

Abstract
Effective systemic delivery of small interfering RNAs (siRNAs) to tissues other than liver remains a challenge. siRNAs are small (∼15 kDa) and therefore rapidly cleared by the kidneys, resulting in limited blood residence times and tissue exposure. Current strategies to improve the unfavorable pharmacokinetic (PK) properties of siRNAs rely on enhancing binding to serum proteins through extensive phosphorothioate modifications or by conjugation of targeting ligands. Here, we describe an alternative strategy for enhancing blood and tissue PK based on dynamic modulation of the overall size of the siRNA. We engineered a high-affinity universal oligonucleotide anchor conjugated to a high-molecular-weight moiety, which binds to the 3′ end of the guide strand of an asymmetric siRNA. Data showed a strong correlation between the size of the PK-modifying anchor and clearance kinetics. Large 40-kDa PK-modifying anchors reduced renal clearance by ∼23-fold and improved tissue exposure area under the curve (AUC) by ∼26-fold, resulting in increased extrahepatic tissue retention (∼3- to 5-fold). Furthermore, PK-modifying oligonucleotide anchors allowed for straightforward and versatile modulation of blood residence times and biodistribution of a panel of chemically distinct ligands. The effects were more pronounced for conjugates with low lipophilicity (e.g., N-Acetylgalactosamine [GalNAc]), where significant improvement in uptake by hepatocytes and dose-dependent silencing in the liver was observed.
Keywords: oligonucleotide, PEGylation, extrahepatic, GalNAc, siRNA conjugates, RNA interference, Gene silencing
Graphical abstract
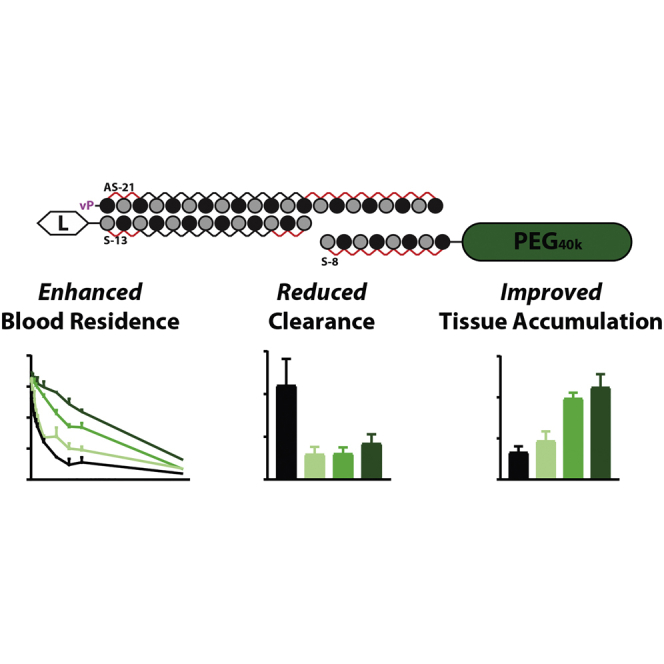
PK-modifying anchors constitute a versatile strategy from modulation of the overall hydrodynamic size of conjugated siRNAs, increasing their blood circulating times and tissue exposure after intravenous administrations. The concept is applicable to virtually any sequence and is of particular interest to expand the therapeutic use of siRNAs beyond liver applications.
Introduction
RNA interference (RNAi)-based therapeutics are on the brink of revolutionizing human medicine by enabling the treatment of a wide range of incurable genetically defined disorders through modulation of disease-associated RNA transcripts. In recent years, the clinical landscape of these drugs has rapidly accelerated resulting in the groundbreaking approvals of four drugs, patisiran, givosiran, lumasiran and vutrisiran, for the treatment of liver disorders.1, 2, 3 Despite the success stories in the liver, developing small interfering RNA (siRNA) constructs for systemic administration still pose great challenges due to their poor pharmacokinetics (PK) proprieties. Indeed, both partially or fully modified unconjugated siRNAs quickly (>90%) clear from the bloodstream after intravenous administration with an elimination half-life of 2–5 min.4, 5, 6, 7, 8, 9 The main reasons for such rapid clearance are believed to be: (i) the inability to bind plasma and cell-surface proteins preventing fast cellular uptake into target tissues and (ii) the relatively small size of the oligonucleotide molecule (7–15 kDa), which is well below the cutoff for kidney filtration.
Widely accepted synthetic strategies to modulate PK of therapeutic oligonucleotides are backbone modifications and conjugation chemistries. For decades, phosphorothioate (PS) linkages have been used in the field of antisense oligonucleotides, not only to improve stability but to enhance non-specific binding to plasma proteins, hence reducing immediate clearance of these compounds. This strategy relies on the unique properties of a single-stranded PS-containing oligonucleotide, with stretches of 10–12 PS being necessary to achieve substantial serum protein binding.10 This approach is not readily translatable to siRNAs, where the rigid structure of the duplex is believed to limit PS-driven interactions.10 Nonetheless, the presence of a single-stranded phosphorothioated overhang on asymmetric siRNAs in the context of conjugated siRNAs has been shown to enhance distribution and efficacy in vivo.11
By far the most successful approach to modify PK of siRNA scaffolds consists of the use of a conjugated ligand. N-Acetylgalactosamine (GalNAc) leverages the asialoglycoprotein (ASGPR) receptor and allows rapid and targeted delivery of siRNAs to hepatocytes.12 The GalNAc delivery platform capitalizes on the high expression and turnover of the ASGPR as well as on the discontinued and fenestrated epithelia, high blood flow, and vascularization of the liver to enable distribution to this organ. Despite their effectiveness, GalNAc-siRNAs are still rapidly eliminated by kidney filtration, especially if administered intravenously, and therefore require careful consideration of the route of administration. Subcutaneous administration of GalNAc-siRNAs adds an important absorption phase that counteracts the rapid initial renal clearance to produce meaningful gene silencing in the liver. Adequate selection of the route administration plays an important role in systemic delivery of GalNAc oligonucleotides and will also most certainly be crucial in the context of other conjugate systems. However, the route of administration alone is not likely to provide a complete solution for the poor PK proprieties of siRNAs in a context of less efficient conjugates and will need to be used in conjunction with other strategies to improve biodistribution. Although the uniqueness of the GalNAc platform is currently unparalleled, other ligand- or antibody-based targeted-delivery approaches have been explored, with varying degrees of success, against glucagon-like peptide receptor (GLP1) for pancreas targeting13 or transferrin receptor for skeletal and cardiac muscle targeting.14 Furthermore, conjugation of lipid moieties has enabled broad functional delivery of therapeutic oligonucleotides to a variety of extrahepatic tissues.11,15 Different lipid configurations enable interactions with a well-defined set of plasma proteins, delaying renal clearance and leading to a preestablished tissue-distribution profile in part defined by the protein binding partner (s).8,9,11 While delivery to many tissues, including muscle, heart, and adipose tissue, has been demonstrated with lipid conjugates, a significant part of the dose (30%–50%) is still rapidly cleared upon administration and/or accumulates in primary clearance tissues. Apart from lipid- and GalNAc-based approaches, the success of most other receptor-ligand pairs of interest has been, in general, hindered by relatively low recycling/turnover time of the receptor, low blood flow at the target tissues, and existence of complex continuous endothelia. Therefore, developing strategies that allow for longer exposure to target tissues without solely relying on protein binding remains a critical requirement.
Modulation of the overall hydrodynamic size by conjugation of a large inert polymer like polyethylene glycol (PEG) has been long explored as a way to improve blood residence of different classes of drugs.16 Size increases above the molecular cutoff for kidney filtration result in delayed renal clearance and longer circulating times.16 Here, we describe and characterize a strategy for enhancing blood residence and biodistribution of therapeutic oligonucleotides based on the dynamic modulation of the overall size of the oligonucleotide drug. To this end, we engineered a universal anchor based on a high-affinity 8-mer oligonucleotide conjugated to PEG, which binds to the 3′ end of the guide strand of an asymmetric siRNA at a single-stranded region. Data show a strong direct correlation between the molecular weight of the PK-modifying anchor (10–40 kDa) and the reduction in clearance kinetics, with 40 kDa anchors improving tissue exposure by ∼23-fold and reducing renal clearance by ∼26-fold correspondingly. As expected, a decrease in clearance rates results in ∼3- to 5-fold improvement in tissue retention (liver, spleen, pancreas, heart, and adrenals) and dose-dependent silencing. The relative contribution of the increase in overall size was more pronounced in a context of non-lipophilic conjugates and paved the path toward enabling discovery and exploration of novel ligands with the mission of achieving selective and targeted delivery to tissues beyond the liver.
Results
Developing an oligonucleotide anchor system for modulation of siRNA biodistribution in vivo
Generally therapeutic oligonucleotides (6–18 kDa) are cleared from the blood within minutes after systemic administration given that their molecular weight (MW) lies below the cutoff for glomerular filtration (40–60 kDa) in the kidney.17 In order to modulate the clearance kinetics of these compounds and improve tissue distribution in vivo, we designed a system to dynamically increase the overall size of the molecule by taking advantage of the asymmetric nature of fully modified siRNA scaffolds (Figure 1A, left panel). We designed an oligonucleotide anchor that is fully complementary to the overhanging tail at the 3′ end of the antisense strand (AS). A PK-modifying unit was conjugated at the 5′ end of the anchor to enable formation of a large multi-strand duplex (∼56 kDa). A high molecular PEG moiety (40 kDa) was used as a proof-of-concept polymer. The small oligonucleotide anchors used were fully phosphorothioated to increase nuclease stability.
