

Proteostasis denotes a cellular state in which protein synthesis, folding, and degradation are maintained at a homeostatic state such that an intact yet dynamic proteome is preserved. Cellular capacity to preserve proteostasis declines with age, which is assumed to contribute to the pathogenesis of age-related diseases. Proteostasis failure manifested as the formation of aberrant protein aggregates, including the amyloid plaques in Alzheimer’s disease (AD), Lewy bodies in Parkinson’s disease, and TAR DNA binging protein 43 inclusions in amyotrophic lateral sclerosis (ALS), is a defining feature of neurodegenerative diseases. The root cause of the proteostasis failure and protein aggregation is still enigmatic. Studies in various systems suggest that cellular co-factors play important roles in “seeding” the aforementioned pathogenic protein aggregation. But the molecular nature of the initial seeding activities remains poorly defined. The pathogenic role of the disease-characterizing, macroscopically visible protein aggregates is also uncertain. Several high-profile clinical trials targeting specific protein aggregates are inconclusive and there is no evidence for a clinically relevant therapeutic effect as of now (Mullane and Williams, 2018), suggesting that the key proteostasis-disruptive, disease-causing events remain to be identified. In this perspective, I will discuss recent evidence supporting that faulty translation products resulting from inadequate quality control of translational stalling and ribosome collision during the translation of certain problematic mRNAs can be the root cause of proteostasis failure and may represent novel therapeutic targets for neurodegenerative diseases.
Studies of protein aggregation have historically been focused on alterations of fully synthesized proteins. Such changes emphasize posttranslational modifications (phosphorylation, ubiquitination, acetylation, oxidation, cleavage, etc.), and significant efforts have been made to link these molecular events to aging-related cellular dysfunctions such as oxidative stress and impairments of the autophagy and ubiquitin-proteosome systems. Recent studies provide compelling evidence that problems of proteostasis can begin with nascent peptide chains still associated with translating ribosomes, necessitating the deployment of ribosome-associated quality control (RQC) to handle faulty translation products (Brandman and Hegde, 2016).
During translation, ribosome slowdown and stalling can occur for various reasons. Some are functional and serve to facilitate cellular dynamics, such as co-translational protein folding, subcellular protein targeting, and mRNA localization. Others are detrimental and can be triggered by damaged mRNAs, mRNA secondary structures, an insufficient supply of aminoacyl-tRNAs, inefficient ribosome termination/recycling, or environmental stresses such as ultraviolet irradiation (Wu et al., 2020). Ribosome slowdown and stalling can result in ribosome collision, which is sensed by the cell as a proxy for aberrant translation and triggers RQC. Key factors involved are the ubiquitin ligase ZNF598 and the 40S subunit protein Rack1, which recognize the distinct 40S-40S interface characterizing collided ribosomes and promote ubiquitination of specific 40S subunit proteins (Juszkiewicz and Hegde, 2017; Sundaramoorthy et al., 2017), and the apoptosis-associated speck-like protein complex that disassembles the leading collided ribosome (Juszkiewicz et al., 2020). This then triggers a series of downstream quality control events, including ribosome subunit splitting and recycling by ABCE1 (an ATPase of the ATP-binding cassette transporters superfamily), modification of the nascent peptide chains still attached to the 60S subunit by a C-terminal alanine and Threonine addition (CAT-tailing) process (Shen et al., 2015), and degradation of the aberrant translation products by the Ltn1/VCP/NEMF complex (Brandman et al., 2012). Although the exact physiological role of the CAT-tails remains to be established, their accumulation under impaired RQC conditions can perturb proteostasis (Choe et al., 2016).
