

Abstract
Microorganisms which can degrade and grow on the purified sheath of a sheathed bacterium Sphaerotilus natans were collected from soil and river water. Two bacterial strains were isolated from the soil and designated strains TB and TK. Both strains are rod shaped, negatively stained by gram staining, facultatively anaerobic, and formed ellipsoidal endospores. These characteristics suggested that the isolates belong to the genus Paenibacillus, according to Ash et al. (C. Ash, F. G. Priest, and M. D. Collins, Antonie Leeuwenhoek 64:253–260, 1993). Phylogenetic analysis based on the 16S rDNA supported this possibility. Purification of the sheath-degrading enzyme was carried out from the culture broth of strain TB. The molecular weight of the enzyme was calculated to be 78,000 and 50,000 by sodium dodecyl sulfate-polyacrylamide electrophoresis and gel filtration chromatography, respectively. Enzyme activity was optimized at pH 6.5 to 7.0 and 30 to 40°C. The reaction was accelerated by the addition of Mg2+, Ca2+, Fe3+, and iodoacetamide, whereas it was inhibited by the addition of Cu2+, Mn2+, and dithiothreitol. The enzyme acted on the polysaccharide moiety of the sheath, producing an oligosaccharide the size of which was between the sizes of maltopentaose and maltohexaose. As the reaction proceeded, the absorbance at 235 nm of the reaction mixture increased, suggesting the generation of unsaturated sugars. Incorporation of unsaturated sugars was also suggested by the thiobarbituric acid reaction. It is possible that the enzyme is not a hydrolytic enzyme but a kind of polysaccharide eliminase which acts on the basic polysaccharide.
Sphaerotilus natans is one of the sheathed bacteria that construct a tube-like sheath surrounding each cell and is often referred to as one of the filamentous bacteria which cause poor settling problems (bulking) in activated sludge (23). There are two genera of sheathed bacteria, Sphaerotilus, which is often found in polluted streams, and Leptothrix, which is a typical inhabitant of metal-rich streams. Because of their similarities in morphological and physiological characteristics, the classification “Sphaerotilus-Leptothrix group” has been proposed (23). Their similarity has been further confirmed by phylogenetic analysis based on 16S rRNA sequences (17, 18). The sheaths of both genera are a complex of protein and polysaccharide (5, 20). The sheath of S. natans is extremely rich in cysteine and contains galactosamine (or N-acetylgalactosamine) and glucose as sugar components (20). The sugar components of the sheath of Leptothrix discophora are N-acetylgalactosamine and uronic acids, and its protein moiety is rich in cysteine (5). The sheath of L. discophora is easily broken down by the addition of disulfide bond-reducing reagents, suggesting that the fibrous matrix composed of heteropolysaccharide and peptide is cross-linked by disulfide bonds and constructs a sheath structure (6). In contrast, the sheath of S. natans is resistant to disulfide bond-reducing reagents (20). Decomposition of the sheath of S. natans is achieved by selective cleavage of amide bonds by the addition of hydrazine releasing heteropolysaccharide composed of glucose and galactosamine (20). It can be assumed that the sheath of S. natans is constructed by covalent cross-linking of the polysaccharide and peptide.
No attempts to elucidate the chemical structures of bacterial sheaths have been carried out. A promising way of analyzing the structure of such complicated macromolecules is to decompose them into the constitutional units by applying specific degrading enzymes. For example, the structure of murein was elucidated by analyzing the enzymatic products of lysozyme. However, no proteases or glucanases which achieve bacterial sheath degradation have ever been known. In the present study, we searched for bacteria capable of growth on the sheath of S. natans and attempted to purify sheath-degrading enzymes potentially applicable for elucidation of the sheath structure.
MATERIALS AND METHODS
Preparation of sheath and sheath polysaccharide.
The sheath of S. natans IFO 13543T was prepared by the method previously described (20). The sheath polysaccharide was prepared by the following procedure. The sheath (20 mg) was suspended in 20 ml of 3 N NaOH and allowed to settle at 30°C for 3 days under an N2 or Ar atmosphere. After the solution was acidified to pH 2 to 3 by the addition of 2 N HCl, it was filtered through a glass filter to remove the residual sheath. Two volumes of ethanol was added to the filtrate, and the released polysaccharide was precipitated. The precipitate was rinsed with cold 70% ethanol, dissolved in distilled water, and dialyzed against distilled water. The dialysate (deacetylated sheath polysaccharide) was lyophilized (about 10 mg), and N-acetylation was performed as follows. The deacetylated sheath polysaccharide was dissolved in 10 ml of saturated NaHCO3 solution. While mixing the solution at room temperature, 2 ml of acetic anhydride was added 5 times at 15-min intervals. After the last addition of acetic anhydride, the solution was further stirred for 30 min to complete the reaction. The solution was passed through a column of Amberlite 50W-X8 (H+) (2.5 by 20 cm) and then dialyzed against distilled water. The N-acetylated sheath polysaccharide was obtained by lyophilization of the dialysate.
Screening and isolation of sheath-degrading bacteria.