Figure 1.

A minimum of eight fully modified nt bases are necessary to support formation of a multi-strand siRNA duplex for in vivo delivery
(A) (Left) Schematic of fully modified fluorescently labeled asymmetric siRNA and of oligonucleotide anchors of varying length (5–8 nt) covalently attached to a polyethylene glycol (PEG) moiety. (Right) Gel-shift assay depicting the ability of each oligonucleotide anchor to hybridize with the parent asymmetric siRNA duplex in a range of molar ratios. The oligonucleotide anchor must have at least 8 nt to enable the shift of the parent duplex to the top of the gel, indicating formation of a multi-strand duplex. (B) (Top) Wild-type FVB/N female mice were treated intravenously (23.5 nmol, ∼12 mg/kg) with cholesterol-conjugated asymmetric siRNA duplex hybridized to a Cy3-labeled 8-nt-long oligonucleotide anchor (cholesterol [Chol] 21-13-8 cy3) or with the anchor alone (8-mer Cy3). (Bottom) Tiled fluorescent images (10× objective) were obtained from tissue sections of the liver and the kidney 48 h post-injection. The shift in biodistribution indicates that the 8-mer anchor enables the formation of a multi-strand duplex that is stable in vivo. n = 2/group. Blue: nuclei (DAPI), red: Cy3-labeled oligonucleotide. Scale bar, 2 mm.
To evaluate the minimal length of the anchor necessary to support efficient formation of a multi-strand duplex, a panel of different size anchors (5- to 8-nt long) were synthesized and conjugated to a 40-kDa PEG (Figure 1A, right panel). Successful hybridization of the anchor to the parent duplex will delay migration of the duplex in the gel, resulting in a visible band at the top (Figure 1A, right panel). No detectable duplex formation was observed with 5- and 6-nt-long anchors, with the 7-mer anchor requiring 4-fold molar excess for hybridization. Thus, a minimum of 8 nucleobases was required for adequate binding of the anchor to the parent asymmetric siRNA scaffold (GC rich, ∼88%) and formation of a multi-strand duplex in vitro.
We then needed to confirm that the complex would be sufficiently stable in vivo, where body temperature (37°C) and the diverse protein interactions might interfere. A cyanine 3 (Cy3)-labeled 8-mer anchor (8-mer Cy3) was synthesized and intravenously (i.v.) administered by itself or annealed to a cholesterol-conjugated asymmetric siRNA (cholesterol [Chol] 21-13-8 Cy3) (Figure 1B, top panel), and tissues were collected for analysis at 48 h. As expected for a short fully phosphorothioated oligonucleotide,10 the 8-mer Cy3 anchor was predominantly retained by the kidney’s proximal epithelia with limited liver exposure (Figure 1B, bottom panel). Chol-conjugated siRNAs preferentially distribute to the liver.11,18,19 Preannealing the 8-mer Cy3 anchor with a Chol-conjugated siRNA led to a major shift in its distribution from the kidney to the liver, suggesting that the multi-strand duplex has sufficient stability in vivo. Together, in vitro and in vivo data demonstrated that an 8-mer oligonucleotide anchor supports stable formation of a multi-strand duplex.
PK-modifying anchors efficiently delay clearance kinetics and enhance tissue distribution of fully modified siRNA duplexes
In order to study the effect of size of the PK-modifying unit in the clearance kinetics and biodistribution of unconjugated asymmetric siRNA duplexes, PEG moieties of different sizes (10–40 kDa) were conjugated to the 5′ end of the 8-mer oligonucleotide anchor (Figure 2A, left panel). All anchors were tested and shown to be able to form a stable multi-strand duplex, producing a shift based on the MW of the PEG moiety in the gel electrophoresis assay (Figure 2A, right panel).
Figure 2.

Enhancement in blood residence and tissue accumulation is strongly correlated to the molecular weight of PK-modifying anchors
(A) (Left) Schematic of fully modified fluorescently labeled asymmetric siRNA and of 8-mer oligonucleotide anchors covalently attached to a variety of high-molecular-weight PEG moieties. (Right) Gel-shift assay depicting binding and hybridization of each variant of the 8-mer anchor to the parent siRNA duplex (21–13), highlighting the differences in migration according to size. (B and C) Wild-type FVB/N female mice treated intravenously (28.5 nmol, ∼13 mg/kg) with siRNA duplexes containing different PEG sizes hybridized to the parent asymmetric compound through an 8-mer anchor. Concentrations of the guide strand in the blood and tissues were assessed by PNA-based hybridization assay and values normalized to the MW of the parent unconjugated siRNA duplex for comparison. (B) (Left) Concentration-time profiles of the parent siRNA using different size PK-modifying anchors. Serial blood samples were collected from the saphenous vein. (Right) Pharmacokinetic parameters calculated based on non-compartmental analysis. (C) Tissue biodistribution profile relative to the parent siRNA when using different size PK-modifying anchors at 48 h post-injection. n = 4–5/group. Mean ± Standard deviation (SD).
Duplexes containing PK-modifying anchors and the parent unconjugated siRNA compound were administered i.v. in mice (28.5 nmol, ∼13 mg/kg). Blood samples were taken at given time points and assayed for the presence of the guide strand by peptide nucleic acid (PNA) hybridization assay to determine the concentration-time profile of each compound in this matrix (Figure 2B). The summary of PK parameters calculated by non-compartmental analyses of the drug profiles in the blood is shown in Figure 2B. As expected, unconjugated siRNAs (21–13) were rapidly cleared from circulation, whereas duplexes containing 10 (21-13-8 PEG10k), 20 (21-13-8 PEG20k), or 40 kDa (21-13-8 PEG40k) PEG moieties displayed delayed clearance rates (3-, 11-, and 23-fold improvement, respectively) (Figure 2B, table). The maximum concentration (CMax) detected for the unconjugated 21–13 parent compound (t = 5 min, 42.3 μg/mL) was significantly lower than what was observed for any of the PEGylated compounds (t = 5 min, 150.1, 192.4, and 273.8 μg/mL, respectively) (Figure 2B, table), suggesting that ∼80%–90% of the unconjugated 21–13 compound was cleared within 5 min post-injection. The area under the curve (AUC) was calculated based on the trapezoidal rule and informed on the total overall exposure to the drug. Even smaller PK-modifying anchors containing PEG10 kDa produced a ∼3-fold increase in the AUC relative to the parent unconjugated siRNA, but significantly more pronounced enhancements were observed with PEG 20 kDa and PEG 40 kDa (12- and 26-fold, respectively) (Figure 2B, table). Together, these data showed that there was a size-dependent decrease in clearance rates (inverse correlation) that translated into significant improvements in overall tissue exposure (direct correlation), with PK-modifying anchors containing 40 kDa PEG moieties providing the best improvements in all PK parameters.
The concentration of compound in the blood is the result of the interplay between distribution (tissue uptake) and elimination (kidney clearance). To investigate if the higher AUCs and lower clearance rates granted by the PK-modifying anchors would result in enhanced tissue distribution, we quantified the presence of siRNA guide strand in multiple tissues (Figure 2C) or assessed tissue accumulation by fluorescent microscopy (Figure 3B) at 48 h post-injection. Overall, PK-modifying anchors enhanced delivery to a wide variety of tissues including liver, spleen, pancreas, heart, and adrenals after single i.v. administration. Although siRNA constructs containing the smaller 10 kDa PEG moiety showed a positive trend compared with the parent compound, this group only achieved statistically significant improvements in tissue accumulation in the liver and heart (Figure 2C). On the other hand, larger PEG moieties (20 and 40 kDa) showed ∼2- to 4-fold statistically significant enhancement in accumulation for most organs when compared with the unconjugated parent siRNA, with 21-13-8 PEG 40 kDa performing the best among the configurations tested. There was a positive correlation between the size of the PEG moiety and the improvement observed for most organs evaluated, except for the kidneys and the lungs. Conjugation of a PEG moiety of any size effectively produced a significant shift in distribution away from the kidney to other tissues with no major effect of size of the PEG. The reduction in kidney accumulation was not only detected by PNA hybridization assay (Figure 2C) but was also noticeable by fluorescent microscopy, where PEGylated constructs showed significantly less Cy3 signal in the cortex of the kidney than the parent siRNA compound (Figure 3B). The abrupt decrease in kidney accumulation with any of the PK-modifying anchors is likely to be a cumulative result of the enhanced distribution to other tissues and the reduction in retention at proximal epithelia in the kidneys due to the large size of the construct. Lastly, in the lungs, no significant benefits on tissue accumulation were observed with the use of PK-modifying anchors, with all compounds presenting a similar level of accumulation to the parent siRNA compound (∼10 ng/mg).
Figure 3.

