

Abstract
BACKGROUND:
High-impact pathogenic variants in more than a thousand genes are involved in Mendelian forms of neurodevelopmental disorders (NDD).
METHODS:
This study describes the molecular and clinical characterization of 28 probands with NDD harboring heterozygous AGO1 coding variants, occurring de novo for all those whose transmission could have been verified (26/28).
RESULTS:
A total of 15 unique variants leading to amino acid changes or deletions were identified: 12 missense variants, two in-frame deletions of one codon, and one canonical splice variant leading to a deletion of two amino acid residues. Recurrently identified variants were present in several unrelated individuals: p.(Phe180del), p.(Leu190Pro), p.(Leu190Arg), p.(Gly199Ser), p.(Val254Ile) and p.(Glu376del). AGO1 encodes the Argonaute 1 protein, which functions in gene-silencing pathways mediated by small non-coding RNAs. Three-dimensional protein structure predictions suggest that these variants might alter the flexibility of the AGO1 linkers domains, which likely would impair its function in mRNA processing. Affected individuals present with intellectual disability of varying severity, as well as speech and motor delay, autistic behavior and additional behavioral manifestations.
CONCLUSION:
Our study establishes that de novo coding variants in AGO1 are involved in a novel monogenic form of NDD, highly similar to the recently reported AGO2-related NDD.
Keywords: AGO1, microRNA, intellectual disability, autism, argonaute
INTRODUCTION
Neurodevelopmental disorders (NDD), such as intellectual disability (ID) and autism spectrum disorder (ASD), have important genetic contributions characterized by extreme heterogeneity. More than a thousand genes have now been implicated in monogenic forms of NDD[1] and many more have been identified as candidates for ID/ASD, including genes showing enrichment of rare de novo variants in large-scale sequencing studies of affected individuals[2]. In individuals affected by ID or ASD, few de novo missense variants have been reported in AGO1 (or EIF2C1), a gene encoding a protein from the argonaute family, which participates in RNA silencing pathways, suggesting that AGO1 could be a promising candidate gene for NDD[3–6].
The argonaute protein family, identified originally in plants, includes AGO and PIWI proteins, and is involved in gene-silencing pathways guided by small non-coding RNAs (sncRNA, including short interfering RNAs, microRNAs, Piwi-interacting RNAs)[7]. PIWI proteins are involved in transposon repression in germinal cells whereas AGO proteins are involved in translation repression and degradation of targeted mRNA[8]. In addition to their role in mRNA post-transcriptional regulation in the cytoplasm, AGO proteins have also been shown to have nuclear activities, playing a role in the regulation of transcription, chromatin remodeling, alternative splicing regulation, and even in DNA double-strand break repair[8–12].
Large deletions at the 1p34.3 loci including AGO1 together with AGO3 (and sometimes AGO4) among other genes were previously reported in five children with psychomotor developmental delay as well as additional non-specific features (feeding difficulty, language impairment, facial dysmorphy)[13,14]. In addition, AGO2 was very recently implicated in NDD[15]: 21 individuals with heterozygous de novo variants in AGO2 were reported, including 11 missense variants, one in-frame deletion and one 235 kb deletion involving the first three exons of AGO2. Functional studies revealed that those variants hampered correct sncRNA-mediated silencing, having a loss-of-function effect.
We here report 28 individuals from 26 families affected by NDD carrying heterozygous amino acid substitutions or deletions predicted-damaging in AGO1, thus, causally-linking coding variants in this gene to ID. These variants distributed along different domains of the protein, are predicted to affect the flexibility of AGO1 structure, which might impair its role in sncRNA-induced gene regulation.
MATERIALS AND METHODS
Identification of AGO1 variants
AGO1 variants were identified by laboratories from Europe, Canada, and USA using various high throughput sequencing strategies (targeted gene panels, exome sequencing or whole genome sequencing) and retrieved through a worldwide collaborative network connected notably via GeneMatcher[16] (see Table 1 and Supplementary text). Variants are annotated according to the NM_012199.4 transcript, were confirmed by independent Sanger sequencing and concordance of the trio was checked. Predictions of missense variant effects were performed using in silico tools such as Combined Annotation Dependent Depletion (CADD)[17], SIFT[18] and Polyphen-2[19]. Effects on splicing were analyzed using SpliceAI[20], and via Alamut® Visual (Interactive Biosoftware) and confirmed by cDNA analysis from blood mRNA.
Table 1:
List of missense changes identified in AGO1 in individuals with intellectual disability
| Family | Add. families prev. reported | Genomic position (GRCh37) - chr 1 | cDNA | Protein | SIFT | PPH2 (HumVar) | CADD | Domain | ΔΔGfold† | Context‡ | Dynamics Changeα |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1, 2, 3, 4, 5 | - | g.36359301_36359303del | c.539_541del | p.(Phe180del) | NA | NA | NA | L1 linker | NA | Core | NA |
| 6 | - | g.36359328C>T | c.566C>T | p.(Pro189Leu) | Deleterious (score: 0.02) | Possibly Damaging (score: 0.605) | 28.3 | L1 linker | 1.0±0.4 | PAZ | PC1 −0.4, PC2 −1.6 |
| 7, 8 | - | g.36359331T>G | c.569T>G | p.(Leu190Arg) | Deleterious (score: 0) | Probably Damaging (score: 1.000) | 29.2 | L1 linker | 9.5±0.6 | PAZ | NA |
| 9 | Rauch et al.[3] | g.36359331T>C | c.569T>C | p.(Leu190Pro) | Deleterious (score: 0) | Probably Damaging (score: 1.000) | 29.3 | L1 linker | 5.6±0.4 | PAZ | PC1 −0.3, PC2 −1.1 |
| - | Martinez et al.[21] | g.36359345G>A | c.583G>A | p.(Glu195Lys) | Deleterious (score: 0) | Probably Damaging (score: 0.999) | 30 | L1 linker | 2.5±0.3 | PAZ | PC1 0.1, PC2 −0.7 |
| 10, 11, 12 (Hamdan et al.)5, 13, 14 | Sakaguchi et al.[6] | g.36359357G>A | c.595G>A | p.(Gly199Ser) | Deleterious (score: 0) | Probably Damaging (score: 1.000) | 29.6 | L1 linker | 17.9±0.7 | Near Core | PC1 0.6, PC2 −0.9 |
| - | DDD Study[22] | g.36359409A>T | c.647A>T | p.(Asp216Val) | Deleterious (score: 0) | Probably Damaging (score: 0.996 ) | 32 | L1 linker | 2.5±0.7 | Bridge R712 | PC1 −0.4, PC2 −0.6 |
| 15 | - | g.36359636A>G | c.650–2A>G (r.650_655del) | p.Val217_Ser218del | NA | NA | NA | L1 linker | NA | Linker and RNA H-bond | NA |
| 16 | - | g.36359746G>A | c.758G>A | p.(Arg253His) | Deleterious (score: 0.03) | Benign (score: 0.173) | 25.6 | PAZ | 0.6±0.6 | Helix Cap | PC1 0.2, PC2 −1.5 |
