

Summary
Soybean oil is one of the most consumed vegetable oils worldwide. Genetic improvement of its concentration in seeds has been historically pursued due to its direct association with its market value. Engineering attempts aiming to increase soybean seed oil presented different degrees of success that varied with the genetic design and the specific variety considered. Understanding the embryo’s responses to the genetic modifications introduced, is a critical step to successful approaches. In this work, the metabolic and transcriptional responses to AtWRI1 and AtDGAT1 expression in soybean seeds were evaluated. AtWRI1 is a master regulator of fatty acid (FA) biosynthesis, and AtDGAT1 encodes an enzyme catalysing the final and rate‐limiting step of triacylglycerides biosynthesis. The events expressing these genes in the embryo did not show an increase in total FA content, but they responded with changes in the oil and carbohydrate composition. Transcriptomic studies revealed a down‐regulation of genes putatively encoding for oil body packaging proteins, and a strong induction of genes annotated as lipases and FA biosynthesis inhibitors. Novel putative AtWRI1 targets, presenting an AW‐box in the upstream region of the genes, were identified by comparison with an event that harbours only AtWRI1. Lastly, targeted metabolomics analysis showed that carbon from sugar phosphates could be used for FA competing pathways, such as starch and cell wall polysaccharides, contributing to the restriction in oil accumulation. These results allowed the identification of key cellular processes that need to be considered to break the embryo’s natural restriction to uncontrolled seed lipid increase.
Keywords: lipid, Glycine max, transcription factor, metabolic engineering
Introduction
Soybean (Glycine max [L.] Merr.) is the largest feedstock for protein and the second largest source of vegetable oil in the world with an estimated 2020/2021 production of over 363 million metric tonnes (USDA: www.nass.usda.gov). Commodity soybean seeds accumulate close to 40% protein and 20% oil. Soybean protein is primarily used as feed and food, while its oil is broadly incorporated into food, feed, and industrial products (Clemente and Cahoon, 2009). The demand for soybean oil is projected to continue to increase in the next 10 years (Dohlman et al., 2021). Soybean seed oil is composed almost exclusively of triacylglycerols (TAGs), which contains three acyl groups from five dominant common fatty acids: palmitic acid, stearic acid, oleic acid, linoleic acid, and linolenic acid (Clemente and Cahoon, 2009). Optimizing carbon flux towards fatty acid (FA) synthesis, incrementing nutritional properties, and expanding end‐use functionality products have been among the foci of soybean breeding and genetics improvement programmes.
TAG biosynthesis is conserved among plant species: it involves de novo FA biosynthesis in plastids and TAG assembly in the endoplasmic reticulum (ER) (Bates, 2016; Xu and Shanklin, 2016). De novo FA biosynthesis is initiated with the carboxylation of acetyl‐CoA by the heteromeric acetyl‐CoA carboxylase (pACC). The product of this reaction, malonyl‐CoA, is the substrate for consecutive rounds of two carbon additions catalysed by the FA synthase complex. The growing acyl chains are esterified to an acyl carrier protein (ACP) and proceed up to FA chain length of 18 carbons with either no or one unsaturation. The acyl groups are subsequently cleaved from ACP by acyl‐ACP thioesterases and released as free FAs. The FAs are activated to CoA esters by long chain acyl CoA synthases (LACS) located about the outer plastid envelope, and transported to the ER. Then, the newly synthesized acyl‐CoA mixes with other recycled FA and can be used in four identified acylation reactions. The acyl‐receptors include: (1) lysophosphatidylcholine (lyso‐PC) generating phosphatidylcholine (PC) by the lyso‐PC acyltransferase (LPCAT); (2) glycerol‐3‐phosphate generating lysophosphatidic acid (lyso‐PA) by the glycerol‐3‐phosphate acyltransferase (GPAT); (3) lyso‐PA through a second round of acylation producing phosphatidic acid (PA) by the lyso‐PA acyltransferase (LPAAT); and (4) diacylglycerol (DAG) generating TAG by the DAG acyltransferase (DGAT). Using 14C‐pulse labelling of cultured soybean embryos, it was estimated that approximately 57% of newly synthesized FAs were directly esterified to the sn‐1 or sn‐2 position of PC (Bates et al., 2009). PC acyl chains can be desaturated, exchanged with other FA from the acyl‐CoA pool (aka PC editing), and/or transacylated to the sn‐3 position of the DAG, as another path for generating TAGs catalysed by the phosphatidylcholine:diacylglycerol acyltransferase (PDAT) (Vanhercke et al., 2013; Zhang et al., 2009). The DAG backbone for TAG biosynthesis could potentially be derived from two consecutive acylations of glycerol‐3‐phosphate and posterior dephosphorylation of the PA (Kennedy pathway), or derived from the interconversion of PC:DAG. In soybean embryos, the DAG pool used for TAG synthesis was shown to be mostly derived from PC backbone (Bates et al., 2009). Each step across the TAG biosynthetic pathway can be targeted for optimization as a way to boost oil accumulation; however, determination of rate limiting steps, along the pathway, remains a challenge (Bates, 2016).
Recent metabolic engineering efforts aiming to enhance seed oil have focused on both increasing the synthesis of FAs (PUSH) and their incorporation into TAGs (PULL). The most studied ‘PUSH’ approach utilizes the overexpression of a global regulator of lipid biosynthesis WRINKLED1 (WRI1). WRI1 is a member of the APETALA2/ethylene responsive element‐binding protein (AP2/EREBP) transcription factor family (Baud et al., 2007; Cernac and Benning, 2004; Focks and Benning, 1998). In Arabidopsis, AtWRI1 protein participates in the transcriptional regulation of glycolytic and FA biosynthetic genes by binding to a consensus motif, AW‐Box ‘CnTnG(n)7CG’, in the upstream region of the target genes (Baud et al., 2009; Chen et al., 2018; Maeo et al., 2009). In many cases, overexpression of WRI1, by either constitutive or tissue‐specific promoters, resulted in increased lipid accumulation in seeds and vegetative tissues in various plant species, including Arabidopsis, maize, rice, camelina, and soybean (An and Suh, 2015; Cernac and Benning, 2004; Chen et al., 2020; Guo et al., 2020; Liu et al., 2010; Shen et al., 2010; Sun et al., 2017). Analyses of downstream regulation of various WRI1 homologs showed that they shared regulation partners (Grimberg et al., 2015). The ‘PULL’ strategies investigated, include the overexpression of DGAT which also resulted in higher accumulation of oil, and showed a synergistic effect when stacked with WRI1 expression cassette. For example, transient co‐expression of Arabidopsis WRI1 and DGAT1 genes in Nicotiana benthamiana leaves resulted in TAG levels five‐fold higher than that observed from transient expression of individual genes (Vanhercke et al., 2013). Higher oil levels were also observed in seeds when AtWRI1 and AtDGAT1 were overexpressed in Arabidopsis under seed‐specific promoters (van Erp et al., 2014).
Two WRI1 homologs, GmWRI1a (Glyma.15G221600) and GmWRI1b (Glyma.08G227700), are best characterized in soybean. Both genes have been successfully overexpressed in soybean, rendering seeds with increased total seed oil, changes in FA composition, and yield (Chen et al., 2018; Guo et al., 2020). GmWRI1a and GmWRI1b bind directly to AW‐box elements and regulate FA production, elongation, desaturation, and export from plastid (Chen et al., 2018; Guo et al., 2020; Maeo et al., 2009). Soybean events harbouring these transgenic alleles also showed an increase in seed yield without affecting the protein level. Introducing a transgenic AtWRI1 into soybean, however, did not translate to an increase in the total oil but rather a doubling of the palmitate percentage in the oil, likely due to an induction of a specific homolog of an acyl–acyl carrier protein thioesterases (FATB) gene (Vogel et al., 2019). The contrasting observations in soybean events carrying an ectopic GmWRI1 expression allele and AtWRI1 cassette, in regards to seed oil accumulation highlights the need to gain insight on the underlying biology leading to these differing phenotypic outcomes.
The introduction of other DGAT variants into soybean has also led to an increase in seed TAG levels. For example, introducing a transgenic DGAT2A allele from the oil‐accumulating fungus Umbelopsis ramanniana or DGAT1 from Sesamum indicum L. in soybean during seed development led to 1.5% and 1.75% increase in total oil content, respectively (Lardizabal et al., 2008; Wang et al., 2014). Expression of DGAT appears to enhance carbon flux towards oil since the increase in total oil was associated with reduced soluble carbohydrate content in mature seeds (Roesler et al., 2016). Despite the evidence of DGAT improving oil content and its synergy with WRI1 observed in other species, there have been no communicated investigations into the metabolic and transcriptomic changes that occur during seed development in soybean events carrying a transgene stack of AtDGAT and AtWRI1.
The work described herein communicates the research outcomes on embryo transcriptomic and metabolic responses to the stable co‐expression of AtWRI1 and AtDGAT1 in soybean. The data highlighted the induction of processes at both transcription and post transcriptional levels that offset the positive flux towards in FA and TAG accumulation, triggered by AtWRI1 and AtDGAT1 transgene stack. In addition, we identified competing end‐products, which may contribute to the restricted oil biosynthesis. Employing transcriptomic and metabolic analyses, we observed that gene expression could explain the steady state levels of metabolites associated with sucrose metabolism and those associated with the initial steps of glycolysis, but not the levels of glycolytic intermediates downstream of triose phosphates. Furthermore, using transcriptomics datasets gathered on soybean events carrying either AtWRI1/AtDGAT1 stack or AtWRI1 alone (Vogel et al., 2019), we catalogued the putative targets for AtWRI1 in soybean. The identification of embryo’s responses and adaptations to AtWRI1 and AtDGAT1 expression allows guidance for further engineering approaches aimed at increased oil content in soybean.
Results
Arabidopsis WRI1 and DGAT1 were successfully expressed in soybean
In order to evaluate whether the expression of AtWRI1 and AtDGAT1 could direct carbon flow towards lipid accumulation, three independent soybean events, hereafter designated as WD‐1, WD‐2, and WD‐3, were characterized. The two expression cassettes were regulated by the seed‐specific beta‐conglycinin promoter (Allen et al., 1989) and cloned into a T‐DNA element (Figure S1). Integration of the T‐DNA element in the genome of the selected events was confirmed by Southern blot analysis. The results showed that events WD‐2 and ‐3 carry a single transgenic locus, while WD‐1 harbours two (Figure S2). The selected events were carried down to homozygosity. The homozygous lineages were evaluated under field conditions in 2018, 2019, and 2020, as well controlled greenhouse environments.
Mature seeds of transgenic WD presented different fatty acid and carbohydrate composition
To assess whether the expression of the AtWRI1 and AtDGAT1 transgenic alleles altered seed reserve accumulation, the composition of the mature seeds was determined from plants grown under both greenhouse and field conditions (Figure 1). WD‐3 displayed the same lipid content and FA composition as the WT (data not shown), and, therefore, was not included for further analyses. Under greenhouse conditions, the transgenic events WD‐1 and ‐2 showed no significant differences with the WT regarding total FA and protein contents at maturity (Figure 1a). However, their FA composition was significantly affected. An increase in the levels of palmitic and linoleic acids, and a decrease in stearic and oleic acids were observed in the transgenic events (Figure 1a). Similar results of total FA content and composition in mature seeds were obtained when these events were grown under field environments, but with a reduced accumulation of linolenic acid (Figure 1b). The presence of a DGAT1 transgenic allele was expected to enhance the assembly of TAGs, however, no difference in TAG content was observed between the transgenic events and WT plants (Figure 1c). Quantification of carbohydrates in mature seeds, harvested under greenhouse conditions, revealed an increase in the total content of soluble sugars and sugar alcohols in the transgenic events. Specifically, WD‐1 and‐2 events had higher levels of tetraols, pentitols, and raffinose relative to the control (Figure 1d,e). Interestingly, the only soluble sugar with decreased levels in the transgenics was maltose. In addition, starch content at maturity was significantly higher than WT in WD‐2 (Figure 1f). The levels of the matrix polysaccharides, hemicelluloses, and crystalline cellulose, were determined by the quantification of individual monosaccharides after hydrolysis. WT and transgenic events presented a similar profile of hemicellulose subunit, with only WD‐2 showing a significant decrease in rhamnose, galactose, and total hemicellulose (Figure 1g). Finally, no significant change in the levels of crystalline cellulose was observed (Figure 1h). These data demonstrated that the expression of AtWRI1 and AtDGAT1 affected mature seed carbon partition, especially the composition of FA and soluble sugars.
Figure 1.