High-molecular-weight PK-modifying anchors delay kidney clearance and enhance distribution of fully modified asymmetric siRNA duplexes to the liver after single intravenous administration
(A) Schematic of fully modified Cy3-labeled asymmetric siRNA and of 8-mer oligonucleotide anchors covalently attached to a variety of high-molecular-weight (MW) PEG moieties. (B) Wild-type FVB/N female mice were treated intravenously (single dose, 28.5 nmol) with siRNA duplexes containing different MW PEGs hybridized to the parent asymmetric compound through an 8-mer anchor. Tissue biodistribution was assessed 48 h post-injection. (Top) Tiled fluorescent images of sections of the liver. (Bottom) Tiled fluorescent images of the kidney (5× objective, scale bar, 2 mm). n = 4–5/group.
PK-modifying anchors containing a 40-kDa PEG moiety also delayed clearance kinetics and enhanced tissue distribution of fully modified asymmetric siRNAs after a single subcutaneous administration. In this experiment, the Cy3 label was removed, and both compounds contained a 5’-(E)-vinylphosphonate moiety at the 5′ end of the guide strand (Figure S1A), a modification that was previously shown to stabilize terminal phosphate and interactions with the RNA-induced silencing complex (RISC). Subcutaneous (s.c.) administration of unconjugated siRNAs resulted in peak concentrations in the blood at ∼45 min, whereas PK-modifying anchors significantly delayed time to peak to ∼8 h (Figure S1B, table). Similar to what was observed after i.v. injection, with s.c. administration, PK-modifying anchors significantly enhanced blood circulating times, as reflected by a bigger AUC (8.7-fold), as well as a reduced clearance rate (6.5-fold), when compared with unconjugated siRNAs. Biodistribution analyses showed that PK-modifying anchors containing a 40-kDa PEG significantly reduced clearance and delivery to the kidney (5.6-fold lower than unconjugated) (Figure S1C). Distribution was significantly enhanced to the pancreas (1.6-fold), heart (4.3-fold), spleen (1.6-fold), liver (4.3-fold), and skin (4.2-fold) but not in the lungs and adrenals. While overall results of the impact of PK-modifying anchors on siRNA distribution were similar after i.v. and s.c. administration, the magnitude of the effects were significantly more profound after i.v. administration.
The largest PEG (40 kDa) moiety consistently showed the highest impact on clearance, and it was selected for all follow-up experiments.
PK-modifying anchors allow efficient modulation of blood circulating times and biodistribution of chemically distinct siRNA conjugates
The clinical success of conjugate-mediated delivery has encouraged the search and evaluation of a large number of ligand modalities for targeted or broad functional delivery of therapeutic oligonucleotides.20 In this context, the conjugated modality has been shown to play an important role determining serum protein interactions, which ultimately shape the distribution profile of these drugs.11 Thus, here we sought to investigate the utility of the PK-modifying anchor concept to modulate distribution of chemically distinct siRNAs conjugates. For this, we selected several known ligands, such as N-Acetylgalactosamine (GalNAc), docosahexanoic acid (DHA), docosanoic acid (DCA), and Chol, with significantly different physicochemical proprieties and varying degrees of hydrophobicity (Figures 4A and S2). Rank ordered from high to low in a relative scale of hydrophobicity: Chol > DCA > DHA > GalNAc > unconjugated (Figure S2). In these studies, no fluorescent label was used, and the guide strand contained a vinylphosphonate moiety at the 5′ end.
Figure 4.

PK-modifying anchors allow efficient modulation of blood circulating times and biodistribution of chemically distinct siRNA conjugates
(A) (Top) Schematic of fully modified asymmetric siRNAs conjugated to sugar (N-Acetylgalactosamine [GalNAc]) or lipid moieties (docosahexaenoic acid [DHA], docosanoic acid [DCA], Chol) at the 3′ end of the sense strand. (Bottom) Schematic of an 8-mer oligonucleotide anchor conjugated to a 40-kDa PEG moiety binding to a parent asymmetric siRNA containing a ligand. (B) Wild-type FVB/N female mice treated intravenously (28.5 nmol, ∼13 mg/kg) with parent asymmetric siRNA duplex (21–13) containing a ligand (sugar or lipid) or a PEGylated variant (21-13-8 PEG40k) of the parent compound. Concentrations of the guide strand in the blood and tissues were assessed by PNA-based hybridization assay, and values were normalized to the MW of an unconjugated 21–13 asymmetric siRNA duplex for comparison. (B) (Left) Concentration-time profiles for each parent asymmetric siRNA and corresponding PEGylated version using PK-modifying anchors. Serial blood samples were collected from the saphenous vein. (Right) Tissue biodistribution profile for each parent asymmetric siRNA and corresponding PEGylated version at 48 h post-injection. n = 4–5/group. Mean ± Standard deviation (SD).
PK-modifying anchors improved blood circulating times for all siRNA conjugates tested after a single i.v. administration (Figure 4B, left panel). Results showed that less hydrophobic conjugates, such as GalNAc and DHA, when modulated using PK-modifying anchors, display a substantial enhancement in AUC (∼20- and ∼9-fold, respectively), as well as lower clearance rates (∼16- and ∼9-fold, respectively), when compared with their respective parent siRNA counterpart (Table S2)Table S1. Although more hydrophobic scaffolds also presented improvement in blood AUCs (∼5-fold for both DCA and Chol) and reduction in clearance rate (∼4-fold for DCA; ∼5-fold for Chol) when PEGylated, the differences against the respective parent compound were less pronounced than for less lipophilic conjugates (Table S2)Table S1.
Overall, PK-modifying anchors reduced kidney clearance/delivery and improved tissue retention in target organs of accumulation for all siRNA conjugates tested (Figure 4B, right panel). In the kidney, the effect was once again more significant for less lipophilic compounds (GalNAc, ∼4-fold; DHA, ∼5-fold) than for hydrophobic conjugates (DCA, ∼2-fold; Chol, ∼2-fold). GalNAc and DHA conjugates, when PEGylated using the oligonucleotide anchor system, displayed higher fold enhancements (with statistical significance) than hydrophobic conjugates for a variety of different organs. DCA and Chol parent siRNA compounds achieved higher baseline levels of delivery compared with GalNAc and DHA parent compounds, and the use of the PK-modifying anchors only modestly improved delivery for these hydrophobic scaffolds (Figure 4B, right panel).
Approved GalNAc-conjugated siRNA drugs are administered s.c. given their fast clearance kinetics profile if administered i.v. In this study, PK-modifying anchors improved delivery of GalNAc conjugates to the liver by ∼3-fold after a single i.v. injection. This result encouraged us to further investigate the utility of the PK-modifying anchor system to improve delivery of GalNAc conjugates for therapeutic gene silencing in the liver after i.v. administration.
Development of a standardized GC-rich region for effective modulation of AU-rich sequences using PK-modifying oligonucleotide anchors
PK-modifying anchors remarkably improved distribution of parent GalNAc siRNAs to the liver by ∼3-fold after a single i.v. administration (Figure 4B, right panel). This result prompted us to study the applicability of the oligonucleotide anchor concept to other siRNA sequences against gene targets with relevant expression in the liver. To this end, we designed and tested an 8-mer anchor system in the context of a previously selected huntingtin (HTT)-targeting siRNA.
The HTT-targeting siRNA lead in this study has an AU-rich 3′ end tail (GC content ∼25%) on the AS strand (Figure 5A, left panel; Table S1). Thus, unsurprisingly, the 8-mer oligonucleotide anchor conjugated to a 40 kDa PEG failed to efficiently bind and produce the expected shift of the duplex on the gel, even when the molar amounts of the anchor used was 4-fold higher than the parent duplex (Figure 5A, right panel). Based on initial experiments (Figure 1A), a shorter 6-mer control was included in the study, which was also equally unsuccessful in binding and hybridizing to the parent siRNA (Figure 5A, right panel). This experiment confirmed a somewhat foreseeable challenge that should be addressed in order to expand the applicability of the concept to virtually any siRNA sequence.
Figure 5.

Optimization of a standardized GC-rich PK-modifying anchor
(A) (Left) Schematics of siRNA duplex containing AU-rich region at the tail of the guide strand and corresponding AU-rich oligonucleotide anchors (6- and 8-nt long) covalently attached to a PEG moiety. Legend applies only to oligo schematics in (A) and (C). (Right) Gel-shift assay illustrating poor binding for both 6- and 8-mer oligonucleotide anchors to the parent asymmetric siRNA duplex in a range of molar ratios. Dashed line box indicates the expected shift on the 21–13 duplex if successful hybridization of the anchor occurred (B) (Right) Schematics of Chol-conjugated siRNA duplexes of different lengths (23–15 and 25–17). White circles represent nucleotides that have full complementarity to the mRNA target and passenger strand. Blue circles represent a GC-rich conserved region that is not complementary to the mRNA target. The target binding region (nt 2–17) of the guide strand is indicated by the red bracket. (Left) Concentration-response curves assessed in HeLa cells using RNAiMax. After 72 h, incubation samples were analyzed by Quantigene bDNA assay. Data were normalized to housekeeping gene (Hprt) and displayed as a percentage of untreated control cells. n = 3. Mean ± Standard deviation (SD). (C) (Top) Schematics of GalNAc-conjugated siRNAs with 25-mer antisense strands, containing a GC-rich conserved region (region between arrows) from nt 18 through 23-25. The first 17 nt are fully complementary to the respective mRNA target (dashed box), and the GC-rich tail allows anchoring of an 8-mer covalently attached to a PEG moiety. (Bottom) Gel-shift assay demonstrating successful hybridization of the standard GC-rich 8-mer anchor to HTT-targeting and to APOE-targeting 25–17 siRNA duplexes. Gels were stained with SYBR gold.
