

Abstract
The liver x receptors LXRα (NR1H3) and LXRβ (NR1H2) are members of the nuclear hormone receptor superfamily of ligand dependent transcription factors that regulate transcription in response to the direct binding of cholesterol derivatives. Studies using genetic knockouts and synthetic ligands have defined the LXRs as important modulators of lipid homeostasis throughout the body. This review focuses on the control of cholesterol and fatty acid metabolism by LXRs in the liver and how modifying LXR activity can influence the pathology of liver diseases.
Keywords: LXR, nuclear receptors, cholesterol, fatty acids, liver, NASH
1. Introduction
The liver x receptors LXRα (NR1H3) and LXRβ (NR1H2) are members of the nuclear hormone receptor superfamily of ligand regulated transcription factors. The original identification of both LXR subtypes was based upon DNA sequence homology to other superfamily members with no knowledge of their ligands [1, 2]. Clones encoding LXRα were isolated from a liver cDNA library and the mRNA was found to be highly expressed in the liver, hence the name [2]. Although the liver is a critical site of LXR activity, and the focus of this review, the name liver x receptor is somewhat of a misnomer. In humans and mice LXRα is expressed at relatively high levels in the liver, intestine, adipose, muscle, spleen, lung, adrenal gland, kidney, and in myeloid cells of the immune system. LXRβ is ubiquitously expressed. The two LXR subtypes are encoded by separate genes and the protein sequences are 61% identical/80 % similar when human sequences are compared. The largest differences between subtypes are found in the unstructured amino terminal regions while the DNA binding domains are highly conserved (77% identical/92% similar). The human and mouse LXRα proteins are 92% identical while the identity between human and mouse LXRβ is 85%.
Transient transfection assays and in vitro ligand binding experiments demonstrated that a subset of oxidized cholesterol derivatives (oxysterols) including 22(R)-hydroxycholesterol, 24(S)-hydroxycholesterol, 24(S), 25-epoxycholesterol, 25-hydroxycholesterol and 27-hydroxycholeserol bind directly to LXRs and can function as agonists [3–6]. Cholesterol precursors such as FF-MAS (14-demethyl-14-dehydrolanosterol), zymosterol, and desmosterol have also been reported as LXR agonists [4, 5, 7, 8]. Pharmacological approaches that inhibit cholesterol synthesis and block sterol uptake in cultured cells support a role from cholesterol derivatives as LXR ligands [9, 10]. Furthermore, genetic knockouts that deplete oxysterols or that increase the levels of cholesterol precursors are consistent with these molecules functioning as bona fide LXR ligands in vivo [11, 12]. Nevertheless, if individual cholesterol derivatives function as LXR ligands in specific tissues or during unique physiological or pathological responses remains an unanswered question in the field. In the liver cholesterol derived LXR ligands are likely to be continuously present and it is not known if ligand access to receptors is regulated or if liver LXRs ever exist in a non-liganded state. Future studies that disrupt the ability of LXRs to bind cholesterol-derived ligands could be an approach used to define ligand-dependent and ligand-independent LXR activities.
2. DNA Binding
As described in the introduction, the DNA binding domains of LXRα and LXRβ are highly conserved and both bind to DNA as heterodimers with retinoid x receptors (RXRs) serving as the dimeric partner [2]. In vitro experiments indicate that LXR-RXR heterodimers prefer to bind to direct repeats of the nuclear hormone receptor half site (AGGTCA) separated by 4 nucleotides (DR4) or inverted repeats separated by 1 nucleotide (IR1) [2, 13]. The binding site preferences determined in vitro have been largely confirmed by chromatin immunoprecipitation sequencing (ChIP-Seq) experiments using cells and in mouse liver tissue [7, 14–16]. Interestingly, treatment of mice with the potent synthetic agonist T0901317 results in significant increases in genome-wide LXR binding in the liver. T0901317 not only increases LXR binding to pre-existing sites detected in untreated livers but also promotes binding to more than 11,000 new locations [14]. The functional activity of most of the agonist-dependent binding sites, however, has not been determined. Similarly, the DNA sequence features that distinguish agonist-dependent from agonist-independent binding sites are not defined. For many nuclear receptors treatment with agonists increases dimerization with RXR [17, 18]. LXRα, the major LXR subtype in the liver, is also a relatively unstable protein that is rapidly degraded by ubiquitin-dependent proteolysis. Treatment with strong agonists, however, increase the half-life of LXRα [9]. Therefore, increases in dimerization and in the amount of LXR protein may contribute to the increased genome-wide DNA binding observed in livers of agonist treated mice. Synthetic agonists like T0901317 are often an order of magnitude more potent and significantly more efficacious than endogenous cholesterol derived LXR ligands [19]. Currently it is not known if endogenous ligands contribute to genome-wide LXR binding. Future studies using tissues depleted of endogenous ligands or expressing LXR mutants that cannot bind these molecules will be needed to determine if genome-wide binding can be regulated by endogenous ligands in physiological settings.
3. LXR and Cholesterol Sensing
Cholesterol modulates membrane fluidity, functions as a signaling molecule and serves as a precursor for steroid hormones and bile acids. Excess intracellular cholesterol, however, is toxic necessitating tight regulation of uptake, excretion, and synthesis. The liver is the main location for regulating whole body cholesterol homeostasis and LXRα is the predominant LXR subtype expressed in this tissue. Analysis of individual genetic knockouts of each subtype indicates that the liver-specific LXR functions described in this review are largely LXRα dependent [20–22]. By regulating transcription in response to the direct binding of cholesterol derivatives that track intracellular cholesterol levels, LXRs function in a positively acting feed-forward pathway that promotes cholesterol excretion and catabolism (Figure 1). The details of the individual LXR-dependent pathways that contribute to feed-forward control will be described in later sections of this review. Cholesterol biosynthesis, on the other hand, is controlled by a classic negative feedback mechanism (Figure 1) [23, 24]. Expression of genes encoding enzymes required for cholesterol synthesis is controlled at the level of transcription by the sterol regulatory element binding protein 2 (SREBP2) transcription factor. SREBP2 is produced as a membrane bound inactive precursor imbedded in the endoplasmic reticulum (ER). Upon translocation to the Golgi, proteolytic processing releases mature SREBP2 that migrates to the nucleus and activates transcription. High levels of cholesterol in the ER inhibits the movement of SREBP2 to the Golgi leading to decreased expression of cholesterol synthesis enzymes [23, 24]. Interestingly, both the LXR and SREBP2 pathways converge at the low density lipoprotein receptor (LDLR) which functions as the major mediator for cholesterol uptake in the liver (Figure 1). SREBP2 controls expression of the LDLR gene [23, 24] while activation of LXR increases transcription of the inducible degrader of LDLR (IDOL). IDOL, also known as myosin regulatory light chain interacting protein (MYLIP), is an E3 ubiquitin ligase that targets LDLR for degradation [25, 26].
Figure 1. Cholesterol sensing in the liver.
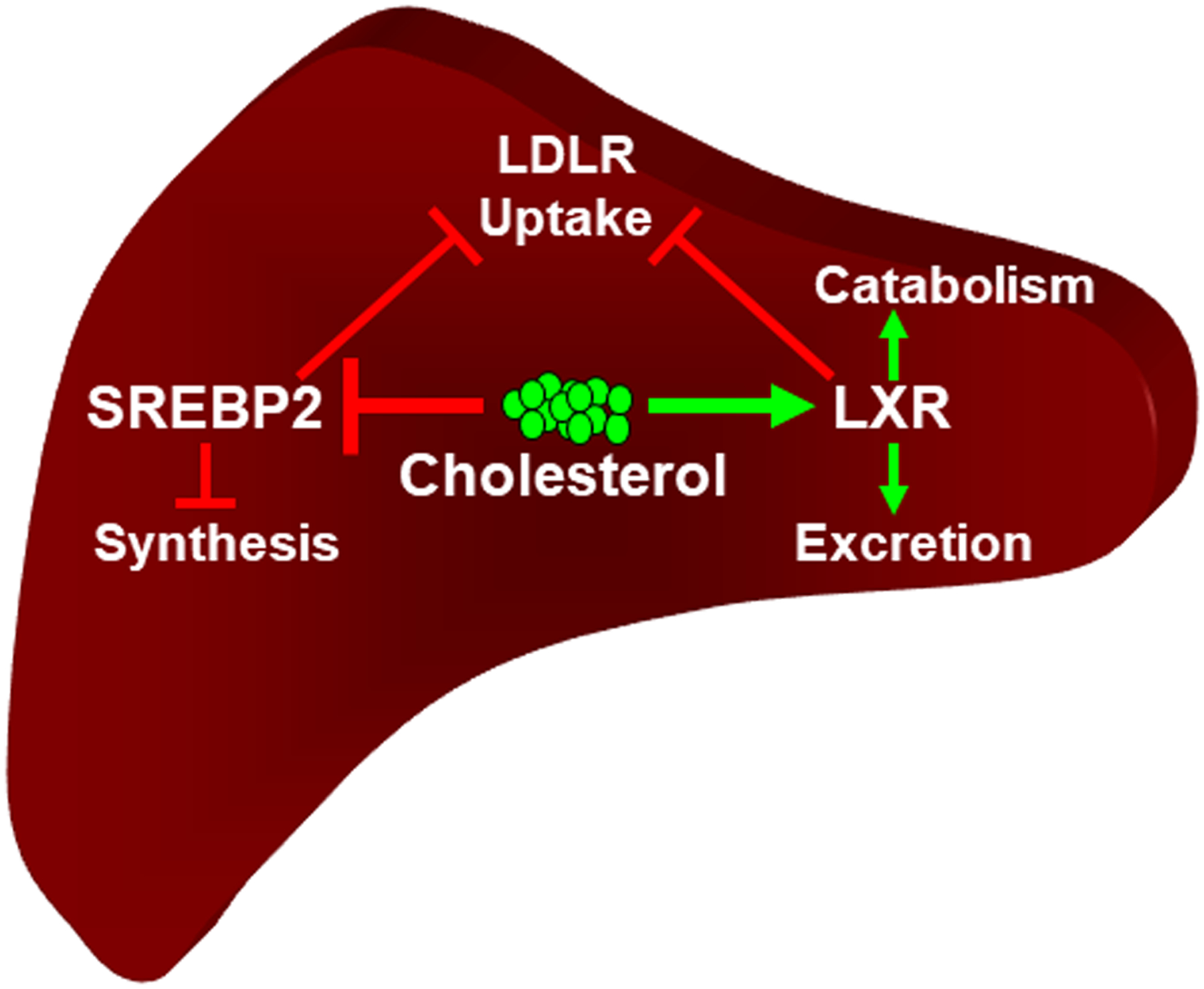
Figure illustrates the response to elevated cholesterol levels. LXRα mediates a feed forward pathway that increases cholesterol catabolism to bile acid and cholesterol excretion while decreasing cholesterol uptake via expression of the LDL receptor. Negative feedback control is mediated by cholesterol dependent inhibition of the transcriptional activity of SREBP2 leading to decreases in cholesterol synthesis and cholesterol uptake. Green arrows indicate positive activation. Red bars indicate inhibition. See the text for details.
4. Cholesterol Transport and HDL
The process of transferring cholesterol from peripheral cells to high-density lipoprotein particles (HDL) followed by transport to the liver for excretion is known as reverse cholesterol transport (RCT; Figure 2) [27–29]. In peripheral cells LXRs regulate expression of the genes encoding the ATP-binding cassette transporters ABCA1 and ABCG1 that transfer intracellular cholesterol to HDL [30, 31], the first step in the RCT pathway (Figure 2). ABCA1 and ABCG1 are expressed in many tissues and can be induced by LXR agonists in these locations [30, 32–35]. Nevertheless, most studies examining the ability of LXRs to regulate cholesterol efflux to HDL have been carried out in macrophages using either primary cells ex vivo or established monocyte/macrophage-derived cell lines. By phagocytosing damaged and dying cells macrophages are often exposed to acute changes in cholesterol levels. Human genetics also suggests an important role for macrophage cholesterol efflux to HDL. Patients with null mutations in the gene encoding ABCA1 have a genetic syndrome named Tangier Disease and often present with accumulation of lipid loaded macrophages in lymph tissues [36]. Importantly, treatment of macrophages in culture with LXR agonists increases efflux of intracellular cholesterol to HDL [37–40]. Conversely, combined genetic knockout of ABCA1 and ABCG1 significantly decreases macrophage cholesterol efflux [41]. Cholesterol accumulation by macrophages in blood vessel walls is an essential step in the pathogenesis of atherosclerotic cardiovascular disease [29, 42]. The ability of LXR agonists to increase macrophage cholesterol efflux to HDL suggested potential roles for LXRs in limiting atherosclerosis by reducing the cholesterol burden within atherosclerotic plaque. Indeed, treatment with LXR agonists decreases or even promotes regression of atherosclerosis in animal models of cardiovascular disease while genetic knockout of LXRs increase disease burden [21, 43–46]. Even though LXR agonists stimulate macrophage cholesterol efflux in vitro, Breevoort et al. [37] demonstrated that macrophage LXR activity makes little or no contribution to the ability of LXR agonists to stimulate RCT in vivo using an assay that measures the movement of cholesterol from macrophages to liver and ultimately to the feces. In contrast, selective deletion of LXRα in the liver significantly impairs the ability of LXR agonists to increase the movement of macrophage derived cholesterol [37]. These studies suggest that activity of LXRα in the liver plays a major role in controlling the movement of cholesterol out of macrophages and perhaps from other organs and tissues throughout the body.
