

Abstract
Type 2 diabetes (T2D) is highly associated with obesity. However, the factors that drive the transition from excessive weight gain to glucose metabolism disruption are still uncertain and seem to revolve around systemic immune disorder. Mucosal-associated invariant T (MAIT) cells, which are innate-like T cells that recognize bacterial metabolites, have been reported to be altered in obese people and to lead to metabolic dysfunction during obesity. By studying the immunophenotypes of blood MAIT cells from a cross-sectional cohort of obese participants with/without T2D, we found an elevation in CD27-negative (CD27−) MAIT cells producing a high level of IL-17 under T2D obese conditions, which could be positively correlated with impaired glucose metabolism in obese people. We further explored microbial translocation caused by gut barrier dysfunction in obese people as a triggering factor of MAIT cell abnormalities. Specifically, accumulation of the bacterial strain Bacteroides ovatus in the peripheral blood drove IL-17-producing CD27− MAIT cell expansion and could be associated with T2D risk in obese individuals. Overall, these results suggest that an aberrant gut microbiota–immune axis in obese people may drive or exacerbate T2D. Importantly, CD27− MAIT cell subsets and Bacteroides ovatus could represent targets for novel interventional strategies. Our findings extend current knowledge regarding the clinical relevance of body mass index (BMI)-associated variation in circulating MAIT cells to reveal the role of these cells in obesity-related T2D progression and the underlying cellular mechanisms.
Keywords: type 2 diabetes, obesity, MAIT cells, Bacteroides ovatus, bacterial translocation
Subject terms: Predictive markers, Chronic inflammation
Introduction
Excess weight is an established risk factor for type 2 diabetes (T2D), and the global epidemic of obesity largely explains the dramatic increases in the incidence and prevalence of T2D over the past 30 years [1]. Even though most patients with diabetes are overweight or obese, not all obese individuals develop T2D [2]. Accumulating evidence points to chronic inflammation caused by immune dysfunction as the biological basis linking obesity to glucose metabolic dysregulation [3–5]. While extensive experimentation in animal models has demonstrated that both the innate and adaptive arms of the immune system play critical roles in the regulation of glucose metabolism, with the participation of multiple immune cell types [5], studies on the immune system in humans with obesity-associated metabolic syndromes are limited. Furthermore, many recent studies have focused on the identification of previously unrecognized immune cell subpopulations to determine their contributions in the context of obesity and related T2D [6]. However, an in-depth understanding of the triggering factors that drive the dysfunction of certain immune cell populations is largely lacking. Therefore, investigating the distinguishing features of immune cells that uniquely characterize nondiabetic versus diabetic obese individuals, as well as addressing the underlying initiating factors, is greatly needed not only to determine a comprehensive mechanism of how immune dysregulation in obesity relates to T2D but also to help advance more tailored therapies for these diseases [7].
In fact, early animal and clinical studies have already pointed out that gut-derived endotoxin may contribute to low-grade systemic inflammation in obesity states. Specific gut bacteria seem to serve as lipopolysaccharide (LPS) sources, and several reports have claimed a role in increased intestinal permeability in the pathogenesis of metabolic disorders [8]. These seminal papers pioneered the concept of bacterial translocation in metabolic diseases; however, this concept remained debated [9] until Massier et al. provided compelling evidence to demonstrate that translocation of enteric bacteria to other tissues is a true phenomenon, not simply spurious results [10, 11]. Given that a balanced relationship between the gut microbiota and the immune system is crucial to prevent excessive inflammation [12, 13], in obesity-related T2D, while gut microbiota dysbiosis and metabolic endotoxemia have become more evident, translocated microbial signatures, as well as how they shape immune responses, and, most importantly, the contribution to disease progression in humans remain to be addressed.
Mucosal-associated invariant T (MAIT) cells have attracted increasing attention over the last few years as a potent unconventional T-cell subset, not only because of their high abundance in the blood and tissue of humans [14] but also because of their well-known close relationship with the intestinal microbiome; therefore, they are sensitive to this inflammatory trigger that impacts health and disease [15]. Magalhaes et al. revealed that MAIT cells exhibit several defects in T2D and obese patients [16]. Recent results from the same group showed that altered MAIT cells promoted metabolic dysfunction during obesity by exacerbating inflammation leading to T2D in mice [17]. All these intriguing observations prompted us to hypothesize that enteric microbiome translocation that occurs with excessive weight gain may lead to MAIT cell abnormalities and impact glycemic control.
To test this hypothesis in humans, we collected peripheral blood samples from a cohort of age-, sex- and body mass index (BMI)-matched participants, including 31 nondiabetic overweight/obese (ObH) participants and 33 type 2 diabetic (ObT2D) counterparts. Twenty-four healthy normal-weight volunteers (HCs) were also included as controls. We first analyzed whether MAIT cells act differently in these metabolic conditions and then further deciphered the specific enteric bacterial strains that drive MAIT cell alterations in these contexts. Together, our results revealed that the well-known feature of obesity-associated gut barrier dysfunction could act as the triggering factor that facilitates “leakage” of specific bacterial components into the periphery to initiate MAIT cell abnormalities associated with the risk of T2D progression. The unique translocated bacterial strain and subsequent MAIT cell changes discovered in our study could be potential targets for the prediction of and/or intervention in obesity-related T2D.
Materials and methods
Study participants
The participants were recruited from our outpatient “3 + N” multidisciplinary weight loss program. Peripheral blood samples were collected from 24 healthy (HC group), 31 overweight/obese without diabetes (ObH group), and 33 overweight/obese with newly diagnosed T2D (new onset or within one year after onset, ObT2D group) adults. T2D was diagnosed according to the American Diabetes Association (ADA) criteria [18]. In participants without diabetes, normal glucose tolerance was defined as a fasting plasma glucose (FPG) concentration of less than 100 mg/dL (5.6 mmol/L), a 2-h oral glucose tolerance test (OGTT-2 h) plasma glucose concentration of less than 140 mg/dL (7.8 mmol/L), and an HbA1c of less than 5.7% (39 mmol/mol). Demographic and clinical characteristics, including age, sex, BMI, FPG, OGTT-2 h result, and HbA1c, were obtained (Table 1). HOMA-IR = fasting plasma insulin (FPI, mIU/L) × FPG (mmol/L)∕22.5. HOMA-β = (20 × FPI)∕(FPG − 3.5) [19]. Overweight and obesity were diagnosed according to the 2000 criteria of the World Health Organization [20]. The exclusion criteria were as follows: autoimmune disease, infectious disease, cancer, secondary obesity (including endocrine disorders, hypothalamic disorders, and some congenital conditions), the use of medications that cause clinically significant weight gain or loss in the last 6 months, severe renal impairment, and severe hepatic impairment. Written informed consent was obtained from the participants in the study. This study was approved by the Third Affiliated Hospital of Sun Yat-sen University Network Ethics Committee.
Table 1.
Clinical characteristics of study participants
| HC | ObH | ObT2D | |
|---|---|---|---|
| n | 24 | 31 | 33 |
| Age, years | 29.9 ± 6.1 | 29.6 ± 6.1 | 31.8.± 5.4 |
| Sex, M/F | 10/14 | 14/17 | 15/18 |
| BMI, kg/m2 | 20.7 ± 2.5 | 32.3 ± 4.4 | 31.4 ± 3.4 |
| FPG, mmol/L | 4.4 ± 0.2 | 5.0 ± 0.4 | 7.1 ± 2.7 |
| OGTT-2 h, mmol/L | - | 6.3 ± 1.1 | 12.1 ± 4.5 |
| HbA1c, mmol/mol | 31.2 ± 2.8 | 34.0 ± 4.6 | 63.4 ± 26.1 |
| HbA1c, % | 5.0 ± 0.3 | 5.3 ± 0.4 | 8.0 ± 2.4 |
| HOMA-IR | 1.7 ± 0.6 | 5.2 ± 2.2 | 5.3 ± 3.6 |
| HOMA-β | 194.3 ± 64.6 | 359.2 ± 211.0 | 152.1 ± 126.2 |
Data are presented as the mean ± SD
HC normal-weight healthy participants, ObH overweight/obese participants without T2D, ObT2D overweight/obese participants with T2D, T2D type 2 diabetes, M male; F female, BMI body mass index, FPG fasting plasma glucose, OGTT oral glucose tolerance test, HOMA-IR homeostatic model assessment for insulin resistance, HOMA-β homeostatic model assessment of β-cell function
PBMC preparation
PBMCs were isolated from 6-ml fresh venous blood samples collected from participants by density gradient centrifugation with Lymphoprep (Axis-Shield). PBMCs were resuspended in cryopreservation medium containing dimethyl sulfoxide (DMSO, Millipore Sigma) and fetal bovine serum (FBS, Thermo Fisher Scientific) and were then stored in liquid nitrogen until needed. Cryopreserved PBMCs were used for cell culture, immunostaining, and cell sorting experiments.
Flow cytometry analysis
PBMCs were incubated with a blue LIVE/DEAD fixable dead cell stain (Thermo Fisher Scientific) for 15 min in the dark at room temperature to distinguish live cells from dead cells. Then, the cells were subsequently washed and incubated with a human Fc receptor blocking solution (BioLegend) for 15 min on ice. The PBMCs were surface stained for 30 min in the dark on ice with the following anti-human monoclonal antibodies (mAbs): anti-CD3 (clone OKT3), anti-CD161 (clone HP-3G10), anti-TCR Vα7.2 (clone 3C10), anti-PD-1 (clone EH12.2H7), anti-CD69 (clone FN50), anti-CD27 (clone LG.3A10), anti-CD5 (clone L17F12), anti-CD4 (clone OKT4) and anti-CD8a (clone RPA-T8) from BioLegend.
