

Abstract
The high kinetic barrier to amide bond formation has historically placed narrow constraints on its utility in reversible chemistry applications. Slow kinetics has limited the use of amides for the generation of diverse combinatorial libraries and selection of target molecules. Current strategies for peptide-based dynamic chemistries require the use of nonpolar co-solvents or catalysts or the incorporation of functional groups that facilitate dynamic chemistry between peptides. In light of these limitations, we explored the use of depsipeptides: biorelevant copolymers of amino and hydroxy acids that would circumvent the challenges associated with dynamic peptide chemistry. Here, we describe a model system of N-(α-hydroxyacyl)-amino acid building blocks that reversibly polymerize to form depsipeptides when subjected to two-step evaporation–rehydration cycling under moderate conditions. The hydroxyl groups of these units allow for dynamic ester chemistry between short peptide segments through unmodified carboxyl termini. Selective recycling of building blocks is achieved by exploiting the differential hydrolytic lifetimes of depsipeptide amide and ester bonds, which we show are controllable by adjusting the solution pH, temperature, and time as well as the building blocks’ side chains. We demonstrate that the polymerization and breakdown of the depsipeptides are facilitated by cyclic morpholinedione intermediates, and further show how structural properties dictate half-lives and product oligomer distributions using multifunctional building blocks. These results establish a cyclic mode of ester-based reversible depsipeptide formation that temporally separates the polymerization and depolymerization steps for the building blocks and may have implications for prebiotic polymer chemical evolution.
Keywords: depsipeptide, dynamic covalent chemistry, abiotic chemistry, green chemistry, peptidomimetic
Introduction
The proteins and peptides of biology exhibit innumerable variations as functional polymers and scaffolds for supramolecular assemblies. Nevertheless, chemists have yet to fully access the favorable properties of peptides in dynamic combinatorial synthesis presumably because of the significant kinetic barrier to amide bond formation.1,2 The few strategies reported to access reversible peptide bond formation involve relatively high temperatures, enzymes, or nonpolar co-solvents, approaches that can impose constraints on the scope of the target peptides and applications that can be addressed with these methods.3−6 For example, several groups have demonstrated dynamic covalent amide chemistry using metal-catalyzed transamidations in nonaqueous solvents, producing peptides with properties not readily translatable to water-based, biocompatible applications.7−9 Alternative approaches to reversible peptide formation involve the incorporation of additional functional groups to allow for a more rapid dynamic chemistry than that of the amide bond. In this fruitful vein, the use of thioesters (e.g., in native chemical ligation) or hydrazide moieties has been shown to allow for a rapid exchange of peptide fragments in both aqueous and nonaqueous solutions to generate dynamic peptide-like libraries.10−18
The presence of amino acids in meteorites and model prebiotic reactions implicates peptides as potential participants in the early evolution of life,19−22 which provides another motivation to investigate the potential for reversible chemistry with peptides in aqueous media. However, over half a century of investigation focused on generating peptides from amino acids under prebiotically plausible conditions has demonstrated only short peptide production, with much of the starting amino acids often being lost as relatively unreactive diketopiperazine (DKP) cyclic dimers.23,24 The more productive approaches have utilized dry-heating at temperatures above the boiling point of water,25,26 activating agents of questionable prebiotic plausibility,23,27,28 or controlled stepwise addition of cross-reactive redox agents.29 More recently, peptide bond formation has been shown to be catalyzed when α-hydroxy acids are dried from solutions with amino acids.30 This robust and plausibly prebiotic mechanism for peptide bond formation primarily produces depsipeptides (oligomers with both peptide and ester linkages) or HO-terminated peptides.
The challenge of prebiotic peptide oligomerization and kinetic barriers to amide bond formation present obstacles to the application and usefulness of peptides before the advent of coded protein synthesis.2,31 An efficient mechanism for the incorporation and removal of amino acids into and from protopeptides could have been important for the selection of stable protopeptides.32,33 These long-standing limitations of peptide chemistry and the observation of depsipeptide formation in model prebiotic reactions stimulated our inquiry into the use of ester bond formation, hydrolysis, and exchange to substitute for dynamic peptide chemistry.
Recently, we and others have demonstrated that dry–wet cycling of mixtures of hydroxy acids and amino acids produces peptides and depsipeptides with length distributions that can be controlled by the feeding of additional hydroxy and amino acids over the course of the reactions.30,34 We reasoned here that the lability and potential for transacylation of ester linkages within depsipeptides, compared to the amide linkages, could be harnessed to achieve a reversible bond formation. In particular, cycling between an evaporative dry phase (here termed ″dry-heating″ or ″drydown″) and a rehydrated wet phase could be used to promote ester formation in the dry phase and cleavage in the wet phase. This process would move materials between smaller amide-linked building block depsipeptides in a manner similar to what has been demonstrated with hydroxy acids.35−37 Such a system of dynamic ester chemistry would be advantageous for enabling dynamic chemistry through a two-step process in water with biocompatible and plausibly prebiotic building blocks. Moreover, depsipeptides are of growing interest in the field of biodegradable materials,38−41 so a method for reversible depsipeptide formation might also prove useful for the discovery of polymers with practical applications.
Here, we demonstrate that synthetic N-(α-hydroxyacyl)-amino acid units (amide-linked hydroxy acid–amino acid heterodimers) function as building blocks in a two-step dynamic polymerization when subjected to hydration–dehydration ″environmental″ cycles.42 The hydroxyl groups of these building blocks allow for dynamic ester exchange in water, while the amide linkages provide hydrogen bond donors to support structure formation by the depsipeptide condensation polymers. This approach is similar to the established peptide fragment exchange via dynamic thioester and amino acid hydrazide chemistry10,11,15 but with functional groups and linkages that may prove more suitable for some applications (e.g., less redox sensitive; peptidomimetic). Along these lines, we have recently shown that bifacial nucleobase-functionalized N-(α-hydroxyacyl)-amino acid building blocks oligomerize spontaneously when dried-down to form depsipeptides that are capable of self-assembly.43 Incorporating a variety of side chain functionalities, we elucidate structural and steric factors that govern the kinetics of depsipeptide polymerization and depolymerization, and further show how these differences can be harnessed to drive the selectivity of specific depsipeptide sequence motifs by varying environmental cycling parameters, thereby achieving water-compatible reversible chemistry between peptide fragments without chemical activation.
