

Abstract
Benign recurrent intrahepatic cholestasis (BRIC) is an autosomal recessive disorder characterized by recurrent cholestasis. ATPase class I, type 8B, member 1 (ATP8B1) encodes familial intrahepatic cholestasis 1 (FIC1), which acts as a phosphatidylserine reversing enzyme in the tubule membrane of hepatocytes to mediate the inward translocation of phosphatidylserine (PS). At present, dozens of ATP8B1 pathogenic mutations have been identified that mainly cause BRIC1 and progressive familial intrahepatic cholestasis 1 (PFIC1). The diagnosis of BRIC1 is based on symptoms, laboratory tests, imaging, liver histology, and genetic testing. BRIC1 treatment seeks to prevent recurrence and reduce disease severity. At present, the main treatment methods include ursodeoxycholic acid (UDCA), rifampin, cholestyramine and haemofiltration, and endoscopic nasobiliary drainage (ENBD). Here, we report a 17-year-old patient with cholestasis who has a rare heterozygous ATP8B1 gene mutation (p.T888K). The patient was treated with UDCA, glucocorticoids and haemofiltration, after which bilirubin levels gradually returned to normal. This case was thought to be caused by an ATP8B1 heterozygous mutation, which may be related to haploinsufficiency (HI).
Keywords: benign recurrent intrahepatic cholestasis, ATP8B1, haploinsufficiency, cholestasis, targeted therapy
Introduction
Familial intrahepatic cholestasis (FIC) is a group of autosomal recessive liver diseases characterized by intrahepatic cholestasis. FIC is a rare disease with an overall estimated incidence of 1 per 50,000 to 1 per 100,000 (1, 2). Benign recurrent intrahepatic cholestasis (BRIC) is characterized by recurrent jaundice, pruritus and malabsorption. The first appearance of jaundice in BRIC can occur at any age, usually < 20 years. BRIC-related symptoms generally last from weeks to months. Elevated serum bilirubin and BS levels were observed during cholestasis episodes, but gamma-glutamyltransferase (GGT) activity tended to be normal. BRIC episodes can be spontaneous or triggered by certain factors. Infection, pregnancy and medications are common triggers (3). BRIC does not progress to liver failure, and the associated symptoms resolve spontaneously. Symptoms may not occur in the intermittent period, and liver function and pathological findings are normal which increases the difficulty of diagnosis.
We report a case of jaundice. Laboratory tests showed significant increase in bilirubin. Biopsy and genetic testing were performed after common diseases were excluded. We found that the patient carried a rare ATP8B1 gene mutation, which was consistent with the diagnosis of BRIC1.
Case Report
A 17-year-old boy presented with jaundice, white clay stool, nausea, and loss of appetite for 1 month. There was no history of rash 3 months before admission. Chinese medicine was externally applied, and the patient took anti-allergy drugs orally. Later, the rash improved, and fever occurred intermittently. The patient was a student with no history of other drug use, blood transfusions, allergies, smoking, or alcohol. His parents are in good health. Physical examination found that his skin and sclera were yellow. No other abnormalities were noted upon physical examination. The liver-related laboratory examination revealed cholestasis, and GGT levels were normal. Aspartate aminotransferase (AST), 36 U/L; alanine aminotransferase (ALT), 35 U/L; alkaline phosphatase (ALP), 157 U/L; Total bile acid (TBA), 150 μmol/L; total bilirubin (TBIL), 298.4 μmol/L; and direct bilirubin (DBIL), 227.4 μmol/L. Serological and laboratory results excluded autoimmune hepatitis, primary biliary cholangitis (PBC), viral hepatitis, Wilson disease and a1-antitrypsin deficiency (Table 1). Ultrasound examination showed no cholelithiasis or bile duct dilatation. The liver stiffness measurement was 11.4 kPa. Computed tomography (CT) shows no abnormality in the liver, and the gallbladder is collapsed without bile filling. Magnetic resonance cholangiopancreatography (MRCP) shows suspected stenosis at the beginning of the common hepatic duct. Endoscopic ultrasonography showed that the extrahepatic bile duct was normal without dilation, and no definite obstruction was observed. Gastroscopy showed the size and morphology of the duodenal papilla were normal (Figure 1).