Figure 2. Hepatic LXR controls RCT.
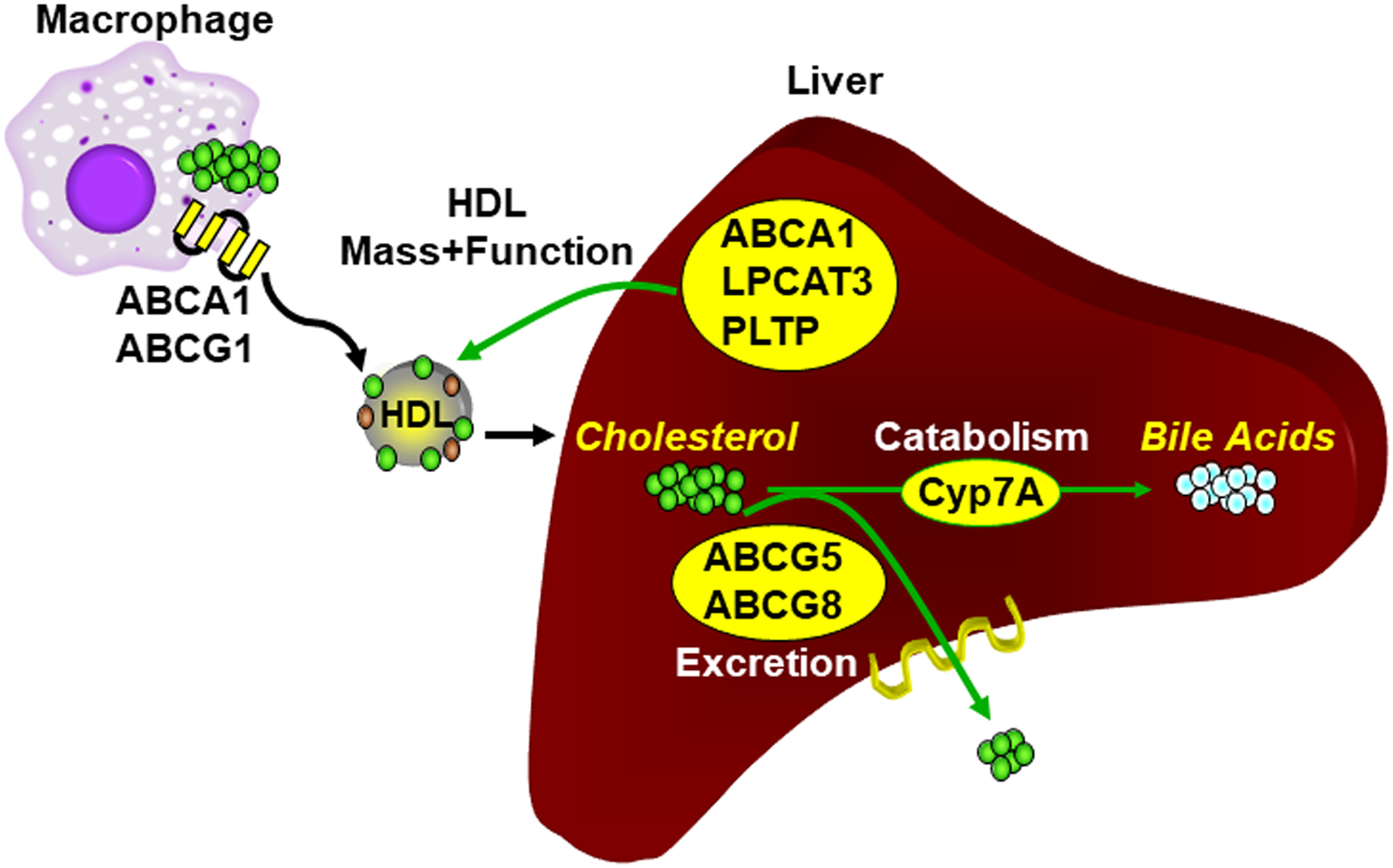
In the liver LXRα controls the flux through the RCT pathway by regulating cholesterol catabolism to bile acids and cholesterol excretion. Hepatic LXRα also controls the ability of HDL to accept cholesterol from macrophages by controlling the number of HDL particles and the functional activity HDL on a per particle basis. See the text for details.
Liver LXR activity modulates RCT in vivo by at least 3 mechanisms (Figure 2). First, as described in the followings section, by regulating cholesterol excretion and catabolism LXRα controls the overall flux through the RCT pathway. Second, studies by Zhang et al. [46] and Breevoort et al. [37] indicate that LXRs modulate the ability of HDL particles to accept cholesterol from macrophages. The LXR-dependent regulation of HDL cholesterol acceptor activity correlates with changes in HDL phospholipid composition which are described in section 6 [37]. Finally, LXR regulates the overall number of HDL particles at least in part by controlling expression of ABCA1 in the liver and intestine [37, 47]. By transferring intracellular cholesterol to newly synthesized lipid-poor HDL particles ABCA1 is also necessary for the biogenesis of HDL [29, 48, 49]. The observation that Tangier Disease patients have little or no circulating HDL is also consistent with a role for ABCA1 in HDL biogenesis [36]. Tissue-specific knockouts of ABCA1 and LXRα indicate that LXR agonists primarily increase HDL levels by regulating ABCA1 expression in the intestine in mice maintained on normal chow diets [47]. Interestingly, when mice are placed on high cholesterol diets a role for liver LXRα in regulating HDL particle number becomes apparent [37]. How dietary cholesterol uncovers a role for liver LXR activity in controlling HDL remains to be determined. Despite strong evidence for LXR-dependent regulation of HDL cholesterol levels, HDL particle number and HDL function in mice, a single LXR agonist had no effect on HDL cholesterol levels after multiple dosing in humans [50]. HDL particle number and function were not reported. In humans LXRs regulate expression of the gene encoding the cholesterol ester transport protein (CETP) an enzyme that transfers cholesterol from HDL to LDL particles [51–53]. CETP is not expressed in rodents and this species-specific difference in lipoprotein remodeling may contribute to the failure to detect increases in HDL cholesterol in humans treated with LXR agonists [50]. Importantly, studies in humans have failed to show beneficial effects of agents that raise HDL cholesterol [54–56] making it unlikely that LXR agonists will be developed for this purpose.
Scavenger receptor-B1 (SR-B1) is the major cell surface receptor for HDL cholesterol, however, cholesterol uptake from HDL does not require clathrin-dependent uptake or lysosomal targeting [57]. In the plasma membrane cholesterol can exist in inaccessible and accessible pools [58] and recently a family of endoplasmic reticulum (ER) anchored proteins, the Gram domain containing 1 proteins (GRAMD1A, 1B and 1C) also referred to as Aster proteins, have been shown to transfer accessible cholesterol from the plasma membrane to the ER [59, 60]. Cholesterol levels in the ER define the level of cholesterol synthesis by controlling the proteolytic processing of SREBP2 to an active transcription factor [23]. The Aster proteins therefore may function to link cholesterol arriving at the membrane from HDL to the regulation of cholesterol synthesis in the ER. In macrophages the gene encoding GRAMD1B was shown to be induced by LXR agonists and ChIP-Seq studies identified a binding site at this locus [60]. The regulation of GRAMD1B suggest that LXRs may act to facilitate the intracellular movement of cholesterol from the plasma membrane and provides a possible link between the feed-forward regulation of cholesterol transport/excretion by LXRs and the negative feedback control of cholesterol synthesis by SREBP2 (Figure 1). In the liver GRAMD1C, which is not regulated by LXR, appears to be the predominant GRAMD1 protein [60]. Wang et al., however, found that GRAMD1B is induced in the livers of mice fed a high cholesterol diet although the contribution of LXR to the diet-dependent regulation was not examined [61].
5. Cholesterol Excretion and Catabolism
Regulation of the ATP binding cassette transporters ABCG5 and ABCG8 in the liver plays a critical role in the ability of LXR agonists to stimulate biliary cholesterol excretion (Figure 2) [31, 62, 63]. ABCG5 and ABCG8 are half transporters that heterodimerize to form functional units [64]. Genetic knockouts and over expression studies indicate that ABCG5/G8 is required for the secretion of cholesterol into bile [62, 63, 65]. ABCG5/G8 also mediates the excretion of plant sterols from the intestine and mutations in the human ABCAG5/G8 genes leads to an inappropriate accumulation of plant sterols in the plasma referred to as sitosterolemia [64]. The genes encoding both subunits are expressed in opposite orientations from the same chromosomal locus and are controlled by a common bi-directional promoter in both mice and humans. Binding sites for LXR-RXR heterodimers have been functionally confirmed within the promoter and additional LXR binding sites throughout the locus have been identified by ChIP-Seq [14, 31]. Importantly, the ability of synthetic LXR agonists or high cholesterol diets to increase expression of ABCG5/G8 and enhance cholesterol excretion is lost in LXRα liver-specific knockout mice [46].
The catabolism of cholesterol to bile acids in the liver serves as an additional mechanism for controlling cholesterol levels. In rodents, LXRα directly regulates the classical pathway of bile acid synthesis by controlling expression of the gene encoding the rate limiting enzyme cholesterol 7α-hydroxylase (Cyp7a1; Figure 2) [46, 66]. As observed for ABCG5/G8, a well characterized LXR response element has been identified in the mouse and rat Cyp7a1 promoters [66]. Treatment of mice with LXR agonists also decreases expression of the genes encoding 12α-hydroxylase (Cyp8b1) and oxysterol 7α-hydroxylase (Cyp7b1) [46, 66, 67]. 12α-hydroxylase sits at a branch point in the bile synthesis pathway and is necessary for the synthesis of cholic acid. The parallel arm in the pathway generates muricholic acid (Figure 3) [68]. Among bile acids tested, muricholic acid promotes the lowest amount of intestinal cholesterol absorption while cholic acid promotes the greatest amount [69, 70]. Oxysterol 7α-hydroxylase, on the other hand, functions in the alternative bile acid synthesis pathway to generate bile acids from oxysterols including those that function as endogenous LXR ligands (Figure 3) [68]. Combining LXR-dependent up-regulation of Cyp7a1 with repression of Cyp8b1 favors the catabolism of cholesterol to muricholic acid and limits the absorption of cholesterol in the intestine. Simultaneous repression of Cyp7b1 maintains the hepatic pool of endogenous LXR ligands (Figure 3). The molecular mechanism(s) describing LXR agonist-dependent repression of Cyp7b1 and Cyp8b1 has not yet been determined. Importantly, there is little evidence for LXR-dependent regulation of bile acid synthesis in humans. The LXRE found in the mouse Cyp7a1 promoter is not conserved in the human gene [71]. Humans also make chenodeoxycholic acid in place of muricholic acid [68] and chenodeoxycholic acid is better at promoting intestinal cholesterol absorption [69, 70].
Figure 3. Control of bile acid synthesis by LXR.
