

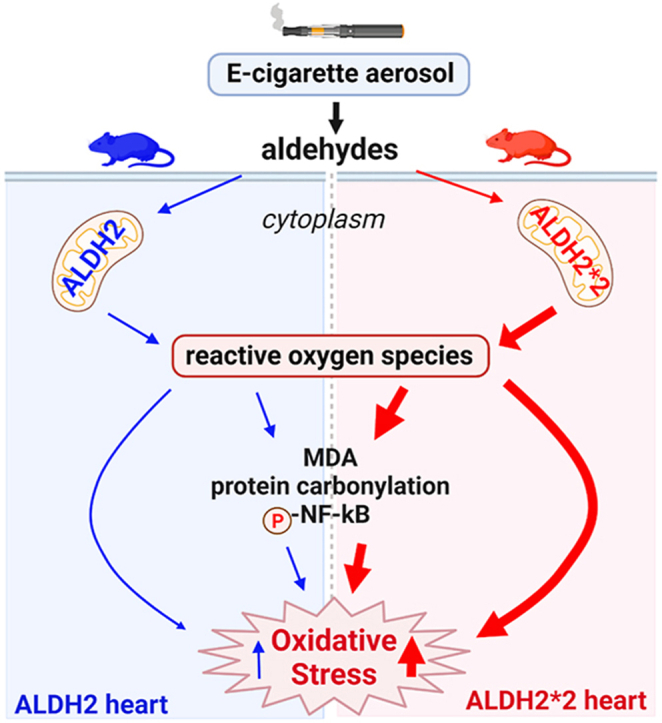
Abstract
Background
E-cigarette aerosol containing aldehydes, including acetaldehyde, are metabolized by the enzyme aldehyde dehydrogenase 2 (ALDH2). However, little is known how aldehyde exposure from e-cigarettes, when coupled with an inactivating ALDH2 genetic variant, ALDH2*2 (present in 8% of the world population), affects cardiovascular oxidative stress.
Objectives
The study was to determine how e-cigarette aerosol exposure, coupled with genetics, impacts cardiovascular oxidative stress in wild type ALDH2 and ALDH2*2 knock-in mice.
Methods
Using selective ion flow mass spectrometry, we determined e-cigarette aerosol contains acetaldehyde levels 10-fold higher than formaldehyde or acrolein. Based on this finding, we tested how isolated ALDH2*2 primary cardiomyocytes respond to acetaldehyde and how intact ALDH2*2 knock-in rodents instrumented with telemeters respond physiologically and at the molecular level to 10 days of e-cigarette aerosol exposure relative to wild type ALDH2 rodents.
Results
For ALDH2*2 isolated cardiomyocytes, acetaldehyde (1 μM) caused a 4-fold greater peak calcium influx, 2-fold increase in ROS production and 2-fold increase in 4-HNE-induced protein adducts relative to wild-type ALDH2 cardiomyocytes. The heart rate in ALDH2*2 mice increased ∼200 beats/min, while, heart rate in ALDH2 mice increased ∼150 beats/min after 10 days of e-cigarette exposure, relative to air-exposed mice. E-cigarette aerosol exposure triggered ∼1.3 to 2-fold higher level of protein carbonylation, lipid peroxidation, and phosphorylation of NF-κB for both strains of mice, with this response exacerbated for ALDH2*2 mice.
Conclusions
Our findings indicate people carrying an ALDH2*2 genetic variant may be more susceptible to increases in cardiovascular oxidative stress from e-cigarette aerosol exposure.
Keywords: E-Cigarette aerosol, Aldehydes, ALDH2*2 variant, Cardiomyocytes, Oxidative stress
Graphical abstract
Highlights
-
•
~540 million people have a genetic variant in aldehyde dehydrogenase 2 (ALDH2*2) that limits aldehyde metabolism.
-
•
Little is known how e-cigarette exposure, when coupled with the ALDH2*2 variant, impacts cardiovascular oxidative stress.
-
•
ALDH2*2 cardiomyocytes and rodents vs. wild type have higher oxidative stress levels after aldehyde or e-cigarette exposure.
-
•
People with an ALDH2*2 variant may be more susceptible to cardiovascular oxidative stress from e-cigarette exposure.
1. Introduction
Electronic cigarettes (e-cigarettes) are popular among teens and young adults [1]. E-cigarette aerosol, although containing less chemicals compared to conventional tobacco cigarette smoke, contains aldehydes produced from the combustion of the primary chemical components of e-liquid propylene glycol (PG) and vegetable glycerin (VG) [2]. Importantly, aldehyde exposure is estimated to contribute to over 92% of the cardiopulmonary disease risk from tobacco smoke [3] and recognized by the Institute of Medicine as one of the most significant cardiovascular toxins within tobacco smoke [4]. However, whether the aldehydes within e-cigarette aerosol are detrimental to the cardiovascular system has not been extensively studied.
Generally, aldehydes are deleterious to the cell and can form aldehyde-induced protein adducts leading to cellular dysfunction and cell death [5]. These aldehydes are metabolized by the enzyme, aldehyde dehydrogenase 2 (ALDH2). In particular, ALDH2 has a Km for acetaldehyde that is 900-fold lower relative to cytosolic ALDH1 with ALDH2 selective and specific for acetaldehyde metabolism [6]. In the cardiovascular system, ALDH2 activation is important in limiting mast cell renin release and mitigating cellular injury from ischemic events by protecting the heart from aldehyde-induced injury [7,8].
However, an inactivating ALDH2 genetic variant, known as ALDH2*2, severely limits aldehyde metabolism. The ALDH2*2 genetic variant is present in approximately 30% of people of East Asian descent (540 million people or 8% of the world population) [9]. This genetic variant leads to an increased cardiomyocyte cell death during ischemia-reperfusion injury [[10], [11], [12], [13]], increased risk of coronary artery disease [14,15], as well as alcohol-induced heart disease [16]. However, it is unknown how aldehydes present within e-cigarette aerosol impact the cardiovascular system. This is particularly important since Asian-American users of e-cigarettes, particularly among young adults [17,18], have increased in recent years, from 2% reported in 2013 to 10% in 2018 [19]. For these reasons, the aim for this study is to determine whether in ALDH2*2 variant mice e-cigarette exposure exacerbates cardiovascular oxidative stress relative to wild type ALDH2 mice.
2. Materials and methods
2.1. Quantification of aldehydes and nicotine in e-cigarette aerosol
Blu, Halo, and JUUL e-cigarettes were tested for aldehyde and nicotine aerosol content. Aerosols were generated by drawing from an e-cigarette using a Masterflex peristaltic pump and collected into Tedlar Bags (Zefon International). Aldehydes and nicotine levels were quantified in real time using selective ion flow tube mass spectrometry (SIFT-MS, Syft).