Soils and river waters were collected from 20 and 10 sites, respectively, in Kanagawa Prefecture, Japan. The screening medium was composed of the following: sheath, 0.1 g/liter; NaNO3, 2 g/liter; K2HPO4, 0.1 g/liter; MgSO4 · 7H2O, 0.5 g/liter; KCl, 0.5 g/liter; and FeSO4 · 7H2O, 0.01 g/liter. To the flasks (100 ml) containing 20 ml of screening medium, about 0.2 g of soil or 1 ml of water was added, and the mixture was allowed to settle at 30°C for 2 weeks. In one culture inoculated with wet garden soil, sheath flocs disappeared. A portion of this culture (0.1 ml) was transferred into a flask containing fresh sheath medium and incubated for 1 week, with this procedure being repeated eight times. A loopful of culture was then streaked on agar plates composed of 0.05 g of beef extract/liter and incubated at 30°C. Colonies that formed on the plates were reinoculated into the screening medium to test sheath degradation. A bacterial strain capable of sheath degradation was obtained and was designated strain TB.
Another sheath-degrading bacterium was isolated from the soil by the following procedure. Soils were collected from 10 sites in Yamanashi Prefecture, Japan. One soil sample exhibited sheath decomposition in the screening medium. After reinoculation into the same medium five times, the culture was heat treated at 50°C for 24 h. A sheath-degradable bacterium isolated on the beef extract agar plates was designated strain TK.
Cultivation and characterization of the isolates.
Strains TB and TK were cultured in the sheath medium composed of the following: sheath, 0.1 g/liter; Proteose Peptone no. 3 (Difco), 2 g/liter; K2HPO4, 0.1 g/liter; MgSO4 · 7H2O, 0.5 g/liter; KCl, 0.5 g/liter; and FeSO4 · 7H2O, 0.01 g/liter. Unless otherwise described, the strains were inoculated into 20 ml of the medium contained in a 100-ml flask and were statically cultured at 30°C. To test the utilization of several organic compounds as sole carbon and nitrogen sources, liquid media and solid media (solidified with 1.5% agar) composed of 0.05% beef extract, tryptone, yeast extract, Bacto Peptone, Proteose Peptone no. 3, Casamino Acids, or malt extract were prepared. Utilization of carbohydrates (including sheath polysaccharide) was tested using media which had the same composition as that of the sheath medium, except that the sheath was replaced with 0.1 g of carbohydrate/liter. Utilization of the sheath polysaccharide was also examined using two other media. One was omitted from Proteose Peptone but supplied with 0.2 g of (NH4)2SO4/liter in substitution, and another was not supplied with any nitrogen sources. Anaerobic cultivation was performed in a BBL GasPak pouch (Becton Dickinson Microbiology Systems, Cockeysville, Md.) packed with a flask containing the sheath medium inoculated with strain TB or TK just before packing.
Phylogenetic analysis.
The DNA of strains TB and TK grown on the sheath medium was prepared using the GenomicPrep cells and tissue DNA isolation kit (Amersham Pharmacia Biotech, Little Chalfont, Buckinghamshire, United Kingdom). Amplification of the 16S rRNA gene (28-1391, Escherichia coli numbering) was performed using primers designed to bind 8 to 27 (5′AGAGTTTGATCATGGCTCAG3′) and 1406 to 1392 (5′ACGGGCGGTGTGTAC3′). The product was cloned into pCR2.1 with the TA cloning method (Invitrogen, San Diego, Calif.), and the nucleotide sequences were determined using a 373S DNA sequencer (Applied Biosystems, Foster City, Calif.) with dye dideoxynucleotide terminators. Homology searches were carried out by the BLASTN program. Multiple alignments were done by Clustal W, and a phylogenetic tree was drawn by a TREECON package (22) based on the neighbor-joining method.
Quantification of cell and sheath.
Because strain TB does not form distinctive colonies on any solid media, it is difficult to estimate its cell densities by colony counting. The growth of the bacterium was estimated by counting the number of cells in the culture with an epifluorescence microscope (Zeiss Axioskop). Culture (10 μl) was mixed with 1 μl of 0.02% acridine orange solution, applied onto an agar slide, and covered with a 1.8- by 1.8-mm glass. Average cell concentration was calculated from observations of 20 to 50 sites.
The sheath amount was estimated by measuring the amount of glucose incorporated in the sheath. Cell and sheath were precipitated by centrifugation. The pellet was suspended in 5 ml of 30 mM Tris-HCl buffer (pH 8.0) supplemented with 0.5 g of EDTA disodium salt/liter, 5 mg of lysozyme was added, and it was incubated at 37°C for 1 h. To destroy the cells, 0.5 ml of 10% sodium dodecyl sulfate (SDS) solution was added, mixed, and then heated at 110°C for 10 min. Because the sheath resists these treatments, it was harvested by centrifugation and washed with distilled water. The glucose amount in the sheath pellet was determined by phenol-sulfuric acid reaction. The glucose content of the sheath was calculated to be around 30% (wt/wt) by the phenol-sulfuric acid reaction (20). The sheath amount was estimated based on the glucose amount detected.
Sheath degradation by crude enzyme.
Strain TB was grown on sheath medium at 20°C for 6 days; cells and residual sheath were removed by centrifugation, and the culture fluid was passed through a 0.22-μm-pore-size membrane filter. The filtrate was dialyzed against distilled water at 4°C for 24 h and filtered through a sterile 0.22-μm filter. The filtrate was used as crude enzyme. The crude enzyme (15 ml) was aseptically added to 20 ml of fresh sheath medium and incubated at 30°C. A portion (1 ml) of the reaction mixture was recovered every 24 h and filtered through a membrane filter, and then the degree of sheath degradation was monitored by determining the amounts of released saccharide (phenol-sulfuric acid reaction) and protein. The amount of residual sheath was also measured.