For the detection of cytokines, PBMCs were treated with a 500× cell stimulation cocktail (plus protein transport inhibitors) containing 40.5 µM PMA, 670 µM ionomycin, 5.3 mM brefeldin A and 1 mM monensin (Thermo Fisher Scientific) for 6 h at 37 °C in RPMI 1640 medium supplemented with 10% FBS. After surface staining, the PBMCs were treated with a fixation/permeabilization kit (BD Biosciences) according to the manufacturer’s instructions and then stained with the following anti-human mAbs: anti-IL-17A (clone BL168), anti-TNF-α (clone Mab11), and anti-IFN-γ (clone 4S. B3) from BioLegend.
For intracellular transcription factor staining, a Foxp3/transcription factor staining buffer set (Thermo Fisher Scientific) was used to fix and permeabilize surface-stained PBMCs according to the manufacturer’s instructions, and then the following anti-human mAbs were used for staining: anti-T-bet (clone 4B10) from BioLegend and anti-RORγt (clone AFKJS-9) and anti-Nur77 (clone 12.14) from Thermo Fisher Scientific.
According to the number of isolated PBMCs, total and CD27-positive/negative MAIT cell staining was always performed, and when possible, phenotypic marker, cytokine production, or transcription factor staining was also performed. Data were acquired with a BD LSRFortessa (BD Biosciences) and analyzed using FlowJo v10.4 (BD).
Quantification of IL-17
Plasma concentrations of IL-17 were quantified using the Human IL-17A ELISA Kit (BioLegend) according to the manufacturer’s instructions.
RNA-seq
CD27-positive or CD27-negative MAIT cells were stained and sorted as CD27+ or CD27− CD3+ CD161+ TCR Vα7.2+ cells on a BD FACSAria III cell sorter (BD Biosciences). RNA from 10,000 sorted cells was used to prepare an RNA-seq library according to the single-cell Smart-seq2 protocol with modification [21]. In brief, RNA was purified after cell lysis with 0.2% Triton X-100, followed by reverse transcription as described previously [21]. cDNA was amplified using 12 PCR cycles. The size distribution of the PCR products was checked on an Agilent high-sensitivity DNA chip before library preparation using the TruePrep DNA Library Prep Kit V2 for Illumina (Vazyme) according to the manufacturer’s instructions, and the library was sequenced as paired-end 150-bp reads on an Illumina NovaSeq.
RNA-seq reads were quantified, and gene expression levels were calculated using Salmon v0.13.1 with the GRCh37 reference genome after raw reads were trimmed with Cutadapt v0.5.0. Subsequently, raw counts were processed with DESeq2 to identify differentially expressed genes. Gene set enrichment analysis (GSEA) was performed on preranked gene lists according to the DESeq2 fold change. We used 1,000 gene set permutations and the classic enrichment statistic. Gene sets were obtained from MSigDB (http://software.broadinstitute.org/gsea/msigdb) and the Gene Ontology (GO) database (GO: 0042775, GO: 0009617). Moreover, we used Reactome categories to perform gene enrichment analysis. Raw RNA-seq data are available from the Gene Expression Omnibus (GEO) under the accession number GSE187349. Differentially expressed genes were defined as genes with a|log2FoldChange(TPM)| ≥ 1.
ZO-1 measurement
The plasma ZO-1 concentration was determined using a ZO-1 ELISA Kit (CUSABIO) as previously described [22].
Metagenomic analysis
Metagenomic analyses were performed on blood samples from 26 participants, including 6 HC donors, 13 ObH participants, and 7 ObT2D participants. DNA was extracted using a MasterPure Complete DNA and RNA Purification kit (Lucigen) according to the manufacturer’s instructions. Sequencing libraries were prepared using the VAHTS Universal DNA Library Prep Kit for MGI (Vazyme). The sequencing libraries were sequenced to a depth of 10–50 million paired-end 2 × 150-bp reads per sample on an MGI SEQ 2000RS instrument. Negative blank controls were processed alongside the samples from DNA extraction through sequencing. All reads were first evaluated by FastQC for quality control for preprocessing. Raw sequencing data were quality filtered using Trimmomatic v0.36 and PRINSEQ v0.20.4. Low-quality reads, duplicated reads, potential adaptor sequences, and reads containing more than 10% ambiguous bases were removed. Human reads were removed from the subsequent analysis using KneadData v0.6.1. For taxonomic profiling, the filtered nonhuman reads were mapped against our custom microbial genome collection using Kraken 2 v2.0.9. Our genome collection included complete bacterial, archaeal, and viral genomes downloaded from the RefSeq database and complete fungal genomes downloaded from the GenBank database. The results of taxonomic classification were filtered using a confidence score of 0.20. Decontamination was performed at the species level. All species detected in blank negative controls were removed.
B. ovatus DNA extraction and quantification
Blood samples were collected from participants into EDTA-coated sterile blood collection tubes, and plasma was collected in sterile Eppendorf tubes. DNA was extracted from plasma samples with the ZymoBIOMICS DNA Miniprep Kit (Zymo Research) according to the manufacturer’s instructions. All the operating steps were performed in a sterile biosafety cabinet to avoid contamination. Quantification of bacterial DNA was performed by real-time PCR according to the protocol by Mishra et al. [23]. The primer pair used for B. ovatus DNA was 967F: 5′-CAGGCTCAAGGAAGCACAAG-3′ and 1100R: 5′-CAAAGAACAGCAGGCCATGT-3′, located on the Endo-1,4-beta-xylanase Z precursor gene [24]. The analysis was performed on a QuantStudio 5 Real-Time PCR system (Thermo Fisher Scientific). Ct values for each reaction were obtained and used for data analysis. Given the low amount of bacterial DNA in the blood, the Ct value of distilled water was used as a negative control to rule out potential contamination from reagents, handling, and the environment. The absolute number of bacteria measured through real-time PCR was determined using a standard curve generated with genomic B. ovatus DNA.
Coculture of PBMCs and B. ovatus or E. coli
B. ovatus ATCC 8483 was obtained from the Guangdong Microbial Culture Collection Center (GDMCC). E. coli DH5α was obtained from Vazyme. PBMCs were cocultured with heat-killed bacteria at a ratio of 1 to 100 for 1 day at 37 °C in a tissue culture incubator with 5% CO2. Brefeldin A Solution (1000×, BioLegend) was added 6 h before harvest. In these cultures, cells were stimulated in the presence of blocking mAb MR1 (clone 26.5, BioLegend) or its isotype control (mouse IgG2a, κ, BioLegend). The PBMCs were then harvested, and MAIT cells were analyzed by flow cytometry analysis as described above.
Statistical analyses
Statistical analyses were performed using Prism 9 (GraphPad). Specific tests for determining statistical significance are indicated in the corresponding figure legends. P < 0.05 was considered statistically significant.
Results
Frequency and functional alterations of circulating MAIT cells in overweight/obese patients without or with T2D
We first analyzed the frequency of MAIT cells in peripheral blood mononuclear cells (PBMCs) isolated from peripheral blood samples collected from the study cohort. MAIT cells were identified as CD3+ TCR Vα7.2+ CD161+ (Fig. 1A). Consistent with the findings of another research group [16], the circulating MAIT cell frequency was lower in overweight/obese (ObH and ObT2D) participants than in HCs in general. Importantly, when comparing the two overweight/obese groups, we found that the MAIT cell frequency was significantly lower in ObT2D participants than in their BMI-matched ObH counterparts (Fig. 1A, B). This further decline in the MAIT cell frequency in ObT2D patients did not seem to be due to BMI because even though we found a negative correlation between BMI and the MAIT cell frequency in normal weight and overweight/obese participants (Fig. 1C), no significant correlation could be found if just focusing on the overweight/obese participants (Fig. 1D). Additionally, although the MAIT cell frequency further decreased in T2D patients, we did not find a predictive potential of the MAIT cell frequency alone for the glucose metabolism status in overweight/obese patients, as indicated by correlation analyses with hemoglobin A1c (HbA1c), homeostatic model assessment for insulin resistance (HOMA-IR), and homeostatic model assessment of β-cell function (HOMA-β) (Supplementary Fig. 1A–C). In addition to the cell frequency change, we then investigated activation marker expression among the groups. Figure 1E and F shows no difference in the expression of the early activation marker CD69 on MAIT cells among the groups; however, the expression of the activation/exhaustion marker PD-1 was increased in the ObT2D group, suggesting that MAIT cells were overactivated in ObT2D patients. Therefore, we next investigated the function of MAIT cells by evaluating cytokine production. After simulating cells with phorbol 12-myristate 13-acetate (PMA) and ionomycin, in general, MAIT cells from overweight/obese participants produced more inflammatory cytokines, including IL-17, TNF-α, and IFN-γ (Fig. 1G, H), than those from healthy participants. Here, higher amounts of IL-17 and TNF-α production could be seen in ObT2D participants than in ObH participants, while IFN-γ production showed no change between these two groups. Of note, IL-17 exhibited an almost threefold increase in ObT2D participants compared to HCs and a 1.5-fold increase compared to ObH participants (Fig. 1H). We also tested IL-17 production by other cell types in addition to MAIT cells in PBMCs. Compared to the HC group, both the ObH and ObT2D groups produced higher levels of IL-17 in total PBMCs. However, there was no further increase in IL-17 production in the ObT2D group (Supplementary Fig. 1D). Among the IL-17-producing PBMCs, MAIT cells accounted for ~15%, while the other 75% were non-MAIT T cells (Supplementary Fig. 1E, F). Non-T cells only contributed a small portion (~10%) of IL-17 (Supplementary Fig. 1G). To validate these findings, the blood IL-17 content was detected by ELISA in our study cohort. Unfortunately, IL-17 from most of the plasma samples was undetectable, which was consistent with the literature [25]. Of note, two samples were above the limit of detection (0.8 pg/mL), and those two were from ObT2D patients (Supplementary Fig. 1H).
