

Keywords: Alzheimer’s disease, Alzheimer’s disease-related neuropathologies, amyloid-β, pathology, APP/PS1 mice, cognitive deficits, damage of synapses and nerve cells, high-throughput RNA sequencing, lamotrigine, neuroinflammation, tau protein hyperphosphorylation
Abstract
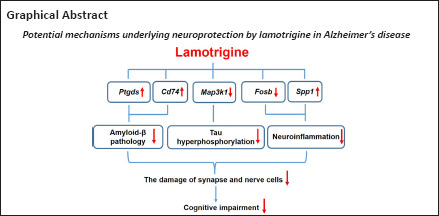
Lamotrigine (LTG) is a widely used drug for the treatment of epilepsy. Emerging clinical evidence suggests that LTG may improve cognitive function in patients with Alzheimer’s disease. However, the underlying molecular mechanisms remain unclear. In this study, amyloid precursor protein/presenilin 1 (APP/PS1) double transgenic mice were used as a model of Alzheimer’s disease. Five-month-old APP/PS1 mice were intragastrically administered 30 mg/kg LTG or vehicle once per day for 3 successive months. The cognitive functions of animals were assessed using Morris water maze. Hyperphosphorylated tau and markers of synapse and glial cells were detected by western blot assay. The cell damage in the brain was investigated using hematoxylin and eosin staining. The levels of amyloid-β and the concentrations of interleukin-1β, interleukin-6 and tumor necrosis factor-α in the brain were measured using enzyme-linked immunosorbent assay. Differentially expressed genes in the brain after LTG treatment were analyzed by high-throughput RNA sequencing and real-time polymerase chain reaction. We found that LTG substantially improved spatial cognitive deficits of APP/PS1 mice; alleviated damage to synapses and nerve cells in the brain; and reduced amyloid-β levels, tau protein hyperphosphorylation, and inflammatory responses. High-throughput RNA sequencing revealed that the beneficial effects of LTG on Alzheimer’s disease-related neuropathologies may have been mediated by the regulation of Ptgds, Cd74, Map3k1, Fosb, and Spp1 expression in the brain. These findings revealed potential molecular mechanisms by which LTG treatment improved Alzheimer’s disease. Furthermore, these data indicate that LTG may be a promising therapeutic drug for Alzheimer’s disease.
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disease (Xu et al., 2016) and is characterized by amyloid-β (Aβ) accumulation, tau protein hyperphosphorylation, and inflammation in the brain (Jellinger, 1998; Macchi et al., 2014). These major neuropathologies affect neuronal and synaptic integrity in the brain and ultimately lead to cognitive deficits (Wu et al., 2015; Kent et al., 2020). To date, there is still no effective medication available for halting Alzheimer’s disease progression.
Lamotrigine (LTG) is a widely-used, broad-spectrum antiepileptic drug (Malik et al., 2006). Interestingly, aside from treating epilepsy, LTG has been found to improve cognitive functions in patients with AD (Tekin et al., 1998). LTG attenuated cognitive deficits in animal models of AD via amelioration of amyloid pathology and suppression of inflammatory responses (Zhang et al., 2014; Wang et al., 2016). However, the signaling pathways responsible for these protective properties remain largely unknown. In this study, we used amyloid precursor protein/presenilin 1 (APP/PS1) double transgenic mice, which represent a model of AD, to test the efficacy of LTG on cognitive deficits, neuronal and synaptic integrity, and AD-related neuropathologies. Specifically, we investigated the potential signaling pathways underlying LTG-induced protection using a high-throughput RNA sequencing strategy.
Methods
Animals and treatments
In this study, only male animals were used to avoid the effect of estrogen on cognitive function, neuronal and synaptic integrity, and AD-related neuropathologies (Hara et al., 2015; Pike, 2017). Five-month-old APP/PS1 mice (official strain name is B6.Cg-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax, MGI ID: 3611279, RRID: MMRRC_034832-JAX; n = 40) and their age-matched wild-type (WT) mice (n = 14) were purchased from Beijing HFK Bioscience Company (license No. SCXK (Jing) 2021-0010). According to our previous findings, APP/PS1 mice begin to develop amyloid plaques in the brain and present early spatial cognitive deficits at the age of 5 months (Jiang et al., 2014c). Mice were provided food and water ad libitum and were maintained in cages under standard housing conditions, including a 12-hour light/dark cycle, 23 ± 2°C, and 60 ± 5% humidity. The animal experiments were reported in accordance with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines (Percie du Sert et al., 2020), and the procedure was reviewed and approved by the Ethics Committee of Nanjing First Hospital (approval No. DWSY-2001336) on October 18, 2020.
APP/PS1 mice were randomly allocated into two groups and administered LTG (30 mg/kg; Sigma-Aldrich; St. Louis, MO, USA) or vehicle (0.2 mL, 0.5% carboxymethyl cellulose sodium) intragastrically once per day for 3 consecutive months. LTG was dissolved in 0.5% carboxyl methyl cellulose sodium solution, and the dose of LTG was selected based on previous studies (Zhang et al., 2014; Wang et al., 2016).
Morris water maze test
The Morris water maze (MWM) was used to test the effect of LTG on cognitive deficits in APP/PS1 mice (Jiang et al., 2014b). The MWM test was conducted during the last 5 days before the mice were sacrificed at 8 months of age (Jiang et al., 2014a; Fu et al., 2019; Duan et al., 2020). The platform (Jiangsu SANS Biotechnology Company, Nanjing, China) was submerged 0.5–1 cm below the surface of the water, and the test was performed four times per day. The mice were placed randomly in one quadrant of the pool and allowed 60 seconds to find the platform. If the mice failed to find the platform within 60 seconds, they were guided to the platform and permitted to stay for 15 seconds. The platform remained in the same position during the test. The swimming speeds and path lengths to the hidden platform were recorded using a video tracking system (Jiangsu SANS Biotechnology Company). The average values for the path lengths in four trials were calculated.