To circumvent this limitation and enable modulation of sequences with high AU content, a conserved GC-rich region (GC content ∼88%) was introduced at the 3′ end of the AS strand (Figure 5B, right panel, blue circles denote conserved region). For convenience, this region consisted of the 8 last nt of the soluble vascular endothelial growth factor receptor 1 (sFLT1) sequence used in the original 8-mer anchor system design. Given that at least 8 nt were previously shown to be required for efficient binding, the length of the AS strand was increased to 23 and 25 nucleotides to reduce interference of the conserved region with the mRNA target binding region. Evaluation of efficacy in HeLa cells (7-point concentration-response study) showed that introducing the conserved region from nt position 16 of the AS strand had a significant detrimental effect on efficacy and potency when compared with fully complementary siRNAs (half maximal effective concentration [EC50] ∼225 and ∼6 pM, respectively) (Figure 5B, left panel). On the other hand, when the conserved region was only introduced from nt position 18 of the AS strand, similar potency to fully complementary siRNA was observed (EC50 ∼13 and ∼10 pM, respectively). In this case, even though the standardized region at the tail of the guide strand was not complementary to the HTT mRNA target, it did not significantly impact efficacy of the compound. Thus, a longer 25-nt guide strand may be the most suited for modulation of AU-rich siRNA sequences through standardized GC-rich PK-modifying anchors.
Standardized GC-rich PK-modifying anchors were then tested in vitro for binding to 25–17 duplexes of two independent siRNA sequences, HTT and apolipoprotein E (APOE) (Figure 5C). Due to the presence of the GC-rich region, a PEGylated 8-mer anchor was able to hybridize to the asymmetric duplex and produce a shift in the gel-electrophoresis assay (Figure 5C, bottom panel). Given the interest in developing a system for gene modulation in the liver, this experiment was conducted in the context of GalNAc-conjugated siRNAs; however, we believe the concept should be readily applicable and provide greater benefit to other conjugated modalities and virtually any sequence.
Standardized GC-rich PK-modifying anchors enhance delivery and efficacy of GalNAc conjugates in the liver
Preliminary distribution studies showed that PK-modifying anchors significantly enhance delivery of GalNAc conjugates to the liver after a single i.v. administration. These studies were performed using GalNAc 21-13-8 siRNA constructs that were fully complementary to sFLT1, a target with low expression in the liver. To study efficacy in the liver, we have optimized a hybrid APOE-targeting siRNA that contains a standardized region at the 3′ end of the guide strand that allows binding of the PK-modifying anchor (Figure 6A).
Figure 6.

Standardized GC-rich PK-modifying anchors enhance delivery of GalNAc conjugates to the liver
(A) Schematics depict Cy3-labeled GalNAc-conjugated siRNA duplexes containing a GC-rich conserved region hybridizing to an 8-mer oligonucleotide anchor (with or without a PEG moiety). (B) Wild-type FVB/N female mice treated intravenously (28.5 nmol, ∼13 mg/kg) with Cy3-labeled GalNAc-conjugated siRNA duplexes as depicted above. (Top) Tiled fluorescent images of sections of the liver (5× objective, scale bar, 2 mm) imaged at 48 h post-injection. (Bottom) High-magnification images (63× objective, scale bar, 25 μm) with unfilled arrow heads indicating perinuclear localization of GalNAc-conjugated siRNAs within hepatocytes. n = 3/group. Blue: nuclei (DAPI), red: Cy3-labeled oligonucleotide.
Biodistribution studies were carried out to evaluate if hybrid GalNAc 25-17-8 scaffolds would present similar distribution patterns observed in initial studies performed with GalNAc 21-13-8 constructs (Figure 4B). Tiled fluorescent images (5×) of the liver showed that after a single i.v. injection (28.5 nmol, ∼13 mg/kg), hybrid GalNAc 25-17-8 constructs successfully distributed to the liver. These data also confirmed that standardized GC-rich PK-modifying anchors significantly enhance delivery of GalNAc conjugates to this organ (Figure 6B, top row). Additionally, high-resolution images (63×) demonstrated that PK-modifying anchors significantly improve delivery of GalNAc-siRNAs to a variety of cell types in the liver, including hepatocytes (Figure 6B, bottom row of images, hepatocytes indicated by hollow arrowhead).
Efficacy studies were conducted to assess the benefit from using PK-modifying anchors for i.v. administration of GalNAc conjugates targeting mouse Apoe in the liver. Test molecules in this study contained the conserved 8-mer region but did not contain the Cy3 label (Figure 7A). By enhancing delivery to the liver (Figure 7B, center), standardized GC-rich PK-modifying anchors enabled significant Apoe mRNA downregulation in the mouse liver 7 days post-injection (Figure 7B, left). APOE-targeting GalNAc siRNA compounds delivered through PK-modifying anchors (GalNAc 25-17-8 PEG) displayed significantly better efficacy than control GalNAc 25-17-8 at both dose levels tested. The maximum silencing achieved at 15 mg/kg was of ∼78.2% for GalNAc 25-17-8 PEG and of ∼65.5% for GalNAc 25-17-8 control (Figure 7B, left). No significant reduction in Apoe mRNA expression was observed with an HTT-targeting GalNAc 25-17-8 PEG siRNA injected at the highest dose level tested. Furthermore, and interestingly, Ago2 pull-downs showed that the presence of the large PEG moiety does not seem to disrupt loading of the guide strand into RISC, and in fact, the higher concentrations achieved in the tissue lead to higher amounts of antisense being loaded in Ago2 (Figure 7B, right).
Figure 7.

Standardized GC-rich PK-modifying anchors enhance gene knockdown by GalNAc conjugates in the liver and display superior Ago-2 loading after intravenous (i.v.) administrations
(A) Schematics depict GalNAc-conjugated siRNA duplexes containing a GC-rich conserved region hybridizing to an 8-mer oligonucleotide anchor (with or without a PEG moiety). (B) Wild-type FVB/N female mice were treated with a single i.v. injection (23.7 nmol, ∼15 mg/kg, or 4.7 nmol, ∼3 mg/kg) of APOE-targeting GalNAc-conjugated siRNAs (Gal 25-17-8 PEG APOE) with or without PK-modifying anchor (Gal 25-17-8 APOE). Huntingtin (HTT)-targeting siRNA (Gal 25-17-8 PEG HTT) was used as negative control for Apoe silencing. Animals were necropsied 7 days post-injection. (Left) Gene expression was assessed from tissue punch biopsies by Quantigene bDNA assay. Data were normalized to housekeeping gene (Cyclophilin B) and presented as a percentage of saline-treated control. (Center) Guide strand of the siRNA was quantified by miqPCR. (left) Ago2 loading was ascertained by protein pulldown followed by miqPCR for quantification of guide strands associated with Ago2. n = 4–5/group. ∗p < 0.05 by two-tailed t test. Mean ± Standard deviation (SD).
We also investigated the utility of standardized GC-rich PK-modifying anchors to enhance delivery of GalNAc conjugates to the liver after s.c. administration (Figure S3A). Tiled fluorescent arrays (10×) of the liver 48 h after injection showed that both PEGylated and parent GalNAc 25-17-8 scaffolds distributed better than the original GalNAc 21-13-8 constructs (Figure S3B). Although in this biodistribution study GalNAc 25-17-8 PEG still presented improved uptake in the liver compared with the parent GalNAc 25-17-8 scaffold, the differences were less pronounced than after i.v. administration (Figure 7A).
Efficacy studies demonstrated that s.c. administration of APOE-targeting GalNAc 25-17-8 PEG significantly reduced plasma APOE, with the highest reduction (∼90%) observed by 7 days post-injection (Figure S4). The knockdown was sustained below 50% for 28 days. However, in this time course study, no statistically significant differences were observed between GalNAc conjugates delivered using the PK-modifying anchors system and the parent GalNAc 25-17-8 compound (Figure S4B). In addition, no significant reductions in plasma APOE were observed in any of the control groups, including PBS and GalNAc 25-17-8 and GalNAc 25-17-8 PEG constructs targeting HTT.
Discussion
Modulation of the overall hydrodynamic size, through conjugation of large inert polymers, has been a widely used strategy to alter clearance kinetics and aid tissue distribution of a variety of drugs, including small molecules, antibodies, and aptamers. Here, we provide a first demonstration of a system that utilizes a PK-modifying oligonucleotide anchor to increase the overall hydrodynamic size of an siRNA drug, efficiently slowing its clearance kinetics and enhancing tissue accumulation and productive gene silencing in vivo.