Previous studies of ribosome stalling, RQC, and CAT-tailing have made extensive use of artificial substrates such as mRNAs with no stop codons or mRNAs encoding long-stretches of Lysine, and such studies are largely done in single-cell systems including yeast and mammalian cell culture. Our studies in the Drosophila PINK1 model of Parkinson’s disease showed, for the first time in metazoans, that aberrant RQC can lead to CAT-tailing of mitochondrial proteins, leading to impairment of proteostasis and contributing to neurodegeneration (Wu et al., 2019). PINK1 encoded a mitochondrial serine/threonine kinase that works together with Parkin, an E3-ubiquitin ligase, to regulate mitochondrial function and mitochondrial quality control by mitophagy. One of the functions of the PINK1/Parkin pathway is to promote the co-translational import of complex-I 30 kDa subunit protein (C-I30). We found reduced C-I30 protein abundance in PINK1 or Parkin mutant flies or when wild-type animals were exposed to mitochondrial toxins. Detailed analysis of C-I30 protein revealed the generation of an upshifted C-I30 band (C-I30-u) under these conditions. After excluding posttranslational modifications (ubiquitination, phosphorylation, mitochondrial targeting sequence-cleavage) and alternative splicing as possible sources of C-I30-u generation, and after demonstration of C-terminal extension (CTE) as a possible mechanism of C-I30-u formation, we discovered that C-I30 was modified by a CAT-tailing like phenomenon we termed mitochondrial-stress-induced translational termination impairment and protein carboxyl terminal extension (MISTERMINATE) (Wu et al., 2019). Moreover, we demonstrated that some CAT-tailed C-I30 proteins were imported into mitochondria, became assembled into respiratory chain complex-I, and caused mitochondrial energy deficit. Other CAT-tailed C-I30 proteins were released into the cytosol where they formed protein aggregates and disrupted proteostasis. Interestingly, distinct from the CAT-tails in yeast that consists exclusively of alanine and Threonine, CTEs of C-I30 in PINK1 mutant also contains other amino acids (e.g., Serine, tyrosine, cysteine, and glutamic acid/proline). CTE in C-I30-u is added to C-I30 protein by ribosomes stalled at the canonical stop codon site. This stalling is caused by mitochondrial stress-induced impairment of translation termination factor eRF1 and the ribosome recycling factor ABCE1. Thus, although the CTE in C-I30-u in metazoans is generated by a mechanism thematically similar to CAT-tailing in yeast, there are fundamental differences. CTE of C-I30 under mitochondrial stress was observed both in vivo in flies and in mammalian cell culture. Genetic manipulations boosting the activities of ABCE1/eRF1 and other RQC factors, or inhibiting factors involved in CAT-tailing, including NEMF or alanine and threonine tRNA synthetases effectively rescued the neuromuscular degeneration in PINK1 flies (Wu et al., 2019), supporting the contribution of aberrant RQC of stalled translation and CAT-tailing to PINK1 pathogenesis. This RQC-related function of PINK1 and its well-established role in mitophagy likely reflect different aspects of PINK1 function in the continuum of mitochondrial homeostasis maintenance. Their mechanistic link will be the focus of future studies.
Expansion of G4C2 repeats in C9orf72 accounts for ~40% of familial and 5–10% of sporadic ALS cases, with repeat numbers ranging from a few dozen to thousands. Possible mechanisms of disease pathogenesis by G4C2 repeat expansion include haploinsufficiency of C9orf72, toxicity associated with RNA foci formed by sense and anti-sense RNAs, or proteotoxicity induced by dipeptide repeat (DPR) proteins translated from G4C2 repeat-carrying transcripts. Increasing evidence emphasize the contribution of DPR toxicity in C9-amyotrophic lateral sclerosis/frontotemporal dementia, especially arginine-containing DPR proteins (glycine-arginine (GR) and proline-arginine), with diverse cellular processes having been implicated. In our studies of poly(GR) pathogenesis, we discovered that CAT-tailing-like modification of poly(GR) contributes to disease (Li et al., 2020). We found that poly(GR) can mimic a mitochondria-targeting signal, causing some poly(GR) to be co-translationally imported into mitochondria. However, poly(GR) translation on mitochondrial surface is frequently stalled, presumably caused by electrostatic interaction between the positively-charged poly(GR) with the negatively-charged residues lining the ribosome exit tunnel. This triggered RQC and CAT-tailing-like CTE, which promotes poly(GR) stabilization, aggregation, and toxicity. To understand the regulatory mechanism governing the RQC of poly(GR) translation, we performed genetic modifier screens in Drosophila. Our genetic studies uncovered a mitochondria-associated non-canonical Notch signaling pathway that impinges on the RQC machinery to restrain poly(GR) accumulation, in part through the AKT/VCP axis. The non-canonical Notch signaling pathway and other genetic modifiers uncovered from the Drosophila studies perform similar roles in patient cells (Li et al., 2020), supporting their fundamental involvement in amyotrophic lateral sclerosis/frontotemporal dementia.