Western blot analysis
Western blots were used to detect the levels of hyperphosphorylated tau and synaptic and glial cell markers. Mice were sacrificed at 8 months of age by cervical dislocation after inhalation anesthesia with an overdose of isoflurane (5%). To extract total protein, whole brain tissues were homogenized with lysis buffer containing a complete protease inhibitor cocktail. The homogenates were centrifuged for 30 minutes (14,000 × g, 4°C), the supernatants were collected, and the protein concentrations were determined with a bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA; Cat# 23227). Equal amounts of protein from different samples were separated on sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinylidene fluoride membranes (Roche Diagnostics GmbH, Mannheim, Germany; Cat# 03010040001). Then, the membranes were blocked with 5% skim milk for 1.5 hours at room temperature and incubated with one of the following primary antibodies overnight at 4°C: rabbit anti-hyperphosphorylated tau (p-Tau at Ser214; 1:1000, Cell Signaling Technology, Danvers, MA, USA, Cat# 77348, RRID: AB_2799895), rabbit anti-p-Tau at Thr205 (1:1000, Abcam, Boston, MA, USA, Cat# ab254410, RRID: AB_2894401), rabbit anti-p-Tau at Thr231 (1:1000, Abcam, Cat# ab151559, RRID: AB_2893278), rabbit anti-Tau (1:1000, Abcam, Cat# ab254256, RRID: AB_2894402), rabbit anti-synaptophysin (1:1000, Cell Signaling Technology; Cat# 4329, RRID: AB_1904154), rabbit anti-ionized calcium binding adapter molecule-1 (IBA-1; 1:1000, Abcam, Cat# ab178847, RRID: AB_2832244), rabbit anti-glial fibrillary acidic protein (GFAP; 1:1000, Cell Signaling Technology, Cat# 80788, RRID: AB_2799963), and anti-β-actin antibody (rabbit monoclonal antibody; 1:1000, Cell Signaling Technology, Cat# 4970, RRID: AB_2223172). The membranes were washed three times for 10 minutes each with Tris-buffered saline containing Tween-20 and incubated with horseradish peroxidase-conjugated anti-rabbit secondary antibody (1:5000, Cell Signaling Technology, Cat# 7074S, RRID: AB_2099233) for 2 hours at room temperature. Finally, the protein bands were detected with a chemiluminescent kit (Thermo Fisher Scientific, Cat# 32209), and the optical density ratios of the target protein bands per β-actin were analyzed using Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA).
Hematoxylin and eosin staining
Hematoxylin and eosin (H&E) staining was used to detect cell damage in the brain. At 8 months of age, mice were anesthetized by 5% isoflurane inhalation and transcardially perfused with phosphate-buffered saline followed by 4% paraformaldehyde. Intact brains were removed from the skulls, fixed in 10% paraformaldehyde for 72 hours, dehydrated, and embedded in paraffin. Five-μm-thick sections of brain tissues were fixed on glass for subsequent H&E staining as previously described (Xie et al., 2020). The morphology of nerve cells in the cerebral cortex and hippocampal CA1 region was observed using an optical microscope (Chongqing Optec Instrument Company; Chongqing, China). After H&E staining, healthy cells were light blue with prominent nuclei, whereas diseased cells appeared darker in color because of morphological changes associated with cellular pyknosis (Wang et al., 2020). The percentages of diseased cells in the cerebral cortex and the hippocampal CA1 region were calculated.
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) was used to detect the concentrations of inflammatory cytokines in the brain. At 8 months of age, the whole brain tissues were homogenized in Tris-buffered saline containing 5 mM ethylenediaminetetraacetic acid, phosphatase inhibitor, ethylenediaminetetraacetate-free protease inhibitor cocktail, and 2 mM 1, 10-phenanthroline as described (Jiang et al., 2016b). The soluble Aβ1–42 levels in the tissue supernatants were assayed with an ELISA kit (Thermo Fisher Scientific, Cat# KHB3544). The levels of interleukin (IL)-1β (Cat# ab197742), IL-6 (Cat# ab100713), and tumor necrosis factor α (TNF-α; Cat# ab208348) in brain tissues were measured using commercial ELISA kits purchased from Abcam. All experiments were performed strictly in accordance with the manufacturer’s instructions.
High-throughput RNA sequencing
Brain tissues were removed from skulls at 8 months of age and immediately frozen in liquid nitrogen. Total RNA was extracted using a TRIzol™ reagent kit (Invitrogen, Carlsbad, CA, USA; Cat# A33254) in accordance with the manufacturer’s protocol. RNA quality was assessed on an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) and visualized using RNase-free agarose gel electrophoresis. Afterward, ribosomal RNAs were removed. The enriched mRNAs were fragmented using a fragmentation buffer and reverse transcribed into complementary DNAs with random primers. Second-strand cDNA was synthesized in a reaction mixture containing DNA polymerase I, RNase H, deoxynucleotide triphosphates, and second-strand synthesis reaction buffer as previously described (Liu et al., 2019). Next, the cDNA fragments were purified with a QiaQuick polymerase chain reaction (PCR) extraction kit (Qiagen, Venlo, Netherlands), the ends were repaired, a base was added, and the fragments were then ligated to Illumina sequencing adapters (Illumina, San Diego, CA, USA). Uracil-N-glycosylase was used to digest the second-strand cDNA. The digested products were size selected by agarose gel electrophoresis, PCR amplified, and sequenced using the Illumina Novaseq 6000 Sequencing System.
The reads obtained from the Illumina Novaseq 6000 sequencer included raw reads containing adapters or low-quality bases, which affected the following assembly and analysis. Thus, to obtain high-quality, clean reads, the reads were filtered using the fastp tool (Chen et al., 2018), which included removing reads containing adapters, more than 10% unknown nucleotides (N), and greater than 50% low quality bases (Q-value ≤ 20). High-quality, clean reads were mapped to the ribosomal RNA database using the short read alignment tool Bowtie2 (Langmead and Salzberg, 2012), and the ribosomal RNA-mapped reads were then removed. The transcript abundances were quantified with StringTie software (Trapnell et al., 2010; Pertea et al., 2015), which uses a reference-based approach. For each transcription region, a fragment per kilobase of transcript per million mapped reads value was calculated to quantify the expression abundance and variation using RSEM software (Li and Dewey, 2011). This method was used to directly compare the differences in transcript expression in the brains of APP/PS1 mice after LTG treatment. The differentially expressed genes (DEGs) were analyzed using DESeq2 software (Love et al., 2014). Genes with fold change ≥ 1.5 (P < 0.001) were considered DEGs.
