

ABSTRACT
Candida glabrata is poised to adapt to drug pressure rapidly and acquire antifungal resistance leading to therapeutic failure. Given the limited antifungal armamentarium, there is an unmet need to explore new targets or therapeutic strategies for antifungal treatment. The lysine acetyltransferase Gcn5 has been implicated in the pathogenesis of C. albicans. Yet how Gcn5 functions and impacts antifungal resistance in C. glabrata is unknown. Disrupting GCN5 rendered C. glabrata cells more sensitive to various stressors, partially reverted resistance in drug-resistant mutants, and attenuated the emergence of resistance compared to wild-type cells. RNA sequencing (RNA-seq) analysis revealed transcriptomic changes involving multiple biological processes and different transcriptional responses to antifungal drugs in gcn5Δ cells compared to wild-type cells. GCN5 deletion also resulted in reduced intracellular survival within THP-1 macrophages. In summary, Gcn5 plays a critical role in modulating the virulence of C. glabrata and regulating its response to antifungal pressure and host defense.
IMPORTANCE As an important and successful human pathogen, Candida glabrata is known for its swift adaptation and rapid acquisition of resistance to the most commonly used antifungal agents, resulting in therapeutic failure in clinical settings. Here, we describe that the histone acetyltransferase Gcn5 is a key factor in adapting to antifungal pressure and developing resistance in C. glabrata. The results provide new insights into epigenetic control over the drug response in C. glabrata and may be useful for drug target discovery and the development of new therapeutic strategies to combat fungal infections.
KEYWORDS: Candida glabrata, GCN5, antifungal resistance, virulence, transcriptomics
INTRODUCTION
Invasive candidiasis is an important fungal infection caused by Candida species with high morbidity and mortality rates (1, 2). Although Candida albicans remains the predominant pathogen for invasive candidiasis, infections due to non-C. albicans Candida species have increased significantly in the past 2 decades (3, 4), among which Candida glabrata is the most frequently isolated species in the SENTRY antifungal surveillance program (5). The global emergence of C. glabrata is worrisome because this yeast species is poised to adapt to drug pressure and acquire antifungal resistance leading to therapeutic failure (1, 6). The combination of this feature and the highly limited antifungal armamentarium currently available in the clinical setting forms a perfect storm plaguing global public health. Thus, there is an urgent need to explore new targets or therapeutic strategies for antifungal development.
Antifungal resistance studies have focused largely on genetic mutations involving the target site and transcription factors regulating drug efflux pumps, aneuploidy, the upregulation of stress response pathways, and biofilm formation (7). Yet an increasing body of evidence has shown that epigenetic pathways may be important factors contributing to drug resistance via existing or novel mechanisms (8–10). Broadly speaking, epigenetic mechanisms can be either RNA based or chromatin based. The latter consists of both chemical and structural modifications that change the accessibility of transcription factors to specific genomic regions, therefore regulates global transcription (9). Among the most studied chromatin modifications, histone acetylation has been suggested to be critical in dictating the mutational landscape of yeast cells, thus contributing to the development of drug resistance in different fungal pathogens (10–12). Histone acetylation is one of the well-characterized posttranslational modifications (PTMs) involving the addition/removal of acetyl groups to/from lysine residues in the amino tails of histones mediated by lysine acetyltransferases (KATs) and lysine deacetylases (KDACs), respectively (13, 14). It has been suggested that close cooperation of KATs/KDACs with dedicated transcriptional regulators forms a dual-layer network of chromatin-mediated transcriptional control in the major fungal pathogen C. albicans (11). Gcn5 is a pleiotropic KAT constituting the catalytic subunit of the SAGA (Spt-Ada-Gcn5-acetyltransferase) complex that is conserved in eukaryotes (15, 16). Previous studies have shown that Gcn5 plays roles in morphogenesis, pathogenesis, virulence, and the stress response in multiple fungal organisms, including C. albicans (17–21). However, other than two recent studies (22, 23) showing that inhibition of Gcn5 attenuated the emergence of azole resistance in C. glabrata in vitro and that a C. glabrata Δgcn5 mutant was less virulent than the wild-type (WT) strain in a Galleria mellonella infection model, little is known about how Gcn5 functions and becomes involved in antifungal resistance in this major human fungal pathogen. Hence, we employed multiple strategies and systematically investigated the impact of Gcn5 on the virulence and drug response of C. glabrata.
RESULTS
Phenotypic and epigenetic profiles and changes in stress responses and drug susceptibility associated with GCN5 in C. glabrata.
The growth of the WT, the genetically deleted knockout (KO) strain gcn5Δ, and reconstituted wild-type strain gcn5Δ::GCN5 was monitored in yeast extract-peptone-dextrose (YPD) broth at 37°C. gcn5Δ cells showed slightly slower growth, with an ~10-min-longer doubling time than that of the WT. The growth curve of the gcn5Δ::GCN5 complementary strain is similar to that of the WT (see Fig. S1 in the supplemental material). In limited histone acetylation profiling, acetylated lysine residue 14 of histone 3 (H3K14) and H3K9 in the gcn5Δ strain were observed to be decreased to ~46% and 60% of the levels in the WT, respectively (Fig. S2), consistent with the known function of Gcn5. Spotting assays demonstrated that the deletion of GCN5 conferred increased susceptibility to various cell stress conditions, including oxidative stress, cell wall and cell membrane perturbation, as well as antifungal agents (Fig. 1A). In vitro susceptibility testing determined that the MICs of the gcn5Δ strain modestly (~2- to 4-fold), yet consistently, decreased for triazole and glucan synthase inhibitor antifungals compared to the WT and complemented strains (Table 1). Interestingly, the gcn5Δ strain displayed hypersensitivity to the new antifungal manogepix (formerly APX001A), with an MIC of <0.008 μg/mL. To gain a better understanding of the impact of Gcn5 on echinocandin resistance, we next disrupted GCN5 from clinically relevant FKS1 and FKS2 mutants constructed in the ATCC 2001 background, including Fks1-625delF, Fks1-S629P, Fks2-659delF, and Fks2-S663P. Upon the deletion of GCN5, all FKS mutants had a 4-fold-lower MIC for micafungin, except for that of the S663P mutant, which decreased only 2-fold (Table 2). This result was largely consistent with what was observed in the spotting assay, where the effect of reduced resistance associated with GCN5 deletion was seemingly more pronounced in FKS1 mutants than in FKS2 mutants (Fig. 1B), suggesting that GCN5 plays a role in modulating FKS expression levels, with differential impacts over FKS1 and FKS2 possibly via divergent interactions with other regulators or pathways involved in the regulation network. Given that FKS2 expression is dependent upon calcineurin signaling (24), we tested whether GCN5 disruption influences the sensitivity to the calcineurin inhibitor FK506 as well as the effect of the combination of the GCN5 deletion and FK506 on the cells’ susceptibility to echinocandins. We found that all GCN5 deletion strains, including both the WT and FKS mutants, displayed a 2- to 4-fold increase in susceptibility to FK506, with the F659del_gcn5Δ strain demonstrating hypersusceptibility (Table 2). While the addition of FK506 is known to reverse Fks2-mediated echinocandin resistance (24), we found even greater increased susceptibility to micafungin in the presence of FK506 upon GCN5 deletion. Strikingly, this reversal of resistance was not only relegated to the FKS2 mutants but also observed with the FKS1 mutants.
FIG 1.

(A) Spotting assay to evaluate stress response changes associated with GCN5 deletion in C. glabrata. Five microliters of 10-fold serial dilutions of the indicated cells was spotted onto plain YPD plates and YPD plates supplemented with hydrogen peroxide (H2O2) (10 mM), SDS (0.01%), calcofluor white stain (10 μg/mL), Congo red (50 μg/mL), fluconazole (16 μg/mL), caspofungin (0.03 μg/mL), and micafungin (0.008 μg/mL). Plates were incubated at 37°C for 24 h. (B) Spotting assay with fks mutants (Fks1-625delF, Fks1-S629P, Fks2-659delF, and Fks2-S663P) with or without Gcn5 function to assess the impact of GCN5 on echinocandin resistance. Micafungin was tested at 0.03 μg/mL, and caspofungin was tested at 0.25 μg/mL.
TABLE 1.