AD is a looming public health crisis. This progressive, neurodegenerative disorder and the most common form of dementia is affecting more than 40 million people worldwide, with the number projected to double by 2025 if no effective treatment becomes available. While a small portion of AD cases is caused by familial genetic mutations, the majority is sporadic with no known cause. At the neuropathological level, AD is characterized by the presence of extracellular plaques composed of beta-amyloid (Aβ) and neurofibrillary tangles composed of hyperphosphorylated tau. Aβ is derived from amyloid precursor protein (APP). Human genetics studies indicate that aberrant APP synthesis or processing is central to AD pathogenesis, but the key etiological driver of disease remains elusive, despite a tremendous amount of effort having been focused on Aβ. Inconclusive results of clinical trials targeting Aβ peptides suggest that the key pathogenic species remain to be identified. Previous studies in mammalian cell culture, mouse transgenic models, and human induced pluripotent stem cell-derived neuron culture suggested that APP C-terminal fragment (APP.C99) can cause disease in an Aβ-independent manner. The mechanism of APP.C99 pathogenesis is incompletely understood. We used Drosophila models expressing APP.C99 with the native ER-targeting signal of human APP, and models expressing full-length human APP only or co-expressing full-length human APP and β-secretase to investigate mechanisms of APP.C99 pathogenesis (Rimal et al., 2021). Key findings from Drosophila models are validated in mammalian cell culture models, mouse 5×FAD model, and postmortem AD patient brain materials. We find that ribosomes stall at the ER membrane during co-translational translocation of APP.C99. The stall sites are mapped to the stop codon site and an internal site between the transmembrane domain and the stop codon site. Moreover, stalling at the stop codon site was shown to be due to decreased levels of ribosome recycling and termination factors ABCE1 and eRF1/eRF3, whereas the internal stalling was caused by a combination of factors including ER-targeting, translocon-gating, and subsequent protein folding and membrane insertion (Rimal et al., 2021), events intrinsic to APP.C99 biogenesis and membrane topogenesis and likely to slow down translation and cause ribosome collision. Ribosome stalling activated RQC to resolve ribosome collision and stalled translation. Stalled APP.C99 species resulting from inadequate RQC are prone to aggregation, causing endolysosomal and autophagy defects and seeding the aggregation of Aβ peptides, the main component of amyloid plaques. Genetic manipulations that remove stalled and CAT-tailed APP.C99, such as ABCE1 overexpression or knockdown of ZNF598, rescued proteostasis failure, endolysosomal/autophagy dysfunction, neuromuscular degeneration, and cognitive deficits in AD models (Rimal et al., 2021). Importantly, many RQC factors such as NEMF, ANKZF1, ZNF598, and Rack1, but not ABCE1 or eRF1, were found deposited at the core of amyloid plaques from AD patient brains (Rimal et al., 2021), a finding that further supports the central role of defective RQC of ribosome collision and stalled translation in AD pathogenesis. These findings demonstrate that amyloid plaque formation is the consequence and manifestation of a deeper level proteostasis failure caused by inadequate RQC of translational stalling and the resultant aberrantly modified APP.C99 species, previously unrecognized etiological drivers of AD. It should be pointed out that translational stalling is clearly not the only way by which abnormal proteins can be generated in neurodegenerative diseases. For example, we showed that the RQC pathway has little effect on tau protein level and toxicity (Li et al., 2020), suggesting that toxic tau species associated with AD and other tauopathies are likely generated by other mechanisms.