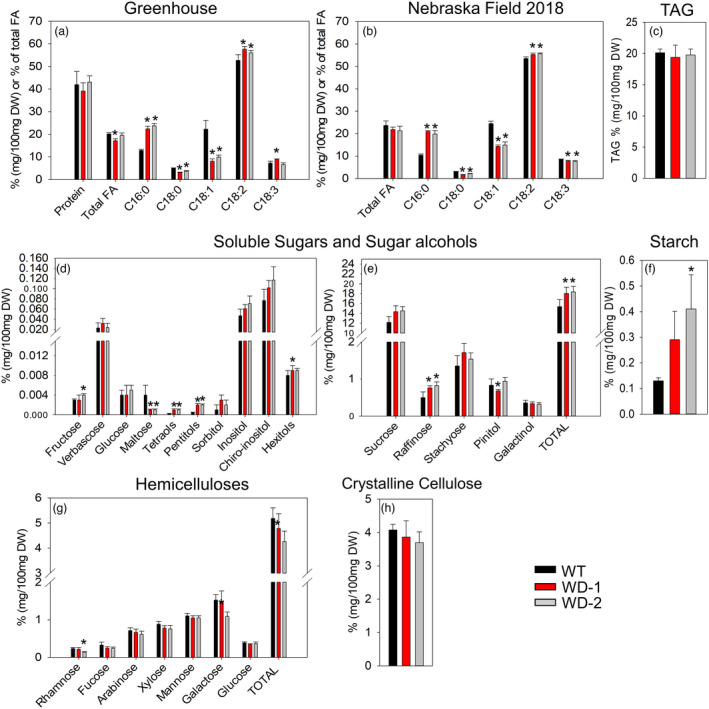
Mature seed composition of soybean. Results from soybean grown under greenhouse conditions (a, d, e, f, g, and h) or in Nebraska fields in 2018 (b and c) are shown. With the exception of the FA composition that was expressed as a percentage of the total FA (a and b), all the other variables measured were expressed in % (mg/100 mg DW). C16:0, C18:0, C18:1, C18:2, C18:3 correspond to palmitic, stearic, oleic, linoleic, and linolenic acids, respectively. For all pairwise multiple comparison procedures, the Fisher LSD or Student–Newman–Keuls method was used as parametric or non‐parametric tests, respectively. Error bars are the standard deviation of three or more biological replicates and the presence of an asterisk denotes a significant difference with the WT (P < 0.05). FA: Fatty acid. TAG: triacylglycerides. DW: Dry weight. WD‐1 and ‐2: independent transgenic events expressing AtWRI1 and AtDGAT1.
Seed yield and germination were decreased in WD events
The transgenic events grown under field conditions in 2018, 2019, and 2020 did not show a significant difference in agronomic parameters, days to flowering and to maturity (Table S1). However, a roughly 15–25% decrease in yield per square meter was observed, without a significant change in 100 seed weight (Table S1). Mature seeds collected from the field showed a mild wrinkled‐seed off‐type, that was markedly enhanced in seeds collected from the greenhouse (Figure S3). To evaluate if the differences in seed composition and phenotype affected seed viability, germination assays were performed in the field (Table S1). The results were consistent in each field trial, showing reduced germination in the transgenic events compared to the WT (Table S1). These data show that both changes in the biomass composition and agronomic parameters were affected by the expression of AtWRT1 and AtDGAT genes in soybean.
Starch and hemicellulose accumulation competed with fatty acid synthesis in WD events during seed filling
To identify earlier processes altered by AtWRI1 and AtDGAT1 expression, and responsible for the changes that ultimately give rise to the mature seed composition, we monitored the storage of the different biomass components in immature embryos, across six developmental stages under controlled greenhouse conditions. The selected points (21–36 days after pollination, DAP) span a period of linear reserve accumulation in the embryo (Figure S4). The results showed that the embryo’s DW did not differ among WT and transgenic events at the developmental points after 27 DAP (Figure 2a). However, the production of protein and FA was reduced in the transgenic events at nearly all developmental points (Figure 2b,c), despite showing similar synthesis rates of protein and FA (Figure S4) and reaching similar concentrations at maturity (Figure 1a). The reduction in stearic and oleic acids, as well as the increase of palmitic and linoleic acids in the WD‐1 and ‐2 events, observed at maturity were detected at the immature stages as well (Figure 3). At some developmental stages, linolenic acid was higher, following the same trend as observed in mature seeds from the greenhouse (Figures 1a and 3). In addition, the transgenic embryos had a 3‐fold increase in the starch accumulation rate (Figure S4), doubling WT levels in the last stages evaluated (Figure 2d). Interestingly, the resulting ratio of FA accumulation rate to starch accumulation rate was 3.48, 0.93, and 0.89 in the WT, WD‐1, and WD‐2, respectively. Therefore, besides the well‐known competition for carbon with protein accumulation, FA synthesis in WD events was also competing with a higher rate of starch production (Figure S4), which likely impacted final FA concentration.
Figure 2.
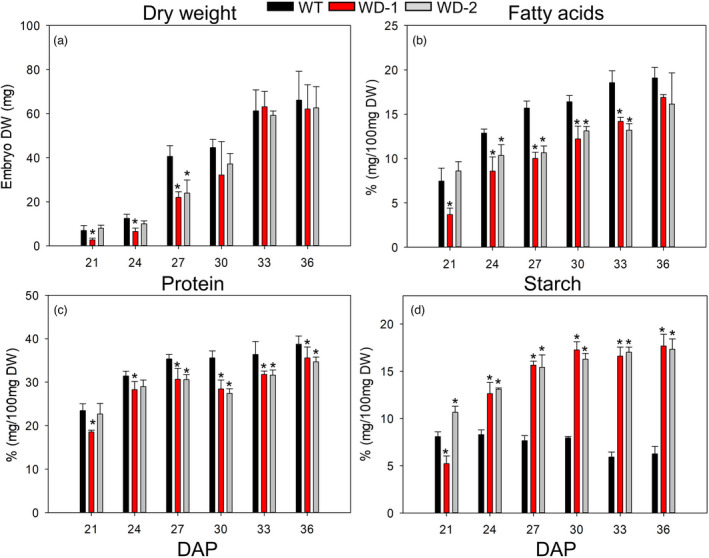
DW, protein, FA, and starch accumulation during development. Results from soybean grown under greenhouse conditions. The embryo’s DW (a), the content of FA (b), protein (c) and starch (d) are shown in % (mg /100 mg DW). For all pairwise multiple comparison procedures, Fisher LSD method or Student–Newman–Keuls method was used as parametric or non‐parametric test, respectively. Error bars are the standard deviation of four biological replicates and the presence of an asterisk denotes a significant difference with the WT (p < 0.05). DW: Dry weight. DAP: days after pollination. WD‐1 and −2: independent transgenic events expressing AtWRI1 and AtDGAT1.
Figure 3.
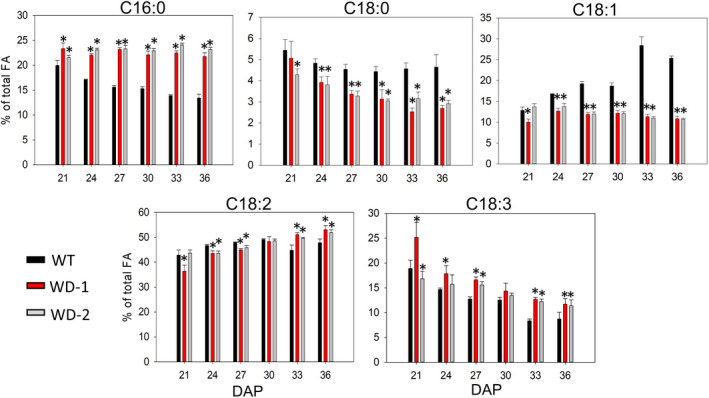
Fatty acid composition during development. The content of each FA is expressed in relation to the total content of FA at each time point. C16:0, C18:0, C18:1, C18:2, C18:3 correspond to palmitic, stearic, oleic, linoleic, and linolenic acids, respectively. For all pairwise multiple comparison procedures, Fisher LSD method or Student–Newman–Keuls method was used as parametric or non‐parametric test, respectively. Error bars are the standard deviation of four biological replicates and the presence of an asterisk denotes a significant difference with the WT (P < 0.05). FA: Fatty acid. DAP: days after pollination. WD‐1 and ‐2: independent transgenic events expressing AtWRI1 and AtDGAT1.
Given the observed changes in the rates of starch accumulation in the WD‐1 and ‐2 events, we subsequently monitored for alterations in levels of cell wall polysaccharides (Figure S5). Two additional time points were added at later stages for this analysis. The results revealed an increase in the content of glucose in the matrix polysaccharides, probably derived from the hemicellulose xyloglucan and amorphous cellulose. The rates of glucose accumulation as hemicelluloses in WD‐1 and ‐2 were 1.50 and 1.25 times higher than the WT, respectively (Figure S6). Besides, the level of total hemicelluloses in the WD events was increased relative WT until 33 DAP, after which they were not significantly different from the WT, and, at maturity, WD‐2 showed a significant decrease (Figure 1g). The levels of crystalline cellulose during development, was significantly increased only in event WD‐2. These results highlight that; besides starch, the production of cell wall polysaccharides was also competing for carbon with FA synthesis in WD events.
AtWRI1 and AtDGAT1 expression increased during the development in WD events
To assess changes in the transcriptome during seed development in the WD‐1 and‐2 events, an RNAseq analysis was performed in immature embryos of both transgenic events grown under greenhouse conditions. Despite both expression cassettes were regulated by the same β‐conglycinin promoter (Figure S1), which in its native genomic context, is active starting at 14 days after flowering (DAF) and peaks at approximately 44 DAF (Figure S7), the expression of AtWRI1 was approximately 10 times higher than AtDGAT1 (Figure 4a,d). The GmWRI1 homologs (Glyma.08G227700 and Glyma.15G221600) were down‐regulated in the embryos of transgenic events during development (Figure 4b,c). The GmDAGT1 gene calls were only slight changed in their expression pattern (Figure 4e–n), suggestive that AtWRI1 is not regulating their transcription as it is for other lipid‐related genes (Figure 5).
Figure 4.
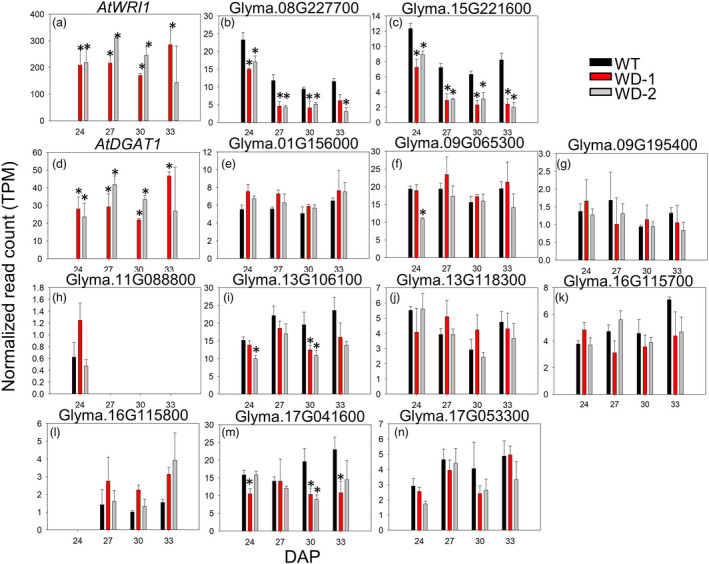
Expression of WRI1 and DGAT1 from Arabidopsis and soybean in the transgenic events grown under greenhouse conditions. The expression of the transgenes and endogenous versions are shown at four developmental points: 24, 27, 30, and 33 DAP. Error bars are the standard deviation of three biological replicates and the presence of an asterisk denotes a significant difference with the WT (FDR<0.05). TPM, transcripts per million. DAP: days after pollination. WD‐1 and ‐2: independent transgenic events expressing AtWRI1 and AtDGAT1.
Figure 5.
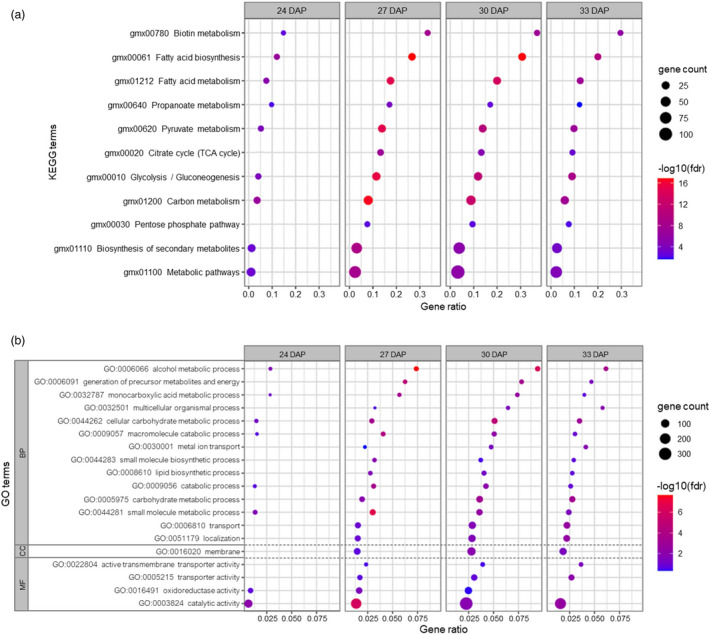
KEGG pathway enrichment and GO functional classification of differentially expressed genes in WD events grown in a greenhouse. Statistically significantly enriched (FDR < 0.05) KEGG pathways in (a) and Gene Ontology terms in (b) are shown for the DEGs identified at each developmental stage. Three GO categories are presented, including Biological Processes (BP), Cellular Compartment (CC), and Molecular Function (MF). The gene count represents the number of non‐unique DEGs enriched in a pathway. The absence of a dot indicates the pathway was not significantly enriched in that developmental stage. Gene ratio was calculated as the number of DEGs involved in a pathway divided by the total number of soybean genes in the pathway. Enriched GO terms resulting from AgriGOv2 were consolidated into fewer non‐redundant GO terms using Revigo (http://revigo.irb.hr) for visualization purposes in (b) (FDR < 0.05). DAP: days after pollination. TCA: tricarboxylic acid cycle.
Gene calls annotated for function in glycolysis and fatty acid metabolism were enriched among the differentially expressed genes in the WD events during seed development
DEGs were identified from each time point in which the transcriptome was assessed via RNAseq analysis, for each transgenic event (Table S2). Common DEGs among the events were selected for further analysis, discarding those that presented opposite behaviour (Table S2). With 969 native genes being differentially expressed, 30 DAP represented the stage with most DEGs (Table S2). Among the 134 gene calls differentially expressed across all four developmental stages and both transgenic events, only 12 were down‐regulated (Figure S8).