Detection of protease activity.
Azocasein was dissolved in 100 mM Tris-HCl (pH 8.0) with 2 mM CaCl2 to be 5 mg/ml. To this solution (0.5 ml), 0.1 ml of culture filtrate of strain TB was added, followed by incubation at 30°C. After 18 h of incubation, 0.4 ml of 15% (wt/vol) trichloroacetic acid was added to terminate the reaction, the mixture was centrifuged, and 1 ml of the supernatant was recovered. After neutralization by the addition of 50 μl of 10 M NaOH, the absorbance at 440 nm was measured.
Purification of sheath polysaccharide-degrading enzyme.
Cultivation of strain TB for enzyme preparation was done in 500-ml flasks containing 100 ml of medium with shaking at 30°C for 72 h. The supernatant of the culture broth (3 liters) was obtained by centrifugation and was brought to 90% saturation with ammonium sulfate. The precipitate formed was collected by centrifugation, suspended in 10 mM Tris-HCl buffer (pH 8.0), and dialyzed against the same buffer at 4°C. To the dialysate, ammonium sulfate was added to 40% saturation, and the result was filtered through a glass filter. The filtrate was loaded on a column (2.5 by 17 cm) of phenyl-Toyopearl 650M (Tosoh, Tokyo, Japan) preequilibrated with 10 mM Tris-HCl buffer (pH 8.0) supplemented with ammonium sulfate (40% saturation). After the column was washed with the equilibration buffer, it was washed with a linear gradient of ammonium sulfate (40 to 0% saturation) in the buffer. Sheath polysaccharide-degrading activity was eluted by washing further with the final buffer.
Assay of sheath polysaccharide-degrading activity.
Sheath polysaccharide-degrading activity was assayed by measurement of the rate of the increase in reducing power in the reaction mixture. The reaction mixture consisted of 0.7 ml of 50 mM Tris-HCl buffer (pH 8.0) containing 0.2 mg/ml of N-acetylated sheath polysaccharide and an appropriate amount (usually 0.1 ml) of the enzyme. The enzyme reaction was carried out for 18 h at 30°C. After the reaction, the amount of reducing sugars was determined by the 3,6-dinitrophthalic acid method (10). One unit of enzyme activity was defined as the quantity of the enzyme which produces a reducing power equal to 1 μmol of glucose per min.
Protein assays.
The amount of protein was determined by the Bio-Rad DC protein assay kit (Bio-Rad, Hercules, Calif.) with bovine serum albumin as a standard. SDS-polyacrylamide gel electrophoresis (PAGE) was performed on a 5 to 20% gradient gel for assessment of the purity and estimation of the molecular weight of the enzyme. The gel was stained with Coomassie brilliant blue R-250. Isoelectric focusing in polyacrylamide was performed using an Ampholine PAGplate, pH 3.5 to 9.5 (Pharmacia, Uppsala, Sweden). The molecular mass of the native enzyme was determined by gel filtration chromatography with Sephacryl S-300 (Pharmacia). The size of the column was 1.5 by 50 cm, and the mobile phase was 66 mM phosphate buffer (pH 7.0) supplemented with 250 mM NaCl.
Amino acid sequence analysis.
The N-terminal amino acid sequence was determined with a Shimadzu PPSQ-10 protein sequencer (Shimadzu, Kyoto, Japan). The enzyme was subjected to SDS-PAGE and then blotted onto a polyvinylidene difluoride membrane. The enzyme on the membrane was visualized with Coomassie brilliant blue G-250 and applied to the protein sequencer. To determine the internal amino acid sequence, the purified enzyme was subjected to SDS-PAGE together with V8 protease (about 1/50 mol). Digestion was done at room temperature for 6 h in the stacking gel (5%) at a low current of 2 to 5 mA, and then the separation of the digests was done in the running gel (20%) at 25 mA. After SDS-PAGE, the digests were blotted onto a polyvinylidene difluoride membrane. A major fragment of 18 kDa was utilized for amino acid sequence analysis.
Analysis of the enzymatic products.
The molecular mass of the enzymatic digest of deacetylated sheath polysaccharide was determined by gel filtration chromatography. Deacetylated sheath polysaccharide (5 mg) was suspended (deacetylated sheath polysaccharide is insoluble in aqueous solutions under neutral and alkali conditions) (20) in 0.5 ml of 66.6 mM phosphate buffer (pH 7.0), and the reaction was then initiated by 0.1 ml of purified enzyme. After incubation for 18 h at 30°C, the reaction mixture was applied to a Toyopearl HW-40S column (1.5 by 70 cm). The flow rate was set at 0.5 ml/min. The mobile phase was 10 mM HCl, and the elution of the product was detected by a refractive index detector.
The monosaccharide composition of the enzymatic digest of sheath polysaccharide was performed by gas liquid chromatography. The digest (originated from 5 mg of sheath polysaccharide) eluted from the Toyopearl column was dried under reduced pressure and used for analysis. The sample preparation and analytical conditions were the same as in the method previously described (20).
The formation of unsaturated sugars during enzymatic digestion of the sheath polysaccharide was detected by monitoring the absorbance at 235 nm based on the generation of double bonds. The existence of unsaturated sugars was also confirmed by the thiobarbituric acid reaction (24).
Nucleotide sequence accession numbers.
