

ABSTRACT
Environmental change, especially frequent droughts, is predicted to detrimentally impact the North American perennial grasslands. Consistent dry spells will affect plant communities as well as their associated rhizobiomes, possibly altering the plant host performance under environmental stress. Therefore, there is a need to understand the impact of drought on the rhizobiome, and how the rhizobiome may modulate host performance and ameliorate its response to drought stress. In this study, we analyzed bacterial and fungal communities in the rhizospheres of three ecotypes (dry, mesic, and wet) of dominant prairie grass, Andropogon gerardii. The ecotypes were established in 2010 in a common garden design and grown for a decade under persistent dry conditions at the arid margin of the species’ range in Colby, Kansas. The experiment aimed to answer whether and to what extent do the different ecotypes maintain or recruit distinct rhizobiomes after 10 years in an arid climate. In order to answer this question, we screened the bacterial and fungal rhizobiome profiles of the ecotypes under the arid conditions of western Kansas as a surrogate for future climate environmental stress using 16S rRNA and ITS2 metabarcoding sequencing. Under these conditions, bacterial communities differed compositionally among the A. gerardii ecotypes, whereas the fungal communities did not. The ecotypes were instrumental in driving the differences among bacterial rhizobiomes, as the ecotypes maintained distinct bacterial rhizobiomes even after 10 years at the edge of the host species range. This study will aid us to optimize plant productivity through the use of different ecotypes under future abiotic environmental stress, especially drought.
IMPORTANCE In this study, we used a 10-year long reciprocal garden system, and reports that different ecotypes (dry, mesic, and wet) of dominant prairie grass, Andropogon gerardii can maintain or recruit distinct bacterial but not fungal rhizobiomes after 10 years in an arid environment. We used both 16S rRNA and ITS2 amplicons to analyze the bacterial and fungal communities in the rhizospheres of the respective ecotypes. We showed that A. gerardii might regulate the bacterial community to adapt to the arid environment, in which some ecotypes were not adapted to. Our study also suggested a possible tradeoff between the generalist and the specialist bacterial communities in specific environments, which could benefit the plant host. Our study will provide insights into the plant host regulation of the rhizosphere bacterial and fungal communities, especially during frequent drought conditions anticipated in the future.
KEYWORDS: rhizobiome, bacteria, fungi, drought, ecotypes, grass
INTRODUCTION
The rhizosphere, which plays a pivotal role in plant function by facilitating elemental and water cycling, and uptake of nutrients (1), is densely populated by diverse microbial communities – the rhizobiome. A wide range of complex interactions ranging from symbiotic to competitive among the rhizobiome microorganisms governs the carbon, nitrogen, and phosphorus uptake and transformations (2, 3). These interactions exist not only among the microorganisms but also between the plant hosts, and their associated rhizobiome. For example, plant hosts may selectively attract and/or repel specific soil microbial communities through their root exudates (4, 5). Furthermore, these microorganisms may establish symbiotic relationships with the host plants, safeguarding the host against pathogens (6, 7). These interactions between the plant host and its rhizobiome are likely to be highly specific and ultimately important for community stability, ecosystem functioning, and maintaining soil biodiversity (8).
Global change can have adverse effects on microbe-microbe and plant-microbe interactions, which in turn, can impact the ecology of the rhizosphere and ecosystem function (2). Some studies have focused on the impacts of climate change on rhizospheres (9–12). However, more concerted efforts are needed to fill the knowledge gaps between how rhizobiomes may be influenced by the interactive effects of the plant host and the ever-changing climate. Ultimately, this will help us to maximize plant growth and survival in stressful environments (13–15), especially impacts from drought.
Microorganisms in the rhizosphere are sensitive to environmental conditions (16) and can be good indicators of soil quality (17). Dissecting the rhizosphere bacterial and fungal communities, their functional roles, and their interactions with the plant hosts are crucial to developing future methods to improve drought tolerance and plant productivity. The root-associated microbes can elevate the drought tolerance mechanisms in plants through the physiological and biochemical pathways in the plants (18). Plant-microbe interactions in the rhizosphere have been shown to enhance host resistance to environmental stress and support plant growth. Some of these plant-associated microbial mechanisms include biofilm formation (19), osmotic adjustments (20), changes in phytohormonal levels (18), increase in antioxidant enzymes (18), increase in nutrient and water uptake (21, 22), and optimization of gas exchanges (21, 23). Climate change, characterized by rising temperature and shifted precipitation patterns, has caused the increase in drought frequency (24, 25) and severity (26, 27). Thus, comprehending the extent of the variability among the dominant species in an ecosystem, and how this variability interacts under the predicted arid conditions is important (28). This is especially critical because the dominant species greatly impact ecosystem processes such as carbon assimilation, nutrient cycling, etc. (29). The motivation to understand plant host-microbe interaction in dominant grasses is therefore extremely crucial in the case of tallgrass prairies, in which grasses are responsible for the majority of the carbon fixing, nutrient cycling and biomass (30, 31).
Our studies focus on Big Bluestem, Andropogon gerardii, the dominant native grass species in the tallgrass prairies of central North America (32). A. gerardii is widely distributed across the Midwest and Northeastern USA (USDA database). Our study focuses on the Central grasslands where this grass dominates, stretching from western Kansas to southern Illinois (33). This precipitation gradient includes a semiarid environment, a region of intermediate precipitation, and a region of heavy rainfall (34). Galliart and colleagues have demonstrated that within this steep precipitation gradient, three genetically distinct A. gerardii regional climate ecotypes (dry, mesic, and wet) exist (28, 35), in terms of leaf area, height, and blade width (36), allocation to roots (37), and chlorophyll abundance (38). Climate change models have also predicted a strong phenotypic cline in A. gerardii across this longitudinal precipitation gradient (28, 36, 39). However, we have limited understanding on how the three ecotypes and their rhizobiomes are differently affected by a semiarid environment to which some ecotypes may be more adapted to than others. As such, understanding the shifts and interaction between the A. gerardii plant host and its associated microbiome at Colby Kansas will provide insights into exploring the impact of future climate change on grasslands and the microbiome.
The goal of this study was to analyze the composition of bacterial and fungal communities in the rhizobiomes of the dry, mesic, and wet A. gerardii ecotypes originating in Hays Kansas (rainfall ~500 mm/year), Manhattan Kansas (rainfall ~870 mm/year) and Carbondale Illinois (rainfall ~1,200 mm/year), respectively. All ecotypes were planted in Colby Kansas (rainfall ~500 mm/year) and grown for 10 years prior to sampling. We asked to what extent do ecotypes of a dominant prairie grass maintain or recruit distinct rhizobiomes after 10 years of growth in a semi-arid climate where precipitation is lower than where the ecotypes originated. We were specifically interested in deciphering the extent of ecotypic variation and/or pressure of a semiarid environment on the dominant tall-grass prairie grass rhizobiome. We postulated that the plant host would exert their ecotypic influences on the rhizobiome even under environmental stress, and thus would observe differences in the ecotypic microbial community. We hypothesized that: (1) because the taxonomic traits are driven by the recruitment of the plant host (40–45), we would be able to identify a core rhizobiome that was associated with the different ecotypes; and (2) because of the semi-arid environment of Colby, we would identify microbial populations which might be more resilient to environmental abiotic stress. This study aims to provide insights necessary to preserve the prairie ecosystems under climate change pressures.
RESULTS AND DISCUSSION
We dissected the rhizosphere bacterial and fungal communities associated with the dominant tallgrass prairie species, A. gerardii. We recovered an average of 17,181 ± 4,935 counts per sample for bacteria, and 37,015 ± 7,394 counts for fungi after primer trimming (Table S1 in the supplemental material). Of the recovered counts, an average of 71.45% bacterial counts and 61.55% fungal counts were annotated to the species level on SILVA and UNITE respectively. Any unknown or unclassified amplicon sequence variants (ASVs) were removed from downstream analyses.
No differences in bacterial or fungal α-diversity among host ecotypes.