Results and Discussion
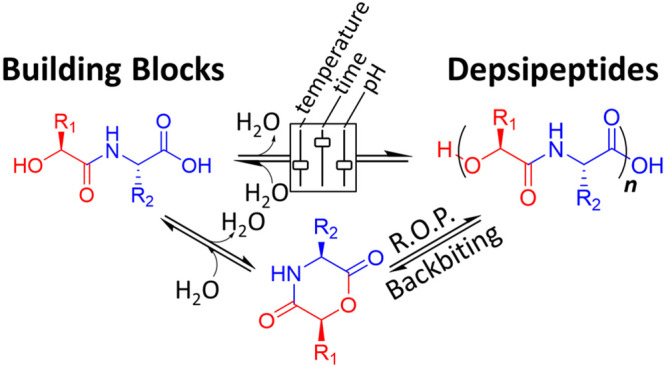
We hypothesized that because the building blocks used here have a six-atom repeat, oligomerization can take place through ring opening polymerization (ROP), with a 2,5-morpholinedione (2,5-MD) intermediate, similar to reported depsipeptide synthetic procedures.44,45 In Scheme 1, we show the dynamic equilibrium between amide-linked heterodimeric building blocks and depsipeptides that we sought to drive with dry–wet environmental cycles. We anticipated that the product depsipeptide esters would break down through a backbiting (sometimes referred to as ″tail-biting″) depolymerization pathway that involves transesterification to produce a 2,5-MD intermediate, as has recently been suggested to occur for similar depsipeptides43 and analogously demonstrated in peptide and polyester degradation pathways.46−48
Scheme 1. Reversible Depsipeptide Formation Enabled by Dry–Wet Cycling.

We prepared a library of amide-linked hydroxy acid–amino acid heterodimeric units, designated by two-letter codes as ″xX″,49 where the uppercase letter represents an amino acid and the lowercase letter represents the hydroxy acid analogue of an amino acid:
| amino acids | G | A | F | D | E | βA |
| hydroxy acids | g | a | f | d |
Heating of 25 μL aqueous solutions of xX building blocks (gG, gA, aG, and aA; unbuffered reaction pH 3) led to a visual dry state in less than 1 h. Applying continued drying at 65 °C for 1 week resulted in over 65% conversions of building blocks to depsipeptide products, with a distribution of oligomer lengths of up to eight heterodimer units (8-mers) (Figure 1A and Figure S1). This length of time for the reaction was chosen based on the observation that building block consumption reached steady-state levels between 3 and 7 days (Figure S1). Infrared spectroscopy confirmed the presence of ester bonds following the dry-state reaction (Figure S1). 2,5-MD formation was evident following dry-state reactions of various building blocks, as determined by LCMS and coinciding HPLC retention times of synthetic standards for certain cyclic building blocks (″c(xX)″, Figure 1A and Figure S2). We found that increasing the dry-state temperature above 65 °C provided slight increases in polymerization but with an increase in side product formation and building block degradation primarily due to amide bond hydrolysis (Figure S3). Polymerization was not significantly enhanced upon drying for periods longer than 1 week, and increases in the initial solution pH from 3 (unbuffered reaction) to 7 prior to drydown generally led to decreases in overall polymerization (Figures S4 and S5). Subjecting depsipeptides produced from dry-heating reactions to rehydration and wet-phase incubations resulted in the recovery of the building blocks by ester bond hydrolysis (Figure 1B). Increasing the wet-phase incubation pH to 6 and the temperature to 65 °C each greatly accelerated the rate of ester depolymerization but did not result in significant building block degradation (Figures S6–S8).
Figure 1.

Depsipeptide formation following dry-heating and depsipeptide depolymerization (building block recycling) following a wet phase. (A) HPLC-UV chromatograms for building blocks dried from unbuffered solutions for 7 days at 65 °C to form depsipeptides. ″c(xX)″ denotes the 2,5-MD of a building block when observed. Numbers: polymer length, e.g., 4 = (gG)4. (B) Time-dependent recovery of building blocks gG, gA, aG, and aA during unbuffered (pH 3) aqueous incubations at 65 °C of 10 mM building block samples previously dried for 7 days at 65 °C.
Variations in the depsipeptide depolymerization rates for the various units illustrate that residue side chains and sequences impact depsipeptide recyclability rates (Figure 1B), as have previously been observed in oligoester and depsipeptide biodegradation studies.38,50 Qualitatively, the relative rates of depolymerization for depsipeptides, derived from the slopes of the plots in Figure 1B, are summarized as:
To reveal possible differences in depsipeptide formation and depolymerization rates in mixed sequences, we dried an equimolar mixture of gG and aA for 7 days at 65 °C. The oligomers of these two units had the greatest difference in aqueous lifetimes among the homogeneous oligomerization products (Figure 1B). The reaction mixture resulted in a heterogeneous population of depsipeptides that includes various gG/aA compositions and lengths (Figure 2A). Among these products were the four possible ester-linked 2-mers, (gG)2, (aA)2, gG-aA, and aA-gG, which formed in varying abundances and whose chromatographic peaks were identified by LCMS and alignment with synthetic standards in HPLC (Figure S9). Aqueous incubation of these mixtures at 65 °C, pH 3 (unbuffered), showed that the xX-gG 2-mers were recycled back to building blocks much more rapidly than the xX-aA 2-mers, demonstrating that the sequence motif xX-aA affords longer aqueous lifetimes to depsipeptides (Figure 2B,C). The (aA)2 and gG-aA species exhibited increases in abundance at earlier degradation times likely due to the liberation of these sequence elements from abundant, longer oligomers (Figure 2C). Moreover, the degradation profiles of the various 3-mers formed (eight possibilities) followed similar trends, with four species (corresponding to xX-aA-xX) exhibiting relative increases in aqueous persistence, a phenomenon that we attribute to the interior positioning of the aA moiety within these mixed polymers reducing the backbiting rate due to increased steric factors (Figure S10). Finally, continuous dry–wet cycling of a gG + aA mixture demonstrated how the depsipeptide product distribution could shift over the course of multiple cycles, as indicated by a gradual increase in the relative amount of gG-aA present accompanied by a decrease in aA-gG (Figure S11). These results suggest that the system is not at thermodynamic equilibrium; rather, these depsipeptides are kinetically controlled products, and one can adjust the parameters of the dry or wet phases to change their extents of formation and breakdown.
Figure 2.
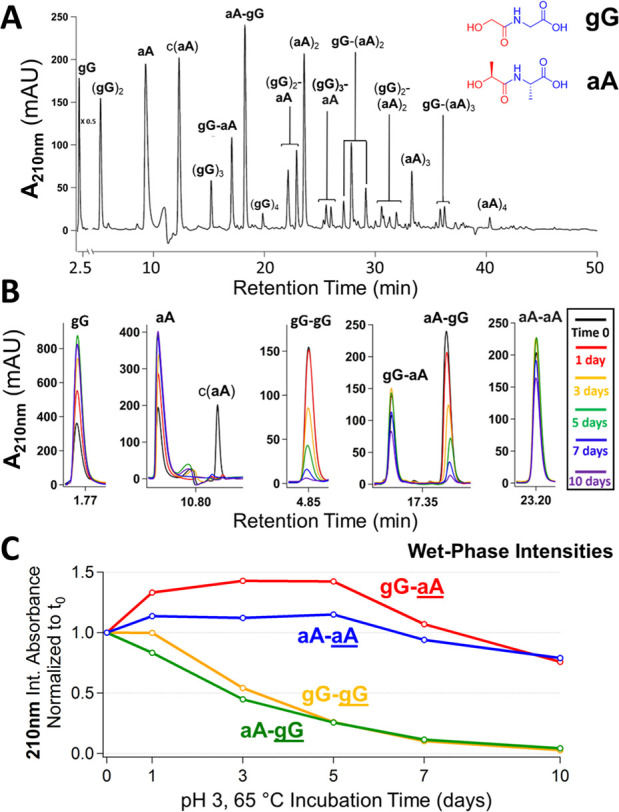
Differential aqueous persistence of depsipeptide sequence motifs. (A) HPLC-UV chromatograms of a mixture of gG and aA (1:1 molar ratio) dried for 7 days at 65 °C, unbuffered (pH 3). (B) Abundances of the four possible 2-mers formed in panel A are shown following up to 10 days of 65 °C unbuffered wet-phase incubation. (C) Integrated absorbances over incubation time with each normalized to the respective initial integrated absorbances in panel B indicate xX-aA motif persistence over xX-gG.