The nucleotide sequences of the 16S rRNA gene of strains TB and TK have been deposited in the DDBJ, EMBL, and GenBank DNA databases under the respective accession numbers of AB041720 and AB041721.
RESULTS
Isolation and some properties of sheath-degrading bacteria.
Sheath degradation was not observed in any cultures inoculated with river water. In a culture supplemented with soils from Kanagawa Prefecture, the sheath was broken down. From the culture, a bacterial strain (TB) capable of degrading the sheath was isolated. The colonies of strain TB on the beef extract agar plate did not exceed 1 mm in diameter even after incubation for 2 weeks. It took more than a 5-day incubation to form colonies on the plate. The colony was colorless, thin, and translucent with a ragged edge and surface. Some of the cells of TB in the colonies had an ellipsoidal endospore with the sporangium being swollen (Fig. 1). TB was still viable after heating at 85°C for 1 h or at 50°C for 24 h, indicating its spore-forming capability. Most of the cells in the colonies were slightly curved rods, and their length and width were 1.0 to 10 μm and 0.4 to 0.5 μm, respectively. However, cells grown on sheath medium were shorter (1.0 to 2.5 μm in length) and were mostly straight rods. A few spores were observed in sheath medium.
FIG. 1.
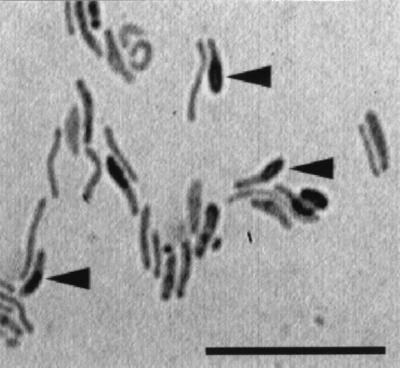
Microscopic observation of strain TB. Strain TB was cultured on a 0.05% beef extract agar plate and incubated at 30°C for 7 days. Cells were stained with saffronin for microscopic observation. Note that most rod-shaped cells were irregularly curved, and some bore thick ellipsoidal spores (see arrowheads). The bar indicates 10 μm.
Considering this heat stability, other sheath-degrading bacteria which have characteristics similar to those of strain TB were searched. Only one type of bacterial colony was formed on the isolation plate, and the isolate was designated strain TK. The colony appearance and cell morphology of strain TK were the same as those of strain TB.
Strains TB and TK grown on sheath medium were negatively stained in a Gram stain even in an early stage of cultivation, and ellipsoidal endospores were formed in old cultures, suggesting they belong to the genus Paenibacillus (1). No pigments were produced. A few motile cells were observed in younger cultures (24 to 48 h). Both strains are facultative anaerobes because cell growth and sheath degradation were observed under strictly anaerobic conditions. They did not form colonies anaerobically on plates of beef extract, tryptone, or yeast extract.
A homology search of the GenBank/EMBL/DDBJ databases revealed that the 16S rDNA sequences of strains TB and TK were similar to those of bacteria belonging to the genus Paenibacillus (Fig. 2). A 22-base sequence specific for the genus Paenibacillus (1), TCGATACCCTTGGTGCCGAAGT, exists in the 16S rDNA of both strains.
FIG. 2.

Phylogenetic network based on 16S rDNA sequences showing the interrelationships of sheath-degrading bacteria and Paenibacillus spp. Bootstrap values (100 replications) are shown near the branch points. Database accession numbers of the reference sequences are in parentheses. The scale bar indicates 0.05 changes per nucleotide position.
Effects of culture conditions on the growth of strain TB.
Typical time courses of the degradation of the sheath and the growth of strain TB in a medium mainly composed of sheath and Proteose Peptone are shown in Fig. 3. The sheath was broken down within 100 h, and cell concentration increased in inverse proportion to the concentration of the residual sheath. Even when Proteose Peptone was omitted from the medium, strain TB was able to grow utilizing the sheath as a sole source of carbon and nitrogen. In this case, more than a 7-day cultivation was required to break down the same amount of sheath. It must be emphasized here that cell growth did not occur when the sheath was omitted from the medium. Both cell growth and sheath degradation were most active at pH 7 and at 30°C.
FIG. 3.

Typical time courses of sheath degradation by and growth of strain TB. Strain TB was statically cultured at 30°C in a series of 500-ml flasks containing 100 ml of a medium composed of sheath (0.01%), peptone (0.2%), and K2HPO4 (0.1%). At intervals of 24 h, one flask was taken out and utilized for determinations of sheath and cells. Symbols: ■, amount of sheath; ●, cell concentration.
Strain TB could grow on both N-acetylated and deacetylated sheath polysaccharide, utilizing them as sole carbon sources in the presence of Proteose Peptone or (NH4)2SO4 as a nitrogen source. Growth was not observed on either glucose, galactosamine, or N-acetylgalactosamine, even when these saccharides were added together, though these monosaccharides are components of the sheath polysaccharide (20). Sheath polysaccharides (whether N-acetylated or deacetylated) were applicable only as carbon sources but did not serve as nitrogen sources because the addition of Proteose Peptone or (NH4)2SO4 was required for cell growth on these polysaccharides.
Enzymatic degradation of sheath.
The time-dependent changes in concentrations of soluble saccharide and protein in the sheath medium supplemented with the cell-free culture filtrate of strain TB are shown in Fig. 4. Both saccharide and protein concentrations increased with time. The sheath was almost broken down in 3 days. The heat-treated culture filtrate did not break down the sheath and released neither saccharide nor protein. The optimum temperature and pH for sheath degradation by the crude enzyme were 30°C and 7 to 8, respectively. Additionally, no protease activity was detected in the crude enzyme (culture filtrate) when azocasein was used as a substrate.