The molecular size of the siRNA has profound impact on blood residence time and systemic tissue accumulation. In this study, and in agreement with previous reports,4,8,9,19 80%–90% of unconjugated siRNA was cleared within 2–3 min after i.v. administration, with the remainder of the injected dose mostly retained in the proximal tubules of the kidney. The largest siRNA configuration tested, a scaffold containing a 40-kDa PEG anchor, displayed the best enhancement in kinetics, improving blood circulating times by ∼20-fold and tissue accumulation by ∼3- to 4-fold. The reduced clearance was likely due to the combined size of the molecule being close to the MW cutoff for glomerular filtration (∼40–60 kDa17). Consistent with this interpretation, the 40-kDa duplex showed reduced accumulation in the kidney. While the observed differences in blood residence time were remarkable compared with the unmodified counterpart, duplexes containing the large PEG anchor still cleared much faster than our expectations based on published data for other oligonucleotide modalities, such as aptamers.21 With aptamers, approximately half of the injected dose continues to circulate in a blood stream 22 h post-injection, while with siRNAs, more than 90% is cleared by 6–7 h. We initially hypothesized that the difference could be explained by the dynamic nature of this PK modulation strategy; however, covalent attachment of PEG directly to the sense strand of the siRNA resulted in a similar blood profile (data not shown), discrediting this potential explanation. Given that the overall size of the molecular structure (modification) was similar between both siRNA and aptamer configurations, the different blood kinetic profile is likely driven by the unique chemical and structural properties that define each oligonucleotide scaffold. This question is of great interest and requires further detailed examination.
Interestingly, in this study, PK-modifying anchors also profoundly influenced clearance kinetics after s.c. administration. The duplexes containing the large 40-kDa anchor displayed a slow-release profile to the blood compartment, with a delayed time to peak. Most likely, this occurred due to a greater retention of this scaffold at the injection site, resulting in slower rates of absorption. Similar effects have also been observed with pegaptanib, a PEGylated RNA aptamer.22, 23, 24 Conjugation of a 40-kDa PEG slowed down absorption rates and delayed clearance from the intravitreal space, improving the residence time of the RNA aptamer pegaptanib in the eye.22, 23, 24 Despite the dramatic improvements observed for both i.v. and s.c. administrations in the current study, it is likely that a further increase and/or optimization of the overall size could help delay clearance further. This could potentially be investigated by increasing the size and/or changing the type of polymer used in the anchor, by exploring other approaches such as increasing the siRNA valency, or by conjugation of an antibody. Significant advances have been reported with antibody-siRNA conjugates, which allow for long circulating times and enable enhanced delivery to cardiac and skeletal muscle after systemic administration.14 The preclinical successes with the antibody conjugate strategy have propelled this technology to the clinic.
The dynamic PK-modifying anchor system provided a good platform for modulation of clearance and biodistribution of a variety of chemically diverse conjugated siRNAs. The positive effects were more profound in the context of hydrophilic conjugates (GalNAc) and moderately hydrophobic (DHA) than with highly lipophilic conjugates (Chol and DCA). This was not surprising, since immediately after i.v. administration, lipid conjugates are known to bind a predetermined set of plasma proteins, which enable longer circulating times and generate a higher baseline for improvement.8,9,11,19 Intriguingly, other preclinical studies investigating the effects of hydrophobic side chains in PEGylated DNA aptamers (28- to 32-nt long; 40-kDa PEG) have found an opposite trend, with higher hydrophobicity enhancing blood clearance after a bolus i.v. dose in rats.25 This suggests, yet again, a potential contribution of oligonucleotide structure and chemical composition in the unique PK of these molecules.
GalNAc conjugates were selected for a follow-up proof-of-concept study since they displayed a considerable improvement in blood circulating times and biodistribution when applying the PK-modifying anchor concept. After i.v. injection, GalNAc conjugates modulated by the 40-kDa anchor showed a significant improvement in AUC and distribution to the liver when compared with their parent GalNAc-siRNA. Other studies have also shown similar results, with GalNAc-conjugated PEGylated ASOs also displaying a significant ∼7-fold improvement in liver delivery after i.v. administration in rats.16 This likely occurs due to a combination of a temporary saturation of the ASGPR after i.v. injection and fast glomerular clearance of non-PEGylated GalNAc conjugates from the blood. Although to a lesser extent than observed after i.v. administrations, large dynamic PK-modifying anchors enhance distribution of GalNAc conjugates to hepatocytes in the liver after s.c. administration. We hypothesize that this is a result of a slower release from the site of injection that precludes the rapid initial kidney filtration observed after i.v. administration. Indeed, the s.c. route is preferred for the administration of GalNac-conjugated siRNAs and ASOs in the clinic to aid reduce clearance and facilitate self-administration of the drug.
Despite the biodistribution improvements enabled by the PK-modifying anchors on GalNAc conjugates, the enhancement in activity was somewhat modest. We hypothesize that this could be due to partial entrapment of siRNAs in an unproductive pathway. On the other hand, the high efficiency of parent GalNAc-siRNAs and the high potency of the Apoe-targeting sequence used may have also, to some extent, masked the true potential of this approach. Even very small amounts of this highly potent compound resulted in potent and sustained knockdown in the liver. Alternatively, this strategy may be better suited for other ligands for which expression of their cognate receptors is low, or with a slow turnover, by enabling longer exposures to target tissues. Furthermore, the high levels of delivery achieved with this technology may constitute a competitive advantage that could potentially translate to longer duration of action. Both of these aspects now warrant further investigations.
Besides the size of the PEG polymer, the conjugation strategy is of much importance. In the chemical architecture presented here, conjugation of PEG to the 8-mer did not disrupt binding of the anchor to the asymmetric siRNA to form a multi-duplex. Similarly, others have found that attachment of PEGs with different sizes (PEG12 up to 40 kDa) does not majorly affect hybridization of short 10- to 18-mer duplexes. No significant differences between melting temperatures (Tms) between unconjugated and conjugated were observed.26, 27, 28 Although the 3′ and 5′ ends of sense strand of the siRNA are amenable to modification, previous studies showed that direct conjugation of bulky entities, including PEG moieties, might interfere with RISC complex assembly or/and Dicer processing and thus efficacy7,29, 30, 31, 32. These effects might be in part explained by steric hindrance and in part by the poor metabolic stability of these early siRNA scaffolds. In this study, no detectable impact on siRNA potency was observed when using PK-modifying anchors in 7-point concentration-response curves in vitro, nor did this strategy have a detrimental effect in gene silencing in vivo. Furthermore, in vivo data show that GalNAc-conjugated siRNAs delivered to the liver with PK-modifying anchors displayed higher levels of Ago2 loading compared with parent compounds. Although it is unlikely that PEGylated oligonucleotides are more efficient loading into Ago2, together these data suggest that the PEGylation strategy used does not negatively impact RISC loading and activity. We hypothesize that this could be potentially due to the dynamic nature of the construct that can lead to the release of the PEGylated anchor to allow for tissue uptake and silencing.
Given that PEG is widely used in the biomedical/pharmaceutical industry, it was used in this study as a model polymer; however, it has been well documented that large PEG molecules can potentially induce immunogenicity.33 Therefore, other inert polymers, such as polyaxamers, polycarbonates, and others, should be evaluated and tested in various sizes and structural architectures to enable clinical translation.
In this work, we designed a multi-modal system that uses a conserved 8-mer anchor that can be applied in the context of any given siRNA sequence. The conserved region was introduced away from the mRNA target recognition positions 2–17 of the guide strand34 to avoid any negative impact in efficacy. Indeed, no detrimental impact in gene silencing was observed in vitro or in vivo when the conserved region was placed at positions 18–25 of the guide strand. Although no binding to the mRNA target is expected within the conserved anchor region,34 further studies are still warranted to confirm the lack of major off-target liabilities. This chemical architecture can be potentially used to modulate the PK of a variety of siRNA sequences, without requirement for individual optimization of the anchor, minimizing synthetic costs. Nonetheless, a systematic evaluation of the sequence of the conserved region is now warranted to ensure that there are no sequence-specific toxicity liabilities.
This study provides proof of concept for a dynamic PK-modifying anchor system that can be used to effectively modulate the hydrodynamic size of siRNAs, reducing their clearance kinetics and enhancing biodistribution. The concept can be readily employed to screen a multitude of novel ligands in vivo. By maximizing tissue exposure, PK-modifying anchors may help the identification of unique ligands that, given their lower binding affinities, limited receptor numbers, or slower recycling, have not yet being flagged for further assessments. In addition, this strategy holds great potential for therapeutic applications enabling modulation of virtually any sequences with relative simplicity.