Fig. 1.

Frequency and functional alterations of circulating MAIT cells in overweight/obese patients without or with T2D. PBMCs were collected from healthy donors (HC, n = 24) and overweight/obese adults without T2D (ObH, n = 31) or with T2D (ObT2D, n = 33) and were analyzed by flow cytometry. A, B Frequencies of circulating MAIT cells (CD3+ CD161+ TCR Vα7.2+). Correlations between the frequency of circulating MAIT cells and BMI of adults in the HC, ObH, and ObT2D groups (C) or the ObH and ObT2D groups (D). Frequencies of CD69+ (E) and PD-1+ (F) MAIT cells among the total MAIT cells in the HC (n = 23 and 19 for (E) and (F), respectively), ObH (n = 28 and 26 for E and F, respectively) and ObT2D (n = 28 and 26 for (E) and (F), respectively) groups. G, H Flow cytometry analysis of IL-17+, TNF-α+ and IFN-γ+ MAIT cells among the total MAIT cells after PMA and ionomycin stimulation in the HC (n = 22), ObH (n = 29), and ObT2D (n = 27) groups. Each symbol represents a single individual (B–D, F and H), and error bars indicate the mean with the SD (B, F and H). ns P > 0.05, *P < 0.05, **P < 0.01 and ***P < 0.001; two-tailed unpaired Student’s t test (B, F and H). Pearson’s correlation test (C, D).
Overall, from HCs to ObT2D patients, we found a progressively decreased frequency of circulating MAIT cells but an increased frequency of cells producing inflammatory cytokines, especially IL-17, which suggests that the alteration in MAIT cells might contribute to obesity-related T2D progression.
The CD27-negative MAIT cell subset is associated with T2D progression in overweight/obese patients
Since we observed an elevation in IL-17 in ObT2D patients, we next wanted to determine which MAIT cell compartment contributes to IL-17 production. It has been reported that in type 1 diabetes patients, the CD27− MAIT cell subset appears to produce more IL-17 than the CD27+ subset [26]. Therefore, we compared the capacity for inflammatory cytokine production between CD27-negative and CD27-positive subsets among the groups. We first found that IL-17 produced by these two MAIT cell populations was not different under normal weight healthy or nondiabetic overweight/obese conditions. However, CD27− MAIT cells produced significantly higher levels of IL-17 than the CD27+ subset in diabetic overweight/obese conditions (Fig. 2A, B). In contrast with IL-17, TNF-α and IFN-γ were mainly produced by the CD27+ MAIT cell subset in all three groups (Supplementary Fig. 2A, B), which suggested that the CD27− MAIT cell subset might be unique for its IL-17 production under the obesity-related T2D status. To reveal the underlying mechanism of this unique pattern of cytokine production, we then evaluated the expression of the transcription factors RORγt and T-bet in MAIT cells and found that the expression ratio of RORγt to T-bet was significantly higher in the ObT2D group than in the HC or ObH group, indicating that MAIT cells were skewed toward an IL-17-producing but not an IFN-γ-producing phenotype under the obesity-related T2D status (Fig. 2C). Furthermore, under the normal weight healthy state, no difference in the ratio was found between the CD27− and CD27+ MAIT cell subsets. However, the ratio was significantly higher in the CD27− MAIT cell population under overweight or obese conditions (Fig. 2D), which is consistent with the previous finding that the CD27− MAIT cell subset has a greater capacity for IL-17 production under obesity-related T2D conditions. Notably, the expression of both RORγt and T-bet in MAIT cells was decreased in overweight or obese patients compared to HCs (Supplementary Fig. 2C, D). In addition, we also found that the CD27− MAIT cell subset expressed higher levels of PD-1, while the CD27+ subset expressed higher levels of CD69 among the different participant groups (Fig. 2E, F), which potentially indicated that CD27+ MAIT cells represented an early activation subset, while CD27− MAIT cells were probably in an overactivated to exhausted state.
Fig. 2.

The CD27-negative MAIT cell subset is associated with T2D progression in overweight/obese patients. A, B Flow cytometry analysis of IL-17+ MAIT cells among CD27− or CD27+ MAIT cells after PMA and ionomycin stimulation in the HC (n = 22), ObH (n = 29) and ObT2D (n = 27) groups or without stimulation (medium). Mean fluorescence intensity (MFI) ratio of RORγt to T-bet in total MAIT cells (C) and CD27− and CD27+ MAIT cell subsets (D) in the HC (n = 19), ObH (n = 20) and ObT2D (n = 21) groups. Frequencies of CD69+ (E) and PD-1+ (F) MAIT cells among the CD27− or CD27+ MAIT cell subsets in the HC (n = 23 and 19 for E and F, respectively), ObH (n = 28 and 26 for (E) and (F), respectively) and ObT2D (n = 28 and 26 for (E) and (F), respectively) groups. G, H Frequency of the CD27− MAIT cell subset in the HC (n = 24), ObH (n = 31) and ObT2D (n = 33) groups. Frequencies of CD27− T (I) and CD27− CD8+ T (J) cells in the HC (n = 24 and 24 for (I) and (J), respectively), ObH (n = 31 and 28 for (I) and (J), respectively) and ObT2D (n = 33 and 30 for (I) and (J), respectively) groups. Correlations between the frequency of circulating CD27− MAIT cells and the HbA1c level (K), HOMA-IR score (L) and HOMA-β score (M) in adults in the ObH (n = 30, 27, and 27 for K, L, and M, respectively) and ObT2D (n = 32, 30, and 30 for (K), (L), and (M), respectively) groups. Each symbol represents a single individual (B–F, H–M), and error bars indicate the mean with the SD (B–F and H–J). ns P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001; two-tailed paired Student’s t test (B, D–F), two-tailed unpaired Student’s t test (C, H–J). Spearman’s correlation test (K–M).
To determine the association of IL-17-producing CD27− MAIT cells with obesity-related T2D progression, we compared the CD27− MAIT cell proportion within our study cohort. As shown in Fig. 2G and H, a twofold increase in the CD27− MAIT cell frequency was observed in the ObT2D group compared to the HC group. Interestingly, there was no difference in the CD27− MAIT cell proportion between the ObH and HC groups, while ObT2D patients had a significantly higher frequency of CD27− MAIT cells than ObH patients. Notably, the increase in the CD27− proportion was unique to MAIT cells, since we did not find a frequency change in the CD27− population of either total T cells or CD4 and CD8 T cells (Fig. 2I, J; Supplementary Fig. 2E). Furthermore, to identify whether the increased frequency of CD27− MAIT cells could serve as a biomarker of impaired glucose metabolism in overweight/obese patients, we analyzed its correlations with HbA1c, HOMA-IR, and HOMA-β. As shown in Fig. 2K, the CD27− MAIT cell frequency was positively correlated with HbA1c in overweight/obese participants. Moreover, although the CD27− MAIT cell frequency did not correlate with insulin resistance status, as suggested by HOMA-IR, the increase predicted poor islet β-cell function (Fig. 2L, M).
In conclusion, we found that the CD27− MAIT cell subset harbored a greater proportion of IL-17 producers than the CD27+ subset under obesity-related T2D conditions, which was associated with the risk of T2D development in overweight/obese participants and might serve as a biomarker of glucose metabolism status in overweight/obese patients.
CD27-negative MAIT cells exhibit an increased mitochondrial mass and decreased response to bacterial transcriptional patterns
To identify the difference in the functional properties between CD27-negative and CD27-positive MAIT subsets under obesity-related T2D conditions, we isolated each population from the PBMCs of three diabetic obese (ObT2D) participants and performed RNA sequencing (RNA-seq) analysis. Principal component analysis revealed two distinct clusters, namely, CD27− MAITs (blue) and CD27+ MAITs (red), suggesting that these two subsets of MAIT cells established completely different transcriptional programs in ObT2D participants (Fig. 3A). Next, we examined the gene signature of CD27-negative and CD27-positive MAIT cells and found that except for a small fraction of genes that were upregulated, most genes were downregulated in CD27− MAIT cells compared to the CD27+ MAIT compartments (Fig. 3B). Of note, among the most differentially expressed genes (Fig. 3C), the top enriched Reactome pathways in upregulated genes of CD27− MAIT cells were linked to mitochondrial ATP synthesis (Fig. 3D, E). Interestingly, it has been recently reported that the increase in IL-17-producing MAIT cells in obesity could be due to dysfunctional mitochondria [27], and we found that CD27− MAIT cells had a significantly higher number of mitochondria than CD27+ MAIT cells in ObT2D participants by performing MitoTracker staining (Fig. 3F, G), suggesting increased mitochondrial mass. Moreover, we noticed that some downregulated genes in CD27− MAIT cells were involved in the response to bacteria, such as S100A8, S100A9, S100A12, and LYZ (Fig. 3H, I), which suggests an attenuation of the bacterial response under obesity-related T2D conditions. We also compared the mRNA levels of IL17A, TNF, IFNG, CD69, and PDCD1 and the ratio of RORC∕TBX21 between the CD27− and CD27+ MAIT subsets and found the same trend as that for the protein level detected by flow cytometry (Supplementary Fig. 3A–F).