Drydown reactions carried out with building blocks other than α-hydroxy acid−α-amino acid linear heterodimers provide insights into the polymerization and depolymerization pathways operable in our system. Specifically, the inclusion of a β-alanine (βA) residue in the building block gβA resulted in reduced polymerization relative to the analogous gA, which we ascribed to the inability of gβA to form the six-membered ring of a 2,5-MD (Figure 3A). We anticipate that due to the structural similarity of having a seven-atom backbone, potential building blocks of the form β-hydroxypropionic acid–amino acid would have polymerization rates similar to those of gβA due to their inability to form a 2,5-MD. Further, heterotrimer and heterotetramer building blocks, such as gAG and gAGA (also unable to form 2,5-MDs), exhibited sharp decreases in the degree of polymerization with increasing building block length (Figure S12). On the other hand, the polymerization in drydown reactions of either gG or gA was accelerated by the addition of a synthetic 2,5-MD (c(gG)), which also indicates that ROP is a predominant factor in polymerization during drydown (Figures S13–S14). The results of a drydown reaction starting with the synthetic 2-mer (gA)2 were also informative, as the product distribution was similar to that of a reaction starting with gA, including the presence of the gA monomer, implying that during sample drying and possibly in the dry state, hydrolysis is happening in addition to esterification (Figure S15).
Figure 3.
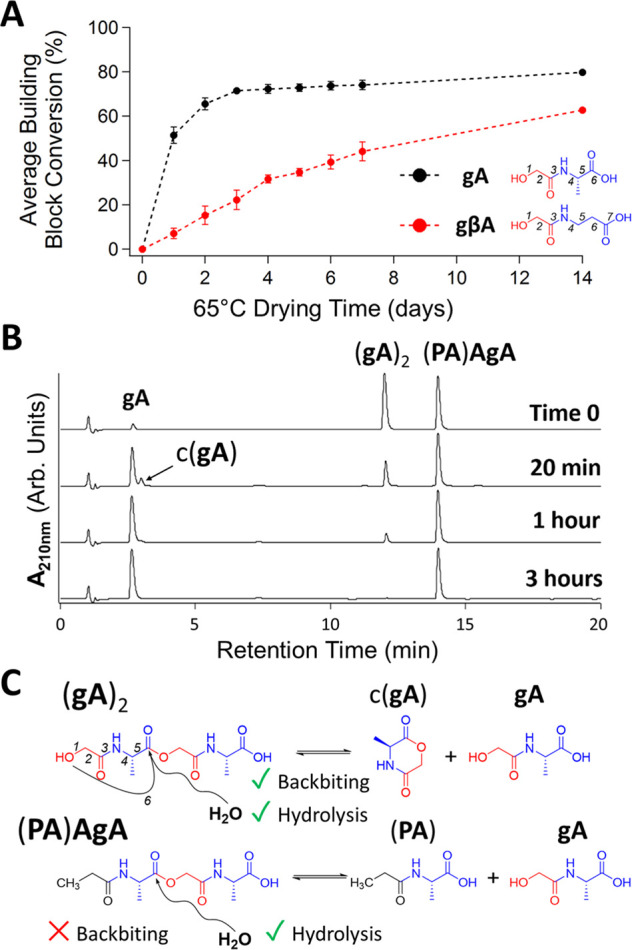
Structural factors influence building block polymerization and breakdown. (A) Time-dependent comparison of building block conversion to depsipeptides between gA and gβA, which can in principle form six- and seven-atom rings upon dehydration, respectively. Dashed lines were added to guide the eye. Bars: SD; n = 2. (B) Standards (gA)2 and (PA)AgA subject to pH 6, 65 °C aqueous incubation. (C) Structures of molecules used in panel B depicting depolymerization routes.
Through intramolecular transesterification, it is possible for the oligomers of xX, with a six-atom backbone repeat, to degrade though backbiting, a depolymerization mechanism previously established for both peptides and polyesters.46−48 To probe this possibility, we compared the degradation of synthetic (gA)2 to that of (PA)AgA, which is isosteric with (gA)2 but bears a terminal CH3 (of PA) in place of the OH of g (Figure 3C). Both molecules can depolymerize via hydrolysis, but only (gA)2 can depolymerize through backbiting, as (PA)AgA lacks a terminal nucleophile (Figure 3C). After co-incubation in a solution for 3 h at 65 °C and pH 6, over 99% of (gA)2 had depolymerized to gA units, while only 7% of (PA)AgA had depolymerized (Figure 3B). The 2,5-MD of gA, c(gA), was observed transiently in this (gA)2 incubation study, supporting our expectation that backbiting, with the cyclic intermediate formation, plays a dominant role in the depolymerization of depsipeptides of the form (xX)n (Figure 3B). We observed similar depolymerization characteristics for these molecules in 65 °C solutions at pH 3 and also at RT at pH 3 and 6 (Figure S16). Additionally, we found that depsipeptides of the form (gβA)n, with a seven-atom backbone repeat, exhibited almost no depolymerization over a 2-day incubation at 65 °C (pH 3), further underscoring the dominance of backbiting through a 2,5-MD intermediate cleavage pathway compared to direct backbone ester hydrolysis (Figure S17).
Esters are ideal for applications where finite material lifetimes and biocompatible monomers are desirable.38 Literature reports on the rates of hydrolysis of esters at 25 °C in neutral solutions show that their half-lives are on the order of days to years, depending on steric and structural factors.51 Quantitative assessment of the degradation rates of (gA)2 and (PA)AgA in water allowed a critical comparison of the difference between the single depsipeptide ester that is highly susceptible to intramolecular transesterification via OH terminal backbiting and the slower rates of cleavage of other, internal depsipeptide esters. For this comparison, we analyzed (gA)2 and (PA)AgA depolymerization data across a wide range of pH and temperatures to obtain the respective rate constants and half-lives under various conditions (see Supporting Information). For example, we found that at pH 2 and 25 °C, the half-life of (gA)2 is about 1 month and the half-life of (PA)AgA is about twice as long (Table 1). In contrast, at pH 7, we observed a 2-orders of magnitude difference in aqueous lifetime, with a half-life of around 4 days for (PA)AgA and only 37 min for (gA)2 (Table 1). These results exemplify that depsipeptide esters, in the absence of backbiting, can survive for substantial periods of time in water, and variations in their sequences (as in (gβA)n oligomers) can affect the lifetime of the depsipeptides considerably when pH conditions are slightly acidic or near-neutral.
