

Abstract
Amines containing bridged bicyclic carbon skeletons are desirable building blocks for medicinal chemistry. Herein, we report the conversion of bicyclo[1.1.1]pentan-1-amines to a wide range of poly-substituted bicyclo[3.1.1]heptan-1-amines through a photochemical, formal (4+2)-cycloaddition of an intermediate imine diradical. To our knowledge, this is the first reported method to convert the bicyclo[1.1.1]pentane skeleton to the bicyclo[3.1.1]heptane skeleton. Hydrolysis of the imine products gives complex, sp3-rich primary amine building blocks.
Graphical Abstract
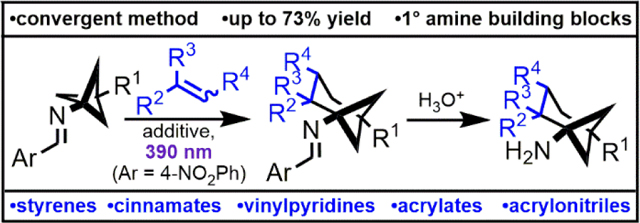
The fraction of carbon atoms that are sp3-hybridized in a drug candidate is positively correlated with the molecule’s advancement through development and clinical trials.1 Growing recognition of this trend among medicinal chemists has amplified interest in building blocks containing bridged polycyclic skeletons. These substructures, like aromatic rings, can provide a conformationally restricted framework upon which to append substituents, but often retain the pharmacological benefits of being aliphatic.2 All—carbon ring systems such as the bicyclo[1.1.1]pentane system are of particular interest due to their inert nature and potential phenyl bioisosterism.3 However, despite recent innovations in the synthesis of 2-substituted bicyclo[1.1.1]pentanes (Qin, Pfizer, Merck, and academic collaborators) and (oxa)bicyclo[2.1.1]hexanes (Enamine), most commercial compounds in this class are mono- or “para”-disubstituted.4–9 Furthermore, commercial Csp3-rich polycyclic building blocks are often achiral, despite stereochemical complexity’s negative correlation with promiscuity and positive correlation with drug candidate success.10 The invention of new synthetic methods to access poly-substituted bicyclic cores with uniquely disposed exit vectors will allow for a more complete biological evaluation of low molecular weight, sp3-rich chemical space.11
To this end, our group has developed several methods to synthesize 1-aminonorbornanes.12,13 In 2019, we reported the photochemical intramolecular formal (3+2)-cycloaddition of cyclopropyl imines bearing a pendant alkene to generate (hetero)aryl-fused 1-aminonorbornanes (Figure 1A).12 Mechanistically, irradiating the imine with 390 nm lamps generates an excited state with N-centered radical character, facilitating the homolytic cleavage of a bond in the adjacent cyclopropane ring, which in turn triggers an intramolecular radical cyclization cascade. Our method intercepts reactive intermediates previously described in Sampedro’s reports of the photoinduced rearrangement of cyclopropylimines to 1-pyrrolines (Figure 1B).14–16 Based on ultraviolet-visible spectroscopy, we have proposed the reaction proceeds via the imine S1(n, 𝜋*) excited state.12 Our work was among the first use of the N-centered radical character of an imine excited state to initiate a radical cyclization cascade. This is a testament to the relatively underexplored nature of the photochemistry of imines compared to that of carbonyls and alkenes.17,18
Figure 1.

Precedent for (3+2)-cycloaddition reactivity and bicyclo[1.1.1]pentane ring opening informed our reactivity hypothesis
Inspired by the pioneering work of Zheng and related photoredox approaches to access N-centered radicals for formal (3+2)- and (4+2)- cycloadditions of strained-ring amines, our group has recently extended our imine-based strategy to the intermolecular context (Figure 1C).19–29 Though cyclopropane ring-opening was consistently observed, we have detected only trace amounts of cyclobutane ring-opening products under these conditions, despite both species containing ostensibly identical chromophores. This difference in reactivity appears consistent with the relative ring-opening kinetics of cyclopropylaminyl radicals and cyclobutylaminyl radicals.30
We reasoned that cyclobutane-containing ring systems with abnormally rapid radical ring-opening kinetics might be more amenable to our reaction design. We were encouraged by the known rate constants for the sequential ring openings of 1r, which suggested intermediates related to 1s could be both rapidly generated and long-lived enough to engage in intermolecular reactivity (Figure 1D).31,32 Specifically, we hypothesized bicyclo[1.1.1]pentan-1-amine 1v may undergo the desired ring cleavage, generating diradical intermediate 1×, the primary radical moiety of which could add to an alkene. Subsequent cyclization would afford a compound containing a relatively inert cyclobutylamine, namely racemic (or achiral) bicyclo[3.1.1]heptan-1-amine 1z (Figure 1E). Alternatively, the diradical alkene adduct might undergo a 1,5-hydrogen atom transfer (HAT) to produce racemic cyclobutene 1aa. If alkene capture of 1× did not occur, 1ab would be expected.