Real-time PCR
We screened previous publications between January 1, 2010 and December 31, 2020 using PubMed (https://pubmed.ncbi.nlm.nih.gov/) and identified five DEGs that were closely related to the AD-associated neuropathologies ameliorated by LTG (Table 1). Real-time PCR (RT-PCR) was performed to confirm the expression of these five DEGs. Total RNA was extracted from whole brains at 8 months of age with TRIzol™ reagent (Thermo Fisher Scientific, Cat# 15596018) as described (Jiang et al., 2016a; Duan et al., 2020). Equal amounts of total RNA were reverse transcribed using the PrimeScript™ RT Reagent Kit (Takara Bio; Kusatsu, Shiga, Japan; Cat# RR047A) under standard conditions. The PCR reactions were carried out with TB Green Premix Ex Taq™ (Takara Bio, Cat# RR420A) using the following settings: one cycle at 95°C for 30 seconds, 40 cycles of 95°C for 5 seconds and 60°C for 34 seconds, and one final cycle of 95°C for 15 seconds, 60°C for 60 seconds, and 95°C for 15 seconds. The specific PCR primers were: Ptgds, forward: 5′-GAA GGC GGC CTC AAT CTC AC-3′, reverse: 5′-CGT ACT CGT CAT AGT TGG CCT C-3′; Cd74, forward: 5′-CGC GAC CTC ATC TCT AAC CAT-3′, reverse: 5′-ACA GGT TTG GCA GAT TTC GGA-3′; Map3k1, forward: 5′-AGC ACG AGT GGT TGG AGA G-3′, reverse: 5′-CTG GGG ATT CCG GCT TCA C-3′; Fosb, forward: 5′-CCT CCG CCG AGT CTC AGT A-3′, reverse: 5′-CCT GGC ATG TCA TAA GGG TCA-3′; Spp1, forward: 5′-ATC TCA CCA TTC GGA TGA GTC T-3′, reverse: 5′-TGT AGG GAC GAT TGG AGT GAA A-3′; Gapdh (reference gene), forward: 5′-CAA CAG CAA CTC CCA CTC TTC-3′, reverse: 5′-GGT CCA GGG TTT CTT ACT CCT T-3′. Relative quantification of the genes was performed by determining the n-fold differential expression using the 2–ΔΔCt method (Schmittgen and Livak, 2008) and was shown as percentage of control.
Table 1.
Differentially expressed genes involved in Alzheimer's disease-related neuropathology
| AD-related neuropathology | Differentially expressed genes |
|---|---|
| Aβ metabolism | Ptgds, Cd74 |
| Tau hyperphosphorylation | Map3k1 |
| Microglia-mediated neuroinflammation | Fosb, Spp1 |
Statistical analysis
No statistical methods were used to predetermine sample sizes; however, our sample sizes are similar to those reported in other publications (Jiang et al., 2014a, b, c, 2016a). No animals or data points were excluded from the analysis. The evaluators were blinded to the different groups. Data were analyzed by GraphPad Prism software (version 7.00; GraphPad Software; San Diego, CA, USA; www.graphpad.com) and are expressed as means ± standard deviation (SD). Data from the MWM test were analyzed among the three groups using two-way repeated measures analysis of variance followed by Bonferroni’s multiple comparisons. For other data, the differences among the three groups were analyzed using one-way analysis of variance followed by Tukey’s post hoc test. Significant differences between two groups were analyzed using Student’s t-tests. P < 0.05 was considered statistically significant.
Results
LTG rescued cognitive deficits in APP/PS1 mice
In the MWM test, no significant difference was observed for swimming speed between the WT controls and APP/PS1 mice, and the swimming speed of APP/PS1 mice was not significantly affected by LTG treatment (Figure 1A). Overall, APP/PS1 mice performed worse than WT controls during the entire MWM test (Fgenotype(1, 110) = 21.52, P < 0.001; Figure 1B). APP/PS1 mice showed a significant increase in path length on days 4 (P < 0.01) and 5 (P < 0.001) of the MWM test compared with that of the WT controls, which confirmed that APP/PS1 mice exhibited spatial cognitive deficits at 8 months of age. As shown in Figure 1B, LTG treatment rescued the cognitive impairment in APP/PS1 mice (Ftreatment(1, 110) = 6.514, P < 0.05). The APP/PS1 + LTG group manifested shorter path lengths than the APP/PS1 group on days 4 (P < 0.05) and 5 (P < 0.01).
Figure 1.
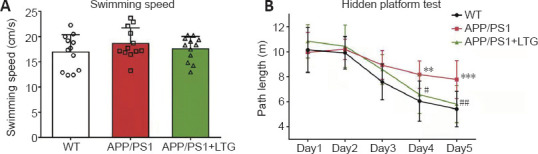
LTG ameliorated the cognitive deficits of APP/PS1 mice observed in the Morris water maze test.
(A) The swimming speeds of the mice before the test. (B) The path lengths of the mice during the hidden platform test over 5 consecutive days. Data are expressed as means ± SD (n = 12). **P < 0.01, ***P < 0.001, vs. WT controls; #P < 0.05, ##P < 0.01, vs. APP/PS1 mice (one-way analysis of variance followed by Tukey’s post hoc test in A, two-way repeated measures analysis of variance followed by Bonferroni’s multiple comparisons in B). APP/PS1: Amyloid precursor protein/presenilin 1; LTG: lamotrigine; WT: wild-type.
LTG alleviated synapse and nerve cell damage in the brains of APP/PS1 mice
Western blots demonstrated that the levels of synaptophysin in the brains of APP/PS1 mice were lower than those in WT controls (P < 0.001), indicating obvious synaptic injury in the brains of APP/PS1 mice at 8 months of age (Figure 2A and B). LTG treatment reversed the decrease of synaptophysin in APP/PS1 mouse brains (P < 0.01), indicating that LTG treatment attenuated synaptic injury in the APP/PS1 mice.
Figure 2.
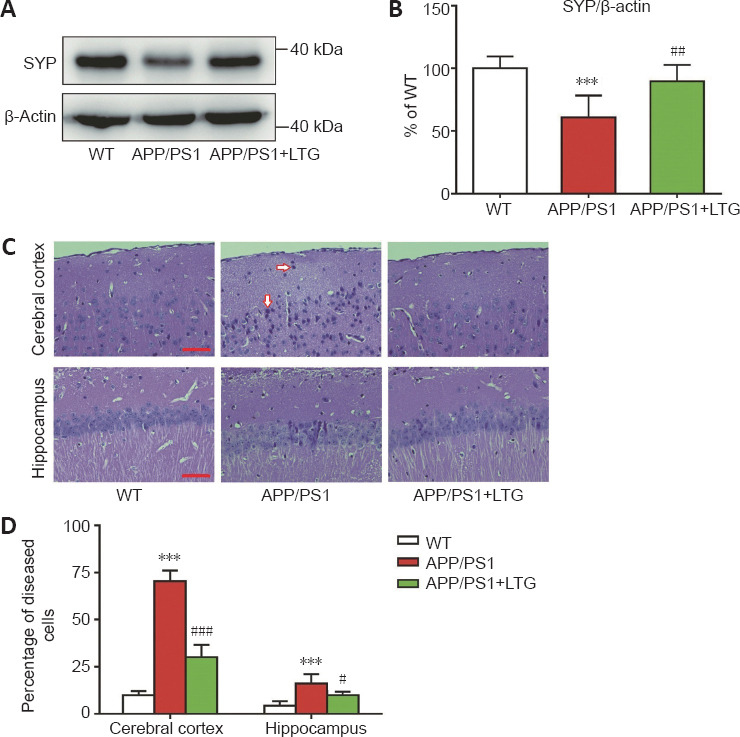
LTG alleviated synapse and nerve cell damage in the brains of APP/PS1 mice.