To assign association with metabolic pathways and biological processes to the gene calls over‐represented in the total pool of DEGs, an enrichment analysis was performed with both KEGG pathways and Gene Ontology (GO) terms databases. The outcome of this enrichment analysis revealed the categories of FA biosynthesis, FA metabolism, and glycolysis/gluconeogenesis were among the top enriched pathways across all stages (Figure 5a), indicating that AtWRI1 is capable of activating the expression of its known targets in the genome of soybean. In accordance with the KEGG pathway enrichment, GO terms of lipid biosynthesis, carbohydrate metabolism, and generation of precursor metabolites and energy processes were identified as the top enriched biological processes (BP). For molecular functions (MF), the enriched GO term with the highest gene count was catalytic activities (Figure 5b). Interestingly, the membrane was the only enriched GO term for cellular compartments (CC) and this is in alignment with the enriched GO terms of transport in BP and transporter activity in MF (Figure 5b).
Annotated gene calls assigned to de novo fatty acid synthesis were up‐regulated while those for oil body structural proteins down‐regulated in WD events during seed development
To evaluate what specific steps in lipid biosynthesis were affected by AtWRI1 and AtDGAT1 co‐expression, we investigated the processes that takes place in the chloroplast and the ER (Figure 6). One or more genes involved in each step of the plastidic de novo FA synthesis pathway were up‐regulated in two or more developmental stages, including subunits of the ACC and the FA synthase complex (Figure 6). Moreover, gene calls annotated to function in ER FA elongation and desaturation, such as ketoacyl‐CoA synthase and reductase, were also induced in the transgenic events (Figure 6). In addition to AtDGAT1 expression, only one enzyme from TAG biosynthesis, a putative glycerol 3‐phosphate acyltransferase, was found in the DEG list (Figure 6). Finally, genes encoding putative oleosins and caleosins, structural proteins required for oil body (OB) stabilization, were down‐regulated up to 10 times in the transgenic events (Figure 6). Therefore, despite the induction of FA synthesis genes and the expression of AtDGAT1, the reduced expression of the lipid‐droplet packaging proteins may limit oil accumulation.
Figure 6.
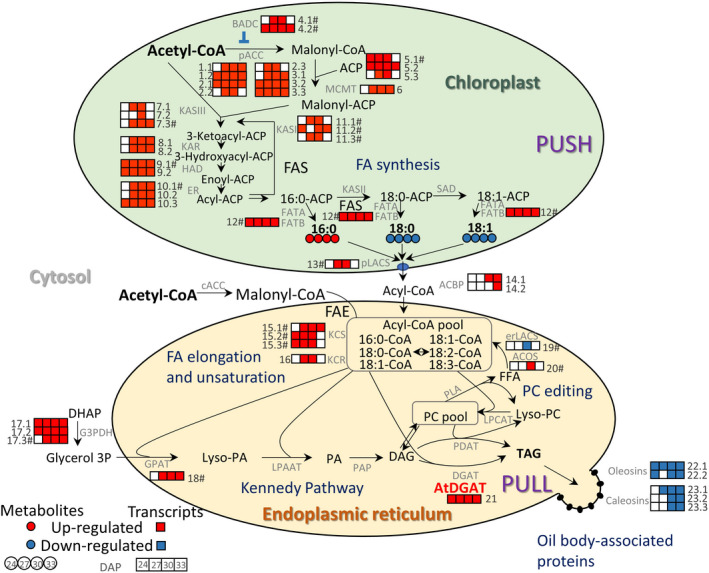
Alteration of lipid biosynthesis in WD embryos between 24 and 33 DAP. Red and blue circles were used to account for significantly increased or decreased levels of each FA at each stage, respectively (n = 4, P < 0.05). Red and blue boxes were used to show the significantly up‐ or down‐regulated expression of each enzyme at each stage under greenhouse conditions, respectively (n = 3, P < 0.05, |log2(Fold Change)| ≥ 1). The symbol # next to the corresponding enzyme number denotes the presence of the AW‐box motif in the promoter of the enzyme’s gene. ACP, plastidic acyl carrier protein; FAS, fatty acid synthase; pACC, heteromeric acetyl CoA carboxylase, biotin carboxylase subunit: (1.1) Glyma.05G221100 and (1.2) Glyma.08G027600, biotin carboxyl carrier protein: (2.1) Glyma.13G057400, (2.2) Glyma.18G265300 and (2.3) Glyma.19G028800, carboxyltransferase alpha subunit: (3.1) Glyma.18G195700, (3.2) Glyma.18G195900 and (3.3) Glyma.18G196000; BADC, biotin/lipoyl attachment domain‐containing protein: (4.1) Glyma.11G233700# and (4.2) Glyma.18G023300#; ACP, acyl carrier protein: (5.1) Glyma.13G214600#, (5.2) Glyma.15G098500 and (5.3) Glyma.20G230100; MCMT, malonyl‐CoA: ACP malonyltransferase: (6) Glyma.11G164500; KASIII, ketoacyl‐ACP synthase III: (7.1) Glyma.09G277400, (7.2) Glyma.15G003100 and (7.3) Glyma.18G211400#; KAR, ketoacyl‐ACP reductase: (8.1) Glyma.11G248000 and (8.2) Glyma.18G009200; HAD, hydroxyacyl‐ACP dehydrase: (9.1) Glyma.08G179900# and (9.2) Glyma.15G052500; ER, enoyl‐ACP reductase: (10.1) Glyma.08G345900#, (10.2) Glyma.12G027300, and (10.3) Glyma.18G156100; KASI, ketoacyl‐ACP synthase I: (11.1) Glyma.05G129600#, (11.2) Glyma.08G024700# and (11.3) Glyma.08G084300#; SAD, stearoyl‐ACP desaturase; FATA, fatty acyl thioesterase A; FATB, fatty acyl thioesterase B: (12) Glyma.04G151600#; pLACS, plastidic long‐chain acyl‐ CoA synthetase: (13) Glyma.06G112900#; ACBP, acyl CoA‐binding protein: (14.1) Glyma.13G152900 and (14.2) Glyma.11G014900; ACCc, homomeric acetyl CoA carboxylase; FAE, fatty acid elongase; KCS, ketoacyl‐CoA synthase: (15.1) Glyma.06G012500#, (15.2) Glyma.04G149300# and (15.3) Glyma.06G214800#; KCR, ketoacyl‐CoA reductase: (16) Glyma.11G245600; DHAP, dihydroxyacetone phosphate; G3PDH, glycerol 3 phosphate dehydrogenase: (17.1) Glyma.19G053500, (17.2) Glyma.02G186600 and (17.3) Glyma.10G107100#; GPAT, glycerol‐3‐phosphate acyltransferase: (18) Glyma.20G070400; Lyso‐PA, lysophosphatidic acid; LPAAT, 2‐lysophosphatidic acid acyltransferase; PA, phosphatidic acid; PAP, phosphatidate phosphatase; DAG, diacylglycerol; TAG, triacylglycerol; DGAT, acyl‐CoA: diacylglycerol acyltransferase; PDAT, phospholipid:diacylglycerol acyltransferase; PC, phosphatidylcholine; LPCAT, lysophosphatidylcholine acyltransferase; PLA, phospholipase A; erLACS, ER long‐chain acyl‐ CoA synthetase: (19) Glyma.10G010800#; ACOS, acyl‐CoA synthase: (20) Glyma.08G329700; FFA, free fatty acid; AtDGAT, transgen expressed (21); Oleosin: (22.1) Glyma.05G004300 and (22.2) Glyma.19G004800; and Caleosins: (23.1) Glyma.10G189900, (23.2) Glyma.20G200900, (23.3) Glyma.20G201000. DAP: days after pollination.
Putative AtWRI1 targets were identified
Many of the WRI1 targets described in the literature were found to be induced in WD events grown under greenhouse conditions, including genes encoding subunits of the plastid pyruvate kinase (PK), pyruvate dehydrogenase (PDH) and pACC, and versions of the ketoacyl‐ACP synthase I, hydroxyacyl‐ACP dehydrase, and enoyl‐ACP reductase (Figure 6). To complement the list of AtWRI1 putative targets and evaluate transcript behaviour under field conditions, we enlarged the RNA‐seq study and analysed the transcriptome of immature seeds collected from the field at R5, R5/6, and R6 stages of development (equivalent to 24, 30, and 38 DAP, respectively). The biologicals utilized in this analysis were WT, WD‐1 event along with an event, designated here as W‐1, previously described (Vogel et al., 2019). W‐1 only carries the AtWRI1 cassette, which will allow for the discrimination between the indirect changes in gene expression due to the presence of AtDGAT1 from those directly related to the expression of AtWRI1. Compared to the greenhouse RNA‐seq experiments, substantially fewer DEGs were identified in the field (Table S2). AtWRI1 expression was detected in field samples (Figure S9a); however, the down‐regulation of GmWRI1 homologs was less evident than under greenhouse conditions (Figure S9b,c). The expression of AtDGAT1 and GmDGAT1 homologs in the field, on the other hand, are consistent with the greenhouse analyses, reaching comparable expression levels for most of the homologs (Figure S9d,n). Comparisons between the DEGs identified from the field and the greenhouse for WD‐1, revealed 25 and 212 common DEGs at R5‐24 DAP and R5/6‐30 DAP stages, respectively (Figure S10a). Furthermore, in the field experiment, among WD‐1 and W‐1 DEGs, we identified 531 common ones along the development (Figure S10b). Including data with a higher degree of variability, such as the samples from plants grown in the field, allows a more robust identification of true targets.
To identify putative targets of AtWRI1, we vetted calls using three criteria: (1) genes up‐regulated in at least one stage in the field W‐1 and WD‐1 events and at least one stage in the greenhouse WD‐1 and ‐2 events; (2) genes with the AW‐box motif in their 2 kb upstream sequence; and (3) genes whose expression correlates with the expression of AtWRI1 with a Pearson correlation coefficient higher than 0.85 (Zaborowski and Walther, 2020). This vetting process identified 82 genes, including transcription factors, kinase receptors, transporters, genes related to lipid and central metabolism, and some with unknown function (Table S3). Among the vetted gene calls with annotated functions associated with FA metabolism that have not been previously identified as AtWRI1 targets, were some putatively encoding for FA amide hydrolases, a long chain acyl‐CoA synthetase, and monoacylglycerol and GDSL‐motif lipases (Table 1). Interestingly, besides PK and PDH regulation, other central metabolism genes were detected as putative AtWRI1 targets, including genes annotated as glucose 6‐phosphate/phosphate translocator, UDP‐glucose pyrophosphorylase, fructose bisphosphate aldolase, plastidic glyceraldehyde 3‐phosphate dehydrogenase, and plastidic malate dehydrogenase (Table 1). Some gene calls that are not directly assigned function in lipid metabolism or carbon or energy supply were also identified. These include a putative actin subunit, abscisic acid, jasmonates, ethylene, and auxin‐associated genes (Table 1), potentially linking the AtWRI1 regulon beyond the FA metabolism and glycolysis in developing soybean seeds.
Table 1.
