

Abstract
Frontotemporal lobar degeneration (FTLD) includes clinically similar FTLD-tau or FTLD-TDP proteinopathies which lack in vivo markers for accurate antemortem diagnosis. To identify early distinguishing sites of cortical atrophy between groups, we retrospectively analyzed in vivo volumetric MRI from 42 FTLD-Tau and 21 FTLD-TDP patients and validated these findings with postmortem measures of pathological burden. Our frequency-based staging model revealed distinct loci of maximal early cortical atrophy in each group, including dorsolateral and medial frontal regions in FTLD-Tau and ventral frontal and anterior temporal regions in FTLD-TDP. Sørenson-Dice calculations between proteinopathy groups showed little overlap of phases. Conversely, within-group subtypes showed good overlap between 3R- and 4R-tauopathies, and between TDP-43 Types A and C for early regions with subtle divergence between subtypes in subsequent phases of atrophy. Postmortem validation found an association of imaging phases with pathologic burden within FTLD-tau (F(4, 238)=17.44, p<0.001) and FTLD-TDP (F(4,245) = 42.32, p<0.001). These results suggest that relatively early, distinct markers of atrophy may distinguish FTLD proteinopathies during life.
Keywords: Frontotemporal lobar degeneration, magnetic resonance imaging, percent area occupied, tau, TDP-43, sporadic
Graphical abstract
1. Introduction
Frontotemporal lobar degeneration (FTLD) is a heterogenous set of pathologic disorders due to the misfolding of either tau (FTLD-tau), a microtubule-stabilizing protein, or transactive response DNA-binding protein of ∼43kD (TDP-43), known as FTLD-TDP (MacKenzie et al., 2010)(Neumann et al., 2006). These disorders are associated with various clinical frontotemporal dementia (FTD) phenotypes, including behavioral variant FTD (bvFTD) and primary progressive aphasia (PPA) that present with or without an accompanying motor impairment. While there is a statistical association between some clinical phenotypes and the associated pathology, these associations vary in their reliability. For example, variants of clinically defined PPA are often associated with specific underlying pathologies, yet clinical findings are not always confirmed at autopsy (Grossman, 2010). Also, studies of corticobasal degeneration (CBD) and progressive supranuclear palsy (PSP) have revealed that clinical features do not always correlate with the presumed deficit of anatomical structure. This clinical and pathological heterogeneity is a barrier to evaluation and treatment of FTLD. Therefore, investigation of pathological phenotype independent of clinical phenotype may be very informative.
As with many neurodegenerative diseases (Brettschneider et al., 2015a; Guo and Lee, 2014), FTLD is believed to progress and spread via a prion-like mechanism and axon-mediated cell-cell transmission (Boluda et al., 2015; Brettschneider et al., 2015b; Guo and Lee, 2014; David J. Irwin et al., 2016b; Kovacs et al., 2020a; Vatsavayai et al., 2019). However, it is unclear whether the anatomic patterns of initial and subsequent disease are similar across patients with FTLD-tau and FTLD-TDP pathology (Arnold et al., 2013; Kim et al., 2012; Nana et al., 2019; Vatsavayai et al., 2019). Identifying early loci of disease and patterns of spread in FTLD-tau and FTLD-TDP and their subtypes in vivo is crucial, as they could potentially serve as early diagnostic markers and dynamic treatment targets. In the present study, we identify likely anatomic sites of early disease and phases of progression by evaluating regional cortical atrophy in patients with sporadic, autopsy-confirmed FTLD-tau or FTLD-TDP using whole-brain, antemortem MRI imaging to calculate phases of disease progression in a frequency-based manner similar to that employed in autopsy staging studies (Braak and Braak, 1991; Phillips et al., 2018), and we compare the results of the MRI analyses with objective digital assessments of regional postmortem pathologic burden in available tissue samples (David J Irwin et al., 2016b).
Few studies in FTLD have quantitatively tested the assumption that disease phase correlates with severity of pathology. Unlike selective pathology sampling, in vivo brain MRI can easily obtain an image of the entire cerebrum, allowing us to identify the presumed early loci and subsequent spread of gray matter degeneration throughout the entire cortical mantle. These results provide a measure of presumed underlying pathology, but more data are needed to fully inform this relationship. Previous reports have described MRI changes in partially overlapping frontal and temporal regions in patients with known FTLD spectrum pathology (Brown et al., 2019a; Frings et al., 2012; Kassubek et al., 2014; Whitwell and Josephs, 2012) (Whitwell et al., 2007; Whitwell and Josephs, 2012). Postmortem studies of pathological burden have shown early regional differences between FTLD-tau and FTLD-TDP patients with clinically similar phenotypes (Giannini et al., 2019a; Irwin et al., 2018). However, direct comparisons of imaging studies with regional pathologic burden are very rare. In this study, we hypothesized that there would be distinct early regions of cortical atrophy between FTLD-tau and FTLD-TDP groups. We tested this hypothesis by examining antemortem MRI in patients with well-characterized pathology (Giannini et al., 2019a; Jonkman et al., 2019). Although subcortical regions have been staged early in subtypes of FTLD-tau and FTLD-TDP (Kovacs et al., 2020a), we limited our analysis to cortical regions with the goal of capturing areas with the greatest overlap of proteinopathy subtypes that would best generalize to the overall proteinopathy group. We then related the results of the macroscopic antemortem MRI analyses with regional parametric measures of postmortem microscopic digital pathologic burden (Irwin et al., 2016b) by directly correlating %AO (percentage of area occupied) for individuals within each pathological sampled region to phase assignment for that region as determined by imaging indexed by a pathology-to-imaging atlas (Spotorno et al., 2020) (see supplementary figure A.7).
Within both FTLD-tau and FTLD-TDP, moreover, there is heterogeneity in the associated molecular pathology. Patients with a three-repeat (3R) tauopathy frequently have dementia with Pick bodies (PiD), and these are pathologically distinct from those with a four-repeat (4R) tauopathy; 4R tauopathies are divided into, among others, cases with progressive supranuclear palsy (PSP) pathology and corticobasal degeneration (CBD) (Forrest et al., 2019; Mackenzie and Neumann, 2016). For TDP-43 proteinopathies, five subtypes have been identified – Type A to Type E(Lee et al., 2017; Neumann and Mackenzie, 2019). While postmortem pathology studies suggest that there may be common brain areas where each proteinopathy class accumulates pathology, differential patterns of disease have been observed in the subtypes of both tau (Forrest et al., 2019; David J. Irwin et al., 2016a; Irwin et al., 2015; Kovacs et al., 2020b) and TDP-43 (Brettschneider et al., 2014; Giannini et al., 2021; Irwin et al., 2015) pathologies. Epicenters of pathologic disease have been hypothesized (Kim et al., 2012), and diversity of pathologic distribution also has been seen within FTLD spectrum pathology (Vatsavayai et al., 2019). There is evidence of subtle differences in postmortem distribution of pathology between proteinopathy subtypes that present with different clinical presentations during life (Giannini et al., 2021; Nana et al., 2019; Whitwell et al., 2020). However, these differences between clinical groups are relatively modest compared to the dissociation between pathological groups characterized at autopsy.
Based on these typical outcomes, we further hypothesized that phases of regional cortical atrophy would partially overlap between subtypes within FTLD-tau and FTLD-TDP groups and that we would see divergent anatomic patterns of MRI atrophy between subtypes of FTLD-tau and FTLD-TDP in association with subsequent disease progression. To test these hypotheses, we evaluated antemortem MRI comparatively among the subtypes of FTLD-tau and FTLD-TDP. To assess the validity of our MRI results, we directly compared inferred phases of disease based on MRI atrophy with available digital quantitative analyses of tissue pathology.
2. Methods
2.1. Patients
We retrospectively analyzed data from 63 autopsy-confirmed patients (42 with tau inclusions, 21 with TDP-43 inclusions) and 170 healthy controls matched for education, sex and age, using a propensity matching package in R (Ho et al., 2011) from all available participants in the Penn Integrated Neurodegenerative Disease Database (INDD) (Toledo et al., 2014) at the Penn Frontotemporal Degeneration Center (FTDC). Pathological diagnosis was made by expert pathologists (EBL, JQT) according to published criteria using well established procedures (Toledo et al., 2014). Inclusion into the study required that patients must have participated in at least one antemortem T1-weighted structural MRI scan. All patients had been evaluated by an experienced cognitive neurologist (MG, DJI, LM) at the Penn Cognitive Neurology Outpatient Clinic, had received clinical evaluation within 1 year of scan date, and were evaluated at a weekly multidisciplinary consensus meeting. Patients were excluded if they had AD neuropathologic scores greater than minimal by the ABC metric (Montine et al., 2012) or had any other postmortem diagnosed co-pathology. All patients were genotyped based on pedigree analysis (Wood et al., 2013), and those patients with FTLD mutations were excluded to minimize potential confounding results from clinical and pathological differences that have been noted between mutation carriers and sporadic cases (Capozzo et al., 2017; Janssen et al., 2002). Diagnostic groupings were based solely on pathological diagnosis regardless of clinical diagnosis. However, clinical diagnoses for all participants are provided in the supplement (see supplementary tables A3-A5). The FTLD-tau cohort included three pathological subtypes: PSP, CBD, and PiD. The FTLD-TDP cohort included TDP-43 Types A, B, C, and E pathological subtypes. We excluded patients with amyotrophic lateral sclerosis (ALS) that did not have cognitive deficits at any time during life based on either a documented cognitive or social difficulty in the clinical note or by a low score on the Edinburgh Cognitive and Behavioral ALS Screen (ECAS)(Abrahams et al., 2014), thus excluding 20 patients with only a motor disorder. We also excluded cases with any clinical or pathological evidence of stroke, significant head injury, infection, immune-mediated disorder, or intracranial mass or hydrocephalus. Controls were matched to patients for age, education and sex (Ho et al., 2011), and were negative for neurological or psychiatric history. All available matching controls were used to estimate the coefficients from the linear model for the prediction term of the w-score formula (Brown et al., 2019b; La Joie et al., 2012) so that the w-score denominator term would best reflect the general population for age, sex, education level. All procedures for patients and controls were performed following an informed consent procedure according to guidelines approved by the Institutional Review Board at the University of Pennsylvania. Table 1 summarizes the characteristics of the patient groups and provides comparisons for demographic characteristics.