We performed the Kruskal-Wallis statistical analyses and found no support for differences in bacterial α-diversity (SObs, Shannon’s H’index: H = 6.374, P = 0.041, or Faith’s PD index: H = 3.626, P = 0.163 and observed ASVs index: H = 3.959, P = 0.138) among A. gerardii ecotypes (Fig. 1A). Venn diagrams of the shared bacterial and archaeal ASVs reveal 1,703 shared ASVs (98.66%) among the three ecotypic rhizobiomes (Fig. 1A). These shared ASVs were Actinobacteria, Proteobacteria, Acidobacteria, Verrucomicrobia, Bacteroidetes, Thaumarchaeota, Chloroflexi, Firmicutes, Patescibacteria, Planctomycetes, Armatimonadetes, Gemmatimonadetes, Latescibacteria, Cyanobacteria, Rokubacteria, Entotheonellaeota, Nitrospirae, BRC1, Chlamydiae, Dependentiae, FBP, Elusimicrobia, Deinococcus-Thermus, Fibrobacteres, and WS2. Similar to the bacterial α-diversity, we observed no support for differences in the fungal rhizobiome α-diversity among the three A. gerardii ecotypes when we performed the Kruskal-Wallis statistical analysis (SObs, Shannon’s H’index: H = 3.759, P = 0.153, or Faith’s PD index: H = 4.798, P = 0.091 and observed ASVs index: H = 3.393, P = 0.183) (Fig. 1B). There was one unique fungal ASV belonging to the wet ecotypes, and none in the other ecotypes. There were 829 (99.28%) overlapping ASVs among the three ecotypes (Fig. 1B). The rhizobiome ASVs that were shared among the dry, wet and mesic ecotypes belonged to Basidiomycota, Ascomycota, Mortierellomycota, Glomeromycota, Kickxellomycota, Chrytridiomycota, Rozellomycota, Aphelidiomycota, and Entomophthoromycota.
FIG 1.

A: Bacterial α-Diversity indices among the dry, mesic, and wet ecotypes. (a) Shannon index, (b) faith-pd index, and (c) observed ASVs index, and (d) Venn diagrams represent the overlapping bacterial and archaeal ASVs among the dry, mesic, and wet rhizobiomes. The bacterial α-Diversity was not significantly different among the samples. B: Fungal α-Diversity indices among the dry, mesic, and wet ecotypes. (a) Shannon index (b) faith-pd index and (c) observed ASVs index (d) Venn diagrams representing the overlapping fungal ASVs among the dry, mesic, and wet rhizobiomes. The fungal α-Diversity was not significantly different among the samples.
Bacterial composition differed among host ecotypic rhizobiome.
We performed PERMANOVA statistical analyses and showed that bacterial composition at the phylum level differed among the three A. gerardii ecotypes (PERMANOVA: Pseudo-F = 4.1963, p[permutation(perm)] = 0.001, p[Monte Carlo(MC)] = 0.002, NMDS; stress = 0.13). We also observed a difference in bacterial composition among the ecotypes at the genus level (PERMANOVA: Pseudo-F = 2.1014, p(perm) = 0.001, p(MC) = 0.003). We analyzed the samples based on Bray-Curtis similarity, and observed that the mesic data cloud dispersion was smaller than that of the dry and wet ecotypes (Fig. 2A). In contrast to bacteria, we did not observe any evidence for differences in the fungal rhizobiome composition among the dry, wet and mesic ecotypic rhizobiomes when we performed the PERMANOVA statistical analyses (PERMANOVA: Pseudo-F = 2.0827, p(perm) = 0.071, p(MC) = 0.08, NMDS; stress = 0.11; Fig. 2B).
FIG 2.

Bacterial and fungal composition between the dry, mesic, and wet ecotypes. NMDS ordinations were obtained from Bray-Curtis similarity matrix. The matrix was calculated from square-root transformed relative abundance of 16S and ITS 2 rRNA amplicon sequences (A) bacterial community compositions are separated between the three ecotypes (B) fungal community compositions are not separated between the three ecotypes.
Significant differences in ecotypic bacterial composition.
Based on pairwise Kruskal-Wallis test, bacterial rhizobiome communities were distinct among all three ecotypes (dry v wet: p(MC) = 0.034, dry v mesic: p(MC) = 0.001, wet v mesic: p(MC) = 0.005). The top seven bacterial phyla present in all the three ecotypes are Proteobacteria, Actinobacteria, Acidobacteria, Chloroflexi, Bacteroidetes, Verrucomicrobia, Planctomycetes, and one archaeal taxa, Thaumarchaeota (Fig. 3, Fig. S1 in the supplemental material). Acidobacteria, Bacteroidetes, and Proteobacteria are ubiquitous in soil, suggesting their importance in our samples (46). Similarly, Chloroflexi, Verrucomicrobia, Planctomycetes, and Thaumarchaeota are common soil dwellers, reported to contribute to diverse soil processes (47–50), which also corroborates with the detection of the bacteria in our analyses regardless of the ecotypes. We used ANOVA followed by Tukey post hoc test (51), and showed that the relative abundance of Proteobacteria (F = 7.292, P = 0.004) and Thaumarchaeota (F = 4.451, P = 0.020) differed between the dry and wet ecotypes. Proteobacteria were more abundant in the dry than in the wet ecotype, whereas Thaumarchaeota abundance was the opposite (Fig. 3). Some Proteobacteria may improve plant performance and growth, and can increase in abundance under drought conditions (52, 53), suggesting that Proteobacteria might be important for the sustainable growth of A. gerardii under the challenging environmental conditions in Colby. Thaumarchaeota are the dominant archaea in soil systems (54), and well-known ammonia oxidizers (55). We surmise that Thaumarchaeota in our study might have the potential to enhance the resilience of the A.gerardii wet ecotype under abiotic stressful conditions through the transformation of ammonia into nitrate (56).
FIG 3.

The relative abundance of the top seven bacterial and one archaeal taxa present in all the three ecotypes. Proteobacteria and Thaumarchaeota were significantly different between the dry and wet ecotypes. Letters in a box plot are significantly different at P < 0.05 (D, M, W = significantly different from dry, mesic, and wet ecotypes, respectively).
Post-hoc SIMPER analyses at the Phylum level showed that Proteobacteria, Actinobacteria, Acidobacteria, Chloroflexi, Bacteroidetes and Verrucomicrobia contributed most to the differences among the dry, mesic and wet ecotypes (Table S2 in the supplemental material). Using post hoc SIMPER analyses, we observed that Actinobacteria, Acidobacteria, and Proteobacteria contributed to the greatest differences between the dry and mesic ecotypes, dry and wet ecotypes, and mesic and wet ecotypes. Verrucomicrobia and Bacteroidetes contributed to the greatest differences between the dry and mesic ecotypes (Table S2). Planctomycetes and Thaumarchaeota contributed to the differences between all the ecotypes (Table S2). Comparing the dry and wet ecotypes, we observed that Bacteroidetes, and Thaumarchaeota contributed to their greatest differences. Verrucomicrobia, and Thaumarchaeota contributed most to the differences between the mesic and wet ecotypes. Putting it all together, despite the different ecotypic rhizobiome sharing 98.66% bacterial and archaeal ASVs, the relative abundances of the bacterial populations in the different ecotypes resulted in the ecotypic bacterial compositional differences (Table S2).