Fig. 3.

CD27-negative MAIT cells exhibit an increased mitochondrial mass and decreased response to bacterial transcriptional patterns. Circulating CD27− and CD27+ MAIT cells were isolated from 3 obese adults with T2D by fluorescence-activated cell sorting (FACS) and were subjected to RNA-seq analysis. A Principal component analysis (PCA) showed that the gene expression pattern of CD27− MAIT cells was different from that of CD27+ MAIT cells. B Heatmap revealing the clustering of differentially expressed genes between CD27− and CD27+ MAIT cells. C Volcano plot showing the differentially expressed genes (adjusted P value of RNA-seq < 0.05) between CD27− and CD27+ MAIT cells. Significantly differentially expressed genes (DEGs, adjusted P value < 0.05) are highlighted in red (upregulated, log2FoldChange ≥ 1) and blue (downregulated, log2FoldChange ≤ −1). Selected genes related to mitochondria and the response to bacteria are indicated. D Reactome pathways enriched among the genes upregulated in CD27− (blue) and CD27+ MAIT cells (red). E Bar plots showing the corresponding expression profiles of genes related to mitochondrial function (GO: 0042775) between CD27− and CD27+ MAIT cells. F, G MitoTracker Green staining of circulating CD27− and CD27+ MAIT cells from the ObT2D group was quantified by flow cytometry. The gray line in F shows the fluorescence minus one (FMO) control staining. The numbers in F represent the MFI for each group. H Gene set enrichment analysis (GSEA) showing the response to bacterial signatures depleted in CD27− MAIT cells. I Heatmap depicting the corresponding expression profiles of genes involved in the response to bacteria (GO: 0009617) between CD27− and CD27+ MAIT cells. Each symbol represents a single individual, and error bars indicate the mean with the SD (G). **P < 0.01; two-tailed paired Student’s t test (G)
Overall, in parallel with the previously observed expansion of CD27− MAIT cell subsets and its association with T2D progression in obese participants, these data further addressed the distinct gene signature of these cells and indicated that imbalanced cell-bacteria interactions might contribute to T2D progression in overweight/obese participants.
Unique microbial signature in the peripheral blood of obese patients with T2D
Next, we investigated the trigger for MAIT cell alteration under obesity-related T2D status. Given that MAIT cells have been reported to be uniquely activated by microbial-derived vitamin B metabolites, activation or increased differentiation among MAIT cells should be microbially based. Furthermore, gut microbiome translocation was observed in many metabolic diseases due to gut barrier dysfunction, and we hypothesized that translocated enteric bacteria might contribute to T2D by activating MAIT cells. Thus, we first examined intestinal barrier function among the three groups by determining the plasma levels of Tight junction protein 1 (ZO-1). As shown in Fig. 4A, we observed an increasing trend of the ZO-1 concentration in both the ObH and ObT2D groups compared to HCs, indicating an increase in gut permeability that could facilitate enteric commensal microbiota leakage into the bloodstream when people gain weight. However, a further increase in ZO-1 levels in obese participants with T2D compared to their nondiabetic compartments did not reach significance. To further assess the existence of microorganisms in the peripheral blood, we performed metagenomics analysis with 26 peripheral blood samples (6 HC, 13 ObH and 7 ObT2D). As expected, we found 37 microorganisms that were detected in at least one blood sample. We next analyzed the composition of the blood bacterial community within the HC, ObH, and ObT2D groups. As shown in Fig. 4B and C, both the phylum and species of bacteria in the blood were much more diverse in overweight/obese participants than in normal-weight healthy participants. Of note, the Bacteroidetes phylum of bacteria, which was barely detected in the blood of healthy participants (0.2%), was present in the overweight/obese participants and occupied a small segment of the bacterial composition in the ObH group (6.8%) and, importantly, dominated in the ObT2D group (41.3%). In detail, 11 bacterial species were found in blood specimens from the HC group, while 12 and 19 bacterial species were found in the ObH and ObT2D groups, respectively (Fig. 4C). To reveal the unique microbial signature in peripheral blood, we compared the bacterial species from each group (Fig. 4D) and found 16 species of bacteria that were specifically present in the blood of ObT2D participants (Fig. 4E). Within this group, Bacteroides ovatus (B. ovatus), which belongs to the Bacteroidetes phylum, was the most prevalent bacterial species present in three out of seven blood specimens from ObT2D participants.
Fig. 4.

Unique microbial signature in peripheral blood of obese patients with T2D. A Enzyme-linked immunosorbent assay (ELISA) was used to analyze the tight junction protein ZO-1 level in plasma from the HC (n = 10), ObH (n = 10) and ObT2D (n = 11) groups. B–E Comparative metagenomic analysis was performed for peripheral blood specimens from 6 HC donors and 13 ObH and 7 ObT2D participants. B Relative abundance of the major phyla of microbes in the metagenomes of blood samples from the HC, ObH, and ObT2D groups. C Number of unique or common microbial species among the HC, ObH, and ObT2D groups. D Relative abundance of the microbial species in the metagenomes of blood samples from the HC, ObH, and ObT2D groups. Species within the same color column belong to the corresponding phylum in (B). E Relative abundance of the unique microbial species in the ObT2D group. F, G Comparisons of Bacteroides ovatus Endo-1,4-beta-xylanase Z precursor (xynZ) gene detection using qPCR. Cycle threshold (Ct) values (F) and B. ovatus number per 10 ml of plasma determined by absolute quantification using diluted B. ovatus as a standard curve (G) are shown for each sample from the HC (n = 30, including six newly recruited lean healthy donors), ObH (n = 26) and ObT2D (n = 30) groups. Water served as a negative control (n = 8). Each symbol represents a single individual, and error bars indicate the mean with SD (A, F–G). ns P > 0.05, *P < 0.05; two-tailed unpaired Student’s t test.
To further validate the prevalence of B. ovatus in the blood of overweight/obese participants under diabetic conditions, we performed real-time PCR analysis of the plasma collected from participants of our previous study cohort. We first compared the cycle threshold (Ct) value of B. ovatus among the three groups, and distilled water was used as a blank control to exclude possible contamination, which is a common way to assess low abundance microorganisms in the tissue [23]. As shown in Fig. 4F, the Ct value of B. ovatus was compared among 30 HC (including 6 newly recruited normal weight healthy blood donors), 26 ObH and 30 ObT2D participants. A significantly lower Ct value that corresponded to a higher content of DNA being amplified could be seen in all three groups compared to the water control, indicating that the presence of B. ovatus was real and not a false-positive result. Among these, the Ct value showed no difference between HC and ObH conditions, while a significant reduction could be seen in the ObT2D group, which confirmed an association between the accumulation of B. ovatus and obesity-related T2D development. In addition, the absolute number of B. ovatus present in the blood plasma was estimated by using B. ovatus strain ATCC8483 as a standard template. As shown in Fig. 4G, the average number of B. ovatus in both the HC and ObH groups was approximately 9 bacteria per 10 ml of plasma, while in the ObT2D group, this number increased to 14 bacteria per 10 ml of plasma, which confirmed an association between the increasing abundance of B. ovatus in peripheral blood and obesity-related T2D progression.
Altogether, we discovered that Bacteroides spp. preferentially inhabited the blood of overweight/obese participants, especially those who had T2D, in parallel with defects in gut barrier function due to excessive weight. Of note, B. ovatus, which belongs to the phylum Bacteroidetes, was highly abundant in the blood of diabetic obese participants, which indicated the potential deleterious role of these commensal bacterial translocations in participants from the overweight/obese transition to T2D.
Immune response of circulating MAIT cells to Bacteroides ovatus stimulation in vitro reproduces the alteration of MAIT cells in diabetic obese people in vivo
To further assess whether translocated commensal B. ovatus could contribute to obesity-related T2D progression by triggering IL-17-producing MAIT cells, we performed an in vitro bacterial stimulation assay by coculturing PBMCs with heat-killed B. ovatus to mimic the immune response of MAIT cells in vivo (Fig. 5A). After one day of coculture, we found an elevation in the CD27− MAIT cell subset, which showed an increase in the average frequency from 35% in the unstimulated medium control group to 45% in the post-B. ovatus stimulation group (Fig. 5B, C). In addition, both IL-17 and TNF-α production by MAIT cells was dramatically increased after B. ovatus stimulation, while IFN-γ production did not change (Fig. 5D–I). In parallel, the expression of CD69 and PD-1 was upregulated after coculturing with B. ovatus (Fig. 5L–O). Consistent with the in vivo results, we found that the CD27− MAIT cell subset was the main producer of IL-17 and TNF-α, indicating that the expansion of the blood CD27− MAIT cell subset was stimulated by the translocated commensal bacterium B. ovatus (Fig. 5J, K). Interestingly, this alteration of MAIT cells seems to be specific to B. ovatus stimulation, since the induction of the CD27− MAIT subset and the production of IL-17 and TNF-α were significantly lower when stimulated with E. coli, which is also a gram-negative bacteria but was not detectable in the patient plasma by metagenomic analyses (Supplementary Fig. 4A–D). To verify that the above cytokine production by MAIT cells occurred through TCR activation, we evaluated the TCR signal strength markers Nur77 and CD5 on MAIT cells in the different experimental settings. As shown in Fig. 5P–S, significant increases in both Nur77 and CD5 expression could be seen after B. ovatus stimulation compared to medium control treatment. Moreover, MR1 blockade significantly reduced the increase in Nur77 or CD5 expression upon stimulation (Supplementary Fig. 4E, F), which excluded the possibility that this phenotype could be due to a general effect of bacterial endotoxin stimulation.