Table 1. Patient Demographics for FTLD-tau and FTLD-TDP groups.
Descriptive statistics across patient groups for baseline variables. Median and interquartile range (median[IQR]) are provided. Mann-Whitney-Wilcox was performed for continuous variables and chi-squared test was performed for categorical variables; p value is based on comparison between groups.
| FTLD-tau | FTLD-TDP | p | |
|---|---|---|---|
|
| |||
| n | 42 | 21 | |
| Age at MRI (years) | 67.0 [61.0, 72.0] | 64.0 [58.0, 69.0] | 0.048 |
| Disease Duration at MRI (years) | 5.0 [2.0, 6.0] | 3.0 [2.0, 5.0] | 0.038 |
| MRI to Autopsy Interval (years) | 3.0 [2.0, 4.0] | 3.0 [2.0, 4.0] | 0.959 |
| Disease Duration at Autopsy (years) | 8.0 [5.0, 10.0] | 6.0 [4.0, 9.0] | 0.186 |
| Age at Autopsy (years) | 72.0 [66.0, 74.0] | 68.0 [62.0, 73.0] | 0.092 |
| Sex = Male (%) | 27 (64.3) | 10 (47.6) | 0.320 |
| Education (years) | 16.0 [15.0, 18.0] | 16.0 [12.0, 18.0] | 0.308 |
2.2. Neuroimaging Acquisition and Processing
We retrospectively selected 231 T1-weighted, high resolution, MPRAGE MRI scans. Patients and healthy controls were limited to 1 scan per individual. For participants with multiple scans available, we chose the earliest scan date available within a range in years (2004–2017) to capture the earliest phases of disease. Scans were acquired using three major sequences listed below. A small subset (tau: n=6, TDP: n=2) were acquired using slightly different sequences than those listed below (see Supplementary methods). Staging analysis was unchanged when these scans were removed and were consequently included in the final analysis.
3.0 Tesla SIEMENS TIM Trio scanner, 8 channel head coil, axial plane with repetition time = 1620 ms, echo time = 3.87 ms, slice thickness = 1.0 mm, flip angle = 15 degrees, matrix = 192 × 256, in-plane resolution = 0.98 × 0.98 mm.
3.0 Tesla SIEMENS TIM Trio scanner, 64 channel head coil, sagittal plane with repetition time = 2300 ms, echo time = 2.95 ms, slice thickness = 1.2 mm, flip angle = 0 degrees, matrix = 256 × 240, in-plane resolution = 1.05 × 1.05 mm.
3.0 Tesla SIEMENS Prisma scanner, 64 channel head coil, sagittal plane with repetition time 2400 ms, echo time = 1.96 ms, flip angle = 8 degrees, matrix = 320 × 320, slice thickness = 0.8 mm, in-plane resolution = 0.8 × 0.8 mm.
We processed images using the Advanced Normalization Tools (ANTs) antsCorticalThickness.sh pipeline (Avants et al., 2014; Tustison et al., 2014). Through this process, low frequency intensity nonuniformity was corrected with N4 bias correction (Tustison et al., 2010). Segmentation used template-based priors to divide images into 6 classes (cortical gray matter (GM), subcortical GM, white matter (WM), cerebrospinal fluid (CSF), brainstem, and cerebellum). We selected regions of interest (ROI) from the publicly available Lausanne atlas (Hagmann et al., 2008) with parcellations of 250 labels per hemisphere. This label set was limited to cortical regions only. Labels were transformed to native space by using the warps made with antsCorticalThickness to transform a set of ROIs in Oasis template space into the native space. We used volume rather than thickness or density since preliminary studies showed volume to be a more reliable measure to index cortical atrophy in correlations with autopsy data (Schwarz et al., 2016). Scans for both patients and controls underwent quality control by two independent raters who were blinded to group membership and to each other’s judgment (SEB, CAO). Scans were eliminated for excessive head motion or poor segmentation. Only scans with an average rating of ≥2 on a 3-point scale using these criteria were accepted for inclusion. Scans with a rating of 1 failed and were excluded, while a rating of 2 required judgment from the rater, and a rating of 3 passed without issue.
2.3. Phase model
2.3.1. W-score calculation
To quantify atrophy, GM volume was calculated for patients and healthy controls from the output of T1-weighted images. To calculate W-scores (La Joie et al., 2012), actual GM volume estimates, intracranial volume (ICV) and age were collected for each participant. A linear regression analysis, including age and intracranial volume as independent variables, was performed at each ROI for all healthy controls. From this regression analysis, we extracted the coefficients (betas) for calculation of predicted GM volume adjusting for age and ICV. For each ROI within each patient, the coefficients for age and ICV were multiplied by patient age and ICV. These values, along with the model intercept, were subtracted from the patient GM volume. This value was divided by the standard deviation of the residuals from the healthy control model according to the equation:
Individual W-scores calculated for each patient were binarized, such that any ROI was considered atrophied if it fell below threshold (W-score < −0.5), chosen to capture the varying degrees of atrophy that have been found among subtypes in specific brain regions(Josephs et al., 2008; Whitwell and Josephs, 2012) while optimizing sensitivity to atrophy and minimizing potential ceiling and floor effects. The number of patients with atrophy at threshold or below in each region was tabulated.
2.3.2. Frequency-based calculations
After w-score calculation of ROIs in the entire patient data set, the regions were sorted into phases independently for the FTLD-tau and FTLD-TDP groups, and then the subtypes of each pathology group. These phases were calculated based on Braak’s seminal approach to pathologic disease progression in Alzheimer’s disease (AD) (Braak and Braak, 1991), by which staging studies of autopsied brains infer a pattern of progressive spread of disease through a qualitative examination of the anatomic distribution of relative pathologic burden. Areas determined to have greater pathologic burden are thought to represent anatomic regions where pathology has been accumulating for a longer period of time, and therefore are thought to have occurred earlier in the disease process; anatomic regions with less pathologic burden, according to this reasoning, are thought to have been accumulating pathology over less time, and therefore are thought to have been involved in spreading pathology later in the time course of the disease. Within a group, phases were defined by the ratio Na(r) / Nm, where Na(r) is the number of patients in the group with atrophy in region r, and Nm = max(Na(r), r = 1,…,448) is the maximum Na(r) in the group over all regions. Phase 1 regions were defined as those with Na(r) within 90–100% (inclusive) of Nm. Those within 80–89% were Phase 2; 70–79%, were Phase 3; 60–69% were Phase 4; 50–59% were Phase 5. The remainder of regions were considered to be below staging due to minimal frequency of atrophy.
2.3.3. Subtype analysis
Pathological subtypes within FTLD-tau and FTLD-TDP cohorts were analyzed, and phases were calculated within each respective subtype. Tau subtypes included PiD, CBD, and PSP, and FTLD-TDP subtypes included those with TDP-43 Type A and Type C inclusions. Type B (n=5) and Type E (n=4) inclusion subgroups were grouped together for analysis. Phases for each subtype were calculated by the same process described in the larger model. Subtypes were then compared for overlapping ROIs within a phase.
2.4. Pathology regional anatomic analysis
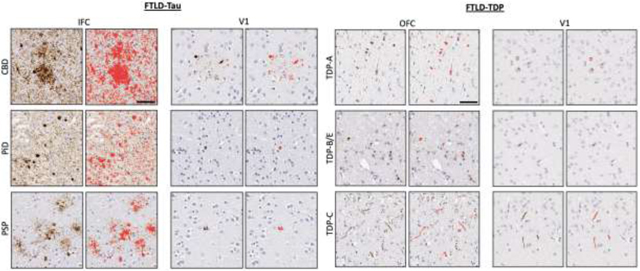
We studied burden of pathology from a random hemisphere at autopsy according to standardized NIA/AA diagnostic guidelines (Hyman et al., 2012) in a group of autopsied patients (n=134), in each proteinopathy group (FTLD-tau=63, FTLD-TDP=71) (see supplemental table S2). Bilateral sampling was done by well-established and validated methods(Giannini et al., 2019b). Only a subset of participants had bilateral sampling in a limited number of regions, and no brain region had bilateral sampling in all participants. Instead, most participants were randomly sampled in either the right or left hemisphere per region. For these reasons, brain regions were averaged across hemispheres when sampled bilaterally. Available number of patient samples in each group included: the anterior cingulate (aCING, FTLD-tau=48, FTLD-TDP=32), middle frontal cortex (MFC, FTLD-tau=62, FTLD-TDP=30), orbitofrontal cortex (OFC, FTLD-tau=60, FTLD-TDP=32), anterior insula (aINS, FTLD-tau=21, FTLD-TDP=8), inferior frontal cortex (IFC, FTLD-tau=20, FTLD-TDP=3), superior parietal cortex (SPC, FTLD-tau=30, FTLD-TDP=9), superior-middle temporal cortex (SMTC, FTLD-tau=55, FTLD-TDP=29), angular gyrus (ANG, FTLD-tau=46, FTLD-TDP=31) and primary visual cortex (V1, FTLD-tau=20, FTLD-TDP=33). Data from the subset of imaging participants in this study are provided in the supplement (supplementary figures A.4 & A.5), and the results of these two analyses are very similar.