(A) The protein levels of SYP in the brains of mice were detected using western blot assay. (B) Quantitation of SYP protein levels. (C) Hematoxylin and eosin staining of the cerebral cortex and hippocampal CA1 region in mice. Healthy cells are light blue with prominent nuclei. In contrast, damaged cells (arrows) are darker in color with morphological changes from cellular pyknosis. The number of diseased cells in the cerebral cortex and the hippocampus of APP/PS1 mice was elevated significantly when compared with that of WT controls. LTG treatment significantly reduced the number of diseased cells in the cerebral cortex and the hippocampus of APP/PS1 mice. Scale bar: 50 μm. (D) Quantitation of the percentage of diseased cells in the three groups. Data are expressed as means ± SD (n = 6). ***P < 0.001, vs. WT controls; #P < 0.05, ###P < 0.001, vs. APP/PS1 mice (one-way analysis of variance followed by Tukey’s post hoc test). APP/PS1: Amyloid precursor protein/presenilin 1; LTG: lamotrigine; SYP: synaptophysin; WT: wild-type.
H&E staining was performed to determine the protective effect of LTG on nerve cells in the brains of APP/PS1 mice. The percentage of diseased cells in the cerebral cortex and the hippocampal CA1 region of APP/PS1 mice was elevated significantly when compared with that of WT controls (P < 0.001), indicating marked damage of nerve cells in the brains of APP/PS1 mice at 8 months of age. LTG treatment significantly reduced the percentage of diseased cells in the cerebral cortex (P < 0.001) and the hippocampus (P < 0.05) of APP/PS1 mice (Figure 2C and D), indicating a protective effect of LTG against nerve cell damage.
LTG reduced the Aβ1–42 levels in the brains of APP/PS1 mice
Abundant Aβ1–42, one of the pathological hallmarks of AD (Kent et al., 2020), was detected by ELISA in the brains of APP/PS1 mice. LTG treatment significantly decreased Aβ1–42 levels in the brains of APP/PS1 mice compared with that in vehicle-treated mice (P < 0.001; Figure 3).
Figure 3.
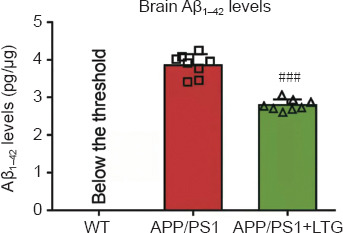
LTG reduced Aβ1–42 levels in the brains of APP/PS1 mice.
The concentration of Aβ1–42 in the brains was measured using an enzyme-linked immunosorbent assay. Data are expressed as means ± SD (n = 8). ###P < 0.001, vs. APP/PS1 mice (Student’s t-test). APP/PS1: Amyloid precursor protein/presenilin 1; Aβ: amyloid-β; LTG: lamotrigine; WT: wild-type.
LTG ameliorated tau hyperphosphorylation in the brains of APP/PS1 mice
Hyperphosphorylated tau protein represents another pathological hallmark of AD (Kent et al., 2020). Compared with those of WT controls, the levels of hyperphosphorylated tau at Ser214, Thr205 (Figures 4A–C), and Thr231 (Figure 4A and D) sites were elevated in the brains of APP/PS1 mice (P < 0.001). LTG treatment substantially ameliorated tau hyperphosphorylation at Ser214, Thr205, and Thr231 sites in the brains of APP/PS1 mice (P < 0.01 for each; Figures 4A–D).
Figure 4.
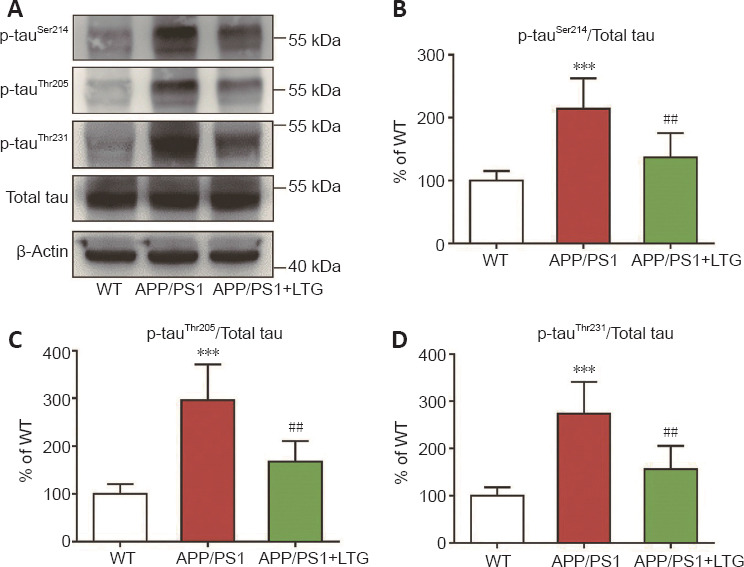
LTG ameliorated tau hyperphosphorylation in the brains of APP/PS1 mice.
(A) Hyperphosphorylated tau proteins at Ser214, Thr205, and Thr231 sites were detected by western blot assay. (B–D) Quantitation of hyperphosphorylated tau proteins at Ser214 (B), Thr205 (C), and Thr231 (D) sites. Data are expressed as means ± SD (n = 6). ***P < 0.001, vs. WT controls; ##P < 0.01, vs. APP/PS1 mice (one-way analysis of variance followed by Tukey’s post hoc test). APP/PS1: Amyloid precursor protein/presenilin 1; LTG: lamotrigine; P-tau: hyperphosphorylated tau; WT: wild-type.