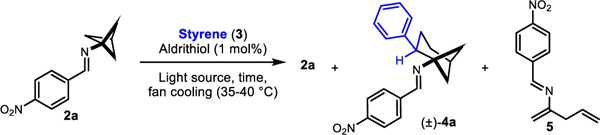
In our initial attempt to realize this reactivity, we drew conditions from our intermolecular formal (3+2)-cycloaddition work.19 Gratifyingly, irradiating 2a, styrene (3), and 2,2’-dipyridyl disulfide (Aldrithiol™) in ethyl acetate with 390 nm lamps afforded the desired product 4a (Table 1, Entry 1). However, 4a was the minor product of the reaction, and skipped diene 5, which results from unimolecular background reaction, was the major product. Changing the solvent from ethyl acetate to acetonitrile had little effect on the reaction profile (Table 1, Entry 2). Increasing the concentration led to a significant improvement in yield and selectivity, (Table 1, Entries 3 and 4). Running the reaction in neat styrene further favored the desired intermolecular reactivity over the intramolecular pathway (Table 1, Entries 5 and 6). All other light sources investigated gave inferior conversion and yield of 4a at 3 hours when compared to the 390 nm lamp, though lamps with emission maxima spanning from 370 nm to 456 nm showed useful levels of reactivity (Table 1, Entries 7–11). A thermal control showed no background reactivity (Table 1, Entry 12), supporting our assertion that the formal cycloaddition is indeed a photochemical process. Degassing (3 freeze-pump-thaw cycles) and inclusion of Aldrithiol™ only modestly improved the reaction profile at 3 hours, encouraging features for operational simplicity and future scaling of the reaction (Table 1, Entries 13 and 14). However, at the prolonged reaction times necessary to achieve full conversion when using 7.5 equivalents of alkene (~16 hours), excluding Aldrithiol™ or including air had a small but noticeable deleterious effect (Table 1, Entries 15–17). The cyclobutene of type 1aa was not identified as a significant contributor to the crude reaction mixtures.
Table 1.
Reaction optimization
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | time (h) | 3 (eq) | Lamp (nm) | Solvent | 2ah % | 4ah % | 5h % |
| 1a,f | 3 | 7.5 | 390 | EtOAc | 6 | 15 | 59 |
| 2 a,f | 3 | 7.5 | 390 | CH3CN | 5 | 16 | 57 |
| 3 b,f | 3 | 7.5 | 390 | EtOAc | 7 | 29 | 40 |
| 4 a,f | 3 | 7.5 | 390 | CH3CN | 6 | 26 | 37 |
| 5 g | 3 | 7.5 | 390 | neat | 18 | 38 | 23 |
| 6 f | 3 | 15 | 390 | neat | 6 | 47 | 28 |
| 7 g | 3 | 7.5 | 370 | neat | 44 | 26 | 19 |
| 8 g | 3 | 7.5 | 427 | neat | 40 | 30 | 17 |
| 9 g | 3 | 7.5 | 440 | neat | 58 | 21 | 11 |
| 10 g | 3 | 7.5 | 456 | neat | 76 | 12 | 7 |
| 11 g | 3 | 7.5 | 525 | neat | 98 | 0 | 0 |
| 12 e,g | 3 | 7.5 | -- | neat | 99 | 0 | 0 |
| 13 c,g | 3 | 7.5 | 390 | neat | 20 | 33 | 20 |
| 14 d,g | 3 | 7.5 | 390 | neat | 22 | 37 | 22 |
| 15 c,g | 16 | 7.5 | 390 | neat | -- | 36 | 8 |
| 16 d,g | 16 | 7.5 | 390 | neat | -- | 36 | 9 |
| 17 g | 16 | 7.5 | 390 | neat | -- | 48 | 9 |
0.65 mL.