Together, these studies showed that certain mRNAs encoding disease-associated proteins are physiological substrate of the RQC pathway and that aberrant protein species containing CAT tail-like CTEs resulting from defective RQC of stalled translation perturb proteostasis and contribute to cognitive dysfunction and neuromuscular degeneration in animal models. Remarkably, evidence so far indicates that RQC plays a particularly important role in maintaining mitochondrial homeostasis. Thus, defective RQC offers a mechanistic link between proteostasis failure and mitochondrial dysfunction, two pathological hallmarks of neurodegenerative diseases (Figure 1). It is worth noting that we are only scratching the surface in terms of understanding the biological consequence of translational stalling and ribosome collision on cellular homeostasis, as recent studies suggest that collided ribosomes may present cell signaling platforms that can activate innate immune and stress response pathways that impart cell survival or cell death decisions (Wu et al., 2020; Wan et al., 2021). Future studies, preferably done in human induced pluripotent stem cell-derived models that express disease-causing proteins at physiological levels and thus obviate drawbacks of non-physiological overexpression commonly associated with animal models, will address: 1) the molecular mechanisms underlying RQC failure in different diseases; 2) the composition of the CTEs added to different stalled translation products; 3) the mechanisms of neurotoxicity induced by collided ribosomes and proteins containing aberrant CTEs, especially their link to stress response pathways such as the integrated stress response; 4) how is the RQC process linked to environmental stresses and the aging process, which underlie the pathogenesis of sporadic neurodegenerative disease (Frisoni et al., 2021); and 5) possible link between RQC and translational accuracy, which has been associated with lifespan extension and neuroprotection (Martinez-Miguel et al., 2021). Our results also suggested novel therapeutic strategies. For example, in the case of AD, restoring endolysosomal or autophagy function impaired by APP.C99, or modulating RQC to get rid of aberrant APP.C99 RQC products deserve serious consideration. Moreover, boosting ER and mitochondrial function, which are expected to indirectly reduce the ribosome stalling of endoplasmic reticulum translocon-engaged APP.C99, may also prove to be beneficial. Research efforts in these directions have the potential to lead to paradigm shift in our understanding of neurodegenerative disease mechanisms and ultimately to the development of therapeutic strategies applicable to a broad spectrum of diseases.
Figure 1.
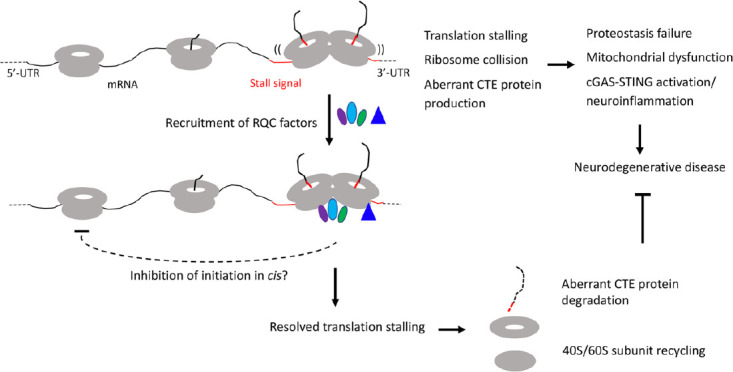
Diagram depicting the effect of RQC recruitment on resolving stalled translation and restoring cellular homeostasis, and how inadequate quality control of stalled translation and collided ribosomes may be linked to various hallmarks of neurodegenerative diseases, including proteostasis failure, mitochondrial dysfunction, and neuroinflammation.
CTE: C-terminal extension; cGAS-STING: the cyclic GMP-AMP synthase-stimulator of interferon genes; RQC: ribosome-associated quality control; UTR: untranslated regions.
Additional file: Open peer review report 1 (84.7KB, pdf) .
Footnotes
Availability of data and materials: All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open peer reviewer: Xinjie Chen, The State University of New York, USA.