| 17, 18 | - | g.36359748G>A | c.760G>A | p.(Val254Ile) | Deleterious (score: 0) | Benign (score: 0.092) | 22.9 | PAZ | −1.0±0.0 | Helix Center | PC1 −0.7, PC2 −1.5 |
| 19 | - | g.36360821C>T | c.971C>T | p.(Pro324Leu) | Deleterious (score: 0) | Probably Damaging (score: 1.000) | 31 | PAZ | 2.1±0.1 | PAZ | NA |
| - | Sanders et al.[4] | g.36367118C>T | c.1064C>T | p.(Thr355Ile) | Deleterious (score: 0) | Probably Damaging (score: 0.973) | 24.8 | PAZ | 0.4±0.6 | Helix Cap | NA |
| 20 | - | g.36367127A>G | c.1073A>G | p.(Gln358Arg) | Deleterious (score: 0) | Probably Damaging (score: 0.996) | 26.8 | PAZ | 0.3±0.2 | Linker | NA |
| 21, 22 | - | g.36367180_36367182del | c.1126_1128del | p.(Glu376del) | NA | NA | NA | L2 linker | NA | Linker | NA |
| 23 | - | g.36367661A>T | c.1253A>T | p.(Tyr418Phe) | Deleterious (score: 0) | Benign (score: 0.053) | 24.4 | L2 linker | 0.3±0.1 | Linker | PC1 0.7, PC2 −0.9 |
| 24 | - | g.36384011A>T | c.2252A>T | p.(His751Leu) | Deleterious (score: 0) | Probably Damaging (score: 0.982) | 29.1 | PIWI | −1.1±0.8 | RNA H-bond | PC1 0.6, PC2 −1.2 |
| 25 | - | g.36384732C>T | c.2342C>T | p.(Thr781Met) | Deleterious (score: 0) | Probably Damaging (score: 1.000) | 28 | PIWI | 1.8±0.9 | PIWI core | NA |
| 26 | - | g.36384779A>T | c.2389A>T | p.(Ile797Phe) | Deleterious (score: 0) | Probably Damaging (score: 0.969) | 28.3 | PIWI | 3.1±0.5 | PIWI near RNA | PC1 1, PC2 −0.8 |
Version of the programs used: SIFT version v6.2.0; Polyphen2 (PPH2) HumVar version v2.2.2r398; CADD version GRCh37-v1.4
Change in folding free energy upon each mutation, using a 3D structure-based algorithm. See Methods for details.
3D molecular context of the amino acid position.
See Results for details.
MD simulations were summarized using PC analysis; we summarized the change in PC1 and PC2 using the number of standard deviations away from the average WT conformation that each mutation sampled on average.
Collection of clinical information
Physicians with experience in clinical dysmorphology examined a posteriori all 28 individuals of this cohort, including the individual carrying the previously described variant in Hamdan et al[5] (F12). Photographs were collected from research participants after informed consent had been obtained. Clinical data for additional individuals with de novo variants previously reported[3–6,21,22] were retrieved from published data. Face2Gene Research application (FDNA Inc., Boston, MA) using DeepGestalt technology (algorithm 19.1.9)[23] was used to compare 14 frontal facial photographs of individuals with AGO1 variants compared to 14 control individuals matched for age, sex, and ethnicity provided by Face2Gene. To estimate the power of DeepGestalt in distinguishing affected individuals from controls, a cross validation scheme was used, including a series of binary comparisons for which the data were split randomly multiple times into training sets and test sets (10 times). The results of the comparisons are reported using the receiver operating characteristic (ROC) curve and area under the curve (AUC).
Building 3D Model for AGO1
The protein structure of human AGO1 has been experimentally determined (PDB 4kxt[24]). We used the available experimental structure with homology-based methods[25] to fill in short sections of the protein that were not resolved experimentally. Structures were solved with simple (poly A) guide RNAs that were also partially resolved. We added data from the rhotabacter sphaeroides Argonaute experimental structure (PDB 5awh[26]) and used Discovery Studio (BIOVIA. Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 4.5, San Diego: Dassault Systèmes; 2015) to complete coordinates of a 21-base (A19U2) guide RNA. We used BioR[27] to compile protein annotations from multiple sources including dbNSPF[28]. We used FoldX[29] v4.0 for mutagenesis and structure-based calculations of ΔΔGfold. We visualized protein structures using PyMOL (Molecular Graphics System, Version 1.9 Schrödinger, LLC).
Molecular Dynamics Simulations
Generalized Born implicit solvent molecular dynamics (isMD) simulations were carried out using NAMD[30] and the CHARMM36 force field, using a similar procedure to our previous work. Briefly, we utilized an interaction cutoff of 12Å with strength tapering (switching) beginning at 10Å, a simulation time step of 1fs, conformations recorded every 2ps. Each initial conformation was used to generate 6 replicates, and each was energy minimized for 5,000 steps, followed by heating to 300K over 300ps via a Langevin thermostat. A further 13ns of simulation trajectory was generated and the final 10ns (60ns per variant) were analyzed.
Analysis of Protein Structures and Simulations
All trajectories were aligned to the initial wild type conformation using Cα atoms. Then, Root Mean-Square Deviation (RMSD) values were reported for each. We calculated per-domain RMSDs by first re-aligning all trajectories using each domain individually, then measuring the RMSD within that domain, alone, providing a measure of distortion within each domain and across MD simulation time. Residue-level Root Mean-Square Fluctuation (RMSF) values were calculated for the whole protein across trajectories. Variances were computed using median absolute difference (MAD) and Z-scores computed by MAD-scaling the observation-median differences. We used Cα cartesian space Principal Component (PC) analyses across simulations to define the dynamics of AGO1. Variant data was projected onto the PCs to compare how essential motion is altered by each genomic variant. Individual PCs were visualized using porcupine plots where a cone represents the direction and relative magnitude of each residue’s motion. We calculated free-energy landscapes (FELs) of MD trajectories using the approach of Karamzadeh, et al.[31]. We show topologic lines from the FEL where each line indicates a specific conformational sampling probability and matched by a corresponding color gradient where each color also indicates a specific conformational sampling probability. We measure alignment-free conformational changes using internal distances[32]. We calculated the ensemble-averaged median pairwise distance between residues on the last half of each trajectory to summarize internal distance changes. To simplify visualization of these median changes, we averaged information across groups of three consecutive amino acids. To conservatively estimate statistical significance, we used a permutation procedure where data were sub-sampled to 100 points and compared using a t-test. This was repeated 1000 times and the median p-value across repeats reported. The analysis was carried out using custom scripts, leveraging VMD and the Bio3D R package.