Table 1. Kinetic Depolymerization Data for (gA)2 and (PA)AgA for a Range of Conditionsa.
| (gA)2t1/2 (h) |
(PA)AgAt1/2 (h) |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (°C) | pH: 2 | 3 | 4 | 5 | 6 | 7 | 8 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| 25 | 674 | 647 | 146 | 43.2 | 2.78 | 0.63 | 0.18 | 1394 | 2528 | 1655 | 1027 | 806 | 88.6 | 45.0 |
| 37 | 308 | 298 | 62.6 | 12.6 | 1.34 | 0.12 | 0.02 | 758 | 1300 | 826 | 583 | 432 | 51.9 | 23.9 |
| 50 | 76.8 | 53.5 | 26.1 | 2.91 | 0.76 | 0.03 | 0.01 | 175 | 232 | 281 | 157 | 83.2 | 14.1 | 3.43 |
| 65 | 34.4 | 27.9 | 5.63 | 0.82 | 0.17 | 0.01 | 0.01 | 93.3 | 90.6 | 94.0 | 49.8 | 29.4 | 4.63 | 0.88 |
| (gA)2 depolymerization, (ksc + kbb) (h–1) | (PA)AgA depolymerization, ksc (h–1)*10–3 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 25 | 0.001 | 0.001 | 0.005 | 0.016 | 0.249 | 1.11 | 3.96 | 0.497 | 0.27 | 0.419 | 0.675 | 0.86 | 7.83 | 15.4 |
| 37 | 0.002 | 0.002 | 0.011 | 0.055 | 0.516 | 5.86 | 31.1 | 0.914 | 0.533 | 0.839 | 1.19 | 1.60 | 13.4 | 29.0 |
| 50 | 0.009 | 0.013 | 0.027 | 0.238 | 0.914 | 22.7 | 66.4 | 3.95 | 2.98 | 2.47 | 4.42 | 8.33 | 49.2 | 202 |
| 65 | 0.020 | 0.025 | 0.123 | 0.843 | 4.21 | 62.3 | 100 | 7.43 | 7.64 | 7.38 | 13.9 | 23.5 | 150 | 792 |
See Supporting Information for calculations and parameters as well as raw data.
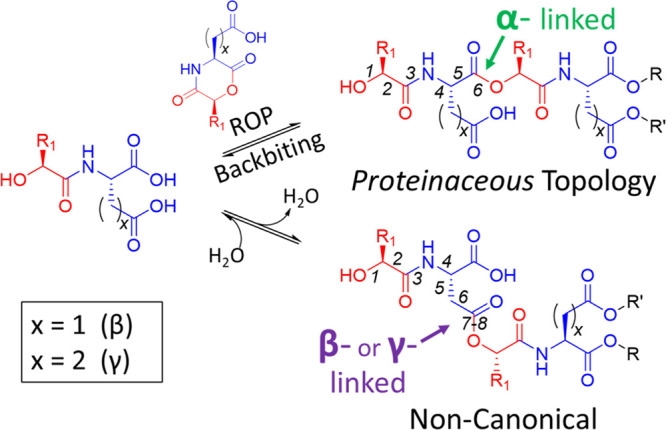
Having D (Asp) or E (Glu) as the amino acid building block allows for variations in depsipeptide regiochemistry due to the availability of the additional carboxylic acid to form ester linkages (Scheme 2). We used this attribute to investigate differences in aqueous persistence between the various possible topologies and to determine if depsipeptide product distributions could be skewed toward linkages with lower rates of hydrolysis. For example, homogeneously α-linked (xD)n and (xE)n depsipeptides should form and depolymerize via 2,5-MD intermediates, while side-chain-linked regiochemistries (e.g., β and γ linkages) would be less favorably formed but would have a substantially longer backbone linkage lifetime than α linkages (Scheme 2). It is also possible that transesterification occurs within the oligomers through anhydride intermediates, allowing for further sequence shuffling. Branching would also be possible for these bifunctional units as the drydown products elongate to 3-mers and beyond (Figure S18). Consistent with these possibilities, dry-heating samples of xD or xE units unbuffered at 65 °C gave rise to a great variety of sequence isomers for all polymer sizes, as revealed by LCMS (Figure S18). Within the gE drydown products, we observed up to 16-mer depsipeptides, the largest depsipeptides formed in our studies (Figure S19).
Scheme 2. Bifunctional Building Blocks Allow for Variations in Depsipeptide Regiochemistry.
By adjusting the pH of drying, the (aD)n product distributions could be made to favor particular depsipeptide topologies over others (Figure S20), an effect that we attributed to the increased rate of backbiting at near-neutral pH hindering the accumulation of native α-linked polymers. Applying continuous dry–wet cycling of aD at a pH of 5.5, we found that the product distribution could be skewed to select for side chain (β) linked species in a highly controllable manner (Figure 4 and Figures S21–S22). Coelution of the aD 2-mers with synthetic standards confirmed that β-(aD)2 was the species that survived the wet phases, while α-(aD)2 degraded rapidly (Figure 4C). We infer that the wet-phase-surviving (aD)3–(aD)5 species were predominantly O-terminated with a β-linkage to an aD unit, which disallows backbiting (Figure 4A,B). The reduced yields observed for the various 3- to 5-mers may be due to the dominant backbiting depolymerization of the α-terminated species, which could also have contributed to the observed increases in β-(aD)2 yields following the wet phases (Figure 4B). The 2-mers of gD, gE, and aE were each expected to form two regioisomers in dry–wet cycles, similar to (aD)2. The gD and aE 2-mers each exhibited two distinct HPLC peaks that fluctuated in intensity in a similar fashion to those of α- and β-(aD)2, allowing us to make putative assignments for the native and side-chain-linked (gD)2 and (aE)2 species (Figures S23–S24). The gE depsipeptides, on the other hand, only displayed one peak corresponding to the 2-mers in the HPLC chromatograms, which may be due to the overlap of the α and γ regioisomers (Figure S25). The inseparability of the gE 2-mers is exemplified in Figure S19, which shows two very closely eluting peaks corresponding to the mass of (gE)2 in the selected ion-monitoring traces.
Figure 4.

pH-controlled selectivity for side-chain-linked depsipeptides using bifunctional building blocks. (A) aD dry–wet cycled in pH 5.5 TEAOAc showed comparable abundances of both (aD)2 isomers formed following 24 h dry-heating phases (orange traces) and preferential preservation of β-(aD)2 following 18 h wet phases (blue traces). Peaks labeled [D/a(aD)2] displayed a mass consistent with a dehydration product from D/a + (aD)2. A stack plot of the chromatograms in panel A is provided in Figure S22 for clarity. (B) Integrated abundances of (aD)2 regioisomers and higher-order oligomer peaks (total integrated areas) from panel A over cycling time. Yellow regions indicate drying and heating at 75 °C (24 h). Blue regions indicate rehydration and incubation in the solution state at 65 °C (18 h). (C) Alignment with synthetic standards of α- and β-(aD)2 confirms regioisomer identities.