We used DeSEQ2 (P < 0.05, Fig. 4) and detected that Rhizobium had significant differences between the dry and wet ecotypic rhizobiomes. Rhizobium had higher relative abundances in the dry ecotype. Comparing dry and mesic ecotypes, we noticed that Rhizobium, Pseudomonas, Cellulomonas, Rhodococcus, Parviterribacter, Parasegetibacter, Flavihumibacter, Cellvibrio, and Candidatus Berkiella were more dominant in dry ecotype than in mesic ecotype. On the opposite side, Microbispora, Sorangium, Zavarzinella, and Candidatus Udaeobacter had more dominance in mesic than in wet ecotype. In other studies, Rhizobium has been found to be drought-stress tolerant (57), and well-known to aid plants during drought conditions (58). Putting it all together, our study suggested that Rhizobium, being the most predominant in the dry ecotype, might have the potential influence to benefit the host in the dry environments. This may also help to explain the higher leaf nitrogen concentrations and higher chlorophyll absorbance we observed in the dry ecotype, regardless of planting location (36, 38). We compared the differences between mesic and wet ecotypes as well, and observed that Parafrigoribacterium, Ktedonobacter, Streptosporangium, Acidicapsa, and Pseudoduganella were more dominant in the mesic ecotype. On the contrary, the top genera that were more predominant in wet ecotypes compared to the mesic belong to Candidatus Berkiella, Cellvibrio, Flavihumibacter, Terrabacter, Parasegetibacter, Parviterribacter, Cellulomonas, Solitalea, Sphaerisporangium, Nonomuraea, Achromobacter, Acinetobacter, Pseudorhodoferax, Nocardia, Leucobacter, among others (Table S3 in the supplemental material). Leucobacter has been identified to grow in wet, low-light environments (59), and we observed that Leucobacter had higher relative abundance in the wet ecotype (60) suggesting that Leucobacter might be better adapted to the wet environment.
FIG 4.
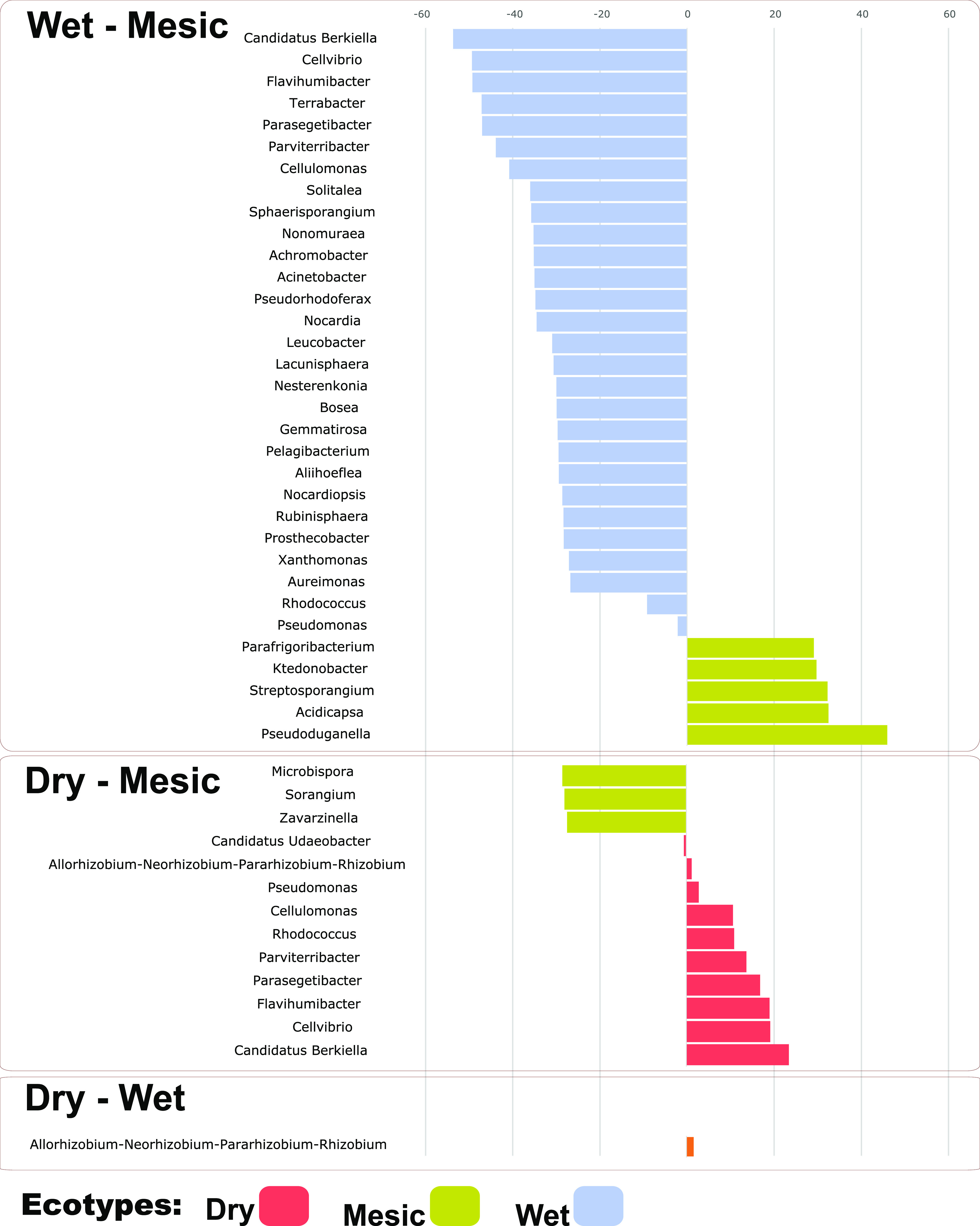
DeSEQ2 analysis to reveal the bacterial genera that were significantly different in relative abundance between the dry, mesic, and wet ecotypes (P < 0.05). Pairwise comparisons were performed between wet-mesic, dry-mesic, and dry-wet ecotypes.
No differences in ecotypic fungi composition.
Unlike the observed differences in rhizobiome bacterial composition among ecotypes, we did not notice the same pattern in the ecotypic fungi composition. We observed that the ecotypic fungal compositions were not significantly different from each other at the phylum level when we performed the PERMANOVA statistical analyses (PERMANOVA: Pseudo-F = 2.0827, p(perm) = 0.071, p(MC) = 0.08). Pairwise Kruskal-Wallis tests also indicated no significant differences in the rhizosphere fungal community composition between the ecotypes (dry v wet: p(MC) = 0.190, dry v mesic: p(MC) = 0.117, wet v mesic: p(MC) = 0.072). The top eight fungal phyla present in all three ecotypes were Ascomycota, Basidiomycota, Mortierellomycota, Mucoromycota, Glomeromycota, Chytridiomycota, Kickxellomycota, and Rozellomycota (Fig. 5, Fig. S2 in the supplemental material). Besides that Ascomycota and Basidiomycota had the highest relative abundance, post hoc SIMPER analyses at the phylum level also indicated that Ascomycota and Basidiomycota were the top phyla contributing to the similarities in all the three ecotypes (Fig. 5, Table S2). Consistent with the results shown in our study, Ascomycota dominate the rhizosphere fungal communities (61–63). Basidiomycota has been reported to be isolated from soil as well, and is well-known for rhizobiome dwellers (64, 65). At the genus level, Phallus and Cladosporium (SIMPER analysis) were among the top genera contributing to the similarities between the ecotypes (Table S2).
FIG 5.

The relative abundance of Ascomycota and Basidiomycota in the dry, mesic and wet ecotypes. Ascomycota and Basidiomycota are also the most abundant phyla in all the ecotypes.
Soil carbon and nitrogen ratio differences between the ecotypes.
We performed soil physicochemical properties and measured soil %Carbon (%C) and %Nitrogen (%N) (Table S4 in the supplemental material). There was no significant difference between ratio of %Carbon and %Nitrogen (C/N) of dry and mesic rhizospheric soil (ANOVA: F = 0.802, P = 0.373). However, we observed significant (at P < 0.1 level) differences between soil C/N of dry v wet (ANOVA: F = 3.032, P = 0.085), and mesic v wet ecotypes (ANOVA: F = 5.323, P = 0.024) (Fig. 6). We surmised that while the plant host and its associated rhizobiome had an influence on the soil biochemistry (66), more insights could be gained based on the microbial function in addition to the taxonomic identity. The huge diversity in the rhizosphere is known to perform multiple microbial functions, with high functional redundancy among the microbial members (67).
FIG 6.