RESULTS
Identification of de novo variants in AGO1 in individuals with intellectual disability
A total of 15 different heterozygous variants in AGO1, including 12 amino acid substitutions, two single amino acid deletions, and one splice variant leading to a two-residue deletion were identified in 26 unrelated families (Table 1, Figure 1, Figure 2A). For all individuals for whom parental DNA was available (all except F23 and F25), variants were found to occur de novo. No other genetic diagnoses potentially explaining the clinical manifestations were identified (Supplementary text). Two variants we identified had already been reported in individuals with NDD: p.(Leu190Pro) (Rauch et al.[3]) and p.(Gly199Ser) (Hamdan et al.[5] and Sakaguchi et al.[6]). Thirteen variants were novel to this study. The proband from family F15 has a de novo c.650–2A>G variant predicted to impair the use of the canonical acceptor splice site (MaxEnt: −100.0%, NnSplice: −100.0%) and to activate a cryptic acceptor site (Splice AI prediction). Activation of the cryptic acceptor site was subsequently confirmed by mRNA Sanger sequencing (Supplementary Figure S1), leading to the in-frame deletion of two amino acids (r.650_655del, p.Val217_Ser218). Six variants were recurrent: p.(Gly199Ser) (six individuals), p.(Phe180del) (five individuals), p.(Leu190Arg) (two individuals), p.(Val254Ile) (two individuals) and p.(Glu376del) (two individuals). Including the three previously reported (p.(Glu195Lys)[21], p.(Asp216Val)[22] and p.(Thr355Ile)[4]), a total of 18 variants were identified in individuals with NDD (Table 1, Figure 2A). All variants were absent from the general population (gnomAD) and all but one affect residues that are highly conserved until C. elegans (Supplementary Figure S1). In silico predictions (SIFT and Polyphen2) predict all these missense variants to be deleterious except p.(Arg253His) and p.(Val254Ile). AGO1 is constrained in the population for missense variation as observed in gnomADv2.1.1 (Z-score = 5.68, observed/expected = 0.31). The CADD scores of missense variants identified in individuals with NDD (mean = 27.8) are significantly higher than those of missense variants reported in gnomAD (mean: 24.3, t-test p-value = 0.0001).
Figure 1: Facial characteristics of individuals with AGO1 variants.
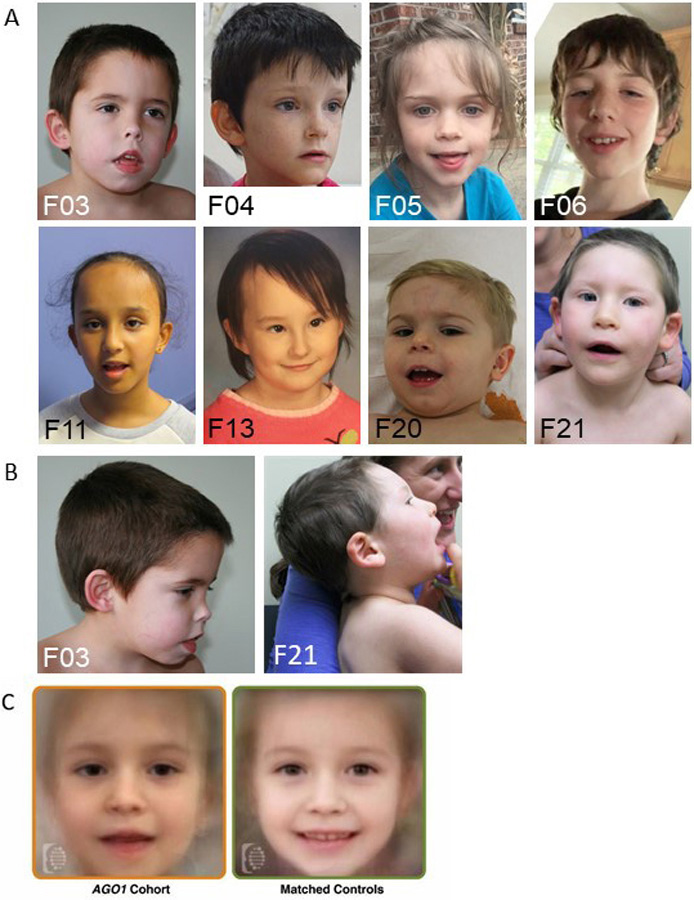
(A) Front faces, (B) profile faces from families who consented for photographs publication (C) Face2Gene Facial analysis using Face2Gene Research application (FDNA Inc. Boston, MA) of unrelated individuals with AGO1-associated disorder (n = 14) compared to controls matched (n= 14) for sex, age, and ethnicity.
Figure 2: Schematic representation of AGO1 protein with its functional domains showing the locations of the coding variants identified in individuals with ID.

(A) AGO1 protein (NP_036331.1) with its functional domains: N-terminal domain (34–164), Linker 1 (L1) domain (173–225), PAZ domain (226–368), Liker 2 (372–418), Mid domain (427–508), and PIWI domain (515–816). PAZ and PIWI domains have motifs of interaction with RNA guide and PIWI domain has motif of RNA blocked access to the active site. The arrows show de novo variants identified in individuals from this cohort or previously reported (P: previously reported in literature); T: found in monozygotic twins, and in underlined those that are recurrent. (B) Protein structure of AGO1 colored by protein domain and with the sites of variants indicated by spheres. The linker domains, designated L1 and L2, are separated in sequence by the PAZ domain, but intertwine in 3D, forming common interfaces between the N-terminal, PAZ, and PIWI domains (C) Many of the variants of interest are within the first linker domain. This domain forms a narrow beta sheet with three strands. Variants occur in the middle of these strands at the closest point between domain L1 and the guide RNA backbone, and towards the end of L1 and near the PAZ domain interface (D) D216 is within L1 and makes specific contacts with R712 in the PIWI domain. R712 also interacts directly with the guide RNA.