Antifungal susceptibility changes associated with GCN5 deletion
| Antifungal agent | Time of readout (h) | MIC (μg/mL) |
||
|---|---|---|---|---|
| WT | gcn5Δ | gcn5Δ::GCN5 | ||
| Itraconazole | 24 | 0.25 | 0.125 | 0.25 |
| 48 | 1 | 0.25 | 1 | |
| Posaconazole | 24 | 0.5 | 0.125 | 0.5 |
| 48 | 1 | 0.5 | 1 | |
| Voriconazole | 24 | 0.5 | 0.25 | 0.5 |
| 48 | 1 | 0.5 | 1 | |
| Fluconazole | 24 | 16 | 8 | 16 |
| 48 | 32 | 16 | 32 | |
| Anidulafungin | 24 | 0.03 | 0.015 | 0.03 |
| Caspofungin | 24 | 0.06 | 0.06 | 0.06 |
| Micafungin | 24 | 0.03 | 0.008 | 0.015 |
| Manogepix | 24 | 0.064 | ≤0.008 | 0.03 |
| Ibrexafungerp | 24 | 0.5 | 0.25 | 0.5 |
TABLE 2.
Deletion of GCN5 increases echinocandin susceptibility and reverses Fks1-mediated echinocandin resistance by FK506
| Strain | 24-h MIC (μg/mL) |
||
|---|---|---|---|
| Micafungin | FK506 | Micafungin + FK506 (4 μg/mL) | |
| WT | 0.03 | 32 | 0.015 |
| gcn5Δ | 0.008 | 8 | ≤0.001 |
| S629P | 0.5 | 32 | 0.25 |
| S629P_gcn5Δ | 0.125 | 16 | ≤0.03 |
| F625del | 0.5 | 32 | 0.125 |
| F625del_gcn5Δ | 0.125 | 8 | ≤0.03 |
| S663P | 4 | 32 | 1 |
| S663P_gcn5Δ | 2 | 16 | 0.5 |
| F659del | 2 | 16 | 0.25 |
| F659del_gcn5Δ | 0.5 | ≤0.125 | ≤0.03 |
GCN5 disruption leads to reduced drug tolerance and resistance development in C. glabrata.
Antifungal tolerance and resistance, impeding effective antifungal therapy and leading to unfavorable clinical outcomes, are two different but relevant phenotypes that fungal pathogens display in response to antifungal agents. Tolerance to fungicidal drugs is defined as the ability of fungal cells to survive at drug concentrations above the MIC (25), and tolerance is considered a key prerequisite for echinocandin resistance in C. glabrata (26, 27). As for fungistatic drugs such as azoles, the concept can be adjusted, and it is reasonable to consider that Candida spp. are generally tolerant to azoles (25). To better understand how Gcn5 shapes the antifungal response in C. glabrata, we carried out time-kill studies comparing the survival of the WT and gcn5Δ strains in the absence or presence of micafungin or fluconazole, with concentrations ranging from 0.03 to 1.92 μg/mL or from 16 to 1,024 μg/mL, respectively. Enhanced fungicidal activity of micafungin was visualized on killing curves at all levels tested against the gcn5Δ strain compared to the WT (Fig. 2A), where a greater colony count reduction was observed with the gcn5Δ strain than with the WT at all time points prior to 48 h, and most strikingly, the lowest level of micafungin (0.03 μg/mL) was also fungicidal against the gcn5Δ strain yet only fungistatic against the WT. Similarly, the antifungal activity of fluconazole was largely improved upon GCN5 deletion (Fig. 2B). Despite being considered fungistatic, fluconazole at 1,024 μg/mL resulted in a 2.44-log10 CFU/mL reduction of gcn5Δ cells after 24 h of treatment.
FIG 2.

Time-kill curve of micafungin and fluconazole against C. glabrata wild-type (WT) and gcn5Δ cells. The cells were incubated at 37°C in RPMI 1640 in the presence or absence of micafungin (MCF) and fluconazole (FLC) at the indicated concentrations. CFU counting was carried out at predetermined time points (0, 2, 4, 8, 24, and 48 h) after drug treatment. (A) Micafungin was significantly more potent against gcn5Δ than against WT cells. The 0.03-μg/mL micafungin treatment was fungicidal to gcn5Δ but not to WT cells. (B) Fluconazole was more potent against gcn5Δ than against WT cells, with significantly enhanced antifungal activity observed with treatment for 24 h or longer at concentrations of 256 and 1,024 μg/mL. Fungal population size change comparisons between WT and gcn5Δ cells at each time point are listed underneath the killing curves for micafungin and fluconazole, respectively.
To determine the effects of GCN5 on C. glabrata resistance development, we harvested drug-treated cells at the 24- and 48-h time points of the time-kill study and plated them onto drug-containing plates (0.25 μg/mL micafungin or 512 μg/mL fluconazole) to measure the resistance frequency. Due to the very potent killing of micafungin, resistant colonies were obtained only from WT cells treated with the MIC (0.03 μg/mL) of micafungin, and gcn5Δ cells were completely devoid of resistance development within 48 h of exposure to micafungin at all levels tested (Fig. 3A). In the next performed experiment using an extremely low level of micafungin of 8 ng/mL, a low level of resistance (2.23 × 10−8) was acquired by gcn5Δ cells after 48 h but not 24 h of drug exposure (Fig. 3A and Table 3). Under this condition, WT cells developed micafungin resistance at both 24 and 48 h postexposure at comparable frequencies. Resistant colonies from both the WT and gcn5Δ strains were selected for FKS sequencing, and all contained Fks2-659delF (Table 3). The gcn5Δ strain also exhibited a lower frequency of resistance than that of the WT to fluconazole across all levels tested at both time points (Fig. 3B). Unexpectedly, no PDR1 mutation was identified from 10 resistant colonies selected from the 256- and 1,024-μg/mL fluconazole treatment groups (Table 3). Together, these data revealed that the deletion of GCN5 in C. glabrata leads to a significant decrease in antifungal tolerance and resistance development.
FIG 3.

Deleting GCN5 in C. glabrata leads to reduced resistance development under antifungal pressure. Micafungin resistance (A) and fluconazole resistance (B) were measured in both WT and gcn5Δ cells at the 24-h and 48-h time points of the time-kill assay. Cells were plated onto YPD plates containing micafungin (0.25 μg/mL) and fluconazole (512 μg/mL), respectively. The plots show mean resistant colony frequencies ± standard deviations (SD) from ≥3 independent experiments.
TABLE 3.
Deletion of GCN5 leads to decreases in frequencies of resistant colonies and resistance-conferring mutations
| Antifungal agent | Concn (μg/mL) | Strain | Time (h) | Avg resistant colony frequency ± SD | No. of mutations identified/total no. of colonies sequenceda | Protein mutation (nucleotide change) |
|---|---|---|---|---|---|---|
| Micafungin | 0.03 | WT | 24 | 1.32E−07 ± 5.09E−09 | 5/5 | Fks2-659delF (1971_1973delTTC) |
| 48 | 1.32E−07 ± 1.94E−08 | 12/12 | Fks2-659delF (1971_1973delTTC) | |||
| 0.03 | gcn5Δ | 24 | 0 | |||
| 48 | 0 | |||||
| 0.008 | WT | 24 | 1.57E−08 ± 4.32E−09 | NT | ||
| 48 | 1.73E−08 ± 1.64E−10 | NT | ||||
| 0.008 | gcn5Δ | 24 | 0 | |||
| 48 | 2.23E−08 ± 2.23E−08 | 4/4 | Fks2-659delF (1971_1973delTTC) | |||
| Fluconazole | 16 | WT | 24 | 3.95E−04 ± 7.56E−06 | NT | |
| 48 | 6.72E−05 ± 1.29E−06 | NT | ||||
| 16 | gcn5Δ | 24 | 2.52E−04 ± 1.71E−05 | NT | ||
| 48 | 3.90E−05 ± 2.64E−06 | NT | ||||
| 64 | WT | 24 | 2.99E−03 ± 5.73E−05 | NT | ||
| 48 | 1.60E−03 ± 1.34E−03 | NT | ||||
| 64 | gcn5Δ | 24 | 2.84E−03 ± 1.68E−04 | NT | ||
| 48 | 9.33E−05 ± 8.09E−05 | NT | ||||
| 256 | WT | 24 | 2.67E−02 ± 5.12E−04 | 0/2 | No PDR1 mutation identified | |
| 48 | 3.98E−02 ± 7.62E−04 | 0/1 | ||||
| 256 | gcn5Δ | 24 | 4.26E−03 ± 2.88E−04 | 0/1 | ||
| 48 | 8.95E−03 ± 8.77E−03 | 0/1 | ||||
| 1,024 | WT | 24 | 2.08E−03 ± 3.98E−05 | 0/1 | ||
| 48 | 2.26E−01 ± 7.22E−02 | 0/2 | ||||
| 1,024 | gcn5Δ | 24 | 0 | |||
| 48 | 1.37E−04 ± 9.28E−06 | 0/2 | ||||
NT, not tested.