0.172 mL.
no Aldrithiol.
no freeze-pump-thaw.
60 °C and shielded from light.
0.2 mmol imine.
0.4 mmol imine.
yield based on Q1HNMR analysis with 1,3,5-trimethoxybenzene internal standard
5 decomposes under the reaction conditions upon prolonged irradiation, forming solid deposits in the reaction vessel. Polymeric styrene-derived material (presumably polystyrene) also forms under such conditions. Though these solids reduce the photon flux entering the reaction solution, thereby slowing conversion, they are easily removed by chromatography and can simplify otherwise challenging chromatographic separations of the desired bicyclo[3.1.1]heptane-containing products from 5. To reduce alkene waste and simplify purification, we adopted the ~16-hour reaction time with 7.5 equivalents of neat alkene as the standard conditions for our scope study. Clearly, solid alkenes are incompatible with these conditions and necessitate the inclusion of an inert solvent. Furthermore, alkene classes differ in their rates of radical trapping, leading to drastically different levels of by-product formation, changing the calculus for optimal concentration and alkene stoichiometry. Since one set of conditions could not fully capture the potential of the reaction, several modifications were examined (vide infra).
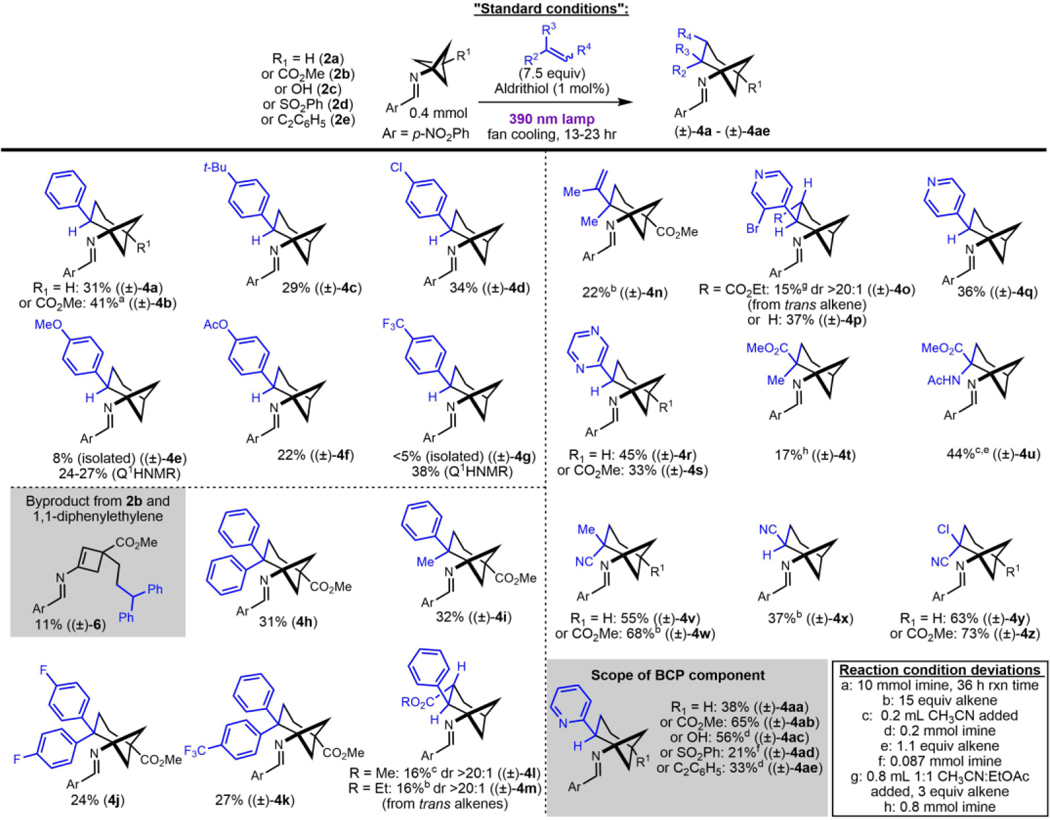
With suitable conditions in hand, the reaction scope of alkenes was studied broadly (Table 2). Gratifyingly, electron-rich and electron-poor styrenes showed the desired reactivity, albeit with modest yields. Reaction of 2b with styrene gave product 4b in 41% yield on 10 mmol scale (1.56 g product). 1,1-Diarylethylenes underwent the desired ring closure to form sterically hindered bicyclo[3.1.1]heptan-1-amines, but also produced a significant amount of cyclobutene byproducts. For instance, cyclobutene 6 was isolated in 11% yield alongside a 31% yield of 4h. ɑ-Methyl styrene was well-tolerated, giving 4i in 32% yield. Though relatively unreactive, methyl cinnamate and ethyl cinnamate provided 4l and 4m, respectively, with high diastereoselectivity. As with other styrenes, cinnamates proceed with regioselectivity that suggests the intermediacy of a benzylic radical. Since a thermodynamic control argument invoking the reversibility of alkene addition would be inconsistent the competitive formation of 4l and 4m versus 5, we attribute the regioselectivity to kinetic control.