2.2. ALDH2 and ALDH2*2 mouse expression and activity
The ALDH2*2 knock-in mouse was created on a C57/BL6J background followed by editing and inserting a single copy of the ALDH2*2 variant gene (E504K) by homologous recombination [20]. When challenged with alcohol, the mice accumulate acetaldehyde similar to the levels seen in humans carrying the ALDH2*2 genetic variant [20]. All animals were maintained in a constant 12-h dark/12-h light cycle in an AAALAC-accredited Veterinary Service Center at Stanford University. Food and water were available ad libitum.
The ALDH2 protein expression for heart, lung, and liver homogenates were analyzed by Western blot as previously described using an ALDH2 primary antibody (Abcam, ab103892, 1:1000) [21]. The ALDH enzymatic activity was measured as previously described using 200 μg of mitochondrial fraction for each organ homogenate (μmol NADH/min/mg protein) [8].
2.3. Primary mouse cardiac myocyte studies
Adult male cardiac myocytes were isolated from wild type ALDH2 and ALDH2*2 mice as previously described [22] and were plated on laminin (Sigma) coated-plates for calcium imaging or ROS production.
2.3.1. Calcium influx
Cover slips containing cardiac myocytes were mounted on a Zeiss Axiovert inverted fluorescent microscope. Cardiac myocytes were superfused continuously with DMEM serum free media by a peristaltic pump at a flow rate of 2 ml/min. After collection of baseline data, cells were superfused with a pulse of acetaldehyde (0.1, and 1 μM) for 30 s to mimic an exposure when inhaling an e-cigarette. Real-time calcium levels were acquired using an alternating excitation wavelength (340 and 380 nm), as previously described [23].
2.3.2. Reactive oxygen species production
Reactive oxygen species generation was measured by Amplex red (50 μM with 0.1 U/mL horseradish peroxidase) or Dichlorofluorescein diacetate (DCF-DA, 10μM) in the presence and absence of acetaldehyde (0.1 and 1 μM) or 4-HNE (20 μM) and measured for 2 h using a microplate reader (TECAN, Switzerland) [24].
2.4. Rodent e-cigarette aerosol exposure studies
2.4.1. Telemeter implantation for EKG recording
Telemeters (KAHA Sciences) for EKG recording were implanted under isoflurane anesthesia with EKG leads tunneled and placed in a lead II configuration. Heart rate was measured by placing rodents on a wireless digital receiver t-Base (KAHA Sciences) and recorded by LabChart (AD Instruments). After confirmation of the telemeter functionality, heart rate was measured while rodents were exposed to room air or e-cigarette aerosol.
2.4.2. E-cigarette aerosol exposure and tissue collection
After telemeter implantation, rodents were divided into 4 groups: room air exposed wild type ALDH2 mice, room air exposed homozygous ALDH2*2 mice, e-cigarette exposed wild type mice, and e-cigarette exposed ALDH2*2 mice. Before exposure, the baseline heart rates were recorded for 5 min. A wild type ALDH2 mouse with an ALDH2*2 mouse were then paired and exposed to e-cigarette aerosol or room air for 4 sessions a day for 10 days. Each session (30 min) contained 7 min of exposure phase (a total of 14 puffs) followed by 23 min of a smoke-free recovery phase. Heart rate was recorded daily. The puff volume and exposure duration were based on prior studies [[25], [26], [27], [28]]. After completing 10 days of exposure, hearts were homogenized for molecular studies.
2.4.3. Protein carbonylation
Carbonyl groups introduced into protein side chains were measured by an OxyBlot protein oxidation detection kit (Millipore). Specifically, 15–20 μg of protein was added into either positive DNPH (2,4-dinitrophenylhydrazine) solution or negative derivatization-control solution. DNP-derivatized protein were separated and detected by Western blot.
2.4.4. Free MDA production
Free malondialdehyde (MDA) was measured by reacting with thiobarbituric acid (TBAR) to generate a MDA-TBAR adduct according to the manufacturer's instructions (Abcam). The absorbance of MDA-TBA was measured at 532 nm using a Synergy 2 plate reader (BioTek).
2.4.5. 4-HNE protein adducts
The formation of 4-HNE-induced protein adducts was measured by a 4-HNE primary antibody (Alpha Diagnostic, HNE51-5, 1:1000), as described [20].
2.4.6. NF-κB analysis
Nuclear factor kappa B (NF-κB/p65) in heart homogenates were analyzed by Western blot as previously described using primary antibody anti-phospho-NFκB (ThermoFisher, MA5-15160, 1:1000) or anti-NFκB (ThermoFisher, PA1-186, 1:1000). Membranes were imaged by using a gel imaging system (Azure Biosystems).
3. Statistics
Data analysis were performed using Graph Pad Prism 7.0 software with one and two-way ANOVA with Bonferroni correction performed for multiple comparisons between groups. For direct comparison between two groups, a Students t-test was performed. Data were expressed as mean ± SEM with ^denoting significance between genotypes and +denoting significance within genotypes at p<0.05, &at p<0.01, and *at p<0.001.
4. Results
4.1. Acetaldehyde as the primary aldehyde within e-cigarette aerosol
Three e-cigarette brands (Blu, Halo and JUUL) were quantified for nicotine and aldehydes (Fig. 1A, Supplemental Table 1). The reagent ions, reaction ratio, and mass used to detect nicotine and aldehydes by SIFT-MS were summarized (Supplemental Table 2). Aerosolized nicotine, acetaldehyde, formaldehyde, and acrolein for Blu (24 mg/ml), Halo (24 mg/ml), and JUUL (50 mg/ml) e-cigarettes were measured relative to air (Fig. 1B and C). Acetaldehyde levels were the highest in all e-cigarettes relative to formaldehyde and acrolein (Fig. 1C: Acetaldehyde: Blu, 10.7±4.6* ppm, Halo, 8.6±3.4* ppm, JUUL, 5.3±1.4* ppm versus formaldehyde: Blu, 0.8±0.6 ppm, Halo, 0.7±0.5 ppm, JUUL, 0.2±0.08 ppm or acrolein: Blu: 1.2±0.9 ppm, Halo: 0.6±0.4 ppm, JUUL: 0.09±0.06 ppm, respectively, n=20/group versus other aldehydes and air). Additionally, the total aldehyde load (acetaldehyde, formaldehyde, acrolein) varied between the 3 e-cigarette brands and linearly correlated with the ratio of propylene glycol to vegetable glycerin (Fig. 1D, JUUL (30/70): 5.6±0.3 ppm, Halo (50/50): 9.9±0.9 ppm, Blu (60/40): 12.6±1.3 ppm, R2= 0.99, p=0.0025). Together, these results identify that e-cigarette aerosol primarily contains acetaldehyde with other aldehydes several-fold less within e-cigarette aerosol.