Perturbed cell wall architecture and reduced adhesion upon GCN5 deletion revealed by transcriptional profiling.
To understand transcriptomic changes associated with GCN5 disruption in C. glabrata, we performed RNA sequencing (RNA-seq) analysis using WT and gcn5Δ cells harvested at logarithmic phase. Totals of 101 and 98 genes were up- and downregulated at least 2-fold with a significant P value (P ≤ 0.01), respectively, in the gcn5Δ strain relative to the WT strain (Fig. 4A; Data Set S1). Gene Ontology (GO) enrichment analysis found that genes differentially expressed upon GCN5 deletion are mainly enriched in cell transmembrane activities and cell wall assembly, as shown in Fig. 4B. Even though the transcriptomic changes seemed to be balanced by the comparable numbers of up- and downregulated genes, ranking of the significant expression differences (less than or equal to −2-fold or greater than or equal to 2-fold with a P value of ≤0.01) showed that the top 20 most significantly changed genes upon GCN5 deletion were predominantly downregulated (Data Set S1). Noticeably, most of the downregulated genes play important roles in cell wall and cell membrane functions, including the cell wall adhesin genes EPA6 and EPA13 and the β-mannosyltransferase gene BMT5, etc. To validate the RNA-seq findings, a selective set of genes with the most significant changes in transcriptomic profiling were subjected to quantitative real-time reverse transcription-PCR (qRT-PCR) for mRNA expression quantification, and consistent results were observed (Fig. S3). Given that associations between adhesins, particularly EPA6, and biofilm formation are well established and that the capacity to strongly adhere to many different surfaces is an important virulence factor of C. glabrata (28, 29), we characterized the adhesion of gcn5Δ cells to polystyrene (early stage of biofilm formation) and compared it with those of the parental WT and complemented strains (Fig. 5). Corroborating the transcriptional profiling results, adherence to polystyrene after 24 h of incubation was diminished to ~75% of that of the WT upon the deletion of GCN5 (P = 0.019), whereas it was restored to the WT level when GCN5 was complemented to the deletion mutant.
FIG 4.

Transcriptional profiling of the gcn5Δ strain. Shown are a volcano plot of differentially expressed genes (DEGs) (A) and GO enrichment analysis of DEGs associated with GCN5 deletion (B). NS, not significant.
FIG 5.
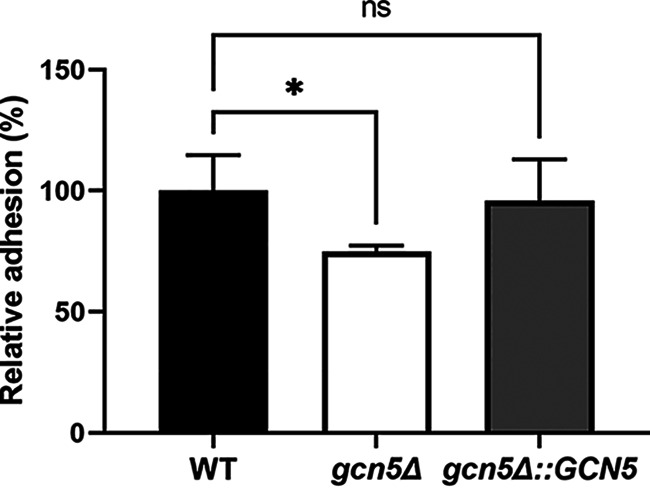
Adhesion to polystyrene after 24 h measured by crystal violet staining. Adherence was normalized to that of the WT. Data represent the means ± SD obtained from 4 technical and 2 biological replicates (*, P < 0.05; ns, not significant).
To better understand antifungal susceptibility changes associated with GCN5 deletion, we quantified the expression levels of several key genes known to impact drug tolerance/resistance by qRT-PCR, including FKS1, FKS2, CDR1, CDR2, and CRZ1 (Fig. S3). The lack of GCN5 resulted in a 1.6-fold drop in FKS1 expression (P = 0.007), accompanied by a possibly compensatory 1.4-fold increase in FKS2 expression (P = 0.09), indicating that GCN5-governed transcriptional regulation has a positive modulating effect on FKS1 but not FKS2 expression. Given that FKS1 and FKS2 in ATCC 2001 are expressed at a roughly 2:1 ratio based on our previous report (30) as well as the current study (data not shown), it is reasonable to envision that the net effect of such opposite expression changes of FKS1 and FKS2 in the gcn5Δ strain is an overall decrease in the production of β-glucan synthase, explaining the moderately increased echinocandin sensitivity upon GCN5 deletion. Interestingly, differential regulation was also observed for drug efflux pumps in gcn5Δ cells, where CDR2 expression decreased 3.2-fold (P = 0.03) but the CDR1 mRNA level was not significantly changed compared to those of the WT. Examining the expression of CRZ1, a key transcription factor of the calcineurin signaling pathway (31), found that GCN5 disruption had a modest repression effect (1.5-fold decrease [P = 0.18]) on CRZ1 transcription. Taken together, the genetic ablation of GCN5 alters the transcriptional landscape, impacting multiple biological processes, primarily cell wall biosynthesis, cell membrane integrity, and transmembrane transport.
Transcriptomic analysis of WT and gcn5Δ cells under antifungal pressure.
To uncover the role of Gcn5 in the antifungal response, we subjected both WT and gcn5Δ cells to low levels of fluconazole and micafungin, 8 μg/mL and 15 ng/mL, respectively, and analyzed transcriptomic changes induced by the antifungal drug in both cell types after a 2-h exposure. Compared to the no-drug control, fluconazole pressure triggered a globally upregulated transcriptome in WT cells, with 63 genes being significantly upregulated and only 16 being significantly downregulated (Fig. 6A). In comparison, gcn5Δ cells displayed a very blunt transcriptional response to the same level of fluconazole stress, with only 24 genes in total being upregulated over 2-fold and no gene being downregulated more than 2-fold (Fig. 6C). A comparison of the two sets of upregulated genes in WT and gcn5Δ cells found that most of the significantly induced genes in the gcn5Δ strain (18/24) were also upregulated in the WT (Fig. S4). Enriched GO mapping (Fig. 6B and D) also showed that WT and gcn5Δ cells shared some similarities in transcriptional responses to fluconazole stress, including oxidation-reduction processes, the stress response, cell wall structure, and ergosterol biosynthesis. To validate the RNA-seq findings, we next selected several genes involved in these biological processes and performed RT-PCR to quantify their expression levels (Fig. S5). Three genes associated with transmembrane transporter activity, STR3 (putative cystathionine β-lyase), CAGL0L06776g (predicted to have DNA-binding transcription factor activity), and CAGL0L03828g (orthologs of which have electron transfer activity), were significantly upregulated by ~6- to 15-fold in WT cells upon fluconazole stress but were only slightly induced in gcn5Δ cells under the same pressure. Likewise, the expression levels of the putative glycosylphosphatidylinositol (GPI)-linked cell wall protein-encoding genes CAGL0H09614g and AWP7 in fluconazole-treated gcn5Δ cells were significantly lower than those in the fluconazole-treated WT strain due to lower increased expression levels in the knockout cells than in the WT under the same drug pressure, consistent with the RNA-seq results. To gain mechanistic insights into the increased sensitivity to azoles observed with gcn5Δ cells, we profiled the expression levels of key genes known to influence fluconazole tolerance and resistance, including ERG11, CDR1, CDR2, and PDR1, for both strains under fluconazole pressure (Fig. S5). As a result, ERG11, PDR1, and CDR2 were all expressed at a significantly lower level in fluconazole-treated gcn5Δ cells than in fluconazole-treated WT cells, largely agreeing with the reduced fluconazole tolerability phenotype of the deletion mutant. However, CDR1 was more highly expressed in gcn5Δ than in WT cells upon fluconazole exposure, presumably a compensatory result of the suppressed CDR2. Overall, these findings suggest that Gcn5 mediates transcriptome activation involving multiple pathways to resist fluconazole activity; hence, the removal of GCN5 suppressed an effective transcriptional response, conferring increased sensitivity of knockout cells to fluconazole.