Putative WRI1 targets
| Line | log2FC_Greenhouse | log2FC_Field | AW site | PCC ATWRI1 | Top Arabidopsis BLASTP Hit | ATH hit annotations | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WD‐1 | WD‐2 | WD‐1 | WD‐2 | WD‐1 | WD‐2 | WD‐1 | WD‐2 | WD‐1 | WD‐1 | WD‐1 | W‐1 | W‐1 | W‐1 | |||||
| Gene ID | 24 DAP | 27 DAP | 30 DAP | 33 DAP | R5 | R5/6 | R6 | R5 | R5/6 | R6 | ||||||||
| Glyma.16G173100 | 1.5 | 1.3 | 2.1 | 2.4 | 2.1 | 2.3 | 2.2 | 1.7 | 2.1 | 2.0 | 526 | 0.99 | AT5G52920 | Pyruvate kinase beta subunit 1 | ||||
| Glyma.18G023300 | 1.2 | 1.1 | 1.9 | 2.1 | 1.8 | 2.0 | 1.9 | 1.4 | 1.7 | 1.6 | 913 | 0.99 | AT3G56130 | Biotin/lipoyl attachment domain‐containing protein | ||||
| Glyma.05G195500 | 4.4 | 4.5 | 4.1 | 4.7 | 4.5 | 4.8 | 4.7 | 4.3 | 2.5 | 4.0 | 2.5 | 4.0 | 1089 | 0.98 | AT5G64440 | Fatty acid amide hydrolase | ||
| Glyma.03G168700 | 1.4 | 1.2 | 1.4 | 1.7 | 1.3 | 1.4 | 1.8 | 1.1 | 1.7 | 1.9 | 1896 | 0.98 | AT5G43940 | Alcohol dehydrogenase 2 | ||||
| Glyma.17G231400 | 1.3 | 1.1 | 1.8 | 2.0 | 1.7 | 1.7 | 1.8 | 1.1 | 1.8 | 1.9 | 1608 | 0.97 | AT2G34590 | Pyruvate dehydrogenase 1b | ||||
| Glyma.08G024700 | 2.0 | 2.1 | 4.9 | 1.6 | 3.5 | 2.7 | 4.8 | 1.8 | 1.8 | 1876 | 0.97 | AT5G46290 | Ketoacyl‐ACP Synthase I | |||||
| Glyma.18G211400 | 1.2 | 1.8 | 1.8 | 1.8 | 1.7 | 1.8 | 1.2 | 1.6 | 1.6 | 2 | 0.96 | AT1G62640 | Ketoacyl‐ACP synthase III | |||||
| Glyma.07G033000 | 1.0 | 1.2 | 1.2 | 1.6 | 1.3 | 1.4 | 2.4 | 2.2 | 1587 | 0.96 | AT1G80280 | Alpha/beta‐hydrolases | ||||||
| Glyma.20G122500 | 1.1 | 1.1 | 1.5 | 1.6 | 1.3 | 1.4 | 1.8 | 1.2 | 1.7 | 1.6 | 586 | 0.96 | AT2G01140 | Fructose bisphosphate aldolase 3 | ||||
| Glyma.08G179900 | 1.4 | 1.1 | 1.9 | 1.9 | 2.1 | 2.2 | 2.3 | 1.6 | 1.7 | 1.7 | 306 | 0.96 | AT2G22230 | Hydroxyacyl‐ACP dehydrase | ||||
| Glyma.01G081900 | 3.1 | 2.6 | 5.0 | 4.9 | 5.0 | 4.8 | 6.0 | 5.3 | 1.7 | 4.0 | 1.8 | 4.1 | 1552 | 0.95 | AT3G25860 | Pyruvate dehydrogenase E2 | ||
| Glyma.17G015500 | 1.9 | 1.6 | 2.9 | 2.8 | 2.9 | 2.8 | 3.2 | 2.2 | 1.1 | 3.9 | 1.1 | 4.1 | 244 | 0.95 | AT1G61800 | Glucose 6 phosphate/phosphate translocator 2 | ||
| Glyma.15G276300 | 1.6 | 1.8 | 1.4 | 1.6 | 1.6 | 1.1 | 1.2 | 1.2 | 405 | 0.95 | AT5G13710 | Sterol methyltransferase 1 | ||||||
| Glyma.06G214800 | 1.3 | 1.1 | 1.7 | 1.6 | 1.8 | 1.7 | 3.0 | 3.0 | 1890 | 0.95 | AT2G26640 | Ketoacyl‐CoA synthase 11 | ||||||
| Glyma.13G152500 | 1.0 | 1.3 | 1.5 | 1.1 | 1.3 | 1.5 | 1.2 | 1.6 | 1.5 | 1971 | 0.95 | AT5G17310 | UDP‐glucose pyrophosphorylase 2 | |||||
| Glyma.03G092700 | 1.2 | 1.3 | 1.3 | 1.5 | 1.4 | 1.1 | 1.4 | 1.6 | 58 | 0.94 | AT1G79530 | Glyceraldehyde‐3‐phosphate dehydrogenase 1 | ||||||
| Glyma.05G000900 | 1.1 | 1.9 | 1.8 | 1.7 | 1.6 | 1.8 | 1.2 | 1.7 | 1.6 | 1613 | 0.94 | AT3G12110 | Actin 11 | |||||
| Glyma.04G149300 | 1.6 | 1.4 | 2.3 | 2.3 | 2.7 | 3.1 | 2.9 | 2.7 | 104 | 0.94 | AT2G26640 | Ketoacyl‐CoA synthase 11 | ||||||
| Glyma.08G003100 | 3.8 | 3.9 | 3.5 | 4.3 | 3.9 | 4.3 | 4.6 | 4.6 | 1.6 | 3.6 | 3.0 | 4.7 | 936 | 0.94 | AT5G64440 | Fatty acid amide hydrolase | ||
| Glyma.11G233700 | 1.1 | 1.3 | 1.0 | 1.2 | 1.3 | 1.2 | 1.5 | 800 | 0.93 | AT3G56130 | Biotin/lipoyl attachment domain‐containing protein | |||||||
| Glyma.10G215400 | 1.1 | 1.8 | 1.7 | 2.0 | 1.9 | 1.8 | 1.2 | 1.6 | 1.6 | 121 | 0.93 | AT1G34430 | Pyruvate dehydrogenase E2 | |||||
| Glyma.12G135300 | 3.6 | 3.3 | 3.9 | 4.1 | 4.2 | 4.4 | 4.2 | 3.8 | 1.7 | 5.0 | 2.2 | 4.9 | 1921 | 0.92 | AT3G28345 | ABC transporter | ||
| Glyma.01G190600 | 1.2 | 1.3 | 1.8 | 1.7 | 1.8 | 2.4 | 1909 | 0.92 | AT2G14960 | Auxin‐responsive GH3 family protein | ||||||||
| Glyma.04G151600 | 1.4 | 1.3 | 1.5 | 2.0 | 1.6 | 2.0 | 2.2 | 1.8 | 2.3 | 2.2 | 163 | 0.92 | AT1G08510 | Acyl‐ACP thioesterase B | ||||
| Glyma.20G114500 | 1.2 | 2.0 | 1.7 | 2.0 | 1.7 | 2.0 | 1.1 | 1.6 | 1.8 | 1736 | 0.92 | AT3G25860 | Pyruvate dehydrogenase E2 | |||||
| Glyma.15G178400 | 1.0 | 1.3 | 1.7 | 1.4 | 1.4 | 1.3 | 1.6 | 1.5 | 1895 | 0.92 | AT1G54100 | Aldehyde dehydrogenase 7B4 | ||||||
| Glyma.08G164600 | 1.5 | 1.5 | 2.2 | 2.1 | 2.5 | 2.7 | 3.1 | 2.0 | 2.9 | 3.4 | 1834 | 0.91 | AT3G04940 | Cysteine synthase D1 | ||||
| Glyma.15G182900 | 4.7 | 5.4 | 5.8 | 7.1 | 5.1 | 6.3 | 5.7 | 5.8 | 3.5 | 5.6 | 5.9 | 95 | 0.91 | AT1G53920 | GDSL‐motif lipase 5 | |||
| Glyma.05G026300 | 1.0 | 1.2 | 1.0 | 1.3 | 1.2 | 1.4 | 1.0 | 1.1 | 437 | 0.90 | AT3G47520 | NAD dependent malate dehydrogenase | ||||||
| Glyma.06G112900 | 1.2 | 1.0 | 1.4 | 1.3 | 1.4 | 1.1 | 1.2 | 1379 | 0.90 | AT1G77590 | Long chain acyl‐CoA synthetase 9 | |||||||
| Glyma.15G183000 | 1.5 | 1.7 | 3.1 | 3.9 | 2.7 | 3.8 | 3.5 | 3.5 | 3.9 | 1.5 | 4.3 | 75 | 0.90 | AT1G53920 | GDSL‐motif lipase 5 | |||
| Glyma.07G268100 | 1.0 | 1.2 | 1.4 | 1.2 | 1.2 | 1.3 | 1.3 | 1.4 | 1981 | 0.90 | AT5G47670 | LEC1‐LIKE, NF‐YB6,nuclear factor Y, | ||||||
| Glyma.06G087100 | 1.4 | 1.0 | 1.6 | 1.9 | 1.4 | 1.7 | 2.1 | 1.6 | 2.3 | 2.7 | 495 | 0.89 | AT5G11650 | Monoacylglycerol lipase | ||||
| Glyma.19G206100 | 3.1 | 2.6 | 2.9 | 3.3 | 3.0 | 3.3 | 3.8 | 3.6 | 2.2 | 3.9 | 2.0 | 3.6 | 211 | 0.89 | AT5G62000 | Auxin response factor 2 | ||
| Glyma.13G223800 | 2.4 | 2.4 | 2.8 | 2.5 | 2.4 | 1.9 | 2.2 | 1.3 | 2.5 | 4.1 | 2.9 | 4.5 | 687 | 0.89 | AT1G69850 | Nitrate transporter, ABA importer transporter 1 | ||
| Glyma.09G074000 | 5.2 | 5.9 | 5.3 | 6.7 | 5.1 | 6.4 | 5.8 | 6.4 | 4.4 | 4.9 | 1936 | 0.89 | AT1G53920 | GDSL‐motif lipase 5 | ||||
| Glyma.08G159500 | 1.0 | 1.9 | 1.5 | 2.0 | 2.0 | 2.2 | 1.8 | 1.7 | 1.9 | 1814 | 0.88 | AT1G05590 | Beta‐hexosaminidase 2 | |||||
| Glyma.06G312200 | 1.3 | 1.5 | 2.0 | 2.4 | 2.0 | 2.3 | 1.1 | 2.2 | 2.5 | 483 | 0.87 | AT1G65440 | Global transcription factor group B1 | |||||
| Glyma.07G027000 | 1.2 | 1.7 | 2.3 | 1.5 | 2.2 | 2.4 | 785 | 0.87 | AT1G80580 | Ethylene‐responsive transcription factor | ||||||||
| Glyma.01G198700 | 1.3 | 2.0 | 1.8 | 1.6 | 1.8 | 1.8 | 1.2 | 1.4 | 1.8 | 124 | 0.86 | AT4G17810 | C2H2 domain regulatory protein | |||||
Only genes with a significantly different expression and a |log2(Fold Change)| ≥ 1 with respect to the WT were included. PCC, Pearson correlation coefficient. DAP: days after pollination. WD‐1 and ‐2: independent transgenic events expressing AtWRI1 and ATDGAT1. W‐1 event expressing only AtWRI1 (Vogel et al., 2019).
Among vetted AtWRI1 targets, we identified one putative acyl–acyl carrier protein thioesterase (GmFATB, Glyma.04g151600) in the transgenic events in all the stages, which likely accounts for the observed significant increase in palmitic and decrease in stearic and oleic acid levels during seed development and at maturity (Figures 1, 3, and 6). Gene calls with functional annotations that would negatively affect oil accumulation were also found to be potential targets of AtWRI1. For example, genes putatively encoding lipolytic enzymes, such as GDSL esterase/lipases, α/β hydrolases, and monoacylglycerol lipases (MAGL), were identified as putative AtWRI1 targets and were highly up‐regulated in the transgenic events (Table 1). Particularly, three gene calls, annotated as GDSL esterase/lipase motif type II (homolog to At1G53920), Glyma.09G074000, Glyma.15G182900, and Glyma.15G183000, were expressed up to 64 times higher than in the WT, pattern conserved along with development (Table 1). In addition to these potential AtWRI1 targets, Other genes annotated as lipases and lipid transfer proteins, lacking an AW‐motif in their sequence, were also induced in different stages of development (Figure S11). Lastly, the up‐regulation of two putative genes encoding soybean biotin/lipoyl attachment domain‐containing (BADC) proteins (Glyma.11G233700 and Glyma.18G023300) during development, proposed to act as pACC inhibitors (Salie et al., 2016), was observed (Figure 6). The differential regulation of these gene calls may have contributed to the lack of an observed oil content increase in the transgenic events.
Gene calls with functional annotation in carbohydrate metabolism were differentially regulated
To assess if the observed differences in carbohydrate metabolism were governed by transcriptional or post‐transcriptional mechanisms we analysed the transcript levels in gene calls annotated for functionality in central metabolism during development. The results reveal that transcripts linked with sucrose metabolism were affected in the embryos of the transgenic events. The expression of a gene encoding a putative sucrose 6‐phosphate synthase was down‐regulated, while genes encoding putative degradative enzymes, such as a neutral invertase and two sucrose synthases, were up‐regulated (Figure 7). Changes in expression of gene calls associated with sucrose metabolism were accompanied with the down‐regulation of a gene encoding a putative cell wall/vacuolar invertase inhibitor and the up‐regulation of two genes encoding putative sucrose transporters, especially at the latest stages (Figure 7). The WD events displayed an induction in transcripts assigned function to almost all glycolytic enzymes, with the exception of three genes putatively encoding two phosphofructokinase enzymes and an NADP‐dependent glyceraldehyde 3‐phosphate dehydrogenase that were down‐regulated (Figure 7). These results suggested an overall increase in carbon flux towards precursors for FA synthesis and energy.
Figure 7.