We also found that depsipeptides produced in drydown reactions were enriched in α-linked (proteinaceous) topology with increasing bulkiness of the hydroxy acid side chain substituent on building blocks with D or E as the amino acid (Figure 5). For the fX motif, the percent α-linkage formation of 2-mers reached 93% following 7 day drying at 65 °C (Figure 5). The spiking of α-linked synthetic standards for 2- through 4-mers of both fD and fE units suggested that these depsipeptides were composed primarily of native α-linked topology (Figures S26–S27). We suspect that the observed α-selectivity for the bifunctional units may arise from Thorpe–Ingold effect-type kinetic effects, which would promote the formation of the 2,5-MD (and thus promote subsequent ROP) as the hydroxy acid substituent size increases.52,53 We note that all fX building blocks were dried with 1 equiv of the additive 2-hydroxypyridine (2-HP) to mitigate their precipitation during dry-heating, in a manner similar to recent studies in our laboratory with depsipeptide nucleic acids (see Figure S28 for details).43
Figure 5.
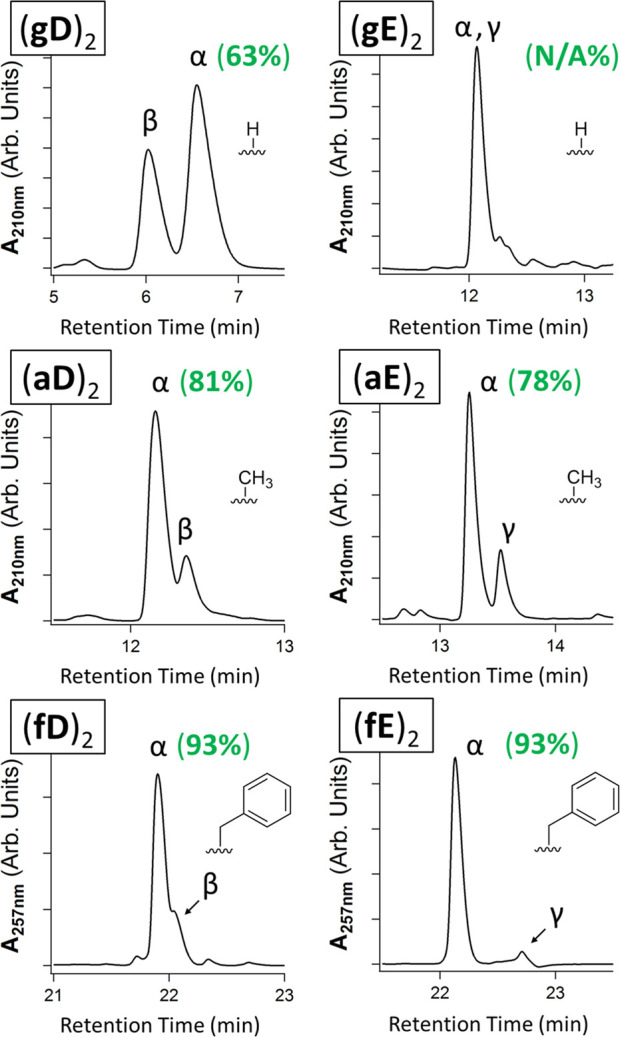
Hydroxy acid side chain size dictates native α-depsipeptide selectivity. Unbuffered 7 day, 65 °C drying of xD and xE building blocks showing 2-mers and percent α-linkage by integration. The α and γ regioisomers of (gE)2 appear to overlap; thus, their relative abundances are indeterminate. Hydroxy acid side chains are shown in each panel. The identities of the α- and γ-linked 2-mers of fE were determined using synthetic standards (Figure S26), while the 2-mers of fD were identified using an α-linked standard and by analysis of the degradation profile of the 2-mers (Figures S26–S27). fD and fE samples underwent drying with 1 equiv of 2-hydroxypyridine to prevent precipitation (Figure S28).
Finally, the results reported here used carboxylic acid side chains exclusively on the amino acid components of heterodimeric building blocks. We expected that similar results would be obtained with building blocks for which the hydroxy acid residue was malic acid (d), which would place the carboxylic acid side chain group on the first residue of the heterodimer (i.e., of the form dX). However, we determined that such building blocks exhibited extensive degradation upon dry-heating, primarily in the form of intramolecular dehydration and amide hydrolysis, which reduced the ability of these building blocks to exhibit the continuous recycling properties we sought for this reversible chemistry application and hampered the formation of orderly alternating amide-ester depsipeptide polymers (Figure S29). Moreover, building blocks incorporating serine (having a reactive side chain hydroxyl group) as the second residue (i.e., xS) displayed a similar propensity for degradation owing to intramolecular dehydration and amide bond hydrolysis (Figure S30).
Conclusions
The results presented here demonstrate a promising mode of water-based dynamic chemistry for biodegradable peptidomimetic polymers and potentially expand the tool set of dynamic chemistry by providing a route to a reversible chemistry with depsipeptides that does not require chemical activation or harsh conditions.2,6−8,11,15 The current system advances from the existing paradigms by expanding the monomer library to include both amino and hydroxy acids in a controlled manner, and further by demonstrating environmentally responsive biomimetic-polymer formation and breakdown, allowing for the sampling of biocompatible sequences using the dynamic ester linkages of depsipeptides. Overall, our results indicate that the monomers g, a, f, D, and E are better suited for the dynamic system at hand, as these monomers in building blocks of the form xX display notable persistence when subjected to repeated dry–wet cycles and with extended heating in the dry phase.
In this system presented here, molecules that are able to backbite (e.g., (gA)2) would have kinetic polymerization and depolymerization advantages over others (conditions permitting), possibly making them able to resample their constituent building blocks on faster time scales. These results may have implications for the manner in which potential building blocks on the early earth may have undergone recycling-like processes and sequence space sampling prior to the emergence of life. Furthermore, the optimal pH ranges for building block polymerization (mildly acidic) differ from those of optimal depsipeptide depolymerization (near-neutral). This disparity may be advantageous for early, prebiotic oligomers as expedient formation and slow degradation under similarly acidic conditions (e.g., in a putative CO2-rich atmosphere or with acidity resulting from the building blocks’ carboxylic groups themselves, as in our ″unbuffered″ studies) allow for the persistence of various depsipeptides, whereas an eventual change in environmental pH could be viewed as a selective pressure that favors the preservation of more resilient species (e.g., in the case of β-(aD)2, etc.). In this respect, the oligomers can undergo a primitive form of natural selection.
The present system utilizes kinetic trapping of depsipeptides by adjusting various parameters of reversible dehydrating and aqueous phases, with the goal of selecting for supramolecular or template-binding structures with peptide-like properties. While existing dynamic covalent chemistry systems utilize a simultaneous polymerization and depolymerization of building blocks, our system temporally separates the two processes that overall constitute a reversible system and simultaneously allows for fine-tuning of (de)polymerization characteristics using temperature, time, pH, and building block side chains. Future studies might employ this system for the dynamic selection of depsipeptides with structural or functional properties.
Methods
Synthetic Procedures
Building blocks were prepared following a previously published base-catalyzed synthesis (see Supporting Information).34 Synthetic depsipeptide standards were prepared using various organic synthesis procedures as detailed in the Supporting Information.