P-Reviewer: Chen X; C-Editors: Zhao M, Liu WJ, Wang Lu; T-Editor: Jia Y
References
- 1.Brandman O, Stewart-Ornstein J, Wong D, Larson A, Williams CC, Li GW, Zhou S, King D, Shen PS, Weibezahn J, Dunn JG, Rouskin S, Inada T, Frost A, Weissman JS. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell. 2012;151:1042–1054. doi: 10.1016/j.cell.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brandman O, Hegde RS. Ribosome-associated protein quality control. Nat Struct Mol Biol. 2016;23:7–15. doi: 10.1038/nsmb.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choe YJ, Park SH, Hassemer T, Korner R, Vincenz-Donnelly L, Hayer-Hartl M, Hartl FU. Failure of RQC machinery causes protein aggregation and proteotoxic stress. Nature. 2016;531:191–195. doi: 10.1038/nature16973. [DOI] [PubMed] [Google Scholar]
- 4.Frisoni GB, Altomare D, Thal DR, Ribaldi F, van der Kant R, Ossenkoppele R, Blennow K, Cummings J, van Duijn C, Nilsson PM, Dietrich PY, Scheltens P, Dubois B. The probabilistic model of Alzheimer disease:the amyloid hypothesis revised. Nat Rev Neurosci. 2021;23:53–66. doi: 10.1038/s41583-021-00533-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Juszkiewicz S, Hegde RS. Initiation of quality control during poly(A) translation requires site-specific ribosome ubiquitination. Mol Cell. 2017;65:743–750. doi: 10.1016/j.molcel.2016.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Juszkiewicz S, Speldewinde SH, Wan L, Svejstrup JQ, Hegde RS. The ASC-1 complex disassembles collided ribosomes. Mol Cell. 2020;79:603–614. doi: 10.1016/j.molcel.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li S, Wu Z, Tantray I, Li Y, Chen S, Dong J, Glynn S, Vogel H, Snyder M, Lu B. Quality-control mechanisms targeting translationally stalled and C-terminally extended poly(GR) associated with ALS/FTD. Proc Natl Acad Sci U S A. 2020;117:25104–25115. doi: 10.1073/pnas.2005506117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez-Miguel VE, Lujan C, Espie-Caullet T, Martinez-Martinez D, Moore S, Backes C, Gonzalez S, Galimov ER, Brown AEX, Halic M, Tomita K, Rallis C, von der Haar T, Cabreiro F, Bjedov I. Increased fidelity of protein synthesis extends lifespan. Cell Metab. 2021;33:2288–2300. doi: 10.1016/j.cmet.2021.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mullane K, Williams M. Alzheimer's disease (AD) therapeutics - 1:Repeated clinical failures continue to question the amyloid hypothesis of AD and the current understanding of AD causality. Biochem Pharmacol. 2018;158:359–375. doi: 10.1016/j.bcp.2018.09.026. [DOI] [PubMed] [Google Scholar]
- 10.Rimal S, Li Y, Vartak R, Geng J, Tantray I, Li S, Huh S, Vogel H, Glabe C, Grinberg LT, Spina S, Seeley WW, Guo S, Lu B. Inefficient quality control of ribosome stalling during APP synthesis generates CAT-tailed species that precipitate hallmarks of Alzheimer's disease. Acta Neuropathol Commun. 2021;9:169. doi: 10.1186/s40478-021-01268-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen PS, Park J, Qin Y, Li X, Parsawar K, Larson MH, Cox J, Cheng Y, Lambowitz AM, Weissman JS, Brandman O, Frost A. Protein synthesis. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains. Science. 2015;347:75–78. doi: 10.1126/science.1259724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sundaramoorthy E, Leonard M, Mak R, Liao J, Fulzele A, Bennett EJ. ZNF598 and RACK1 regulate mammalian ribosome-associated quality control function by mediating regulatory 40S ribosomal ubiquitylation. Mol Cell. 2017;65:751–760. doi: 10.1016/j.molcel.2016.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wan L, Juszkiewicz S, Blears D, Bajpe PK, Han Z, Faull P, Mitter R, Stewart A, Snijders AP, Hegde RS, Svejstrup JQ. Translation stress and collided ribosomes are co-activators of cGAS. Mol Cell. 2021;81:2808–2822. doi: 10.1016/j.molcel.2021.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu CC, Peterson A, Zinshteyn B, Regot S, Green R. Ribosome collisions trigger general stress responses to regulate cell fate. Cell. 2020;182:404–416. doi: 10.1016/j.cell.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Z, Tantray I, Lim J, Chen S, Li Y, Davis Z, Sitron C, Dong J, Gispert S, Auburger G, Brandman O, Bi X, Snider M, Lu B. MISTERMINATE mechanistically links mitochondrial dysfunction with proteostasis failure. Mol Cell. 2019;75:835–848. doi: 10.1016/j.molcel.2019.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.