Fig. 5.

The immune response of circulating MAIT cells to Bacteroides ovatus stimulation in vitro reproduces the alteration of MAIT cells in diabetic obese patients in vivo. A A schematic representation showing the B. ovatus stimulation protocol. PBMCs from the ObT2D group were stimulated with heat-killed B. ovatus (100 bacteria per cell) or were cultured in growth medium (medium) for 1 day in 96-well plates. B–K Flow cytometry analysis showing the frequencies of CD27− MAIT cells (B, C) and IL-17- (D, E), TNF-α- (F, G), and IFN-γ-producing (H, I) MAIT cells after B. ovatus stimulation compared to no stimulation. Frequencies of IL-17+ (J) and TNF-α+ (K) MAIT cells among CD27− or CD27+ MAIT cell subsets without or with bacterial stimulation (n = 13 for C, E, G, I and K) Flow cytometry analysis showing the expression of CD69 (L, M), PD-1 (N, O), Nur77 (P, Q) and CD5 (R, S) on MAIT cells after B. ovatus stimulation compared to no stimulation (n = 9 for M, O, Q and S). The numbers in L, N, P, and R represent the MFI for each group. Each symbol represents a single individual, and error bars indicate the mean with the SD (C, E, G, I, K, M, O, Q, and S). ns P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001; two-tailed paired Student’s t test.
In conclusion, we confirmed that B. ovatus could activate human circulating MAIT cells through TCR signaling and induce IL-17-producing CD27− MAIT cell expansion in vitro, which indicated that the increased abundance of B. ovatus in the peripheral blood of ObT2D participants could be the triggering factor driving MAIT cell abnormalities associated with T2D development in overweight/obese people.
Discussion
One consensus report from an international conference jointly sponsored by the Endocrine Society, the ADA and the European Association for the Study of Diabetes in 2011 addressed a major question related to linking obesity to T2D: “Why don’t all patients with obesity develop T2D?”, which provides a future direction for research to benefit prevention, interventions, and overall patient care [28]. Here, we identified the factors uniquely associated with T2D versus nondiabetic overweight/obesity by analyzing a study cohort of 88 participants. Our study addressed distinguishing features suggestive of an aberrant gut microbiota–immune system–glucose metabolism axis in ObT2D patients compared to ObH individuals (Fig. 6).
Fig. 6.
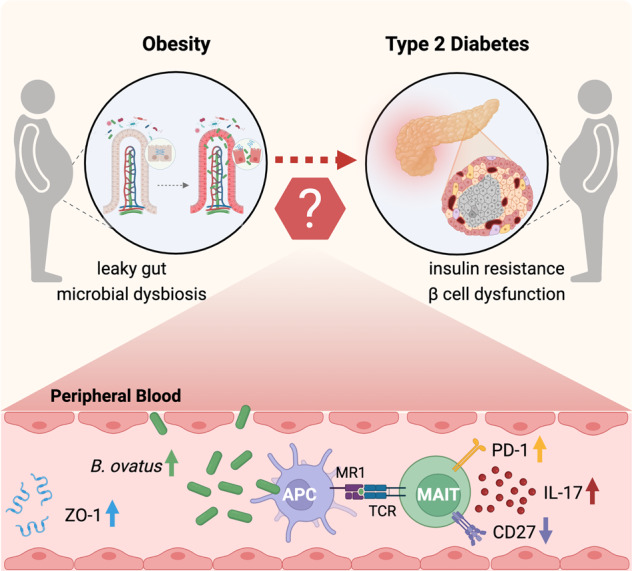
Aberrant gut microbiota–immune system–glucose metabolism axis is associated with the risk of T2D in overweight/obese people. Disturbed gut barrier function caused by excessive weight leads to translocation of the gut microbiota, which facilitates leakage of a specific enteric bacterial strain, namely, Bacteroides ovatus, into the periphery and serves as a triggering factor of IL-17-producing CD27− MAIT cell expansion, which is positively correlated with impaired glucose metabolism.
The influence of the gut microbiome on metabolic health is a rapidly emerging area of research [29, 30]. Many studies have provided compelling evidence related to the composition and diversity of the gut microbiota, as well as gut barrier function to prevent leakage of bacteria and bacterial products across the intestine, to support the roles of the microbiome in obesity and T2D [31]. However, the exact mechanisms that link specific variations in the gut microbiota with the development of obesity and related T2D are still obscure [32]. In our study, we tried to unravel this missing connection from an immune perspective. Recent advances in understanding the pathophysiology of obesity and related T2D have highlighted the involvement of the immune system [16], while altered gut microbial ecosystems have been identified to be associated with immune disorders in many disease conditions [33].
From an evolutionary point of view, the immune system and metabolism are connected and cross-regulated by common proinflammatory molecules [34, 35]. As it has been well accepted that T2D is not only a metabolic disease but also an inflammatory disorder, IL-17, which is mainly produced by Th17 cells, has been reported to play a crucial role in the creation of inflammation in both obese individuals [36, 37] and animal models [38, 39]. Importantly, obese people who have insulin resistance showed elevated blood levels of IL-17 [37], which means that IL-17 can serve as a marker for inflammation that accompanies obesity. Additionally, treatment with anti-IL-17 neutralizing antibodies elevated serum adiponectin concentrations, reduced serum levels of TNF-α, which is a well-known cytokine that inhibits insulin signaling, and enhanced adipocyte differentiation markers [40]. These data indicate that IL-17 could enhance the induction of classical proinflammatory cytokines and chemokines (such as TNF-α, IL-6, IL-1β, IFN-γ, and CCL20) mechanistically through the JAK1, JAK2, PI3K, and NF-κB pathways [41–43], thus contributing to the induction of insulin resistance and finally leading to the development of T2D.
Considering the dysfunction of the gut microbiota in T2D patients and the tight relationship between IL-17/Th17 and insulin sensitivity, increasing evidence suggests that certain gut microbes, such as segmented filamentous bacteria, can induce the development and activation of IL-17-producing T cells [44–46]. Following the gut microbiota–immune system axis hypothesis, MAIT cells, which are predominant innate-like lymphocytes in humans and are characterized by their recognition of microbe-derived intermediates of vitamin B2 (riboflavin) synthesis [47], are particularly important in this rationale. In the obesity and T2D field, Agnès Lehuen’s and Andrew E. Hogan’s groups have already performed a series of excellent studies characterizing MAIT cell phenotypic changes in humans and have determined their deleterious role in the development of metabolic dysfunction in animal models [16, 17, 27]. In addition to the consistent observations regarding the decreased frequency but increased cytokine production, especially IL-17 production by peripheral MAIT cells in ObT2D participants in our cohort, the significance of our study is that we found that the proportion of CD27− MAIT cells correlating with an increased level of HbA1c and decreased HOMA-β score in overweight/obese participants were sharply elevated in those with T2D, thus indicating the immune biomarker potential of T2D risk in obese people. CD27 is known as a T-cell early activation marker [48–50] and could be used as a marker, together with CD161, to define MAIT cell development in the human thymus: stage 1 (CD161− CD27−), stage 2 (CD161− CD27+) and stage 3 (CD161+ CD27pos–lo) [51]. Here, we found that the CD27− MAIT cell subset harbored a greater proportion of IL-17 producers than the CD27+ subset, specifically in the ObT2D group of participants, while this difference was absent in the other groups. Interestingly, similar induction of CD27− MAIT cells was also reported in type 1 diabetes [15, 26], asthma [52], and multiple myeloma [53] patients, which indicates that the expansion of this unique subset may represent a general feature of chronic inflammatory diseases caused by an unexplored mechanism, thereby prompting us to further investigate the gene signature and molecular pathway by comparing CD27-negative and CD27-positive MAIT cells under T2D conditions through an RNA-seq approach. We obtained two hints from analyzing the most differentially expressed genes in CD27− MAIT cells. The upregulated mitochondrial ATP synthesis genes, as well as the increased mitochondrial mass, indicated that the greater IL-17 production by CD27− MAIT cells might be due to previously reported mitochondrial dysfunction in obesity [27]. Another suggestive clue was that bacterial response-related genes were downregulated in CD27− MAIT cells. Given that this subset of cells was elevated sharply in the blood of ObT2D participants, all these results led us to explore whether an imbalance between bacteria and MAIT cells occurred in the periphery under obesity and T2D conditions.