FIG. 4.

Release of saccharide and protein during enzymatic degradation of sheath. Sheath medium supplemented with crude enzyme was incubated at 30°C. At intervals of 24 h, the amounts of soluble saccharide (●, based on glucose), protein (■, based on bovine serum albumin), and residual sheath (▴) were measured. Heat-denatured (95°C, 30 min) enzyme was prepared and incubated with sheath in the same manner as the control, with the amounts of saccharide (○), protein (□), and sheath (▵) being measured.
Purification and molecular weight of the sheath polysaccharide-degrading enzyme.
In consideration of convenience and sensitivity, the N-acetylated sheath polysaccharide was used as a substrate for measuring the sheath-degrading activity. In the culture of strain TB, the activity of sheath degradation was not detected at the beginning of cultivation, until 24 h; then it rose to a maximum at 84 h. Because sufficient activity was already obtained at 50 h, cultivation for the preparation of sheath-degrading enzyme was carried out for 72 h. Table 1 shows the recoveries and purities of the enzyme at each purification step. The enzyme was purified 984-fold with a 1.5% yield with these purification steps. The purity of the enzyme effectively increased by hydrophobic chromatography. A typical elution profile of the sheath polysaccharide-degrading activity on a phenyl-Toyopearl column is shown in Fig. 5. The purity of the enzyme recovered from the peak eluted with the final buffer was confirmed by SDS-PAGE, giving one protein band in the position of about 78 kDa (Fig. 6). The molecular mass of the enzyme was determined to be about 50 kDa by gel filtration chromatography. Rather low molecular mass in gel filtration may be caused by the interaction between the enzyme and the resin. The isoelectric point of the enzyme was determined to be 6.7 by isoelectric focusing. The N-terminal amino acid sequence of the purified enzyme was determined to be NH2-ATVYEVGPGKTYTSIGSVPF. The internal sequence of NH2-EGNYIYGNGN was obtained from the major V8 protease digest of the TB enzyme. No relative protein was indicated by a BLASTp search based on these two partial amino acid sequences.
TABLE 1.
Purification of sheath polysaccharide-degrading enzyme from strain TB
| Purification step | Total protein (mg) | Total activity (mU) | Sp act (mU/mg) | Yield (%) | Fold purification |
|---|---|---|---|---|---|
| Culture | 1,935 | 1,770 | 0.915 | 100 | 1 |
| Dialysis | 269 | 941 | 3.50 | 53 | 3.8 |
| Phenyl-Toyopearl | 0.03 | 27 | 900 | 1.5 | 984 |
FIG. 5.

Elution of sheath polysaccharide-degrading activity on hydrophobic interaction chromatography. Proteins were eluted by decreasing the concentration of ammonium sulfate in the mobile phase (······). Fractions of 200 drops were collected to measure protein concentration (■) and enzyme activity (●).
FIG. 6.
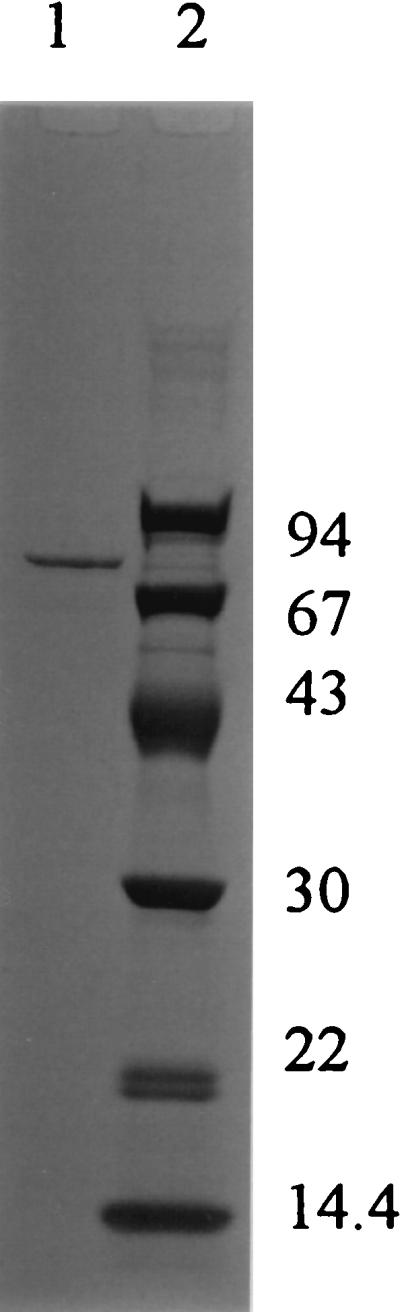
SDS-PAGE analysis of sheath-degrading enzyme from strain TB. A denatured sheath-degrading enzyme eluted and pooled from a phenyl-Toyopearl column was separated on a 5 to 20% gradient polyacrylamide gel and stained with Coomassie brilliant blue. Lane 1, sheath-degrading enzyme; lane 2, molecular mass standards (kDa).
Factors affecting the activity of the sheath polysaccharide-degrading enzyme.