Dry-Heating (Drydown) Phases
Concentrated stocks of building blocks were diluted to 10 mM using doubly distilled water (Milli-Q, 25 μL volume). Buffers were incorporated into the solutions in 10× equivalency from concentrated filtered stocks as needed for pH-dependent experiments. Samples were dried in PCR tubes (VWR) with caps open in ovens (65 and 85 °C) or on a BioRad PCR Cycler heating block (75, 95, and 105 °C) for the stated time periods. 2,5-MD seeding experiments were performed from lyophilized solids to prevent hydrolysis during dry-heating. For analysis, samples were resuspended with distilled water back to 10 mM in total building block concentration. Samples were briefly vortexed and sonicated to ensure the complete dissolution of waxy pellets.
Wet-Phase Incubation Phases
Crude depsipeptide samples were initially prepared by dry-heating building blocks to generate product mixtures. These samples were resuspended back to their initial starting concentration (typically in 25 μL volume) in building blocks with distilled water for unbuffered experiments or with 10 equiv of buffer as needed. All pH values were verified using pH strips. RT (25 °C) incubations were performed by sealing the cap on the PCR tube for the specified time period. High-temperature incubations (65 °C) required the addition of ∼150 μL of mineral oil to prevent evaporation in samples, which were sealed and enclosed in a BioRad PCR cycler set to 65 °C with the lid temperature set to 100 °C. Samples were recovered out from under the oil by pipetting repeatedly onto a clean laboratory parafilm to separate the oil from the aqueous phase. Controls were run to verify that there was no observable mass loss due to partitioning into the oil phase for these aqueous incubations by using a high-accuracy analytical balance (Mettler Toledo). Unbuffered samples were sent for analysis, while pH-adjusted samples were quenched with HCl to restore nascent pH. Experiments comparing (gA)2 and (PA)AgA degradation were executed similarly (in 40 μL volumes) but required no initial dry-heating.
Continuous Dry–Wet Cycling
Samples were subjected to iterations of the above dry-heating and wet-phase incubation procedures as needed. For gG + aA, xD, and xE dry–wet cycles, initial volumes were always 150 μL of 10 mM total building block solutions. For xD and xE, 24 h dry-heating phases took place at 75 °C, and 18 h aqueous incubations took place at 65 °C under oil. Buffer solutions were replenished following each dry phase to restore the buffer pH, as TEA buffer volatility otherwise caused the pH to return to 3.0 upon rehydration. Following each dry or wet phase, 25 μL of the solution was removed and frozen for analysis, with the volume gradually decreasing to total depletion by the end of the third wet phase. All phases of the gG + aA mixed experiment took place in the 65 °C oven, with no mineral oil layer added. Following daily rehydration, samples were placed back in the 65 °C oven with caps open to allow the solution to evaporate slowly for 24 h, resulting in cycles of ∼6 h wet phase and ∼18 h dry phase.
High-Performance Liquid Chromatography
Reverse phase HPLC analyses were conducted using an Agilent 1260 quaternary pump and Agilent 1260 autosampler with a DAD UV–vis detector with a path length of 1.0 cm. Samples were separated using a Phenomenex Kinetex 2.6 mm XB-C18 100 Å LC column 150 × 2.1 mm. The column temperature was 25 °C. Injection volumes were 10 μL with a 100 μL/s injection speed and H2O needle wash. The binary solvent system was: (A) 0.1% formic acid in LCMS-grade water and (B) LCMS-grade acetonitrile. The flow rate was 0.3 mL/min, with binary solvent gradient: 5 min 100% A, 0% B; 20 min ramp to 45% A, 55% B; 10 min 0% A, 100% B; 1 min ramp 100% A, 0% B; and 14 min 100% A, 0% B. Wavelengths printed were 210 and 257 nm, with an entire spectrum (180–400 nm) detected in 2 nm steps. Analysis of the gG + aA mixed samples was done similarly using a longer, more shallow gradient reaching 45% A, 55% B after 40 min followed by a 100% solvent B wash.
Liquid Chromatography–Mass Spectrometry
Species were identified by mass using an Agilent 1290 HPLC pump and thermostat with an Agilent 1260 autosampler and DAD UV–vis detector with a path length of 0.6 cm. The reverse phase separation apparatus involved the identical column and HPLC gradient described above. This system was coupled to an electrospray ionization mass spectrometry system, an Agilent 6130 single quad MS, using the following parameters: scanning ±65–±2000 m/z, capillary voltage: 2.0 kV, and fragmentor voltage: 70 V.
Acknowledgments
We thank David M. Fialho and Cesar Menor-Salvan for discussions, and Bradley T. Burcar and Jay G. Forsythe technical assistance.
Glossary
ABBREVIATIONS
- 2,5-MD
2,5-morpholinedione
- DKP
diketopiperazine
- ESI-MS
electrospray ionization mass spectrometry
- LCMS
liquid chromatography–mass spectrometry
- PLA
polylactic acid
- ROP
ring opening polymerization
- RT
room temperature (25 °C)
- SD
standard deviation
- TEAOAc
triethylammonium acetate buffer
- TEAB
triethylammonium bicarbonate buffer
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.2c00087.
Synthetic procedures, spectral characterization, additional figures, as well as ESI-MS, HRMS, NMR, LCMS-SIM, and FTIR methods (PDF)
Author Present Address
∇ Institute of Chemistry, The Hebrew University of Jerusalem, Jerusalem 91904, Israel
Author Contributions
∥ M.C. and M.F.P. contributed equally. The manuscript was written through contributions of several authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest. This research was supported by the NSF and the NASA Astrobiology Program under the NSF Center for Chemical Evolution (CHE-1504217). M.F.P. acknowledges the NASA postdoctoral program.
The authors declare no competing financial interest.