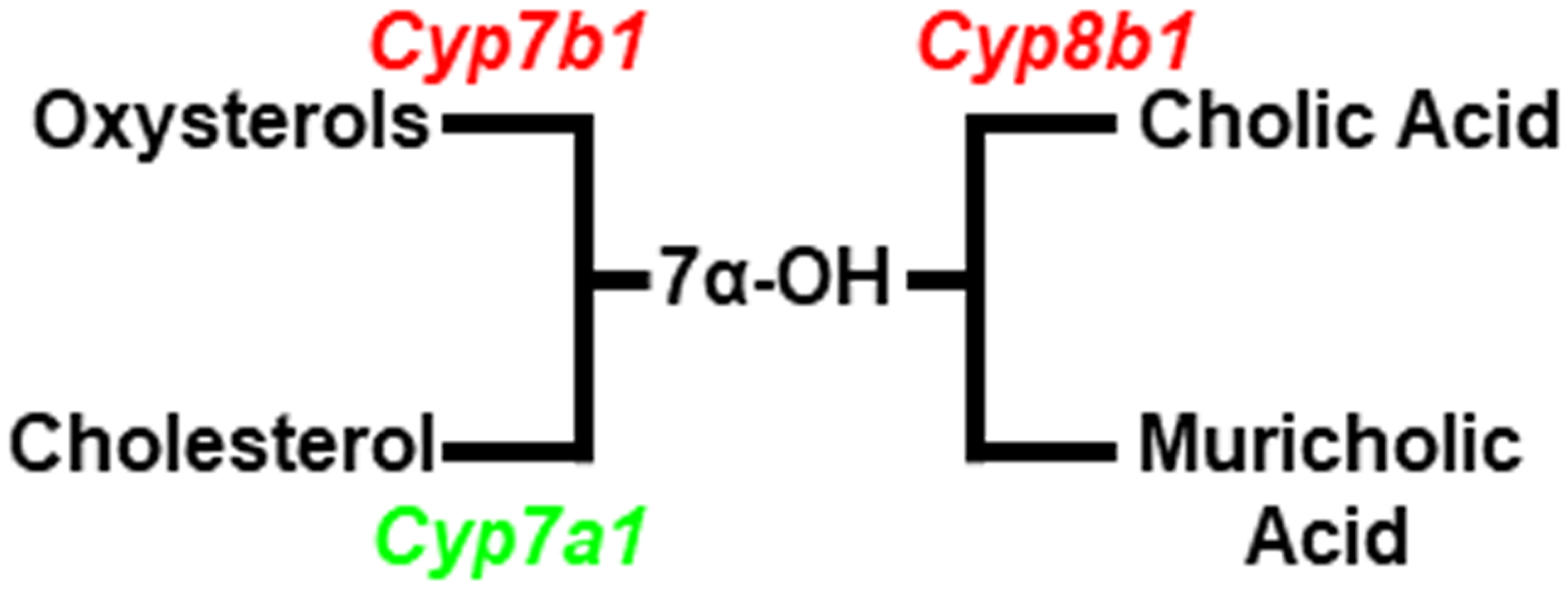
By increasing expression of Cyp7a1 and decreasing expression of Cyp7b1 LXR activation drives the catabolism of cholesterol to bile acids while preserving oxysterol levels. LXR dependent repression of Cyp8b1 drives the composition of bile acids to muricholic acid which decreases the intestinal absorption of cholesterol.
Cholesterol and bile acid synthesis follow a circadian rhythm with peaks in the early part of the dark phase in rodents when feeding and activity begin to increase [72]. Nevertheless, little is known about how LXR activity interfaces with the circadian clock. Retinoid-related orphan receptor alpha (RORα) is an additional cholesterol regulated nuclear receptor that positively regulates expression of brain and muscle ARNT-like protein 1 (Bmal1) a core component of the circadian clock [73–75]. Studies by Wada et al. indicate that RORα and LXRs mutually suppress each other in vivo [76, 77]. LXRα has also been shown to positively regulate expression of the gene encoding basic helix-loop-helix family member e40 (BHLHE40, also known as DEC1) another transcription factor implicated in circadian control [78]. Studies examining circadian rhythms and lipid metabolism in liver-specific LXR knockout mice will be needed to understand how LXR activity interacts with the hepatic clock.
6. Fatty Acids, Triglycerides and Phospholipids
Along with regulating cholesterol transport, analysis of genetic knockouts and synthetic ligands identified critical roles for LXRs in the regulation of fatty acid and triglyceride synthesis [79, 80]. Early studies identified the gene encoding sterol regulatory element binding protein 1c (SREBP1c), itself a master transcriptional regulator of fatty acid synthesis, as a direct LXR target gene [19, 81]. Nevertheless, synthetic LXR agonists still increase fatty acid and triglyceride synthesis in SREBP1c knockout mice [82] suggesting, at least in response to strong pharmacological agonists, that LXRα can control fatty acid synthesis independent of SREBP1c. Subsequent studies have identified binding sites for LXRs in regulatory regions of genes encoding enzymes involved fatty acid synthesis, fatty acid chain elongation and fatty acid desaturation [66, 83–85]. LXR was also shown to regulate expression of the carbohydrate response element binding protein (ChREBP) a third lipogenic transcription factor [86–88]. It is likely that SREBP1c, ChREBP and LXRα function to coordinately regulate hepatic fatty acid synthesis in response to in-coming signals such as insulin, glucose and changing cholesterol levels.
Storing excess cholesterol as fatty acid-esters protects cells from the toxic effects of free cholesterol. This protective mechanism has been suggested as one reason for coupling cholesterol transport and fatty acid synthesis via LXR activity [80]. Treatment with synthetic LXR agonists, however, also increases the secretion of the triglyceride rich very low density lipoprotein particles (VLDL) leading to significant increases in plasma triglycerides [89–91]. In rodents which mostly carry plasma cholesterol in HDL particles, increasing VLDL secretion may be of little consequence. Indeed, LXR agonists strongly reduce atherosclerosis in mouse models of cardiovascular even in the face of large increases in plasma triglycerides [21, 43, 45, 92]. On the other hand, in non-human primates and in humans LXR agonists lead to increases in low density lipoprotein (LDL) cholesterol [25, 50, 51] that most likely arises from the remodeling of VLDL in the blood. The hyperlipidemic effects of synthetic LXR agonists have proved to be a large hurdle slowing the clinical development of these agents. Not surprisingly there have been large drug discovery efforts focused on identifying LXR agonists that maintain the beneficial effects on cholesterol transport while limiting hyperlipidemia. Unfortunately, while LXR agonists with improved therapeutic profiles in rodents and non-human primates have been identified the most advanced of these compounds failed to maintain separation of hyperlipidemia from cholesterol transport upon repeated dosing in humans [50]. In an elegant series of experiments using hydrogen-deuterium exchange Belorusova et al. [93] recently determined that LXR ligands which dissociate hyperlipidemia from cholesterol transport in animal models preferentially stabilize a region of the ligand binding domain (LBD) located in helix 3. These dissociated ligands, however, do not stabilize helix 12 also referred to as activation function 2 (AF2). Stabilization of helix 12 by packing on the surface of the LBD is thought to be the major mechanism of ligand-dependent activation of nuclear receptors [94]. The work of Belorusova suggests identifying ligands that stabilize helix 3 without impacting the dynamics of helix 12 may provide a rational approach for the identification of LXR ligands with improved therapeutic profiles in vivo.
LXRα directly regulates expression of several fatty acid elongases and desaturases in the liver and treatment with agonists leads to increases in long chain unsaturated fatty acids [83, 85, 95, 96]. LXRs are also critical regulators of the gene encoding lysophosphatidylcholine acyltransferase 3 (LPCAT3) an enzyme that mediates the incorporation of fatty acids at the sn2 position of phospholipids [97, 98]. Thus, activation of LXRs leads to remodeling of phospholipid composition by increasing the presence of long chain desaturated fatty acids particularly arachidonoyl containing phospholipids. Importantly, mice lacking LPCAT3 in the liver exhibit reduced plasma triglycerides, hepatic steatosis, and secrete lipid-poor VLDL lacking arachidonoyl phospholipids [99–101]. Mechanistic studies indicate that arachidonoyl phospholipids enhance the lipidation of VLDL particles as they are produced in the ER. LXRα is also responsible for hepatic expression of the gene encoding the phospholipid transport protein (PLTP) [102, 103], a second enzymes that plays a role in the lipidation and secretion of VLDL [104–108]. Thus, along with promoting the excretion and catabolism of cholesterol, activation of LXRs also drives the production and secretion of VLDL providing an additional mechanism to move cholesterol out of the liver. Although not directly demonstrated, it is likely that regulating the expression of LPCAT3 and PLTP modulates the phospholipid composition of HDL and contributes to the ability of hepatic LXRα to control HDL cholesterol acceptor activity described in section 4 [37].
Increased levels of phospholipids with desaturated fatty acids in the ER enhances vesicular transport to the Golgi facilitating movement of unprocessed SREBP1c [109]. In the Golgi, proteolytic processing of SREBP1c releases the active transcription factor which migrates to nucleus and increases expression of genes involved in fatty acid synthesis [23]. Thus, LXRα regulates hepatic fat synthesis at multiple levels (Figure 4). First, LXRα directly regulates expression of genes encoding enzymes involved in fat synthesis. Second, LXRα directly regulates expression of SREBP1c; itself a transcription factor regulating fatty acid synthesis. Third, by altering phospholipid composition LXRα indirectly increases SREBP1c-dependent transcription by increasing ER to Golgi transport. Finally, LXRα facilitates the lipidation and secretion of triglyceride rich VLDL particles. LXR mediated changes in phospholipid composition have also been shown to reduce ER stress and inflammation which may function to protect the ER when cholesterol levels rise [98].
Figure 4. Control of fatty acid synthesis and VLDL secretion by LXR.
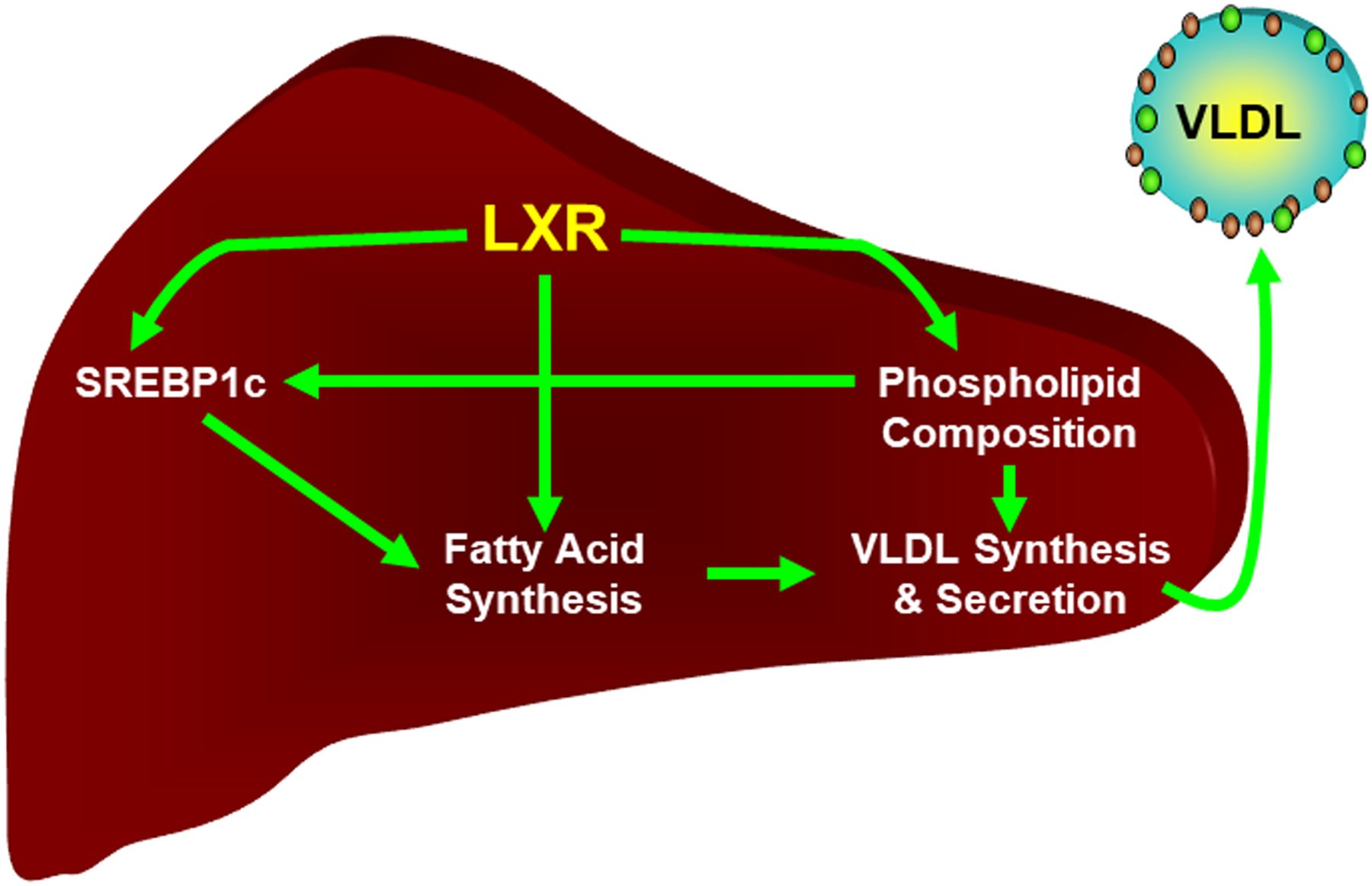
LXR directly regulates the expression of genes encoding enzymes required for fatty acid synthesis as well the gene encoding SREBP1c, a second lipogenic transcription factor. Regulation of phospholipid composition indirectly influences the lipidation and secretion of VLDL and the activity of SREBP1c. See the text for details.
The ability of synthetic LXR ligands and high cholesterol diets to increase bile acid synthesis and biliary cholesterol excretion is lost in LXR knockout mice. Nevertheless, no difference in these parameters or in expression of the relevant genes are detected when normal chow fed LXR positive and LXR knockout mice are compared [46, 66]. In contrast, as described in section 6, hepatic fatty acid synthesis is significantly down-regulated in normal chow fed LXR knockout mice compared to controls [21, 22, 46, 66, 110]. These observations suggest that different LXR-dependent gene networks can differ in their modes of regulation. Fatty acid synthesis genes are sensitive to genetic deletion of LXRs under all conditions. Cholesterol excretion/catabolism genes only appear to require LXR to respond to high dietary cholesterol or to potent synthetic LXR ligands. We suggest that there is a subset of genes with relatively low affinity LXR binding sites (e.g., cholesterol excretion/catabolism) which are only significantly occupied by LXRs when relatively high concentrations of ligands are achieved (see section 2). Genes with high affinity binding sites (e.g., fatty acid synthesis), however, are regulated under both low and high ligand concentrations (Figure 5).