The effects of metal ions on the polysaccharide-degrading activity are summarized in Table 2. K+, Na+, Ni2+, and Al3+ had little effect on the activity. Cu2+, Mn2+, and Zn2+ inhibited the enzyme activity by 10 to 94%. In contrast, Mg2+, Ca2+, and Fe3+ enhanced the enzyme activity, causing a 23 to 30% stimulation. The effects of chemical reagents on the enzyme activity were examined, with the results being shown in Table 3. Sodium azide and EDTA had no influence on the activity, suggesting that metal ions were not necessarily required. The serine residue was not supposed to be important to exhibit the enzyme activity because phenylmethylsulfonyl fluoride and diisopropylfluorophosphate showed little effect. Strong inhibition was observed with the addition of dithiothreitol (DTT), revealing the importance of disulfide bonds to maintain the activity. Carboxylation of sulfhydryl groups by monoiodoacetic acid resulted in a 27% decrease in activity. In contrast, carboxyamidation of sulfhydryl groups by the addition of iodoacetamide extremely accelerated the reaction, causing a 140% increase in activity. Sulfhydryl groups may play an important role in sheath depolymerase activity.
TABLE 2.
Effect of metal ions on sheath polysaccharide-degrading activitya
| Metal salt | Relative activity (%) |
|---|---|
| None | 100 |
| KCl | 110 |
| NaCl | 118 |
| MgSO4 | 128 |
| CuSO4 | 6 |
| ZnSO4 | 90 |
| MnCl2 | 29 |
| CaCl2 | 123 |
| NiCl2 | 109 |
| AlCl3 | 109 |
| FeCl3 | 130 |
Sheath polysaccharide-degrading activity was assayed under standard conditions, with 1 mM as the final concentration of metal salt. The activity without the addition of any metal salt was defined as 100%.
TABLE 3.
Effect of inhibitors on sheath polysaccharide-degrading activitya
| Inhibitorb | Relative activity (%) |
|---|---|
| None | 100 |
| MIA | 73 |
| IAA | 240 |
| DTT | 0 |
| PMSF | 107 |
| NaN3 | 108 |
| DFP | 97 |
| EDTA | 108 |
Sheath polysaccharide-degrading activity was assayed under standard conditions, with 1 mM as the final concentration of inhibitors. The activity without the addition of any inhibitors was defined as 100%.
Abbreviations: MIA, monoiodoacetic acid; IAA, iodoacetamide; DTT, dithiothreitol; PMSF, phenylmethylsulfonyl fluoride; NaN3, sodium azide; DFP, diisopropylfluorophosphate.
The optimum pH for the sheath polysaccharide-degrading activity was in the range of 7 to 8. The enzyme was most stable at about pH 7. The optimum temperature for the enzyme activity was 30 to 40°C. The enzyme was stable up to 40°C, but it was immediately inactivated at 70°C. The apparent Km value at 30°C (pH 8.0) for the N-acetylated sheath polysaccharide generated from a Lineweaver-Burk double-reciprocal plot was 43.5 mg/ml. The enzyme depolymerizes both N-acetylated and deacetylated sheath polysaccharides but not starch, dextran, cellulose, polygalactosamine, chitin, or chitosan, showing a strict substrate specificity.
Mode of action of the sheath polysaccharide-degrading enzyme.
Polysaccharide-degrading enzymes can be classified into hydrolytic enzymes and eliminative enzymes (19). The eliminase activity which generates unsaturated oligosaccharides by cleaving glycosidic linkages through an elimination mechanism is known to be detected by measurement at 235 nm or by interaction with thiobarbituric acid after periodate oxidation (19). During the degradation of sheath polysaccharide by the enzyme produced by strain TB, the increase in absorbance at 235 nm and thiobarbituric acid reaction were observed (Fig. 7). These increases were observed whether the N-acetylated or deacetylated sheath polysaccharide was supplied as an enzyme substrate. From these results, the sheath polysaccharide-degrading enzyme of strain TB is suggested to be a kind of eliminase but not a hydrolytic enzyme.
FIG. 7.

Generation of unsaturated sugar residues during enzymatic depolymerization of the sheath polysaccharide. N-acetylated sheath polysaccharide (1 mg) was dissolved in 0.5 ml of water, and 0.5 ml of the purified degrading enzyme (originated from 12.5 ml of culture) in 10 mM Tris-HCl buffer (pH 8.0) was added. To investigate the generation of unsaturated sugars, the increase in the absorbance at 235 nm (○) of the reaction mixture was monitored with respect to incubation time. A thiobarbituric acid reaction was also performed, and the generation of unsaturated sugars was detected by the absorbance at 550 nm (●).
The elution profile of the enzymatic product on gel filtration chromatography is shown in Fig. 8. Buffer components were eluted at 110 to 125 min. The negative detector output at 127 to 138 min was probably due to the elution of water in the reaction mixture. A peak at 89 min was found to be the enzymatic product, because the fraction was positive to thiobarbituric acid reaction exhibiting maximum absorbance at 550 nm. The molecular mass of the product was assumed to be a pentamer or hexamer by comparing the elution time with those of maltooligosaccharides.
FIG. 8.

Gel filtration of sheath polysaccharide digested with purified enzyme. Deacetylated sheath polysaccharide was digested by purified enzyme and applied onto a column of Toyopearl HW-40S. Chromatographic conditions are described in Materials and Methods.