Table 2.
Reaction scope study
|
Alkene classes besides styrenes also undergo this chemistry. Vinyl-substituted heteroaromatics gave modest to moderate yields of the desired products (Table 2, examples 4o-4s and 4aa-4ae). Unsaturated carboxylic acid derivatives also participated in this reaction. Reaction of 2b with 2-chloroacrylonitrile gave 4z in 73% yield. The formation of skipped diene byproducts was completely suppressed by 2-chloroacrylonitrile based on crude NMR analysis, suggesting some alkenes may be sufficiently reactive to compete with the unimolecular background reaction in the solution phase and/or at decreased loadings. Indeed, in acetonitrile solution, the solid alkene 2-acetamidoacrylate showed comparable performance using the standard 7.5 equivalents versus 1.1 equivalents of alkene (Table 2, example 4u).33
The reaction also shows good functional group tolerance with respect to “para”- substitution of the bicyclo[1.1.1]pentane component. The easily accessed 2b was compared to 2a across a variety of alkenes and generally showed comparable isolated yields (Table 2). A broader comparison was performed when employing 2-vinylpyridine as the alkene (Table 2, examples 4aa-4ae). The free hydroxyl group of 2c did not interfere in the reaction. The benzene-sulfonyl group of 2d, though a potential leaving group via homolysis of the C-S bond prior intermolecular reactivity, was tolerated in the reaction to give 4ad. Alkyne functionality was likewise tolerated to give 4ae.
We next turned our attention to synthetic manipulations of the formal cycloadducts, focusing on reactions most relevant to medicinal and agrochemical applications. Primary amines 7a, 7e, and 7f were each produced on multi-millimole scale by hydrolysis of their corresponding crude cycloaddition reaction mixtures (Figure 2A). Acid-base extraction operations purged excess alkene and the carbonyl-containing hydrolysis byproducts, affording crude bicyclo[3.1.1]pentan-1-amines, often in good purity. This was particularly attractive as the chromatographic separation of byproducts of type 1aa and 1ab, as well as trace 4-nitrobenzaldehyde (resulting from imine hydrolysis by adventitious water), from several of the bicyclo[3.1.1]heptane-containing imines, was challenging. For example, amines 7b, 7c, and 7d, were easily isolated in yields approaching the Q1HNMR assay yields of the corresponding imines 4e, 4g, and 4a, respectively, which had presented purification challenges.
Figure 2.

Application of the formal (4+2)-cycloaddition chemistry to the synthesis of analogues of biologically-relevant molecules. Full experimental details can be found in the supporting information.
Inspired by previous work from our laboratory, primary amines prepared in this manner were used to synthesize analogues of the succinate dehydrogenase inhibitor (SDHI) fungicide Boscalid, wherein Boscalid’s ortho-aryl aniline core was replaced with bicyclo[3.1.1]heptan-1-amines (Figure 2A).34,35 A library mimicking related pyrazole carboxamide SDHI fungicides was similarly prepared via amine-acid coupling mediated by EDC•HCl.36 Sulfonylation of the primary amines was also simple and high-yielding. For instance, 7d was converted in 97% yield to sulfonamide 10, which is an analogue of 11, an anti-mitotic compound reported to decrease viability and inhibit growth in several ovarian cancer cell lines (Figure 2B).37
Next, imine 4b was elaborated to several protected ɣ-amino acid building blocks (Figure 2C). First, 4b was hydrolyzed to give 7g, which was advanced over two steps to 12. 7g was similarly converted to 14. Complementarily, 2a was carried through formal cycloaddition and hydrolysis in a telescoped fashion to yield 7h, with comparable yields observed when employing batch or flow processing during the photochemical step.38 7h was converted to carbamate 15 and saponified in a telescoped fashion to yield 16, a fellow ɣ-amino acid isomer of 14.
