. [DOI] [PubMed] [Google Scholar]
- 187.Rovin BH, Teng YKO, Ginzler EM, Arriens C, Caster DJ, Romero-Diaz J, et al. Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2021;397(10289):2070–80. Epub 2021/05/11. doi: 10.1016/s0140-6736(21)00578-x. [DOI] [PubMed] [Google Scholar]
- 188.Pons-Estel GJ, Alarcón GS, McGwin G Jr., Danila MI, Zhang J, Bastian HM, et al. Protective effect of hydroxychloroquine on renal damage in patients with lupus nephritis: LXV, data from a multiethnic US cohort. Arthritis Rheum. 2009;61(6):830–9. Epub 2009/05/30. doi: 10.1002/art.24538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kasitanon N, Fine DM, Haas M, Magder LS, Petri M. Hydroxychloroquine use predicts complete renal remission within 12 months among patients treated with mycophenolate mofetil therapy for membranous lupus nephritis. Lupus. 2006;15(6):366–70. Epub 2006/07/13. doi: 10.1191/0961203306lu2313oa. [DOI] [PubMed] [Google Scholar]
- 190.Cea M, Cagnetta A, Fulciniti M, Tai Y-T, Hideshima T, Chauhan D, et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood. 2012;120(17):3519–29. Epub 2012/09/05. doi: 10.1182/blood-2012-03-416776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Macri C, Wang F, Tasset I, Schall N, Page N, Briand JP, et al. Modulation of deregulated chaperone-mediated autophagy by a phosphopeptide. Autophagy. 2015;11(3):472–86. Epub 2015/02/27. doi: 10.1080/15548627.2015.1017179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Page N, Gros F, Schall N, Décossas M, Bagnard D, Briand JP, et al. HSC70 blockade by the therapeutic peptide P140 affects autophagic processes and endogenous MHCII presentation in murine lupus. Ann Rheum Dis. 2011;70(5):837–43. Epub 2010/12/22. doi: 10.1136/ard.2010.139832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Oaks Z, Winans T, Huang N, Banki K, Perl A. Activation of the Mechanistic Target of Rapamycin in SLE: Explosion of Evidence in the Last Five Years. Curr Rheumatol Rep. 2016;18(12):73. Epub 2016/11/05. doi: 10.1007/s11926-016-0622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Warner LM, Adams LM, Sehgal SN. Rapamycin prolongs survival and arrests pathophysiologic changes in murine systemic lupus erythematosus. Arthritis Rheum. 1994;37(2):289–97. Epub 1994/02/01. doi: 10.1002/art.1780370219. [DOI] [PubMed] [Google Scholar]
- 195.Piranavan P, Perl A. Improvement of renal and non-renal SLE outcome measures on sirolimus therapy - A 21-year follow-up study of 73 patients. Clin Immunol. 2021;229:108781. Epub 2021/06/19. doi: 10.1016/j.clim.2021.108781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lui SL, Tsang R, Chan KW, Zhang F, Tam S, Yung S, et al. Rapamycin attenuates the severity of established nephritis in lupus-prone NZB/W F1 mice. Nephrol Dial Transplant. 2008;23(9):2768–76. Epub 2008/05/01. doi: 10.1093/ndt/gfn216. [DOI] [PubMed] [Google Scholar]
- 197.Yap DY, Ma MK, Tang CS, Chan TM. Proliferation signal inhibitors in the treatment of lupus nephritis: preliminary experience. Nephrology (Carlton). 2012;17(8):676–80. Epub 2012/07/24. doi: 10.1111/j.1440-1797.2012.01646.x. [DOI] [PubMed] [Google Scholar]
- 198.Li X, Zhang X, Pan Y, Shi G, Ren J, Fan H, et al. mTOR regulates NLRP3 inflammasome activation via reactive oxygen species in murine lupus. Acta Biochim Biophys Sin (Shanghai). 2018;50(9):888–96. Epub 2018/07/31. doi: 10.1093/abbs/gmy088. [DOI] [PubMed] [Google Scholar]
- 199.Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. 2006;177(12):8338–47. Epub 2006/12/05. doi: 10.4049/jimmunol.177.12.8338. [DOI] [PubMed] [Google Scholar]
- 200.Yap DYH, Tang C, Chan GCW, Kwan LPY, Ma MKM, Mok MMY, et al. Longterm Data on Sirolimus Treatment in Patients with Lupus Nephritis. J Rheumatol. 2018;45(12):1663–70. Epub 2018/10/03. doi: 10.3899/jrheum.180507. [DOI] [PubMed] [Google Scholar]
- 201.Canaud G, Bienaime F, Tabarin F, Bataillon G, Seilhean D, Noel LH, et al. Inhibition of the mTORC pathway in the antiphospholipid syndrome. N Engl J Med. 2014;371(4):303–12. doi: 10.1056/NEJMoa1312890. [DOI] [PubMed] [Google Scholar]