FIG 6.

Fluconazole-triggered transcriptional changes. (A and B) Volcano plot of DEGs (A) and GO enrichment analysis of DEGs (B) in WT cells. (C and D) Volcano plot of DEGs (C) and GO enrichment analysis of DEGs (D) in gcn5Δ cells.
Unlike fluconazole, the fungicidal drug micafungin exerted much more severe stresses on cells, resulting in a remarkably higher number of genes differentially expressed in both WT and gcn5Δ cells than in the corresponding no-drug controls (Fig. 7A and C). Interestingly, the transcriptomic changes occurred at a greater scale in gcn5Δ than in WT cells, with ~4 to 5 times more genes altering expression levels over 2-fold in the knockout cells, indicating that cell wall destruction may be a more severe menace for cells lacking GCN5; hence, a more extensive transcriptomic adaptation is needed. A Venn diagram (Fig. S6) overlaying the differentially expressed gene sets triggered by micafungin in WT and gcn5Δ cells shows that about 60% of the upregulated genes and 2/3 of the downregulated genes in the WT had significant transcriptional changes with the same direction in the gcn5Δ strain. However, these overlapping genes account for only <20% of the transcriptomic changes found in the gcn5Δ strain. Consistently, enriched GO mapping (Fig. 7B and D) displayed that the cellular pathways involved in the response to micafungin varied in the WT versus the gcn5Δ strain. The transcriptional regulation of the WT in the face of micafungin was focused mainly on adjusting the fungal cell wall organization and cell membrane transport activity. Under the same pressure, the gcn5Δ strain elicited more extensive transcriptional alterations, a large part of which was to rewire RNA composition, processing, and transcription machinery. The top 10 most significantly changed genes in WT cells in response to micafungin treatment were predominantly upregulated (Data Set S1), whereas downregulation was observed in 8 out of the 10 most significantly changed genes in gcn5Δ cells (Data Set S1). To verify these findings, we performed qRT-PCR to quantify the expression levels of a few representative genes (CAGL0M03377g, SUT2, CAGL0K10626g, and CAGL0I00484g) (Fig. S7). Not surprisingly, all tested genes had significantly lower expression levels in gcn5Δ than in WT cells under micafungin pressure. However, this is not the case for FKS genes. The expression level of FKS2 was 1.78-fold higher in micafungin-treated gcn5Δ cells than in micafungin-treated WT cells, although FKS1 was expressed at comparable levels in both types of cells. These results suggest that gcn5Δ cells experienced a higher level of cell wall destruction than WT cells from micafungin exposure, which triggered a more extensive compensatory induction of FKS expression.
FIG 7.

Transcriptional profiling of WT and gcn5Δ cells in response to micafungin pressure. (A and B) Volcano plot of DEGs (A) and GO enrichment analysis of DEGs (B) in the WT triggered by micafungin. (C and D) Volcano plot of DEGs (C) and GO enrichment analysis of DEGs (D) in the gcn5Δ strain in relation to micafungin treatment. LSU, large subunit; SSU, small subunit; ITS1, internal transcribed spacer 1; ETS, external transcribed spacer.
Lack of GCN5 reduces intracellular replication in macrophages.
To study the role of Gcn5 in host-pathogen interactions, we set up an in vitro infection system using the human monocytic cell line THP-1. Phorbol-12 myristate 13-acetate (PMA)-differentiated THP-1 cells were infected with C. glabrata cells at a multiplicity of infection (MOI) of 1:10; thereafter, phagocytosis and intracellular replication were measured at 2 h and 24 h postinfection, respectively. During the initial interaction with macrophages, gcn5Δ cells elicited seemingly enhanced phagocytosis, but lacking statistical significance (Fig. 8A), compared to the WT and the complemented strains (115.2% for gcn5Δ versus 92.5% for WT and 100.8% for gcn5Δ::GCN5 cells [P = 0.18]). However, significantly reduced intracellular replication at 24 h postinfection was observed with gcn5Δ cells (Fig. 8B) compared to cells with Gcn5 function (223.1% for gcn5Δ versus 519.6% for WT cells [P = 0.037]; 223.1% for gcn5Δ versus 530.7% for gcn5Δ::GCN5 cells [P = 0.023]).
FIG 8.

In vitro host-pathogen interaction alterations associated with GCN5 deletion in C. glabrata. (A) Phagocytosis of C. glabrata cells by THP-1 macrophages at 2 h postinfection. (B) The gcn5Δ strain had significantly decreased intracellular survival at 24 h postinfection compared to the WT and complemented strains. Data represent the means ± SD from at least 3 independent experiments (*, P < 0.05).
DISCUSSION
Despite advances achieved in the treatment of fungal infections, antifungal resistance arises at faster paces in multiple human fungal pathogens than that of antifungal drug development, menacing global health. As we deepen our understanding of mechanisms of antifungal resistance, chromatin modification and relevant gene expression regulation start to be recognized as playing pivotal roles in the adaptation of fungal species to antifungal stress, suggesting a potential avenue to tackle antifungal resistance (10, 14). Yet studies in this perspective are highly limited, and a full appreciation of which and how chromatin modifications are involved in the antifungal drug response and resistance is lacking. In light of recent findings of Gcn5 being critical for controlling virulence in C. albicans (20), we aimed to understand how this pleiotropic chromatin modifier shapes the antifungal response and impacts resistance development in C. glabrata, an important human fungal pathogen notorious for the rapid acquisition of antifungal resistance.
C. glabrata lacks hypha formation, a key virulence trait of C. albicans and through which Gcn5 was previously found to impact the stress response and modulate the virulence of C. albicans (19, 20). In our study, deletion of GCN5 in C. glabrata reduced cell growth in rich medium to only a modest level, but it rendered increased sensitivity of cells to various stressors, including antifungal agents. Using in vitro susceptibility testing, we found that the MICs of the gcn5Δ strain were consistently decreased modestly by 1 to 2 2-fold dilutions for the azole and echinocandin drugs tested, whereas the hypersensitivity to the new investigational antifungal drug manogepix, which targets GPI anchor biosynthesis maturation, was unexpected. Considering the different targets and mechanisms of action of the drug classes included in our tests, these results suggest that Gcn5 acts as a master regulator coordinating the synthesis of both cell wall and cell membrane structural components. The drastic change in susceptibility to manogepix indicates that Gcn5 deletion-induced cell wall/membrane dysfunction may be synergistic with manogepix blockage of the GPI biosynthesis pathway, which leads to more effective cell killing. In fact, a perturbed cell wall architecture upon GCN5 deletion was revealed by transcriptional profiling, presenting as markedly downregulated levels of adhesins (EPA6 and EPA13) and β-mannosyltransferase (BMT5 and BMT2) in knockout cells. Such an altered cell wall of the gcn5Δ strain may have facilitated the actions of all classes of antifungal agents but largely favored manogepix. Further investigation is warranted to unravel the exact mechanism underlying this phenomenon. Another phenotypic feature stemming from the cell wall changes, particularly the decreased expression of adhesins, associated with GCN5 deletion was the significantly diminished adherence capacity and biofilm formation of the gcn5Δ strain on a polystyrene surface. As adhesion and biofilm formation are well-known virulence factors, this observation is an affirmative testimony to the involvement of Gcn5 in the virulence of C. glabrata. Interestingly, a previous study reported that disrupting ADA2, another component of the SAGA complex, conferred mutant hypervirulence, owing at least in part to the highly induced expression of adhesins in the deletion mutant (32). It is worth noting that Gcn5 and Ada2 are interacting partners with distinct functions. Gcn5 mainly serves as a histone acetyltransferase, while Ada2 is more of a transcriptional adaptor (33). The contrasting adhesin expression changing mode from inactivating these two elements individually may reflect the divergent roles of these two proteins in gene regulation.