Figure 5. Model for the differential regulation of LXR target genes in hepatocytes.
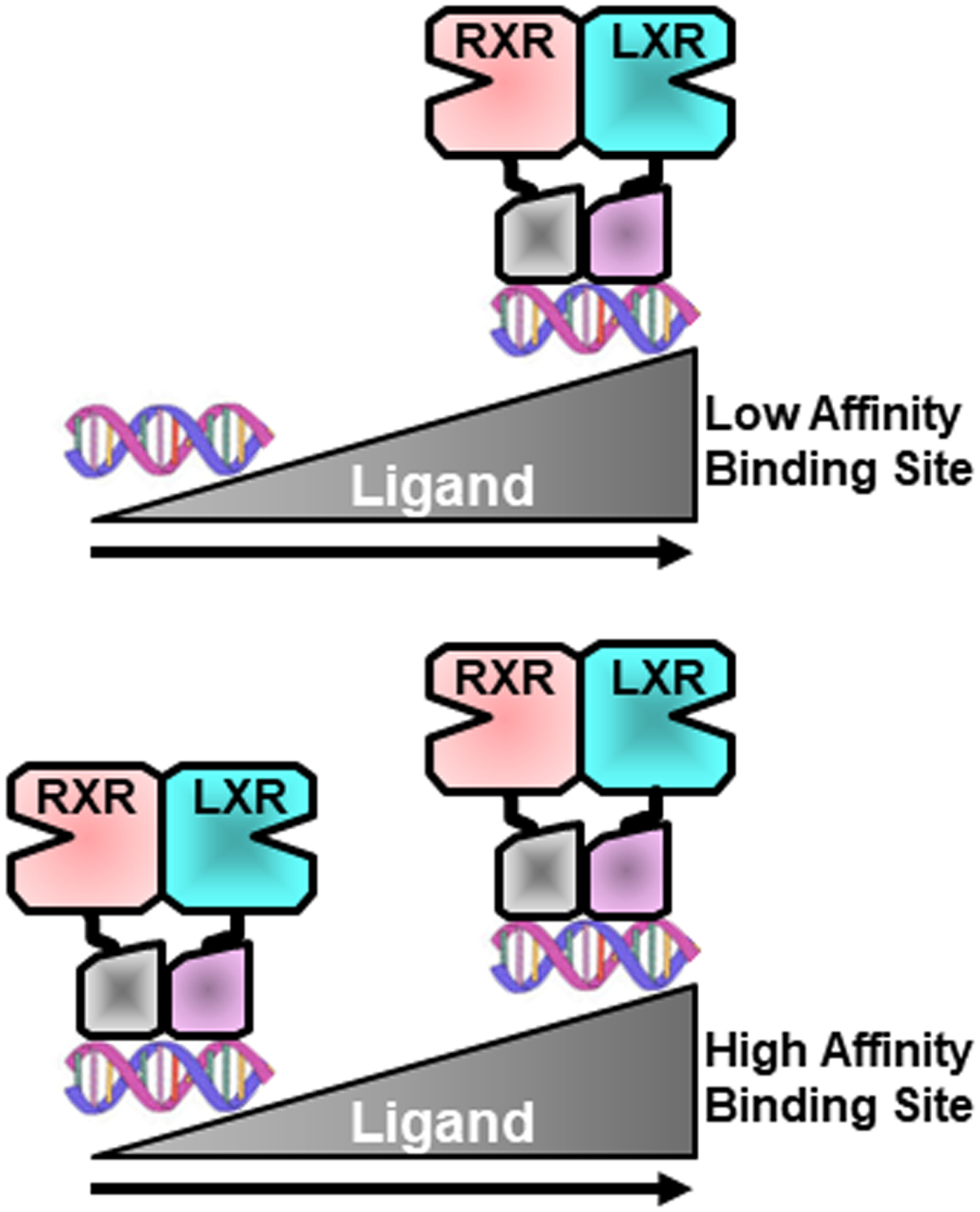
We suggest that genes with relatively low affinity LXR binding sites (top) are only regulated by LXR when ligand concentrations are high such as after feeding with high cholesterol diets or after treatment with potent synthetic ligands. Genes with relatively high affinity binding sites ae regulated by LXRs even when ligand concentrations are relatively low. See text for details.
Hepatic fatty acid synthesis is reported to be elevated in patients with non-alcoholic fatty liver disease (NAFLD) [111, 112]. NAFLD is currently estimated to affect 25% of the adult population in the United States and is linked to the increasing world-wide obesity epidemic [111, 113]. Synthetic LXR antagonists decrease hepatic fatty acid synthesis and reduce hepatic triglyceride accumulation in animal models of NAFLD suggesting potential therapeutic roles for such molecules [114, 115]. Nevertheless, the etiology of NAFLD is complicated. Approximately 25% of NAFLD patients progress to non-alcoholic steatohepatitis (NASH). NASH is characterized by immune infiltration of the liver, large lipid filled hepatocytes (hepatocyte ballooning), and fibrosis. Importantly, NASH increases the risks for cirrhosis, hepatocellular carcinoma, and liver failure [113]. The processes triggering the progression of NAFLD to NASH or even if there is a true stepwise progression from one pathological state to the other remain to be determined. Elevated liver cholesterol is also observed in patients with NASH and non-esterified (free) cholesterol correlates with disease severity [116–119]. Furthermore, clinical studies indicate that patients with NASH can benefit from inhibiting cholesterol synthesis with statins [120–123]. High intracellular cholesterol is toxic leading to endoplasmic reticulum stress, inflammation, and cell death all of which have been suggested to contribute to NASH [113]. Studies implicating elevated hepatic cholesterol in NASH raise questions regarding the potential utility of inhibiting LXR activity with small molecule antagonists that may also increase hepatic cholesterol (see section 5). Consistent with these concerns are studies with LXR knockouts suggesting that decreasing LXR activity increases liver fibrosis [124, 125]. Thus, genetic deletion of LXRs and pharmacological inhibition of LXR activity paradoxically leads to opposite and conflicting results in liver disease models.
7. Carbohydrate Metabolism and Type II Diabetes
Type II diabetes is often associated with elevated hepatic fatty acid synthesis and increases in plasma triglyceride levels. Since synthetic LXR agonists can promote profound hypertriglyceridemia it was quite surprising that these same molecules were shown to have significant anti-diabetic activity in models of type II diabetes including db/db mice and high fat fed animals [126–129]. Several mechanisms and sites of action for the anti-diabetic activity of LXR agonists have been described. Importantly, Commerford et al. used euglycemic-hyperinsulinemic clamp measurements of high fat fed rats to conclude that inhibition of hepatic glucose production accounts for a large majority of LXR anti-diabetic activity [127]. Consistent with the results of Commerford et al., treatment with LXR agonists also decreases expression of phosphoenolpyruvate carboxykinase (PCK) and other hepatic gluconeogenic enzymes [126, 129]. ChIP-Seq studies have identified potential LXR binding sites in the promoter regions of PCK and glucose-6-phosphatase, however, it is not known if LXRs directly repress gluconeogenic gene expression or if the inhibitory effect of activating LXR is indirect [14]. Hypoxic inducible factor 1 alpha (HIF1α) is known to induce glycolysis under condition of low oxygen. Recent studies suggest that LXRs can induce the gene encoding HIF1α in macrophages [130, 131]. If an LXR-HIF1α pathway contributes to hepatic carbohydrate metabolism, however, has not been determined. By simultaneously promoting fatty acid synthesis and inhibiting hepatic glucose production, potent LXR agonists partially mimic the activity of insulin in the liver. Consistent with an “insulin-like” function, in adipose LXRs have been shown to regulate expression of the gene encoding GLUT4 [128, 129], the major insulin stimulated glucose transporter. Therefore, when viewed in the context of the established roles for LXRs in regulating hepatic cholesterol and fatty acid metabolism, the ability to coordinately regulate carbohydrate metabolism suggests a broader role for LXRs as integrators of metabolic signals that identify the fed-state. In that regard Mitro et al. provided a crystal structure indicating that glucose can directly bind to the LXR ligand binding pocket and suggested that LXRs may directly sense glucose levels [132]. There has, however, been little follow up on this study.
8. Kupffer Cells and Hepatic Stellate Cells
Kupffer cells are tissue resident macrophages of the liver and lineage tracing experiments identified LXRα as a transcription factor required to determine and to maintain Kupffer cell fate [133, 134]. The Kupffer cell fate-determining LXR gene network includes genes such as Cdh5, Pcolce2, Kcna2, and Il18bp and is distinct from the well characterized LXR-dependent networks controlling cholesterol and fatty acid metabolism that are also LXR regulated in these cells [134–137]. Kupffer cells are derived from the yolk sac and invade the embryo at the onset of organogenesis [133]. Following selective depletion of Kupffer cells in adult animals, however, bone marrow derived monocytes migrate to the perisinusoidal space of the liver where the combinatorial activation of the Notch and transforming growth factor beta (TGFβ) pathways leads to rapid induction of LXRα and the expression of Kupffer cell-specific genes in these cells [135, 136]. Similar Notch and TGFβ dependent pathways appear to act during normal Kupffer cell development [135]. Interestingly, exposure of mice to a high fat/high fructose/high cholesterol diet reported to promote NASH leads to decreased expression of Kupffer cell-specific genes in enriched populations of liver myeloid cells. Genome-wide analysis suggests decreased binding of LXRα at regulatory regions controlling Kupffer cell fate determining genes and relocalization to regions controlling genes associated with increased lipid burden and tissue scarring [137]. Therefore, as has been seen with other macrophage populations, Kupffer cells can assume distinct phenotypes in response to changing environmental signals. The main driver of this phenotypic switch appears to be a diet-dependent increase in the levels of activating transcription factor 3 (ATF3) which functions to drive LXRα to new regulatory regions [137]. Fibrosis and an inappropriate tissue scarring/wound healing response are associated with NASH raising the possibility that diet-dependent alterations in Kupffer cell LXR activity contribute to rising incidence of this disease.
Hepatic stellate cells are an additional cell type in the liver that plays critical roles in the pathology of liver disease [113, 138]. Stellate cells reside in the space between sinusoidal endothelial cells and the surface of hepatocytes referred to as the Space of Disse. In normal liver physiology these cells function as the major storage site for vitamin A [138]. Upon insult or injury stellate cells become activated, assume a myofibroblast phenotype and secrete numerous extracellular matrix proteins driving fibrosis. Importantly, the activation of stellate cells plays a critical role in the transformation of NAFLD to NASH [113, 138]. Signals from lipid filled and/or apoptotic hepatocytes as well as from pro-inflammatory immune cells including TGFβ, osteopontin, platelet derived growth factor, hedgehog ligands, chemokines and danger associated molecular patterns (DAMPS) all can activate stellate cells leading to increased extracellular matrix production and fibrosis [113]. Cholesterol accumulation has been shown to sensitize hepatic stellate cells to the pro-fibrotic action of TGFβ [139], raising the possibility that LXR dependent regulation of cholesterol metabolism may be important in this cell type. Consistent with a role for LXR in stellate cells, Beaven et al. [124] demonstrated that LXR agonists suppress markers of fibrosis in hepatic stellate cells ex vivo while stellate cells isolated from LXR knockout mice express higher levels of fibrotic genes. Increased liver fibrosis was also observed in LXR knockout mice compared to control mice when the carbon tetrachloride and methionine-choline deficient diet models of liver damage were examined [124]. If the ability of LXRs to limit fibrosis is dependent on promoting cholesterol efflux from hepatic stellate cells, however, has not been addressed.
A single base polymorphism in the gene encoding patatin-like phospholipase domain-containing protein 3 (PNPLA3) that changes isoleucine at position 148 to methionine is genetically associated with large spectrum of a liver diseases including NAFLD and NASH [140]. Nevertheless, the function of PNPLA3 and how it contributes to liver disease remains to be determined. Bruschi et al. found that LXR transcriptional activity is impaired in cells expressing the PNPLA3 methionine 148 variant and that these cells accumulate more cholesterol in vitro compared to cells expressing PNPLA3 isoleucine 148 [141]. How PNPLA3 influences LXR function remains an open question. The work of Bruschi et al. does, however, raises the important possibility that LXR activity in stellate cells contributes to the susceptibility to liver disease.