LTG attenuated the inflammatory responses in the brain of APP/PS1 mice
As shown in Figure 5A–C, concentrations of the inflammatory cytokines IL-1β (P < 0.001), IL-6 (P < 0.001), and TNF-α (P < 0.01) were elevated in the brains of APP/PS1 mice compared with that in WT controls, indicating an obvious neuroinflammatory response in the brains of APP/PS1 mice at 8 months of age. LTG treatment dramatically reduced the concentrations of IL-1β (P < 0.01), IL-6 (P < 0.01), and TNF-α (P < 0.05) in the brains of APP/PS1 mice, demonstrating that LTG attenuated neuroinflammation in these mice.
Figure 5.
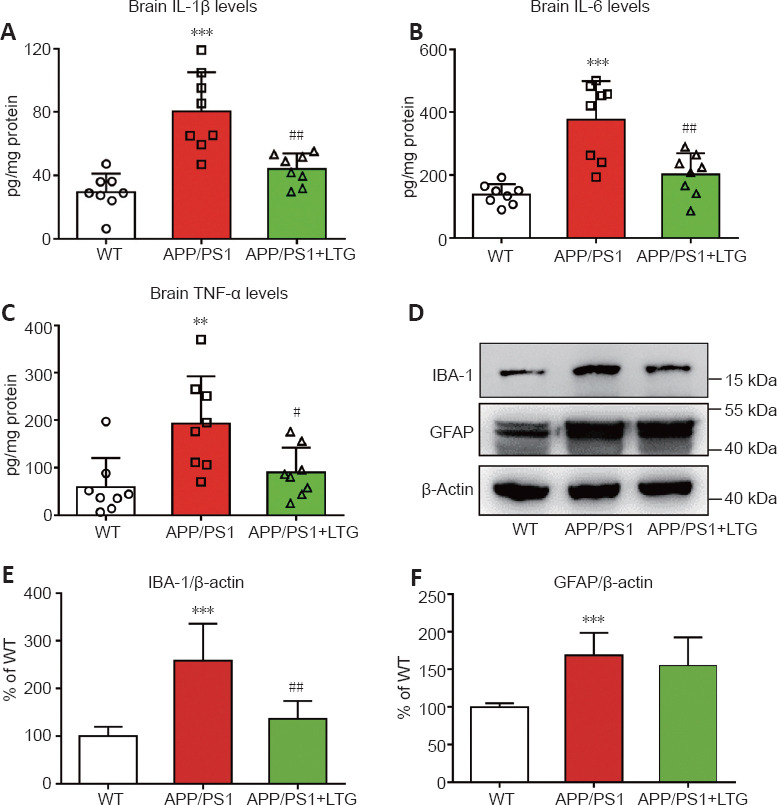
LTG inhibited inflammatory responses in the brains of APP/PS1 mice.
(A–C) Quantitative analysis of IL-1β (A), IL-6 (B), and TNF-α (C) levels by enzyme-linked immunosorbent assays. (D) The levels of IBA-1 and GFAP proteins were detected by western blot assay. (E, F) Quantitative analysis of IBA-1 (E) and GFAP (F) protein levels. Data are expressed as means ± SD (n = 8 in A–C, n = 6 in D–F). **P < 0.01, ***P < 0.001, vs. WT controls; #P < 0.05, ##P < 0.01, vs. APP/PS1 mice (one-way analysis of variance followed by Tukey’s post hoc test). APP/PS1: Amyloid precursor protein/presenilin 1; GFAP: glial fibrillary acidic protein; IBA-1: ionized calcium binding adapter molecule-1; IL: interleukin; LTG: lamotrigine; TNF-α: tumor necrosis factor α; WT: wild-type.
Activation of microglia and astrocytes plays a crucial role in mediating neuroinflammation in AD (Fakhoury, 2018). As shown in Figures 5D–F, levels of the microglial marker IBA-1 (Feng et al., 2019) and astrocyte marker GFAP (Li et al., 2020) were increased in the brains of APP/PS1 mice when compared with that of WT controls (P < 0.001), indicating activation of microglia and astrocytes. LTG treatment significantly decreased the levels of IBA-1 in the brains of APP/PS1 mice (P < 0.01; Figure 5D and E), suggesting that LTG inhibited microglial activation in APP/PS1 mice. The levels of GFAP were unaltered in the brains of APP/PS1 mice following LTG treatment (Figure 5D and F).
LTG altered the expression of genes that are closely related to AD neuropathologies
To further investigate the molecular mechanisms by which LTG ameliorated these AD-related neuropathologies, high-throughput RNA sequencing was performed. As indicated in Figure 6A and B, LTG treatment significantly altered the expression of 47 genes (11 downregulated and 36 upregulated) in the brains of APP/PS1 mice. We screened five DEGs that were closely related to the AD-related neuropathologies ameliorated by LTG. Among them, Ptgds and Cd74 are involved in Aβ metabolism, Map3k1 is related to tau hyperphosphorylation, and Fosb and Spp1 are associated with microglia-mediated neuroinflammation. The changes in expression of these five DEGs in the brain following LTG treatment was further confirmed by RT-PCR (Figure 6C).
Figure 6.
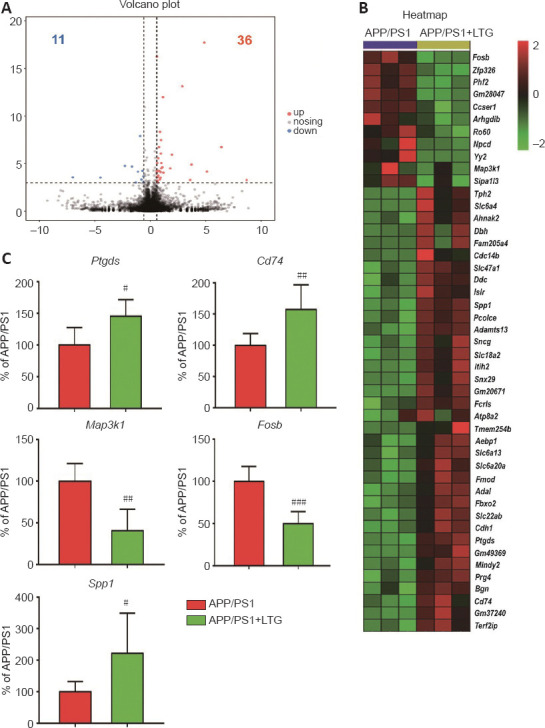
LTG altered the expression of genes that are closely related to Alzheimer’s disease neuropathology.