Given that echinocandins are first-line therapies against invasive Candida infections, we attempted to dissect further the relationship between Gcn5 and echinocandin resistance. Therefore, we knocked out GCN5 from prominent FKS1 and FKS2 mutants that are all echinocandin resistant. While all FKS mutants demonstrated reduced resistance to echinocandin upon the deletion of GCN5, the reversal of resistance was somewhat more pronounced in FKS1 mutants yet less efficient in FKS2 mutants, especially the S663P mutant. These results are consistent with FKS expression changes found with the gcn5Δ strain in the WT background, which showed that the level of the FKS1 transcript in the gcn5Δ strain was less than 60% of the level in the WT strain, accompanied by an ~40% compensatory increase in FKS2 expression relative to that in the WT. These data imply that the attenuated resistance to echinocandin in FKS1 mutants upon GCN5 disruption was at least partially gained through FKS1 suppression, which outcompeted the compensatory increase in FKS2 expression. However, the net effect of the restoration of echinocandin susceptibility upon GCN5 deletion on FKS2 mutants was more diluted because under the same level of FKS2 mRNA, mutated Fks2 protein produces resistance, but the WT Fks2 protein does not. Knowing that expression regulation of FKS2 in C. glabrata is dependent upon calcineurin signaling, we tested the calcineurin inhibitor FK506 for susceptibility in all parental strains (WT and FKS mutants) and the corresponding GCN5 knockout strains. As a result, a 2- to 4-fold decrease in the FK506 MIC was observed in all strains upon GCN5 deletion, except for the Fks2-659delF mutant being hypersusceptible. Using a sub-MIC of FK506 to block the partial function of Fks2, we found that a greater reversal of micafungin resistance was achieved in all FKS mutants, further confirming that Gcn5 mediated FKS modulation primarily toward the positive regulation of FKS1.
Another key feature associated with GCN5 deletion was the attenuated resistance acquisition under drug pressure. In the time-kill assay, while WT cells were tolerant to a low level of micafungin (0.03 μg/mL) and developed resistance via FKS mutation as quickly as 24 h after exposure, the gcn5Δ strain under the same pressure rendered cell death of the majority of the inoculated cell population and was therefore devoid of resistance within 48 h of treatment. Only when micafungin exposure dropped to 0.008 μg/mL did FKS-mediated resistance emerge from gcn5Δ cells at a frequency comparable to, yet at a pace slower than, that of the WT. Similarly, gcn5Δ cells displayed a consistently lower frequency of phenotypic resistance than the WT strain under fluconazole pressure, although we did not identify a PDR1 mutation from any of the selectively sequenced resistant colonies. These results not only echo the recent findings by Usher and Haynes (22) but also provide an additional layer to understand how GCN5 impacts echinocandin resistance. To understand how GCN5 regulates the cell response to these two most common antifungal classes, we profiled the transcriptomes of both the WT and gcn5Δ strains in the presence and absence of modest levels of fluconazole and micafungin. As expected, we found that a 2-h exposure to a sub-MIC of fluconazole led to a largely induced transcriptome in the WT strain, particularly involving membrane, transmembrane transporter activity, and fungal cell wall organization pathways. However, the gcn5Δ strain displayed a markedly rigid transcriptomic profile in response to fluconazole exposure, with only 22 genes in total being upregulated over 2-fold. Notably, even though the WT and gcn5Δ strains showed similar patterns of differential expression upon fluconazole exposure, the gcn5Δ strain tended to be less responsive to cell damage caused by fluconazole; therefore, the expression changes of the gcn5Δ strain were mostly smaller than those of the WT. These results suggest that Gcn5 in C. glabrata plays a role in prompting swift cell defense and activating effective adaptation to fluconazole. Intriguingly, when drug exposure was switched to micafungin, a highly disturbed transcriptome of the gcn5Δ strain was observed, with approximately 4 times more genes being up- or downregulated >2-fold than in the WT. Enriched GO mapping also showed highly divergent transcriptomic responses in gcn5Δ and WT cells. A considerable number of differentially expressed genes in gcn5Δ cells were relevant to ribosome biogenesis, which may be a snapshot of the cells’ last line of defense against life-threatening stress from micafungin treatment. In comparison, transcriptomic changes in WT cells were more focused on upregulating cell wall integrity pathway genes as well as transmembrane transporter activity, suggesting that the cells were operating a concerted machinery to resist and repair cell wall damage induced by micafungin. This observation is also consistent with the most recently published study investigating transcriptomic alterations in C. glabrata cells surviving micafungin treatment (26).
As a successful human pathogen, C. glabrata is highly adapted to interaction with host cells and the immune response. Macrophages are professional phagocytes that act as part of the innate immune system of the host, contributing to antifungal defense via phagocytosis and clearance of invading fungal pathogens (34). Phagocytosis of C. glabrata starts with pattern recognition of fungal cell wall components such as β-glucan and mannan by the C-type lectin receptors dectin-1 and dectin-2 on macrophages (35, 36). Hence, a disturbed cell wall architecture is deemed to alter C. glabrata-macrophage interactions, as observed in previous studies involving deletion mutants lacking cell surface-associated aspartyl proteases and those with defects in protein glycosylation (37, 38). In view of both phenotypic and transcriptomic data suggestive of an altered cell wall composition upon GCN5 deletion, we attempted to determine whether the loss of Gcn5 has an impact on host-pathogen interactions using in vitro-differentiated THP-1 macrophages. Indeed, the gcn5Δ strain demonstrated significantly reduced intracellular survival compared to the WT, while the phagocytosis of mutant cells by macrophages was slightly more efficient. The production of toxic reactive oxygen species (ROS) is one central aspect of the macrophage antimicrobial response. Despite evidence that C. glabrata possesses robust and redundant antioxidant systems conferring high-level resistance to oxidative stress, ROS production in macrophages at least partially contributes to the intracellular killing of this fungus (39). Taking the phenotypic testing results into consideration, the reduced survival of gcn5Δ cells within THP-1 macrophages is somewhat expected and may be due partly to the increased sensitivity of the deletion mutant to oxidative stress and/or other stresses encountered in the macrophage internal milieu. The fact that gcn5Δ cells were phagocytosed at a slightly higher rate than WT cells suggests that cell wall disturbance, presumably improper construction and/or assembly of GPI-anchored proteins, especially adhesins (Epa6 and Epa13), may have resulted in a modestly increased exposure of the skeletal components to macrophages. Previous studies have shown that Epa6 is a significant virulence factor of C. glabrata that contributes strongly to both biofilm formation and adherence to epithelial cells to establish experimental urinary tract infection (28, 40). In these backgrounds, our results suggest that blocking the function of Gcn5 may be a potential way to not only attenuate the virulence of C. glabrata but also facilitate host cells clearing infections, although more in-depth studies are needed.
Taken together, the results of our study show that the histone acetyltransferase Gcn5 plays a critical role in modulating the virulence of C. glabrata and regulating its response to antifungal pressure and host defense. Despite great interest raised in the past in using chromatin modification targets to aid in antifungal discovery (14), there is a lack of appreciation for how epigenetic mechanisms, such as histone acetylation, are involved in the antifungal response and resistance development. The findings of the present study provide insights into this understudied topic and demonstrate the possibility of limiting resistance in C. glabrata as well as enhancing the efficacy of existing antifungal therapy and promoting host defense by inactivating Gcn5 function. In theory, chemical inhibition should mimic some of the phenotypes obtained by genetic ablation. Therefore, future studies exploring and evaluating specific Gcn5 inhibitors as a potential adjunct to existing antifungal therapy to improve clinical outcomes are warranted.
MATERIALS AND METHODS
Strains and growth conditions.
C. glabrata strain ATCC 2001 was obtained from the American Type Culture Collection (Manassas, VA). FKS1 and FKS2 mutants (Fks1-625delF, Fks1-S629P, Fks2-659delF, and Fks2-S663P) were constructed in the ATCC 2001 background (see Table S1 in the supplemental material). All C. glabrata strains were grown at 37°C in yeast extract-peptone-dextrose (YPD) medium (1% yeast extract, 2% peptone, and 2% dextrose) with shaking at 150 rpm.
GCN5 disruption and complementation.