Materials and methods
Oligonucleotide synthesis
Oligonucleotides were synthesized using modified 2′-F, 2′-O-Me phosphoramidites with standard protecting groups (Chemgenes). Cy3 phosphoramidite (Quasar 570) and 5′-vinyl-phosphonate (VP) phosphoramidite were purchased from GenePharma and Chemgenes, respectively. Phosphoramidite solid-phase synthesis was made on a MerMade12 (BioAutomation) using modified protocols. Unconjugated oligonucleotides were synthesized on controlled pore glass (CPG) functionalized with a long-chain alkyl amine (LCAA) and Unylinker terminus (Chemgenes). Chol-conjugated oligonucleotides were grown with the Chol moiety bound to a tetraethylenglycol (TEG) attached through a succinate linker to LCAA-CPG support (Chemgenes). GalNAc-conjugated oligonucleotides were synthesized on custom 3′-GalNAc-CPG as described in Sharma et al.35 DCA- and DHA-conjugated oligonucleotides were synthesized on custom CPGs as described in Biscans et al.15 Phosphoramidites were prepared at 0.1 M in anhydrous acetonitrile (ACN), with an added dry 15% dimethylformamide (DMF) in the 2′-OMe U amidite. 5-(Benzylthio)-1H-tetrazole (BTT) was used as the activator at 0.25 M. Detritylations were performed using 3% trichloroacetic acid in dichloromethane (DCM). Capping was done with non-tetrahydrofuran-containing reagents CAP A, 20% n-methylimidazole in ACN and CAP B, 20% acetic anhydride (Ac2O), and 30% 2,6-lutidine in ACN (synthesis reagents were purchased at AIC). Sulfurization was performed with 0.1-M solution of 3-[(dimethylaminomethylene)amino]-3H-1,2,4-dithiazole-5-thione (DDTT) in pyridine (ChemGenes) for 3 min. Phosphoramidite coupling times were 3 min for all amidites used.
Deprotection and purification of oligonucleotides
Unconjugated, Cy3-labeled, and conjugated oligonucleotides (DCA, DHA, GalNAc, Chol) were cleaved and deprotected with a solution of 30% ammonium hydroxide/40% methylamine (AMA) 1:1 (v/v) for 2 h at room temperature (RT). VP-containing oligonucleotides were cleaved and deprotected as described previously.36 Briefly, VP-containing oligonucleotides, while still on solid support, were treated post-synthesis with an anhydrous mixture of trimethylsilyl bromide/ACN/DMF/pyridine (3:9:9:1) for 1 h at RT on a rotary device. The reaction was then quenched with water, and the CPG was then rinsed with can and DCM and allowed to dry before base deprotection. CPG-containing VP oligonucleotides were treated with a solution of 3% diethylamine (DEA) in aqueous ammonia at 35°C for 20h.
The solutions containing deprotected oligonucleotides were filtered to remove the CPG and dried under vacuum. The resulting pellets were resuspended in 5% ACN in water. The purification of unconjugated and VP- and GalNAc-containing oligonucleotides was performed by ion exchange on an Agilent 1290 Infinity II HPLC system equipped with a column packed in house with Source 15Q anion exchange resin (GE Healthcare). Buffer conditions were as follows: eluent A, 10-mM sodium acetate (pH 7) in 20% can, and eluent B, 1-M sodium perchlorate in 20% ACN. Purification of Cy3-labeled, DHA-, DCA-, and Chol-conjugated oligonucleotides was done by reverse phase using a PRP-C18 column (Hamilton Company). Buffer conditions were as follows: eluent A, 50-mM sodium acetate in 5% ACN, and eluent B, 100% ACN. The temperature was 60°C in both cases. Peaks were monitored at 260 and, for Cy3-labeled oligonucleotides, at 550 nm. Fractions were analyzed by liquid chromatography-mass spectrometry (LC-MS), and pure fractions were dried under vacuum. Oligonucleotides were resuspended in 5% can, desalted through fine Sephadex G-25 media (GE Healthcare), and lyophilized.
PEGylation of the oligonucleotide anchor
A C6 amino modifier linker (Genepharma) was previously coupled at the 5′ of the oligonucleotide using a 15-min coupling time, no capping step, and Monomethoxy trityl (MMT)-ON. The oligonucleotide was deprotected with AMA 1:1 v/v for 2 h at RT and dried down. MMT was cleaved with treatment for 1 h at RT with 80% acetic acid. The oligonucleotide was then precipitated with sodium acetate in isopropanol. The pellet was dried under vacuum, resuspended in 5% ACN, desalted by size exclusion using Sephadex G25, and dried down under vacuum. The PEG was attached to the oligonucleotide via an NHS ester reaction. Briefly, 1 equiv of the oligonucleotide was dissolved in 0.1-M sodium bicarbonate buffer (pH 8.5) and mixed with 5 equiv of the PEG-NHS ester dissolved in dry DMF. The reaction was incubated in the dark overnight using a rotatory device. The mixture was purified by anion exchange with the conditions described above. Fractions were analyzed by LC-MS and PAGE. Pure fractions with pegylated oligonucleotides were combined and lyophilized. PEG-NHS esters were purchased from (JenKem Technology).
Quality control (QC) analysis of oligonucleotide strands and duplexes
The identities of oligonucleotides were verified by LC-MS analysis on an Agilent 1260 HPLC coupled with an Agilent 6530 accurate mass quadrupole time of flight (Q-TOF) using the following conditions: buffer A, 100-mM hexafluoroisopropanol (HFIP)/9-mM triethylamine (TEA) in LC-MS-grade water; buffer B, 100-mM HFIP/9-mM TEA in LC-MS-grade methanol; column, Agilent AdvanceBio oligonucleotides C18; temperature, 45°C; flow rate, 0.5 mL/min. LC peaks were monitored on UV at 260 nm. MS parameters were as follows: source, electrospray ionization; ion polarity, negative mode; range, 100–3,200 m/z; scan rate, 2 spectra/s; capillary voltage, 4,000; fragmentor, 180 V.
Duplex formation and correct annealing of the oligonucleotide anchor with the asymmetric duplex was assessed by non-denaturing gel electrophoresis. Unless otherwise stated, strands were mixed in equimolar amounts and heated to 95°C and cooled down to RT. Novex precast Bis-Tris 20% gels (Invitrogen) were loaded with 25-pmol oligonucleotide per lane and run at 180 V for ∼60 min. When assaying duplexes containing Cy3 label, gels were imaged for the fluorescent tag before incubating with the nucleic acid stain. Gels were treated with SYBR Gold (Invitrogen, S11494) for 10 min. Gels were imaged using a Biorad VersaDoc imaging system, and images were analyzed through the QuantiOne software.
Preparation of oligonucletoides for in vitro and in vivo studies
Oligonucleotides for in vivo were dried down and resuspended in saline (0.9% NaCl) for injections.
Cell culture and in vitro dose response
HeLa cells were maintained in DMEM (Cellgro, 10-013-CV) supplemented with 10% fetal bovine serum (FBS) (Gibco, 26140) and 100 U/mL pen/strep (Invitrogen, 15140) and expanded at 37°C and 5% CO2. Seven-point dose-response curves were generated by treating HeLa cells with various concentrations of siRNA formulated with RNAiMax (Invitrogen, 13778–150) for 72 h at 37°C and 5% CO2. Transfection was carried out in 50:50 DMEM/OptiMEM (Gibco, 31985–070) and 3% FBS with no antibiotics. Cells were lysed by 30-min incubation at 55°C with diluted QuantiGene (QG) lysis mixture (Invitrogen, QP0524) containing proteinase K (Invitrogen, 25530–049). Gene silencing was assessed by QG branched DNA (bDNA) assay as per manufacturer’s instructions (see brief description below) using the following probe sets: mouse Htt (SB-14150) and mouse Hprt (SB-15463). Htt data was normalized to housekeeping Hprt and represented as the percentage of untreated control. n = 3.
In vivo studies
Experimental protocols involving animals were approved by the University of Massachusetts Medical School Institutional Animal Care and Use Committee (IACUC Protocol #A-2411) and performed according to the guidelines and regulations therein. All studies were conducted in adult FVB/N females (9–12 weeks old, 22–25 g).
PK and biodistribution studies
For PK studies, both legs of the animal were shaved under brief anesthesia 48 h prior to initiating the study. Compounds were administered i.v. or s.c., and blood samples were collected at given time points from the saphenous vein using Microvette CB300 tubes. Animals were euthanized 48 h after compound administration and perfused with PBS, and tissues were collected for PNA hybridization or microscopy analyses. n = 3–5/group.
PNA hybridization assay
Tissues collected for this purpose were incubated in RNAlater (Sigma, R0901) overnight at 4°C and stored at −80°C until analysis. PNA hybridization assay was used to quantify the accumulation of siRNA guide strand in tissue lysates, and it was conducted as previously described in Godinho et al.19 Briefly, 10-μL blood or 2- to 3-mm tissue punches were lysed using 200- to 300-μL lysis solution (MasterPure EpiCentre, MTC096H) containing proteinase K (1–2 mg/mL) (Invitrogen, 25530–049). Tissue lysates were incubated at 55°C for 1 h. Detergents were precipitated by adding 20-μL KCl (3 M) and by rapid centrifugation of samples at 4,000 × g for 10 to 15 min. Supernatants were then diluted in hybridization buffer (50 mM Tris 10% ACN [pH 8.8]) containing ∼33 nM (5 pmol/150uL of total sample) of Cy3-labeled PNA (PNABio) complementary to the guide strand being quantified. Annealing of guide strands and PNA was carried out on a Biorad c1000 thermal cycler (90°C for 15 min and 50°C for 15 min). Samples were run through an Agilent Technologies 1260 Infinity Quad-pump High-Performance Liquid Chromatography system using a DNAPac PA100 anion exchange column (Thermo Fisher Scientific) for separation and a 1260 FLD fluorescent detector. For details on the mobile phase, please refer to Godinho et al.19 Tissue concentrations were calculated based on the area of integrated peaks in reference to calibration curves generated for each individual compound. In studies where multiple 3′ end conjugates were being assessed, all values were normalized to the MW of an unconjugated 21–13 asymmetric siRNA duplex. PK parameters were calculated based on model independent analysis using PKSolver on group averages per time point.37
Fluorescent imaging
Tissues for fluorescent microscopy were post-fixed in 10% formalin at 4°C overnight and embedded in paraffin blocks for sectioning. Slides with 4-μm sections were incubated in xylene (8 min × 2) in a series of ethanol dilutions (100%, 95%, and 80% for 4 min each). All tissues were stained with 2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI) (2 min) and finally washed with PBS (2 min). Coverslips were mounted using Permafluor mounting medium (ThermoScientific, TA-030-FM).