9. Post-Translational Modifications
Like many nuclear receptors LXRs are post-translationally modified. Interestingly, 3 post-translational modifications appear to act largely through a single lysine residue in LXRα. Li et al demonstrated that acetylation of lysine 434 (K434; K432 in the mouse sequence) in the LXRα LBD decreases transcriptional activity [142]. The responsible acetyltransferase has not been identified, however, Sirtuin 1 (SIRT1) was shown to deacetylate K434 in a ligand-dependent manner. Consistent with an activating role for deacetylation, LXR transcription activity was reduced in livers from SIRT1 knockout mice. Deacetylation of K434 stimulates ubiquitination of this amino acid resulting in a decrease in protein half-life [142]. Several studies have shown that ligand-dependent transcriptional regulation by nuclear receptors is coupled to ubiquitin mediated degradation [143, 144] suggesting a potential link between an acetylation/deacetylation cycle and LXR transcriptional activity. K434 along with K328 of LXRα along with K410 and K448 of LXRβ can also be modified by SUMOylation. Ghisletti et al. [145] and Lee et al. [146] have suggested that SUMOylation is necessary for LXR-dependent anti-inflammatory activity. SUMOylated LXRs directly repress pro-inflammatory gene expression by either blocking the removal of transcriptional repressors gene regulatory elements stimulated nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and/or by inhibiting the DNA binding activity of signal transducer and activated transcription 1 (STAT1) [145, 146]. Taken together, these studies highlight the potential that cross-talk among enzymatic systems mediating the acetylation, SUMOylation and ubiquitination modulates LXR activity. Despite the functional consequences of the LXR modifications described above, Ito et al. found that introducing LXRα with both K328 and K434 changed to arginine into immortalized bone marrow derived macrophages derived from LXR knockout mice completely rescued both the positive regulation of LXR target genes as well as the repression of pro-inflammatory gene expression [147]. Most of the analysis of LXR post-translational modification has been carried out in cell culture systems. The apparent contradictions among these studies emphasizes the importance of using CRIPR and other genome modification approaches to introduce mutations of interest into the endogenous LXR loci to facilitate experiments in vivo.
Serine 198 of LXRα (S198; S196 in the mouse sequence) has been shown to be phosphorylated in a ligand-dependent fashion [148, 149]. Casein kinase 2 and protein kinase A have been suggested to be the responsible enzymes [149, 150]. Mutation of the serine to alanine in mice (S196A) alters the transcriptional response to LXR ligands with a shift to more anti-inflammatory gene expression profile in immune cells [151]. When placed on a high fat/high cholesterol diet female S196A mice exhibit elevated de novo fatty acid synthesis and increased hepatic triglyceride accumulation. The expression of LXR target genes involved in fatty acid synthesis such as SREBP1c is also enhanced in S196A mice relative to controls. Although steatosis is increased in livers from female S196A mice, hepatic inflammation and fibrosis is surprisingly decreased indicating that the progression from fatty liver to NASH is impaired [152]. It is tempting to speculate that the increase in steatosis and decrease in inflammation/fibrosis observed in S196A mice reflects differences in the gene networks regulated by LXRα in hepatocytes, immune cells, and hepatic stellate cells respectively. Future studies employing cell type specific expression of S196A will be needed to test this hypothesis. Less well studied LXR post-translational modifications include poly ADP-ribosylation (inhibitory) [153] and O-linked β-N-acetylglucosamine (O-GlcNAc; stimulatory) [154]. Modification of LXRs by O-GlcNAc increases with rising glucose levels suggesting a mechanism that potentially allows LXRs to function as a glucose sensor [154] (see Section 7).
10. Future Directions
Over the last 10 years the contributions of the LXRs, particularly LXRα, to liver physiology have been well studied. Nevertheless, many important questions related to LXR activity and to the potential therapeutic benefits of LXR ligands are still unanswered. The ability of oxysterols and other sterols to bind directly to LXRs and to regulate LXR dependent transcription has been well established [4, 5, 7, 8, 11, 12]. If individual endogenous LXR ligands are uniquely required during different physiological settings or to activate specific subsets of LXR regulated genes, however, is not known. The use of CRISPR to generate tagged LXRs along with improvements in mass spectrometry suggest that it should be possible to identify endogenous ligands directly bound to LXRs in vivo. The classic model of nuclear receptor activity describes a non-liganded inactive state with receptors bound to transcriptional corepressors. Agonists promote conformational changes that decrease corepressor binding and promote interactions with coactivator proteins that increase transcription [94]. Since it is hard to imagine that the liver is ever “cholesterol free” one can question if hepatic LXRs ever experience a non-liganded state and if the classic model of nuclear receptor activity is even applicable in this context. Future studies exploring the activity of LXR mutants that do not bind endogenous ligands could be used to explore ligand independent LXR activities.
The combination of potent synthetic LXR ligands and genetic knockouts have uncovered roles for LXRs in the transcriptional regulation of cholesterol, fatty acid, and carbohydrate metabolism [80]. LXR is generally considered to a be a cholesterol sensor but it is not obvious why the liver would coordinately regulate cholesterol, fatty acid, and carbohydrate metabolism in response to changing cholesterol levels. Cholesterol is a precursor required for the synthesis of several molecules that function as autocrine, paracrine, and endocrine signals including steroid hormones and bile acids. Perhaps then it is more appropriate to consider one or more endogenous LXR ligands as a classical hormonal signal and not simply as a surrogate marker of intracellular cholesterol levels. In such a system LXRα may function not as a cholesterol-regulated transcription factor but as a sensor of the fed state that like insulin promotes energy storage and inhibits gluconeogenesis. As described above, identifying ligands bound to LXR in cells and in liver tissue could shed light onto how LXR activity is regulated during normal physiology and in pathological settings.
The therapeutic potential of LXR ligands for treating liver diseases remains an important and unanswered question. The hyperlipidemic response to LXR agonists has been a major hurdle to the development of ligands for treating chronic metabolic diseases such as atherosclerosis and type II diabetes [50]. Limiting activity in the liver also may be necessary if LXR ligands are going to prove useful for other indications where they are currently being explored such as Alzheimer’s disease and cancer. In hepatocytes genes encoding proteins involved in fatty acid synthesis and cholesterol excretion respond differentially to genetic knockout of LXRs. Expression of fatty acid synthesis genes such as SREBP1c are strongly decreased in LXRα knockouts while cholesterol excretion genes such as ABCG5 and ABCG8 are not. LXRα, however, is necessary for both subsets of genes to induced by high cholesterol diets or by synthetic LXR agonists [31, 46, 66]. What accounts for the gene-selective response to LXRα knockout is not known? A better understanding of the molecular basis for this selectivity, however, may provide the opportunity to identify small molecules that preferentially regulate one pathway or the other.
The critical role for LXRα in regulating hepatic fatty acid synthesis [19, 46, 66, 81] raises the possibility that small molecule LXR antagonists may have clinical benefits for patients with NAFLD [114, 115]. The incidence of NAFLD is rising in concert with the obesity epidemic, however, the etiology of this disease is complicated. The transition from simple fatty liver to NASH with its associated inflammation and fibrosis appears to be what puts patients at increased risk for cirrhosis, liver failure, and liver cancer [113]. Since elevated hepatic cholesterol levels have been linked to the incidence and severity of NASH studies [116–119] using LXR antagonists should proceed cautiously. LXR antagonists may be useful relatively early during the disease course to reduce fatty liver before patients transition to NASH but could prove detrimental at later stages. Continued study of the role of LXR and cholesterol sensing in hepatocytes, Kupffer cells, infiltrating immune cells, and hepatic stellate cells will be needed to maximize the potential therapeutic activity of LXR ligands for the treatment of liver disease.
Acknowledgements
Research in the Schulman lab is supported by a grant from the NIH/NIDDK (1R01DK119182-01A) to I.G.S.
References
- [1].Shinar DM, Endo N, Rutledge SJ, Vogel R, Rodan GA, Schmidt A, NER, a new member of the gene family encoding the human steroid hormone nuclear receptor, Gene 147(2) (1994) 273–6. [DOI] [PubMed] [Google Scholar]
- [2].Willy PJ, Umesono K, Ong ES, Evans RM, Heyman RA, Mangelsdorf DJ, LXR, a nuclear receptor that defines a distinct retinoid response pathway., Genes Dev. 9 (1995) 1033–1045. [DOI] [PubMed] [Google Scholar]
- [3].Fu X, Menke JG, Chen Y, Zhou G, MacNaul KL, Wright SD, Sparrow CP, Lund EG, 27-hydroxycholesterol is an endogenous ligand for liver X receptor in cholesterol-loaded cells, J Biol Chem 276(42) (2001) 38378–87. [DOI] [PubMed] [Google Scholar]
- [4].Janowski BA, Grogan MJ, Jones SA, Wisely GB, Kliewer SA, Corey EJ, Mangelsdorf DJ, Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta, Proc Natl Acad Sci U S A 96(1) (1999) 266–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ, An oxysterol signalling pathway mediated by the nuclear receptor LXRa., Nature 383 (1996) 728–731. [DOI] [PubMed] [Google Scholar]
- [6].Lehmann JM, Kliewer SA, Moore LB, Smith-Oliver TA, Oliver BB, Su JL, Sundseth SS, Winegar DA, Blanchard DE, Spencer TA, Willson TM, Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway, J Biol Chem 272(6) (1997) 3137–40. [DOI] [PubMed] [Google Scholar]
- [7].Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, Reichart D, Fox JN, Shaked I, Heudobler D, Raetz CR, Wang EW, Kelly SL, Sullards MC, Murphy RC, Merrill AH Jr., Brown HA, Dennis EA, Li AC, Ley K, Tsimikas S, Fahy E, Subramaniam S, Quehenberger O, Russell DW, Glass CK, Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses, Cell 151(1) (2012) 138–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yang C, McDonald JG, Patel A, Zhang Y, Umetani M, Xu F, Westover EJ, Covey DF, Mangelsdorf DJ, Cohen JC, Hobbs HH, Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands, J Biol Chem 281(38) (2006) 27816–26. [DOI] [PubMed] [Google Scholar]
- [9].Ignatova ID, Angdisen J, Moran E, Schulman IG, Differential Regulation of Gene Expression by LXRs in Response to Macrophage Cholesterol Loading, Mol Endocrinol 27(7) (2013) 1036–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liebergall SR, Angdisen J, Chan SH, Chang Y, Osborne TF, Koeppel AF, Turner SD, Schulman IG, Inflammation Triggers Liver X Receptor-Dependent Lipogenesis, Mol Cell Biol 40(2) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen W, Chen G, Head DL, Mangelsdorf DJ, Russell DW, Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice, Cell Metab 5(1) (2007) 73–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Heverin M, Meaney S, Brafman A, Shafir M, Olin M, Shafaati M, von Bahr S, Larsson L, Lovgren-Sandblom A, Diczfalusy U, Parini P, Feinstein E, Bjorkhem I, Studies on the cholesterol-free mouse: strong activation of LXR-regulated hepatic genes when replacing cholesterol with desmosterol, Arterioscler Thromb Vasc Biol 27(10) (2007) 2191–7. [DOI] [PubMed] [Google Scholar]