Soil % carbon and % nitrogen ratio (C/N) between the ecotypes. Letters in a box plot are significant different at P < 0.1. (D, M, and W, significantly different from dry, mesic, and wet soil types, respectively).
Ecotype differences in bacterial diversity evident under arid conditions.
The distribution of microorganisms in an environment is often expressed in the famous tenet “everything is everywhere but the environment selects'' (68). Our 10-year-long reciprocal common garden study provided an excellent opportunity to gain insights into whether and how specifically adapted ecotypic microorganisms will thrive and proliferate under arid conditions. The most ideal situation to understand these differences in bacterial diversity would include the ability to compare and contrast this study with the rhizobiome profiles before planting the A. gerardii ecotypes in Colby. Although this study is limited by the lack of time-series samples throughout the last 10 years, we deduced that after 10 years of planting and maintaining under drought stress conditions, the environmental pressure would alter the rhizobiome composition, especially when the hosts or ecotypes had limited or no influences (69–72). On the contrary, we observed that there were differences in bacterial community composition in perennial grassland ecotypes under drought stress, although there were no effects on the fungal compositions. Even without the before planting rhizobiome profiles, the bacterial community composition between ecotypes did in fact differ in the arid environment, suggesting that the host-mediated adaptation persisted even under arid conditions. The earlier report associated with A. gerardii has indicated that the ecotypic variation was observable in several aspects of growth in the plant. The ecotypic variation not only impacted aboveground features but also had a prominent effect on the belowground ecosystem processes mediated by microbial communities (37). We observed the higher relative abundance of Rhizobium in the dry ecotypic rhizosphere, suggesting that the Rhizobium might be associated with the observed higher leaf nitrogen concentrations and chlorophyll absorbance (36, 38). So, synthesizing results from our study and information from previous publications, we provided more insights to potential microbial populations in our study that might help to help the A. gerardii ecotypes to be more resilient to drought stress. The plant host can implement diverse mechanisms by secreting root exudates, employing defense strategies and structural modifications to recruit a specific and optimized microbiome (73). We demonstrated in our study that the individual plant genotype might influence the bacterial rhizobiome, and other reports showed that these beneficial plant genotypic traits capable of impacting the rhizobiome could also be heritable (35, 74–76). There is a fine margin between the influence by the ecotype host and the environment on the microbiome communities. In our present work, we were able to determine that after 10 years of growing under conditions outside the normal ecotypic environment, the host ecotype might be able to “overcome” environmental pressure to a certain extent and regulate the bacterial community composition. Previous studies have indicated that drought stress can escalate the relative abundance of fungal populations while decreasing the relative abundance of bacterial communities in the same poplar plantation (77). Also, it is known that bacterial networks in soil are less stable than the fungal networks (78). Putting all these together, our study suggested that the ecotypic fungal populations might be more capable at adapting, and could have diverse resistance mechanisms toward drought conditions, resulting in the fungal community composition remaining unchanged. Moving forward, future time series association between plant physiological, genotypic and associated microbial community analyses would provide further insights into the impact of plant-microbe interaction.
In this study, we explored the concept of microbial “generalists'' and “specialists''. Generalist microbial populations are able to adapt to diverse habitats, while microbial populations are referred to as specialists when those can only adapt to specific habitats (79). There are previous reports which acknowledge the contribution of the generalists and specialists impacting the dynamics of the different microbial communities (80, 81). We observed that the differences in bacterial community composition were more prominent between dry and mesic as well as wet and mesic, compared to dry and wet ecotypes. While we were not able to identify specific bacterial populations as generalists and specialists from this study, we postulated that dry and wet ecotypic bacterial populations could be driven by specialists, living on the wet and arid margin of the plant species range. On the other hand, bacterial populations from the intermediate mesic ecotype might be guided more by the generalists. There are clear challenges in identifying specialist and generalist bacterial and fungal populations; understanding the co-existences of the specialists and generalists; and roles these specialists and generalists played in enhancing plant host resistance during environmental stress. Future work in elucidating the identity and functions of plant associated microbial specialists and generalists will provide insights in addressing these challenges. From this study, we surmised that A. gerardii ecotypes had unique bacterial assemblage contributing to the rhizobiome. However, the classification of whether the bacterial populations were generalists or specialists might have an influence on the resultant ecotypic rhizobiome due to host and environmental interaction.
To conclude, our study provided the knowledge that will help tackle the challenges faced by grassland restorative efforts by providing insights into the impact of environmental stress on plant host-associated microbiomes. We showed that bacterial populations were influenced by the respective ecotypes, while the fungal populations were not significantly different between the ecotypes. This study also suggested the existence of host-mediated bacterial community adaptation for A.gerardii’s rhizosphere, and a possible tradeoff between the specialist and generalist bacterial communities in specific environments, that might ultimately benefit the plant host. The plant microbiome and its derived functions can substantially extend the plant hosts’ adaptive capacity and resilience to a variety of environmental stressors (82). In the Great Plains, droughts are common (83) and productivity is limited (84), especially in tallgrass prairies. This study provides novel insights into the understanding of the impact of the rhizobiome on the enhancement of drought tolerance of A. gerardii, and its implications in grassland restoration efforts and management.
MATERIALS AND METHODS
Experimental design and sampling.
The common garden in Colby is located at the Kansas State University Agricultural Research Center in Thomas County (39°23′N, 101°04′W). The common garden was established in 2010‐10 years before our sampling. The seeds of four populations of each of native dry (Hays), mesic (Manhattan, KS), and wet (Carbondale Illinois) A. gerardii ecotypes (36) were germinated and then grown inside the greenhouse in potting mix substrate (Metro-Mix 510). Established 3-4-month-old plants from all the populations were then planted in western Kansas (Colby) as a surrogate for the drier conditions expected in the future (85). The size of the Colby common garden plot was 67.5 m2. Each ecotype was represented by four populations with 12 replicate plants (36). There was a total of 12 plants (4 populations × 3 ecotypes) in a randomized complete block design with 10 blocks (36). Plants were planted 0.75 m apart along each row, and the soil around the plants was covered with a water-penetrable cloth to control unwanted plants. Some plants did not survive through the 10 years in the common garden. We collected a total of 95 rhizosphere soil cores (15 cm deep, 1.25 cm diameter) from the dry (n = 33), mesic (n = 30), and wet (n = 32) ecotypes during the growing season in Summer 2019. We considered the topsoil (0-15cm) to assess the impacts of grass ecotypes on rhizobiome. There are previous reports of using the topsoil to analyze the microbiome composition and diversity since the topsoil is considered to contain the most diverse microorganisms (86, 87). Each soil core was placed in a plastic bag, transported on ice, and stored at −80°C until genomic DNA extraction.
Soil total nitrogen and carbon analyses.
We passed each soil sample through a 4-mm sieve to homogenize soil and remove large roots, and handpicked small roots from each soil sample for 10 min per sample. A 15 g subsample of sieved soil was then dried at 55°C for 1 week, grounded to a fine powder in a mixer mill (SPEX Instruments, Metuchen, NJ), and re-dried at 55°C. Approximately 50 mg ground soil was analyzed for %C and %N using dry combustion followed by gas chromatography on a Thermo Scientific FlashSmart 2000 NC Soil Analyzer (Milan, Italy). We used Analysis of Variance (ANOVA) in R to detect the statistical significance in %Carbon and %Nitrogen ratio (C/N) between the dry, mesic, and wet ecotypes (51).
DNA extraction, metabarcoding, and analyses.
We extracted the genomic DNA from the root samples and soil associated with it (0.150g each) using the Omega E.Z.N.A. Soil DNA Kit (Omega Bio‐Tek, Inc., Norcross, GA, USA), with a slightly modified protocol. Bulk soil was separated from the rhizosphere soil by handshaking the roots gently, and any soil that was attached to the root was considered part of the rhizosphere. Briefly, we modified the protocol using a Qiagen TissueLyser II (Qiagen, Hilden, Germany) for 2 min at 20 rev/s, and eluted the purified DNA with a final volume of 100 μL. The extracted microbial DNA were sequenced on the Illumina MiSeq, with the 16S rRNA V4 region amplified using the primers 515F and 806R with barcodes (88), and the internal transcribed spacer region ITS2 amplified using the primers fITS7 (89–91) and ITS4 (92), at the Kansas State University Integrated Genomics Facility.