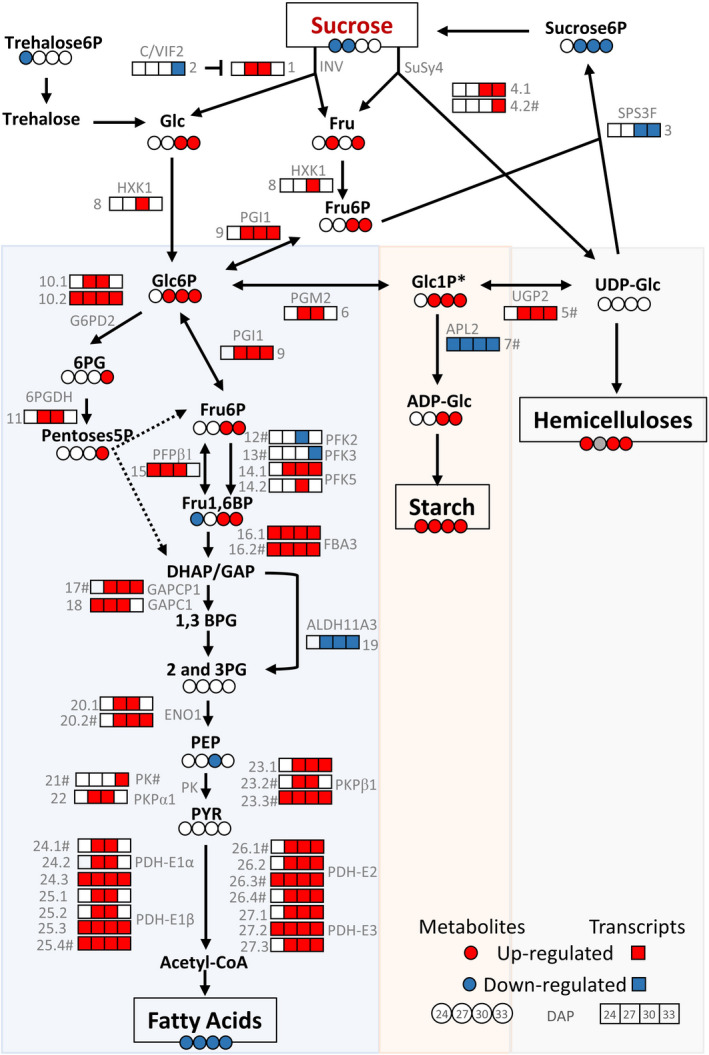
Carbon partitioning in WD embryos between 24 and 33 DAP. Metabolites and end products: red and blue circles were used to account for significantly increased or decreased levels of each metabolite at each stage, respectively (n = 4, P < 0.05) and a grey circle reflects data not measured. The contents of Glc1P are shown with an asterisk (*) as were determined together with the levels of mannose 1‐phosphate. Sucrose6P, sucrose 6‐phosphate; Threhalose6P, trehalose 6‐phosphate; Glc, glucose; Fru, fructose; Glc6P, glucose 6‐phosphate; Glc1P, glucose 1‐phosphate; Fru6P, fructose 6‐phosphate; Fru1,6BP, fructose 1,6‐bisphosphate; UDP‐Glc, UDP‐glucose; ADP‐Glc, ADP‐glucose; 6PG, 6‐phosphogluconate; Pentoses5P, pentoses 5‐phosphate; DHAP, dihydroxyacetone phosphate; GAP, glyceraldehyde 3 phosphate; 1,3BPG, 1,3‐bisphosphoglycerate; 2 and 3PG, 2‐and 3‐phosphoglycerate; PEP, phosphoenolpyruvate and PYR, pyruvate. Enzyme expression: red and blue boxes were used to show the significantly up‐ or down‐regulated expression of each enzyme at each stage under greenhouse conditions, respectively (n = 3, P < 0.05). The enzyme names correspond to the specific homolog isoforms in Arabidopsis. The symbol # denotes the presence of the AW‐box motive in the promoter of the enzyme’s gene. INV, neutral invertase (1) Glyma.04G005700; C/VIF2, cell wall/vacuolar inhibitor of fructosidase 2 (2) Glyma.04G214000; SPS3F, sucrose phosphate synthase 3F (3) Glyma.18G108100; SuSy4, sucrose synthase 4 (4.1) Glyma.09G073600 and (4.2) Glyma.13G114000#; UGP2, UDP‐glucose pyrophosphorylase 2 (5) Glyma.13G152500#; PGM2, phosphoglucomutase (6) Glyma.08G044100; APL2, ADP‐glucose pyrophosphorylase large subunit (7) Glyma.14G072500#; HXK1, hexokinase 1 (8) Glyma.07G015100; PGI1, phosphoglucose isomerase 1 (9) Glyma.06G094300;G6PD2, glucose 6 phosphate dehydrogenase 2 (10.1) Glyma.02G096800 and (10.2) Glyma.18G284600; 6PGDH, 6‐phosphogluconate dehydrogenase (11) Glyma.05G214000; PFK2, phosphofructokinase 2 (12) Glyma.07G269500#;PFK3, phosphofructokinase 3 (13) Glyma.13G353400#; PFK5, phosphofructokinase 5 (14.1) Glyma.08G280700 and (14.2) Glyma.18G145500; PFPβ1, pyrophosphate: fructose 6‐phosphate 1‐phosphotransferase (15) Glyma.18G135100; FBA3, fructose bisphosphate aldolase 3 (16.1) Glyma.10G268500 and (16.2) Glyma.20G122500#; GAPCP1, glyceraldehyde‐3‐phosphate dehydrogenase of plastid 1 (17) Glyma.03G092700#; GAPC1, glyceraldehyde 3 phosphate dehydrogenase C subunit 1 (18) Glyma.11G247600; ALDH11A3, NADP‐dependent glyceraldehyde 3‐phosphate dehydrogenase (19) Glyma.02G202500; ENO1, enolase 1 (20.1) Glyma.04G144300 and (20.2) Glyma.06G208200#; PK, pyruvate kinase family protein (21) Glyma.19G190100;PKPα1, plastidial pyruvate kinase 1 subunit alpha (22) Glyma.20G211200; PKPβ1, plastidial pyruvate kinase 1 subunit beta (23.1) Glyma.09G126300, (23.2) Glyma.10G227800#, (23.3) Glyma.16G173100#; PDH‐E1α, E1a component of pyruvate dehydrogenase complex (24.1) Glyma.03G261000#, (24.2) Glyma.16G018300, (24.3) Glyma.19G260000; PDH‐E1β, E1b component of pyruvate dehydrogenase complex (25.1) Glyma.05G141000, (25.2) Glyma.08G096300, (25.3) Glyma.14G092100, (25.4) Glyma.17G231400#; PDH‐E2, E2 component of pyruvate dehydrogenase complex (26.1) Glyma.10G215400#, (26.2) Glyma.20G176300, (26.3) Glyma.01G081900#, (26.4) Glyma.20G114500# and PDH‐E3, E3 component of pyruvate dehydrogenase complex (27.1) Glyma.15G143100, (27.2) Glyma.07G241600, (27.3) Glyma.17G032300.
We also analysed expression levels of genes putatively functioning within pathways competing with FA synthesis, such as starch and cell wall production. Unexpectedly, the expression of two putative genes encoding ADP‐glucose pyrophosphorylase regulatory subunits were down‐regulated in the WD events along the selected developmental timepoints. This enzyme catalyses the first and rate‐limiting step in starch biosynthesis. However, we also observed an up‐regulation of a gene encoding a phosphoglucomutase that produces the starch biosynthesis substrate, glucose 1‐phosphate (Figure 7), was also observed. Assuming transcript changes, would correlate with post‐translations enzymatic activities, these two opposing key enzymes, coupled with complex pattern of transient starch accumulation, highlights that the seed‐centric phenotypic outcomes observed in the WD events is likely governed at a transcriptional and post‐transcriptional manner. Differences in hemicellulose content also cannot be explained solely at transcript level. Among many of the gene calls functionally annotated as enzymes involved in cell wall metabolism, we could identify DEGs encoding two probable glycosyltransferases (exostosin family proteins) with putative functions in cell wall biosynthesis, four xyloglucan endotransglucosylase/hydrolase, three pectin methylesterases, and six plant invertase/pectin methylesterase inhibitor superfamily. The transcriptome changes among these gene calls could not fully explain the phenotypic characteristics observed in the transgenic events, again suggesting that post‐transcriptional regulations influence soybean’s adaptation to the expression of AtWRI1 and AtDGAT1 during maturation.
Metabolic profile of WD events during reproductive stage of development under greenhouse conditions
To gain insight on the potential post‐transcriptional regulations that are influencing the observed seed phenotypic outcomes in the WD events a metabolic profiling of the developing embryos was carried out targeting the same four developmental stages selected for the transcriptomics analysis. Here, a total of 101 intermediates of central metabolism were quantified, including amino acids, sugars, sugar alcohols, organic acids, and phosphorylated compounds (Table S4). Principal component analysis of the data at each developmental stage showed a clear separation between the metabolomic profiles of WD‐1 and ‐2 and the WT, with the number of metabolites with significantly different levels increasing at later stages (Figure S12). At each time point, the top 15 metabolites with significantly different levels compared to the WT were identified. Among the main differences observed in the transgenic events, included many sugars and sugar phosphates, TCA intermediates, and amino acids such as Glu, Gly, and Lys (Figure S12).
Among the changes observed in the various sugar metabolites, the levels of sucrose and sucrose 6‐phosphate were reduced in the WD events, while many of the intermediates of the preparatory phase of glycolysis were increased (Figure 7). The precursors of starch biosynthesis, on the other hand, including glucose 1‐phosphate (quantified together with mannose 1‐phosphate) and ADP‐glucose, accumulated to higher levels in the transgenic events (Figure 7). The increase in ADP‐glucose, despite the observed down‐regulation of the gene putatively encoding the ADP‐glucose pyrophosphorylase larger subunit (Figure 7), is consistent with the larger amount of starch during development (Figure 2d). How these changes in levels of sugars and sugars phosphates control the flux of carbon towards the different end products: TAG, starch or the cell wall matrix, is unknown. However, the results taken together clearly show post‐transcriptional regulatory processes are contributing to the observed distinctive seed composition phenotypes of the WD events.
Discussion
The identification of novel genetic variation leading to improved yield, protection of yield, and/or quality of the harvest requires a better understanding of a plant’s metabolism and adaptability to perturbations. Progress in this space is a continuous improvement process through a build‐test‐learn cycle. The expression of AtWRI1 and/or AtDGAT1 has been widely used as an approach to increase carbon flux towards oil accumulation, with varied levels of success (Baud et al., 2009; Guo et al., 2020; Jako et al., 2001; Li et al., 2015; Roesler et al., 2016; Vanhercke et al., 2013). In soybean, seed‐specific ectopic expression of GmWRI1a (Glyma.15G221600) in a ‘Williams 82’ background, showed an increase in total oil, namely changes in oleic and linoleic acids (Chen et al., 2018). In contrast, the expression of AtWRI1 under the control of conglycinin promoter in Thorne background, led to negligible changes in total seed oil content, but palmitate levels increased by approximately 20% (Vogel et al., 2019). Therefore, it seems that the effect of transgenic AtWRI1 expression on oil content and profile have a strong dependence on the genetic design, promoter, isoform, tissue, variety, and/or species used. In this work, we analysed specifically Thorne embryos and their response to the localized expression of AtWRI1 and AtDGAT1 during seed development.
AtWri1 and AtDGAT1 were expressed at different levels in Thorne embryos
The two events selected for deeper characterizations that expressed AtWRI1 and AtDGAT1, WD‐1 and WD‐2, shared a similar degree of cellular changes at both the transcriptome and metabolome level. The two‐gene stack was successfully co‐expressed in the events, with relatively high transcript accumulation across the reproductive stages analysed (Figures 4 and S9). AtWRI1 transcripts, however, reached higher levels than the ones of AtDGAT1 (Figures 4 and S9). Multi‐gene engineering strategies that rely on multiple copies of the same promoter are often associated with sub‐optimal co‐expression levels, which can result from DNA instability or transgene silencing (Shockey et al., 2015). Furthermore, the lower levels of AtDGAT1 expression could contribute to the similarity in the phenotype of the WD events with events that expressed only AtWRI1 (Vogel et al., 2019).
Multiple processes restricted uncontrolled fatty acid increase to maintain cellular lipid homeostasis
The analysis of the WD events showed that the introduction of AtWRI1 and AtDGAT1 into Thorne genome provoked changes in the seed composition over development. Protein and total FA contents were lower in the WD events in early seed maturation (Figure 2), but reached WT levels at maturity (Figure 1). In addition, despite the attempt to pull more FA into TAG biosynthesis by the simultaneous co‑expression of AtWRI1 and AtDGAT1, TAG levels were not enhanced in the transgenic events (Figure 1). Nearly all the steps of glycolysis and de novo FA synthesis were up‐regulated (Figure 6), suggesting that other downstream regulatory mechanisms are limiting the ‘PUSH and PULL’ efforts. We identified four processes that were affected in the transgenic events and that are able to restrict oil accumulation. Firstly, WD‐1 and WD‐2 events displayed an up‐regulation of the transcripts annotated as BADC (Figure 6). These proteins were first described as long‐term irreversible inhibitors of the pACC in the presence of excess FA, and consequently FA synthesis inhibitors (Liu et al., 2019; Salie et al., 2016). A contrasting role of BADC on pACC was recently suggested (Shivaiah et al., 2020), where these proteins may facilitate the assembly of its intermediary subcomplexes. The regulatory role of this protein over the activity of pACC needs further investigation to fully address its potential as a bottleneck. Secondly, the WD events displayed altered expression of multiple genes putatively encoding lipases (Figure S11). We identified 16 differentially expressed genes putatively encoding proteins with lipolytic activities, 15 of which were up‐regulated in at least one stage of development in both WD‐1 and ‐2 events (Figure S11). Particularly, the expression of three of these genes putatively encoding GDSL‐motif lipases, Glyma.09G074000, Glyma.15G182900, and Glyma.15G183000, were induced from 4 to 64 times at the developmental stages evaluated. Monitoring accumulation of vegetative lipids in tobacco leaves, Grimberg et al. (2015) postulated the development of a futile cycle of FA synthesis and degradation when transiently expressing different WRI1 homologs in Nicotiana benthamiana. It was suggested that boosting TAG levels in a cell, may trigger the up‐regulation of gene calls with FA degradation function, thereby acting like a cellular rheostat to limit oil accumulation. Thirdly, the strong repression of putative OB proteins, such as oleosins and caleosins (Figure 6), suggests that TAG packaging is altered in WD events, which, ultimately, may affect the oil accumulation capacity of the embryo. In accordance with our results, a recent study, where GmOLE1 (Glyma.20G196600) was overexpressed, suggested that soybean oil content is limited by oleosin expression level (Zhang et al., 2019). Finally, it cannot be excluded that the down‐regulation of the endogenous WRI1 homologs in the transgenic events impacted oil enhancement through the reduction of the expression of specific targets not recognized by AtWRI1 (Figures 4 and S9). It has been previously shown that the ability of the upstream region of WRI1 to initiate transcription in planta is negatively affected by the presence of WRI1 proteins (Snell et al., 2019). This conserved negative autoregulatory mechanism of WRI1 expression is a result of downstream actions of WRI1, as this protein is unable to bind to its upstream region (Snell et al., 2019). This negative loop can explain the down‐regulation of the GmWRI1 homologs when AtWRI1 is strongly expressed (Figure 4). Altogether, these results indicate that soybean embryos respond to AtWRI1 and AtDGAT1 expression by triggering mechanisms, at many levels, to restrict uncontrolled FA increase and maintain cellular lipid homeostasis.
FA composition was altered in mature seed of the transgenic events, showing higher levels of palmitic and linoleic acid and reductions in stearic and oleic acid, evident from early maturation (Figures 1 and 3). These changes may arise, partially, as a consequence of the up‐regulation, at all the studied points of development, of one FATB homolog (Glyma.04g151600), putatively encoding an acyl–acyl carrier protein thioesterase (Figure 6). An accumulation of palmitate in Arabidopsis seeds has been reported in lines overexpressing AtFATB1 (Dörmann et al., 2000). However, contrasting results have been reported in Chen et al. (2018) where an up‐regulation of seven GmFATB homologs (including Glyma.04g151600) with no increase in palmitate levels were reported in GmWRI1a overexpressing seeds. Further studies will be needed to corroborate the impact of each GmFATB homolog in seed FA composition.