We measured %AO by TDP-43 or tau inclusions in selected regions of the samples, as published (David J Irwin et al., 2016b). Briefly, tissue fixed in formalin was immunostained for phosphorylated TDP-43 (rat monoclonal TAR5P-1D3, p409/410; Millipore Sigma) (Neumann et al., 2009) and tau (AT8; Invitrogen) (Mercken et al., 1992). Whole-slide images at 20x magnification were obtained using an Aperio AT2, Leica Biosystem, Wetzlar, Germany) in the Penn Digital Neuropathology Lab at 20x magnification (Giannini et al., 2019a; David J. Irwin et al., 2016b). Digital image analysis was performed with QuPath software (version 0.2.0-m5) to measure the % area occupied (%AO) of proteinopathy in GM on each histopathological section using previously validated methods (Giannini et al., 2019c; David J Irwin et al., 2016b). We determined the Lausanne ROIs corresponding to these sampled regions through consensus discussion of pathologists and image analysts (Spotorno et al., 2020)(see supplementary figure A.7).
2.5. Statistical analyses
Demographic features were compared between groups using a Mann-Whitney U test because they displayed a non-normal distribution, as shown by the Shapiro-Wilk normality test (onset to autopsy interval = FTLD-tau: p<0.001, FTLD-TDP: p=0.11, MRI date to autopsy interval = FTLD-tau: p=0.01, FTLD-TDP: p= 0.06, onset to MRI date interval = FTLD-tau: p<0.001, FTLD-TDP: p=0.001). Categorical variables were tested with chi-square tests. Phases of neuroimaging data were calculated using W-scores, as described above. Phase differences between proteinopathy groups and between subtypes within groups were calculated from neuroimaging data using the Dice similarity coefficient (DSC) (Dice, 1945; Yushkevich et al., 2010). First, binary variables were assigned positive one (the subtype phase agrees with the total proteinopathy group phase) or zero (phase between total group and subtype disagree). Each subtype was contrasted with a second subtype and again with a third subtype until Dice overlap had been calculated between all pairs of subtypes within a proteinopathy group. The DSC between two subtypes, where A and B each represent a subtype, is given by
Between FTLD-tau and FTLD-TDP groups, DSC was calculated using the same formula applied to a set of brain regions for each phase that had been assigned that phase for either group. Neuropathological data were tested using parametric statistics after natural log transformation of %AO occupied for all brain regions sampled. To demonstrate the overlap between groups as phases progress, each DSC was calculated starting with Phase 1 then calculated again to include the next successive phase (Phase 2) until all phases were included. All resulting DSCs for phase sets were reported (Table 2). To test the association of imaging phase with pathological burden at autopsy, within-group analysis was performed through a linear mixed effects (LME) model with random intercepts for individual patients to account for multiple measurements, missing data, and individual differences (Laird and Ware, 1982), and an ANOVA was performed on the LME model. %AO values were first log-transformed to account for the right-skewed distribution (see Supplementary figure A.3). Imaging phase derived from the imaging model was incorporated as an independent factor. Significant differences between group demographic profiles were addressed in imaging and pathology analyses as follows: disease onset to autopsy interval was incorporated as a covariate in LME of pathological burden (%AO); and age at MRI was factored into calculation of patient W-scores in the imaging analysis as a proxy for disease duration at MRI. The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Table 2. Sorenson-Dice Coefficient for FTLD-tau and FTLD-TDP subtypes.
Comparisons between subtypes demonstrating that for FTLD-tau, early phases show overlap but diverge progressively by order of phase. For FTLD-TDP subtypes, the relationship was less linear but did show some divergence as with increasing phase order. Higher values indicate greater overlap.
| FTLD-tau | FTLD-TDP | |||||
|---|---|---|---|---|---|---|
| phase | PSP & CBD | PSP & PiD | CBD & PiD | Type A & Type C | Type A & Types B/E | Type C & Type B/E |
| Phase 1 | 0.86 | 0.86 | 1.00 | 0.67 | 0.40 | 0.40 |
| Phase 2 | 0.73 | 0.60 | 0.73 | 0.62 | 0.22 | 0.29 |
| Phase 3 | 0.62 | 0.50 | 0.62 | 0.62 | 0.40 | 0.31 |
| Phase 4 | 0.57 | 0.43 | 0.50 | 0.60 | 0.40 | 0.29 |
| Phase 5 | 0.57 | 0.40 | 0.47 | 0.52 | 0.35 | 0.27 |
3. Results
3.1. Patient Demographics
Patient groups showed no significant differences in sex (x2=0.99, p=0.32), interval from MRI to autopsy (W=444.5, p=0.96), disease duration at autopsy (W=519, p=0.19), age at autopsy (W=543.5, p=0.09) or years of education (W=497.5, p=0.31). There were differences between age at MRI (W=563, p=0.05), and disease duration at MRI (W=568, p=0.04) (Table 1).
3.2. Phases of FTLD-tau-associated imaging atrophy
Regions of atrophy in the overall FTLD-tau group are illustrated in Figure 1. All Phase 1 regions were confined to the frontal cortex. These regions included IFC, superior frontal cortex (SFC), MFC and precentral gyrus. Since high proportions of the SFC (35/42), IFC (34/42) and precentral regions (33/42) were found to be Phase 1 regions in the entire FTLD-tau cohort and in each of the FTLD-tau subgroups (see below), we considered these regions to be the imaging-based early loci of FTLD-tau pathology. Phase 2 regions included areas that were adjacent to Phase 1 regions in the frontal lobe, including OFC and aINS. Phase 2 regions also included nonadjacent areas outside of the frontal lobe, including posterior cingulate (pCING), and superior temporal gyrus (STG). Phase 3 regions comprised additional areas in the frontal lobe, including aCING and the frontal pole, as well as regions outside of the frontal lobe including the paracentral gyrus, middle temporal gyrus (MTG), ANG, and the supramarginal gyrus (SMG). Phase 4 regions included the banks of the superior temporal sulcus (BANKSSTS), superior parietal cortex (SPC), the postcentral gyrus (PoCG), the parahippocampal gyrus (PHG), the precuneus (PC), the fusiform gyrus (FFG), the transverse temporal gyrus (TTG) and entorhinal cortex (EC). Finally, Phase 5 regions included the visual cortex (V1), the lingual gyrus and the isthmus of the cingulate. Phases of all other cerebral regions were not considered further, as too few individuals showed atrophy.
Fig. 1. Results of the MRI phase assignment algorithm for FTLD-tau (top) and FTLD-TDP (bottom) cohorts.

Phase 1 (black): putative epicenters of disease. Phase 2: maroon: Phase 3: red: Phase 4: orange: Phase 5: yellow. The remainder of brain regions (uncolored) had minimal atrophy and were considered to be below staging.
3.3. FTLD-tau subtype analysis
Regions of cortical atrophy in the three tau pathologic subtypes are illustrated in Supplementary figure A.1 and overlapping regions are illustrated in Figure 2. In the tau subtype analysis, all three subtypes had overlapping Phase 1 brain regions in IFC, SFC, and precentral gyrus. In CBD and PiD, but not PSP, there were overlapping Phase 1 regions in the MFC. Subtypes PSP and CBD, but not PiD, had overlapping Phase 1 regions in the SMG. There were additional Phase 1 regions that were non-overlapping and not found to be Phase 1 for the tau group as a whole, but that emerged upon individual subtype analysis, including the SPC (CBD), precuneus (CBD) and several regions for PiD (see Figure 2). Subsequent phases had varied overlapping patterns (see DSC analysis below).
Fig. 2. Overlapping of phases across FTLD-tau and subtypes.

Results from staging algorithm for FTLD-Tau group overall (first column): PSP (second column); CBD (third column): PID (fourth column). Phase 1 (dark purple): putative epicenters of disease. Phase 2: light purple: Phase 3: magenta: Phase 4: orange; Phase 5: pale orange. The remainder of brain regions (beige) liad minimal atrophy and were considered to be below staging.
The DSC analysis of FTLD-tau subtypes is summarized in Table 2. Pairwise comparisons of FTLD-tau subtypes in this analysis showed high levels of overlapping cortical atrophy in early phases, but progressively decreasing levels of overlapping cortical atrophy with later phases of disease. DSC analysis of FTLD-tau and FTLD-TDP groups showed very little overlap, with a range from scores of 0.00 for phase 1 regions (no overlap) to a maximum value of 0.39 (minimal overlap) in phase 4.