We acquired a total of 1,729,418 bacterial and 3,570,536 fungal sequencing reads before quality check (QC) and trimming (Table S1 in the supplemental material). We used QIIME 2 (v. 2019.7) to process the sequence data and to profile the rhizobiome communities (93). We used QIIME 2 plugin cutadapt (94) to remove the primer sequences; reads with no primer were discarded. Additionally, we used DADA2 (95) for quality control with the same parameters across different runs and truncated the reads to length where the 25th percentile of the reads had a quality score below 15. The pre-trained classifier offered by QIIME 2, using SILVA database (v. 132) was used for taxonomic assignment for bacteria. Similarly, the UNITE classifier was trained on the full reference “develop” sequences (version 8.2, release date 2020-2-20) (96) using QIIME 2 2020.2 before taxonomic assignment of the fungal reads. We rarefied the data set to normalize the differences in sequencing depth between the samples before estimating diversity indices (97). α-diversity was calculated to present the species diversity in each sample (98). We estimated observed richness (SObs), Shannon’s diversity (H’), and Faith’s PD using a rarefied data set (8,023 reads for 16S; 10,258 reads for ITS). Observed richness (SObs) is defined as the species numbers observed in a sample/set of samples (99). Similar to the α-diversity analysis, we used Bray Curtis distances to compare the compositional dissimilarity among the different ecotypes and used non-metric multidimensional scaling (NMDS) to visualize the distance matrices. Differences in the relative abundances of bacterial and fungal phyla among the ecotypes were analyzed using PERMANOVA in R followed by Tukey’s post hoc test (P < 0.05) (51). SIMPER post hoc analyses were performed to identify those community members that contributed to the highest differences among the ecotypes (100). The cutoff used for all SIMPER analyses was 70% to list only the taxonomic groups that contributed highest to the similarity or differences among the ecotypes. We used DeSEQ2 and highlighted marked differences in the disproportionate relative abundance of bacteria taxa among the ecotypes (P < 0.05) (101).
Data availability.
All raw sequence data is available in the NCBI under BioProject accession no. PRJNA772708 and biosamples SAMN22405120 to SAMN22405309. Additional information can be found in the supplementary sections, and the bacterial and fungal taxon assignments along with their counts in each sample are available in figshare https://doi.org/10.6084/m9.figshare.19469846.
Supplementary Material
ACKNOWLEDGMENTS
The study is based upon the work supported by the National Science Foundation EPSCoR Award No. OIA-1656006 and matching support from the State of Kansas Board of Regents. This study was supported by the United States Department of Agriculture, National Institute of Food and Agriculture (USDA NIFA), under the Award Number: 2020-67019-31803. We are thankful to Alina Akhunova of Kansas State Integrated Genomics Facility for the help with 16S and ITS amplicon sequencing.
Footnotes
Supplemental material is available online only.
Contributor Information
Sonny T. M. Lee, Email: leet1@ksu.edu.
Kristen M. DeAngelis, University of Massachusetts Amherst
Ghada Dawwam, Beijing Forestry University.
Xin Jing, Peking University.
REFERENCES
- 1.Adl S. 2016. Rhizosphere, food security, and climate change: a critical role for plant-soil research. Rhizosphere 1:1–3. doi: 10.1016/j.rhisph.2016.08.005. [DOI] [Google Scholar]
- 2.Ahkami AH, White RA, Handakumbura PP, Jansson C. 2017. Rhizosphere engineering: enhancing sustainable plant ecosystem productivity. Rhizosphere 3:233–243. doi: 10.1016/j.rhisph.2017.04.012. [DOI] [Google Scholar]
- 3.Barea J-M, Pozo MJ, Azcón R, Azcón-Aguilar C. 2005. Microbial co-operation in the rhizosphere. J Exp Bot 56:1761–1778. doi: 10.1093/jxb/eri197. [DOI] [PubMed] [Google Scholar]
- 4.Bais HP, Loyola-Vargas VM, Flores HE, Vivanco JM. 2001. Root-specific metabolism: the biology and biochemistry of underground organs. In Vitro Celldevbiol-Plant 37:730–741. doi: 10.1007/s11627-001-0122-y. [DOI] [Google Scholar]
- 5.Estabrook EM, Yoder JI. 1998. Plant-plant communications: rhizosphere signaling between parasitic angiosperms and their hosts. Plant Physiol 116:1–7. doi: 10.1104/pp.116.1.1. [DOI] [Google Scholar]
- 6.Nardi S, Concheri G, Pizzeghello D, Sturaro A, Rella R, Parvoli G. 2000. Soil organic matter mobilization by root exudates. Chemosphere 41:653–658. doi: 10.1016/s0045-6535(99)00488-9. [DOI] [PubMed] [Google Scholar]
- 7.Jones P, Garcia BJ, Furches A, Tuskan GA, Jacobson D. 2019. Plant Host-Associated Mechanisms for Microbial Selection. Front Plant Sci 10:862. doi: 10.3389/fpls.2019.00862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagg C, Hautier Y, Pellkofer S, Banerjee S, Schmid B, van der Heijden MG. 2021. Diversity and asynchrony in soil microbial communities stabilizes ecosystem functioning. Elife 10. doi: 10.7554/eLife.62813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Classen AT, Sundqvist MK, Henning JA, Newman GS, Moore JAM, Cregger MA, Moorhead LC, Patterson CM. 2015. Direct and indirect effects of climate change on soil microbial and soil microbial-plant interactions: what lies ahead. Ecosphere 6:art130. doi: 10.1890/ES15-00217.1. [DOI] [Google Scholar]
- 10.Bei Q, Moser G, Wu X, Müller C, Liesack W. 2019. Metatranscriptomics reveals climate change effects on the rhizosphere microbiomes in European grassland. Soil Biol Biochem 138:107604. doi: 10.1016/j.soilbio.2019.107604. [DOI] [Google Scholar]
- 11.Compant S, Van Der Heijden MGA, Sessitsch A. 2010. Climate change effects on beneficial plant–microorganism interactions. FEMS Microbiol Ecol 73:197–214. [DOI] [PubMed] [Google Scholar]
- 12.Xie J, Dawwam GE, Sehim AE, Li X, Wu J, Chen S, Zhang D. 2021. Drought stress triggers shifts in the root microbial community and alters functional categories in the microbial gene pool. Front Microbiol 12:744897. doi: 10.3389/fmicb.2021.744897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long SP, Zhu X-G. 2009. Can we use evolutionary algorithms to outdo evolution? A computational approach to increasing crop photosynthetic productivity. Comparative Biochemistry and Physiology, Part A 2:S223. [Google Scholar]
- 14.Long SP, Marshall-Colon A, Zhu X-G. 2015. Meeting the global food demand of the future by engineering crop photosynthesis and yield potential. Cell 161:56–66. doi: 10.1016/j.cell.2015.03.019. [DOI] [PubMed] [Google Scholar]
- 15.Ort DR, Merchant SS, Alric J, Barkan A, Blankenship RE, Bock R, Croce R, Hanson MR, Hibberd JM, Long SP, Moore TA, Moroney J, Niyogi KK, Parry MAJ, Peralta-Yahya PP, Prince RC, Redding KE, Spalding MH, van Wijk KJ, Vermaas WFJ, von Caemmerer S, Weber APM, Yeates TO, Yuan JS, Zhu XG. 2015. Redesigning photosynthesis to sustainably meet global food and bioenergy demand. Proc Natl Acad Sci USA 112:8529–8536. doi: 10.1073/pnas.1424031112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rudgers JA, Fox S, Porras-Alfaro A, Herrera J, Reazin C, Kent DR, Souza L, Chung YA, Jumpponen A. 2022. Biogeography of root‐associated fungi in foundation grasses of North American plains. J Biogeogr 49:22–37. doi: 10.1111/jbi.14260. [DOI] [Google Scholar]