- [13].Landrier JF, Grober J, Demydchuk J, Besnard P, FXRE can function as an LXRE in the promoter of human ileal bile acid-binding protein (I-BABP) gene, FEBS Lett 553(3) (2003) 299–303. [DOI] [PubMed] [Google Scholar]
- [14].Boergesen M, Pedersen TA, Gross B, van Heeringen SJ, Hagenbeek D, Bindesboll C, Caron S, Lalloyer F, Steffensen KR, Nebb HI, Gustafsson JA, Stunnenberg HG, Staels B, Mandrup S, Genome-wide profiling of liver X receptor, retinoid X receptor, and peroxisome proliferator-activated receptor alpha in mouse liver reveals extensive sharing of binding sites, Mol Cell Biol 32(4) (2012) 852–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pehkonen P, Welter-Stahl L, Diwo J, Ryynanen J, Wienecke-Baldacchino A, Heikkinen S, Treuter E, Steffensen KR, Carlberg C, Genome-wide landscape of liver X receptor chromatin binding and gene regulation in human macrophages, BMC genomics 13 (2012) 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ramon-Vazquez A, de la Rosa JV, Tabraue C, Lopez F, Diaz-Chico BN, Bosca L, Tontonoz P, Alemany S, Castrillo A, Common and Differential Transcriptional Actions of Nuclear Receptors Liver X Receptors alpha and beta in Macrophages, Mol Cell Biol 39(5) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cheskis B, Freedman LP, Modulation of nuclear receptor interactions by ligands: kinetic analysis using surface plasmon resonance, Biochemistry 35(10) (1996) 3309–18. [DOI] [PubMed] [Google Scholar]
- [18].Cheskis B, Lemon BD, Uskokovic M, Lomedico PT, Freedman LP, Vitamin D3-retinoid X receptor dimerization, DNA binding, and transactivation are differentially affected by analogs of 1,25-dihydroxyvitamin D3, Mol Endocrinol 9(12) (1995) 1814–24. [DOI] [PubMed] [Google Scholar]
- [19].Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B, Role of LXRs in control of lipogenesis, Genes Dev 14(22) (2000) 2831–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Alberti S, Schuster G, Parini P, Feltkamp D, Diczfalusy U, Rudling M, Angelin B, Bjorkhem I, Pettersson S, Gustafsson JA, Hepatic cholesterol metabolism and resistance to dietary cholesterol in LXRbeta-deficient mice, J Clin Invest 107(5) (2001) 565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bischoff ED, Daige CL, Petrowski M, Dedman H, Pattison J, Juliano J, Li AC, Schulman IG, Non-redundant roles for LXRalpha and LXRbeta in atherosclerosis susceptibility in low density lipoprotein receptor knockout mice, J Lipid Res 51(5) (2010) 900–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lund EG, Peterson LB, Adams AD, Lam MH, Burton CA, Chin J, Guo Q, Huang S, Latham M, Lopez JC, Menke JG, Milot DP, Mitnaul LJ, Rex-Rabe SE, Rosa RL, Tian JY, Wright SD, Sparrow CP, Different roles of liver X receptor alpha and beta in lipid metabolism: effects of an alpha-selective and a dual agonist in mice deficient in each subtype, Biochem Pharmacol 71(4) (2006) 453–63. [DOI] [PubMed] [Google Scholar]
- [23].Brown MS, Goldstein JL, The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor, Cell 89(3) (1997) 331–40. [DOI] [PubMed] [Google Scholar]
- [24].Jeon TI, Osborne TF, SREBPs: metabolic integrators in physiology and metabolism, Trends Endocrinol Metab 23(2) (2012) 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hong C, Marshall SM, McDaniel AL, Graham M, Layne JD, Cai L, Scotti E, Boyadjian R, Kim J, Chamberlain BT, Tangirala RK, Jung ME, Fong L, Lee R, Young SG, Temel RE, Tontonoz P, The LXR-Idol axis differentially regulates plasma LDL levels in primates and mice, Cell Metab 20(5) (2014) 910–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zelcer N, Hong C, Boyadjian R, Tontonoz P, LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor, Science 325(5936) (2009) 100–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Adorni MP, Zimetti F, Billheimer JT, Wang N, Rader DJ, Phillips MC, Rothblat GH, The roles of different pathways in the release of cholesterol from macrophages, J Lipid Res 48(11) (2007) 2453–62. [DOI] [PubMed] [Google Scholar]
- [28].Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH, The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis, J Lipid Res 50 Suppl (2009) S189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N, HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis, Cell Metab 7(5) (2008) 365–75. [DOI] [PubMed] [Google Scholar]
- [30].Costet P, Luo Y, Wang N, Tall AR, Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor, J Biol Chem 275(36) (2000) 28240–5. [DOI] [PubMed] [Google Scholar]
- [31].Repa JJ, Berge KE, Pomajzl C, Richardson JA, Hobbs H, Mangelsdorf DJ, Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta, J Biol Chem 277(21) (2002) 18793–800. [DOI] [PubMed] [Google Scholar]
- [32].Chung S, Sawyer JK, Gebre AK, Maeda N, Parks JS, Adipose tissue ATP binding cassette transporter A1 contributes to high-density lipoprotein biogenesis in vivo, Circulation 124(15) (2011) 1663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].de Haan W, Bhattacharjee A, Ruddle P, Kang MH, Hayden MR, ABCA1 in adipocytes regulates adipose tissue lipid content, glucose tolerance, and insulin sensitivity, J Lipid Res 55(3) (2014) 516–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Muscat GE, Wagner BL, Hou J, Tangirala RK, Bischoff ED, Rohde P, Petrowski M, Li J, Shao G, Macondray G, Schulman IG, Regulation of cholesterol homeostasis and lipid metabolism in skeletal muscle by liver X receptors, J Biol Chem 277(43) (2002) 40722–8. [DOI] [PubMed] [Google Scholar]
- [35].Repa JJ, Turley SD, Lobaccaro JA, Medina J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM, Mangelsdorf DJ, Regulation of Absorption and ABC1-Mediated Efflux of Cholesterol by RXR Heterodimers, Science 289(5484) (2000) 1524–1529. [DOI] [PubMed] [Google Scholar]
- [36].Oram JF, Lawn RM, ABCA1. The gatekeeper for eliminating excess tissue cholesterol, J Lipid Res 42(8) (2001) 1173–9. [PubMed] [Google Scholar]
- [37].Breevoort SR, Angdisen J, Schulman IG, Macrophage-Independent Regulation of Reverse Cholesterol Transport by Liver X Receptors, Arterioscler Thromb Vasc Biol 34 (2014) 1650–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ishibashi M, Filomenko R, Rebe C, Chevriaux A, Varin A, Derangere V, Bessede G, Gambert P, Lagrost L, Masson D, Knock-down of the oxysterol receptor LXRalpha impairs cholesterol efflux in human primary macrophages: Lack of compensation by LXRbeta activation, Biochem Pharmacol (2013). [DOI] [PubMed] [Google Scholar]
- [39].Schwartz K, Lawn RM, Wade DP, ABC1 gene expression and ApoA-I-mediated cholesterol efflux are regulated by LXR, Biochem Biophys Res Commun 274(3) (2000) 794–802. [DOI] [PubMed] [Google Scholar]
- [40].Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, Tontonoz P, Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha, Proc Natl Acad Sci U S A 97(22) (2000) 12097–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, Tall AR, Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice, J Clin Invest 117(12) (2007) 3900–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Moore KJ, Tabas I, Macrophages in the pathogenesis of atherosclerosis, Cell 145(3) (2011) 341–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, Chen M, Noh G, Goodman J, Hagger GN, Tran J, Tippin TK, Wang X, Lusis AJ, Hsueh WA, Law RE, Collins JL, Willson TM, Tontonoz P, Synthetic LXR ligand inhibits the development of atherosclerosis in mice, Proc Natl Acad Sci U S A 99(11) (2002) 7604–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tangirala RK, Bischoff ED, Joseph SB, Wagner BL, Walczak R, Laffitte BA, Daige CL, Thomas D, Heyman RA, Mangelsdorf DJ, Wang X, Lusis AJ, Tontonoz P, Schulman IG, Identification of macrophage liver X receptors as inhibitors of atherosclerosis, Proc Natl Acad Sci U S A 99(18) (2002) 11896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Terasaka N, Hiroshima A, Koieyama T, Ubukata N, Morikawa Y, Nakai D, Inaba T, T-0901317, a synthetic liver X receptor ligand, inhibits development of atherosclerosis in LDL receptor-deficient mice, FEBS Lett 536(1–3) (2003) 6–11. [DOI] [PubMed] [Google Scholar]
- [46].Zhang Y, Breevoort SR, Angdisen J, Fu M, Schmidt DR, Holmstrom SR, Kliewer SA, Mangelsdorf DJ, Schulman IG, Liver LXRalpha expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice, J Clin Invest 122(5) (2012) 1688–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Brunham LR, Kruit JK, Pape TD, Parks JS, Kuipers F, Hayden MR, Tissue-specific induction of intestinal ABCA1 expression with a liver X receptor agonist raises plasma HDL cholesterol levels, Circ Res 99(7) (2006) 672–4. [DOI] [PubMed] [Google Scholar]
- [48].Navab M, Reddy ST, Van Lenten BJ, Fogelman AM, HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms, Nat Rev Cardiol 8(4) (2011) 222–32. [DOI] [PubMed] [Google Scholar]
- [49].Ye D, Lammers B, Zhao Y, Meurs I, Van Berkel TJ, Van Eck M, ATP-binding cassette transporters A1 and G1, HDL metabolism, cholesterol efflux, and inflammation: important targets for the treatment of atherosclerosis, Current drug targets 12(5) (2011) 647–60. [DOI] [PubMed] [Google Scholar]
- [50].Kirchgessner TG, Sleph P, Ostrowski J, Lupisella J, Ryan CS, Liu X, Fernando G, Grimm D, Shipkova P, Zhang R, Garcia R, Zhu J, He A, Malone H, Martin R, Behnia K, Wang Z, Barrett YC, Garmise RJ, Yuan L, Zhang J, Gandhi MD, Wastall P, Li T, Du S, Salvador L, Mohan R, Cantor GH, Kick E, Lee J, Frost RJ, Beneficial and Adverse Effects of an LXR Agonist on Human Lipid and Lipoprotein Metabolism and Circulating Neutrophils, Cell Metab 24(2) (2016) 223–33. [DOI] [PubMed] [Google Scholar]
- [51].Groot PH, Pearce NJ, Yates JW, Stocker C, Sauermelch C, Doe CP, Willette RN, Olzinski A, Peters T, d’Epagnier D, Morasco KO, Krawiec JA, Webb CL, Aravindhan K, Jucker B, Burgert M, Ma C, Marino JP, Collins JL, Macphee CH, Thompson SK, Jaye M, Synthetic LXR agonists increase LDL in CETP species, J Lipid Res 46(10) (2005) 2182–91. [DOI] [PubMed] [Google Scholar]
- [52].Luo Y, Tall AR, Sterol upregulation of human CETP expression in vitro and in transgenic mice by an LXR element, J Clin Invest 105(4) (2000) 513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Masson D, Staels B, Gautier T, Desrumaux C, Athias A, Le Guern N, Schneider M, Zak Z, Dumont L, Deckert V, Tall A, Jiang XC, Lagrost L, Cholesteryl ester transfer protein modulates the effect of liver X receptor agonists on cholesterol transport and excretion in the mouse, J Lipid Res 45(3) (2004) 543–50. [DOI] [PubMed] [Google Scholar]
- [54].Rader DJ, Tall AR, The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis?, Nat Med 18(9) (2012) 1344–6. [DOI] [PubMed] [Google Scholar]
- [55].Tall AR, Rader DJ, Trials and Tribulations of CETP Inhibitors, Circ Res 122(1) (2018) 106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tariq SM, Sidhu MS, Toth PP, Boden WE, HDL hypothesis: where do we stand now?, Curr Atheroscler Rep 16(4) (2014) 398. [DOI] [PubMed] [Google Scholar]
- [57].Shen WJ, Asthana S, Kraemer FB, Azhar S, Scavenger receptor B type 1: expression, molecular regulation, and cholesterol transport function, J Lipid Res 59(7) (2018) 1114–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Das A, Brown MS, Anderson DD, Goldstein JL, Radhakrishnan A, Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis, eLife 3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Naito T, Ercan B, Krshnan L, Triebl A, Koh DHZ, Wei FY, Tomizawa K, Torta FT, Wenk MR, Saheki Y, Movement of accessible plasma membrane cholesterol by the GRAMD1 lipid transfer protein complex, eLife 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sandhu J, Li S, Fairall L, Pfisterer SG, Gurnett JE, Xiao X, Weston TA, Vashi D, Ferrari A, Orozco JL, Hartman CL, Strugatsky D, Lee SD, He C, Hong C, Jiang H, Bentolila LA, Gatta AT, Levine TP, Ferng A, Lee R, Ford DA, Young SG, Ikonen E, Schwabe JWR, Tontonoz P, Aster Proteins Facilitate Nonvesicular Plasma Membrane to ER Cholesterol Transport in Mammalian Cells, Cell 175(2) (2018) 514–529 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wang X, Cai B, Yang X, Sonubi OO, Zheng Z, Ramakrishnan R, Shi H, Valenti L, Pajvani UB, Sandhu J, Infante RE, Radhakrishnan A, Covey DF, Guan KL, Buck J, Levin LR, Tontonoz P, Schwabe RF, Tabas I, Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis, Cell Metab 31(5) (2020) 969–986 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yu L, Li-Hawkins J, Hammer RE, Berge KE, Horton JD, Cohen JC, Hobbs HH, Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol, J Clin Invest 110(5) (2002) 671–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Yu L, York J, von Bergmann K, Lutjohann D, Cohen JC, Hobbs HH, Stimulation of cholesterol excretion by the liver X receptor agonist requires ATP-binding cassette transporters G5 and G8, J Biol Chem 278(18) (2003) 15565–70. [DOI] [PubMed] [Google Scholar]