Fig. 1.

E-cigarette aerosol primarily contains acetaldehyde with other aldehydes several-fold less within e-cigarette aerosol. A. Three brands of e-cigarettes. B. Amount of nicotine and C.aldehydes (acetaldehyde, formaldehyde and acrolein) in e-cigarette aerosol or room air. Data was expressed as mean ± SEM (ppm). D. Total aldehyde load (ppm) relative to the percentage of propylene glycol from different e-cigarette brands. R2 was calculated by Pearson correlation. *p < 0.001, comparison between e-cigarette aerosol and air, calculated by one-way ANOVA with Bonferroni correction. n= 20 measurements per e-cigarette.
4.2. ALDH2 expression and activity across organs
As the enzyme responsible for metabolizing acetaldehyde is ALDH2, we used wild type ALDH2 and ALDH2*2 knock-in rodents and questioned whether there were organ-specific differences in ALDH2 expression and activity (Fig. 2A). Interestingly, the relative protein expression in heart homogenates for wild type ALDH2 mice was 2-fold lower relative to the lung and liver by Western blot (Fig. 2B: heart 0.57±0.14+ relative to lung 1.05±0.11 and liver 1.41±0.05, ALDH2/GAPDH relative densitometry units). Further, the protein expression of ALDH2 had similar trends between genders (Supplemental Fig. 1), and ALDH2*2 rodents had a 2-fold lower protein expression for the heart relative to the lung and liver (Fig. 2B: heart 0.23±0.05+ relative to lung 0.53±0.07 and liver 0.67±0.16). The tissue type, protein expression, and point mutation within an integral α-helix of the enzyme caused ALDH activity for the ALDH2*2 heart homogenate to have 2 to 3-fold lower activity when comparing the respective wild type ALDH2 organ (Fig. 2C).
Fig. 2.

Basal level of ALDH2 expression and activity and cardiac myocyte response to acetaldehyde. A. An ALDH2*2 knock-in rodent on a C57/BL6J background was generated with a missense mutation (glutamic acid to lysine at 487) that reflects the human ALDH2*2 genetic variant. B. Top: Representative Western blot of ALDH2 protein (56 kDa) expression for heart, lung, and liver tissue homogenates for ALDH2 and ALDH2*2 mice. Bottom: Quantification of ALDH2 protein expression normalized to GAPDH (36 kDa) (n=6/group). C. ALDH2 enzymatic activity for homogenates of each genotype and organ (n=6/group). D. Calcium influx by 1 μM acetaldehyde. KCl (60 mM) was used as a positive control. E. H2O2 activity by 0.1 or 1 μM acetaldehyde with 4-HNE (20μΜ) as a positive control assessed by Amplex Red (n=8/group). F. Immunostaining of primary cardiomyocytes for 4-hydroxynoneal (4-HNE) protein adducts in vehicle or 1 μM acetaldehyde treated cells (left panel, bar = 20 μm). Green: dystrophin, Red: 4-HNE protein adducts, Blue: DAPI. Right: Quantification of 4-HNE protein adducts (n=8/group). WT= wild type, *2 = ALDH2*2, H= heart, Lu= lung, Liv= liver. Blue represents wild type ALDH2 mice and red represents the ALDH2*2 mice. ^p < 0.05, between genotypes; and +p < 0.05, &p < 0.01, *p < 0.001, within genotypes, respectively, calculated by one-way ANOVA with Bonferroni correction. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
4.3. Primary cardiomyocyte response to acetaldehyde
When challenging ALDH2 and ALDH2*2 primary adult cardiomyocytes with an acetaldehyde pulse to mimic an e-cigarette, a clear difference in the dose-dependent calcium influx occurred between the genotypes. ALDH2*2 myocytes responded with an intracellular calcium influx even at the lowest acetaldehyde dose tested (0.1 μM), as opposed to wild type ALDH2 myocytes (Supplemental Fig. 1B). The difference in calcium influx between the ALDH2 genotypes continued to separate dose-dependently. For ALDH2*2 myocytes, acetaldehyde (1 μM) caused a 4-fold greater peak response to calcium influx versus wild type ALDH2 myocytes (Fig. 2D: 19.1±5.2^ versus 4.0±0.6, % maximal peak response, respectively). The peak maximal response triggered by 0.1 or 1 μM acetaldehyde also varied between genotypes (Fig. 2D). ALDH2*2 myocytes had a higher intracellular ROS level at baseline relative to wild type ALDH2 myocytes as determined by Amplex red (Fig. 2E: 3990.8±326.7^ versus 1726.2±290.8, relative fluorescence units, respectively, n=8). When challenged with acetaldehyde, ROS was exacerbated for the ALDH2*2 cardiac myocytes relative to wild type ALDH2 cardiac myocytes (0.1uM treated: 5193±1406^ vs 2044±21, 1 μM treated: 6605±1769^ vs 2733±406). These findings were also consistent when using DCF-DA for ALDH2*2 and ALDH2 untreated and acetaldehyde-treated cardiac myocytes (Supplemental Fig. 1C). At baseline, ALDH2*2 primary cardiomyocytes also demonstrated higher levels of 4-HNE-induced protein adducts relative to wild type ALDH2 cardiomyocytes. When exposed to 0.1 μM acetaldehyde, 4-HNE protein adducts were not statistically significant (Supplemental Fig. 1D) which was increased by 1 μM acetaldehyde (Fig. 2F).
4.4. E-cigarette exposure impact on heart rate in rodents
As acetaldehyde is highly prevalent in all e-cigarette aerosols tested, we questioned what impact the inactivating genetic variant ALDH2*2 relative to wild type ALDH2, has on e-cigarette aerosol exposure in rodents. We first quantified heart rate physiologically during and after exposure (as alcohol consumption and metabolism to acetaldehyde results in tachycardia for humans carrying the ALDH2*2 variant) [29,30]. A representative EKG waveform in normal sinus rhythm was provided for our mouse model (Supplemental Fig. 2A). Wild type ALDH2 and ALDH2*2 mice were exposed to e-cigarette aerosol or air four times per day for 10 days (Fig. 3A). During exposures, mice were exposed paired by genotype to e-cigarette aerosol or air while continuously monitoring oxygen and carbon monoxide levels within the exposure chamber (Fig. 3B). Concentrations of nicotine and aldehydes for e-cigarette aerosol or air within the exposure chamber were also quantified (Supplemental Figs. 2B and 2C).