The embryos expressing AtWRI1 and AtDGAT1 also responded with an alteration of their carbohydrate metabolism (Figures 1, 2, and S5). An increase in the levels of sugar and sugar phosphates was observed in the WD events along the reproductive stages monitored (Figure 7), with the exception of sucrose, sucrose 6‐phosphate, and trehalose 6‐phosphate. Increased expression of genes encoding sucrose degradative enzymes, such as invertase and sucrose synthase, coupled with a down‐regulation of invertase inhibitor and sucrose biosynthesis genes, likely explains the lower levels of sucrose and higher content of hexoses and hexose phosphates observed in the embryos (Figure 7). Trehalose 6‐phosphate, a signal of cellular sucrose status, can regulate FA biosynthesis by stabilizing WRI1 protein, avoiding its phosphorylation by SnRK1 and posterior degradation by the proteasome (Zhai et al., 2018). Conversely, when the cellular sucrose content is low, levels of trehalose 6‐phosphate decrease, WRI1 becomes phosphorylated and is degraded, with the result of reducing energy‐consuming pathways (Zhai et al., 2018). Even though AtWRI1 targets were induced, the reduction in trehalose 6‐phosphate levels, may have increased AtWRI1 turnover, which in turn, may have lessened the degree of up‐regulation of its target gene calls in the soybean genome. The alteration of the embryo’s hexose metabolic status during development also affected the rate of accumulation of starch and hemicelluloses (Figures S4 and S6), which was not fully accompanied by changes in the expression of genes involved in these biosynthetic pathways. These data further support the notion that post transcriptional processes such as metabolic regulation and carbon redistribution may also have contributed to the restriction in oil accumulation in the events.
Interestingly, there are some similarities between the Arabidopsis embryos' responses to a complete loss of WRI1 (Atwri1 mutants) (Focks and Benning, 1998) and outcomes observed in soybean co‐expressing AtWRI1 and AtDGAT1. Except for the extremely low oil accumulation in the Atwri1, the Arabidopsis mutant and the WD events share some similar physiological and biochemical responses, including a wrinkled seed phenotype, reduced germination, higher starch accumulation, higher glucose and fructose (except sucrose), and a deficient conversion of sucrose into FA (Focks and Benning, 1998). Therefore, perturbations in lipid homeostasis, in both directions, led to similar embryo adaptations.
AtWRI1 potential targets were identified
The systematic prediction of AtWRI1 potential targets in the soybean genotype Thorne, by a comparative analysis of gene calls up‐regulated in WD‐1, WD‐2, versus the W‐1 events, revealed a high correlation with the expression of AtWRI1, and the presence of an AW‐box in their upstream sequence (Table S3). The gene calls identified through this process include those functionally annotated for late glycolytic and FA biosynthetic genes, which is consistent with previous reports (Fei et al., 2020). Among the putative targets with central metabolism functions, gene calls with an AW‐box footprint in their up‐stream region included a putative glucose 6‐phosphate/phosphate translocator (GPT2), UDP‐glucose pyrophosphorylase (UGP2), and plastidic malate dehydrogenase (Table 1). GPT2 is responsible for the transport of glucose 6‐phosphate into the plastids where it can be used as a carbon source for starch and FA biosynthesis or for NADPH generation via the oxidative pentose phosphate pathway (Kunz et al., 2010; Niewiadomski et al., 2005). In addition to having a central role in carbon partitioning between cytosol and plastids, GPT2 transcript abundance correlates with the levels of glucose, and its induction by the sugar‐sensing pathway was proposed as a mechanism to regulate cellular sugar homeostasis (Kunz et al., 2010). The regulation of GPT2 by AtWRI1 may provide another mechanism by which AtWRI1 can direct carbohydrates towards FA synthesis. Similarly, the induction of UGP2 by AtWRI1 could direct UDP‐glucose—the product of sucrose synthase, and other AtWRI1 known target (Fei et al., 2020)—towards the production of glucose 1‐phosphate and glucose 6‐phosphate impacting both glycolysis and FA biosynthesis (Figure 7). However, in soybean embryos co‐expressing AtWRI1 and AtDGAT1, UDP‐glucose carbons were directed towards cell wall polysaccharides, and glucose 1‐phosphate apparently shunted to starch biosynthesis (Figure 7). Finally, plastidic malate dehydrogenase was found to contribute to the production of carbon precursor and NADPH for de novo FA synthesis in developing maize (Alonso et al., 2010; Cocuron et al., 2019) and pennycress embryos (Tsogtbaatar et al., 2020), as well as in the maintenance of the energy homeostasis in developing Arabidopsis thaliana seeds (Selinski et al., 2014). Given these outcomes, it is probable that WRI1 governs additional processes to ensure the cellular environment and carbon flux for FA synthesis.
In addition to the identification of FATB and BADC gene calls that were previously proposed as AtWRI1 targets (Liu et al., 2019; Vogel et al., 2019) four putative lipolytic genes were highlighted in this study (Figure S11). These include, a MAGL and three GDSL‐motif lipases (Figure S11). As it was suggested for BADC FA inhibition (Liu et al., 2019), coordinated regulation of oil biosynthetic and degradative genes by WRI1 help maintain cellular homeostasis of lipids.
Additional putative AtWRI1 targets identified in soybean included hormone‐related genes annotated to encode for auxin‐responsive GH3 family proteins and an auxin response factor protein (Table 1). AtGH3.3, a gene involved in auxin degradation, was found up‐regulated in wri1 Arabidopsis mutants, and AtWRI1 protein was proven to recognize and bind to the AW‐box in AtGH3.3 promoter (Kong et al., 2017). A role for AtWRI1 in root auxin homeostasis was suggested (Kong et al., 2017), however remains to be studied if that function is occurring in embryos.
Novel strategies for oil improvement in soybean
The findings of this study can guide future genetic strategies aiming to improve oil content in soybean seeds. Firstly, the beta‐conglycinin promoter may not be ideal for this effort, as it allows some degree of expression in roots (Figure S7). As mentioned above, AtWRI1 affects auxin responses in roots (Kong et al., 2017) and therefore, root architecture, which may explain the reduced yield in WD events (Table 1). Secondly, if oil levels are to be enhanced, it will require sufficient availability of OB packaging proteins to support an increased TAG biosynthesis. Thirdly, editing of the AW‐box or targeted down‐regulation of the undesired FA synthesis inhibitors and lipolytic targets of AtWRI1 may also need to be considered in order to protect the increased accumulation of oil. Finally, genetic approaches to reduce the flow of hexose phosphates to competing pathways, such as starch and cell wall polysaccharides, should be evaluated.
Methods
Vectors construction and plant transformation
AtDGAT1 (GenBank accession NP_179535.1) and AtWRI1 (GenBank accession NP_191000) sequences were codon optimized for soybean expression and synthesized by GenScript, Piscataway, NJ. Both genes were under the control of the soybean seed‐specific beta‐conglycinin promoter (Allen et al., 1989), fused to the tobacco etch virus translational enhancer element (Carrington and Freed, 1990), and terminated with the 35S cauliflower mosaic virus polyadenylation signal (Figure S1). The two gene cassettes were assembled into a final binary vector, referred as pPTN1248, which harbours a bar gene (Thompson et al., 1987) under control of the Agrobacterium Pnos promoter for herbicide selection (Figure S1). pPTN1248 was mobilized into A. tumefaciens strain EHA101 (Hood et al., 1986) via triparental mating. The transformations were performed on the soybean variety Thorne using the procedures previously described (Xing et al., 2000; Zhang et al., 1999). The identification of transformed plants was carried out by monitoring the expression of the bar gene via leaf painting technique (Zhang et al., 1999). Genomic DNA isolated following a modified CTAB method (Springer, 2010) was used for PCR genotyping in all generations of the transgenic plants. Synthetic AtWRI1 gene was amplified with the primer set 5’‐ GTCTATCTCGGTGCTTACGACTCT‐3’ and 5’‐ CCTGAGGTGGTTCCTCTACATACTG‐3’. Amplification of AtDGAT1 was carried out with the primer set 5’‐ GATCGCTGTTAACTCTAGGCTGA‐3’ and 5’‐AGCAGTAGAACATACAGAGCCACAC‐3’. The PCR reactions were conducted with GoTaq Green Master Mix, following the manufacturer’s instructions (Promega Corporation, Madison, WI). The number of transgenic allele integrations was determined by Southern blot hybridization. For that, genomic DNA was isolated from young leaves according to the protocol described by Dellaporta et al., 1983. Ten micrograms of the genomic DNA were digested with the restriction enzyme SalI, which has a single restriction site within the T‐DNA at the bar ORF. Then, DNA fragments were separated by electrophoresis on 0.8% agarose gels, blotted and UV crossed linked to a nylon membrane (Bio‐Rad cat #162‐0196, Hercules, CA). Membranes were hybridized with a labelled probe generated by amplification of a sequence of either AtWRI1 or AtDGAT1 by the random primer labelling technique using dCT32P (Prime‐It II Cat # 300385; Stratagene, La Jolla, CA) as previously described (Eckert et al., 2006). For homozygous plant identification, genotyping was performed for 24 or 48 plants, for single or double insertion events, respectively, along different generations of soybean in the field.
Field growing conditions and germination and yield analyses
Field trials were conducted on homozygous transgenic events and lineages of T4, and T5 generations of transgenic plants in crop seasons 2018, 2019, and 2020. The soybean variety Thorne was used as a control for all cropping seasons. In 2018, three independent events of Thorne transformation with pPTN1248, namely 970‐1 (WD‐1), 970‐2 (WD‐2), and 978‐3 (WD‐3) were evaluated, while in 2019 and 2020, only the WD‐1 event was analysed. In 2019, the transgenic event 917‐17 (W‐1) which expressed the single gene AtWRI1 (Vogel et al., 2019) was also included in the field trial. All evaluations were conducted in completely randomized plots in three replicates. Each plot consisted of four 10 ft rows in a four‐row plot design (30 inches row to row) in which two border rows were the WT Thorne. Data were collected from the plants in the inner two rows. Either two or three plots per event or lineage were planted each year depending on the seed availability. One hundred seeds were sown in each row. After sowing for two weeks, a total number of plants were counted and germination rates were calculated based on the initial number of seeds sown (200 seeds per plot). Observations were performed for phenology according to a soybean guideline (Pedersen et al., 2004). At harvest, total seed weight per plot and one hundred seed weights were recorded.
Greenhouse growing conditions and sample collection
The seeds were planted in 30 by 26 cm plastic pots (Nursery supplies Inc., Orange, CA, USA) containing Baccto, Horticultural sphagnum peat, and perlite (Michigan peat company, Houston, TX, USA). After planting, the pots were placed in a greenhouse at 30 and 25 °C, day and night, respectively, with a photo‐period of 16 h. The light intensity was maintained at around 450 μmol m–2 s–1. The transgenic events, WD‐1, WD‐2, and the control WT were separated in the greenhouse by pollen screens to avoid cross‐pollination between genotypes. After the first 45 days, the plants were fertilized every 2 weeks with 6 g of 15‐5‐15 fertilizer (Everris NA Inc., Dublin, OH, USA). Open flowers were tagged every day in order to identify the correct developmental stage. Samples for biomass, metabolomic and transcriptomic analysis were collected at different points of development (21, 24, 27, 30, 33, 36, 42, and 48 averaged DAP) and at maturity. Samples from each developmental stage corresponded to three DAP that once averaged matches the given timepoint. For example, the 21 DAP samples were collected from 20, 21, and 22 DAP pods, and 24 DAP samples were collected from 23, 24, and 25 DAP pods, etc. The collected immature seeds were peeled to remove the seed coat and endosperm and frozen under liquid N2. At least four replicates were collected for each time point and genotype, and each replicate consisted of one embryo, with the exception of 21 and 24 DAP, where three embryos were used instead. For the mature stage, four seeds were ground for each replicate and four replicates were analysed. Finally, the samples for biomass and metabolomics were freeze dried in a lyophilizer (Labconco Freezone 12 plus Freeze Dryer) for three days prior to analysis while samples for transcriptomic were kept at −80 °C.
Biomass extraction and quantification
Seven mature seeds collected from two plants from the field (2018) were pooled as one biological replicate and three replicates were used to determine the total FA, FA compositions, and TAG. The seeds were grounded using a coffee grinder and ~200 mg of the seed powder was used for lipid extraction, using chloroform and methanol method (Bligh and Dyer, 1959). For each tube, 10 mg of triheptadecanoin was used as a standard reference for calculation of total lipid. The lipid phase was collected and divided into two parts for total lipid and TAG analyses, then dried at 50 °C under nitrogen. TAG was purified using the Supelco Supelclean LC‐Si SPE column and dried under nitrogen at 50 °C. Both total lipid and total TAG fractions were then methylated using methanol/sulphuric acid 2.5% for 45 min at 90 °C. FAMEs were isolated and analysed on a 6890N gas chromatography‐flame ionization detector (Agilent Technologies, Santa Clara, CA) fitted with a 30 m × 250 µm HP‐INOWAX column (Cat # 19091N‐133; Agilent Technologies).