3.4. Phases of FTLD-TDP-associated imaging atrophy
Regions of atrophy in the FTLD-TDP group as a whole (including subtypes A, B, C, and E) are illustrated in Figure 1. In the FTLD-TDP group as a whole, Phase 1 regions were not restricted to the frontal lobe but were also found in the temporal lobe. Specifically, Phase 1 regions included the STG (19/21), MTG (18/21), OFC (18/21), aCING (18/21) and aINS (18/21). None of these regions overlapped with Phase 1 regions found for FTLD-tau. However, some Phase 1 regions in FTLD-TDP overlapped with Phase 2 regions found in FTLD-tau, including OFC and aINS. Brain regions meeting criteria for Phase 2 in the FTLD-TDP cohort also involved frontal and temporal regions, including IFC, MFC, and SFC in the frontal lobe, and FFG and ITC in the temporal lobe. Phase 2 regions in FTLD-TDP were adjacent to Phase 1 regions. Comparison of the tauopathy group and the TDP group revealed that Phase 2 regions in FTLD-TDP overlapped with some Phase 1 regions in FTLD-tau, including IFC, MFC, and SFC. Phase 3 atrophy extended further posteriorly into the BANKSSTS, PHG, and EC of the temporal lobe. Phase 3 regions also included frontal cortical regions in the frontal pole and precentral gyrus and extended into parietal cortex. Overlapping Phase 3 regions in tau and TDP-43 groups included the frontal pole. By Phase 4 and Phase 5, atrophy expanded in earlier regions and spread further posteriorly into the PoCG and PC. In Phase 4, areas of overlapping atrophy in FTLD-tau and FTLD-TDP included the PoCG, SPC, and TTG. The visual cortex emerged as atrophic for both FTLD-tau and FTLD-TDP groups in Phase 5.
3.5. FTLD-TDP subtype analysis
In the subtype analysis illustrated in Figure 3 and Supplementary Figure A.2, FTLD-TDP Types A and C were found to have overlapping Phase 1 brain regions in aINS, MTG and OFC, all regions found to be atrophic at Phase 1 for the FTLD-TDP group as a whole. In Phase 2, there was overlap of atrophy in Type A and Type C in IFC and MFC, which were also observed in the TDP group as a whole.
Fig. 3. Overlapping of phases across FI1D-TDP and subtypes.

Results from staging algorithm for FTLD-TDP group overall (first column): TDP-43 Type A (second column): TDP-43 Type C (third column): TDP-43 Type B (fourth column). Phase 1 (dark purple): putative epicenters of disease: Phase 2: light purple; Phase 3: magenta: Phase 4: orange: Pliase 5: pale orange. The remainder of brain regions (beige) had minimal atrophy and were considered to be below staging.
The DSC analysis of FTLD-TDP subtypes is summarized in Table 2. Pairwise comparisons of FTLD-TDP subtypes in this analysis showed high levels of overlapping cortical atrophy in early phases between Types A and C, but progressively decreasing levels of overlapping cortical atrophy with later phases of disease. Group B/E showed low overlap in the earlies phases some decreased overlap with subsequent phases.
3.6. Pathology
Figures 4 and 5 summarize the %AO pathology burden in all available tissue samples. Please see the Supplement (figures A.5-A.6) for the %AO pathology burden in the subset of these autopsy cases with imaging data in the above analyses; the results are virtually identical to those of the larger pathology cohort. The pattern of spread determined from the imaging findings generally showed an anterior to posterior directionality that follows the trend of pathological burden in the %AO analysis. For the FTLD-tau group, the IFC region, considered to be in Phase 1 and an early locus of the imaging analysis, showed the highest %AO pathology burden (Figure 4). We ran a linear mixed effect model (LME) to test for the effect of imaging phase on %AO, covarying for disease duration at autopsy. From an ANOVA performed on an LME model, we found an overall effect for imaging phase on %AO, FTLD-tau (F(4, 238)=17.44, p<0.001) and FTLD-TDP (F(4,245) = 42.32, p<0.001) across the targeted brain regions such that earlier imaging phases had greater %AO in the corresponding pathology regions compared to later imaging phases, where there was less %AO in the corresponding pathology regions. The LME model showed a significant effect of phase in the FTLD-tau group for the majority of phases from Phase 1 to Phase 2 (beta=−0.48 SE=0.19, p=0.01), to Phase 3 (beta=−0.41, SE=0.20, p=0.03), to Phase 4 (beta=−0.78, SE=0.23, p<0.001), to Phase 5 (beta=−1.41, SE=0.24, p<0.001). MFC, although found to be a Phase 1 region for two of three FTLD-tau subgroups, was grouped in this analysis with Phase 2 regions (OFC, ANG and SMTC) since only two out of the three subtypes found MFC to be a Phase 1 region when analyzed individually. These Phase 2 regions had less %AO than imaging Phase 1 regions. The aCING and ANG were considered to be Phase 3; they showed less %AO than two out of four of the Phase 2 regions. The SPC was considered to be Phase 4 of the imaging analysis. Region V1, considered to be in Phase 5 in the imaging analysis, had even lower %AO than the regions in earlier imaging phases. The superior parietal region was considered a Phase 4 imaging region but had a higher %AO than prior phases, possibly because although in FTLD-tau group SPL was phase 4, in the CBD subtype SPL was in Phase 1 region in CBD, in part reflecting the divergence that we see in later phases.
Fig. 4. Pathological burden comparison to MR! staging in FTLD-tau.

%AO of tau deposition for brain regions staged through the MRI staging algorithm. Colors represent brain regions. Neuroimaging phases (horizontal axis). Natural log (In) transformed %AO (vertical axis).
Fig. 5. Pathological burden comparison to MR1 staging in FTLD-1DP.

%AO of TDP-43 deposition for brain regions staged through the MRI staging algorithm. Colors represent brain regions. Neuroimaging pluses (horizontal axis). Natural log (In) transformed %AO (vertical axis).
For the FTLD-TDP group (Figure 5), LME analysis in these cases are as follows; Phase 1 to Phase 2 (beta=−0.03, SE=0.10, p=0.47), to Phase 3 (beta=−0.17, SE=0.09, p=0.75), to Phase 4 (beta=−21, SE=0.16, p=0.07), and to Phase 5 (beta=−1.02, SE=0.11, p<0.001). Imaging Phase 1 regions aINS and OFC had higher %AO relative to other imaging regions with the exception of aCING. SMTC was considered a Phase 1 imaging region but was an exception in showing an equivalent %AO pathologic burden to some Phase 2 regions. All Phase 1 and Phase 2 imaging regions had greater pathology burden than the SPC and the occipital cortex (imaging Phase 4), which had the lowest %AO.
4. Discussion
In this study, we analyzed antemortem neuroimaging in a manner paralleling the staging approach used in autopsy studies. We tested the hypotheses that FTLD-tau and FTLD-TDP have somewhat distinct early loci of disease, that subtypes share epicenters of early pathology within pathology groups, and that the anatomic distribution of atrophy becomes increasingly divergent at later phases of disease for pathology subtypes within FTLD-tau and FTLD-TDP. Consistent with these hypotheses, we found an early frontal locus in FTLD-tau and a frontal-temporal early locus in FTLD-TDP, and these were largely non-overlapping. Additional analyses revealed that all subtypes within FTLD-tau and both types A and C in FTLD-TDP had evidence of GM atrophy in a subset of the same early loci found for each proteinopathy group as a whole and that subtypes within FTLD-tau and FTLD-TDP increasingly differed in patterns of spread with presumed disease progression. We found converging evidence with the imaging findings from digital analysis of postmortem pathology samples, where Phase 1 imaging areas were found to have the densest pathology in each pathology group. We were able to identify patterns of disease progression in subsequent phases in this imaging study within the larger FTLD-tau and FTLD-TDP groups that largely corresponded to pathological burden quantified at autopsy in those regions. These results provide promising translational potential to differentiate between these groups for the benefit of early identification of specific molecular targets in therapeutic trials. We discuss each of our hypotheses in turn below for each pathology group.
FTLD syndromes present with clinically similar profiles despite having varied underlying FTLD proteinopathies. This underlying pathology in some individuals is due to misfolded tau but may often be from misfolded TDP-43 in others. Such varied cases of pathology type have been found in individuals diagnosed with bvFTD (Perry et al., 2017; Whitwell et al., 2009) and in forms of PPA, including semantic variant PPA (svPPA) and nonfluent/agrammatic PPA (nfvPPA) (Spinelli et al., 2017). It can also be the case that similar clinical phenotypes arise from different proteinopathy subtypes, such as CBD and PSP within the FTLD-tau group (Ling et al., 2010). The distinct early loci that we were able to identify between the FTLD-tau and FTLD-TDP groups provide a differential marker for use in future studies, as they distinguish FTLD-tau from FTLD-TDP independent of clinical phenotype. Likewise, the differences that we detected in later phases for subtypes within the larger FTLD-tau cohort provide discrimination power because these diverging patterns between CBD and PSP may help in future studies to disambiguate cases of CBS that are often misdiagnosed clinically between these two subtypes (Ling et al., 2010). An interesting finding in this study is that FTLD-tau and FTLD-TDP group subtypes diverge with increasing disease progression, given that typical clinical outcomes show an overlap in symptoms across clinical phenotypes. Our results may speak to the fact that there are likely individual differences, such as cell or brain regional vulnerabilities that contribute to the spread of misfolded proteins that occur over time. This aspect of FTLD is beyond the scope of this study but presents an interesting future direction.
4.1. Imaging phasing paradigm for FTLD-tau and FTLD-TDP
The notion of epicenters in neurodegenerative disease stems from the development of Braak staging of AD pathology (Braak and Braak, 1991). This pathbreaking research established that the characteristic accumulation of insoluble fibrous tau material in AD is not distributed at random, but in fact follows a characteristic pattern of early loci of disease across individuals. This concept of early loci has also been studied in FTLD, in which stereotypical patterns of spread have been found through postmortem pathological analysis (Brettschneider et al., 2014; David J. Irwin et al., 2016b). These postmortem studies suggest distinct epicenters for FTLD-tau and FTLD-TDP cohorts. Another view, however, suggests that there may be a shared epicenter for both FTLD-tau and FTLD-TDP that stems from vulnerable cell types such as fork cells and von Economo neurons in the aINS and the adjacent frontal cortex (Kim et al., 2012; Seeley et al., 2008).