Metabolic inflammation has long been believed to be “sterile” [54]. However, an increasing body of evidence generated in obese mice implicates the gut microbiota as the endotoxin source, while antibiotic treatment reduces systemic LPS levels and improves glucose tolerance [55–58]. These preclinical findings shed light on the extraintestinal and systemic translocation of the microbiome in metabolic diseases [54]. Consistent with the animal model, the first conceptual evidence for bacterial translocation in human T2D was reported in 2011 when Amar et al. showed that the 16S rDNA gene content in the blood was associated with future T2D risk [9, 59]. Recently, multiple tissues, including the blood, adipose tissue, and liver, have been further discovered to have tissue-specific microbial signatures in human obesity and T2D conditions, which provides compelling evidence for enteric bacterial translocation across the intestinal barrier to reach the circulation or even distant organs in metabolic disorders [10, 11, 60, 61]. In our study, we discovered that a phylum of gut commensal bacteria, namely, Bacteroidetes, was barely detected in the blood of HCs but started emerging in the peripheral blood when people put on excessive weight and were dominant in ObT2D participants by metagenomic analysis. Although Bacteroidetes has been reported to be associated with metabolic diseases in several studies, the distinct mechanism by which these bacteria contribute to obesity and T2D development is still uncertain and even controversial [62–67]. Furthermore, most related studies have relied primarily on analysis of stool microbiota, while our findings show for the first time evidence that Bacteroides spp., particularly B. ovatus, which was found to be the most prevalent strain of Bacteroides spp. in the blood of T2D participants, could enter the circulation by penetrating through the impaired intestinal barrier under obesity and T2D conditions. Interestingly, Bacteroides spp. has also been reported to be one of the top bacterial phyla that synthesize vitamin B2 in the body [68, 69]. Given the biological features of MAIT cells and their contribution to obesity-related T2D progression discussed above, a clear connection between B. ovatus and abnormalities in the altered MAIT cell phenotype was revealed by performing an in vitro coculture study in which we found that heat-killed B. ovatus could stimulate an elevation in the CD27− MAIT cell subset in parallel with an increase in IL-17 production. Interestingly, we did not observe the same phenotype of MAIT cells upon E. coli stimulation, which could partially explain why ObT2D patients showed increased IL-17 production by MAIT cells. Moreover, impaired intestinal barrier function has been well defined in metabolic disorders, which might facilitate gut microbiome “leak” into the circulation. In this case, a larger amount of bacteria could be another reason for the higher IL-17 production by MAIT cells in ObT2D patients.
Considering this in vitro assay, one possible speculation is that the elevation of the CD27− MAIT cell subset that we observed in T2D patients might be due to the downregulation of CD27 expression by the CD27+ MAIT compartment, since there was no extra source of fresh CD27− MAIT cells in this assay, and 24-h stimulation is too short for the proliferation of CD27− MAIT cells, as it normally takes 6 days of stimulation to induce MAIT cell proliferation [70].
In addition to all of the findings above, we did observe some “inconsistent” but interesting phenotypes. For example, MAIT cells from diabetic patients produced higher levels of proinflammatory cytokines and had high PD-1 expression, which indicated “impaired” TCR activation. In fact, PD-1 is known as a marker of both T-cell activation and exhaustion. It has been reported that PD-1 can be induced and can limit the overactivation upon T-cell stimulation through a negative feedback mechanism. Furthermore, sustained expression of PD-1 on chronically activated T cells contributes to T-cell exhaustion [71]. Here, we believe that MAIT cells from diabetic patients were in the transition state from overactivation to exhaustion, thus exhibiting both high levels of PD-1 expression and cytokine production, which was identical to the overactivation but not exhaustion phenotype of iNKT cells in our previous paper [72] in which elevated PD-1 expression is actually a protective feedback mechanism to prevent activation-induced cell death. Another interesting observation was that in ObT2D patients, we observed an elevation of CD27− MAIT cells that on the one hand produced more IL-17 but on the other hand downregulated antibacterial immune response-related genes. It is known that IL-17 plays dual roles in protection from microbes to induce proinflammatory diseases. The gut microbiota is a plausible regulator of IL-17 production and functions [73]. During pathogenic exogenous bacterial invasion, IL-17 can be induced and can promote innate inflammation through the induction of chemokines, cytokines, and antimicrobial peptides to protect the host by limiting these pathogenic microbes [74]. However, when facing endogenous bacteria, commensal-specific IL-17 is involved in reinforcing barrier function [75], while dysbiosis of the microbiota is associated with pathological expression of IL-17 and deterioration of diseases [76–79]. Therefore, in our study, we speculated that blood MAIT cells were chronically activated by Bacteroides ovatus in obesity-related T2D conditions, which adopted the strategy of the latter scenario and resulted in elevated IL-17 production by CD27− MAIT cells, thus playing a pathological role in T2D exacerbation.
Overall, we conclude that an imbalanced gut microbiota–immune system–glucose metabolism axis is associated with T2D risk based on a study of the interindividual differences among obese people. Specifically, our data suggest that the gut commensal strain B. ovatus accumulating in the peripheral blood triggers expansion of circulating CD27− MAIT cells that may exacerbate the risk of obesity-related T2D. These findings improve the understanding of obesity heterogeneity and could help advance the development of effective and cost-effective preventive and treatment strategies for obesity-related T2D. However, a longitudinal study needs to be performed in the future to validate the interventional potential of these findings beyond the current cross-sectional analysis.
Regarding to the limitations of this study are as follows: First, we found increased CD27− MAIT cells in the peripheral blood of T2D patients. The unsolved question was where these increased cells originated from and what they target. Parabiosis experiments with mice indicate that MAIT cells are long-term resident cells in the spleen, lymph nodes, lung, liver, and thymus [80, 81]. However, liver and oral mucosa MAIT cells also express a tissue-resident profile in humans [81, 82]. Moreover, Violet et al. showed that MAIT cells in the lymph migrated from tissues and were capable of exiting tissues to recirculate based on a shared TCR repertoire between thoracic duct lymph and blood samples [83]. To date, it is still uncertain to what extent tissue MAIT cells are permanent residents and whether they can leave tissues. Second, we can only speculate that gut commensals are the origin of the detected bacterial DNA in the blood without direct evidence. According to the metagenomics analysis, the detected bacterial species belonged to the phyla usually found in the gastrointestinal tract, and the bacterial composition was reminiscent of the gut microbiome [84]. Moreover, under stringent experimental conditions focusing on sterile handling, bioinformatic control for contaminants, and strict criteria for cohort enrollment (excluding people who have infectious diseases), these blood bacteria should be endogenous and most likely from the gut, since bacterial translocation across the intestinal barrier in metabolic disorders has been well proven by multiple compelling pieces of evidence. However, we could not exclude other sources of bacterial DNA, such as the oral cavity and skin. We did see an elevation of blood ZO-1 in ObT2D patients compared to HC controls, which indicated leakage of gut bacteria due to increased intestinal permeability. However, this was indirect evidence, and direct evidence determining the bacterial source requires a microbial tracing method utilizing fluorescence-labeled bacteria in vivo [85], which we were unable to perform on a human cohort. Another limitation of the present study is that we suggested the presence of bacterial DNA in the blood but could not distinguish whether it was live and dead bacteria. Further investigation using fluorescence in situ hybridization (FISH) and bacterial culture will be helpful to address this point. According to Massier et al., live translocated bacteria have been visualized in adipose tissue from obese and T2D patients through a catalyzed reporter deposition fluorescence in situ hybridization (CARD-FISH) approach [10]. Considering the possible bacterial migration route and the whole body connecting with the bloodstream, it is conceivable that the bacterial DNA in the blood is from live bacteria, since the gut bacteria might take the circulatory route to reach other tissues. However, the other possibility that the bacterial DNA is dead cell fragments could also be true, since according to the principle of the gut ecosystem, translocated commensal bacteria would be arrested and cleared by the immune system [86] in a healthy state, whereas in disease conditions, it is unclear whether the increased level of translocated commensals in the blood is simply a matter of the impaired gut barrier or a result of dysfunctional immune scavenger cells that could not clear these microscopic invaders and remove the dead cell fragments. Therefore, although we elucidated a “gut microbiota–immune system–glucose metabolism” mechanism, it would be interesting to test the possibility of an “immune system–gut microbiota–glucose metabolism” axis in future studies.
Supplementary information
Acknowledgements
This study was funded by the National Key R&D Program of China (2017YFA0105803), the National Natural Science Foundation of China (32000621 and 81770826), the Key Area R&D Program of Guangdong Province (2019B020227003), the Science and Technology Plan Project of Guangzhou City (202102010338 and 202007040003), the 5010 Clinical Research Projects of Sun Yat-sen University (2015015) and the Dengfeng Plan High-level Hospital Construction Opening Project of Foshan Fourth People’s Hospital (FSSYKF-2020009).
Author contributions
YL, YY, JW, PC, LW, YC, and YL designed the research; YL, YY, JW, PC, ML, XT, YT, YW, FZ, XW, QL, YN, and YL performed the research; XW, QL, TC, XP, XH, YZ, GS, and WWC contributed new concepts/reagents/research materials/analytical tools; YL, YY, JW, PC, LW, YC, and YL analyzed the data; YL, YY, JW, WWC, LW, YC, and YL wrote the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Yue Li, Yi Yang, Jin Wang, Peihong Cai.
Contributor Information
Lai Wei, Email: weil9@mail.sysu.edu.cn.
Yanming Chen, Email: chyanm@mail.sysu.edu.cn.
Yan Lu, Email: luyan36@mail.sysu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41423-022-00871-4.