Immature embryos (21–36 DAP) and mature seeds collected under greenhouse conditions were analysed according to their biomass content. FA and proteins, and starch were sequentially extracted and quantified as previously described (Cocuron et al., 2014) with minor modifications. Briefly, 5 mg of dried grounded embryos, supplemented with 100 μg of glyceryl triheptadecanoate as internal standard, were subjected to three consecutive rounds of FA extractions using 1 ml of hexane/isopropanol (2:1). The dried pooled FA supernatant was methylated using methanol/sulphuric acid 2.5% for 120 min at 80 °C. FAMEs were then analysed using an Agilent Technologies 6890N network GC system coupled to a single‐quadrupole 5975B VLMSD mass spectrometer and injector 7683B series (Santa Clara, CA, USA). The oven's initial temperature was set to 120 °C and held for 0.5 min after being raised to 120 °C at 100 °C/min to reach a final temperature of 245 °C which was held for 4.25 min. FAME derivatives were quantified using the known internal standard concentration. Proteins were extracted from the residual pellet after FA extraction by three rounds of 1 ml of extraction buffer [20 mm of Tris–HCl, pH 7.5, 150 mm of NaCl, and 1% (w/v) SDS]. The supernatants were combined and total protein content was quantified using the DC Protein Assay kit from Bio‐Rad and bovine serum albumin (BSA) as a standard. Finally, starch extraction was performed on the pellets after protein extraction by digestion with 10 µL of amyloglucosidase from the Megazyme International Ireland Ltd Total Starch Assay Kit for 1, 5 h at 55 °C. Glucose content was then quantified using the colorimetric method from the Total Starch Assay Kit (Megazyme International Ireland). For all pairwise multiple comparison procedures, Fisher LSD method or Student‐Newman‐Keuls method was used as parametric or non‐parametric test, respectively using Sigmaplot 12.3. GraphPad Prism 9 software was used for slope statistics.
Carbohydrate quantification
Soluble sugars, hemicellulose and cellulose from mature soybean seed and immature embryos (24, 30, 33,36, 42, and 48 DAP) were quantified as described in Moretti et al. (2020).
RNA extraction of greenhouse collected samples and sequencing
Immature embryos of the same greenhouse grown plants as the biomass and metabolomics analyses were used to collect RNA for the transcriptomics analysis. For each of the three genotypes of WD‐1, WD‐2, and WT, RNA was extracted at four timepoints of 24, 27, 30, and 33 DAP. For each replicate, one immature embryo was flash‐frozen with liquid nitrogen and ground with pestle and mortar into fine powder from which RNA was isolated and cleaned using the Macherey‐Nagel NucleoSpin RNA Plant Mini kit (MACHEREY‐NAGEL Inc.). RNA samples were submitted for sequencing via the Illumina NovaSeq 6000 platform with 150 bp paired‐end reads at the Genomics Shared Resource of the Ohio State University Comprehensive Cancer Center.
Field‐grown sample collection, RNA extraction, and sequencing
Immature seeds from WD‐1, W‐1, and WT grown in the field (2019) were collected at R5, R5/6, and R6 developmental stages (Pedersen et al., 2004). Stages R5 and R5/6 are equivalent to the 24 DAP and 30 DAP timepoints in the greenhouse, respectively. For each biological replicate, four seeds from one plant were bulked, and three plants were used as a different replicate. RNA extraction was performed using the QIAGEN RNEAsy Kit (QIAGEN, INC.). The quality of the RNA was evaluated by 1% agarose gels and its concentration was quantified by Nanodrop. The RNA was cleaned from any DNA contamination by DNAse treatment using the Turbo DNA free kit (Invitrogen, INC.). The sequencing of RNA was performed via Illumina NovaSeq 6000 by the University of Nebraska Medical Center.
Sequence alignment and differential expression analysis
Illumina‐associated adapter sequences were trimmed by the sequencing service. Sequence alignment and differential expression analysis were conducted using the kallisto/sleuth pipeline. Reads of all sequenced samples were assessed for quality control using the FastQC program (version 0.11.5). High‐quality reads were loaded to kallisto (version 0.43.1) available as a graphic user interface on Cyverse (www.cyverse.org) with bootstrap value set at 100 and k‐mer length at 31. AtWRI1 and AtDGAT sequences used in the transformation vector were added to the G. max cDNA database corresponding to the Wm82.a2.v1 genome retrieved from Ensembl (http://plants.ensembl.org). This cDNA library served as the transcriptome reference for pseudo‐alignment and read quantification. Results from kallisto were used as input for differential expression analysis at gene level via the sleuth package (version 0.30.0) in R (version 3.6.3). Pairwise comparisons between WD‐1, WD‐2, and W‐1 versus WT plants were conducted at each of the timepoints. Beta values from the Wald test were used as estimators of log2 fold‐change (log2FC) values. Differentially expressed genes (DEG) had likelihood ratio test‐derived FDR‐adjusted P‐values (greenhouse data) or p‐values (field data) below 0.05 and fold change of 2 (or |log2FC| ≥ 1). Due to high variation in field data, unadjusted P‐values, rather than adjusted p‐values, were used as a less stringent cutoff.
Gene set enrichment analysis
Common DEGs found in both pairwise comparisons between WD‐1 versus WT and WD‐2 versus WT were used for gene enrichment analysis at each timepoint. Gene Ontology (GO) analysis was conducted via AgriGOv2 (http://bioinfo.cau.edu.cn) and KEGG pathway via DAVID (http://david.ncifcrf.gov). Significantly enriched KEGG and GO terms had FDR‐adjusted p‐values below 0.05. Detailed annotation of DEG was obtained from SoyBase (http://soybase.org) and Phytozome v12.0 (http://www.phytozome.net).
Gene co‐expression analysis
Greenhouse RNA‐seq data were used to calculate correlation with AtWRI1. Each gene has 12 data points for three genotypes and four time points. The mean values of three replicates on the same time point were used. The Pearson correlation was calculated between AtWRI1 and other 49661 expressed genes with the R function cor(). These expressed genes have a total of TPM > 0.
Targeted metabolomics
For targeted metabolomics analysis, four time points (24, 27, 30, and 33 DAP, four replicates each) collected under greenhouse conditions at a consistent hour of the morning were studied. Intracellular metabolites were extracted from 5 mg of ground freeze‐dried soybean embryos using boiling water, and following the procedure described by Cocuron et al. (2014) and De Souza et al. (2015). Sugars, sugar alcohols, amino acids, organic acids, and phosphorylated compounds were measured using an UHPLC 1290 Infinity II from Agilent Technologies (Santa Clara, CA, USA) coupled with a linear triple‐quadrupole/ion trap mass spectrometer QTRAP 6500+ (AB Sciex Instruments, Framingham, MA, USA) using the methodologies previously described (Cocuron et al., 2014). The quantification was accomplished using (i) [U‐13C6] glucose, [U‐13C4] fumarate, and [U‐13C2] glycine as internal standards for each type of metabolites to account for any loss of material during sample preparation; (ii) unlabelled external standard mix run in parallel to the samples with known concentrations of the different metabolites. All measurements lie within the linear range of the analytical method. Principal component analysis, ANOVA, and Fisher LSD analysis were performed in log transformed data using the free web‐based statistical software MetaboAnalyst 5.0 (P < 0.05) (Pang et al., 2021).
Accession numbers
The RNA‐seq discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE186058 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE186058).
Conflict of interest
The authors declare no conflict of interest.
Author contributions
A.P.A, L.M., and T.E.C. conceived and designed the project. H.N., M.G., and T.Q. designed and generated the transgenic events. T.Q. grew and analysed the transgenic events in the field. A.M., A.R., and C.L.A. grew the plants under greenhouse conditions, collected the samples and performed the biomass, metabolomic, and carbohydrate analysis. T.Q., T.H., and K.V. performed the RNA extraction from the field and greenhouse samples. T.H., Y.S., and C.Z. processed the RNAseq data. C.L.A., T.Q., T.H., and C.Z. contributed to data analysis, interpretation and drafted the manuscript. C.L.A., T.Q., T.H., C.Z., A.P.A, L.M., and T.E.C. reviewed and edited the document. All authors critically revised and agreed to the published version of the manuscript.
Supporting information
Figure S1 T‐DNA of plasmid pPTN1248 harbouring a soybean codon‐optimized AtWRI1 transcription factor and AtDGAT1 from Arabidopsis under control of the beta‐conglycinin promoter.
Figure S2 Southern blot analysis of the transgenic soybean events expressing AtWRI1 and AtDGAT1.
Figure S3 Wrinkled‐seed phenotype.
Figure S4 DW, protein, FA, and starch accumulation rates.
Figure S5 Accumulation of cell wall polysaccharides during development.
Figure S6 Rate of glucose accumulation as hemicelluloses in WD events.
Figure S7 Beta‐conglycinin promoter pattern of expression.
Figure S8 DEGs in transgenic soybeans grown under greenhouse conditions.
Figure S9 Expression of WRI1 and DGAT1 from Arabidopsis and soybean in transgenic soybean grown under field conditions.
Figure S10 Differentially expressed genes in the field experiment and comparison with the greenhouse results.
Figure S11 Changes in lipid transfer protein and lipid degradation enzyme expression in WD events between 24 and 33 DAP under greenhouse conditions.
Figure S12 Principal component analysis and top fifteen changes in metabolites.
Table S1 Soybean field trials in 2018, 2019, and 2020 crop seasons in Nebraska fields.
Table S2 Total number of differential expressed genes for each transgenic event at each developmental stage.
Table S3 Complete list of putative AtWRI1 targets.
Table S4 Metabolite concentrations.
Acknowledgements
The authors thank the BioAnalytical Facility (bdi.unt.edu/baf) and Jean‐Christophe Cocuron at University of North Texas for access to the mass spectrometry equipment. This project was supported by the United Soybean Board (Projects 2020‐162‐0123, 2120‐162‐0119, and 2220‐162‐0115 to L.M., A.P.A. and T.E.C.), the National Science Foundation (Award #: OIA‐1557417 to C.Z.) and the Nebraska Soybean Board (Award 20R‐09‐1/2 #1739 to C.Z. and T.E.C. and Award No. 21R‐03‐19/19#750 to T.E.C).
Arias, C. L. , Quach, T. , Huynh, T. , Nguyen, H. , Moretti, A. , Shi, Y. , Guo, M. , Rasoul, A. , Van, K. , McHale, L. , Clemente, T. E. , Alonso, A. P. and Zhang, C. (2022) Expression of AtWRI1 and AtDGAT1 during soybean embryo development influences oil and carbohydrate metabolism. Plant Biotechnol. J., 10.1111/pbi.13810
Contributor Information
Ana Paula Alonso, Email: anapaula.alonso@unt.edu.
Chi Zhang, Email: zhang.chi@unl.edu.
References
- Allen, R.D. , Bernier, F. , Lessard, P.A. and Beachy, R.N. (1989) Nuclear factors interact with a soybean beta‐conglycinin enhancer. Plant Cell, 1, 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso, A.P. , Piasecki, R.J. , Wang, Y. , LaClair, R.W. and Shachar‐Hill, Y. (2010) Quantifying the labeling and the levels of plant cell wall precursors using ion chromatography tandem mass spectrometry. Plant Physiol. 153, 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An, D. and Suh, M.C. (2015) Overexpression of Arabidopsis WRI1 enhanced seed mass and storage oil content in Camelina sativa . Plant Biotechnol. Rep. 9, 137–148. [Google Scholar]
- Bates, P.D. (2016) Understanding the control of acyl flux through the lipid metabolic network of plant oil biosynthesis. Biochim . Biophys. Acta ‐ Mol. Cell Biol. Lipids, 1861, 1214–1225. [DOI] [PubMed] [Google Scholar]
- Bates, P.D. , Durrett, T.P. , Ohlrogge, J.B. and Pollard, M. (2009) Analysis of acyl fluxes through multiple pathways of triacylglycerol synthesis in developing soybean embryos. Plant Physiol. 150, 55–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud, S. , Mendoza, M.S. , To, A. , Harscoët, E. , Lepiniec, L. and Dubreucq, B. (2007) WRINKLED1 specifies the regulatory action of LEAFY COTYLEDON2 towards fatty acid metabolism during seed maturation in Arabidopsis. Plant J. 50, 825–838. [DOI] [PubMed] [Google Scholar]
- Baud, S. , Wuillème, S. , To, A. , Rochat, C. and Lepiniec, L. (2009) Role of WRINKLED1 in the transcriptional regulation of glycolytic and fatty acid biosynthetic genes in Arabidopsis. Plant J. 60, 933–947. [DOI] [PubMed] [Google Scholar]
- Bligh, E. and Dyer, W. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Carrington, J.C. and Freed, D.D. (1990) Cap‐independent enhancement of translation by a plant potyvirus 5’ nontranslated region. J. Virol. 64, 1590–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cernac, A. and Benning, C. (2004) WRINKLED1 encodes an AP2/EREB domain protein involved in the control of storage compound biosynthesis in Arabidopsis. Plant J. 40, 575–585. [DOI] [PubMed] [Google Scholar]
- Chen, B. , Zhang, G. , Li, P. , Yang, J. , Guo, L. , Benning, C. et al. (2020) Multiple GmWRI1s are redundantly involved in seed filling and nodulation by regulating plastidic glycolysis, lipid biosynthesis and hormone signalling in soybean (Glycine max). Plant Biotechnol. J. 18, 155–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L. , Zheng, Y. , Dong, Z. , Meng, F. , Sun, X. , Fan, X. et al. (2018) Soybean (Glycine max) WRINKLED1 transcription factor, GmWRI1a, positively regulates seed oil accumulation. Mol. Genet. Genomics, 293, 401–415. [DOI] [PubMed] [Google Scholar]
- Clemente, T.E. and Cahoon, E.B. (2009) Soybean oil: genetic approaches for modification of functionality and total content. Plant Physiol. 151, 1030–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocuron, J.C. , Anderson, B. , Boyd, A. and Alonso, A.P. (2014) Targeted metabolomics of Physaria fendleri, an industrial crop producing hydroxy fatty acids. Plant Cell Physiol. 55, 620–633. [DOI] [PubMed] [Google Scholar]
- Cocuron, J.‐C. , Koubaa, M. , Kimmelfield, R. , Ross, Z. and Alonso, A.P. (2019) A combined metabolomics and fluxomics analysis identifies steps limiting oil synthesis in maize embryos. Plant Physiol. 181, 961–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza, A.P. , Cocuron, J.‐C. , Garcia, A.C. , Alonso, A.P. and Buckeridge, M.S. (2015) Changes in whole‐plant metabolism during the grain‐filling stage in sorghum grown under elevated CO2 and drought. Plant Physiol. 169, 1755–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellaporta, S.L. , Wood, J. and Hicks, J.B. (1983) A plant DNA minipreparation: Version II. Plant Mol. Biol. Rep. 1, 19–21. [Google Scholar]
- Dohlman, E. , Hansen, J. & Boussios, D. (2021) USDA Agricultural Projections to 2030. Interagency Agricultural Projections Committee. USDA Long‐Term Projections Report OCE‐2021‐1, 103 pp.