The challenge in investigating these views through a single modality lies with the limitations of each method. Traditional postmortem histopathological staging is very time-consuming, requires extensive tissue sampling and is limited in interpretation without analysis of presymptomatic autopsies, which are exceedingly rare in FTLD (Miki et al., 2014; Sparks et al., 1994; Towfighi, 1972). Neuroimaging, by contrast, is efficient, feasible, and can be obtained during life but lacks postmortem validation. Our study provides evidence from whole-cerebrum, antemortem neuroimaging in patients with available postmortem neuropathology for confirmation of imaging findings. Our approach takes advantage of both modalities to minimize confounds associated with inferences made from each modality alone and has a high potential to lead to more specific treatment targets than currently available by providing a reliable prediction of atrophy pattern between FTLD groups.
4.2. Imaging in cases of FTLD-tau pathology
Through our frequency-based model of imaging phases, we found that the precentral gyrus, MFC, SFC, and IFC were Phase 1 regions for the entire FTLD-tau cohort. Other MRI imaging studies of autopsy-confirmed patients with FTLD-tau pathology have found changes in these cerebral regions, although without considering the impact of the disease progression. For example, a study of FTLD-tau subgroups found overlapping precentral, superior frontal, and inferior frontal disease to varying extents in PSP, CBD, and Pick’s disease (Whitwell and Josephs, 2012). A recent study of Pick’s disease demonstrated progressive MRI frontal disease in a longitudinal, autopsy-confirmed cohort (Whitwell et al., 2020). These findings are also in line with pathology studies that have examined the regional anatomic burden of FTLD-tau pathology, although sampling was limited to a narrow range of anatomic regions in both reports. One study of cases with known FTLD-tau pathology showed %AO in MFC to be higher than other regions in patients diagnosed with PPA (Giannini et al., 2019a). A study of pathological phases for 3R tau also found MFC, among other frontotemporal neocortical and limbic regions, to be a relatively early region in a PiD cohort (David J. Irwin et al., 2016b). While rare, these studies provide converging pathological evidence consistent with our imaging findings in a cohort with known pathology, namely, that specific frontal brain regions may serve as an epicenter for FTLD-tau pathology.
Further evidence for an epicenter of disease comes from our analysis of PSP, CBD, and PiD pathologic subtypes within the FTLD-tau cohort. Although these groups were relatively small, we saw that several areas found to be Phase 1 regions in the primary analysis of the entire cohort were also found to be Phase 1 regions when phases were determined independently for each subtype. The precentral gyrus, SFC, and IFC were Phase 1 regions in PSP, CBD and PiD; MFC was a Phase 1 region in two (CBD and PiD) of these three FTLD-tau subtypes. Thus, atrophy frequency in the FTLD-tau group as a whole was not driven by any one pathologic subtype. We consider IFC, SFC and precentral frontal regions to be the FTLD-tau epicenters because these appeared to qualify as Phase 1 regions in all three FTLD-tau subgroups. Focused analyses of pathologic phases in tau subtypes have implicated early tau deposition in frontal regions in PiD (Irwin et al., 2016a) and in PSP, the earliest stages were frontal, as were subcortical, neocortical motor, frontal, and parietal areas (Kovacs et al., 2020b) and frontal areas in CBD (Ling et al., 2020).
Additional converging evidence for an early locus of disease in specific frontal regions comes from our analysis of %AO within selected cortical regions relevant to FTLD. Although our analysis of pathologic burden is limited to a relatively small number of regions, our statistical model comparing imaging to %AO of pathological samples for the FTLD-tau group revealed that Phase 1 imaging regions had the greatest pathologic burden. These data support our general in vivo observations of MRI measured GM volume. These two modalities are on different anatomical scales, and the relationship between protein aggregation, neurodegeneration and in vivo measures of GM volume are not entirely clear. We and others have previously found an association between postmortem pathology severity and antemortem atrophy in FTLD (Giannini et al., 2019b; Irwin et al., 2018), but more detailed pathology-imaging correlative studies are needed in larger samples to further elucidate these likely complex relationships.
While we identified Phase 1 regions in each subtype of FTLD-tau that corresponded to the epicenter of the entire FTLD-tau group, an analysis of subsequent phases in FTLD-tau subtypes, following the initial epicenter phase, revealed divergent patterns of spreading pathology. For FTLD-tau, only 1–2 subtypes had overlapping brain regions with the overall group in Phases 2–3, and in Phases 4–5 there was no overlap at all. Quantification of Sørenson-Dice overlap revealed a downward trend in ratios from Phase 1 to Phase 5. These findings suggest that, although an epicenter common to all subtypes of FTLD-tau is apparent in the earliest phase of disease, the trajectory of each pathological subtype appears to diverge as disease advances. While this observation must be interpreted cautiously because of the small numbers of cases in each subtype, this finding of increasing diversity of atrophic regions across FTLD-tau subtypes is consistent with our DCS analysis. These results suggest that phenotypic variation seen in these subtypes may be driven in part by the relatively distinct spread of these subtly distinguishing pathologies. Although we did not include clinical phenotype in our subtype analyses, we do show the variation in clinical phenotypes that were diagnosed during life for all pathological subtypes (see Supplementary tables A3, A4, and A5). The clearest evidence for distinguishing patterns of spreading pathology in this study comes from the relatively elevated burden of parietal pathology in CBS, reflecting the higher likelihood of seeing disorders of visuospatial and numerosity functioning in these patients relative to patients with PiD and PSP. Murine models of spreading tau pathology suggest unique anatomic distributions of spreading disease in strains derived from different tauopathies (Narasimhan et al., 2017), and this may also be related in part to recent observations of distinct tertiary folding patterns in each of these tauopathies (Zhang et al., 2020). Comparative studies with larger numbers of cases are needed to confirm this pattern of a common frontal epicenter across all subtypes of FTLD-tau pathology and subsequent divergence of apparent pathologic spread depending on the specific subtype of FTLD-tau pathology.
In our quantitative analysis of digital pathology, we found that %AO pathology was lower in Phase 2 regions relative to Phase 1 regions, and %AO was even lower in regions corresponding to subsequent phases. One exception to this pattern was the relatively elevated pathologic burden in the superior parietal region, which was a Phase 4 imaging region in the overall FTLD-tau analysis. This divergence from the overall trend is consistent with the notion that subtypes are more dissociable in atrophy pattern as disease progresses and may reflect this pattern as CBD was found to be early (Phase 1) in our imaging sub-analysis despite the later phasing found for the group overall. Even though we examined a relatively small sample, the statistically robust association of regional pathologic burden with imaging phase provides strong evidence for the value of imaging phases in our understanding of pathologic progression across the entire cerebrum.
4.3. Imaging associated with FTLD-TDP pathology
In the FTLD-TDP group, we also observed epicenters of disease. The Phase 1 regions found in FTLD-TDP included aCING, OFC, aINS, MTG, and STG. Other imaging studies in autopsy-confirmed cases also observed atrophy in these cerebral regions, although without an indication of the phase of disease when this may have occurred (Whitwell and Josephs, 2012). These findings are also consistent with clinical-pathological research. For example, in one study of autopsy-confirmed PPA cases, %AO for FTLD-TDP deposition was observed in OFC, and was greater than the amount quantified in FTLD-tau patients (Giannini et al., 2019a). In another clinical-pathological study of bvFTD, FTLD-TDP deposition was found to be most severe in OFC and aCING (Irwin et al., 2018). These analyses of postmortem density of pathology align with traditional histopathological analyses suggesting that OFC is among the regions that are likely to serve as an early locus for pathological TDP-43(Brettschneider et al., 2014, 2013; Kril and Halliday, 2011). This study also found early pathology in the amygdala, a deep gray structure that we did not monitor in this study, and future work also should monitor imaging atrophy and pathology burden in deep gray and brain stem structures.
For FTLD-TDP subtypes, we saw overlap in Phase 1 regions across the pathologic subtypes (Type A and Type C). These two subtypes, when analyzed individually, showed overlap with phase 1 regions found in the full cohort overall in aINS, OFC, and MTC, and we believe that these are the epicenters of TDP-43 pathology because of their consistent early emergence in these subtypes of FTLD-TDP.
We found that the apparent epicenters of disease differ somewhat in FTLD-tau compared to FTLD-TDP. While FTLD-tau epicenter regions included IFC, SFC and precentral gyrus, FTLD-TDP epicenters include OFC, aINS and MTC. If confirmed, these findings would suggest an important set of regions on which to focus for studies investigating diagnosis, longitudinal change and treatment trials in FTLD spectrum disorders. However, these findings must be interpreted cautiously. While epicenter regions were observed consistently across subtypes A and C, less consistent overlap was seen with subtype group B/E in subtype analyses, which could result from smaller numbers in the FTLD-TDP subtype groups.
For the FTLD-TDP group, two epicenter regions – aINS and OFC – had greater %AO pathological burden than all other regions. A significant relationship between neuroimaging phase and %AO across all sampled brain regions was significant. A potential limitation of this analysis is that pathological sampling was limited mostly to Phase 1–2 regions from the imaging analysis. Thus, all available brain regions with %AO sampled were observed in neuroimaging Phase 1, Phase 2 or Phase 4. Another limitation is the small number of total cases in the FTLD-TDP cohort. With these caveats in mind, it appears that FTLD-TDP also has an epicenter of disease that can be identified as the earliest phase of imaging atrophy.