We next sought to synthesize ring systems with additional complexity using the formal (4+2)-cycloaddition described herein as a key step. To this end, isolated imine 4p was hydrolyzed to give 7i, which upon copper-catalyzed intramolecular C-N coupling gave 17, a sp3-rich analogue of β-carboline (norharmane, 18), the heterocyclic core of its namesake family of pharmacologically interesting alkaloids (Figure 2D).39,40 Lastly, we engaged 2a in formal cycloaddition with 19 to give the sterically congested polycycle 4af in 11% yield (Figure 2E). 4af contains features representing two distinct medicinal chemistry strategies for imparting rigidity on sp3-rich structures: bridged bicyclic (2) and spiro (1) motifs.41
In summary, we have developed a method of converting bicyclo[1.1.1]pentan-1-amines to bicyclo[3.1.1]heptan-1-amines using an imine-based, photochemical formal (4+2)-cycloaddition. The key kinetic discrepancy exploited in this study, namely the enhanced susceptibility of the bicyclo[1.1.1]pentane system to radical ring-opening relative to simple cyclobutanes (and relative to bicyclo[3.1.1]heptanes), suggests a rich body of bicyclo[1.1.1]pentane formal cycloaddition chemistry yet to be developed. Further results will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge the financial support for this research from the NIH NIGMS (R01-GM127774) and the University of Michigan. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. (DGE 1841052) (for A.S.H.). M.J.S. was supported by the Australian Governments Endeavour Leadership Fellowship. The authors thank James L. Collins III (University of Michigan) for helpful discussions and his assistance in editing this manuscript. The authors thank Dr. Daryl Staveness (Exciplex, Inc.) and Dr. Michael Harmata (University of Missouri – Columbia) for helpful discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information.
Additional experimental details, materials, and methods, including photographs of experimental setup
This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. Journal of Medicinal Chemistry 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- (2).Locke GM; Bernhard SSR; Senge MO Nonconjugated Hydrocarbons as Rigid‐Linear Motifs: Isosteres for Material Sciences and Bioorganic and Medicinal Chemistry. Chemistry – A European Journal 2019, 25, 4590–4647. [DOI] [PubMed] [Google Scholar]
- (3).Levin MD; Kaszynski P; Michl J Bicyclo[1.1.1]pentanes, [n]Staffanes, [1.1.1]Propellanes, and Tricyclo[2.1.0.02,5]pentanes. Chemical Reviews 2000, 100, 169–234. [DOI] [PubMed] [Google Scholar]
- (4).Mykhailiuk PK Saturated bioisosteres of benzene: where to go next? Organic & Biomolecular Chemistry 2019, 17, 2839–2849. [DOI] [PubMed] [Google Scholar]
- (5).Levterov VV; Panasyuk Y; Pivnytska VO; Mykhailiuk PK Water‐Soluble Non‐Classical Benzene Mimetics. Angewandte Chemie International Edition 2020, 59, 7161–7167. [DOI] [PubMed] [Google Scholar]
- (6).Ma X; Han Y; Bennett DJ Selective Synthesis of 1-Dialkylamino-2-alkylbicyclo-[1.1.1]pentanes. Organic Letters 2020, 22, 9133–9138. [DOI] [PubMed] [Google Scholar]
- (7).Anderson JM; Measom ND; Murphy JA; Poole DL Bridge Functionalisation of Bicyclo[1.1.1]pentane Derivatives. Angewandte Chemie International Edition 2021, 60, 24754–24769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yang Y; Tsien J; Hughes JME; Peters BK; Merchant RR; Qin T An intramolecular coupling approach to alkyl bioisosteres for the synthesis of multisubstituted bicycloalkyl boronates. Nature Chemistry 2021, 13, 950–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhao J-X; Chang Y-X; He C; Burke BJ; Collins MR; Del Bel M; Elleraas J; Gallego GM; Montgomery TP; Mousseau JJ; Nair SK; Perry MA; Spangler JE; Vantourout JC; Baran PS 1,2-Difunctionalized bicyclo[1.1.1]pentanes: Long–sought-after mimetics for ortho/meta-substituted arenes. Proceedings of the National Academy of Sciences 2021, 118, e2108881118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lovering F Escape from Flatland 2: complexity and promiscuity. MedChemComm 2013, 4, 515. [Google Scholar]