References
- 1.Pillon NJ, Loos RJF, Marshall SM, Zierath JR. Metabolic consequences of obesity and type 2 diabetes: Balancing genes and environment for personalized care. Cell. 2021;184:1530–44. doi: 10.1016/j.cell.2021.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bramante CT, Lee CJ, Gudzune KA. Treatment of obesity in patients with diabetes. Diabetes Spectr. 2017;30:237–43. doi: 10.2337/ds17-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wondmkun YT. Obesity, insulin resistance, and type 2 diabetes: associations and therapeutic implications. Diabetes Metab Syndr Obes. 2020;13:3611–6. doi: 10.2147/DMSO.S275898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ying W, Lee YS, Dong Y, Seidman JS, Yang M, Isaac R, et al. Expansion of ISLET-resident macrophages leads to inflammation affecting beta cell proliferation and function in obesity. Cell Metab. 2019;29:457–74 e455. doi: 10.1016/j.cmet.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLaughlin T, Ackerman SE, Shen L, Engleman E. Role of innate and adaptive immunity in obesity-associated metabolic disease. J Clin Investig. 2017;127:5–13. doi: 10.1172/JCI88876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu J, Zhao J, Meng H, Zhang X. Adipose tissue-resident immune cells in obesity and type 2 diabetes. Front Immunol. 2019;10:1173. doi: 10.3389/fimmu.2019.01173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith GI, Mittendorfer B, Klein S. Metabolically healthy obesity: facts and fantasies. J Clin Investig. 2019;129:3978–89. doi: 10.1172/JCI129186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jayashree B, Bibin YS, Prabhu D, Shanthirani CS, Gokulakrishnan K, Lakshmi BS, et al. Increased circulatory levels of lipopolysaccharide (LPS) and zonulin signify novel biomarkers of proinflammation in patients with type 2 diabetes. Mol Cell Biochem. 2014;388:203–10. doi: 10.1007/s11010-013-1911-4. [DOI] [PubMed] [Google Scholar]
- 9.Jensen BA, Marette A. Microbial translocation in type 2 diabetes: when bacterial invaders overcome host defence in human obesity. Gut. 2020;69:1724–6. doi: 10.1136/gutjnl-2020-321288. [DOI] [PubMed] [Google Scholar]
- 10.Massier L, Chakaroun R, Tabei S, Crane A, Didt KD, Fallmann J, et al. Adipose tissue-derived bacteria are associated with inflammation in obesity and type 2 diabetes. Gut. 2020;69:1796–806. doi: 10.1136/gutjnl-2019-320118. [DOI] [PubMed] [Google Scholar]
- 11.Anhê FF, Jensen B, Varin TV, Servant F, Van Blerk S, Richard D, et al. Type 2 diabetes influences bacterial tissue compartmentalisation in human obesity. Nat Metab. 2020;2:233–42. doi: 10.1038/s42255-020-0178-9. [DOI] [PubMed] [Google Scholar]
- 12.Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–5. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 13.Seki D, Mayer M, Hausmann B, Pjevac P, Giordano V, Goeral K, et al. Aberrant gut-microbiota-immune-brain axis development in premature neonates with brain damage. Cell Host Microbe. 2021;29:1558–72 e1556. doi: 10.1016/j.chom.2021.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Provine NM, Klenerman P. MAIT cells in health and disease. Annu Rev Immunol. 2020;38:203–28. doi: 10.1146/annurev-immunol-080719-015428. [DOI] [PubMed] [Google Scholar]
- 15.Gazali AM, Schroderus AM, Näntö-Salonen K, Rintamäki R, Pihlajamäki J, Knip M, et al. Mucosal-associated invariant T cell alterations during the development of human type 1 diabetes. Diabetologia. 2020;63:2396–409. doi: 10.1007/s00125-020-05257-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magalhaes I, Pingris K, Poitou C, Bessoles S, Venteclef N, Kiaf B, et al. Mucosal-associated invariant T cell alterations in obese and type 2 diabetic patients. J Clin Investig. 2015;125:1752–62. doi: 10.1172/JCI78941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toubal A, Kiaf B, Beaudoin L, Cagninacci L, Rhimi M, Fruchet B, et al. Mucosal-associated invariant T cells promote inflammation and intestinal dysbiosis leading to metabolic dysfunction during obesity. Nat Commun. 2020;11:3755. doi: 10.1038/s41467-020-17307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.American Diabetes, A. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes-2021. Diabetes Care. 2021;44:S15–S33. doi: 10.2337/dc21-S002. [DOI] [PubMed] [Google Scholar]
- 19.Wallace TM, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care. 2004;27:1487–95. doi: 10.2337/diacare.27.6.1487. [DOI] [PubMed] [Google Scholar]
- 20.Obesity: preventing and managing the global epidemic. Report of a WHO consultation. World Health Organ Tech Rep Ser. 2000;894, i–xii, 1–253. [PubMed]
- 21.Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9:171–81. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 22.Ram AK, Pottakat B, Vairappan B. Increased systemic zonula occludens 1 associated with inflammation and independent biomarker in patients with hepatocellular carcinoma. BMC Cancer. 2018;18:572. doi: 10.1186/s12885-018-4484-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mishra A, Lai GC, Yao LJ, Aung TT, Shental N, Rotter-Maskowitz A, et al. Microbial exposure during early human development primes fetal immune cells. Cell. 2021;184:3394–409 e3320. doi: 10.1016/j.cell.2021.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kollarcikova M, Faldynova M, Matiasovicova J, Jahodarova E, Kubasova T, Z Seidlerova Z, et al. Different bacteroides species colonise human and chicken intestinal tract. Microorganisms. 2020;8:1483. [DOI] [PMC free article] [PubMed]
- 25.Bergin R, Kinlen D, Kedia-Mehta N, Hayes E, Cassidy FC, Cody D, et al. Mucosal-associated invariant T cells are associated with insulin resistance in childhood obesity, and disrupt insulin signalling via IL-17. Diabetologia. 2022. 10.1007/s00125-022-05682-w. Epub ahead of print. [DOI] [PMC free article] [PubMed]
- 26.Harms RZ, Lorenzo KM, Corley KP, Cabrera MS, Sarvetnick NE. Altered CD161 bright CD8+ mucosal associated invariant T (MAIT)-like cell dynamics and increased differentiation states among juvenile type 1 diabetics. PLoS One. 2015;10:e0117335. doi: 10.1371/journal.pone.0117335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brien AO, Kedia-Mehta N, Tobin L, Veerapen N, Besra GS, Shea DO, et al. Targeting mitochondrial dysfunction in MAIT cells limits IL-17 production in obesity. Cell Mol Immunol. 2020;17:1193–5. doi: 10.1038/s41423-020-0375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eckel RH, Kahn SE, Ferrannini E, Goldfine AB, Nathan DM, Schwartz MW, et al. Obesity and type 2 diabetes: what can be unified and what needs to be individualized? J Clin Endocrinol Metab. 2011;96:1654–63. doi: 10.1210/jc.2011-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agus A, Clement K, Sokol H. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut. 2021;70:1174–82. doi: 10.1136/gutjnl-2020-323071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol. 2021;19:55–71. doi: 10.1038/s41579-020-0433-9. [DOI] [PubMed] [Google Scholar]
- 31.Allin KH, Tremaroli V, Caesar R, Jensen B, Damgaard M, Bahl MI, et al. Aberrant intestinal microbiota in individuals with prediabetes. Diabetologia. 2018;61:810–20. doi: 10.1007/s00125-018-4550-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boulange CL, Neves AL, Chilloux J, Nicholson JK, Dumas ME. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016;8:42. doi: 10.1186/s13073-016-0303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, et al. Linking the human gut microbiome to inflammatory cytokine production capacity. Cell. 2016;167:1897. doi: 10.1016/j.cell.2016.11.046. [DOI] [PubMed] [Google Scholar]
- 34.von Stebut E, Boehncke WH, Ghoreschi K, Gori T, Kaya Z, Thaci D, et al. IL-17A in psoriasis and beyond: cardiovascular and metabolic implications. Front Immunol. 2019;10:3096. doi: 10.3389/fimmu.2019.03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 36.Zapata-Gonzalez F, Auguet T, Aragonès G, Guiu-Jurado E, Berlanga A, Martinez S, et al. Interleukin-17A gene expression in morbidly obese women. Int J Mol Sci. 2015;16:17469–81. doi: 10.3390/ijms160817469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sumarac-Dumanovic M, Stevanovic D, Ljubic A, Jorga J, Simic M, Stamenkovic-Pejkovic D, et al. Increased activity of interleukin-23/interleukin-17 proinflammatory axis in obese women. Int J Obes. 2009;33:151–6. doi: 10.1038/ijo.2008.216. [DOI] [PubMed] [Google Scholar]
- 38.Cavallari JF, Denou E, Foley KP, Khan WI, Schertzer JD. Different Th17 immunity in gut, liver, and adipose tissues during obesity: the role of diet, genetics, and microbes. Gut Microbes. 2016;7:82–89. doi: 10.1080/19490976.2015.1127481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Endo Y, Asou HK, Matsugae N, Hirahara K, Shinoda K, Tumes DJ, et al. Obesity drives Th17 cell differentiation by inducing the lipid metabolic kinase, ACC1. Cell Rep. 2015;12:1042–55. doi: 10.1016/j.celrep.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 40.Ohshima K, Mogi M, Jing F, Iwanami J, Tsukuda K, Min LJ, et al. Roles of interleukin 17 in angiotensin II type 1 receptor-mediated insulin resistance. Hypertension. 2012;59:493–9. doi: 10.1161/HYPERTENSIONAHA.111.183178. [DOI] [PubMed] [Google Scholar]
- 41.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14:585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shinjo T, Iwashita M, Yamashita A, Sano T, Tsuruta M, Matsunaga H, et al. IL-17A synergistically enhances TNFalpha-induced IL-6 and CCL20 production in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2016;477:241–6. doi: 10.1016/j.bbrc.2016.06.049. [DOI] [PubMed] [Google Scholar]