- 202.Lieberman LA, Tsokos GC. The IL-2 defect in systemic lupus erythematosus disease has an expansive effect on host immunity. J Biomed Biotechnol. 2010;2010:740619. Epub 2010/07/14. doi: 10.1155/2010/740619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.He J, Zhang R, Shao M, Zhao X, Miao M, Chen J, et al. Efficacy and safety of low-dose IL-2 in the treatment of systemic lupus erythematosus: a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2020;79(1):141–9. Epub 2019/09/21. doi: 10.1136/annrheumdis-2019-215396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Koga T, Mizui M, Yoshida N, Otomo K, Lieberman LA, Crispín JC, et al. KN-93, an inhibitor of calcium/calmodulin-dependent protein kinase IV, promotes generation and function of Foxp3⁺ regulatory T cells in MRL/lpr mice. Autoimmunity. 2014;47(7):445–50. Epub 2014/05/16. doi: 10.3109/08916934.2014.915954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Koga T, Ichinose K, Kawakami A, Tsokos GC. Current Insights and Future Prospects for Targeting IL-17 to Treat Patients With Systemic Lupus Erythematosus. Front Immunol. 2020;11:624971. Epub 2021/02/19. doi: 10.3389/fimmu.2020.624971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Liang B, Gardner DB, Griswold DE, Bugelski PJ, Song XY. Anti-interleukin-6 monoclonal antibody inhibits autoimmune responses in a murine model of systemic lupus erythematosus. Immunology. 2006;119(3):296–305. Epub 2006/10/28. doi: 10.1111/j.1365-2567.2006.02433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wallace DJ, Strand V, Merrill JT, Popa S, Spindler AJ, Eimon A, et al. Efficacy and safety of an interleukin 6 monoclonal antibody for the treatment of systemic lupus erythematosus: a phase II dose-ranging randomised controlled trial. Ann Rheum Dis. 2017;76(3):534–42. Epub 2016/09/28. doi: 10.1136/annrheumdis-2016-209668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Olsen S, Burdick JF, Keown PA, Wallace AC, Racusen LC, Solez K. Primary acute renal failure (“acute tubular necrosis”) in the transplanted kidney: morphology and pathogenesis. Medicine. 1989;68(3):173–87. [DOI] [PubMed] [Google Scholar]
- 209.Kyttaris VC, Zhang Z, Kuchroo VK, Oukka M, Tsokos GC. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J Immunol. 2010;184(9):4605–9. Epub 2010/03/24. doi: 10.4049/jimmunol.0903595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.van Vollenhoven RF, Hahn BH, Tsokos GC, Wagner CL, Lipsky P, Touma Z, et al. Efficacy and safety of ustekinumab, an IL-12 and IL-23 inhibitor, in patients with active systemic lupus erythematosus: results of a multicentre, double-blind, phase 2, randomised, controlled study. Lancet. 2018;392(10155):1330–9. Epub 2018/09/27. doi: 10.1016/s0140-6736(18)32167-6. [DOI] [PubMed] [Google Scholar]
- 211.Schmidt T, Paust HJ, Krebs CF, Turner JE, Kaffke A, Bennstein SB, et al. Function of the Th17/interleukin-17A immune response in murine lupus nephritis. Arthritis & rheumatology (Hoboken, NJ). 2015;67(2):475–87. Epub 2014/11/12. doi: 10.1002/art.38955. [DOI] [PubMed] [Google Scholar]
- 212.Casey KA, Guo X, Smith MA, Wang S, Sinibaldi D, Sanjuan MA, et al. Type I interferon receptor blockade with anifrolumab corrects innate and adaptive immune perturbations of SLE. Lupus Sci Med. 2018;5(1):e000286. Epub 2018/12/13. doi: 10.1136/lupus-2018-000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lee J, Park Y, Jang SG, Hong SM, Song YS, Kim MJ, et al. Baricitinib Attenuates Autoimmune Phenotype and Podocyte Injury in a Murine Model of Systemic Lupus Erythematosus. Front Immunol. 2021;12:704526. Epub 2021/09/10. doi: 10.3389/fimmu.2021.704526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wenderfer SE, Stepkowski SM, Braun MC. Increased survival and reduced renal injury in MRL/lpr mice treated with a novel sphingosine-1-phosphate receptor agonist. Kidney Int. 2008;74(10):1319–26. Epub 2008/09/05. doi: 10.1038/ki.2008.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Okazaki H, Hirata D, Kamimura T, Sato H, Iwamoto M, Yoshio T, et al. Effects of FTY720 in MRL-lpr/lpr mice: therapeutic potential in systemic lupus erythematosus. J Rheumatol. 2002;29(4):707–16. Epub 2002/04/13. [PubMed] [Google Scholar]