- Dörmann, P. , Voelker, T.A. and Ohlrogge, J.B. (2000) Accumulation of palmitate in Arabidopsis mediated by the acyl‐acyl carrier protein thioesterase FATB1. Plant Physiol. 123, 637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert, H. , La Vallee, B. , Schweiger, B. , Kinney, A. , Cahoon, E. and Clemente, T.E. (2006) Co‐expression of the borage Delta 6 desaturase and the Arabidopsis Delta 15 desaturase results in high accumulation of stearidonic acid in the seeds of transgenic soybean. Planta, 224, 1050–1057. [DOI] [PubMed] [Google Scholar]
- Edgar, R. , Domrachev, M. and Lash, A.E. (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Erp, H. , Kelly, A.A. , Menard, G. and Eastmond, P.J. (2014) Multigene engineering of triacylglycerol metabolism boosts seed oil content in Arabidopsis. Plant Physiol. 165, 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei, W. , Yang, S. , Hu, J. , Yang, F. , Qu, G. , Peng, D. and Zhou, B. (2020) Research advances of WRINKLED1 (WRI1) in plants. Funct. Plant Biol, 47, 185–194. [DOI] [PubMed] [Google Scholar]
- Focks, N. and Benning, C. (1998) Wrinkled 1: A novel, low‐seed‐oil mutant of Arabidopsis with a deficiency in the seed‐specific regulation of carbohydrate metabolism. Plant Physiol. 118, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimberg, Å. , Carlsson, A. , Marttila, S. , Bhalerao, R. and Hofvander, P. (2015) Transcriptional transitions in Nicotiana benthamiana leaves upon induction of oil synthesis by WRINKLED1 homologs from diverse species and tissues. BMC Plant Biol. 15, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, W. , Chen, L. , Chen, H. , Yang, H. , You, Q. , Bao, A. et al. (2020) Overexpression of GmWRI1b in soybean stably improves plant architecture and associated yield parameters, and increases total seed oil production under field conditions. Plant Biotechnol. J. 18, 1639–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood, E.E. , Helmer, G.L. , Fraley, R.T. and Chilton, M.D. (1986) The hypervirulence of Agrobacterium tumefaciens A281 is encoded in a region of pTiBo542 outside of T‐DNA. J. Bacteriol, 168, 1291–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jako, C. , Kumar, A. , Wei, Y. , Zou, J. , Barton, D.L. , Giblin, E.M. et al. (2001) Seed‐specific over‐expression of an Arabidopsis cDNA encoding a diacylglycerol acyltransferase enhances seed oil content and seed weight. Plant Physiol. 126, 861–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, Q. , Ma, W. , Yang, H. , Ma, G. , Mantyla, J.J. and Benning, C. (2017) The Arabidopsis WRINKLED1 transcription factor affects auxin homeostasis in roots. J. Exp. Bot. 68, 4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz, H.H. , Häusler, R.E. , Fettke, J. , Herbst, K. , Niewiadomski, P. , Gierth, M. et al. (2010) The role of plastidial glucose‐6‐phosphate/phosphate translocators in vegetative tissues of Arabidopsis thaliana mutants impaired in starch biosynthesis. Plant Biol. 12, 115–128. [DOI] [PubMed] [Google Scholar]
- Lardizabal, K. , Effertz, R. , Levering, C. , Mai, J. , Pedroso, M.C. , Jury, T. et al. (2008) Expression of Umbelopsis ramanniana DGAT2A in seed increases oil in soybean. Plant Physiol. 148, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , Shao, J. , Tang, S. , Shen, Q. , Wang, T. , Chen, W. and Hong, Y. (2015) Wrinkled1 accelerates flowering and regulates lipid homeostasis between oil accumulation and membrane lipid anabolism in Brassica napus . Front. Plant Sci. 6, 1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , Zhai, Z. , Kuczynski, K. , Keereetaweep, J. , Schwender, J. and Shanklin, J. (2019) Wrinkled1 regulates biotin attachment domain‐containing proteins that inhibit fatty acid synthesis. Plant Physiol, 181, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Hua, W. , Zhan, G. , Wei, F. , Wang, X. , Liu, G. and Wang, H. (2010) Increasing seed mass and oil content in transgenic Arabidopsis by the overexpression of wri1‐like gene from Brassica napus . Plant Physiol. Biochem. 48, 9–15. [DOI] [PubMed] [Google Scholar]
- Maeo, K. , Tokuda, T. , Ayame, A. , Mitsui, N. , Kawai, T. , Tsukagoshi, H. et al. (2009) An AP2‐type transcription factor, WRINKLED1, of Arabidopsis thaliana binds to the AW‐box sequence conserved among proximal upstream regions of genes involved in fatty acid synthesis. Plant J. 60, 476–487. [DOI] [PubMed] [Google Scholar]
- Moretti, A. , Arias, C.L. , Mozzoni, L.A. , Chen, P. , McNeece, B.T. , Mian, M.A.R. et al. (2020) Workflow for the quantification of soluble and insoluble carbohydrates in soybean seed. Molecules, 25, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewiadomski, P. , Knappe, S. , Geimer, S. , Fischer, K. , Schulz, B. , Unte, U. et al. (2005) The Arabidopsis plastidic glucose 6‐phosphate/phosphate translocator GPT1 is essential for pollen maturation and embryo sac development. Plant Cell, 17, 760–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, Z. , Chong, J. , Zhou, G. , de Lima Morais, D.A. , Chang, L. , Barrette, M. et al. (2021) MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 49, W388–W396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen, P. , Kumudini, S. , Board, J. and Conley, S. (2004) Soybean Growth and Development. Ames, IA: Iowa State University, University Extension. [Google Scholar]
- Roesler, K. , Shen, B. , Bermudez, E. , Li, C. , Hunt, J. , Damude, H.G. et al. (2016) An improved variant of soybean type 1 diacylglycerol acyltransferase increases the oil content and decreases the soluble carbohydrate content of soybeans. Plant Physiol, 171, 878–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salie, M.J. , Zhang, N. , Lancikova, V. , Xu, D. and Thelen, J.J. (2016) A family of negative regulators targets the committed step of de novo fatty acid biosynthesis OPEN. Plant Cell, 28, 2312–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selinski, J. , König, N. , Wellmeyer, B. , Hanke, G.T. , Linke, V. , Neuhaus, H.E. and Scheibe, R. (2014) The plastid‐localized NAD‐dependent malate dehydrogenase is crucial for energy homeostasis in developing Arabidopsis thaliana seeds. Mol. Plant, 7, 170–186. [DOI] [PubMed] [Google Scholar]
- Shen, B. , Allen, W.B. , Zheng, P. , Li, C. , Glassman, K. , Ranch, J. et al. (2010) Expression of ZmLEC1 and ZmWRI1 increases seed oil production in maize. Plant Physiol. 153, 980–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivaiah, K.K. , Ding, G. , Upton, B. and Nikolau, B.J. (2020) Non‐catalytic subunits facilitate quaternary organization of plastidic acetyl‐CoA carboxylase. Plant Physiol. 182, 756–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shockey, J. , Mason, C. , Gilbert, M. , Cao, H. , Li, X. , Cahoon, E. and Dyer, J. (2015) Development and analysis of a highly flexible multi‐gene expression system for metabolic engineering in Arabidopsis seeds and other plant tissues. Plant Mol. Biol. 891, 113–126. [DOI] [PubMed] [Google Scholar]
- Snell, P. , Grimberg, Å. , Carlsson, A.S. and Hofvander, P. (2019) WRINKLED1 is subject to evolutionary conserved negative autoregulation. Front. Plant Sci. 10, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer, N.M. (2010) Isolation of plant DNA for PCR and genotyping using organic extraction and CTAB. Cold Spring Harb. Protoc. 2010, pdb.prot5515. [DOI] [PubMed] [Google Scholar]
- Sun, R. , Ye, R. , Gao, L. , Zhang, L. , Wang, R. , Mao, T. et al. (2017) Characterization and ectopic expression of CoWRI1, an AP2/EREBP domain‐containing transcription factor from coconut (Cocos nucifera L.) endosperm, changes the seeds oil content in transgenic Arabidopsis thaliana and Rice (Oryza sativa L.). Front. Plant Sci. 8, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, C.J. , Movva, N.R. , Tizard, R. , Crameri, R. , Davies, J.E. , Lauwereys, M. and Botterman, J. (1987) Characterization of the herbicide‐resistance gene bar from Streptomyces hygroscopicus . EMBO J. 6, 2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsogtbaatar, E. , Cocuron, J.C. and Alonso, A.P. (2020) Non‐conventional pathways enable pennycress (Thlaspi arvense L.) embryos to achieve high efficiency of oil biosynthesis. J. Exp. Bot. 71, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhercke, T. , El Tahchy, A. , Shrestha, P. , Zhou, X.R. , Singh, S.P. and Petrie, J.R. (2013) Synergistic effect of WRI1 and DGAT1 coexpression on triacylglycerol biosynthesis in plants. FEBS Lett. 587, 364–369. [DOI] [PubMed] [Google Scholar]
- Vogel, P.A. , Bayon de Noyer, S. , Park, H. , Nguyen, H. , Hou, L. , Changa, T. et al. (2019) Expression of the Arabidopsis WRINKLED 1 transcription factor leads to higher accumulation of palmitate in soybean seed. Plant Biotechnol. J. 17, 1369–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Huang, W. , Chang, J. , Sebastian, A. , Li, Y. , Li, H. et al. (2014) Overexpression of SiDGAT1, a gene encoding acyl‐CoA:diacylglycerol acyltransferase from Sesamum indicum L. increases oil content in transgenic Arabidopsis and soybean. Plant Cell Tissue Organ Cult. 119, 399–410. [Google Scholar]
- Xing, A. , Zhang, Z. , Sato, S. , Staswick, P. and Clemente, T.E. (2000) The use of the two T‐DNA binary system to derive marker‐free transgenic soybeans. Vitr. Cell. Dev. Biol. 36, 456–463. [Google Scholar]
- Xu, C. and Shanklin, J. (2016) Triacylglycerol metabolism, function, and accumulation in plant vegetative tissues. Annu. Rev. Plant Biol. 67, 179–206. [DOI] [PubMed] [Google Scholar]
- Zaborowski, A.B. and Walther, D. (2020) Determinants of correlated expression of transcription factors and their target genes. Nucleic Acids Res. 48, 11347–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai, Z. , Keereetaweep, J. , Liu, H. , Feil, R. , Lunn, J.E. and Shanklin, J. (2018) Trehalose 6‐phosphate positively regulates fatty acid synthesis by stabilizing WRINKLED1. Plant Cell, 30, 2616–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D. , Zhang, H. , Hu, Z. , Chu, S. , Yu, K. , Lv, L. et al. (2019) Artificial selection on GmOLEO1 contributes to the increase in seed oil during soybean domestication. PLOS Genet. 15, e1008267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M. , Fan, J. , Taylor, D.C. and Ohlrogge, J.B. (2009) DGAT1 and PDAT1 acyltransferases have overlapping functions in Arabidopsis triacylglycerol biosynthesis and are essential for normal pollen and seed development. Plant Cell, 21, 3885–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Xing, A. , Staswick, P. and Clemente, T.E. (1999) The use of glufosinate as a selective agent in Agrobacterium‐mediated transformation of soybean. Plant Cell Tissue Organ Cult. 56, 37–46. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 T‐DNA of plasmid pPTN1248 harbouring a soybean codon‐optimized AtWRI1 transcription factor and AtDGAT1 from Arabidopsis under control of the beta‐conglycinin promoter.
Figure S2 Southern blot analysis of the transgenic soybean events expressing AtWRI1 and AtDGAT1.
Figure S3 Wrinkled‐seed phenotype.
Figure S4 DW, protein, FA, and starch accumulation rates.
Figure S5 Accumulation of cell wall polysaccharides during development.
Figure S6 Rate of glucose accumulation as hemicelluloses in WD events.
Figure S7 Beta‐conglycinin promoter pattern of expression.
Figure S8 DEGs in transgenic soybeans grown under greenhouse conditions.
Figure S9 Expression of WRI1 and DGAT1 from Arabidopsis and soybean in transgenic soybean grown under field conditions.
Figure S10 Differentially expressed genes in the field experiment and comparison with the greenhouse results.
Figure S11 Changes in lipid transfer protein and lipid degradation enzyme expression in WD events between 24 and 33 DAP under greenhouse conditions.
Figure S12 Principal component analysis and top fifteen changes in metabolites.
Table S1 Soybean field trials in 2018, 2019, and 2020 crop seasons in Nebraska fields.
Table S2 Total number of differential expressed genes for each transgenic event at each developmental stage.
Table S3 Complete list of putative AtWRI1 targets.
Table S4 Metabolite concentrations.