While there appears to be an epicenter in FTLD-TDP manifested as the earliest phase of imaging, subsequent phases of imaging in Type A and Type C show divergent patterns in later phases. Quantification of dice overlap revealed a downward trend of ratios as phases progressed from Phase 1 to Phase 5. The overall pattern of an early epicenter and subsequent divergence driven in part by underlying pathology thus also appears in FTLD-TDP, but this finding must be interpreted cautiously.
4.4. Study limitations
While this study provides an imaging approach to phases of disease progression in autopsy-confirmed cohorts of FTLD patients, several caveats should be kept in mind when considering our results. First, even though we studied a relatively large sample of a rare condition with both volumetric imaging and digitized histopathology, examination of subtypes led to assessments of relatively small samples. As in staging studies of pathology, our observations rest in part on assumptions regarding the relationship between imaging atrophy and the time course of disease; we assumed that noticeable focal imaging atrophy seen in a large number of participants is associated with earlier disease. We used the earliest timepoint available for our imaging phasing analysis to capture regions that are prominent early in disease and may be difficult to dissociate from later regions when atrophy becomes more widespread. We recognize that this approach leads to differences between groups in duration from imaging to neuropathological exam. We attempted to address this issue by using disease duration as a covariate in our analysis of pathological burden and accounted for age in our imaging analyses. However, confirmation with a longitudinal study would provide a more robust picture of disease spread over time. Another caveat to this study is that we focused our analyses on cortical regions and therefore our label set contained no subcortical regions. Also, our study only included analysis of GM. However, tau and TDP-43 deposition may also differentially affect WM and subcortical GM(Irwin et al., 2018). Future studies may benefit from the addition of WM and subcortical GM analyses. We also note that pathological samples are analyzed for density of protein deposition within a histopathological section (David J Irwin et al., 2016b). Future work will examine histopathological measures of neuronal loss, gliosis and other neurodegenerative features which may also relate to antemortem MRI atrophy as these methods are developed and validated for large scale studies. Another limitation of our study is in our pathology data set and the relative lack of bilateral sampling across hemispheres. We addressed this issue by averaging across hemispheres when bilateral sampling had been acquired. With regard to the association of imaging to pathology, while we did map histopathological regions and neuroimaging labels using a map derived from consensus among pathology and neuroimaging experts (Spotorno et al., 2020), neuroimaging ROIs and pathological samples do vastly differ in scale. It is notable that aINS and aCING are areas associated with von Economo neurons and fork cells (Kim et al, 2012), and thus we cannot rule out the possibility that these cells play a special role in the vulnerability of the FTLD-TDP cohort but would need more detailed microscopic analyses to make this determination. Finally, although the numbers of participants in the imaging cohorts were relatively small, we were able to supplement the imaging findings with a very large autopsy dataset (n=118) with validated digital measurements of pathological burden for postmortem pathological correlation to imaging phases. However, some caution should be employed when interpreting the FTLD-TDP results since the cohort was much smaller, there was less overlap between the subtypes and the relationship between the pathology and imaging data was less clear than in the FTLD-tau group.
5. Conclusions
For clinical trials to succeed in evaluating responses to treatment targets early in the course of a rare disease such as FTLD, the identification of epicenters of pathology is crucial. The concept of epicenter is also central to understanding pathophysiology and spread of FTLD spectrum disease. This study provided neuroimaging and pathology data to address these issues. We found apparent epicenters in both FTLD-tau and FTLD-TDP, but that the epicenters appear to be somewhat distinct in these two pathologies. Two sources of converging evidence provide support for these conclusions. First, we found the presence of similar epicenters across pathologically distinct PSP, CBD and PiD subtypes of FTLD-tau, and across subtypes of FTLD-TDP including Type A and Type C. Second, there was good correspondence between imaging regions contributing to the epicenter for each pathology and digitized pathology of tissue examinations in some overlapping regions. Further, while epicenters may be distinct between FTLD-tau and FTLD-TDP groups, we found that atrophy in pathologic subtypes tends to diverge as disease progresses.
Supplementary Material
Highlights.
FTLD-tau and FTLD-TDP groups have distinct early loci of cortical atrophy
In vivo biomarkers can distinguish early sites of atrophy between FTLD groups
Subtypes within FTLD groups have divergent atrophy patterns as disease progresses
Atrophy patterns from in vivo MRI correlate with postmortem pathology
Acknowledgements
This work was supported in part by grants from NIH (AG066597, AG054519, AG052943, NS109260, AG061277, AG054519)
We express our sincere appreciation to the patients and families who participated in this research.
Abbreviations
- %AO
percent area occupied
- 3R
three-repeat
- 4R
four-repeat
- aCING
anterior cingulate
- AD
Alzheimer’s disease
- ALS
amyotrophic lateral sclerosis
- ANG
angular gyrus
- aINS
anterior insula
- ANTs
Advanced Normalization Tools
- BANKSSTS
banks of the superior temporal sulcus
- bvFTD
behavioral variant frontotemporal dementia
- CBD
corticobasal degeneration
- CBS
corticobasal syndrome
- CSF
cerebrospinal fluid
- DSC
Dice similarity coefficient
- ECAS
Edinburgh Cognitive and Behavioral ALS Screen
- EC
entorhinal cortex
- FFG
fusiform gyrus
- FTD
frontotemporal dementia
- FTDC
Frontotemporal Degeneration Center
- GM
gray matter
- ICV
intracranial volume
- IFC
inferior frontal cortex
- INDDID
Integrated Neurodegenerative Disease Database
- ITC
inferior temporal cortex
- LME
linear mixed effects
- MFC
middle frontal cortex
- MTC
middle temporal cortex
- OFC
orbitofrontal cortex
- pCING
posterior cingulate
- PSP
progressive supranuclear palsy
- PoCG
the postcentral gyrus
- pHG
the parahippocampal gyrus
- PC
precuneus
- PPA
primary progressive aphasia
- ROI
region of interest
- SPC
superior parietal cortex
- SMTC
superior-middle temporal cortex
- SFC
superior frontal cortex
- STG
superior temporal gyrus
- SMTC
superior medial temporal cortex
- TTG
transverse temporal gyrus
- svPPA
semantic variant primary progressive aphasia
- nfvPPA
nonfluent/agrammatic primary progressive aphasia
- V1
primary visual cortex
- WM
white matter
Footnotes
Disclosure statement
The authors report no actual or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahams S, Newton J, Niven E, Foley J, Bak TH, 2014. Screening for cognition and behaviour changes in ALS. Amyotroph. Lateral Scler. Front. Degener. 15, 9–14. 10.3109/21678421.2013.805784 [DOI] [PubMed] [Google Scholar]
- Arnold SE, Toledo JB, Appleby DH, Xie SX, Wang LS, Baek Y, Wolk DA, Lee EB, Miller BL, Lee VMY, Trojanowski JQ, 2013. Comparative survey of the topographical distribution of signature molecular lesions in major neurodegenerative diseases. J. Comp. Neurol. 10.1002/cne.23430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants BB, Tustison NJ, Stauffer M, Song G, Wu B, Gee JC, 2014. The Insight ToolKit image registration framework. Front. Neuroinform. 8, 44. 10.3389/fninf.2014.00044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boluda S, Iba M, Zhang B, Raible KM, Lee VMY, Trojanowski JQ, 2015. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 129, 221–237. 10.1007/s00401-0141373-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E, 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- Brettschneider J, Del Tredici K, Irwin DJ, Grossman M, Robinson JL, Toledo JB, Fang L, Van Deerlin VM, Ludolph AC, Lee VMY, Braak H, Trojanowski JQ, 2014. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol. 127, 423–439. 10.1007/s00401-0131238-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Del Tredici K, Lee VMY, Trojanowski JQ, 2015a. Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 16, 109–120. 10.1038/nrn3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, Suh E, Van Deerlin VM, Wood EM, Baek Y, Kwong L, Lee EB, Elman L, McCluskey L, Fang L, Feldengut S, Ludolph AC, Lee VMY, Braak H, Trojanowski JQ, 2013. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 74, 20–38. 10.1002/ana.23937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Tredici K Del Lee, V.M.-Y., Trojanowski JQ, 2015b. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci. 16, 109–120. 10.1038/nrn3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Deng J, Neuhaus J, Sible IJ, Sias AC, Lee SE, Kornak J, Marx GA, Karydas AM, Spina S, Grinberg LT, Coppola G, Geschwind DH, Kramer JH, Gorno-Tempini ML, Miller BL, Rosen HJ, Seeley WW, 2019a. Patient-Tailored, Connectivity-Based Forecasts of Spreading Brain Atrophy. Neuron. 10.1016/J.NEURON.2019.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Deng J, Neuhaus J, Sible IJ, Sias AC, Lee SE, Kornak J, Marx GA, Karydas AM, Spina S, Grinberg LT, Coppola G, Geschwind DH, Kramer JH, Gorno-Tempini ML, Miller BL, Rosen HJ, Seeley WW, 2019b. Patient-Tailored, Connectivity-Based Forecasts of Spreading Brain Atrophy. Neuron 104, 856–868.e5. 10.1016/j.neuron.2019.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capozzo R, Sassi C, Hammer MB, Arcuti S, Zecca C, Barulli MR, Tortelli R, Gibbs JR, Crews C, Seripa D, Carnicella F, Dell’Aquila C, Rossi M, Tamma F, Valluzzi F, Brancasi B, Panza F, Singleton AB, Logroscino G, 2017. Clinical and Genetic Analyses of Familial and Sporadic Frontotemporal Dementia Patients in Southern Italy. Alzheimers. Dement. 13, 858. 10.1016/J.JALZ.2017.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dice LR, 1945. Measures of the Amount of Ecologic Association Between Species. Ecology 26, 297–302. 10.2307/1932409 [DOI] [Google Scholar]