To disrupt GCN5, NAT1 was amplified from plasmid pCN-PDC1 (41) with primers that contained overhangs homologous to the up- and downstream regions of C. glabrata GCN5 (Table S1). The purified deletion cassette, GCN5 guide RNA (designed online at https://chopchop.cbu.uib.no/), and the Alt-R CRISPR-Cas9 system (Integrated DNA Technologies, Inc.) were transformed into competent WT (or FKS mutant) cells, as previously described (30). Transformants that grew on YPD plates supplemented with 100 μg/mL nourseothricin were PCR screened and sequenced to confirm the correct deletion. To complement the gcn5Δ mutant, we PCR amplified the coding region of GCN5 from ATCC 2001 genomic DNA (Table S1). Guide RNA targeting the NAT1 region was designed using the online software CHOPCHOP. The purified GCN5 repair cassette, NAT1 guide RNA, and the Alt-R CRISPR-Cas9 system were transformed together into competent deletion mutant cells. Transformants grown on plain YPD plates were replica plated onto nourseothricin-containing YPD plates, and those that grew only on plain YPD plates (nourseothricin sensitive) were further PCR screened and sequenced to confirm successful complementation. All primers used for this procedure are listed in Table S1.
Growth curve and doubling time measurement.
Cultures of each C. glabrata strain grown overnight were diluted to an optical density at 600 nm (OD600) of 0.1 with fresh YPD medium. The absorbance was recorded every 15 min for 12 h by a microplate spectrophotometer (VersaMax enzyme-linked immunosorbent assay [ELISA] microplate reader with SoftMax Pro software; Molecular Devices). Each strain was tested in triplicate, and OD600 values were plotted versus time. The doubling times were calculated as previously described (42).
Western blotting.
Gcn5 is known to acetylate multiple histone lysine residues, primarily lysine residue 14 of histone 3 (H3K14) as well as H3K9, H3K18, H3K23, H3K27, H3K36, and other additional lysine residues found in histones H4 and H2B (43, 44). To validate epigenetic changes associated with GCN5 disruption, we employed Western blot analysis to compare histone acetylation levels in WT, gcn5Δ, and gcn5Δ::GCN5 cells using H3K14 and H3K9 as representative targets. Whole-cell lysates were prepared by trichloroacetic acid (TCA) precipitation. Briefly, cell pellets from cultures grown overnight were resuspended in 20% TCA, disrupted by bead beating, and washed twice with 5% TCA. Protein was pelleted and resuspended in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer, followed by incubation at 95°C for 5 min and centrifugation prior to loading onto 16% acrylamide gels. Primary antibodies for immunoblotting were obtained commercially, including histone H3 antibody (catalog no. 4499; Cell Signaling Technology), acetyl-histone H3(Lys9) (H3K9Ac) antibody (catalog no. 9649; Cell Signaling Technology), acetyl-histone H3(Lys14) (H3K14Ac) antibody (catalog no. 7627; Cell Signaling Technology), and β-actin antibody (catalog no. PA5-85271; Thermo Fisher Scientific). The target protein was visualized with Novex ECL chemiluminescent substrates (Thermo Fisher Scientific) according to the manufacturer’s instructions, and band intensities were determined using ImageJ software (https://imagej.nih.gov/ij/).
In vitro susceptibility testing.
Antifungal susceptibility testing was performed at least in duplicate for each strain according to CLSI guidelines (45). Micafungin (Astellas, Deerfield, IL), caspofungin (Merck, Rahway, NJ), anidulafungin (Pfizer, New York, NY), fluconazole (LKT Laboratories, Saint Paul, MN), voriconazole (Pfizer, New York, NY), itraconazole (Sigma, St. Louis, MO), posaconazole (Sigma, St. Louis, MO), FK506 (Tecoland, Edison, NJ), ibrexafungerp (Scynexis, Inc., Jersey City, NJ), and manogepix (formerly Amplyx Pharmaceuticals, Inc., San Diego, CA) were dissolved and diluted according to CLSI standards (45). To determine the functionality of Fks1, we also performed micafungin susceptibility testing in the presence of FK506 at 4 μg/mL.
Spotting assay.
C. glabrata cells grown overnight were washed twice with phosphate-buffered saline (PBS) and adjusted to ~5 × 105 CFU/mL in PBS. Equal volumes (5 μL) of 10-fold serial dilutions of each strain were spotted onto YPD plates containing various cell stress agents, including 10 mM H2O2, 0.01% SDS, calcofluor white stain (10 μg/mL), and Congo red (50 μg/mL), as well as antifungal agents consisting of fluconazole (16 μg/mL), micafungin (0.008 and 0.03 μg/mL), and caspofungin (0.03 and 0.25 μg/mL). Colony growth was compared with that on the plain YPD control plate after 24 h of incubation at 37°C.
Time-kill assay.
Time-kill studies were performed to compare the tolerances of the WT (ATCC 2001) strain and the gcn5Δ strain to micafungin and fluconazole, according to a procedure described previously, with minor modifications (46). Each drug at concentrations of 1×, 4×, 16×, and 64× WT MIC as well as a no-drug control were included in the evaluation. RPMI 1640 buffered with morpholinepropanesulfonic acid (MOPS) was the growth medium. The starting inoculum of each strain was prepared in a total volume of 5 mL at 1 × 106 CFU/mL for the experiment involving micafungin and 5 × 105 CFU/mL for that involving fluconazole. All samples were incubated at 37°C with shaking at 150 rpm, and a 15-μL aliquot was taken from each sample at 0, 2, 4, 8, 24, and 48 h. The removed aliquots were pelleted and washed in PBS, and proper dilutions were then prepared, plated in duplicate onto YPD plates, and incubated at 37°C for 24 h to determine the colony counts. The frequency of resistant colonies was measured at 24 and 48 h for all samples in duplicate, using YPD plates containing 0.25 μg/mL micafungin or 512 μg/mL fluconazole. Hot spot 1 and 2 regions of FKS1 and FKS2 were sequenced for micafungin-resistant colonies, and PDR1 sequencing was performed for fluconazole-resistant colonies, using primers listed in Table S1.
RNA isolation and RNA-seq analysis.
Cultures of the WT and gcn5Δ strains grown overnight were inoculated in liquid YPD medium at an initial OD600 of 0.2 and grown at 37°C with shaking to an OD600 of 0.6. Cells were then treated with fluconazole (8 μg/mL) or micafungin (15 ng/mL) for 2 h. Untreated WT and gcn5Δ cells were grown in parallel. Cells were harvested at the end of the 2-h treatment and subjected to RNA extraction, with two biological replicates for each strain/treatment group. Total RNA was extracted from each sample according to a protocol described previously (30), and RNA samples were stored at −80°C until shipping to Genewiz (South Plainfield, NJ) for RNA sequencing. The RNA-seq data were processed using the Illumina NovaSeq platform with 150-bp paired-end reads. The read data quality was examined using FastQC v.0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and reads were trimmed to remove low-quality bases (average quality per base of <20) using Trimmomatic v.0.39 (47). The filtered reads were then aligned to the Candida glabrata CBS138/ATCC 2001 genome (www.candidagenome.org) using HISAT 2.2.1 (48), and the raw read counts were obtained using HTSeq v.0.12.3 (49). Differentially expressed genes (DEGs), defined as genes with an absolute log2 fold change [|log2FoldChange|] of ≥1 and adjusted P values of ≤0.05, across different groups were estimated using DEseq2 v.1.34.0 (50) in R 4.0.2. Gene Ontology (GO) annotation was downloaded from the Candida Genome Database (CGD) (www.candidagenome.org), and enrichment analysis was done using clusterProfiler 4.0 (51) in R.
Quantitative real-time reverse-transcription-PCR.
To verify the RNA-seq results, the expression levels of representative genes selected from each comparison were measured by reverse transcription-PCR (RT-PCR) using one-step SYBR PrimeScript RT-PCR kit II (TaKaRa, Shiga, Japan). Reaction mixtures were run on an Mx3005P quantitative PCR (qPCR) system (Agilent Technologies, CA, USA) and contained 10 ng RNA sample, 0.4 μM each primer (Table S1), 12.5 μL 2× one-step SYBR RT-PCR buffer, and 1 μL PrimeScript one-step enzyme mix 2 in a volume of 25 μL. Thermal cycling conditions were as follows: RT at 42°C for 5 min; PCR cycling with an initial denaturation step at 95°C for 10 s, followed by 40 cycles of denaturation at 95°C for 5 s and annealing and elongation at 60°C for 20 s; and a post-PCR melting-curve analysis at 95°C for 5 s and 60°C for 1 min and then increasing to 95°C with a ramp rate of 0.5°C/s (30). Each experiment was carried out in duplicate, and negative controls were included in each run. The PGK1 gene was used as a reference gene to normalize the data (52). Relative quantification of gene expression was performed using the 2−ΔΔCT method (53). The fold changes were determined as the mean normalized expression of mutant or treated samples relative to the mean normalized expression of the untreated ATCC 2001 control. Statistical analysis of gene expression was carried out using GraphPad Prism software, and a P value of <0.05 was considered significant.