- 216.La Mantia L, Tramacere I, Firwana B, Pacchetti I, Palumbo R, Filippini G. Fingolimod for relapsing-remitting multiple sclerosis. Cochrane Database Syst Rev. 2016;4:Cd009371. Epub 2016/04/20. doi: 10.1002/14651858.CD009371.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Taniguchi M, Kitatani K, Kondo T, Hashimoto-Nishimura M, Asano S, Hayashi A, et al. Regulation of autophagy and its associated cell death by “sphingolipid rheostat”: reciprocal role of ceramide and sphingosine 1-phosphate in the mammalian target of rapamycin pathway. J Biol Chem. 2012;287(47):39898–910. Epub 2012/10/05. doi: 10.1074/jbc.M112.416552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Furie RA, Aroca G, Cascino MD, Garg JP, Rovin BH, Alvarez A, et al. B-cell depletion with obinutuzumab for the treatment of proliferative lupus nephritis: a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2021. Epub 2021/10/08. doi: 10.1136/annrheumdis-2021-220920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Furie R, Nicholls K, Cheng TT, Houssiau F, Burgos-Vargas R, Chen SL, et al. Efficacy and safety of abatacept in lupus nephritis: a twelve-month, randomized, double-blind study. Arthritis & rheumatology (Hoboken, NJ). 2014;66(2):379–89. Epub 2014/02/08. doi: DOI: 10.1002/art.38260. [DOI] [PubMed] [Google Scholar]
- 220.Cassia MA, Alberici F, Jones RB, Smith RM, Casazza G, Urban ML, et al. Rituximab as Maintenance Treatment for Systemic Lupus Erythematosus: A Multicenter Observational Study of 147 Patients. Arthritis & rheumatology (Hoboken, NJ). 2019;71(10):1670–80. Epub 2019/05/19. doi: 10.1002/art.40932. [DOI] [PubMed] [Google Scholar]
- 221.Ostendorf L, Burns M, Durek P, Heinz GA, Heinrich F, Garantziotis P, et al. Targeting CD38 with Daratumumab in Refractory Systemic Lupus Erythematosus. N Engl J Med. 2020;383(12):1149–55. Epub 2020/09/17. doi: 10.1056/NEJMoa2023325. [DOI] [PubMed] [Google Scholar]
- 222.Friedman B, Corciulo C, Castro CM, Cronstein BN. Adenosine A2A receptor signaling promotes FoxO associated autophagy in chondrocytes. Sci Rep. 2021;11(1):968. Epub 2021/01/15. doi: 10.1038/s41598-020-80244-x. treating osteoarthritis and promoting cartilage formation (US Patent 10,441,541) assigned to NYU School of Medicine, and they are co-founders of Regenosine Inc. BN Cronstein has consulted for Eli Lilly & Co., Horizon Pharmaceuticals and AstraZeneca and has a grant from AstraZeneca. B Friedman & CM Castro declare no competing financial interests. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Castro CM, Corciulo C, Solesio ME, Liang F, Pavlov EV, Cronstein BN. Adenosine A2A receptor (A2AR) stimulation enhances mitochondrial metabolism and mitigates reactive oxygen species-mediated mitochondrial injury. Faseb j. 2020;34(4):5027–45. Epub 2020/02/14. doi: 10.1096/fj.201902459R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Levack RC, Newell KL, Cabrera-Martinez B, Cox J, Perl A, Bastacky SI, et al. Adenosine receptor 2a agonists target mouse CD11c(+)T-bet(+) B cells in infection and autoimmunity. Nat Commun. 2022;13(1):452. Epub 2022/01/23. doi: 10.1038/s41467-022-28086-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Perl A Review: Metabolic Control of Immune System Activation in Rheumatic Diseases. Arthritis & rheumatology (Hoboken, NJ). 2017;69(12):2259–70. Epub 2017/08/26. doi: 10.1002/art.40223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Huang N, Perl A. Metabolism as a Target for Modulation in Autoimmune Diseases. Trends Immunol. 2018;39(7):562–76. Epub 2018/05/10. doi: 10.1016/j.it.2018.04.006. [DOI] [PubMed] [Google Scholar]