Fig. 3.

Heart rate measurements of ALDH2 and ALDH2*2 mice exposed to e-cigarette aerosol. A. Experimental protocol. Wild type ALDH2 mice or ALDH2*2 mice were exposed to 7 min of e-cigarette aerosol exposure four times daily for 10 total days. B. Picture of the e-cigarette aerosol exposure chamber, where rodents were pair-matched (one wild type ALDH2 and one ALDH2*2 rodent) to an exposure. Device in yellow is a multi-gas monitor. C. Baseline heart rate before exposure. D. Mean heart rate changes during the first exposure. E. midway through the 10-day exposure and. F. the last day of the 10-day exposure. Data is expressed as mean ± SEM. Solid colors are rodents exposed to e-cigarette aerosol, dashed line and open circles are rodents exposed to air. Blue is wild type ALDH2 and red is ALDH2*2 rodents. ^p < 0.05, between genotypes; and +p < 0.05, &p < 0.01, *p < 0.001, within genotypes, respectively, calculated by two-way ANOVA with Bonferroni correction. n=7–8 mice/group. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
At baseline, there was no significant difference in the resting heart rate occurring between rodent groups (Fig. 3C). We next quantified the change in heart rate occurring during the time period of the e-cigarette aerosol exposure (Fig. 3D–F). During the first day of e-cigarette aerosol exposure, only the ALDH2*2 mice showed an elevated heart rate immediately after e-cigarette aerosol exposure versus ALDH2 rodents exposed to e-cigarette aerosol or air (Fig. 3D: 679±32^+ bpm versus 602±20 bpm and 548±16 bpm). Midway through the e-cigarette or air exposures, both the ALDH2*2 and wild type ALDH2 mice had heart rate increases immediately after e-cigarette aerosol exposure (Fig. 3E). After 10 days of exposure, the ALDH2*2 and wild type ALDH2 mice had increased heart rates (Fig. 3F, e-cigarette ALDH2*2 743±36* bpm and e-cigarette ALDH2 723±27* bpm versus air ALDH2*2 mice 536±31 bpm and air ALDH2 561±37 bpm).
4.5. E-cigarette aerosol exposure causes cardiovascular oxidative stress in rodent hearts
After e-cigarette aerosol or room air exposure, we quantified cardiovascular oxidative stress by measuring protein carbonylation, total free malondialdehyde (MDA) production, and 4-hydroxynonenal (4HNE)-protein adduct formation in heart homogenates.
Protein carbonylation was elevated for ALDH2*2 mice or wild type ALDH2 mice exposed to e-cigarette aerosol compared to air (Fig. 4A, Supplemental Fig. 2D). Levels of carbonylation were significantly greater for ALDH2*2 and ALDH2 mice for e-cigarette exposed relative to air (Fig. 4B, ALDH2*2: 14.4±3.4+ vs. 8.1±0.6 and ALDH2 10.2±2.5+ vs. 5.2±0.4, ratio of protein carbonylation to negative control). Free MDA in heart homogenates from ALDH2*2 mice were ∼1.8-fold higher relative to homogenates from air exposed ALDH2*2 mice (Figs. 4C and 98.9±11.6& vs 59.4±6.4 nmol/ml MDA, respectively). This was in contrast to wild type ALDH2 which did not show increased levels of free MDA relative to air-treated wild type ALDH2 mice (Figs. 4C and 54.9±13.8 vs 43.4±9.6 nmol/ml MDA, respectively). Further, 4-HNE-induced protein adducts were elevated for ALDH2*2 and wild type ALDH2 mice after e-cigarette aerosol exposure relative to air (Fig. 4D4E, 62.7±8.7^ and 58.3±4.3^ vs 38.6±4.4 and 33.8±4.4, respectively). To understand the mechanism, we initially measured p-NF-κB and NF-κB in untreated rodent heart homogenates, finding that in ALDH2*2 mice the p-NF-κB to NF-κB ratio was ∼2-fold higher when compared to wild type ALDH2 mice (Supplemental Fig. 2E). The elevated basal p-NF-κB in the untreated ALDH2*2 mice may be due to the increased ROS-dependent phosphorylation of NF-κB [31]. For heart homogenates from ALDH2*2 and wild type ALDH2 mice subjected to e-cigarette aerosol, a significant increase in the ratio of p-NF-κB to NF-κB occurred relative to rodents exposed to air (Figs. 4F 0.59±0.14 and 0.47±0.12 versus 0.3±0.15 and 0.15±0.06, respectively).
Fig. 4.

Aldehydic load after e-cigarette aerosol or air exposure. A. Representative blot of protein carbonyl expression. B. Quantification of protein carbonylation (n=6/group). C. Free malondialdehyde (MDA) in heart homogenates. The concentration of MDA was normalized by the total amount of protein (n=6/group). D. Representative blot of 4-HNE-induced protein adducts with GAPDH (36 kDa) as a loading control. E. Quantification of 4-HNE-induced protein adducts (n=6/group). F: phospho- and total nuclear factor kappa B (NF-κB, 65 kDa) in heart homogenates (n=6/group). ^p < 0.05, between genotypes; and +p < 0.05, &p < 0.01, *p < 0.001, within genotypes, respectively, calculated by one-way ANOVA with Bonferroni correction.
5. Discussion
Here we identified acetaldehyde as a primary aldehyde within e-cigarette aerosol. When exposed to e-cigarette aerosol, the ALDH2*2 mice (due to genetics that limit acetaldehyde metabolism) are more susceptible to cardiovascular oxidative stress when compared to wild type ALDH2 rodents. As the ALDH2*2 mice are a knock-in model where a single copy of the ALDH2 gene is replaced with the inactive human ALDH2*2 variant, this study provides valuable insight that people carrying an ALDH2*2 variant may potentially be more susceptible to the elevated cardiovascular oxidative stress that can occur with frequent e-cigarette aerosol exposure. Exposure to aldehydes within e-cigarette aerosol can promote ROS production, increase protein carbonylation, lipid peroxidation, and NF-κB activation leading to elevated levels of cardiovascular oxidative stress that is further exacerbated in the ALDH2*2 variant due to the limited ability to metabolize aldehydes within e-cigarette aerosol. This may pave the way to developing precision medicine strategies towards understanding cardiovascular risk for e-cigarette exposures by considering how genetics influence aldehyde metabolism.