Clinical manifestations observed in individuals with AGO1 variants
We report clinical data for a total of 33 individuals (17 males, 16 females) from 31 unrelated families. We collected clinical data for the 28 affected individuals in this study including updated information for one previously reported[5] and retrieved clinical data from previously published individuals with AGO1 variants[3–6,21,22]. Head circumference, height, and weight measurements were in the normal ranges at birth and postnatally, except for three individuals (including two twins) who showed neonatal and postnatal microcephaly (<−2 SD). All affected individuals showed borderline to severe ID, (Table 2, Supplementary text) and all showed a severe language delay (30/30; 100 %). Most individuals were able to construct a sentence but with limited spontaneous communication. Three did not acquire language and one presented with language regression. Motor developmental delay was also observed in most affected individuals (28/30; 93 %, mean age at onset of independent walking ~25 months) and seven had persistent hypotonia (13/18; 72 %). Almost half of the individuals had documented or history of seizures (13/28; 46 %), fever-induced in two families (F7, F18). Twin girls (F9) have different seizure types: one has photosensitive, drug-resistant seizures and the other has controlled seizures. Interestingly, the three individuals with the most severe epileptic phenotype (as indicated by the history of status epilepticus) share the p.(Gly199Ser) variant (the fourth individual with this variant has had no seizures by 5 years of age). Several behavioral features were observed in this cohort: most presented with autistic features (24/30; 80 %), and 11 showed self-harm behavior and/or hetero-aggressiveness (11/14, 78.5 %). Attention deficit/hyperactivity (15/22, 68 %) and anxiety (7/8, 87.5 %) were also reported. Additionally, a large majority had various sleeping disturbances (17/22, 77 %) including difficulties falling asleep, early awakening, or hypersomnia. Analysis using DeepGestalt technology by Face2Gene did not show significant differences in facial features from matched controls (mean AUC = 0.55 and AUC STD = 0.14, p-value = 0.392) (Figure 1C). However, individuals with AGO1 variants share some subtle common traits such as a thin upper lip, a tall forehead, elongated almond eyes or a small nose (Figure 1A–B). Individuals with feeding difficulties (10/23, 43 %) showed low postnatal weight: six individuals needed a gastrostomy, three individuals had gastro-esophageal reflux, and the twins of family 24 received tube feeding during the first three weeks of life in the context of prematurity. Two individuals had recent swallowing problems (F3 and F25). Finally, hypothyroidism was reported in two of five individuals with the p.(Phe180del) variant. Variable brain MRI anomalies were noticed in 11/24 individuals (46 %), including corpus callosum agenesis or hypoplasia, cerebral atrophy, cortical dysplasia or colpocephaly but also non-specific anomalies (increase in extra-axial fluid, T2 and FLAIR signal anomalies, arachnoid cyst).
Table 2:
Clinical manifestations observed in individuals with variants in AGO1 (compared to individuals with variants in AGO2 reported by Lessel et al. 2020)
| AGO1 | AGO2 | ||
|---|---|---|---|
| total (n=33) | % | % | |
| ID/DD | 31/31 | 100% | 100% |
| Hypotonia | 13/18 | 72% | 57% |
| Seizures | 13/28 | 46% | 44% |
| Motor delay | 28/30 | 93% | 100% |
| Speech impairment | 30/30 | 100% | 100% |
| Autistic behaviour | 24/30 | 80% | 56% |
| Attention deficit /Hyperactivity | 15/22 | 68% | 53% |
| Anxiety | 7/8 | 87.5 % | NA |
| Aggressiveness | 11/24 | 78.5 % | 24% |
| Sleeping disturbance | 17/22 | 77% | NA |
| Cerebral MRI abnormalities | 11/24 | 46% | 56% |
| Neonatal feeding difficulties | 10/23 | 44% | 63% |
| Gastroesophagial reflux | 3/23 | 13% | 37% |
| Skeletal anomalies | 5/21 | 23% | 47% |
| Heart anomalies | NA | NA | 33% |
| Strabism | 4/26 | 15% | 33% |
| Visual impairment | NA | NA | 29% |
| Abnormal respiration | NA | NA | 26% |
| Dental anomalies | NA | NA | 47% |
ID: intellectual disability; DD: developmental delay; M: male; F: female; NA: not reported
Variants spatially cluster in AGO1
The AGO1 protein (NP_036331.1) functional domains include the MID (AA 427–508), PAZ (AA 226–368), and PIWI (AA 515–816) domains and two linker domains L1 and L2 (Figure 2A). The PAZ domain contains the RNA binding module that recognizes the 3′ end of both siRNA and miRNA. The PIWI domain contains the cleavage site that is inactive in AGO1 and is involved in protein/protein interactions, notably with DICER and GW182.
Most of the variants affect amino acids clustered in 3D along the sides of L1 and PAZ domains facing one another (Table 1, Figure 2B). For instance, Arg253 is located within an alpha-helix in the PAZ domain and makes specific contact with the last residue in the helix, Asp250, and the backbone of Arg284. The variant p.(Arg253His) may modify these interactions and affect organization and stability of the PAZ domain. The nearby variant p.(Val254Ile) is conservative, but valine has the lowest helical propensity of the small hydrophobic amino acids[33]. Thus, this variant could over-stabilize the helix and limit motion of the PAZ domain. Three critical amino acid positions are within the L1 domain and in proximity to the PAZ domain: Pro189, Leu190, and Glu195 (Figure 2C). Pro189 is at the base of a long loop and the backbone geometry of proline may be necessary for limiting the loop’s mobility; p.(Pro189Leu) may thus alter loop dynamics. Leu190 packs within a hydrophobic interface made up of amino acids from L1 and L2; p.(Leu190Pro) or p.(Leu190Arg) may distort the interface between the linker domains. Glu195 is close in space to Trp197 and Lys224, and p.(Glu195Lys) may repel these nearby residues and destabilize the L1 domain. Farther down the L1 domain, the most recurrent variant, p.(Gly199Ser), introduces a serine which could clash with the Ser218 side chain and introduce an unfavorable polar contact, likely destabilizing the L1 domain. Phe180 makes contacts across all three strands of the L1 beta sheet and its deletion would significantly alter regional hydrophobic packing. Asp216 makes a salt bridge with Arg712 in the PIWI domain (Figure 2D). Arg712 also makes specific interactions with the guide RNA. Thus, p.(Asp216Val) is likely to destabilize the interface between L1 and PIWI thereby indirectly altering guide RNA interactions. Finally, p.(His751Leu) is one of the few variants within the PIWI domain. His751 makes hydrogen bonds with the guide RNA and p.(His751Leu), therefore, likely alters the stability of the interaction between AGO1 and the guide RNA.