- [64].Hubacek JA, Berge KE, Cohen JC, Hobbs HH, Mutations in ATP-cassette binding proteins G5 (ABCG5) and G8 (ABCG8) causing sitosterolemia, Hum Mutat 18(4) (2001) 359–60. [DOI] [PubMed] [Google Scholar]
- [65].Wilund KR, Yu L, Xu F, Hobbs HH, Cohen JC, High-level expression of ABCG5 and ABCG8 attenuates diet-induced hypercholesterolemia and atherosclerosis in Ldlr−/− mice, J Lipid Res 45(8) (2004) 1429–36. [DOI] [PubMed] [Google Scholar]
- [66].Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro J-MA, Hammer RE, Mangelsdorf DJ, Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXRa., Cell 93 (1998) 693–704. [DOI] [PubMed] [Google Scholar]
- [67].Uppal H, Saini SP, Moschetta A, Mu Y, Zhou J, Gong H, Zhai Y, Ren S, Michalopoulos GK, Mangelsdorf DJ, Xie W, Activation of LXRs prevents bile acid toxicity and cholestasis in female mice, Hepatology 45(2) (2007) 422–32. [DOI] [PubMed] [Google Scholar]
- [68].Russell DW, The enzymes, regulation, and genetics of bile acid synthesis, Annu Rev Biochem 72 (2003) 137–74. [DOI] [PubMed] [Google Scholar]
- [69].Wang DQ, Lammert F, Cohen DE, Paigen B, Carey MC, Cholic acid aids absorption, biliary secretion, and phase transitions of cholesterol in murine cholelithogenesis, Am J Physiol 276(3) (1999) G751–60. [DOI] [PubMed] [Google Scholar]
- [70].Wang DQ, Tazuma S, Effect of beta-muricholic acid on the prevention and dissolution of cholesterol gallstones in C57L/J mice, J Lipid Res 43(11) (2002) 1960–8. [DOI] [PubMed] [Google Scholar]
- [71].Menke JG, Macnaul KL, Hayes NS, Baffic J, Chao YS, Elbrecht A, Kelly LJ, Lam MH, Schmidt A, Sahoo S, Wang J, Wright SD, Xin P, Zhou G, Moller DE, Sparrow CP, A novel liver X receptor agonist establishes species differences in the regulation of cholesterol 7alpha-hydroxylase (CYP7a), Endocrinology 143(7) (2002) 2548–58. [DOI] [PubMed] [Google Scholar]
- [72].Ferrell JM, Chiang JY, Circadian rhythms in liver metabolism and disease, Acta Pharm Sin B 5(2) (2015) 113–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Cook DN, Kang HS, Jetten AM, Retinoic Acid-Related Orphan Receptors (RORs): Regulatory Functions in Immunity, Development, Circadian Rhythm, and Metabolism, Nucl Receptor Res 2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kallen JA, Schlaeppi JM, Bitsch F, Geisse S, Geiser M, Delhon I, Fournier B, X-ray structure of the hRORalpha LBD at 1.63 A: structural and functional data that cholesterol or a cholesterol derivative is the natural ligand of RORalpha, Structure 10(12) (2002) 1697–707. [DOI] [PubMed] [Google Scholar]
- [75].Wang Y, Kumar N, Solt LA, Richardson TI, Helvering LM, Crumbley C, Garcia-Ordonez RD, Stayrook KR, Zhang X, Novick S, Chalmers MJ, Griffin PR, Burris TP, Modulation of retinoic acid receptor-related orphan receptor alpha and gamma activity by 7-oxygenated sterol ligands, J Biol Chem 285(7) 5013–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wada T, Kang HS, Angers M, Gong H, Bhatia S, Khadem S, Ren S, Ellis E, Strom SC, Jetten AM, Xie W, Identification of oxysterol 7alpha-hydroxylase (Cyp7b1) as a novel retinoid-related orphan receptor alpha (RORalpha) (NR1F1) target gene and a functional cross-talk between RORalpha and liver X receptor (NR1H3), Mol Pharmacol 73(3) (2008) 891–9. [DOI] [PubMed] [Google Scholar]
- [77].Wada T, Kang HS, Jetten AM, Xie W, The emerging role of nuclear receptor RORalpha and its crosstalk with LXR in xeno- and endobiotic gene regulation, Exp Biol Med (Maywood) 233(10) (2008) 1191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Noshiro M, Usui E, Kawamoto T, Sato F, Nakashima A, Ueshima T, Honda K, Fujimoto K, Honma S, Honma K, Makishima M, Kato Y, Liver X receptors (LXRalpha and LXRbeta) are potent regulators for hepatic Dec1 expression, Genes to cells: devoted to molecular & cellular mechanisms 14(1) (2009) 29–40. [DOI] [PubMed] [Google Scholar]
- [79].Hong C, Tontonoz P, Liver X receptors in lipid metabolism: opportunities for drug discovery, Nature reviews. Drug discovery 13(6) (2014) 433–44. [DOI] [PubMed] [Google Scholar]
- [80].Schulman IG, Liver X receptors link lipid metabolism and inflammation, FEBS Lett 591(19) (2017) 2978–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ, Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta, Genes Dev 14(22) (2000) 2819–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS, Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c, J Biol Chem 277(11) (2002) 9520–8. [DOI] [PubMed] [Google Scholar]
- [83].Chu K, Miyazaki M, Man WC, Ntambi JM, Stearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activation, Mol Cell Biol 26(18) (2006) 6786–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Joseph SB, Laffitte BA, Patel PH, Watson MA, Matsukuma KE, Walczak R, Collins JL, Osborne TF, Tontonoz P, Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors, J Biol Chem 277(13) (2002) 11019–25. [DOI] [PubMed] [Google Scholar]
- [85].Wang Y, Kurdi-Haidar B, Oram JF, LXR-mediated activation of macrophage stearoyl-CoA desaturase generates unsaturated fatty acids that destabilize ABCA1, J Lipid Res 45(5) (2004) 972–80. [DOI] [PubMed] [Google Scholar]
- [86].Cha JY, Repa JJ, The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR, J Biol Chem 282(1) (2007) 743–51. [DOI] [PubMed] [Google Scholar]
- [87].Fan Q, Norgaard RC, Bindesboll C, Lucas C, Dalen KT, Babaie E, Itkonen HM, Matthews J, Nebb HI, Gronning-Wang LM, LXRalpha Regulates Hepatic ChREBPalpha Activity and Lipogenesis upon Glucose, but Not Fructose Feeding in Mice, Nutrients 9(7) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Fan Q, Norgaard RC, Grytten I, Ness CM, Lucas C, Vekterud K, Soedling H, Matthews J, Lemma RB, Gabrielsen OS, Bindesboll C, Ulven SM, Nebb HI, Gronning-Wang LM, Saether T, LXRalpha Regulates ChREBPalpha Transactivity in a Target Gene-Specific Manner through an Agonist-Modulated LBD-LID Interaction, Cells 9(5) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Basciano H, Miller A, Baker C, Naples M, Adeli K, LXRalpha activation perturbs hepatic insulin signaling and stimulates production of apolipoprotein B-containing lipoproteins, Am J Physiol Gastrointest Liver Physiol 297(2) (2009) G323–32. [DOI] [PubMed] [Google Scholar]
- [90].Grefhorst A, Elzinga BM, Voshol PJ, Plosch T, Kok T, Bloks VW, van der Sluijs FH, Havekes LM, Romijn JA, Verkade HJ, Kuipers F, Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles, J Biol Chem 277(37) (2002) 34182–90. [DOI] [PubMed] [Google Scholar]
- [91].Okazaki H, Goldstein JL, Brown MS, Liang G, LXR-SREBP-1c-phospholipid transfer protein axis controls very low density lipoprotein (VLDL) particle size, J Biol Chem 285(9) (2010) 6801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Levin N, Bischoff ED, Daige CL, Thomas D, Vu CT, Heyman RA, Tangirala RK, Schulman IG, Macrophage liver X receptor is required for antiatherogenic activity of LXR agonists, Arterioscler Thromb Vasc Biol 25(1) (2005) 135–42. [DOI] [PubMed] [Google Scholar]
- [93].Belorusova AY, Evertsson E, Hovdal D, Sandmark J, Bratt E, Maxvall I, Schulman IG, Akerblad P, Lindstedt EL, Structural analysis identifies an escape route from the adverse lipogenic effects of liver X receptor ligands, Commun Biol 2 (2019) 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Weikum ER, Liu X, Ortlund EA, The nuclear receptor superfamily: A structural perspective, Protein Sci 27(11) (2018) 1876–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Qin Y, Dalen KT, Gustafsson JA, Nebb HI, Regulation of hepatic fatty acid elongase 5 by LXRalpha-SREBP-1c, Biochim Biophys Acta 1791(2) (2009) 140–7. [DOI] [PubMed] [Google Scholar]
- [96].Varin A, Thomas C, Ishibashi M, Menegaut L, Gautier T, Trousson A, Bergas V, de Barros JP, Narce M, Lobaccaro JM, Lagrost L, Masson D, Liver x receptor activation promotes polyunsaturated Fatty Acid synthesis in macrophages: relevance in the context of atherosclerosis, Arterioscler Thromb Vasc Biol 35(6) (2015) 1357–65. [DOI] [PubMed] [Google Scholar]
- [97].Ishibashi M, Varin A, Filomenko R, Lopez T, Athias A, Gambert P, Blache D, Thomas C, Gautier T, Lagrost L, Masson D, Liver x receptor regulates arachidonic acid distribution and eicosanoid release in human macrophages: a key role for lysophosphatidylcholine acyltransferase 3, Arterioscler Thromb Vasc Biol 33(6) (2013) 1171–9. [DOI] [PubMed] [Google Scholar]
- [98].Rong X, Albert CJ, Hong C, Duerr MA, Chamberlain BT, Tarling EJ, Ito A, Gao J, Wang B, Edwards PA, Jung ME, Ford DA, Tontonoz P, LXRs Regulate ER Stress and Inflammation through Dynamic Modulation of Membrane Phospholipid Composition, Cell Metab 18(5) (2013) 685–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Hashidate-Yoshida T, Harayama T, Hishikawa D, Morimoto R, Hamano F, Tokuoka SM, Eto M, Tamura-Nakano M, Yanobu-Takanashi R, Mukumoto Y, Kiyonari H, Okamura T, Kita Y, Shindou H, Shimizu T, Fatty acyl-chain remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport, eLife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Li Z, Ding T, Pan X, Li Y, Li R, Sanders PE, Kuo MS, Hussain MM, Cao G, Jiang XC, Lysophosphatidylcholine acyltransferase 3 knockdown-mediated liver lysophosphatidylcholine accumulation promotes very low density lipoprotein production by enhancing microsomal triglyceride transfer protein expression, J Biol Chem 287(24) (2012) 20122–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Rong X, Wang B, Dunham MM, Hedde PN, Wong JS, Gratton E, Young SG, Ford DA, Tontonoz P, Lpcat3-dependent production of arachidonoyl phospholipids is a key determinant of triglyceride secretion, eLife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Cao G, Beyer TP, Yang XP, Schmidt RJ, Zhang Y, Bensch WR, Kauffman RF, Gao H, Ryan TP, Liang Y, Eacho PI, Jiang XC, Phospholipid transfer protein is regulated by liver X receptors in vivo, J Biol Chem 277(42) (2002) 39561–5. [DOI] [PubMed] [Google Scholar]
- [103].Mak PA, Kast-Woelbern HR, Anisfeld AM, Edwards PA, Identification of PLTP as an LXR target gene and apoE as an FXR target gene reveals overlapping targets for the two nuclear receptors, J Lipid Res 43(12) (2002) 2037–41. [DOI] [PubMed] [Google Scholar]
- [104].Jiang XC, Qin S, Qiao C, Kawano K, Lin M, Skold A, Xiao X, Tall AR, Apolipoprotein B secretion and atherosclerosis are decreased in mice with phospholipid-transfer protein deficiency, Nat Med 7(7) (2001) 847–52. [DOI] [PubMed] [Google Scholar]
- [105].Lie J, de Crom R, van Gent T, van Haperen R, Scheek L, Lankhuizen I, van Tol A, Elevation of plasma phospholipid transfer protein in transgenic mice increases VLDL secretion, J Lipid Res 43(11) (2002) 1875–80. [DOI] [PubMed] [Google Scholar]
- [106].Luo Y, Shelly L, Sand T, Reidich B, Chang G, Macdougall M, Peakman MC, Jiang XC, Pharmacologic inhibition of phospholipid transfer protein activity reduces apolipoprotein-B secretion from hepatocytes, J Pharmacol Exp Ther 332(3) (2010) 1100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Manchekar M, Liu Y, Sun Z, Richardson PE, Dashti N, Phospholipid Transfer Protein Plays a Major Role in the Initiation of Apolipoprotein B-containing Lipoprotein Assembly in Mouse Primary Hepatocytes, J Biol Chem 290(13) (2015) 8196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Yazdanyar A, Jiang XC, Liver phospholipid transfer protein (PLTP) expression with a PLTP-null background promotes very low density lipoprotein production, Hepatology (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Rong X, Wang B, Palladino EN, de Aguiar Vallim TQ, Ford DA, Tontonoz P, ER phospholipid composition modulates lipogenesis during feeding and in obesity, J Clin Invest 127(10) (2017) 3640–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Quinet EM, Savio DA, Halpern AR, Chen L, Schuster GU, Gustafsson JA, Basso MD, Nambi P, Liver X receptor (LXR)-beta regulation in LXRalpha-deficient mice: implications for therapeutic targeting, Mol Pharmacol 70(4) (2006) 1340–9. [DOI] [PubMed] [Google Scholar]