Tiled (5–10× objective) and high-magnification (63× objective) fluorescent images were acquired with a Leica DMi8 fluorescent microscope fitted with a Hamamatsu C11440 ORCA-Flash 4.0 camera. All sections within a study were acquired under the same light intensity and exposure settings for each channel. Although DAPI was always acquired during imaging, to enable easy visualization and comparison among groups with relatively low intensity signal, in some instances only the Cy3 channel is shown.
Gene-silencing efficacy studies
Efficacy studies in vivo were conducted with GalNAc-conjugated siRNAs targeting APOE. Compounds were administered subcutaneously at two dose levels: 23.7 (∼15 mg/kg) and 4.7 nmol (∼3 mg/kg). Animals were euthanized after 7 days, and tissues were collected, incubated in RNAlater (Sigma, R0901) overnight at 4°C, and stored at −80°C until analysis.
For mRNA assessments, 2-mm tissue punches (Braintree Scientific, MTP-33 31) were taken from the liver and homogenized using QG homogenizing solution (Affymetrix, QG0517) containing proteinase K (Invitrogen, 25530–049). QIAGEN TissueLyser II (for 2 × 5 min at Freq 30/s) was used to disrupt the tissue. Samples were then incubated at 55°C for 60 min, and mRNA was quantified using QG 2.0 bDNA assay as per manufacturer’s instructions. Briefly, 60-μL mouse Apoe (SB-13611) or Ppib (SB-10002) probe sets were placed on a QG bDNA capture plate followed by 40-μL diluted tissue lysate. After overnight incubation at 55°C, signal amplification was carried out, and luminescence was measured using a SpectraMax M3 microplate reader. All Apoe data were normalized to housekeeping gene and represented as a percentage of PBS-treated control. n = 4–5/group.
To determine Ago2 loading, Ago2 pull-downs were performed based on previously published protocols.38 Tissue punches were homogenized in 500-uL NET buffer (50-mM Tris-HCl [pH 7.5], 150-mM NaCl, 5-mM EDTA, 0.5% v/v NP-40 alternative, 10% w/v glycerol, 1-mM NaF, 0.5-mM DTT, 1-mM AEBSF). Lysates were centrifuged at 20,000 × g for 10 min, and the supernatants were collected. Fifty-uL GSH agarose bead slurry was spun at 500 × g for 2 min and washed three times with 1-mL PBST (PBS, 0.05% Tween 20), and 100-μg GST-T6B was peptide added and bound by rotating at 4°C for 3 h. Beads were washed 3 times in PBS and equilibrated in 1-mL NET buffer. NET buffer was removed, and the tissue lysate was added and rotated overnight at 4°C. The next day, the beads were washed 3 times with 1-mL NET buffer and finally resuspended in 50-uL PBS + 0.25% Triton X-100 and heated to 95°C for 5 min to release the siRNAs. The input samples were diluted 1:100 in PBS + 0.25% Triton X-100 and heated to 95°C for 5 min as well. Quantification of siRNAs was done following a modified version of the miQPCR protocol.39 Briefly, 1 uL of either the pulldowns or the input was ligated to a preadenylated DNA adapter (IDT, Seq./App/NT TCG TAT CCA GTG CGA GGC TTC GAA CTA CGA CCT GCA TAA ACG GC/3AmMO/), at a 1-μM final concentration, using T4 RNL2KQ (NEB, M0373S) with 25% PEG8000 in a 5-μL reaction at RT for 2 h. The ligation reaction was then heated to 95°C for 5 min and then brought down to 42°C, and 15 μL of a reverse transcriptase master-mix containing the mQ-RT primer (IDT, Seq. CCC AGT TAT GGC CGT TTA TGC AGG T) at a final concentration of 1 uM and 100 units of M-MuLV reverse transcriptase (NEB, M0253S) were added to the ligations. The RT reaction was run for 30 min at 42°C and stopped by heating the reaction to 65°C for 20 min. qPCR was then conducted using a universal reverse primer (IDT, Seq. GGT GCC CAG TTA TGG CCG TTT A) and an siRNA-specific forward primer (IDT, Seq. CTA CCG TGG TGT TGG ATA TGG ATG TTG TC) in PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, A25741) following the manufacturer’s specifications. siRNA standards for quantification were prepared by spiking the siRNA into untreated liver lysate and following the same protocol as for the samples.
Acknowledgments
This work was supported by the National Institutes of Health (NIH), grant numbers RO1GM10880302 and RO1NS03819415. B.M.D.C.G. was supported by a Milton-Safenowitz Fellowship (grant ID 17-PDF-363) from the Amyotrophic Lateral Sclerosis Association (ALSA). The authors acknowledge Dr. Emily Haberlin for editorial feedback on the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
B.M.D.C.G., M.R.H., and A.K. conceived the project and contributed to the experimental design. B.M.D.C.G., E.G.K., R.A.H., A.H.C., J.F.A., J.W.G., Z.K., S.H., A.B., J.C., and C.M.F. contributed experimentally. D.E. and A.B. synthesized compounds. B.M.D.C.G. and A.K. critically analyzed the data and wrote the manuscript.
Declaration of interests
B.M.D.C.G., M.R.H., and A.K. have filed a patent application for dynamic PK-modifying oligonucleotide anchors.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.06.005.
Contributor Information
Bruno M.D.C. Godinho, Email: bruno.godinho@umassmed.edu.
Anastasia Khvorova, Email: anastasia.khvorova@umassmed.edu.