- 17.Mendes R, Garbeva P, Raaijmakers JM. 2013. The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol Rev 37:634–663. doi: 10.1111/1574-6976.12028. [DOI] [PubMed] [Google Scholar]
- 18.Poudel M, Mendes R, Costa LAS, Bueno CG, Meng Y, Folimonova SY, Garrett KA, Martins SJ. 2021. The role of plant-associated bacteria, fungi, and viruses in drought stress mitigation. Front Microbiol 12:743512. doi: 10.3389/fmicb.2021.743512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan N, Bano A. 2019. Exopolysaccharide producing rhizobacteria and their impact on growth and drought tolerance of wheat grown under rainfed conditions. PLoS One 14:e0222302. doi: 10.1371/journal.pone.0222302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bresson J, Varoquaux F, Bontpart T, Touraine B, Vile D. 2013. The PGPR strain Phyllobacterium brassicacearum STM196 induces a reproductive delay and physiological changes that result in improved drought tolerance in Arabidopsis. New Phytol 200:558–569. doi: 10.1111/nph.12383. [DOI] [PubMed] [Google Scholar]
- 21.Augé RM. 2001. Water relations, drought and vesicular-arbuscular mycorrhizal symbiosis. Mycorrhiza 11:3–42. doi: 10.1007/s005720100097. [DOI] [Google Scholar]
- 22.Bryla DR, Duniway JM. 1997. Growth, phosphorus uptake, and water relations of safflower and wheat infected with an arbuscular mycorrhizal fungus. New Phytol 136:581–590. doi: 10.1046/j.1469-8137.1997.00780.x. [DOI] [PubMed] [Google Scholar]
- 23.Bárzana G, Aroca R, Paz JA, Chaumont F, Martinez-Ballesta MC, Carvajal M, Ruiz-Lozano JM. 2012. Arbuscular mycorrhizal symbiosis increases relative apoplastic water flow in roots of the host plant under both well-watered and drought stress conditions. Ann Bot 109:1009–1017. doi: 10.1093/aob/mcs007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wanders N, Wada Y. 2015. Human and climate impacts on the 21st century hydrological drought. J Hydrol 526:208–220. doi: 10.1016/j.jhydrol.2014.10.047. [DOI] [Google Scholar]
- 25.Huang Q, Zhang Q, Singh VP, Shi P, Zheng Y. 2017. Variations of dryness/wetness across China: Changing properties, drought risks, and causes. Glob Planet Change 155:1–12. doi: 10.1016/j.gloplacha.2017.05.010. [DOI] [Google Scholar]
- 26.Liu B, Chen C, Lian Y, Chen J, Chen X. 2015. Long-term change of wet and dry climatic conditions in the southwest karst area of China. Glob Planet Change 127:1–11. doi: 10.1016/j.gloplacha.2015.01.009. [DOI] [Google Scholar]
- 27.Ayantobo OO, Li Y, Song S, Yao N. 2017. Spatial comparability of drought characteristics and related return periods in mainland China over 1961–2013. J Hydrol 550:549–567. doi: 10.1016/j.jhydrol.2017.05.019. [DOI] [Google Scholar]
- 28.Galliart M, Bello N, Knapp M, Poland J, St Amand P, Baer S, Maricle B, Smith AB, Johnson L. 2019. Local adaptation, genetic divergence, and experimental selection in a foundation grass across the US Great Plains’ climate gradient. Glob Change Biol 25:850–868. doi: 10.1111/gcb.14534. [DOI] [PubMed] [Google Scholar]
- 29.Grime JP. 1998. Benefits of plant diversity to ecosystems: immediate, filter and founder effects. J Ecol 86:902–910. doi: 10.1046/j.1365-2745.1998.00306.x. [DOI] [Google Scholar]
- 30.Risser PG, Birney EC, Blocker HD. 1981. The true prairie ecosystem.
- 31.Johnson LC, Matchett JR. 2001. Fire and grazing regulate belowground processes in tallgrass prairie. Ecology 82:3377–3389. 2.0.CO;2. doi: 10.1890/0012-9658(2001)082[3377:FAGRBP]2.0.CO;2. [DOI] [Google Scholar]
- 32.Gustafson DJ, Gibson DJ, Nickrent DL. 2004. Competitive relationships of Andropogon gerardii (Big Bluestem) from remnant and restored native populations and select cultivated varieties. Funct Ecol 18:451–457. doi: 10.1111/j.0269-8463.2004.00850.x. [DOI] [Google Scholar]
- 33.Johnson LC, Olsen JT, Tetreault H, DeLaCruz A, Bryant J, Morgan TJ, Knapp M, Bello NM, Baer SG, Maricle BR. 2015. Intraspecific variation of a dominant grass and local adaptation in reciprocal garden communities along a US Great Plains’ precipitation gradient: implications for grassland restoration with climate change. Evol Appl 8:705–723. doi: 10.1111/eva.12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olsen JT, Caudle KL, Johnson LC, Baer SG, Maricle BR. 2013. Environmental and genetic variation in leaf anatomy among populations of Andropogon gerardii (Poaceae) along a precipitation gradient. Am J Bot 100:1957–1968. doi: 10.3732/ajb.1200628. [DOI] [PubMed] [Google Scholar]
- 35.Gray MM, St Amand P, Bello NM, Galliart MB, Knapp M, Garrett KA, Morgan TJ, Baer SG, Maricle BR, Akhunov ED, Johnson LC. 2014. Ecotypes of an ecologically dominant prairie grass (Andropogon gerardii) exhibit genetic divergence across the U.S. Midwest grasslands’ environmental gradient. Mol Ecol 23:6011–6028. doi: 10.1111/mec.12993. [DOI] [PubMed] [Google Scholar]
- 36.Galliart M, Sabates S, Tetreault H, DeLaCruz A, Bryant J, Alsdurf J, Knapp M, Bello NM, Baer SG, Maricle BR, Gibson DJ, Poland J, St Amand P, Unruh N, Parrish O, Johnson L. 2020. Adaptive genetic potential and plasticity of trait variation in the foundation prairie grass Andropogon gerardii across the US Great Plains’ climate gradient: Implications for climate change and restoration. Evol Appl 13:2333–2356. doi: 10.1111/eva.13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mendola ML, Baer SG, Johnson LC, Maricle BR. 2015. The role of ecotypic variation and the environment on biomass and nitrogen in a dominant prairie grass. Ecology 96:2433–2445. doi: 10.1890/14-1492.1. [DOI] [PubMed] [Google Scholar]
- 38.Caudle KL, Johnson LC, Baer SG, Maricle BR. 2014. A comparison of seasonal foliar chlorophyll change among ecotypes and cultivars of Andropogon gerardii (Poaceae) by using nondestructive and destructive methods. Photosynt 52:511–518. doi: 10.1007/s11099-014-0057-2. [DOI] [Google Scholar]
- 39.Smith AB, Alsdurf J, Knapp M, Baer SG, Johnson LC. 2017. Phenotypic distribution models corroborate species distribution models: A shift in the role and prevalence of a dominant prairie grass in response to climate change. Glob Chang Biol 23:4365–4375. doi: 10.1111/gcb.13666. [DOI] [PubMed] [Google Scholar]
- 40.Bulgarelli D, Garrido-Oter R, Münch PC, Weiman A, Dröge J, Pan Y, McHardy AC, Schulze-Lefert P. 2015. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 17:392–403. doi: 10.1016/j.chom.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ofek-Lalzar M, Sela N, Goldman-Voronov M, Green SJ, Hadar Y, Minz D. 2014. Niche and host-associated functional signatures of the root surface microbiome. Nat Commun 5:4950. doi: 10.1038/ncomms5950. [DOI] [PubMed] [Google Scholar]
- 42.Bulgarelli D, Rott M, Schlaeppi K, Ver Loren van Themaat E, Ahmadinejad N, Assenza F, Rauf P, Huettel B, Reinhardt R, Schmelzer E, Peplies J, Gloeckner FO, Amann R, Eickhorst T, Schulze-Lefert P. 2012. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488:91–95. doi: 10.1038/nature11336. [DOI] [PubMed] [Google Scholar]