Aldehydes are generated from e-cigarettes by the incomplete combustion of propylene glycol and vegetable glycerin when exposed to oxygen [32]. Although e-cigarette aerosol contains aldehydes, the levels could be as much as 70-fold less compared to conventional cigarette smoke [2]. Our findings identify within e-cigarette aerosol aldehydes including acrolein, acetaldehyde, and formaldehyde which are consistent with prior studies [2,[33], [34], [35]]. By using real-time mass spectrometry to measure aldehydes, our study identifies acetaldehyde as the primary aldehyde within e-cigarette aerosol. A prior study also identified acetaldehyde as the predominant aldehyde within e-cigarette aerosol relative to formaldehyde and acrolein [2]. The level of acetaldehyde measured within e-cigarette aerosol for our study also reflect those of a prior study identifying that acetaldehyde levels are 10-fold higher than acrolein [33]. Other studies suggest acetaldehyde levels may be at or slightly less than the levels of formaldehyde and acrolein within e-cigarette aerosol [2,34,35]. However, acetaldehyde is quite volatile relative to formaldehyde and acrolein, and the lower levels of acetaldehyde may be attributable to the time for sample preparation causing neutralization and dispersal of acetaldehyde which would account for the lower reading. This is the advantage of using a SIFT MS technique to assess e-cigarette aerosol quantities as the measurements are performed immediately and in real-time. Interestingly, we also observed that the total aldehyde load within e-cigarette aerosol increased with higher percentages of propylene glycol. Together, these findings describe a real-time monitoring technique to quantify levels of aldehydes in e-cigarette aerosol while identifying that acetaldehyde is a predominant aldehyde within e-cigarette aerosol.
Cigarette smoke-induced oxidative stress within the cardiovascular system is an important trigger for the pathogenesis of cardiovascular diseases, including coronary artery disease, atrial fibrillation, heart failure, and cardiac ischemia-reperfusion injury [15,36]. E-cigarette use also elevates oxidized low-density lipoprotein, a marker of oxidative stress, 1.5-fold in humans when compared to non-e-cigarette, non-tobacco users [37]. Additionally, e-cigarette aerosol can trigger lipid peroxidation which form endogenous aldehydes including 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA) [26,38]. As 4-HNE can inhibit ALDH2 enzymatic activity, this can further drive increases in oxidative stress levels from e-cigarette exposures by limiting acetaldehyde metabolism [8]. Importantly, when ALDH2*2 rodents were exposed pair-matched to e-cigarette aerosol exposure with wild type ALDH2 mice, our findings provide new insight that genetics which limit aldehyde metabolism can drive heightened levels of cardiovascular oxidative stress with e-cigarette exposure. This is caused by a lesser capability for the ALDH2*2 enzyme to neutralize aldehydes leading to an imbalance requiring less oxidative stress to cause a more pro-reactive environment.
Our study indicates that the ALDH2 enzyme contributes to the heart rate-mediated responses to e-cigarette aerosol. Heart rate increases after e-cigarette or tobacco cigarette use occurs in humans and is correlated with plasma nicotine concentrations [39]. During e-cigarette use, heart rate may increase ∼5 beats per minute in humans [40]. Human subjects vaping e-cigarettes without nicotine when compared to the same subjects vaping e-cigarettes with nicotine also would suggest the immediate increase in heart rate is only partially caused by nicotine [41]. We observed that e-cigarette aerosol exposure increases heart rate in ALDH2 and ALDH2*2 rodents, with increases in ALDH2*2 rodents earlier and more pronounced relative to wild type ALDH2 rodents. Taken together, these data indicate that aldehydes also contribute to the heart rate response of e-cigarettes. With limited aldehyde metabolism, an increase in heart rate likely occurs bya similar mechanism to how accumulation of acetaldehyde increases heart rate after alcohol consumption for those carrying the ALDH2*2 variant.
Our study also identifies aldehydes from e-cigarette aerosol can induce oxidative stress. Oxidative stress is known to trigger and exacerbate the pathogenesis of atherosclerosis, hypertension, and diastolic dysfunction in humans and animal models [42,43]. Reactive aldehydes from e-cigarette aerosol promote the overproduction of ROS, which exacerbates endogenous lipid peroxidation, protein adduct formation as well as endogenous aldehydes including 4-HNE and MDA, leading to increases in oxidative stress [44]. Kuntic and colleagues identified that reactive aldehydes in e-cigarette aerosol were important mediators of e-cigarette-induced oxidative stress in wild type mice [42]. Our study highlights the role of aldehyde metabolism in e-cigarette-induced oxidative stress and importantly identifies that inefficient aldehyde metabolism, as for the ALDH2*2 mice, causes higher levels of oxidative stress relative to wild type ALDH2 mice. Other studies also report e-liquid can generate the reactive aldehyde MDA [[45], [46], [47]]. In addition, we observed that acetaldehyde as the primary aldehyde in e-cigarette aerosol promotes intracellular Ca2+ influx, with the ALDH2*2 variant further enhanceing the Ca2+ influx. Sussan and colleagues showed that elevated ROS production induced by the e-cigarette exposure is associated with a higher level of lipid peroxidation marker MDA in mouse lung homogenates [26]. We observed similar ROS production and increased levels of lipid peroxidation markers in both wild type ALDH2 mice and ALDH2*2 mice after e-cigarette aerosol exposure. These observations link aldehyde metabolism to e-cigarette-mediated oxidative stress and calcium handling which can eventually lead to cardiovascular dysfunction.
We find that the NF-κB pathway is involved in mediating e-cigarette-induced cardiovascular oxidative stress and at baseline this inflammatory pathway is increased for ALDH2*2 knock-in mice relative to wild type ALDH2 mice. This is likely secondary to increased baseline levels of ROS in the ALDH2*2 knock-in mice. Similar to prior work, this accumulation of intracellular ROS and NF-κB activation by phosphorylation of Ser536 likely leads to downstream activation of pro-inflammatory cytokines which can result in heightened levels of inflammation [48]. For cardiomyocytes isolated from C57BL/6J mice, human pulmonary microvascular endothelial cells, and C57BL/6J mice exposed to the e-cigarette vapor, the proinflammatory cytokines including interleukin-1β, ICAM-1 MCP1, interleukin-6, interleukin-8 and TNF-α, were upregulated by NF-kB activation [49,50]. In human pulmonary artery smooth muscle cells, the balance of NF-κB activation and downstream signaling events of inflammation are also regulated by ALDH2 [51]. E-cigarette aerosol-induced lung injury has also been linked to oxidative stress and the pro-inflammatory NF-κB pathways [52]. In the context of our present study, ALDH2*2 mice demonstrate a higher level of NF-κB phosphorylation after e-cigarette exposure compared to wild type ALDH2 mice. Together, this study suggests that activation of NF-κB can lead to triggering of a pro-inflammatory pathway which may explain how e-cigarette exposure can elevate cardiovascular oxidative stress.