Variants are predicted to affect AGO1 dynamic conformational change
We used molecular dynamic (MD) simulations to assess variant-associated global and local changes to AGO1. Global changes to AGO1 conformation across our MD simulations were assessed using RMSD, RMSF and PC analysis (Figure 3). We found the variants have the largest change in mobility for the N-term and PAZ domains, and more modest mobility changes to the MID domain (Figure 3A). Because RMSF depends on how the trajectories are aligned, and we aligned based on the PIWI domain, we observed relatively small overall fluctuations of the PIWI domain. Therefore, patterns in RMSF indicated that the largest features in our simulations were domain-domain motions, and that these were modified by genomic variants. Next, to assess domain-domain distance, we measured linear distances from specific residues in the PIWI, MID, and linker regions. Certain PAZ and linker variants such as p.(Val254Ile), p.(Gly199Ser), p.(Arg253His), and p.(Pro189Leu) were associated with significantly shorter distances between the PIWI and linker (Figure 3B). The same variants p.(Val254Ile) and p.(Gly199Ser) and additional ones p.(Tyr418Phe) and p.(His751Leu) were associated with significantly shorter distances between the MID and linker (Figure 3C). Because the relative orientation of these domains is likely important for AGO1 function, we also assessed the angles between them. Variants in multiple domains altered the orientation of PAZ with respect to linker (Figure 3D), and the MID-PIWI-PAZ orientation (Figure 3E). Thus, we identified additional and more specific changes in domain-domain orientations associated with genomic variants. Because many conformational changes occur concurrently in MD simulations, we next assessed all dynamics data together using PC analysis (Figure 3F). We visualized the predominant dynamics of AGO1 using a free-energy landscape. We found that the WT protein sampled one side of the landscape, while variants had significant sampling of a different side. PC states are characterized by the relative orientations of the PAZ, Nterm, and MID domains (see Supplemental Animation 1 for PC visualizations). The variants had two effects on these predominant dynamics. First, they shift the average conformation to one favoring a wider angle of the PAZ-PIWI-MID domains. Second, the transition between conformations was blurred with a significant amount of time spent deviated from WT-like confirmations. This indicates that these variants may be leading to dysregulated dynamics. We investigated internal distance changes as a summary of dysregulated dynamics (Supplementary Figure S3) and demonstrated the variants were associated with lower coordination between domains. Changes to domain coordination could be an additional informative criterion for assessing multi-domain enzymes. To further assess the intra-domain change associated with each variant, we calculated the per-domain RMSD (Supplementary Figure S4) and found variants throughout the structure could be associated with alterations of the same or distant domains. For example, p.(Leu190Pro) in the linker domain and p.(Val254Ile) in the PAZ domain both lead to alterations of linker and MID domain internal organization.
Figure 3: Simulations reveal changes in domain orientation associated with de novo missense AGO1 variants.

We used MD simulations to assess how the native structure of AGO1 would respond to the introduction of a subset of identified genomic variants. (A) Variability of each amino acid was quantified using RMSF after aligning each trajectory to the initial WT conformation of the PIWI domain and averaging across replicates. Domains are colored as in Figure 1 and each variant colored according to the domain it is within. (B-E) We monitored selected distances and angles as a simple way to assess conformational changes between the (B) linker and PIWI domains, (C) linker and MID domains, (D) the orientation of the PAZ domain, and (E) the openness of the RNA binding region. (F) We show the free energy landscape across molecular dynamics (MD) simulations as a color gradient from high-energy to low energy. Above, we show one-dimensional PC samplings as a combined violin and boxplot. The left- and right-hand panels summarize all data from the WT and from our novel variants, respectively. Selected variant’s PC1 sampling is shown above the panel, and all variant’s (that underwent MD) PC2 sampling to the side of the panel. Four regions of low energy are indicated by letters A-D. Representative images of AGO1 structure from the simulations taken from these points of low energy are shown below and labeled by the corresponding letters with the WT shown for comparison. To summarize one aspect of the difference between these four regions, we show the angle between the centers of mass of the MID, PIWI, and PAZ domains.
DISCUSSION
This study clinically and molecularly characterizes a cohort of 28 individuals from 26 unrelated families with heterozygous coding variants in AGO1 and establishes that de novo AGO1 variation is responsible for a form of NDD characterized by psychomotor delay, behavioral features and language impairment. Including three previously reported variants, 18 total variants have been described in AGO1 in 33 individuals with NDD, six being recurrent (Table1). All variants lead to substitutions or deletions of one or two amino acids, and no truncating variants were observed. Interestingly, four of the affected AGO1 residues, Phe180, Leu190, Gly199 and Thr355, were also found altered at equivalent residues in AGO2 (p.Phe182del, p.Leu192Pro, p.Gly201Cys or Val, p.Thr357Met) in ID patients (Supplementary Figure S1)[15].
ID and language delay were reported in all affected while most displayed motor delay, seizures, autistic features, and behavior problems. No recurrent structural brain nor other organ malformation was noticeable and if individuals share some common facial traits, no typical specific gestalt was observed. Therefore, the AGO1-associated NDD does not present as a clinically recognizable entity. Moreover, the individuals present differences in phenotypic severity, which do not appear to correlate with the nature or location of the variant. The severity of ID, as well as growth parameters, was for instance highly variable among the three individuals with the p.(Phe180del) variant. We could therefore suggest that additional genetic or environmental factors might play a role in the phenotypic expressivity, modulating the effect of the AGO1 variant. On the contrary, some specific traits seem to correlate with specific variants, such as hypothyroidism reported in two individuals with the p.(Phe180del) variant, or status epilepticus in three individuals with the p.(Gly199Ser) variant. A larger cohort would be necessary to confirm these observations. Individuals with AGO1 or AGO2 variants showed common clinical findings: ID with autistic features and aggressiveness, impaired speech development, motor delay, MRI anomalies and frequent gastrointestinal disorder or reflux. Minor additional clinical features are also reported in both cohorts: skeletal (clinodactyly or bradymetacarpy, scoliosis), vision problems (strabism, myopia/hyperopia, visual impairment), heart, dental or breathing anomalies, and also anxiety and sleeping disturbance. To note, we observed three monozygotic twin sets in AGO1/AGO2 cohorts. This apparent excess of monozygotic twins (6/54 patients) requires further study.