- 43.Lemanceau P, Blouin M, Muller D, Moënne-Loccoz Y. 2017. Let the Core Microbiota Be Functional. Trends Plant Sci 22:583–595. doi: 10.1016/j.tplants.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 44.Yan Y, Kuramae EE, de Hollander M, Klinkhamer PGL, van Veen JA. 2017. Functional traits dominate the diversity-related selection of bacterial communities in the rhizosphere. ISME J 11:56–66. doi: 10.1038/ismej.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Louca S, Polz MF, Mazel F, Albright MBN, Huber JA, O’Connor MI, Ackermann M, Hahn AS, Srivastava DS, Crowe SA, Doebeli M, Parfrey LW. 2018. Function and functional redundancy in microbial systems. Nat Ecol Evol 2:936–943. 10.1038/s41559-018-0519-1. [DOI] [PubMed] [Google Scholar]
- 46.Chowdhury TR, Lee J-Y, Bottos EM, Brislawn CJ, White RA, Bramer LM, Brown J, Zucker JD, Kim Y-M, Jumpponen A, Rice CW, Fansler SJ, Metz TO, McCue LA, Callister SJ, Song H-S, Jansson JK. 2019. Metaphenomic responses of a native prairie soil microbiome to moisture perturbations. mSystems 4. doi: 10.1128/mSystems.00061-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X, Chen Q, Han X. 2013. Soil bacterial communities respond to mowing and nutrient addition in a steppe ecosystem. PLoS One 8:e84210. doi: 10.1371/journal.pone.0084210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Navarrete AA, Soares T, Rossetto R, van Veen JA, Tsai SM, Kuramae EE. 2015. Verrucomicrobial community structure and abundance as indicators for changes in chemical factors linked to soil fertility. Antonie Van Leeuwenhoek 108:741–752. doi: 10.1007/s10482-015-0530-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ivanova AA, Kulichevskaya IS, Merkel AY, Toshchakov SV, Dedysh SN. 2016. High diversity of planctomycetes in soils of two lichen-dominated sub-arctic ecosystems of northwestern Siberia. Front Microbiol 7:2065. doi: 10.3389/fmicb.2016.02065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu X, Seuradge BJ, Neufeld JD. 2017. Biogeography of soil Thaumarchaeota in relation to soil depth and land usage. FEMS Microbiol Ecol 93. doi: 10.1093/femsec/fiw246. [DOI] [PubMed] [Google Scholar]
- 51.Team RC. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 52.Jang S-W, Yoou M-H, Hong W-J, Kim Y-J, Lee E-J, Jung K-H. 2020. Re-analysis of 16S amplicon sequencing data reveals soil microbial population shifts in rice fields under drought condition. Rice (NY) 13:44. doi: 10.1186/s12284-020-00403-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim W-I, Cho WK, Kim S-N, Chu H, Ryu K-Y, Yun J-C, Park C-S. 2011. Genetic diversity of cultivable plant growth-promoting rhizobacteria in Korea. J Microbiol Biotechnol 21:777–790. doi: 10.4014/jmb.1101.01031. [DOI] [PubMed] [Google Scholar]
- 54.Schleper C, Nicol GW. 2010. Ammonia-oxidising archaea: physiology, ecology and evolution. Advances in Microbial Physiology. 10.1016/b978-0-12-381045-8.00001-1. [DOI] [PubMed] [Google Scholar]
- 55.Stieglmeier M, Alves RJE, Schleper C. 2014. The Phylum Thaumarchaeota, p 347–362. In Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (ed), The prokaryotes: other major lineages of bacteria and the archaea. Springer Berlin, Berlin, Germany. [Google Scholar]
- 56.Taffner J, Erlacher A, Bragina A, Berg C, Moissl-Eichinger C, Berg G. 2018. What is the role of archaea in plants? New insights from the vegetation of Alpine bogs mSphere 3. doi: 10.1128/mSphere.00122-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rehman A, Nautiyal CS. 2002. Effect of drought on the growth and survival of the stress-tolerant bacterium Rhizobium sp. NBRI2505 sesbania and its drought-sensitive transposon Tn 5 mutant. Curr Microbiol 45:368–377. doi: 10.1007/s00284-002-3770-1. [DOI] [PubMed] [Google Scholar]
- 58.Staudinger C, Mehmeti-Tershani V, Gil-Quintana E, Gonzalez EM, Hofhansl F, Bachmann G, Wienkoop S. 2016. Evidence for a rhizobia-induced drought stress response strategy in Medicago truncatula. J Proteomics 136:202–213. doi: 10.1016/j.jprot.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 59.Muir RE, Tan M-W. 2007. Leucobacter chromiireducens subsp. solipictus subsp. nov., a pigmented bacterium isolated from the nematode Caenorhabditis elegans, and emended description of L. chromiireducens. Int J Syst Evol Microbiol 57:2770–2776. doi: 10.1099/ijs.0.64822-0. [DOI] [PubMed] [Google Scholar]
- 60.Muir Rachel E, Tan M-W. 2008. Virulence of Leucobacter chromiireducens subsp. solipictus to Caenorhabditis elegans: characterization of a novel host-pathogen interaction. Appl Environ Microbiol 74:4185–4198. doi: 10.1128/AEM.00381-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Egidi E, Delgado-Baquerizo M, Plett JM, Wang J, Eldridge DJ, Bardgett RD, Maestre FT, Singh BK. 2019. A few Ascomycota taxa dominate soil fungal communities worldwide. Nat Commun 10. doi: 10.1038/s41467-019-10373-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Praeg N, Illmer P. 2020. Microbial community composition in the rhizosphere of Larix decidua under different light regimes with additional focus on methane cycling microorganisms. Sci Rep 10:22324. doi: 10.1038/s41598-020-79143-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qin S, Yeboah S, Xu X, Liu Y, Yu B. 2017. Analysis on fungal diversity in rhizosphere soil of continuous cropping potato subjected to different furrow-ridge mulching managements. Front Microbiol 8:845. doi: 10.3389/fmicb.2017.00845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thorn RG, Reddy CA, Harris D, Paul EA. 1996. Isolation of saprophytic basidiomycetes from soil. Appl Environ Microbiol 62:4288–4292. doi: 10.1128/aem.62.11.4288-4292.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang Y, Dou Y, Huang Y, An S. 2017. Links between soil fungal diversity and plant and soil properties on the loess plateau. Front Microbiol 8:2198. doi: 10.3389/fmicb.2017.02198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Y, Dong S, Gao Q, Liu S, Ganjurjav H, Wang X, Su X, Wu X. 2017. Soil bacterial and fungal diversity differently correlated with soil biochemistry in alpine grassland ecosystems in response to environmental changes. Sci Rep 7:43077. doi: 10.1038/srep43077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yin B, Crowley D, Sparovek G, De Melo WJ, Borneman J. 2000. Bacterial functional redundancy along a soil reclamation gradient. Appl Environ Microbiol 66:4361–4365. doi: 10.1128/AEM.66.10.4361-4365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.de Wit R, Bouvier T. 2006. “Everything is everywhere, but, the environment selects”; what did Baas Becking and Beijerinck really say? Environ Microbiol 8:755–758. doi: 10.1111/j.1462-2920.2006.01017.x. [DOI] [PubMed] [Google Scholar]
- 69.Hueso S, Hernández T, García C. 2011. Resistance and resilience of the soil microbial biomass to severe drought in semiarid soils: The importance of organic amendments. Appl Soil Ecol 50:27–36. [Google Scholar]
- 70.Alster CJ, German DP, Lu Y, Allison SD. 2013. Microbial enzymatic responses to drought and to nitrogen addition in a southern California grassland. Soil Biol Biochem 64:68–79. doi: 10.1016/j.soilbio.2013.03.034. [DOI] [Google Scholar]