(A) The volcano plot of DEGs. The red dots represent upregulated genes, and the blue dots represent downregulated genes. (B) The heatmap of DEGs. Red means high expression, and green means low expression (n = 3). (C) Ptgds, Cd74, Map3k1, Fosb, and Spp1 mRNA expression in brains of mice was detected using real-time polymerase chain reaction (n = 6). Data are expressed as means ± SD. #P < 0.05, ##P < 0.01, ###P < 0.001, vs. APP/PS1 mice (Student’s t-test). APP/PS1: Amyloid precursor protein/presenilin 1; DEG: differentially expressed gene; LTG: lamotrigine.
Discussion
In this study, we demonstrated that LTG treatment improved the performance of APP/PS1 mice in the MWM test, implying a protective effect of LTG against AD-related cognitive deficits. This observation was in accordance with a study from Zhang et al. (2014), who showed that chronic LTG treatment rescued learning and memory deficits in APP/PS1 mice. It is noteworthy that the spatial cognitive function of WT controls was not significantly influenced by LTG (Zhang et al., 2014), which suggested that LTG had no direct impact on spatial cognitive functions.
Neuronal and synaptic integrity in the brain is essential for normal cognitive function, while neuronal and synaptic injury is closely linked with cognitive decline in AD (Giannakopoulos et al., 2003; Arendt, 2009). Consistent with this observation, our APP/PS1 mice at 8 months of age exhibited apparent damage of synapse and nerve cells in the brain, which was accompanied by obvious cognitive deficits. The damage to synapses and nerve cells was alleviated by LTG treatment as indicated by the marked increase in synaptophysin levels and reduction in the percentage of diseased nerve cells in the brains of LTG-treated APP/PS1 mice. These results were supported by a recent study in which chronic LTG treatment prevented impairment of synaptic plasticity and neuronal loss of APP/PS1 mice (Zhang et al., 2014). The protective effects of LTG on synapses and nerve cells may contribute to the amelioration of AD-related spatial cognitive deficits.
As main neuropathological hallmarks of AD, Aβ pathology, hyperphosphorylated tau, and chronic neuroinflammation are directly linked to mental dysfunction and even loss of synapses and neurons in the brain (Congdon and Sigurdsson, 2018; Rajendran and Paolicelli, 2018). We investigated whether the protective effect of LTG against synaptic injury and nerve cell damage in the brains of APP/PS1 mice was attributable to improvements in these AD-related neuropathologies. We showed that LTG treatment significantly decreased Aβ levels in the brains of APP/PS1 mice, which was supported by previous findings that chronic LTG treatment reduced the generation of Aβ (Zhang et al., 2014). In our study, we also revealed that LTG treatment ameliorated tau hyperphosphorylation in the brains of APP/PS1 mice as shown by the marked LTG-mediated decreases in the levels of hyperphosphorylated tau at pathological sites, including Ser214, Thr205, and Thr231. To our knowledge, this is the first study that shows LTG inhibits tau hyperphosphorylation. Moreover, we demonstrated that LTG treatment attenuated inflammatory responses in the brains of APP/PS1 mice as indicated by the substantial reductions of IL-1β, IL-6, and TNF-α concentrations after LTG treatment. These observations were compatible with a recent study in which LTG exerted an anti-inflammatory effect in mitogen-induced mice and lipopolysaccharide-induced RAW264.7 cells (Abu-Rish et al., 2018). Interestingly, we found that LTG treatment decreased the levels of IBA-1 but not GFAP, although this finding needs to be further validated by other methods, such as immunofluorescent staining. The change in IBA-1 expression indicated that the anti-inflammatory effect of LTG in AD mice was achieved by inactivation of microglia rather than astrocytes. However, our findings contradict studies by other groups (Zhang et al., 2014; Wang et al., 2016) who found that astrocyte activation was suppressed by LTG. These conflicting results may be attributable to a longer LTG treatment duration in these other studies.
To further explore the underlying molecular mechanisms by which LTG ameliorated AD-related neuropathologies, we performed high-throughput RNA sequencing and screened DEGs in the brains of APP/PS1 mice following LTG treatment. Five DEGs that were strongly linked to AD-related neuropathologies were identified. Specifically, Ptgds and Cd74 are closely associated with Aβ pathology. Ptgds encodes a glutathione-independent prostaglandin D synthase that converts prostaglandin H2 to prostaglandin D2, which in turn decreases Aβ generation by inhibiting PS1 (Lu et al., 2018). Furthermore, the protein encoded by Cd74 has been reported to suppress the production of Aβ by interacting with APP (Matsuda et al., 2009; Kiyota et al., 2015). In the current study, LTG treatment significantly upregulated the expression of Ptgds and Cd74 in the brains of APP/PS1 mice. Based on the above evidence, we speculate that the reduction of Aβ levels in the brain of APP/PS1 mice after LTG treatment may be ascribed to the upregulation of Ptgds and Cd74 expression. Among the five DEGs, Map3k1 encodes a serine/threonine kinase and is closely associated with tau hyperphosphorylation (Ambegaokar and Jackson, 2011; Cuarental et al., 2019). In our study, LTG treatment markedly downregulated Map3k1 expression in the brains of APP/PS1 mice. Therefore, it is possible that the amelioration of tau hyperphosphorylation in the brains of APP/PS1 mice after LTG treatment was a consequence of Map3k1 downregulation. In addition, Fosb and Spp1 are involved in microglia-mediated neuroinflammation. Fosb facilitates the formation of the transcription factor complex AP-1, and AP-1 has been shown to exacerbate inflammatory responses of microglia (Lin et al., 2011). Spp1 has been reported to attenuate microglial activation and, thus, played a protective role in the pathogenesis of neurodegenerative diseases (Comi et al., 2010; Yu et al., 2017). Notably, in this study, we found that LTG treatment substantially downregulated Fosb and upregulated Spp1 in the brains of APP/PS1 mice. Based on these findings, the LTG-mediated inhibition of inflammatory responses in the brains of APP/PS1 mice was likely attributable to the regulation of Fosb and Spp1.
This study also has some limitations. First, the neuroprotective effects of LTG were only evaluated in an AD transgenic mouse model. In the future, cellular models, such as induced pluripotent stem cell-derived neurons and microglia from AD patients, should be employed to further validate the protective effect of LTG and elucidate cell type-specific mechanisms. Second, in this study, whole brain tissues were used for RNA sequencing, which likely masked potential spatial- or cell type-specific alterations in gene expression induced by LTG. In future studies, spatial transcriptome sequencing as well as single-cell sequencing should be employed to further investigate LTG-mediated changes in spatial- and cell type-specific gene expression patterns. Third, in the current study, five DEGs that were strongly linked to AD-related neuropathologies were identified using high-throughput RNA sequencing and RT-PCR after LTG treatment. The spatial distribution and cellular localization of these five DEGs should be further assessed using immunohistochemical methods in the future. Fourth, knockdown and over-expression experiments specific for these DEGs, by creating new model mice, will better verify the roles of these genes.