Supplementary Material
References
- Rowan S. J.; Cantrill S. J.; Cousins G. R. L.; Sanders J. K. M.; Stoddart J. F. Dynamic covalent chemistry. Angew. Chem., Int. Ed. 2002, 41, 898–952. . [DOI] [PubMed] [Google Scholar]
- Corbett P. T.; Leclaire J.; Vial L.; West K. R.; Wietor J. L.; Sanders J. K.; Otto S. Dynamic combinatorial chemistry. Chem. Rev. 2006, 106, 3652–3711. 10.1021/cr020452p. [DOI] [PubMed] [Google Scholar]
- Beste L. F.; Houtz R. C. Amide interchange reactions. J. Polym. Sci. 1952, 8, 395–407. 10.1002/pol.1952.120080404. [DOI] [Google Scholar]
- Isowa Y.; Ohmori M.; Ichikawa T.; Mori K.; Nonaka Y.; Kihara K.-i.; Oyama K.; Satoh H.; Nishimura S. The thermolysin-catalyzed condensation reactions of N-substituted aspartic and glutamic acids with phenylalanine alkyl esters. Tetrahedron Lett. 1979, 20, 2611–2612. 10.1016/S0040-4039(01)86363-2. [DOI] [Google Scholar]
- De Martin L.; Ebert C.; Gardossi L.; Linda P. High isolated yields in thermolysin-catalysed synthesis of Z-l-aspartyl-l-phenylalanine methyl ester in toluene at controlled water activity. Tetrahedron Lett. 2001, 42, 3395–3397. 10.1016/S0040-4039(01)00452-X. [DOI] [Google Scholar]
- Toledano S.; Williams R. J.; Jayawarna V.; Ulijn R. V. Enzyme-triggered self-assembly of peptide hydrogels via reversed hydrolysis. J. Am. Chem. Soc. 2006, 128, 1070–1071. 10.1021/ja056549l. [DOI] [PubMed] [Google Scholar]
- Bon E.; Bigg D. C. H.; Bertrand G. Aluminum chloride-promoted transamidation reactions. J. Org. Chem. 1994, 59, 4035–4036. 10.1021/jo00094a004. [DOI] [Google Scholar]
- Eldred S. E.; Stone D. A.; Gellman S. H.; Stahl S. S. Catalytic transamidation under moderate conditions. J. Am. Chem. Soc. 2003, 125, 3422–3423. 10.1021/ja028242h. [DOI] [PubMed] [Google Scholar]
- Hoerter J. M.; Otte K. M.; Gellman S. H.; Cui Q.; Stahl S. S. Discovery and mechanistic study of AlIII-catalyzed transamidation of tertiary amides. J. Am. Chem. Soc. 2008, 130, 647–654. 10.1021/ja0762994. [DOI] [PubMed] [Google Scholar]
- Woll M. G.; Gellman S. H. Backbone thioester exchange: A new approach to evaluating higher order structural stability in polypeptides. J. Am. Chem. Soc. 2004, 126, 11172–11174. 10.1021/ja046891i. [DOI] [PubMed] [Google Scholar]
- Larsson R.; Pei Z.; Ramström O. Catalytic self-screening of cholinesterase substrates from a dynamic combinatorial thioester library. Angew. Chem., Int. Ed. 2004, 43, 3716–3718. 10.1002/anie.200454165. [DOI] [PubMed] [Google Scholar]
- Ura Y.; Al-Sayah M.; Montenegro J.; Beierle J. M.; Leman L. J.; Ghadiri M. R. Dynamic polythioesters via ring-opening polymerization of 1,4-thiazine-2,5-diones. Org. Biomol. Chem. 2009, 7, 2878–2884. 10.1039/b903612a. [DOI] [PubMed] [Google Scholar]
- Ghosh S.; Ingerman L. A.; Frye A. G.; Lee S. J.; Gagné M. R.; Waters M. L. Dynamic cyclic thiodepsipeptide libraries from thiol-thioester exchange. Org. Lett. 2010, 12, 1860–1863. 10.1021/ol1004752. [DOI] [PubMed] [Google Scholar]
- Carnall J. M. A.; Waudby C. A.; Belenguer A. M.; Stuart M. C. A.; Peyralans J. J.-P.; Otto S. Mechanosensitive self-replication driven by self-organization. Science 2010, 327, 1502–1506. 10.1126/science.1182767. [DOI] [PubMed] [Google Scholar]
- Hirsch A. K. H.; Buhler E.; Lehn J.-M. Biodynamers: self-organization-driven formation of doubly dynamic proteoids. J. Am. Chem. Soc. 2012, 134, 4177–4183. 10.1021/ja2099134. [DOI] [PubMed] [Google Scholar]
- Dadon Z.; Samiappan M.; Shahar A.; Zarivach R.; Ashkenasy G. A high-resolution structure that provides insight into coiled-coil thiodepsipeptide dynamic chemistry. Angew. Chem., Int. Ed. 2013, 52, 9944–9947. 10.1002/anie.201303900. [DOI] [PubMed] [Google Scholar]
- Ruff Y.; Garavini V.; Giuseppone N. Reversible native chemical ligation: a facile access to dynamic covalent peptides. J. Am. Chem. Soc. 2014, 136, 6333–6339. 10.1021/ja4129845. [DOI] [PubMed] [Google Scholar]
- Altay Y.; Tezcan M.; Otto S. Emergence of a new self-replicator from a dynamic combinatorial library requires a specific pre-existing replicator. J. Am. Chem. Soc. 2017, 139, 13612–13615. 10.1021/jacs.7b07346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S. L. A production of amino acids under possible primitive earth conditions. Science 1953, 117, 528–529. 10.1126/science.117.3046.528. [DOI] [PubMed] [Google Scholar]
- Kvenvolden K.; Lawless J.; Pering K.; Peterson E.; Flores J.; Ponnamperuma C.; Kaplan I. R.; Moore C. Evidence for extraterrestrial amino-acids and hydrocarbons in the Murchison meteorite. Nature 1970, 228, 923–926. 10.1038/228923a0. [DOI] [PubMed] [Google Scholar]
- Cronin J. R.; Pizzarello S. Amino acids in meteorites. Adv. Space Res. 1983, 3, 5–18. 10.1016/0273-1177(83)90036-4. [DOI] [PubMed] [Google Scholar]
- Cleaves H. J.; Chalmers J. H.; Lazcano A.; Miller S. L.; Bada J. L. A reassessment of prebiotic organic synthesis in neutral planetary atmospheres. Orig. Life Evol. Biosph. 2008, 38, 105–115. 10.1007/s11084-007-9120-3. [DOI] [PubMed] [Google Scholar]
- Brack A.; Ehler K. W.; Orgel L. E. N, N’-carbonyldiimidazole-induced diketopiperazine formation in aqueous solution in the presence of adenosine-5′-monophosphate. J. Mol. Evol. 1976, 8, 307–310. 10.1007/BF01739255. [DOI] [PubMed] [Google Scholar]
- Cleaves H. J. II The origin of the biologically coded amino acids. J. Theor. Biol. 2010, 263, 490–498. 10.1016/j.jtbi.2009.12.014. [DOI] [PubMed] [Google Scholar]
- Fox S. W.; Harada K. Thermal copolymerization of amino acids to a product resembling protein. Science 1958, 128, 1214–1214. 10.1126/science.128.3333.1214. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Garcia M.; Surman A. J.; Cooper G. J. T.; Suárez-Marina I.; Hosni Z.; Lee M. P.; Cronin L. Formation of oligopeptides in high yield under simple programmable conditions. Nat. Commun. 2015, 6, 8385. 10.1038/ncomms9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman L.; Orgel L.; Ghadiri M. R. Carbonyl sulfide-mediated prebiotic formation of peptides. Science 2004, 306, 283–286. 10.1126/science.1102722. [DOI] [PubMed] [Google Scholar]
- Parker E. T.; Zhou M.; Burton A. S.; Glavin D. P.; Dworkin J. P.; Krishnamurthy R.; Fernández F. M.; Bada J. L. A plausible simultaneous synthesis of amino acids and simple peptides on the primordial Earth. Angew. Chem. 2014, 126, 8270–8274. 10.1002/ange.201403683. [DOI] [PubMed] [Google Scholar]