- 43.Zepp J, Wu L, Li X. IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease. Trends Immunol. 2011;32:232–9. doi: 10.1016/j.it.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Yin Y, Chen X, Zhao Y, Wu Y, Li Y, et al. Induction of intestinal Th17 cells by flagellins from segmented filamentous bacteria. Front Immunol. 2019;10:2750. doi: 10.3389/fimmu.2019.02750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sano T, Kageyama T, Fang V, Kedmi R, Martinez CS, Talbot J, et al. Redundant cytokine requirement for intestinal microbiota-induced Th17 cell differentiation in draining lymph nodes. Cell Rep. 2021;36:109766. doi: 10.1016/j.celrep.2021.109766. [DOI] [PubMed] [Google Scholar]
- 46.Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, et al. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. 2014;40:594–607. doi: 10.1016/j.immuni.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Constantinides MG, Link VM, Tamoutounour S, Wong AC, Perez-Chaparro PJ, Han S-J, et al. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science 2019;366:eaax6624. [DOI] [PMC free article] [PubMed]
- 48.Hendriks J, Gravestein LA, Tesselaar K, van Lier RAW, Schumacher TNM, Borst J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol. 2000;1:433–40. doi: 10.1038/80877. [DOI] [PubMed] [Google Scholar]
- 49.Ahlers JD, Belyakov IM. Memories that last forever: strategies for optimizing vaccine T-cell memory. Blood. 2010;115:1678–89. doi: 10.1182/blood-2009-06-227546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guerra-Maupome M, Palmer MV, Waters WR, McGill JL. Characterization of gammadelta T cell effector/memory subsets based on CD27 and CD45R expression in response to Mycobacterium bovis. Infect Immunohorizons. 2019;3:208–18. doi: 10.4049/immunohorizons.1900032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koay HF, Gherardin NA, Enders A, Loh L, Mackay LK, Almeida CF, et al. A three-stage intrathymic development pathway for the mucosal-associated invariant T cell lineage. Nat Immunol. 2016;17:1300–11. doi: 10.1038/ni.3565. [DOI] [PubMed] [Google Scholar]
- 52.Lezmi G, Abou-Taam R, Garcelon N, Dietrich C, Machavoine F, Delacourt C, et al. Evidence for a MAIT-17-high phenotype in children with severe asthma. J Allergy Clin Immunol. 2019;144:1714–6 e1716. doi: 10.1016/j.jaci.2019.08.003. [DOI] [PubMed] [Google Scholar]
- 53.Gherardin NA, Loh L, Admojo L, Davenport AJ, Richardson K, Rogers A, et al. Enumeration, functional responses and cytotoxic capacity of MAIT cells in newly diagnosed and relapsed multiple myeloma. Sci Rep. 2018;8:4159. doi: 10.1038/s41598-018-22130-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tilg H, Zmora N, Adolph TE, Elinav E. The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol. 2020;20:40–54. doi: 10.1038/s41577-019-0198-4. [DOI] [PubMed] [Google Scholar]
- 55.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–72. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 56.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–81. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 57.Rohr MW, Narasimhulu CA, Rudeski-Rohr TA, Parthasarathy S. Negative effects of a high-fat diet on intestinal permeability: a review. Adv Nutr. 2020;11:77–91. doi: 10.1093/advances/nmz061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scheithauer TPM, Rampanelli E, Nieuwdorp M, Vallance BA, Verchere CB, van Raalte DH, et al. Gut microbiota as a trigger for metabolic inflammation in obesity and type 2 diabetes. Front Immunol. 2020;11:571731. doi: 10.3389/fimmu.2020.571731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amar J, Serino M, Lange C, Chabo C, Iacovoni J, Mondot S, et al. Involvement of tissue bacteria in the onset of diabetes in humans: evidence for a concept. Diabetologia. 2011;54:3055–61. doi: 10.1007/s00125-011-2329-8. [DOI] [PubMed] [Google Scholar]
- 60.Burcelin R, Serino M, Chabo C, Garidou L, Pomié C, Courtney M, et al. Metagenome and metabolism: the tissue microbiota hypothesis. Diabetes Obes Metab. 2013;15(Suppl 3):61–70. doi: 10.1111/dom.12157. [DOI] [PubMed] [Google Scholar]
- 61.Cani PD, Van Hul M. Microbial signatures in metabolic tissues: a novel paradigm for obesity and diabetes? Nat Metab. 2020;2:211–2. doi: 10.1038/s42255-020-0182-0. [DOI] [PubMed] [Google Scholar]
- 62.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 63.Zhou W, Sailani MR, Contrepois K, Zhou Y, Ahadi S, Leopold SR, et al. Longitudinal multi-omics of host-microbe dynamics in prediabetes. Nature. 2019;569:663–71. doi: 10.1038/s41586-019-1236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–4. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102:11070–5. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schwiertz A, Taras D, Schäfer K, Beijer S, Bos NA, Donus C, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity. 2010;18:190–5. doi: 10.1038/oby.2009.167. [DOI] [PubMed] [Google Scholar]
- 67.Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Magnusdottir S, Ravcheev D, de Crecy-Lagard V, Thiele I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front Genet. 2015;6:148. doi: 10.3389/fgene.2015.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yoshii K, Hosomi K, Sawane K, Kunisawa J. Metabolism of dietary and microbial vitamin B family in the regulation of host immunity. Front Nutr. 2019;6:48. doi: 10.3389/fnut.2019.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kurioka A, Ussher JE, Cosgrove C, Clough C, Fergusson JR, Smith K, et al. MAIT cells are licensed through granzyme exchange to kill bacterially sensitized targets. Mucosal Immunol. 2015;8:429–40. doi: 10.1038/mi.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. 2018;18:153–67. doi: 10.1038/nri.2017.108. [DOI] [PubMed] [Google Scholar]
- 72.Lu Y, Zhong MC, Qian J, Calderon V, Cruz Tleugabulova M, Mallevaey T, et al. SLAM receptors foster iNKT cell development by reducing TCR signal strength after positive selection. Nat Immunol. 2019;20:447–57. doi: 10.1038/s41590-019-0334-0. [DOI] [PubMed] [Google Scholar]
- 73.Douzandeh-Mobarrez B, Kariminik A. Gut microbiota and IL-17A: physiological and pathological responses. Probiotics Antimicrob Proteins. 2019;11:1–10. doi: 10.1007/s12602-017-9329-z. [DOI] [PubMed] [Google Scholar]
- 74.McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. 2019;50:892–906. doi: 10.1016/j.immuni.2019.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature. 2015;520:104–8. doi: 10.1038/nature14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang C, Chen J, Wang J, Zhou H, Lu Y, Lou L, et al. Dysbiosis of intestinal microbiota and decreased antimicrobial peptide level in paneth cells during hypertriglyceridemia-related acute necrotizing pancreatitis in rats. Front Microbiol. 2017;8:776. doi: 10.3389/fmicb.2017.00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.López P, de Paz B, Rodríguez-Carrio J, Hevia A, Sánchez B, Margolles A, et al. Th17 responses and natural IgM antibodies are related to gut microbiota composition in systemic lupus erythematosus patients. Sci Rep. 2016;6:24072. doi: 10.1038/srep24072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heimesaat MM, Grundmann U, Alutis ME, Fischer A, Göbel UB, Bereswill S. The IL-23/IL-22/IL-18 axis in murine Campylobacter jejuni infection. Gut Pathog. 2016;8:21. doi: 10.1186/s13099-016-0106-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, et al. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016;8:43. doi: 10.1186/s13073-016-0299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Legoux F, Bellet D, Daviaud C, El Morr Y, Darbois A, Niort K, et al. Microbial metabolites control the thymic development of mucosal-associated invariant T cells. Science. 2019;366:494–9. doi: 10.1126/science.aaw2719. [DOI] [PubMed] [Google Scholar]
- 81.Salou M, Legoux F, Gilet J, Darbois A, du Halgouet A, Alonso R, et al. A common transcriptomic program acquired in the thymus defines tissue residency of MAIT and NKT subsets. J Exp Med. 2019;216:133–51. doi: 10.1084/jem.20181483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sobkowiak MJ, Davanian H, Heymann R, Gibbs A, Emgård J, Dias J, et al. Tissue-resident MAIT cell populations in human oral mucosa exhibit an activated profile and produce IL-17. Eur J Immunol. 2019;49:133–43. doi: 10.1002/eji.201847759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Violet V, Buggert M, Slichter CK, Berkson JD, Mair F, Addison MM, et al. Human MAIT cells exit peripheral tissues and recirculate via lymph in steady state conditions. JCI Insight. 2018;3:e98487. [DOI] [PMC free article] [PubMed]
- 84.Sankar SA, Lagier JC, Pontarotti P, Raoult D, Fournier PE. The human gut microbiome, a taxonomic conundrum. Syst Appl Microbiol. 2015;38:276–86. doi: 10.1016/j.syapm.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 85.Fu WL, Xiao GX, Yue XL, Hua C, Lei MP. Tracing method study of bacterial translocation in vivo. World J Gastroenterol. 2000;6:153–5. doi: 10.3748/wjg.v6.i1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Natividad JM, Verdu EF. Modulation of intestinal barrier by intestinal microbiota: pathological and therapeutic implications. Pharm Res. 2013;69:42–51. doi: 10.1016/j.phrs.2012.10.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.