Adhesion assay.
C. glabrata cells grown overnight in liquid YPD medium were adjusted to an OD600 of 1.0 in fresh YPD broth, 200 μL of which was inoculated into a 96-well microtiter plate. After 24 h of incubation at 37°C without shaking in a humid environment, unattached cells were removed by gentle washing with distilled water three times. Plates were air dried, 100 μL of 0.1% (wt/vol) crystal violet was then added to each well, and the plates were incubated at 37°C for 30 min, followed by gentle washing with distilled water and air drying. Finally, 200 μL of 33% glacial acetic acid was added to each well, and adhesion was quantified by measuring the OD595 using a plate reader (Infinite Pro; Tecan). All strains (WT, gcn5Δ, and gcn5Δ::GCN5) were tested in parallel in four technical and two biological replicates.
THP-1 macrophage infection.
The human monocyte cell line THP-1 (ATCC TIB202) was cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) at 37°C under 5% CO2. The THP-1 macrophage infection process was performed as described previously by Rasheed et al., with minor modifications (37). Briefly, 24-well plates were seeded with 1 × 106 THP-1 cells, and THP-1 cells were differentiated into macrophages for 16 h in the presence of 16 nM phorbol-12 myristate 13-acetate (PMA), followed by a 12-h recovery. On the day of infection, 50 μL of a suspension of 1 × 106 CFU/mL of C. glabrata cells (WT, gcn5Δ, and gcn5Δ::GCN5) was added to each well of the THP-1 cells to obtain a multiplicity of infection (MOI) of 1:10 (fungi to macrophages) and incubated at 37°C with 5% CO2. After 2 and 24 h of coculture, macrophages were washed three times with prewarmed sterile PBS to remove nonphagocytized extracellular C. glabrata cells. Macrophages were then lysed in water, and 100 μL of the properly diluted lysate was plated onto a YPD plate to determine the intracellular CFU counts. The phagocytosis rate for each strain was measured using the 2-h CFU counts divided by the CFU of the inoculum. The intracellular replication of C. glabrata for each strain was calculated by dividing the CFU at the 24-h time point by those at 2 h.
Data availability.
Raw RNA-seq data have been deposited at the Gene Expression Omnibus (accession no. GSE194310).
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) grant R01AI109025 (D.S.P.). William Paterson University (Provost’s Office) provided assigned release time (ART) to K.H. and a graduate assistantship (M.S. biotechnology) to S.E.
Y.Z. and K.H. conceptualized the project. S.Y., P.P., A.L., and S.E. performed experiments. Y.Z. and L.C. performed data analysis. Y.Z., S.Y., K.H., and D.S.P. contributed to manuscript writing. All authors reviewed and edited the article.
We declare that we have no competing interests.
Footnotes
Supplemental material is available online only.
Contributor Information
Yanan Zhao, Email: yanan.zhao@hmh-cdi.org.
Damian J. Krysan, University of Iowa Hospitals and Clinics
REFERENCES
- 1.Pristov KE, Ghannoum MA. 2019. Resistance of Candida to azoles and echinocandins worldwide. Clin Microbiol Infect 25:792–798. doi: 10.1016/j.cmi.2019.03.028. [DOI] [PubMed] [Google Scholar]
- 2.Silva S, Negri M, Henriques M, Oliveira R, Williams DW, Azeredo J. 2012. Candida glabrata, Candida parapsilosis and Candida tropicalis: biology, epidemiology, pathogenicity and antifungal resistance. FEMS Microbiol Rev 36:288–305. doi: 10.1111/j.1574-6976.2011.00278.x. [DOI] [PubMed] [Google Scholar]
- 3.Pappas PG, Lionakis MS, Arendrup MC, Ostrosky-Zeichner L, Kullberg BJ. 2018. Invasive candidiasis. Nat Rev Dis Primers 4:18026. doi: 10.1038/nrdp.2018.26. [DOI] [PubMed] [Google Scholar]
- 4.Friedman DZP, Schwartz IS. 2019. Emerging fungal infections: new patients, new patterns, and new pathogens. J Fungi (Basel) 5:67. doi: 10.3390/jof5030067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfaller MA, Diekema DJ, Turnidge JD, Castanheira M, Jones RN. 2019. Twenty years of the SENTRY Antifungal Surveillance Program: results for Candida species from 1997-2016. Open Forum Infect Dis 6:S79–S94. doi: 10.1093/ofid/ofy358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walker LA, Gow NA, Munro CA. 2010. Fungal echinocandin resistance. Fungal Genet Biol 47:117–126. doi: 10.1016/j.fgb.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar K, Askari F, Sahu MS, Kaur R. 2019. Candida glabrata: a lot more than meets the eye. Microorganisms 7:39. doi: 10.3390/microorganisms7020039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Las Penas A, Juarez-Cepeda J, Lopez-Fuentes E, Briones-Martin-Del-Campo M, Gutierrez-Escobedo G, Castano I. 2015. Local and regional chromatin silencing in Candida glabrata: consequences for adhesion and the response to stress. FEMS Yeast Res 15:fov056. doi: 10.1093/femsyr/fov056. [DOI] [PubMed] [Google Scholar]
- 9.Chang Z, Yadav V, Lee SC, Heitman J. 2019. Epigenetic mechanisms of drug resistance in fungi. Fungal Genet Biol 132:103253. doi: 10.1016/j.fgb.2019.103253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Kane CJ, Weild R, Hyland EM. 2020. Chromatin structure and drug resistance in Candida spp. J Fungi (Basel) 6:121. doi: 10.3390/jof6030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su S, Li X, Yang X, Li Y, Chen X, Sun S, Jia S. 2020. Histone acetylation/deacetylation in Candida albicans and their potential as antifungal targets. Future Microbiol 15:1075–1090. doi: 10.2217/fmb-2019-0343. [DOI] [PubMed] [Google Scholar]
- 12.Shih PY, Liao YT, Tseng YK, Deng FS, Lin CH. 2019. A potential antifungal effect of chitosan against Candida albicans is mediated via the inhibition of SAGA complex component expression and the subsequent alteration of cell surface integrity. Front Microbiol 10:602. doi: 10.3389/fmicb.2019.00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hnisz D, Tscherner M, Kuchler K. 2011. Targeting chromatin in fungal pathogens as a novel therapeutic strategy: histone modification gets infectious. Epigenomics 3:129–132. doi: 10.2217/epi.11.7. [DOI] [PubMed] [Google Scholar]
- 14.Kuchler K, Jenull S, Shivarathri R, Chauhan N. 2016. Fungal KATs/KDACs: a new highway to better antifungal drugs? PLoS Pathog 12:e1005938. doi: 10.1371/journal.ppat.1005938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Dienemann C, Stutzer A, Urlaub H, Cheung ACM, Cramer P. 2020. Structure of the transcription coactivator SAGA. Nature 577:717–720. doi: 10.1038/s41586-020-1933-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee KK, Workman JL. 2007. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol 8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 17.Xue-Franzen Y, Johnsson A, Brodin D, Henriksson J, Burglin TR, Wright AP. 2010. Genome-wide characterisation of the Gcn5 histone acetyltransferase in budding yeast during stress adaptation reveals evolutionarily conserved and diverged roles. BMC Genomics 11:200. doi: 10.1186/1471-2164-11-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez-Soto D, Gonzalez-Prieto JM, Ruiz-Herrera J. 2015. Transcriptomic analysis of the GCN5 gene reveals mechanisms of the epigenetic regulation of virulence and morphogenesis in Ustilago maydis. FEMS Yeast Res 15:fov055. doi: 10.1093/femsyr/fov055. [DOI] [PubMed] [Google Scholar]