Although mitochondria are abundant within the heart relative to other organs, ALDH2 expression within the heart is 2.4-fold less relative to the lung and 2.8-fold to the liver. This finding identifies organ systems that are the first exposed to aldehyde sources having a higher capacity to metabolize aldehydes; likely in order to protect the body from oxidative stress caused by inhaled and consumed substances. The tissue specific distribution of ALDH2 puts the cardiovascular system, with less distribution and enzymatic activity relative to other organs, at a greater susceptibility for the reactive aldehyde-induced damage to e-cigarette aerosol which is amplified by carrying the ALDH2*2 genetic variant. Differences in tissue specific expression and activity of ALDH2 may lead to specific organ systems being more susceptible to organ injury from aldehydes [53,54]. Similar to our findings, a recent non-biased approach to map the proteome of the human body for 32 different organs measured from 14 individuals revealed the liver had ∼2-fold higher ALDH2 protein expression relative to the heart [55]. Taken together, organ specific levels of ALDH2 expression may be an important player in driving the pathophysiology of e-cigarette aerosol. Understanding ALDH2 expression across organ systems may also advance the understanding and provide a common link to how tobacco cigarettes, e-cigarettes and alcohol, as sources of aldehyde exposure, alone or concomitantly can lead to an increased risk of cardiovascular disease.
Our study should be interpreted within the context of potential limitations. Although we did not measure cardiac function and morphology for the ALDH2*2 mice in this study, prior studies identify that baseline cardiac function (measured by heart rate, left ventricular pressure and coronary perfusion pressure) [56], heart weight [57] and the heart histology [57] for ALDH2*2 mice were not different when compared to the wild type ALDH2 mice. Also, we used whole body exposure to e-cigarette aerosol rather than a nose-only exposure. Regardless of the exposure method, our study matched rodents of different ALDH2 genotypes pairwise to e-cigarette exposure to identify what impact the ALDH2*2 variant will have on cardiovascular oxidative stress relative to wild type ALDH2 rodents. In addition, we used homozygote ALDH2*2 rodents whereas most of the human population are heterozygous for the ALDH2*2 variant. Although we report ALDH2 protein expression was similar between genders, a prior study identified that wild type ALDH2 mouse female heart homogenates have a 20–30% increase in phosphorylation causing a 20–30% increase in ALDH2 enzymatic activity relative to male heart homogenates [58]. As these gender differences result in a ∼10% change in enzymatic activity for wild type ALDH2 rodents this is minor when considering the ALDH2*2 variant triggers a 3-fold lower enzymatic activity relative to the wild type ALDH2 rodents. However, more studies are needed to identify how gender, when coupled with differences in ALDH2 genetics, impacts cardiovascular oxidative stress with e-cigarette aerosol exposure. Although we primarily examined the impact of acetaldehyde exposure in vitro, e-cigarette aerosol also contains formaldehyde and acrolein. Therefore, we cannot exclude a potential synergistic effect when rodents are exposed to aldehydes within e-cigarette aerosol concomitantly. Regardless of these limitations, our data provide novel insight that within certain populations carrying genetics which limit aldehyde metabolism, there is a potential concern that oxidative stress is exacerbated with e-cigarette aerosol exposure.
In summary, we identify acetaldehyde as a predominant aldehyde within e-cigarette aerosol and further describe how rodents carrying an inactivating variant of ALDH2, ALDH2*2, are more susceptible to cardiovascular oxidative stress when exposed to e-cigarette aerosol. We also unexpectedly identify that the expression and activity of ALDH2 is lower in the heart relative to the lung and liver. This may provide a basis to understand how the heart as an organ may be more susceptible aldehyde-induced oxidative stress via NF-κB triggering a proinflammatory pathway. Ultimately, this study highlights the importance of considering the impact of genetics to develop a further understanding of the cardiovascular risks with e-cigarette aerosol use [59].
Statements and declarations
The authors have no competing interests to declare that are relevant to the content of this article.
Author contributions
Xuan Yu designed and performed the molecular and cell studies, acquired, analyzed and interpreted the data and wrote the manuscript. Xiaocong Zeng and Pritam Sinharoy designed the animal e-cigarette exposure. Xiaocong Zeng and Feng Xiao conducted the animal e-cigarette exposure studies, and Xiaocong Zeng wrote the method for mice e-cigarette exposure. Ri Chen conducted the e-cigarette aerosol collection and quantification studies, and wrote the method for the aerosol analysis. Feng Xiao conducted the cardiomyocytes isolation. Eric R. Gross design the study, interpreted the data, reviewed and edited manuscript, and supervised the study. All authors read and approved the final manuscript.
Declaration of competing interest
Please check the following as appropriate:
✓ All authors have participated in (a) conception and design, or analysis and interpretation of the data; (b) drafting the article or revising it critically for important intellectual content; and (c) approval of the final version.
✓ This manuscript has not been submitted to, nor is under review at, another journal or other publishing venue.
✓ The authors have no affiliation with any organization with a direct or indirect financial interest in the subject matter discussed in the manuscript.
The following authors have affiliations with organizations with direct or indirect financial interest in the subject matter discussed in the manuscript:
Author's name Affiliation.
No conflict of interest for authors.
Acknowledgments
This work was supported by the Tobacco-Related Disease Research Program (TRDRP) T31FT1392 (X.Y), National Institute of General Medical Sciences and the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number GM119522, GM119522-S01 and HL144388 (E.R.G). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102369.