The monosaccharide composition of the product recovered from the gel filtration column was investigated by gas liquid chromatography. Although the sheath polysaccharide is formed from glucose and galactosamine (or N-acetylgalactosamine) in a molar ratio of 1:4 as previously reported (20), the molecular ratio of glucose and (N-acetylated) galactosamine in the purified product was calculated to be in the range of 1:2 to 1:3, depending on the analysis.
DISCUSSION
A sheath-degrading bacterium strain, TB, can grow on a sheath as the sole carbon and nitrogen source. However, it requires some nitrogen sources when cultured on the polysaccharide prepared from the sheath, suggesting that galactosamine residues cannot be utilized as a nitrogen source. Therefore, the protein moiety of the sheath was proven to be assimilated as a nitrogen source. Unexpectedly, no protease activity was detected in the culture filtrate. Sheath polysaccharide, which is composed of glucose and galactosamine (or N-acetylgalactosamine) (20), is assumed to be digested not into monosaccharides but into oligosaccharide(s) because strain TB cannot utilize either glucose or galactosamine (N-acetylgalactosamine) as carbon sources. Strain TB was revealed to extracellularly digest the sheath polysaccharide into oligosaccharide(s) but not into monosaccharides. Probably, the sheath polysaccharide is constructed by a repeating unit of the pentasaccharide consisting of one glucose and four galactosamine residues, considering that the overall composition of the polysaccharide is glucose and galactosamine in a molar ratio of 1:4 (20). The restricted digestion of the sheath into specific oligomers should be profitable for strain TB to compete with other first-growing bacteria of wide substrate utilization if the specific peptide and oligosaccharide are not easily assimilated by these competitors.
Because the enzymatic product is positive to thiobarbituric acid reaction, it can be assumed that the product is incorporated with a deoxilated sugar residue or an unsaturated sugar residue. An increase in the absorbance at 235 nm during the enzymatic reaction suggests that the generation of an unsaturated sugar residue is more probable. Based on such an assumption, the sheath polysaccharide-degrading enzyme is considered to be not a hydrolytic enzyme but an eliminase-type enzyme. A number of polysaccharide eliminases (lyases) (EC 4.2.2) which depolymerize acidic polysaccharides, such as alginate, hyaluronate, chondroitin, heparin, xanthan, and emulsan, are known (19). Lyases are defined as a class of eliminative enzymes that act on a wide variety of acidic polysaccharides degraded into oligomers incorporated with unsaturated derivatives of uronic acids at the nonreducing terminal (13). According to this description, the TB enzyme reported in this paper is not a lyase but a new type of eliminase specific for basic polysaccharides. Considering the result in which the molar ratio of N-acetylgalactosamine decreased due to enzymatic digestion of the N-acetylated sheath polysaccharide, a double bond is likely to be introduced into the galactosamine residue.
Most bacterial lyases require metal ions such as Mg2+, Ca2+, Na+, and K+ (2, 3, 4, 7, 8, 9, 12, 15, 16, 21) for their activation. In contrast, sheath polysaccharide-degrading activity was not metal ion dependent, because activity was detected in the presence of EDTA. An enhancing effect was observed from the addition of Mg2+, Ca2+, or Fe3+, although Cu2+, Zn2+, and Mn2+ negatively affected the activity. In addition to these three metal ions, DTT strongly inhibited sheath polysaccharide-degrading activity, suggesting the importance of disulfide bonds to exhibiting activity.
According to Ash et al. (1), the cells of Paenibacillus strains have gram-positive structure but usually stain negatively in the laboratory Gram stain. Phenotypic characteristics such as cell shape, gram-negative, motility, formation of swollen sporangia, and growth under anaerobic conditions suggest that the sheath-degrading bacteria isolated in this study are members of the genus Paenibacillus (1). Another typical phenotypic characteristic of the genus Paenibacillus is the ability to degrade macromolecules, including polysaccharides and proteins, by extracellular enzymes (1). Strains belonging to Paenibacillus polymyxa (formerly called Bacillus polymyxa) capable of secreting lyases that act on pectin were isolated, though the lyases secreted by these bacteria were not purified and characterized (11, 14). Every known Paenibacillus strain utilizes glucose accompanied by gas production. However, the isolate (strain TB) hardly utilized glucose. Though some characteristics of the sheath-degrading bacteria we isolated are not identical to those of other known Paenibacillus strains, they might be new members of the genus Paenibacillus. The phylogenetic analysis supported this possibility.