- Canavelli P.; Islam S.; Powner M. W. Peptide ligation by chemoselective aminonitrile coupling in water. Nature 2019, 571, 546–549. 10.1038/s41586-019-1371-4. [DOI] [PubMed] [Google Scholar]
- Forsythe J. G.; Yu S.-S.; Mamajanov I.; Grover M. A.; Krishnamurthy R.; Fernandez F. M.; Hud N. V. Ester-mediated amide bond formation driven by wet-dry cycles: a possible path to polypeptides on the prebiotic earth. Angew. Chem., Int. Ed. 2015, 54, 9871–9875. 10.1002/anie.201503792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggy A. The free energy of formation of the amide bond in polyamides. J. Appl. Chem. 1954, 4, 154–159. 10.1002/jctb.5010040402. [DOI] [Google Scholar]
- Huber C.; Eisenreich W.; Hecht S.; Wächtershäuser G. A possible primordial peptide cycle. Science 2003, 301, 938–940. 10.1126/science.1086501. [DOI] [PubMed] [Google Scholar]
- Raggi L.; Bada J. L.; Lazcano A. On the lack of evolutionary continuity between prebiotic peptides and extant enzymes. Phys. Chem. Chem. Phys. 2016, 18, 20028–20032. 10.1039/C6CP00793G. [DOI] [PubMed] [Google Scholar]
- Yu S.-S.; Solano M. D.; Blanchard M. K.; Soper-Hopper M. T.; Krishnamurthy R.; Fernández F. M.; Hud N. V.; Schork F. J.; Grover M. A. Elongation of model prebiotic proto-peptides by continuous monomer feeding. Macromolecules 2017, 50, 9286–9294. 10.1021/acs.macromol.7b01569. [DOI] [Google Scholar]
- Weber A. L. Thermal synthesis and hydrolysis of polyglyceric acid. Origins Life Evol., Biosphere 1989, 19, 7–19. 10.1007/BF01808284. [DOI] [PubMed] [Google Scholar]
- Mamajanov I.; MacDonald P. J.; Ying J.; Duncanson D. M.; Dowdy G. R.; Walker C. A.; Engelhart A. E.; Fernández F. M.; Grover M. A.; Hud N. V.; Schork F. J. Ester formation and hydrolysis during wet–dry cycles: generation of far-from-equilibrium polymers in a model prebiotic reaction. Macromolecules 2014, 47, 1334–1343. 10.1021/ma402256d. [DOI] [Google Scholar]
- Chandru K.; Guttenberg N.; Giri C.; Hongo Y.; Butch C.; Mamajanov I.; Cleaves H. J. II Simple prebiotic synthesis of high diversity dynamic combinatorial polyester libraries. Commun. Chem. 2018, 1, 30. 10.1038/s42004-018-0031-1. [DOI] [Google Scholar]
- Feng Y.; Guo J. Biodegradable polydepsipeptides. Int. J. Mol. Sci. 2009, 10, 589–615. 10.3390/ijms10020589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Jimenez G. M.; Burgos-Hernandez A.; Ezquerra-Brauer J. M. Bioactive peptides and depsipeptides with anticancer potential: sources from marine animals. Mar. Drugs 2012, 10, 963–986. 10.3390/md10050963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y. F.; Hudalla G. A.; Han H.; Collier J. H. Controllably degradable β-sheet nanofibers and gels from self-assembling depsipeptides. Biomater. Sci. 2013, 1, 1037–1045. 10.1039/c3bm60161g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M. M.; Eckes K. M.; Suggs L. J. Charge and sequence effects on the self-assembly and subsequent hydrogelation of Fmoc-depsipeptides. Soft Matter 2014, 10, 2693–2702. 10.1039/C4SM00009A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav N.; White D.; Chang S. Peptide formation in the prebiotic era: thermal condensation of glycine in fluctuating clay environments. Science 1978, 201, 67–69. 10.1126/science.663639. [DOI] [PubMed] [Google Scholar]
- Fialho D. M.; Karunakaran S. C.; Greeson K. W.; Martínez I.; Schuster G. B.; Krishnamurthy R.; Hud N. V. Depsipeptide nucleic acids: prebiotic formation, oligomerization, and self-assembly of a new proto-nucleic acid candidate. J. Am. Chem. Soc. 2021, 13525. 10.1021/jacs.1c02287. [DOI] [PubMed] [Google Scholar]
- Janneke H.; Frits E. K.; Shuji S.; Johannes W. V. B.; Jan F. Synthesis of poly [oxyethylidenecarbonylimino (2-oxoethylene)][poly (glycine-d, l-lactic acid)] by ring opening polymerization. Makromol. Chem. Rapid Commun. 1985, 6, 9–14. 10.1002/marc.1985.030060103. [DOI] [Google Scholar]
- in’t Veld P. J.; Dijkstra P. J.; Van Lochem J. H.; Feijen J. Synthesis of alternating polydepsipeptides by ring-opening polymerization of morpholine-2, 5-dione derivatives. Die Makromol. Chem.: Macromol. Chem. Phys. 1990, 191, 1813–1825. 10.1002/macp.1990.021910808. [DOI] [Google Scholar]
- Steinberg S.; Bada J. L. Diketopiperazine formation during investigations of amino acid racemization in dipeptides. Science 1981, 213, 544–545. 10.1126/science.213.4507.544. [DOI] [PubMed] [Google Scholar]
- De Jong S. J.; Arias E. R.; Rijkers D. T. S.; Van Nostrum C. F.; Kettenes-Van Den Bosch J. J.; Hennink W. E. New insights into the hydrolytic degradation of poly(lactic acid): Participation of the alcohol terminus. Polymer 2001, 42, 2795–2802. 10.1016/S0032-3861(00)00646-7. [DOI] [Google Scholar]
- Van Nostrum C. F.; Veldhuis T. F. J.; Bos G. W.; Hennink W. E. Hydrolytic degradation of oligo(lactic acid): a kinetic and mechanistic study. Polymer 2004, 45, 6779–6787. 10.1016/j.polymer.2004.08.001. [DOI] [Google Scholar]
- Forsythe J. G.; Petrov A. S.; Millar W. C.; Yu S.-S.; Krishnamurthy R.; Grover M. A.; Hud N. V.; Fernández F. M. Surveying the sequence diversity of model prebiotic peptides by mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E7652–E7659. 10.1073/pnas.1711631114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile P.; Chiono V.; Carmagnola I.; Hatton P. V. An overview of poly(lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. 10.3390/ijms15033640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabey W.; Mill T. Critical review of hydrolysis of organic compounds in water under environmental conditions. J. Phys. Chem. Ref. Data 1978, 7, 383–415. 10.1063/1.555572. [DOI] [Google Scholar]
- Beesley R. M.; Ingold C. K.; Thorpe J. F. CXIX.—The formation and stability of spiro-compounds. Part I. spiro-Compounds from cyclo hexane. J. Chem. Soc., Trans. 1915, 107, 1080–1106. 10.1039/CT9150701080. [DOI] [Google Scholar]
- Levine M. N.; Raines R. T. Trimethyl lock: a trigger for molecular release in chemistry, biology, and pharmacology. Chem. Sci. 2012, 3, 2412–2420. 10.1039/c2sc20536j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.