- [111].Engin A, Non-Alcoholic Fatty Liver Disease, Adv Exp Med Biol 960 (2017) 443–467. [DOI] [PubMed] [Google Scholar]
- [112].Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, Nakagawa T, Kuwabara M, Sato Y, Kang DH, Tolan DR, Sanchez-Lozada LG, Rosen HR, Lanaspa MA, Diehl AM, Johnson RJ, Fructose and sugar: A major mediator of non-alcoholic fatty liver disease, J Hepatol 68(5) (2018) 1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Schwabe RF, Tabas I, Pajvani UB, Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis, Gastroenterology 158(7) (2020) 1913–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Griffett K, Welch RD, Flaveny CA, Kolar GR, Neuschwander-Tetri BA, Burris TP, The LXR inverse agonist SR9238 suppresses fibrosis in a model of non-alcoholic steatohepatitis, Mol Metab 4(4) (2015) 353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Huang P, Kaluba B, Jiang XL, Chang S, Tang XF, Mao LF, Zhang ZP, Huang FZ, Liver X Receptor Inverse Agonist SR9243 Suppresses Nonalcoholic Steatohepatitis Intrahepatic Inflammation and Fibrosis, Biomed Res Int 2018 (2018) 8071093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Caballero F, Fernandez A, De Lacy AM, Fernandez-Checa JC, Caballeria J, Garcia-Ruiz C, Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH, J Hepatol 50(4) (2009) 789–96. [DOI] [PubMed] [Google Scholar]
- [117].Ioannou GN, The Role of Cholesterol in the Pathogenesis of NASH, Trends Endocrinol Metab 27(2) (2016) 84–95. [DOI] [PubMed] [Google Scholar]
- [118].Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, Kellum J, Warnick R, Contos MJ, Sanyal AJ, Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease, Cell Metab 15(5) (2012) 665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, Sanyal AJ, A lipidomic analysis of nonalcoholic fatty liver disease, Hepatology 46(4) (2007) 1081–90. [DOI] [PubMed] [Google Scholar]
- [120].Athyros VG, Boutari C, Stavropoulos K, Anagnostis P, Imprialos KP, Doumas M, Karagiannis A, Statins: An Under-Appreciated Asset for the Prevention and the Treatment of NAFLD or NASH and the Related Cardiovascular Risk, Curr Vasc Pharmacol 16(3) (2018) 246–253. [DOI] [PubMed] [Google Scholar]
- [121].Doumas M, Imprialos K, Dimakopoulou A, Stavropoulos K, Binas A, Athyros VG, The Role of Statins in the Management of Nonalcoholic Fatty Liver Disease, Curr Pharm Des 24(38) (2018) 4587–4592. [DOI] [PubMed] [Google Scholar]
- [122].Nascimbeni F, Pellegrini E, Lugari S, Mondelli A, Bursi S, Onfiani G, Carubbi F, Lonardo A, Statins and nonalcoholic fatty liver disease in the era of precision medicine: More friends than foes, Atherosclerosis 284 (2019) 66–74. [DOI] [PubMed] [Google Scholar]
- [123].Sigler MA, Congdon L, Edwards KL, An Evidence-Based Review of Statin Use in Patients With Nonalcoholic Fatty Liver Disease, Clin Med Insights Gastroenterol 11 (2018) 1179552218787502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Beaven SW, Wroblewski K, Wang J, Hong C, Bensinger S, Tsukamoto H, Tontonoz P, Liver X receptor signaling is a determinant of stellate cell activation and susceptibility to fibrotic liver disease, Gastroenterology 140(3) (2011) 1052–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Xing Y, Zhao T, Gao X, Wu Y, Liver X receptor alpha is essential for the capillarization of liver sinusoidal endothelial cells in liver injury, Scientific reports 6 (2016) 21309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Cao G, Liang Y, Broderick CL, Oldham BA, Beyer TP, Schmidt RJ, Zhang Y, Stayrook KR, Suen C, Otto KA, Miller AR, Dai J, Foxworthy P, Gao H, Ryan TP, Jiang XC, Burris TP, Eacho PI, Etgen GJ, Antidiabetic action of a liver x receptor agonist mediated by inhibition of hepatic gluconeogenesis, J Biol Chem 278(2) (2003) 1131–6. [DOI] [PubMed] [Google Scholar]
- [127].Commerford SR, Vargas L, Dorfman SE, Mitro N, Rocheford EC, Mak PA, Li X, Kennedy P, Mullarkey TL, Saez E, Dissection of the insulin-sensitizing effect of liver X receptor ligands, Mol Endocrinol 21(12) (2007) 3002–12. [DOI] [PubMed] [Google Scholar]
- [128].Grefhorst A, van Dijk TH, Hammer A, van der Sluijs FH, Havinga R, Havekes LM, Romijn JA, Groot PH, Reijngoud DJ, Kuipers F, Differential effects of pharmacological liver X receptor activation on hepatic and peripheral insulin sensitivity in lean and ob/ob mice, American journal of physiology. Endocrinology and metabolism 289(5) (2005) E829–38. [DOI] [PubMed] [Google Scholar]
- [129].Laffitte BA, Chao LC, Li J, Walczak R, Hummasti S, Joseph SB, Castrillo A, Wilpitz DC, Mangelsdorf DJ, Collins JL, Saez E, Tontonoz P, Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue, Proc Natl Acad Sci U S A 100(9) (2003) 5419–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Menegaut L, Thomas C, Jalil A, Julla JB, Magnani C, Ceroi A, Basmaciyan L, Dumont A, Le Goff W, Mathew MJ, Rebe C, Derangere V, Laubriet A, Crespy V, Pais de Barros JP, Steinmetz E, Venteclef N, Saas P, Lagrost L, Masson D, Interplay between Liver X Receptor and Hypoxia Inducible Factor 1alpha Potentiates Interleukin-1beta Production in Human Macrophages, Cell reports 31(7) (2020) 107665. [DOI] [PubMed] [Google Scholar]
- [131].Sohrabi Y, Sonntag GVH, Braun LC, Lagache SMM, Liebmann M, Klotz L, Godfrey R, Kahles F, Waltenberger J, Findeisen HM, LXR Activation Induces a Proinflammatory Trained Innate Immunity-Phenotype in Human Monocytes, Frontiers in immunology 11 (2020) 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Mitro N, Mak PA, Vargas L, Godio C, Hampton E, Molteni V, Kreusch A, Saez E, The nuclear receptor LXR is a glucose sensor, Nature 445(7124) (2007) 219–23. [DOI] [PubMed] [Google Scholar]
- [133].Mass E, Ballesteros I, Farlik M, Halbritter F, Gunther P, Crozet L, Jacome-Galarza CE, Handler K, Klughammer J, Kobayashi Y, Gomez-Perdiguero E, Schultze JL, Beyer M, Bock C, Geissmann F, Specification of tissue-resident macrophages during organogenesis, Science 353(6304) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Scott CL, T’Jonck W, Martens L, Todorov H, Sichien D, Soen B, Bonnardel J, De Prijck S, Vandamme N, Cannoodt R, Saelens W, Vanneste B, Toussaint W, De Bleser P, Takahashi N, Vandenabeele P, Henri S, Pridans C, Hume DA, Lambrecht BN, De Baetselier P, Milling SWF, Van Ginderachter JA, Malissen B, Berx G, Beschin A, Saeys Y, Guilliams M, The Transcription Factor ZEB2 Is Required to Maintain the Tissue-Specific Identities of Macrophages, Immunity 49(2) (2018) 312–325 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Bonnardel J, T’Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L, Vanneste B, De Prijck S, Nedospasov SA, Kremer A, Van Hamme E, Borghgraef P, Toussaint W, De Bleser P, Mannaerts I, Beschin A, van Grunsven LA, Lambrecht BN, Taghon T, Lippens S, Elewaut D, Saeys Y, Guilliams M, Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche, Immunity 51(4) (2019) 638–654 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Sakai M, Troutman TD, Seidman JS, Ouyang Z, Spann NJ, Abe Y, Ego KM, Bruni CM, Deng Z, Schlachetzki JCM, Nott A, Bennett H, Chang J, Vu BT, Pasillas MP, Link VM, Texari L, Heinz S, Thompson BM, McDonald JG, Geissmann F, Glass CK, Liver-Derived Signals Sequentially Reprogram Myeloid Enhancers to Initiate and Maintain Kupffer Cell Identity, Immunity 51(4) (2019) 655–670 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, Bruni CM, Ouyang Z, Li RZ, Sun X, Vu BT, Pasillas MP, Ego KM, Gosselin D, Link VM, Chong LW, Evans RM, Thompson BM, McDonald JG, Hosseini M, Witztum JL, Germain RN, Glass CK, Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis, Immunity 52(6) (2020) 1057–1074 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Trivedi P, Wang S, Friedman SL, The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells, Cell Metab (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, Okada Y, Kurihara C, Irie R, Yokoyama H, Shimamura K, Usui S, Ebinuma H, Saito H, Watanabe C, Komoto S, Kawaguchi A, Nagao S, Sugiyama K, Hokari R, Kanai T, Miura S, Hibi T, Free cholesterol accumulation in hepatic stellate cells: mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice, Hepatology 59(1) (2014) 154–69. [DOI] [PubMed] [Google Scholar]
- [140].Eslam M, Valenti L, Romeo S, Genetics and epigenetics of NAFLD and NASH: Clinical impact, J Hepatol 68(2) (2018) 268–279. [DOI] [PubMed] [Google Scholar]
- [141].Bruschi FV, Claudel T, Tardelli M, Starlinger P, Marra F, Trauner M, PNPLA3 I148M Variant Impairs Liver X Receptor Signaling and Cholesterol Homeostasis in Human Hepatic Stellate Cells, Hepatol Commun 3(9) (2019) 1191–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L, SIRT1 deacetylates and positively regulates the nuclear receptor LXR, Mol Cell 28(1) (2007) 91–106. [DOI] [PubMed] [Google Scholar]
- [143].Lonard DM, Nawaz Z, Smith CL, O’Malley BW, The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation, Mol Cell 5(6) (2000) 939–48. [DOI] [PubMed] [Google Scholar]
- [144].Osburn DL, Shao G, Seidel HM, Schulman IG, Ligand-dependent degradation of retinoid X receptors does not require transcriptional activity or coactivator interactions, Mol Cell Biol 21(15) (2001) 4909–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK, Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma, Mol Cell 25(1) (2007) 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Lee JH, Park SM, Kim OS, Lee CS, Woo JH, Park SJ, Joe EH, Jou I, Differential SUMOylation of LXRalpha and LXRbeta mediates transrepression of STAT1 inflammatory signaling in IFN-gamma-stimulated brain astrocytes, Mol Cell 35(6) (2009) 806–17. [DOI] [PubMed] [Google Scholar]
- [147].Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN, Gratton E, Parks J, Tontonoz P, LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling, eLife 4 (2015) e08009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Chen M, Bradley MN, Beaven SW, Tontonoz P, Phosphorylation of the liver X receptors, FEBS Lett 580(20) (2006) 4835–41. [DOI] [PubMed] [Google Scholar]
- [149].Torra IP, Ismaili N, Feig JE, Xu CF, Cavasotto C, Pancratov R, Rogatsky I, Neubert TA, Fisher EA, Garabedian MJ, Phosphorylation of liver X receptor alpha selectively regulates target gene expression in macrophages, Mol Cell Biol 28(8) (2008) 2626–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Yamamoto T, Shimano H, Inoue N, Nakagawa Y, Matsuzaka T, Takahashi A, Yahagi N, Sone H, Suzuki H, Toyoshima H, Yamada N, Protein kinase A suppresses sterol regulatory element-binding protein-1C expression via phosphorylation of liver X receptor in the liver, J Biol Chem 282(16) (2007) 11687–95. [DOI] [PubMed] [Google Scholar]
- [151].Voisin M, Gage MC, Becares N, Shrestha E, Fisher EA, Pineda-Torra I, Garabedian MJ, LXRalpha Phosphorylation in Cardiometabolic Disease: Insight From Mouse Models, Endocrinology 161(7) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Becares N, Gage MC, Voisin M, Shrestha E, Martin-Gutierrez L, Liang N, Louie R, Pourcet B, Pello OM, Luong TV, Goni S, Pichardo-Almarza C, Roberg-Larsen H, Diaz-Zuccarini V, Steffensen KR, O’Brien A, Garabedian MJ, Rombouts K, Treuter E, Pineda-Torra I, Impaired LXRalpha Phosphorylation Attenuates Progression of Fatty Liver Disease, Cell reports 26(4) (2019) 984–995 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Shrestha E, Hussein MA, Savas JN, Ouimet M, Barrett TJ, Leone S, Yates JR 3rd, Moore KJ, Fisher EA, Garabedian MJ, Poly(ADP-ribose) Polymerase 1 Represses Liver X Receptor-mediated ABCA1 Expression and Cholesterol Efflux in Macrophages, J Biol Chem 291(21) (2016) 11172–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Anthonisen EH, Berven L, Holm S, Nygard M, Nebb HI, Gronning-Wang LM, Nuclear receptor liver X receptor is O-GlcNAc-modified in response to glucose, J Biol Chem 285(3) (2010) 1607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]