The 3D conformational model of AGO revealed a flexible protein with two mobile domains, the L1 and L2 linker domains. The global structure contains four globular domains (PAZ, MID, PIWI and N terminal), and a deep cleft bordered by L1 and L2 linkers. L1 and PAZ domains seemed to be more flexible than the L2, PIWI and MID domain, as shown in Figure 2. The flexibility of the protein seems to be important to permit transitions between the different phases of the RISC process: sncRNA loading and processing, mRNA target clip, helper protein recruitment, and finally RISC complex release. Opening and closure of the PAZ/L1 jaw seems necessary for the proper function of AGO protein. Among the 18 AGO1 variants reported to date the majority clustered within the L1, PAZ and PIWI domains. We analyzed the effects of variants located in several domains using structural biology as an interpretive framework. Our assessment indicated that they were deeply buried in the cleft region and close to the RNA guide molecule, but did not affect the residues involved in RNA guide anchorage; in addition, dynamical simulations showed these variants narrowed the angulation between the PAZ/L1 domains, suggesting these variants may hamper AGO1 flexibility during the phases of mRNA processing. Recently, the AGO1x protein isoform was described, product of the translational stop read-through of AGO1 transcripts induced by the expression of the Let7a miRNA. Ectopic expression of AGO1x is a negative competitor of the miRNA pathway[34]. We could hypothesize that mutated AGO1 may act as the AGO1x isoform through competitive inhibition of wild type AGO proteins. Functional studies will be necessary to investigate this hypothesis.
The absence of truncating AGO1 variants in our cohort is surprising and is not in favor of a loss-of-function (LoF) as the unique molecular mechanism involved in AGO1-related ID. However, the observation of five children with large heterozygous deletions including AGO1 together with AGO3 (and sometimes AGO4 among other genes), and a deletion encompassing the AGO2 first 3 exons challenges this observation[13–15]. AGO2 variant functional analysis revealed a complex cellular deregulation: a decrease in mRNA silencing was observed as expected for a LoF mechanism but with a variable impact, depending on the mRNA target and the tested AGO2 variant. Reduced target release and reduced phosphorylation of the serine cluster at residues 824 to 834 in AGO2 was observed for most of the tested variants due to probable protein dynamics perturbation, likely leading to AGO2 deregulation and reduced functions [15].
A better understanding of how AGO1 functions in the brain will be essential to resolving how pathogenic AGO1 variants cause this NDD. In the developing mouse brain, although ubiquitously expressed, Ago1 was shown to be upregulated in neurogenic progenitors and mature neurons[35]. This expression pattern correlates with neuroblastoma SH-SY-5Y cell studies showing human AGO1 participates in promoting differentiation after neuronal induction[35]. Homozygous inactivation of Ago1 in mice showed postnatal lethality as reported in IMPC (International Mouse Phenotyping Consortium, mousephenotype.org): only 9 % of homozygotes Ago1tm1a/tm1a mice are obtained from heterozygous mating suggesting a prenatal lethality. The surviving homozygotes (both males and females) showed decreased body weight and increased anxiety (abnormal behavior in the open field). Pathogenic variants in several other genes encoding protein partners of AGO1/AGO2 involved in the regulation of mRNA decay and translation, such as CNOT1–3[36],[37],[38] or DDX6[39], reportedly cause NDDs[40]. Apart from their role in post-transcriptional regulation, human AGO1 and AGO2 are also involved in the transcriptional regulation and splicing[9]. Therefore, alterations of these nuclear functions could contribute to the pathophysiology of the neurodevelopmental disorders caused by pathogenic variants in these genes.
In conclusion, this collaborative study, reporting molecular and clinical data from 26 families with heterozygous de novo coding variants in AGO1, confirms the involvement of rare coding variants in this gene in NDD. Future studies investigating transcriptional and posttranscriptional regulation defects or other dysfunction stemming from AGO1 pathogenic variants will be important for elucidating the precise mechanisms of disease that may inform potential therapeutic strategies.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all the families for their participation in the study. We thank the Strasbourg Hospital molecular diagnostic lab, especially the team working on diagnosis of Intellectual Disability and all the bioinformatics team, the Cambridge Broad Institute of MIT and Harvard, especially the team working on clinic and diagnostic of patients, the Centre National de Génotypage for their participation in library preparation and DNA sequencing, and the Center for Individualized Medicine at Mayo Clinic for their support.
FUNDINGS
Research reported in this publication was supported by the National Institute Of Mental Health of the National Institutes of Health under Award Number U01MH119689. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also thank the CREGEMES for its financial support.
Footnotes
DATASHARING
The variants have been submitted to ClinVar.
COMPETING INTEREST
Kirsty McWalter, Erin Torti, Francisca Millan, Amy Dameron, Mari J. Tokita are employees of GeneDx. Zoe Powis and Kelly Minks are employees of Ambry Genetics.
ETHICS APPROVAL STATEMENT Affected individuals were referred by clinical genetic services and a genetic testing was done as part of routine clinical care. All patients enrolled and/or their legal representative have signed informed consent for research use and authorization for publication, including photograph publication authorization for those included in Figure 1. All the institutions received local IRB approval to use these data in research purpose. The main IRB approval was obtained from the Ethics Committee of the Strasbourg University Hospital (CCPPRB).
DATA AVAILABILITY
Variants were submitted to ClinVar database. Additional data are available upon request.