Supplementary material
Additional experimental procedures and data analyses were provided in supplementary materials.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Mirbolouk M., et al. Prevalence and distribution of E-cigarette use among U.S. Adults: behavioral risk factor surveillance system, 2016. Ann. Intern. Med. 2018;169(7):429–438. doi: 10.7326/M17-3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogunwale M.A., et al. Aldehyde detection in electronic cigarette aerosols. ACS Omega. 2017;2(3):1207–1214. doi: 10.1021/acsomega.6b00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haussmann H.J. Use of hazard indices for a theoretical evaluation of cigarette smoke composition. Chem. Res. Toxicol. 2012;25(4):794–810. doi: 10.1021/tx200536w. [DOI] [PubMed] [Google Scholar]
- 4.Medicine I.o. National Academies Press; Washington (DC): 2010. Secondhand Smoke Exposure and Cardiovascular Effects: Making Sense of the Evidence. [PubMed] [Google Scholar]
- 5.Grimsrud P.A., et al. Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem. 2008;283(32):21837–21841. doi: 10.1074/jbc.R700019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klyosov A.A., et al. Possible role of liver cytosolic and mitochondrial aldehyde dehydrogenases in acetaldehyde metabolism. Biochem. 1996;35(14):4445–4456. doi: 10.1021/bi9521093. [DOI] [PubMed] [Google Scholar]
- 7.Koda K., et al. Aldehyde dehydrogenase activation prevents reperfusion arrhythmias by inhibiting local renin release from cardiac mast cells. Circulation. 2010;122(8):771–781. doi: 10.1161/CIRCULATIONAHA.110.952481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen C.H., et al. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321(5895):1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen C.H., et al. Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol. Rev. 2014;94(1):1–34. doi: 10.1152/physrev.00017.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ebert A.D., et al. Characterization of the molecular mechanisms underlying increased ischemic damage in the aldehyde dehydrogenase 2 genetic polymorphism using a human induced pluripotent stem cell model system. Sci. Transl. Med. 2014;6(255) doi: 10.1126/scitranslmed.3009027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ueta C.B., et al. Cardioprotection induced by a brief exposure to acetaldehyde: role of aldehyde dehydrogenase 2. Cardiovasc. Res. 2018;114(7):1006–1015. doi: 10.1093/cvr/cvy070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma H., et al. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur. Heart J. 2011;32(8):1025–1038. doi: 10.1093/eurheartj/ehq253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X., Sun A. Aldehyde dehydrogenase-2 roles in ischemic cardiovascular disease. Curr. Drug Targets. 2017;18(15):1817–1823. doi: 10.2174/1389450117666160912174417. [DOI] [PubMed] [Google Scholar]
- 14.Mizuno Y., et al. Variant aldehyde dehydrogenase 2 (ALDH2*2) is a risk factor for coronary spasm and ST-segment elevation myocardial infarction. J. Am. Heart Assoc. 2016;5(5) doi: 10.1161/JAHA.116.003247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buchanan N.D., et al. Cardiovascular risk of electronic cigarettes: a review of preclinical and clinical studies. Cardiovasc. Res. 2020;116(1):40–50. doi: 10.1093/cvr/cvz256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ren J., Wold L.E. Mechanisms of alcoholic heart disease. Ther Adv Cardiovasc Dis. 2008;2(6):497–506. doi: 10.1177/1753944708095137. [DOI] [PubMed] [Google Scholar]
- 17.Wong D.N., Fan W. Ethnic and sex differences in E-cigarette use and relation to alcohol use in California adolescents: the California Health Interview Survey. Publ. Health. 2018;157:147–152. doi: 10.1016/j.puhe.2018.01.019. [DOI] [PubMed] [Google Scholar]
- 18.Chen X., Yu B., Wang Y. Initiation of electronic cigarette use by age among youth in the U.S. Am. J. Prev. Med. 2017;53(3):396–399. doi: 10.1016/j.amepre.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 19.Creamer M.R., et al. Tobacco product use and cessation indicators among adults - United States, 2018. MMWR Morb. Mortal. Wkly. Rep. 2019;68(45):1013–1019. doi: 10.15585/mmwr.mm6845a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zambelli V.O., et al. Aldehyde dehydrogenase-2 regulates nociception in rodent models of acute inflammatory pain. Sci. Transl. Med. 2014;6(251) doi: 10.1126/scitranslmed.3009539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Y., et al. Transient receptor potential ankyrin 1 activation within the cardiac myocyte limits ischemia-reperfusion injury in rodents. Anesthesiology. 2016;125(6):1171–1180. doi: 10.1097/ALN.0000000000001377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Connell T.D., Rodrigo M.C., Simpson P.C. Isolation and culture of adult mouse cardiac myocytes. Methods Mol. Biol. 2007;357:271–296. doi: 10.1385/1-59745-214-9:271. [DOI] [PubMed] [Google Scholar]
- 23.Andrei S.R., et al. TRPA1 is functionally co-expressed with TRPV1 in cardiac muscle: Co-localization at z-discs, costameres and intercalated discs. Channels. 2016;10(5):395–409. doi: 10.1080/19336950.2016.1185579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griendling K.K., et al. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: a scientific statement from the American heart association. Circ. Res. 2016;119(5):e39–75. doi: 10.1161/RES.0000000000000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glynos C., et al. Comparison of the effects of e-cigarette vapor with cigarette smoke on lung function and inflammation in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2018;315(5):L662–L672. doi: 10.1152/ajplung.00389.2017. [DOI] [PubMed] [Google Scholar]
- 26.Sussan T.E., et al. Exposure to electronic cigarettes impairs pulmonary anti-bacterial and anti-viral defenses in a mouse model. PLoS One. 2015;10(2) doi: 10.1371/journal.pone.0116861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dawkins L., et al. 'Vaping' profiles and preferences: an online survey of electronic cigarette users. Addiction. 2013;108(6):1115–1125. doi: 10.1111/add.12150. [DOI] [PubMed] [Google Scholar]
- 28.Noel A., et al. Generation of electronic cigarette aerosol by a third-generation machine-vaping device: application to toxicological studies. J Vis Exp. 2018;138 doi: 10.3791/58095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eng M.Y., Luczak S.E., Wall T.L. ALDH2, ADH1B, and ADH1C genotypes in Asians: a literature review. Alcohol Res. Health. 2007;30(1):22–27. [PMC free article] [PubMed] [Google Scholar]
- 30.Harada S., Agarwal D.P., Goedde H.W. Aldehyde dehydrogenase deficiency as cause of facial flushing reaction to alcohol in Japanese. Lancet. 1981;2(8253):982. doi: 10.1016/s0140-6736(81)91172-7. [DOI] [PubMed] [Google Scholar]