- (11).Tajabadi FM; Campitelli MR; Quinn RJ Scaffold Flatness: Reversing the Trend. Springer Science Reviews 2013, 1, 141–151. [Google Scholar]
- (12).Staveness D; Collins JL; McAtee RC; Stephenson CRJ Exploiting Imine Photochemistry for Masked N‐Centered Radical Reactivity. Angewandte Chemie International Edition 2019, 58, 19000–19006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Staveness D; Sodano TM; Li K; Burnham EA; Jackson KD; Stephenson CRJ Providing a New Aniline Bioisostere through the Photochemical Production of 1-Aminonorbornanes. Chem 2019, 5, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Sampedro D; Soldevilla A; Rodríguez MA; Campos PJ; Olivucci M Mechanism of the N-Cyclopropylimine-1-pyrroline Photorearrangement. Journal of the American Chemical Society 2005, 127, 441–448. [DOI] [PubMed] [Google Scholar]
- (15).Soldevilla A; Sampedro D; Campos PJ; Rodríguez MA The N-Cyclopropylimine-1-pyrroline Photorearrangement as a Synthetic Tool: Scope and Limitations. The Journal of Organic Chemistry 2005, 70, 6976–6979. [DOI] [PubMed] [Google Scholar]
- (16).Campos PJ; Soldevilla A; Sampedro D; Rodríguez MA N-Cyclopropylimine-1-pyrroline Rearrangement. A Novel Photochemical Reaction. Organic Letters 2001, 3, 4087–4089. [DOI] [PubMed] [Google Scholar]
- (17).Kandappa SK; Valloli LK; Ahuja S; Parthiban J; Sivaguru J Taming the excited state reactivity of imines - from non-radiative decay to aza Paterno-Buchi reaction. Chem Soc Rev 2021, 50, 1617–1641. [DOI] [PubMed] [Google Scholar]
- (18).Latrache M; Hoffmann N Photochemical radical cyclization reactions with imines, hydrazones, oximes and related compounds. Chemical Society Reviews 2021, 50, 7418–7435. [DOI] [PubMed] [Google Scholar]
- (19).Sowden MJ; Collins JL III; Staveness D; Stephenson CRJ A One Pot Photochemical Method for the Generation of Functionalized Aminocyclopentanes. ChemRxiv 2020, DOI: 10.26434/chemrxiv.13079159.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Maity S; Zhu M; Shinabery RS; Zheng N Intermolecular [3+2] Cycloaddition of Cyclopropylamines with Olefins by Visible-Light Photocatalysis. Angewandte Chemie International Edition 2012, 51, 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nguyen TH; Morris SA; Zheng N Intermolecular [3+2] Annulation of Cyclopropylanilines with Alkynes, Enynes, and DiynesviaVisible Light Photocatalysis. Advanced Synthesis & Catalysis 2014, 356, 2831–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wang J; Zheng N The Cleavage of a C-C Bond in Cyclobutylanilines by Visible-Light Photoredox Catalysis: Development of a [4+2] Annulation Method. Angewandte Chemie International Edition 2015, 54, 11424–11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wang J; Mao C; Feng P; Zheng N Visible‐Light‐Mediated [4+2] Annulation of N‐Cyclobutylanilines with Alkynes Catalyzed by Self‐Doped Ti3+ @TiO2. Chemistry – A European Journal 2017, 23, 15396–15403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wang Q; Zheng N A Photocatalyzed Synthesis of Naphthalenes by Using Aniline as a Traceless Directing Group in [4 + 2] Annulation of Amino-benzocyclobutenes with Alkynes. ACS Catalysis 2017, 7, 4197–4201. [Google Scholar]