REFERENCES
- [1].Vissers LELM, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nature Reviews Genetics 2016;17:9–18. doi: 10.1038/nrg3999 [DOI] [PubMed] [Google Scholar]
- [2].Lelieveld SH, Wiel L, Venselaar H, Pfundt R, Vriend G, Veltman JA, Brunner HG, Vissers LELM, Gilissen C. Spatial Clustering of de Novo Missense Mutations Identifies Candidate Neurodevelopmental Disorder-Associated Genes. Am J Hum Genet 2017;101:478–84. doi: 10.1016/j.ajhg.2017.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, Dufke A, Cremer K, Hempel M, Horn D, Hoyer J, Joset P, Röpke A, Moog U, Riess A, Thiel CT, Tzschach A, Wiesener A, Wohlleber E, Zweier C, Ekici AB, Zink AM, Rump A, Meisinger C, Grallert H, Sticht H, Schenck A, Engels H, Rappold G, Schröck E, Wieacker P, Riess O, Meitinger T, Reis A, Strom TM. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. The Lancet 2012;380:1674–82. doi: 10.1016/S0140-6736(12)61480-9 [DOI] [PubMed] [Google Scholar]
- [4].Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M,Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Šestan N, Lifton RP, Günel M, Roeder K, Geschwind DH, Devlin B, State MW. De novo mutations revealed by whole exome sequencing are strongly associated with autism. Nature 2012;485:237–41. doi: 10.1038/nature10945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hamdan FF, Srour M, Capo-Chichi J-M, Daoud H, Nassif C, Patry L, Massicotte C, Ambalavanan A, Spiegelman D, Diallo O, Henrion E, Dionne-Laporte A, Fougerat A, Pshezhetsky AV, Venkateswaran S, Rouleau GA, Michaud JL. De Novo Mutations in Moderate or Severe Intellectual Disability. PLoS Genetics 2014;10:e1004772. doi: 10.1371/journal.pgen.1004772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sakaguchi A, Yamashita Y, Ishii T, Uehara T, Kosaki K, Takahashi T, Takenouchi T. Further evidence of a causal association between AGO1, a critical regulator of microRNA formation, and intellectual disability/autism spectrum disorder. European Journal of Medical Genetics 2019;62:103537. doi: 10.1016/j.ejmg.2018.09.004 [DOI] [PubMed] [Google Scholar]
- [7].Sasaki T, Shiohama A, Minoshima S, Shimizu N. Identification of eight members of the Argonaute family in the human genome☆. Genomics 2003;82:323–30. doi: 10.1016/S0888-7543(03)00129-0 [DOI] [PubMed] [Google Scholar]
- [8].Meister G Argonaute proteins: functional insights and emerging roles. Nat Rev Genet 2013;14:447–59. doi: 10.1038/nrg3462 [DOI] [PubMed] [Google Scholar]
- [9].Allo M, Agirre E, Bessonov S, Bertucci P, Gomez Acuna L, Buggiano V, Bellora N, Singh B, Petrillo E, Blaustein M, Minana B, Dujardin G, Pozzi B, Pelisch F, Bechara E, Agafonov DE, Srebrow A, Luhrmann R, Valcarcel J, Eyras E, Kornblihtt AR. Argonaute-1 binds transcriptional enhancers and controls constitutive and alternative splicing in human cells. Proceedings of the National Academy of Sciences 2014;111:15622–9. doi: 10.1073/pnas.1416858111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shuaib M, Parsi KM, Thimma M, Adroub SA, Kawaji H, Seridi L, Ghosheh Y, Fort A, Fallatah B, Ravasi T, Carninci P, Orlando V. Nuclear AGO1 Regulates Gene Expression by Affecting Chromatin Architecture in Human Cells. Cell Systems 2019;9:446–458.e6. doi: 10.1016/j.cels.2019.09.005 [DOI] [PubMed] [Google Scholar]
- [11].Ameyar-Zazoua M, Rachez C, Souidi M, Robin P, Fritsch L, Young R, Morozova N, Fenouil R,Descostes N, Andrau J-C, Mathieu J, Hamiche A, Ait-Si-Ali S, Muchardt C, Batsché E, Harel-Bellan A. Argonaute proteins couple chromatin silencing to alternative splicing. Nature Structural & Molecular Biology 2012;19:998–1004. doi: 10.1038/nsmb.2373 [DOI] [PubMed] [Google Scholar]
- [12].Wei W, Ba Z, Gao M, Wu Y, Ma Y, Amiard S, White CI, Rendtlew Danielsen JM, Yang Y-G, Qi Y. A role for small RNAs in DNA double-strand break repair. Cell 2012;149:101–12. doi: 10.1016/j.cell.2012.03.002 [DOI] [PubMed] [Google Scholar]
- [13].Tokita MJ, Chow PM, Mirzaa G, Dikow N, Maas B, Isidor B, Le Caignec C, Penney LS, Mazzotta G, Bernardini L, Filippi T, Battaglia A, Donti E, Earl D, Prontera P. Five children with deletions of 1p34.3 encompassing AGO1 and AGO3. European Journal of Human Genetics 2015;23:761–5. doi: 10.1038/ejhg.2014.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jacher JE, Innis JW. Interstitial microdeletion of the 1p34.3p34.2 region. Mol Genet Genomic Med 2018;6:673–7. doi: 10.1002/mgg3.409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lessel D, Zeitler DM, Reijnders MRF, Kazantsev A, Hassani Nia F, Bartholomäus A, Martens V,Bruckmann A, Graus V, McConkie-Rosell A, McDonald M, Lozic B, Tan E-S, Gerkes E, Johannsen J,Denecke J, Telegrafi A, Zonneveld-Huijssoon E, Lemmink HH, Cham BWM, Kovacevic T, Ramsdell L, Foss K, Le Duc D, Mitter D, Syrbe S, Merkenschlager A, Sinnema M, Panis B, Lazier J, Osmond M, Hartley T, Mortreux J, Busa T, Missirian C, Prasun P, Lüttgen S, Mannucci I, Lessel I, Schob C, Kindler S, Pappas J, Rabin R, Willemsen M, Gardeitchik T, Löhner K, Rump P, Dias K-R, Evans C-A, Andrews PI, Roscioli T, Brunner HG, Chijiwa C, Lewis MES, Jamra RA, Dyment DA, Boycott KM, Stegmann APA, Kubisch C, Tan E-C, Mirzaa GM, McWalter K, Kleefstra T, Pfundt R, Ignatova Z, Meister G, Kreienkamp H-J. Germline AGO2 mutations impair RNA interference and human neurological development. Nat Commun 2020;11:5797. doi: 10.1038/s41467-020-19572-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: A Matching Tool for Connecting Investigators with an Interest in the Same Gene. Human Mutation 2015;36:928–30. doi: 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics 2014;46:310–5. doi: 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Adzhubei I, Jordan DM, Sunyaev SR. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr Protoc Hum Genet 2013;0 7:Unit7.20. doi: 10.1002/0471142905.hg0720s76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, Chow ED, Kanterakis E, Gao H, Kia A, Batzoglou S, Sanders SJ, Farh KK-H. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019;176:535–548.e24. doi: 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- [21].Martinez J, Patkaniowska A, Urlaub H, Lührmann R, Tuschl T. Single-Stranded Antisense siRNAs Guide Target RNA Cleavage in RNAi. Cell 2002;110:563–74. doi: 10.1016/S0092-8674(02)00908-X [DOI] [PubMed] [Google Scholar]