- 31.Lingappan K. NF-kappaB in oxidative stress. Curr Opin Toxicol. 2018;7:81–86. doi: 10.1016/j.cotox.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaur G., Muthumalage T., Rahman I. Mechanisms of toxicity and biomarkers of flavoring and flavor enhancing chemicals in emerging tobacco and non-tobacco products. Toxicol. Lett. 2018;288:143–155. doi: 10.1016/j.toxlet.2018.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tayyarah R., Long G.A. Comparison of select analytes in aerosol from e-cigarettes with smoke from conventional cigarettes and with ambient air. Regul. Toxicol. Pharmacol. 2014;70(3):704–710. doi: 10.1016/j.yrtph.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 34.Uchiyama S., et al. Determination of carbonyl compounds generated from the E-cigarette using coupled silica cartridges impregnated with hydroquinone and 2,4-dinitrophenylhydrazine, followed by high-performance liquid chromatography. Anal. Sci. 2013;29(12):1219–1222. doi: 10.2116/analsci.29.1219. [DOI] [PubMed] [Google Scholar]
- 35.Goniewicz M.L., et al. Levels of selected carcinogens and toxicants in vapour from electronic cigarettes. Tob Control. 2014;23(2):133–139. doi: 10.1136/tobaccocontrol-2012-050859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aune D., et al. Tobacco smoking and the risk of atrial fibrillation: a systematic review and meta-analysis of prospective studies. Eur J Prev Cardiol. 2018;25(13):1437–1451. doi: 10.1177/2047487318780435. [DOI] [PubMed] [Google Scholar]
- 37.Moheimani R.S., et al. Increased cardiac sympathetic activity and oxidative stress in habitual electronic cigarette users: implications for cardiovascular risk. JAMA Cardiol. 2017;2(3):278–284. doi: 10.1001/jamacardio.2016.5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hasan K.M., et al. Electronic cigarettes cause alteration in cardiac structure and function in diet-induced obese mice. PLoS One. 2020;15(10) doi: 10.1371/journal.pone.0239671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan X.S., D'Ruiz C. Effects of using electronic cigarettes on nicotine delivery and cardiovascular function in comparison with regular cigarettes. Regul. Toxicol. Pharmacol. 2015;71(1):24–34. doi: 10.1016/j.yrtph.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 40.Vansickel A.R., Eissenberg T. Electronic cigarettes: effective nicotine delivery after acute administration. Nicotine Tob. Res. 2013;15(1):267–270. doi: 10.1093/ntr/ntr316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chaumont M., et al. Differential effects of E-cigarette on microvascular endothelial function, arterial stiffness and oxidative stress: a randomized crossover trial. Sci. Rep. 2018;8(1) doi: 10.1038/s41598-018-28723-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuntic M., et al. Short-term e-cigarette vapour exposure causes vascular oxidative stress and dysfunction: evidence for a close connection to brain damage and a key role of the phagocytic NADPH oxidase (NOX-2) Eur. Heart J. 2020;41(26):2472–2483. doi: 10.1093/eurheartj/ehz772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Farsalinos K.E., et al. Acute effects of using an electronic nicotine-delivery device (electronic cigarette) on myocardial function: comparison with the effects of regular cigarettes. BMC Cardiovasc. Disord. 2014;14:78. doi: 10.1186/1471-2261-14-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uchida K. Role of reactive aldehyde in cardiovascular diseases. Free Radic. Biol. Med. 2000;28(12):1685–1696. doi: 10.1016/s0891-5849(00)00226-4. [DOI] [PubMed] [Google Scholar]
- 45.Menicagli R., Marotta O., Serra R. Free radical production in the smoking of E-cigarettes and their possible effects in human Health. Int. J. Prev. Med. 2020;11:53. doi: 10.4103/ijpvm.IJPVM_424_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.El Golli N., et al. Impact of e-cigarette refill liquid with or without nicotine on liver function in adult rats. Toxicol. Mech. Methods. 2016;26(6):419–426. doi: 10.3109/15376516.2016.1160963. [DOI] [PubMed] [Google Scholar]
- 47.Suryadinata R.V., Wirjatmadi B., Lorensia A. The time pattern of selenomethionine administration in preventing free radicals due to exposure to electric cigarette smoke. J Public Health Res. 2021;10(2) doi: 10.4081/jphr.2021.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kwon H.J., et al. Stepwise phosphorylation of p65 promotes NF-kappaB activation and NK cell responses during target cell recognition. Nat. Commun. 2016;7 doi: 10.1038/ncomms11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Masso-Silva J.A., Byun M.K., Alexander L.E.C. Acute and chronic effects of vaping electronic devices on lung physiology and inflammation. Curr Opin Physiol. 2021;22 doi: 10.1016/j.cophys.2021.06.001. ARTN 100447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang X.Q., et al. Cardiomyocyte-specific p65 NF-kappaB deletion protects the injured heart by preservation of calcium handling. Am. J. Physiol. Heart Circ. Physiol. 2013;305(7):H1089–H1097. doi: 10.1152/ajpheart.00067.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu T., et al. Aldehyde dehydrogenase 2 protects against oxidative stress associated with pulmonary arterial hypertension. Redox Biol. 2017;11:286–296. doi: 10.1016/j.redox.2016.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ma T., et al. Electronic cigarette aerosols induce oxidative stress-dependent cell death and NF-kappaB mediated acute lung inflammation in mice. Arch. Toxicol. 2021;95(1):195–205. doi: 10.1007/s00204-020-02920-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McAllister S.L., et al. Aberrant reactive aldehyde detoxification by aldehyde dehydrogenase-2 influences endometriosis development and pain-associated behaviors. Pain. 2021;162(1):71–83. doi: 10.1097/j.pain.0000000000001949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gross E.R., et al. A personalized medicine approach for Asian Americans with the aldehyde dehydrogenase 2*2 variant. Annu. Rev. Pharmacol. Toxicol. 2015;55:107–127. doi: 10.1146/annurev-pharmtox-010814-124915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang L., et al. A quantitative proteome map of the human body. Cell. 2020;183(1):269–283. doi: 10.1016/j.cell.2020.08.036. e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pan G., et al. Effects of intracardiac delivery of aldehyde dehydrogenase 2 gene in myocardial salvage. Gene Ther. 2022 doi: 10.1038/s41434-022-00345-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pan G., Roy B., Palaniyandi S.S. Diabetic aldehyde dehydrogenase 2 mutant (ALDH2*2) mice are more susceptible to cardiac ischemic-reperfusion injury due to 4-hydroxy-2-nonenal induced coronary endothelial cell damage. J. Am. Heart Assoc. 2021;10(18) doi: 10.1161/JAHA.121.021140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lagranha C.J., et al. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ. Res. 2010;106(11):1681–1691. doi: 10.1161/CIRCRESAHA.109.213645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neczypor E.W., et al. E-cigarettes and cardiopulmonary Health: review for clinicians. Circulation. 2022;145(3):219–232. doi: 10.1161/CIRCULATIONAHA.121.056777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.