- Forrest SL, Kril JJ, Halliday GM, 2019. Cellular and regional vulnerability in frontotemporal tauopathies. Acta Neuropathol. 10.1007/s00401-019-02035-7 [DOI] [PubMed] [Google Scholar]
- Frings L, Mader I, Landwehrmeyer BG, Weiller C, Hüll M, Huppertz H-J, 2012. Quantifying change in individual subjects affected by frontotemporal lobar degeneration using automated longitudinal MRI volumetry. Hum. Brain Mapp. 33, 1526–1535. 10.1002/hbm.21304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini LAA, Peterson C, Ohm D, Xie SX, McMillan CT, Raskovsky K, Massimo L, Suh ER, Van Deerlin VM, Wolk DA, Trojanowski JQ, Lee EB, Grossman M, Irwin DJ, 2021. Frontotemporal lobar degeneration proteinopathies have disparate microscopic patterns of white and grey matter pathology. Acta Neuropathol. Commun. 9, 30. 10.1186/s40478-021-01129-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini LAA, Xie SX, McMillan CT, Liang M, Williams A, Jester C, Rascovsky K, Wolk DA, Ash S, Lee EB, Trojanowski JQ, Grossman M, Irwin DJ, 2019a. Divergent patterns of TDP-43 and tau pathologies in primary progressive aphasia. Ann. Neurol. 85, 630–643. 10.1002/ana.25465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini LAA, Xie SX, McMillan CT, Liang M, Williams A, Jester C, Rascovsky K, Wolk DA, Ash S, Lee EB, Trojanowski JQ, Grossman M, Irwin DJ, 2019b. Divergent patterns of TDP-43 and tau pathologies in primary progressive aphasia. Ann. Neurol. 85, 630–643. 10.1002/ana.25465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini LAA, Xie SX, Peterson C, Zhou C, Lee EB, Wolk DA, Grossman M, Trojanowski JQ, McMillan CT, Irwin DJ, 2019c. Empiric methods to account for preanalytical variability in digital histopathology in frontotemporal lobar degeneration. Front. Neurosci. 13. 10.3389/fnins.2019.00682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman M, 2010. Primary progressive aphasia: Clinicopathological correlations. Nat. Rev. Neurol. 10.1038/nrneurol.2009.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Lee VMY, 2014. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 20, 130–138. 10.1038/nm.3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagmann P, Cammoun L, Gigandet X, Meuli R, Honey CJ, Wedeen VJ, Sporns O, 2008. Mapping the structural core of human cerebral cortex. PLoS Biol. 6, e159. 10.1371/journal.pbio.0060159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho DE, Imai K, King G, Stuart EA, 2011. MatchIt: Nonparametric preprocessing for parametric causal inference. J. Stat. Softw. 42, 1–28. 10.18637/jss.v042.i08 [DOI] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ, 2012. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers. Dement. 8, 1–13. 10.1016/j.jalz.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin David J., Brettschneider J, McMillan CT, Cooper F, Olm C, Arnold SE, Van Deerlin VM, Seeley WW, Miller BL, Lee EB, Lee VM-Y, Grossman M, Trojanowski JQ, 2016a. Deep clinical and neuropathological phenotyping of Pick disease. Ann. Neurol. 79, 272–287. 10.1002/ana.24559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin David J, Brettschneider J, McMillan CT, Cooper F, Olm C, Arnold SE, Van Deerlin VM, Seeley WW, Miller BL, Lee EB, Lee VM-Y, Grossman M, Trojanowski JQ, 2016a. Deep clinical and neuropathological phenotyping of Pick disease. Ann. Neurol. 79, 272–87. 10.1002/ana.24559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin David J., Brettschneider J, McMillan CT, Cooper F, Olm C, Arnold SE, Van Deerlin VM, Seeley WW, Miller BL, Lee EB, Lee VMY, Grossman M, Trojanowski JQ, 2016b. Deep clinical and neuropathological phenotyping of Pick disease. Ann. Neurol. 79, 272–287. 10.1002/ana.24559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin David J, Byrne MD, McMillan CT, Cooper F, Arnold SE, Lee EB, Van Deerlin VM, Xie SX, Lee VM-Y, Grossman M, Trojanowski JQ, 2016b. Semi-Automated Digital Image Analysis of Pick’s Disease and TDP-43 Proteinopathy. J. Histochem. Cytochem. 64, 54–66. 10.1369/0022155415614303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, Cairns NJ, Grossman M, McMillan CT, Lee EB, Van Deerlin VM, Lee VM-Y, Trojanowski JQ, 2015. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol. 129, 469–91. 10.1007/s00401-014-1380-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, McMillan CT, Xie SX, Rascovsky K, Van Deerlin VM, Coslett HB, Hamilton R, Aguirre GK, Lee EB, Lee VMY, Trojanowski JQ, Grossman M, 2018. Asymmetry of post-mortem neuropathology in behavioural-variant frontotemporal dementia. Brain 141, 288–301. 10.1093/brain/awx319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen JC, Warrington EK, Morris HR, Lantos P, Brown J, Revesz T, Wood N, Khan MN, Cipolotti L, Fox NC, Rossor MN, 2002. Clinical features of frontotemporal dementia due to the intronic tau 10+16 mutation. Neurology 58, 1161–1168. 10.1212/WNL.58.8.1161 [DOI] [PubMed] [Google Scholar]
- Jonkman LE, Kenkhuis B, Geurts JJG, van de Berg WDJ, 2019. Post-Mortem MRI and Histopathology in Neurologic Disease: A Translational Approach. Neurosci. Bull. 10.1007/s12264-019-00342-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Dickson DW, Boeve BF, Knopman DS, Petersen RC, Parisi JE, Jack CR, 2008. Voxel-based morphometry in autopsy proven PSP and CBD. Neurobiol. Aging 29, 280–289. 10.1016/j.neurobiolaging.2006.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassubek J, Müller HP, Del Tredici K, Brettschneider J, Pinkhardt EH, Lulé D, Böhm S, Braak H, Ludolph AC, 2014. Diffusion tensor imaging analysis of sequential spreading of disease in amyotrophic lateral sclerosis confirms patterns of TDP-43 pathology. Brain 137, 1733–1740. 10.1093/brain/awu090 [DOI] [PubMed] [Google Scholar]
- Kim EJ, Sidhu M, Gaus SE, Huang EJ, Hof PR, Miller BL, DeArmond SJ, Seeley WW, 2012. Selective frontoinsular von economo neuron and fork cell loss in early behavioral variant frontotemporal dementia. Cereb. Cortex 22, 251–259. 10.1093/cercor/bhr004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Lukic MJ, Irwin DJ, Arzberger T, Respondek G, Lee EB, Coughlin D, Giese A, Grossman M, Kurz C, McMillan CT, Gelpi E, Compta Y, van Swieten JC, Laat LD, Troakes C, Al-Sarraj S, Robinson JL, Roeber S, Xie SX, Lee VMY, Trojanowski JQ, Höglinger GU, 2020a. Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol. 140, 99–119. 10.1007/s00401-020-02158-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Lukic MJ, Irwin DJ, Arzberger T, Respondek G, Lee EB, Coughlin D, Giese A, Grossman M, Kurz C, McMillan CT, Gelpi E, Compta Y, van Swieten JC, Laat LD, Troakes C, Al-Sarraj S, Robinson JL, Roeber S, Xie SX, Lee VMY, Trojanowski JQ, Höglinger GU, 2020b. Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol. 140, 99–119. 10.1007/s00401-020-02158-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kril JJ, Halliday GM, 2011. Pathological staging of frontotemporal lobar degeneration, in: Journal of Molecular Neuroscience. Springer, pp. 379–383. 10.1007/s12031011-9528-0 [DOI] [PubMed] [Google Scholar]
- La Joie R, Perrotin A, Barré L, Hommet C, Mézenge F, Ibazizene M, Camus V, Abbas A, Landeau B, Guilloteau D, de La Sayette V, Eustache F, Desgranges B, Chételat G, 2012. Region-specific hierarchy between atrophy, hypometabolism, and β-amyloid (Aβ) load in Alzheimer’s disease dementia. J. Neurosci. 32, 16265–73. 10.1523/JNEUROSCI.2170-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird NM, Ware JH, 1982. Random-Effects Models for Longitudinal Data. Biometrics. 10.2307/2529876 [DOI] [PubMed] [Google Scholar]