In conclusion, we demonstrated that the widely-used, broad-spectrum antiepileptic drug LTG rescued cognitive deficits, alleviated synapse and nerve cell damage, and ameliorated AD-related neuropathologies in APP/PS1 mice. Importantly, we provide the first evidence that the beneficial effects of LTG on AD-related neuropathologies may be achieved by regulating the expression of Ptgds, Cd74, Map3k1, Fosb, and Spp1 in the brain. These findings highlight that LTG may be a promising therapeutic agent for AD in addition to its antiepileptic effects.
Additional file: Open peer review report 1 (81.5KB, pdf) .
Footnotes
Funding: This work was supported by the National Natural Science Foundation of China, No. 81771140 (to YDZ); the Natural Science Foundation of Jiangsu Province of China, No. BK20201117 (to YDZ); and Jiangsu “Six One Project” for Distinguished Medical Scholars of China, No. LGY2020013 (to TJ).
Conflicts of interest: The authors have declared that no conflict of interest, financial or otherwise exists.
Availability of data and materials: All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open peer reviewer: Agustin Cota-Coronado, Centro de Investigación y Asistencia en Tecnología y Diseño del Estado de Jalisco, Mexico.
P-Reviewer: Cota-Coronado A; C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Zunino S, Yu J, Song LP; T-Editor: Jia Y
References
- 1.Abu-Rish EY, Dahabiyeh LA, Bustanji Y, Mohamed YS, Browning MJ. Effect of lamotrigine on in vivo and in vitro cytokine secretion in murine model of inflammation. J Neuroimmunol. 2018;322:36–45. doi: 10.1016/j.jneuroim.2018.06.008. [DOI] [PubMed] [Google Scholar]
- 2.Ambegaokar SS, Jackson GR. Functional genomic screen and network analysis reveal novel modifiers of tauopathy dissociated from tau phosphorylation. Hum Mol Genet. 2011;20:4947–4977. doi: 10.1093/hmg/ddr432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arendt T. Synaptic degeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:167–179. doi: 10.1007/s00401-009-0536-x. [DOI] [PubMed] [Google Scholar]
- 4.Chen S, Zhou Y, Chen Y, Gu J. fastp:an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Comi C, Carecchio M, Chiocchetti A, Nicola S, Galimberti D, Fenoglio C, Cappellano G, Monaco F, Scarpini E, Dianzani U. Osteopontin is increased in the cerebrospinal fluid of patients with Alzheimer's disease and its levels correlate with cognitive decline. J Alzheimers Dis. 2010;19:1143–1148. doi: 10.3233/JAD-2010-1309. [DOI] [PubMed] [Google Scholar]
- 6.Congdon EE, Sigurdsson EM. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2018;14:399–415. doi: 10.1038/s41582-018-0013-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuarental L, Sucunza-Sáenz D, Valiño-Rivas L, Fernandez-Fernandez B, Sanz AB, Ortiz A, Vaquero JJ, Sanchez-Niño MD. MAP3K kinases and kidney injury. Nefrologia. 2019;39:568–580. doi: 10.1016/j.nefro.2019.03.004. [DOI] [PubMed] [Google Scholar]
- 8.Duan R, Xue X, Zhang QQ, Wang SY, Gong PY, E Y, Jiang T, Zhang YD. ACE2 activator diminazene aceturate ameliorates Alzheimer's disease-like neuropathology and rescues cognitive impairment in SAMP8 mice. Aging (Albany NY) 2020;12:14819–14829. doi: 10.18632/aging.103544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fakhoury M. Microglia and astrocytes in Alzheimer's disease:implications for therapy. Curr Neuropharmacol. 2018;16:508–518. doi: 10.2174/1570159X15666170720095240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng X, Zhao Y, Yang T, Song M, Wang C, Yao Y, Fan H. Glucocorticoid-driven NLRP3 inflammasome activation in hippocampal microglia mediates chronic stress-induced depressive-like behaviors. Front Mol Neurosci. 2019;12:210. doi: 10.3389/fnmol.2019.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu X, Qin T, Yu J, Jiao J, Ma Z, Fu Q, Deng X, Ma S. Formononetin ameliorates cognitive disorder via PGC-1αpathway in neuroinflammation conditions in high-fat diet-induced mice. CNS Neurol Disord Drug Targets. 2019;18:566–577. doi: 10.2174/1871527318666190807160137. [DOI] [PubMed] [Google Scholar]
- 12.Giannakopoulos P, Herrmann FR, Bussière T, Bouras C, Kövari E, Perl DP, Morrison JH, Gold G, Hof PR. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 13.Hara Y, Waters EM, McEwen BS, Morrison JH. Estrogen effects on cognitive and synaptic health over the lifecourse. Physiol Rev. 2015;95:785–807. doi: 10.1152/physrev.00036.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jellinger KA. The neuropathological diagnosis of Alzheimer disease. J Neural Transm Suppl. 1998;53:97–118. doi: 10.1007/978-3-7091-6467-9_9. [DOI] [PubMed] [Google Scholar]
- 15.Jiang T, Yu JT, Zhu XC, Tan MS, Gu LZ, Zhang YD, Tan L. Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol Aging. 2014a;35:1243–1251. doi: 10.1016/j.neurobiolaging.2013.11.026. [DOI] [PubMed] [Google Scholar]
- 16.Jiang T, Tan L, Zhu XC, Zhang QQ, Cao L, Tan MS, Gu LZ, Wang HF, Ding ZZ, Zhang YD, Yu JT. Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer's disease. Neuropsychopharmacology. 2014b;39:2949–2962. doi: 10.1038/npp.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang T, Yu JT, Zhu XC, Tan MS, Wang HF, Cao L, Zhang QQ, Shi JQ, Gao L, Qin H, Zhang YD, Tan L. Temsirolimus promotes autophagic clearance of amyloid-βand provides protective effects in cellular and animal models of Alzheimer's disease. Pharmacol Res. 2014c;81:54–63. doi: 10.1016/j.phrs.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 18.Jiang T, Zhang YD, Chen Q, Gao Q, Zhu XC, Zhou JS, Shi JQ, Lu H, Tan L, Yu JT. TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology. 2016a;105:196–206. doi: 10.1016/j.neuropharm.2016.01.028. [DOI] [PubMed] [Google Scholar]