- [22].Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017;542:433–8. doi: 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gurovich Y, Hanani Y, Bar O, Nadav G, Fleischer N, Gelbman D, Basel-Salmon L, Krawitz PM,Kamphausen SB, Zenker M, Bird LM, Gripp KW. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med 2019;25:60–4. doi: 10.1038/s41591-018-0279-0 [DOI] [PubMed] [Google Scholar]
- [24].Nakanishi K, Ascano M, Gogakos T, Ishibe-Murakami S, Serganov AA, Briskin D, Morozov P, Tuschl T, Patel DJ. Eukaryote-Specific Insertion Elements Control Human ARGONAUTE Slicer Activity. Cell Reports 2013;3:1893–900. doi: 10.1016/j.celrep.2013.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pieper U, Webb BM, Dong GQ, Schneidman-Duhovny D, Fan H, Kim SJ, Khuri N, Spill YG, Weinkam P, Hammel M, Tainer JA, Nilges M, Sali A. ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Research 2014;42:D336–46. doi: 10.1093/nar/gkt1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Miyoshi T, Ito K, Murakami R, Uchiumi T. Structural basis for the recognition of guide RNA and target DNA heteroduplex by Argonaute. Nature Communications 2016;7. doi: 10.1038/ncomms11846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kocher J-PA, Quest DJ, Duffy P, Meiners MA, Moore RM, Rider D, Hossain A, Hart SN, Dinu V. The Biological Reference Repository (BioR): a rapid and flexible system for genomics annotation. Bioinformatics 2014;30:1920–2. doi: 10.1093/bioinformatics/btu137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liu X, Jian X, Boerwinkle E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Human Mutation 2011;32:894–9. doi: 10.1002/humu.21517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Research 2005;33:W382–8. doi: 10.1093/nar/gki387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem 2005;26:1781–802. doi: 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Karamzadeh R, Karimi-Jafari MH, Sharifi-Zarchi A, Chitsaz H, Salekdeh GH, Moosavi-Movahedi AA. Machine Learning and Network Analysis of Molecular Dynamics Trajectories Reveal Two Chains of Red/Ox-specific Residue Interactions in Human Protein Disulfide Isomerase. Scientific Reports 2017;7. doi: 10.1038/s41598-017-03966-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rashin AA, Domagalski MJ, Zimmermann MT, Minor W, Chruszcz M, Jernigan RL. Factors correlating with significant differences between X-ray structures of myoglobin. Acta Crystallogr D Biol Crystallogr 2014;70:481–91. doi: 10.1107/S1399004713028812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nick Pace C, Martin Scholtz J. A Helix Propensity Scale Based on Experimental Studies of Peptides and Proteins. Biophysical Journal 1998;75:422–7. doi: 10.1016/S0006-3495(98)77529-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Singh A, Manjunath LE, Kundu P, Sahoo S, Das A, Suma HR, Fox PL, Eswarappa SM. Let‐7a‐regulated translational readthrough of mammalian AGO1 generates a microRNA pathway inhibitor. EMBO J 2019;38. doi: 10.15252/embj.2018100727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Parisi C, Giorgi C, Batassa EM, Braccini L, Maresca G, D’agnano I, Caputo V, Salvatore A, Pietrolati F, Cogoni C, Catalanotto C. Ago1 and Ago2 differentially affect cell proliferation, motility and apoptosis when overexpressed in SH-SY5Y neuroblastoma cells. FEBS Lett 2011;585:2965–71. doi: 10.1016/j.febslet.2011.08.003 [DOI] [PubMed] [Google Scholar]
- [36].De Franco E, Watson RA, Weninger WJ, Wong CC, Flanagan SE, Caswell R, Green A, Tudor C, Lelliott CJ, Geyer SH, Maurer-Gesek B, Reissig LF, Lango Allen H, Caliebe A, Siebert R, Holterhus PM, Deeb A, Prin F, Hilbrands R, Heimberg H, Ellard S, Hattersley AT, Barroso I. A Specific CNOT1 Mutation Results in a Novel Syndrome of Pancreatic Agenesis and Holoprosencephaly through Impaired Pancreatic and Neurological Development. Am J Hum Genet 2019;104:985–9. doi: 10.1016/j.ajhg.2019.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Uehara T, Takenouchi T, Yamaguchi Y, Daimon Y, Suzuki H, Sakaguchi Y, Kosaki K. CNOT2 as the critical gene for phenotypes of 12q15 microdeletion syndrome. Am J Med Genet A 2019;179:659–62. doi: 10.1002/ajmg.a.61068 [DOI] [PubMed] [Google Scholar]
- [38].Martin R, Splitt M, Genevieve D, Aten E, Collins A, de Bie CI, Faivre L, Foulds N, Giltay J, Ibitoye R, Joss S, Kennedy J, Kerr B, Kivuva E, Koopmans M, Newbury-Ecob R, Jean-Marçais N, Peeters E a. J,Smithson S, Tomkins S, Tranmauthem F, Piton A, van Haeringen A. De novo variants in CNOT3 cause a variable neurodevelopmental disorder. Eur J Hum Genet 2019;27:1677–82. doi: 10.1038/s41431-019-0413-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Balak C, Benard M, Schaefer E, Iqbal S, Ramsey K, Ernoult-Lange M, Mattioli F, Llaci L, Geoffroy V, Courel M, Naymik M, Bachman KK, Pfundt R, Rump P, Ter Beest J, Wentzensen IM, Monaghan KG, McWalter K, Richholt R, Le Béchec A, Jepsen W, De Both M, Belnap N, Boland A, Piras IS, Deleuze J-F, Szelinger S, Dollfus H, Chelly J, Muller J, Campbell A, Lal D, Rangasamy S, Mandel J-L, Narayanan V, Huentelman M, Weil D, Piton A. Rare De Novo Missense Variants in RNA Helicase DDX6 Cause Intellectual Disability and Dysmorphic Features and Lead to P-Body Defects and RNA Dysregulation. Am J Hum Genet 2019;105:509–25. doi: 10.1016/j.ajhg.2019.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Weil D, Piton A, Lessel D, Standart N. Mutations in genes encoding regulators of mRNA decapping and translation initiation: links to intellectual disability. Biochem Soc Trans 2020;48:1199–211. doi: 10.1042/BST20200109 [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB RESOURCES
- CADD, https://cadd.gs.washington.edu/snv
- GeneMatcher, https://genematcher.org/
- GnomAD browser, https://gnomad.broadinstitute.org/
- OMIM, https://www.omim.org/
- SySID, https://sysid.cmbi.umcn.nl/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Variants were submitted to ClinVar database. Additional data are available upon request.