- 19.Chang P, Fan X, Chen J. 2015. Function and subcellular localization of Gcn5, a histone acetyltransferase in Candida albicans. Fungal Genet Biol 81:132–141. doi: 10.1016/j.fgb.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 20.Shivarathri R, Tscherner M, Zwolanek F, Singh NK, Chauhan N, Kuchler K. 2019. The fungal histone acetyl transferase Gcn5 controls virulence of the human pathogen Candida albicans through multiple pathways. Sci Rep 9:9445. doi: 10.1038/s41598-019-45817-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Meara TR, Hay C, Price MS, Giles S, Alspaugh JA. 2010. Cryptococcus neoformans histone acetyltransferase Gcn5 regulates fungal adaptation to the host. Eukaryot Cell 9:1193–1202. doi: 10.1128/EC.00098-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Usher J, Haynes K. 2019. Attenuating the emergence of anti-fungal drug resistance by harnessing synthetic lethal interactions in a model organism. PLoS Genet 15:e1008259. doi: 10.1371/journal.pgen.1008259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filler EE, Liu Y, Solis NV, Wang L, Diaz LF, Edwards JE, Jr, Filler SG, Yeaman MR. 2021. Identification of Candida glabrata transcriptional regulators that govern stress resistance and virulence. Infect Immun 89:e00146-20. doi: 10.1128/IAI.00146-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katiyar SK, Alastruey-Izquierdo A, Healey KR, Johnson ME, Perlin DS, Edlind TD. 2012. Fks1 and Fks2 are functionally redundant but differentially regulated in Candida glabrata: implications for echinocandin resistance. Antimicrob Agents Chemother 56:6304–6309. doi: 10.1128/AAC.00813-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delarze E, Sanglard D. 2015. Defining the frontiers between antifungal resistance, tolerance and the concept of persistence. Drug Resist Updat 23:12–19. doi: 10.1016/j.drup.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Rubio R, Jimenez-Ortigosa C, DeGregorio L, Quinteros C, Shor E, Perlin DS. 2021. Multifactorial role of mitochondria in echinocandin tolerance revealed by transcriptome analysis of drug-tolerant cells. mBio 12:e01959-21. doi: 10.1128/mBio.01959-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Healey KR, Perlin DS. 2018. Fungal resistance to echinocandins and the MDR phenomenon in Candida glabrata. J Fungi (Basel) 4:105. doi: 10.3390/jof4030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iraqui I, Garcia-Sanchez S, Aubert S, Dromer F, Ghigo JM, d’Enfert C, Janbon G. 2005. The Yak1p kinase controls expression of adhesins and biofilm formation in Candida glabrata in a Sir4p-dependent pathway. Mol Microbiol 55:1259–1271. doi: 10.1111/j.1365-2958.2004.04475.x. [DOI] [PubMed] [Google Scholar]
- 29.Timmermans B, De Las Peñas A, Castaño I, Van Dijck P. 2018. Adhesins in Candida glabrata. J Fungi (Basel) 4:60. doi: 10.3390/jof4020060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hou X, Healey KR, Shor E, Kordalewska M, Ortigosa CJ, Paderu P, Xiao M, Wang H, Zhao Y, Lin LY, Zhang YH, Li YZ, Xu YC, Perlin DS, Zhao Y. 2019. Novel FKS1 and FKS2 modifications in a high-level echinocandin resistant clinical isolate of Candida glabrata. Emerg Microbes Infect 8:1619–1625. doi: 10.1080/22221751.2019.1684209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyazaki T, Yamauchi S, Inamine T, Nagayoshi Y, Saijo T, Izumikawa K, Seki M, Kakeya H, Yamamoto Y, Yanagihara K, Miyazaki Y, Kohno S. 2010. Roles of calcineurin and Crz1 in antifungal susceptibility and virulence of Candida glabrata. Antimicrob Agents Chemother 54:1639–1643. doi: 10.1128/AAC.01364-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu SJ, Chang YL, Chen YL. 2018. Deletion of ADA2 increases antifungal drug susceptibility and virulence in Candida glabrata. Antimicrob Agents Chemother 62:e01924-17. doi: 10.1128/AAC.01924-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grant PA, Duggan L, Côté J, Roberts SM, Brownell JE, Candau R, Ohba R, Owen-Hughes T, Allis CD, Winston F, Berger SL, Workman JL. 1997. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev 11:1640–1650. doi: 10.1101/gad.11.13.1640. [DOI] [PubMed] [Google Scholar]
- 34.Vazquez-Torres A, Balish E. 1997. Macrophages in resistance to candidiasis. Microbiol Mol Biol Rev 61:170–192. doi: 10.1128/mmbr.61.2.170-192.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ifrim DC, Bain JM, Reid DM, Oosting M, Verschueren I, Gow NA, van Krieken JH, Brown GD, Kullberg BJ, Joosten LA, van der Meer JW, Koentgen F, Erwig LP, Quintin J, Netea MG. 2014. Role of dectin-2 for host defense against systemic infection with Candida glabrata. Infect Immun 82:1064–1073. doi: 10.1128/IAI.01189-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen SM, Shen H, Zhang T, Huang X, Liu XQ, Guo SY, Zhao JJ, Wang CF, Yan L, Xu GT, Jiang YY, An MM. 2017. Dectin-1 plays an important role in host defense against systemic Candida glabrata infection. Virulence 8:1643–1656. doi: 10.1080/21505594.2017.1346756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rasheed M, Battu A, Kaur R. 2018. Aspartyl proteases in Candida glabrata are required for suppression of the host innate immune response. J Biol Chem 293:6410–6433. doi: 10.1074/jbc.M117.813741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.West L, Lowman DW, Mora-Montes HM, Grubb S, Murdoch C, Thornhill MH, Gow NA, Williams D, Haynes K. 2013. Differential virulence of Candida glabrata glycosylation mutants. J Biol Chem 288:22006–22018. doi: 10.1074/jbc.M113.478743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seider K, Gerwien F, Kasper L, Allert S, Brunke S, Jablonowski N, Schwarzmuller T, Barz D, Rupp S, Kuchler K, Hube B. 2014. Immune evasion, stress resistance, and efficient nutrient acquisition are crucial for intracellular survival of Candida glabrata within macrophages. Eukaryot Cell 13:170–183. doi: 10.1128/EC.00262-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Domergue R, Castaño I, De Las Peñas A, Zupancic M, Lockatell V, Hebel JR, Johnson D, Cormack BP. 2005. Nicotinic acid limitation regulates silencing of Candida adhesins during UTI. Science 308:866–870. doi: 10.1126/science.1108640. [DOI] [PubMed] [Google Scholar]
- 41.Zordan RE, Ren Y, Pan SJ, Rotondo G, De Las Penas A, Iluore J, Cormack BP. 2013. Expression plasmids for use in Candida glabrata. G3 (Bethesda) 3:1675–1686. doi: 10.1534/g3.113.006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen YL, Yu SJ, Huang HY, Chang YL, Lehman VN, Silao FG, Bigol UG, Bungay AA, Averette A, Heitman J. 2014. Calcineurin controls hyphal growth, virulence, and drug tolerance of Candida tropicalis. Eukaryot Cell 13:844–854. doi: 10.1128/EC.00302-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuo YM, Andrews AJ. 2013. Quantitating the specificity and selectivity of Gcn5-mediated acetylation of histone H3. PLoS One 8:e54896. doi: 10.1371/journal.pone.0054896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suka N, Suka Y, Carmen AA, Wu J, Grunstein M. 2001. Highly specific antibodies determine histone acetylation site usage in yeast heterochromatin and euchromatin. Mol Cell 8:473–479. doi: 10.1016/s1097-2765(01)00301-x. [DOI] [PubMed] [Google Scholar]
- 45.CLSI . 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts, 3rd ed., M27-A3. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 46.Pfaller MA, Sheehan DJ, Rex JH. 2004. Determination of fungicidal activities against yeasts and molds: lessons learned from bactericidal testing and the need for standardization. Clin Microbiol Rev 17:268–280. doi: 10.1128/CMR.17.2.268-280.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, Fu X, Liu S, Bo X, Yu G. 2021. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation (Camb) 2:100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li QQ, Skinner J, Bennett JE. 2012. Evaluation of reference genes for real-time quantitative PCR studies in Candida glabrata following azole treatment. BMC Mol Biol 13:22. doi: 10.1186/1471-2199-13-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download spectrum.00963-22-s0001.pdf, PDF file, 0.5 MB (507KB, pdf)
Supplemental material. Download spectrum.00963-22-s0002.xlsx, XLSX file, 0.1 MB (138.6KB, xlsx)
Data Availability Statement
Raw RNA-seq data have been deposited at the Gene Expression Omnibus (accession no. GSE194310).