- (25).White DH; Noble A; Booker-Milburn KI; Aggarwal VK Diastereoselective Photoredox-Catalyzed [3 + 2] Cycloadditions of N-Sulfonyl Cyclopropylamines with Electron-Deficient Olefins. Organic Letters 2021, 23, 3038–3042. [DOI] [PubMed] [Google Scholar]
- (26).Uraguchi D; Kimura Y; Ueoka F; Ooi T Urea as a Redox-Active Directing Group under Asymmetric Photocatalysis of Iridium-Chiral Borate Ion Pairs. Journal of the American Chemical Society 2020, 142, 19462–19467. [DOI] [PubMed] [Google Scholar]
- (27).Yin Y; Li Y; Gonçalves TP; Zhan Q; Wang G; Zhao X; Qiao B; Huang K-W; Jiang Z All-Carbon Quaternary Stereocenters α to Azaarenes via Radical-Based Asymmetric Olefin Difunctionalization. Journal of the American Chemical Society 2020, 142, 19451–19456. [DOI] [PubMed] [Google Scholar]
- (28).Muriel B; Gagnebin A; Waser J Synthesis of bicyclo[3.1.0]hexanes by (3 + 2) annulation of cyclopropenes with aminocyclopropanes. Chemical Science 2019, 10, 10716–10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).De Nanteuil F; De Simone F; Frei R; Benfatti F; Serrano E; Waser J Cyclization and annulation reactions of nitrogen-substituted cyclopropanes and cyclobutanes. Chem. Commun. 2014, 50, 10912–10928. [DOI] [PubMed] [Google Scholar]
- (30).Musa OM; Horner JH; Shahin H; Newcomb M A Kinetic Scale for Dialkylaminyl Radical Reactions. Journal of the American Chemical Society 1996, 118, 3862–3868. [Google Scholar]
- (31).Branchaud BP; Glenn AG; Stiasny HC Round-trip radical probes: ring cleavage of the bicyclo[1.1.1]pentylcarbinyl radical. The Journal of Organic Chemistry 1991, 56, 6656–6659. [Google Scholar]
- (32).Walton JC 3-Methylenecyclobutyl, cyclopent-3-enyl, and 3-methylenecyclobutylmethyl radicals; absence of homoallylic conjugation. Journal of the Chemical Society, Perkin Transactions 2 1987, 231. [Google Scholar]
- (33). [See supporting information for a more complete comparision of reaction conditions for the synthesis of 4u]
- (34).Sierotzki H; Scalliet G A Review of Current Knowledge of Resistance Aspects for the Next-Generation Succinate Dehydrogenase Inhibitor Fungicides. Phytopathology® 2013, 103, 880–887. [DOI] [PubMed] [Google Scholar]
- (35).Staveness D; Breunig M; Ortiz V; Sang H; Collins JL; McAtee RC; Chilvers MI; Stephenson CRJ Photochemically derived 1-aminonorbornanes provide structurally unique succinate dehydrogenase inhibitors with in vitro and in planta activity. Cell Reports Physical Science 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36). [See supporting information]
- (37).Huang H-L; Chao M-W; Li Y-C; Chang L-H; Chen C-H; Chen M-C; Cheng C-C; Liou J-P; Teng C-M; Pan S-L MPT0G066, a novel anti-mitotic drug, induces JNK-independent mitotic arrest, JNK-mediated apoptosis and potentiates antineoplastic effect of cisplatin in ovarian cancer. Scientific Reports 2016, 6, 31664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38). [See supporting information for more complete comparison of batch and flow protocols]
- (39).Li S; Cheng X; Wang C A review on traditional uses, phytochemistry, pharmacology, pharmacokinetics and toxicology of the genus Peganum. Journal of Ethnopharmacology 2017, 203, 127–162. [DOI] [PubMed] [Google Scholar]
- (40).Zhang X; Anderson JC A Divergent Synthetic Route to the Vallesamidine and Schizozygine Alkaloids: Total Synthesis of (+)‐Vallesamidine and (+)‐14,15‐Dehydrostrempeliopine. Angewandte Chemie International Edition 2019, 58, 18040–18045. [DOI] [PubMed] [Google Scholar]
- (41).Hiesinger K; Dar’In D; Proschak E; Krasavin M Spirocyclic Scaffolds in Medicinal Chemistry. Journal of Medicinal Chemistry 2021, 64, 150–183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.