REFERENCES
- 1.Ash C, Priest F G, Collins M D. Molecular identification of rRNA group 3 bacilli (Ash, Farrow, Wallbanks and Collins) using a PCR probe test. Proposal for the creation of a new genus Paenibacillus. Antonie Leeuwenhoek. 1993;64:253–260. doi: 10.1007/BF00873085. [DOI] [PubMed] [Google Scholar]
- 2.Bekri M A, Desair J, Keijers V, Proost P, Searle-van Leeuwen M, Vanderrleyden J, Broek A V. Azospirillum irakense produces a novel type of pectate lyase. J Bacteriol. 1999;181:2440–2447. doi: 10.1128/jb.181.8.2440-2447.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyen A, Ghosh M, May T B, Shinaberger D, Koegh R, Chakrabarty A M. Sequence of the algL gene of Pseudomonas aeruginosa and purification of its alginate lyase product. Gene. 1993;131:1–8. doi: 10.1016/0378-1119(93)90662-m. [DOI] [PubMed] [Google Scholar]
- 4.Chatterjee A K, Buchanan G E, Behrens M K, Starr K P. Synthesis of excretion of polygalacturonic acid trans-eliminase in Erwinia, Yersinia, and Klebsiella species. Can J Microbiol. 1978;25:94–102. doi: 10.1139/m79-014. [DOI] [PubMed] [Google Scholar]
- 5.Emerson D, Ghiorse W C. Ultrastructure and chemical composition of the sheath of Leptothrix discophora SP-6. J Bacteriol. 1993;175:7808–7818. doi: 10.1128/jb.175.24.7808-7818.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emerson D, Ghiorse W C. Role of disulfide bonds in maintaining the structural integrity of the sheath of Leptothrix discophora SP-6. J Bacteriol. 1993;175:7819–7827. doi: 10.1128/jb.175.24.7819-7827.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gacesa P. Enzymic degradation of alginates. Int J Biochem. 1992;24:545–552. doi: 10.1016/0020-711x(92)90325-u. [DOI] [PubMed] [Google Scholar]
- 8.Hansen J B, Doublet R S, Ram J. Alginase production by Bacillus circulans. Appl Environ Microbiol. 1984;47:704–709. doi: 10.1128/aem.47.4.704-709.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haraguchi K, Kodama T. Purification and properties of poly(β-d-mannuronate) lyase from Azotobacter chroococcum. Appl Microbiol Biotechnol. 1996;44:576–581. [Google Scholar]
- 10.Iwasaki M, Ota M, Otani Y. Microdetermination of serum amylase activity utilizing the 3,6-dinitrophthalic acid method of determining reducing sugar. Yakugaku Zasshi. 1974;94:138–144. doi: 10.1248/yakushi1947.94.1_138. . (In Japanese.) [DOI] [PubMed] [Google Scholar]
- 11.Karbassi A, Vaughn R H. Purification and properties of polygalacturonic acid trans-eliminase from Bacillus stearothermophilus. Can J Microbiol. 1980;26:377–384. doi: 10.1139/m80-061. [DOI] [PubMed] [Google Scholar]
- 12.Lange B, Wingender J, Winkler U K. Isolation and characterization of an alginate lyase from Klebsiella aerogenes. Arch Microbiol. 1989;152:302–308. doi: 10.1007/BF00409667. [DOI] [PubMed] [Google Scholar]
- 13.Linhardt R J, Galliher P M, Cooney C L. Polysaccharide lyases. Appl Biochem Biotechnol. 1986;12:135–176. doi: 10.1007/BF02798420. [DOI] [PubMed] [Google Scholar]
- 14.Obi S K C, Umerzurike G M. Pectic enzyme activities of bacteria associated with rotted onions (Allium cepa) Appl Environ Microbiol. 1981;42:585–589. doi: 10.1128/aem.42.4.585-589.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peciña A, Pascual A, Paneque A. Cloning and expression of the algL gene, encoding the Azotobacter chroococcum alginate lyase: purification and characterization of the enzyme. J Bacteriol. 1999;181:1409–1414. doi: 10.1128/jb.181.5.1409-1414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Romeo A, Perston J F. Purification and structural properties of an extracellular (1→4)-β-D-mannuronan specific alginate lyase from a marine bacterium. Biochemistry. 1986;25:8385–8391. [Google Scholar]
- 17.Siering P L, Ghiorse W C. Phylogeny of the Sphaerotilus-Leptothrix group inferred from morphological comparisons, genomic fingerprinting, and 16S ribosomal DNA sequence analysis. Int J Syst Bacteriol. 1996;46:173–182. doi: 10.1099/00207713-46-1-173. [DOI] [PubMed] [Google Scholar]
- 18.Siering P L, Ghiorse W C. Development and application of 16S rRNA-targeted probes for detection of iron- and manganese-oxidizing sheathed bacteria in environmental samples. Appl Environ Microbiol. 1997;63:644–651. doi: 10.1128/aem.63.2.644-651.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sutherland I W. Lyases (eliminases) In: Baddiley Sir J, Carey N H, Higgins I J, Potter W G., editors. Biotechnology of microbial exopolysaccharides. Cambridge, United Kingdom: Cambridge University Press; 1990. pp. 47–53. [Google Scholar]
- 20.Takeda M, Nakano F, Nagase T, Iohara K, Koizumi J. Isolation and chemical composition of the sheath of Sphaerotilus natans. Biosci Biotechnol Biochem. 1998;62:1138–1143. doi: 10.1271/bbb.62.1138. [DOI] [PubMed] [Google Scholar]
- 21.Tardy F, Nasser W, Robert-Baudouy J, Hugouvieux-Cotte-Pattat N. Comparative analysis of the five major Erwinia chrysanthemi pectate lyases: enzyme characteristics and potential inhibitors. J Bacteriol. 1997;179:2503–2511. doi: 10.1128/jb.179.8.2503-2511.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van de Peer Y, De Wachter R. TREECON: a software package for the construction and drawing of evolutionary trees. Comput Appl Biosci. 1993;9:177–182. doi: 10.1093/bioinformatics/9.2.177. [DOI] [PubMed] [Google Scholar]
- 23.van Veen W L, Mulder E G, Deinema M H. The Sphaerotilus-Leptothrix groups of bacteria. Microbiol Rev. 1978;42:329–356. doi: 10.1128/mr.42.2.329-356.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weissbach A, Hurwitz J. The formation of 2-keto-3-deoxyheptonic acid in extracts of Escherichia coli B. I and II. J Biol Chem. 1958;234:705–712. [PubMed] [Google Scholar]