- 19.Jiang T, Zhang YD, Gao Q, Zhou JS, Zhu XC, Lu H, Shi JQ, Tan L, Chen Q, Yu JT. TREM1 facilitates microglial phagocytosis of amyloid beta. Acta Neuropathol. 2016b;132:667–683. doi: 10.1007/s00401-016-1622-5. [DOI] [PubMed] [Google Scholar]
- 20.Kent SA, Spires-Jones TL, Durrant CS. The physiological roles of tau and Aβ:implications for Alzheimer's disease pathology and therapeutics. Acta Neuropathol. 2020;140:417–447. doi: 10.1007/s00401-020-02196-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiyota T, Zhang G, Morrison CM, Bosch ME, Weir RA, Lu Y, Dong W, Gendelman HE. AAV2/1 CD74 gene transfer reduces β-amyloidosis and improves learning and memory in a mouse model of Alzheimer's disease. Mol Ther. 2015;23:1712–1721. doi: 10.1038/mt.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li B, Dewey CN. RSEM:accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li D, Liu X, Liu T, Liu H, Tong L, Jia S, Wang YF. Neurochemical regulation of the expression and function of glial fibrillary acidic protein in astrocytes. Glia. 2020;68:878–897. doi: 10.1002/glia.23734. [DOI] [PubMed] [Google Scholar]
- 25.Lin HY, Tang CH, Chen JH, Chuang JY, Huang SM, Tan TW, Lai CH, Lu DY. Peptidoglycan induces interleukin-6 expression through the TLR2 receptor, JNK, c-Jun, and AP-1 pathways in microglia. J Cell Physiol. 2011;226:1573–1582. doi: 10.1002/jcp.22489. [DOI] [PubMed] [Google Scholar]
- 26.Liu W, Wang Z, Wang C, Ai Z. Long non-coding RNA MIAT promotes papillary thyroid cancer progression through upregulating LASP1. Cancer Cell Int. 2019;19:194. doi: 10.1186/s12935-019-0913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu CD, Ma JK, Luo ZY, Tai QX, Wang P, Guan PP. Transferrin is responsible for mediating the effects of iron ions on the regulation of anterior pharynx-defective-1α/βand Presenilin 1 expression via PGE(2) and PGD(2) at the early stage of Alzheimer's Disease. Aging (Albany NY) 2018;10:3117–3135. doi: 10.18632/aging.101615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macchi B, Marino-Merlo F, Frezza C, Cuzzocrea S, Mastino A. Inflammation and programmed cell death in Alzheimer's disease:comparison of the central nervous system and peripheral blood. Mol Neurobiol. 2014;50:463–472. doi: 10.1007/s12035-014-8641-9. [DOI] [PubMed] [Google Scholar]
- 30.Malik S, Arif H, Hirsch LJ. Lamotrigine and its applications in the treatment of epilepsy and other neurological and psychiatric disorders. Expert Rev Neurother. 2006;6:1609–1627. doi: 10.1586/14737175.6.11.1609. [DOI] [PubMed] [Google Scholar]
- 31.Matsuda S, Matsuda Y, D'Adamio L. CD74 interacts with APP and suppresses the production of Abeta. Mol Neurodegener. 2009;4:41. doi: 10.1186/1750-1326-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, Emerson M, Garner P, Holgate ST, Howells DW, Karp NA, Lazic SE, Lidster K, MacCallum CJ, Macleod M, Pearl EJ, et al. The ARRIVE guidelines 2.0:Updated guidelines for reporting animal research. PLoS Biol. 2020;18:e3000410. doi: 10.1371/journal.pbio.3000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pike CJ. Sex and the development of Alzheimer's disease. J Neurosci Res. 2017;95:671–680. doi: 10.1002/jnr.23827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajendran L, Paolicelli RC. Microglia-mediated synapse loss in Alzheimer's disease. J Neurosci. 2018;38:2911–2919. doi: 10.1523/JNEUROSCI.1136-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 37.Tekin S, Aykut-Bingöl C, TanridağT, Aktan S. Antiglutamatergic therapy in Alzheimer's disease--effects of lamotrigine. Short communication. J Neural Transm (Vienna) 1998;105:295–303. doi: 10.1007/s007020050059. [DOI] [PubMed] [Google Scholar]
- 38.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang K, Fernandez-Escobar A, Han S, Zhu P, Wang JH, Sun Y. Lamotrigine reduces inflammatory response and ameliorates executive function deterioration in an Alzheimer's-like mouse model. Biomed Res Int. 2016;2016:7810196. doi: 10.1155/2016/7810196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang W, Zhang Y, Yu W, Gao W, Shen N, Jin B, Wang X, Fang C, Wang Y. Bushenhuoxue improves cognitive function and activates brain-derived neurotrophic factor-mediated signaling in a rat model of vascular dementia. J Tradit Chin Med. 2020;40:49–58. [PubMed] [Google Scholar]
- 41.Wu Y, Dissing-Olesen L, MacVicar BA, Stevens B. Microglia:dynamic mediators of synapse development and plasticity. Trends Immunol. 2015;36:605–613. doi: 10.1016/j.it.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie YX, Zhang M, Zhang CR, Chen F. Relationship between NogoA/NgR1/RhoA signaling pathway and the apoptosis of cerebral neurons after cerebral infarction in rats. Eur Rev Med Pharmacol Sci. 2020;24:295–303. doi: 10.26355/eurrev_202001_19924. [DOI] [PubMed] [Google Scholar]
- 43.Xu L, He D, Bai Y. Microglia-mediated inflammation and neurodegenerative disease. Mol Neurobiol. 2016;53:6709–6715. doi: 10.1007/s12035-015-9593-4. [DOI] [PubMed] [Google Scholar]
- 44.Yu H, Liu X, Zhong Y. The effect of osteopontin on microglia. Biomed Res Int. 2017;2017:1879437. doi: 10.1155/2017/1879437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang MY, Zheng CY, Zou MM, Zhu JW, Zhang Y, Wang J, Liu CF, Li QF, Xiao ZC, Li S, Ma QH, Xu RX. Lamotrigine attenuates deficits in synaptic plasticity and accumulation of amyloid plaques in APP/PS1 transgenic mice. Neurobiol Aging. 2014;35:2713–2725. doi: 10.1016/j.neurobiolaging.2014.06.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.