- Lee EB, Porta S, Michael Baer G, Xu Y, Suh ER, Kwong LK, Elman L, Grossman M, Lee VMY, Irwin DJ, Van Deerlin VM, Trojanowski JQ, 2017. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 10.1007/s00401-017-1679-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H, Gelpi E, Davey K, Jaunmuktane Z, Mok KY, Jabbari E, Simone R, R’Bibo L, Brandner S, Ellis MJ, Attems J, Mann D, Halliday GM, Al-Sarraj S, Hedreen J, Ironside JW, Kovacs GG, Kovari E, Love S, Vonsattel JPG, Allinson KSJ, Hansen D, Bradshaw T, Setó-Salvia N, Wray S, de Silva R, Morris HR, Warner TT, Hardy J, Holton JL, Revesz T, 2020. Fulminant corticobasal degeneration: a distinct variant with predominant neuronal tau aggregates. Acta Neuropathol. 139, 717–734. 10.1007/s00401-019-02119-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H, O’Sullivan SS, Holton JL, Revesz T, Massey LA, Williams DR, Paviour DC, Lees AJ, 2010. Does corticobasal degeneration exist? A clinicopathological reevaluation. Brain 133, 2045–2057. 10.1093/brain/awq123 [DOI] [PubMed] [Google Scholar]
- Mackenzie IRA, Neumann M, 2016. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J. Neurochem. 10.1111/jnc.13588 [DOI] [PubMed] [Google Scholar]
- MacKenzie IRA, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJM, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DMA, 2010. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update, in: Acta Neuropathologica. Springer, pp. 1–4. 10.1007/s00401-009-0612-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercken M, Vandermeeren M, Lübke U, Six J, Boons J, Van de Voorde A, Martin JJ, Gheuens J, 1992. Monoclonal antibodies with selective specificity for Alzheimer Tau are directed against phosphatase-sensitive epitopes. Acta Neuropathol. 84, 265–272. 10.1007/BF00227819 [DOI] [PubMed] [Google Scholar]
- Miki Y, Mori F, Tanji K, Kurotaki H, Kakita A, Takahashi H, Wakabayashi K, 2014. An autopsy case of incipient Pick’s disease: Immunohistochemical profile of early-stage Pick body formation. Neuropathology 34, n/a-n/a. 10.1111/neup.12104 [DOI] [PubMed] [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT, 2012. National Institute on Aging– Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123, 1–11. 10.1007/s00401011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nana AL, Sidhu M, Gaus SE, Hwang JHL, Li L, Park Y, Kim EJ, Pasquini L, Allen IE, Rankin KP, Toller G, Kramer JH, Geschwind DH, Coppola G, Huang EJ, Grinberg LT, Miller BL, Seeley WW, 2019. Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol. 137, 27–46. 10.1007/s00401-018-1942-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV, He Z, Zhang B, Gathagan RJ, Trojanowski JQ, Lee VMY, 2017. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J. Neurosci. 37, 11406–11423. 10.1523/JNEUROSCI.1230-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y, Forman MS, Troost D, Kretzschmar HA, Trojanowski JQ, Lee VMY, 2009. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 117, 137–149. 10.1007/s00401-0080477-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Mackenzie IRA, 2019. Review: Neuropathology of non-tau frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol. 10.1111/nan.12526 [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VMY, 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (80-. ). 314, 130–133. 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- Perry DC, Brown JA, Possin KL, Datta S, Trujillo A, Radke A, Karydas A, Kornak J, Sias AC, Rabinovici GD, Gorno-Tempini ML, Boxer AL, De May M, Rankin KP, Sturm VE, Lee SE, Matthews BR, Kao AW, Vossel KA, Tartaglia MC, Miller ZA, Seo SW, Sidhu M, Gaus SE, Nana AL, Vargas JNS, Hwang JHL, Ossenkoppele R, Brown AB, Huang EJ, Coppola G, Rosen HJ, Geschwind D, Trojanowski JQ, Grinberg LT, Kramer JH, Miller BL, Seeley WW, 2017. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain 140, 3329–3345. 10.1093/brain/awx254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JS, Da Re F, Dratch L, Xie SX, Irwin DJ, McMillan CT, Vaishnavi SN, Ferrarese C, Lee EB, Shaw LM, Trojanowski JQ, Wolk DA, Grossman M, 2018. Neocortical origin and progression of gray matter atrophy in nonamnestic Alzheimer’s disease. Neurobiol. Aging 63, 75–87. 10.1016/j.neurobiolaging.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz CG, Gunter JL, Wiste HJ, Przybelski SA, Weigand SD, Ward CP, Senjem ML, Vemuri P, Murray ME, Dickson DW, Parisi JE, Kantarci K, Weiner MW, Petersen RC, Jack CR, 2016. A large-scale comparison of cortical thickness and volume methods for measuring Alzheimer’s disease severity. NeuroImage Clin. 11, 802–812. 10.1016/j.nicl.2016.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley WW, Crawford R, Rascovsky K, Kramer JH, Weiner M, Miller BL, Gorno-Tempini ML, 2008. Frontal paralimbic network atrophy in very mild behavioral variant frontotemporal dementia. Arch. Neurol. 65, 249–255. 10.1001/archneurol.2007.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks DL, Danner, fred W, Davis DG, Hackney C, , M.S., Landers T, Coyne CM, 1994. Neurochemical and Histopathologic Alterations Characteristic of Pickʼs Disease in a Non-demented Individual. J. Neuropathol. Exp. Neurol. 53, 37–42. 10.1097/00005072-199401000-00005 [DOI] [PubMed] [Google Scholar]
- Spinelli EG, Mandelli ML, Miller ZA, Santos-Santos MA, Wilson SM, Agosta F, Grinberg LT, Huang EJ, Trojanowski JQ, Meyer M, Henry ML, Comi G, Rabinovici G, Rosen HJ, Filippi M, Miller BL, Seeley WW, Gorno-Tempini ML, 2017. Typical and atypical pathology in primary progressive aphasia variants. Ann. Neurol. 81, 430–443. 10.1002/ana.24885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spotorno N, Coughlin DG, Olm CA, Wolk D, Vaishnavi SN, Shaw LM, Dahodwala N, Morley JF, Duda JE, Deik AF, Spindler MA, Chen-Plotkin A, Lee EB, Trojanowski JQ, McMillan CT, Weintraub D, Grossman M, Irwin DJ, 2020. Tau pathology associates with in vivo cortical thinning in Lewy body disorders. Ann. Clin. Transl. Neurol. 7, 2342–2355. 10.1002/acn3.51183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo JB, Van Deerlin VM, Lee EB, Suh E, Baek Y, Robinson JL, Xie SX, McBride J, Wood EM, Schuck T, Irwin DJ, Gross RG, Hurtig H, McCluskey L, Elman L, Karlawish J, Schellenberg G, Chen-Plotkin A, Wolk D, Grossman M, Arnold SE, Shaw LM, Lee VMY, Trojanowski JQ, 2014. A platform for discovery: The University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimer’s Dement. 10.1016/j.jalz.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towfighi J, 1972. Early Pick’s disease - A light and ultrastructural study. Acta Neuropathol. 21, 224–231. 10.1007/BF00688501 [DOI] [PubMed] [Google Scholar]
- Tustison NJ, Avants BB, Cook PA, Zheng Y, Egan A, Yushkevich PA, Gee JC, 2010. N4ITK: Improved N3 bias correction. IEEE Trans. Med. Imaging 29, 1310–1320. 10.1109/TMI.2010.2046908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tustison NJ, Cook PA, Klein A, Song G, Das SR, Duda JT, Kandel BM, van Strien N, Stone JR, Gee JC, Avants BB, 2014. Large-scale evaluation of ANTs and FreeSurfer cortical thickness measurements. Neuroimage. 10.1016/j.neuroimage.2014.05.044 [DOI] [PubMed] [Google Scholar]
- Vatsavayai SC, Nana AL, Yokoyama JS, Seeley WW, 2019. C9orf72-FTD/ALS pathogenesis: evidence from human neuropathological studies. Acta Neuropathol. 10.1007/s00401-018-1921-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Jack CR, Parisi JE, Knopman DS, Boeve BF, Petersen RC, Ferman TJ, Dickson DW, Josephs KA, 2007. Rates of cerebral atrophy differ in different degenerative pathologies. Brain. 10.1093/brain/awm021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Josephs KA, 2012. Neuroimaging in frontotemporal lobar degeneration - Predicting molecular pathology. Nat. Rev. Neurol. 10.1038/nrneurol.2012.7 [DOI] [PubMed] [Google Scholar]
- Whitwell JL, Przybelski SA, Weigand SD, Ivnik RJ, Vemuri P, Gunter JL, Senjem ML, Shiung MM, Boeve BF, Knopman DS, Parisi JE, Dickson DW, Petersen RC, Jack CR, Josephs KA, 2009. Distinct anatomical subtypes of the behavioural variant of frontotemporal dementia: A cluster analysis study. Brain 132, 2932–2946. 10.1093/brain/awp232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Tosakulwong N, Schwarz CC, Senjem ML, Spychalla AJ, Duffy JR, Graff-Radford J, Machulda MM, Boeve BF, Knopman DS, Petersen RC, Lowe VJ, Jack CR, Dickson DW, Parisi JE, Josephs KA, 2020. Longitudinal anatomic, functional, and molecular characterization of Pick disease phenotypes. Neurology 95, e3190–e3202. 10.1212/WNL.0000000000010948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood EM, Falcone D, Suh ER, Irwin DJ, Chen-Plotkin AS, Lee EB, Xie SX, Van Deerlin VM, Grossman M, 2013. Development and validation of pedigree classification criteria for frontotemporal lobar degeneration. JAMA Neurol. 70, 1411–1417. 10.1001/jamaneurol.2013.3956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yushkevich PA, Wang H, Pluta J, Das SR, Craige C, Avants BB, Weiner MW, Mueller S, 2010. Nearly automatic segmentation of hippocampal subfields in in vivo focal T2-weighted MRI. Neuroimage 53, 1208–1224. 10.1016/j.neuroimage.2010.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Tarutani A, Newell KL, Murzin AG, Matsubara T, Falcon B, Vidal R Garringer HJ, Shi Y, Ikeuchi T, Murayama S, Ghetti B, Hasegawa M, Goedert M, Scheres SHW, 2020. Novel tau filament fold in corticobasal degeneration. Nature 580, 283–287. 10.1038/s41586-020-2043-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.