Supplemental information
References
- 1.Godinho B.M., Khvorova A. The era of RNA interference medicines: the clinical landscape of synthetic gene silencing drugs. Saúde Tecnol. 2019;21:05–17. [Google Scholar]
- 2.Adams D., Gonzalez-Duarte A., O’Riordan W.D., Yang C.-C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L., et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 3.Balwani M., Sardh E., Ventura P., Peiró P.A., Rees D.C., Stölzel U., Bissell D.M., Bonkovsky H.L., Windyga J., Anderson K.E., et al. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N. Engl. J. Med. 2020;382:2289–2301. doi: 10.1056/NEJMoa1913147. [DOI] [PubMed] [Google Scholar]
- 4.Morrissey D.V., Lockridge J.A., Shaw L., Blanchard K., Jensen K., Breen W., Hartsough K., Machemer L., Radka S., Jadhav V., et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005;23:1002–1007. doi: 10.1038/nbt1122. [DOI] [PubMed] [Google Scholar]
- 5.Christensen J., Litherland K., Faller T., Van de Kerkhof E., Natt F., Hunziker J., Krauser J., Swart P. Metabolism studies of unformulated internally [3H]-labeled short interfering RNAs in mice. Drug Metab. Dispos. 2013;41:1211–1219. doi: 10.1124/dmd.112.050666. [DOI] [PubMed] [Google Scholar]
- 6.Godinho B.M., Ogier J.R., Quinlan A., Darcy R., Griffin B.T., Cryan J.F., O'driscoll C.M. PEGylated cyclodextrins as novel siRNA nanosystems: correlations between polyethylene glycol length and nanoparticle stability. Int.J. Pharm. 2014;473:105–112. doi: 10.1016/j.ijpharm.2014.06.054. [DOI] [PubMed] [Google Scholar]
- 7.Iversen F., Yang C., Dagnæs-Hansen F., Schaffert D.H., Kjems J., Gao S. Optimized siRNA-PEG conjugates for extended blood circulation and reduced urine excretion in mice. Theranostics. 2013;3:201–209. doi: 10.7150/thno.5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soutschek J., Akinc A., Bramlage B., Charisse K., Constien R., Donoghue M., Elbashir S., Geick A., Hadwiger P., Harborth J., et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 9.Gao S., Dagnaes-Hansen F., Nielsen E.J.B., Wengel J., Besenbacher F., Howard K.A., Kjems J. The effect of chemical modification and nanoparticle formulation on stability and biodistribution of siRNA in mice. Mol. Ther. 2009;17:1225–1233. doi: 10.1038/mt.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crooke S.T., Vickers T.A., Liang X.-h. Phosphorothioate modified oligonucleotide–protein interactions. Nucleic Acids Res. 2020;48:5235–5253. doi: 10.1093/nar/gkaa299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osborn M.F., Coles A.H., Biscans A., Haraszti R.A., Roux L., Davis S., Ly S., Echeverria D., Hassler M.R., Godinho B.M.D.C., et al. Hydrophobicity drives the systemic distribution of lipid-conjugated siRNAs via lipid transport pathways. Nucleic Acids Res. 2018;47:1070–1081. doi: 10.1093/nar/gky1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Springer A.D., Dowdy S.F. GalNAc-siRNA conjugates: leading the way for delivery of RNAi therapeutics. Nucleic Acid Ther. 2018;28:109–118. doi: 10.1089/nat.2018.0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ämmälä C., Drury W.J., 3rd, Knerr L., Ahlstedt I., Stillemark-Billton P., Wennberg-Huldt C., Andersson E.M., Valeur E., Jansson-Löfmark R., Janzén D., et al. Targeted delivery of antisense oligonucleotides to pancreatic β-cells. Sci. Adv. 2018;4:eaat3386. doi: 10.1126/sciadv.aat3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugo T., Terada M., Oikawa T., Miyata K., Nishimura S., Kenjo E., Ogasawara-Shimizu M., Makita Y., Imaichi S., Murata S., et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release. 2016;237:1–13. doi: 10.1016/j.jconrel.2016.06.036. [DOI] [PubMed] [Google Scholar]
- 15.Biscans A., Coles A., Haraszti R., Echeverria D., Hassler M., Osborn M., Khvorova A. Diverse lipid conjugates for functional extra-hepatic siRNA delivery in vivo. Nucleic Acids Res. 2018;47:1082–1096. doi: 10.1093/nar/gky1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris J.M., Chess R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 17.Lawrence M.G., Altenburg M.K., Sanford R., Willett J.D., Bleasdale B., Ballou B., Wilder J., Li F., Miner J.H., Berg U.B., et al. Permeation of macromolecules into the renal glomerular basement membrane and capture by the tubules. Proc. Natl. Acad. Sci. USA. 2017;114:2958. doi: 10.1073/pnas.1616457114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolfrum C., Shi S., Jayaprakash K.N., Jayaraman M., Wang G., Pandey R.K., Rajeev K.G., Nakayama T., Charrise K., Ndungo E.M., et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 2007;25:1149–1157. doi: 10.1038/nbt1339. [DOI] [PubMed] [Google Scholar]
- 19.Godinho B.M., Gilbert J.W., Haraszti R.A., Coles A.H., Biscans A., Roux L., Nikan M., Echeverria D., Hassler M., Khvorova A. Pharmacokinetic profiling of conjugated therapeutic oligonucleotides: a high-throughput method based upon serial blood microsampling coupled to peptide nucleic acid hybridization assay. Nucleic Acid Ther. 2017;27:323–334. doi: 10.1089/nat.2017.0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Godinho B.M., Coles A.H., Khvorova A. In: Advances in Nucleic Acid Therapeutics. Agrawal S., Gait M.J., editors. RCS; 2019. pp. 206–232. [Google Scholar]
- 21.Haruta K., Otaki N., Nagamine M., Kayo T., Sasaki A., Hiramoto S., Takahashi M., Hota K., Sato H., Yamazaki H. A novel PEGylation method for improving the pharmacokinetic properties of anti-interleukin-17a RNA aptamers. Nucleic Acid Ther. 2016;27:36–44. doi: 10.1089/nat.2016.0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tucker C.E., Chen L.-S., Judkins M.B., Farmer J.A., Gill S.C., Drolet D.W. Detection and plasma pharmacokinetics of an anti-vascular endothelial growth factor oligonucleotide-aptamer (NX1838) in rhesus monkeys. J. Chromatogr. B Biomed. Sci. Appl. 1999;732:203–212. doi: 10.1016/s0378-4347(99)00285-6. [DOI] [PubMed] [Google Scholar]
- 23.Ng E.W.M., Shima D.T., Calias P., Cunningham E.T., Guyer D.R., Adamis A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006;5:123–132. doi: 10.1038/nrd1955. [DOI] [PubMed] [Google Scholar]
- 24.Drolet D.W., Nelson J., Tucker C.E., Zack P.M., Nixon K., Bolin R., Judkins M.B., Farmer J.A., Wolf J.L., Gill S.C., et al. Pharmacokinetics and safety of an anti-vascular endothelial growth factor Aptamer (NX1838) following injection into the vitreous humor of rhesus monkeys. Pharm. Res. 2000;17:1503–1510. doi: 10.1023/a:1007657109012. [DOI] [PubMed] [Google Scholar]
- 25.Gupta S., Drolet D.W., Wolk S.K., Waugh S.M., Rohloff J.C., Carter J.D., Mayfield W.S., Otis M.R., Fowler C.R., Suzuki T., et al. Pharmacokinetic properties of DNA aptamers with base modifications. Nucleic Acid Ther. 2017;27:345–353. doi: 10.1089/nat.2017.0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonora G.M., Ivanova E., Zarytova V., Burcovich B., Veronese F.M. Synthesis and characterization of high-molecular mass polyethylene glycol-conjugated oligonucleotides. Bioconjug. Chem. 1997;8:793–797. doi: 10.1021/bc970082p. [DOI] [PubMed] [Google Scholar]
- 27.Jia F., Lu X., Tan X., Wang D., Cao X., Zhang K. Effect of PEG architecture on the hybridization thermodynamics and protein accessibility of PEGylated oligonucleotides. Angew. Chem. Int. Ed. Engl. 2017;56:1239–1243. doi: 10.1002/anie.201610753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shokrzadeh N., Winkler A.-M., Dirin M., Winkler J. Oligonucleotides conjugated with short chemically defined polyethylene glycol chains are efficient antisense agents. Bioorg. Med. Chem. Lett. 2014;24:5758–5761. doi: 10.1016/j.bmcl.2014.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaziova Z., Baumann V., Winkler A.-M., Winkler J. Chemically defined polyethylene glycol siRNA conjugates with enhanced gene silencing effect. Bioorg. Med. Chem. 2014;22:2320–2326. doi: 10.1016/j.bmc.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung S., Lee S.H., Mok H., Chung H.J., Park T.G. Gene silencing efficiency of siRNA-PEG conjugates: effect of PEGylation site and PEG molecular weight. J. Control. Release. 2010;144:306–313. doi: 10.1016/j.jconrel.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 31.Zhu L., Mahato R.I. Targeted delivery of siRNA to hepatocytes and hepatic stellate cells by bioconjugation. Bioconjug. Chem. 2010;21:2119–2127. doi: 10.1021/bc100346n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kow S.C., McCarroll J., Valade D., Boyer C., Dwarte T., Davis T.P., Kavallaris M., Bulmus V. Dicer-labile PEG conjugates for siRNA delivery. Biomacromolecules. 2011;12:4301–4310. doi: 10.1021/bm201199c. [DOI] [PubMed] [Google Scholar]
- 33.d'Avanzo N., Celia C., Barone A., Carafa M., Di Marzio L., Santos H.A., Fresta M. Immunogenicity of polyethylene glycol based nanomedicines: mechanisms, clinical implications and systematic approach. Adv. Ther. 2020;3:1900170. [Google Scholar]
- 34.Kamola P.J., Nakano Y., Takahashi T., Wilson P.A., Ui-Tei K. The siRNA non-seed region and its target sequences are auxiliary determinants of off-target effects. PLoS Comput. Biol. 2015;11:e1004656. doi: 10.1371/journal.pcbi.1004656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma V.K., Osborn M.F., Hassler M.R., Echeverria D., Ly S., Ulashchik E.A., Martynenko-Makaev Y.V., Shmanai V.V., Zatsepin T.S., Khvorova A., et al. Novel cluster and monomer-based GalNAc structures induce effective uptake of siRNAs in vitro and in vivo. Bioconjug. Chem. 2018;29:2478–2488. doi: 10.1021/acs.bioconjchem.8b00365. [DOI] [PubMed] [Google Scholar]
- 36.O'Shea J., Theile C.S., Das R., Babu I.R., Charisse K., Manoharan M., Maier M.A., Zlatev I. An efficient deprotection method for 5′-[O,O-bis(pivaloyloxymethyl)]-(E)-vinylphosphonate containing oligonucleotides. Tetrahedron. 2018;74:6182–6186. [Google Scholar]
- 37.Zhang Y., Huo M., Zhou J., Xie S. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010;99:306–314. doi: 10.1016/j.cmpb.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 38.Hauptmann J., Schraivogel D., Bruckmann A., Manickavel S., Jakob L., Eichner N., Pfaff J., Urban M., Sprunck S., Hafner M., et al. Biochemical isolation of Argonaute protein complexes by Ago-APP. Proc. Natl. Acad. Sci. USA. 2015;112:11841–11845. doi: 10.1073/pnas.1506116112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benes V., Collier P., Kordes C., Stolte J., Rausch T., Muckentaler M.U., Häussinger D., Castoldi M. Identification of cytokine-induced modulation of microRNA expression and secretion as measured by a novel microRNA specific qPCR assay. Sci. Rep. 2015;5:11590. doi: 10.1038/srep11590. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.