- 71.Bachar A, Al-Ashhab A, Soares MIM, Sklarz MY, Angel R, Ungar ED, Gillor O. 2010. Soil microbial abundance and diversity along a low precipitation gradient. Microb Ecol 60:453–461. doi: 10.1007/s00248-010-9727-1. [DOI] [PubMed] [Google Scholar]
- 72.Fierer N, Schimel JP, Holden PA. 2003. Influence of drying–rewetting frequency on soil bacterial community structure. Microb Ecol 45:63–71. doi: 10.1007/s00248-002-1007-2. [DOI] [PubMed] [Google Scholar]
- 73.Pascale A, Proietti S, Pantelides IS, Stringlis IA. 2019. Modulation of the root microbiome by plant molecules: the basis for targeted disease suppression and plant growth promotion. Front Plant Sci 10:1741. doi: 10.3389/fpls.2019.01741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schweitzer JA, Bailey JK, Fischer DG, LeRoy CJ, Lonsdorf EV, Whitham TG, Hart SC. 2008. Plant-soil microorganism interactions: heritable relationship between plant genotype and associated soil microorganisms. Ecology 89:773–781. doi: 10.1890/07-0337.1. [DOI] [PubMed] [Google Scholar]
- 75.Peiffer JA, Spor A, Koren O, Jin Z, Tringe SG, Dangl JL, Buckler ES, Ley RE. 2013. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci USA 110:6548–6553. doi: 10.1073/pnas.1302837110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walters WA, Jin Z, Youngblut N, Wallace JG, Sutter J, Zhang W, González-Peña A, Peiffer J, Koren O, Shi Q, Knight R, Glavina Del Rio T, Tringe SG, Buckler ES, Dangl JL, Ley RE. 2018. Large-scale replicated field study of maize rhizosphere identifies heritable microbes. Proc Natl Acad Sci USA 115:7368–7373. doi: 10.1073/pnas.1800918115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sun Y, Chen HYH, Jin L, Wang C, Zhang R, Ruan H, Yang J. 2020. Drought stress induced increase of fungi:bacteria ratio in a poplar plantation. Catena 193:104607. doi: 10.1016/j.catena.2020.104607. [DOI] [Google Scholar]
- 78.de Vries FT, Griffiths RI, Bailey M, Craig H, Girlanda M, Gweon HS, Hallin S, Kaisermann A, Keith AM, Kretzschmar M, Lemanceau P, Lumini E, Mason KE, Oliver A, Ostle N, Prosser JI, Thion C, Thomson B, Bardgett RD. 2018. Soil bacterial networks are less stable under drought than fungal networks. Nat Commun 9:3033. doi: 10.1038/s41467-018-05516-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sriswasdi S, Yang C-C, Iwasaki W. 2017. Generalist species drive microbial dispersion and evolution. Nat Commun 8:1162. doi: 10.1038/s41467-017-01265-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pandit SN, Kolasa J, Cottenie K. 2009. Contrasts between habitat generalists and specialists: an empirical extension to the basic metacommunity framework. Ecology 90:2253–2262. doi: 10.1890/08-0851.1. [DOI] [PubMed] [Google Scholar]
- 81.Székely AJ, Langenheder S. 2014. The importance of species sorting differs between habitat generalists and specialists in bacterial communities. FEMS Microbiol Ecol 87:102–112. doi: 10.1111/1574-6941.12195. [DOI] [PubMed] [Google Scholar]
- 82.Trivedi P, Batista BD, Bazany KE, Singh BK. 2022. Plant-microbiome interactions under a changing world: responses, consequences and perspectives. New Phytol. 10.1111/nph.18016. [DOI] [PubMed] [Google Scholar]
- 83.Tollerud H, Brown J, Loveland T, Mahmood R, Bliss N. 2018. Drought and land-cover conditions in the Great Plains. Earth Interact 22:1–25. doi: 10.1175/EI-D-17-0025.1.31097909 [DOI] [Google Scholar]
- 84.He M, Kimball JS, Yi Y, Running S, Guan K, Jensco K, Maxwell B, Maneta M. 2019. Impacts of the 2017 flash drought in the US Northern plains informed by satellite-based evapotranspiration and solar-induced fluorescence. Environ Res Lett 14:e074019. doi: 10.1088/1748-9326/ab22c3. [DOI] [Google Scholar]
- 85.Masson-Delmotte V, Zhai P, Pirani A, Connors SL, Péan C, Berger S, Caud N, Chen Y, Goldfarb L, Gomis MI, Huang M, Leitzell K, Lonnoy E. 2021. Climate Change 2021: the physical science basis. Working Group I Contribution to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 86.Hao J, Chai YN, Lopes LD, Ordóñez RA, Wright EE, Archontoulis S, Schachtman DP. 2020. The effects of soil depth on the structure of microbial communities in agricultural soils in Iowa, USA. Appl Environ Microbiol 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wright AH, Harrison WA, Ali S, Migicovsky Z, Douglas GM, Yurgel S, Bunbury-Blanchette A, Franklin J, Adams SJ, Walker AK. 2022. A characterization of a cool-climate organic vineyard’s microbiome. Phytobiomes J 6:69–82. doi: 10.1094/PBIOMES-03-21-0019-R. [DOI] [Google Scholar]
- 88.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ferrer C, Colom F, Frasés S, Mulet E, Abad JL, Alió JL. 2001. Detection and identification of fungal pathogens by PCR and by ITS2 and 5.8S ribosomal DNA typing in ocular infections. J Clin Microbiol 39:2873–2879. doi: 10.1128/JCM.39.8.2873-2879.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kumeda Y, Asao T. 1996. Single-strand conformation polymorphism analysis of PCR-amplified ribosomal DNA internal transcribed spacers to differentiate species of Aspergillus section Flavi. Appl Environ Microbiol 62:2947–2952. doi: 10.1128/aem.62.8.2947-2952.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ihrmark K, Bödeker ITM, Cruz-Martinez K, Friberg H, Kubartova A, Schenck J, Strid Y, Stenlid J, Brandström-Durling M, Clemmensen KE, Lindahl BD. 2012. New primers to amplify the fungal ITS2 region–evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol Ecol 82:666–677. doi: 10.1111/j.1574-6941.2012.01437.x. [DOI] [PubMed] [Google Scholar]
- 92.White TJ, Bruns T, Lee S, Taylor J, et al. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols: a Guide to Methods and Applications 18:315–322. [Google Scholar]
- 93.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 95.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.UNITE Community. 2019. UNITE QIIME release for Fungi. Version 18.11.2018. https://forum.qiime2.org/t/unite-v-8-0-2018-11-18-classifiers-for-qiime2-available-here/8750. [Google Scholar]
- 97.Kleine Bardenhorst S, Vital M, Karch A, Rübsamen N. 2022. Richness estimation in microbiome data obtained from denoising pipelines. Comput Struct Biotechnol J 20:508–520. doi: 10.1016/j.csbj.2021.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.The Human Microbiome Project Consortium. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gotelli NJ, Colwell RK, Magurran AE, McGill BJ. 2011. Estimating species richness, p 39–54. In Magurran AE, McGill BJ. (ed), Biological diversity: frontiers in measurement and assessment. Oxford University Press, New York. [Google Scholar]
- 100.Clarke KR, Gorley RN. 2015. Getting started with PRIMER v7. PRIMER-E: Plymouth. PRIMER-E, Devon, UK. [Google Scholar]
- 101.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download spectrum.02391-21-s001.pdf, PDF file, 1 MB (1.1MB, pdf)
Data Availability Statement
All raw sequence data is available in the NCBI under BioProject accession no. PRJNA772708 and biosamples SAMN22405120 to SAMN22405309. Additional information can be found in the supplementary sections, and the bacterial and fungal taxon assignments along with their counts in each sample are available in figshare https://doi.org/10.6084/m9.figshare.19469846.