TABLE 1.
Laboratory data on admission at previous hospital.
| Biochemistry | Reference range | Peripheral blood | Reference range | ||
| TP (g/L) | 58.7 | 65.0–85.0 | WBC (×109/L) | 5.92 | 4.0–10.0 |
| Albumin (g/L) | 32.7 | 40.0–55.0 | Neutrophil (%) | 47.9 | 40.0–75.0 |
| TBIL (μmol/L) | 294.8 | 3.4–20.5 | Lymphocyte (%) | 38.9 | 20.0–50.0 |
| DBIL (μmol/L) | 227.4 | 0.0–6.8 | Monocyte (%) | 10 | 3.0–10.0 |
| AST (U/L) | 36 | 15–40 | Eosinophil (%) | 2.2 | 0.4–8.0 |
| ALT (U/L) | 35 | 9–50 | Basophile (%) | 1.0 | 0.0–1.0 |
| ALP (U/L) | 157 | 45–125 | Lymphocyte (%) | 38.9 | 20.0–50.0 |
| GGT (U/L) | 19 | 10–60 | RBC (×1012/L) | 4.51 | 4.0–4.5 |
| Urea (mmol/L) | 3.68 | 2.85–7.14 | Hb (g/L) | 137 | 120–140 |
| Cr (μmol/L) | 56 | 29–104 | Hct (L/L) | 0.396 | 0.4–0.5 |
| Cys-C (mg/L) | 1.04 | 0.53–0.95 | PLT (×109/L) | 310 | 100–300 |
|
|
|||||
| Na (mmol/L) | 136.5 | 137.0–147.0 | Coagulation | ||
|
|
|||||
| Cl (mmol/L) | 103.7 | 99.0–110.0 | PT (s) | 13.5 | 11–13.7 |
| K (mmol/L) | 4.13 | 3.50–5.30 | INR | 1.02 | 0.9–1.1 |
| CRP (mg/L) | < 1.00 | 0.00–5.00 | PTA (%) | 97.0 | 80.0–120.0 |
|
|
|||||
| TBA (μmol/L) | 150 | 0–10 | Serology | ||
|
|
|||||
| TSH (mIU/L) | 1.137 | 0.35–4.94 | ANA | (–) | (–) |
| FT3 (pmol/L) | 2.48 | 2.63–5.7 | AMA/AMA-M2 | (–) | (–) |
| FT4 (pmol/L) | 10.77 | 9.01–19.05 | α1 globulin (%) | 3.9 | 2.68–5.03 |
| IgG4 (g/L) | 0.301 | 0.049–1.985 | γ globulin (%) | 14.5 | 9.1–19.81 |
|
|
|||||
| IgG (g/L) | 10.18 | 7.00–17.00 | Viral markers | ||
|
|
|||||
| IgA (g/L) | 2.01 | 0.70–3.80 | Anti-HAV (S/CO) | 0.11 | 0.00–0.80 |
| IgM (g/L) | 0.73 | 0.60–2.50 | HBsAg (IU/mL) | 0 | < 0.05 |
| Ferritin (μg/L) | 722 | 30.00–400.00 | HCVAb (S/CO) | 0.28 | < 1.00 |
| Fe (μmol/L) | 24.7 | 8.3–28.3 | HEV-IgM (S/CO) | 0.02 | 0.00–1.00 |
| AFP (ng/mL) | < 0.91 | 0.00–7.00 | Anti-CMV | (–) | (–) |
| CA19-9 (U/mL) | 28.50 | 0.00–27.00 | EBV DNA (copies/mL) | < 5.00E3 | < 5.00E3 |
| Ceruloplasmin (mg/L) | 347.00 | 200.00–600.00 | Anti-Sarkozy virus | (–) | (–) |
TP, total protein; TBIL, total bilirubin; DBIL, direct bilirubin; AST, aspartate aminotransferase; ALT, alanine transaminase; ALP, alkaline phosphatase; GGT, gamma-glutamyl transferase; Cr, creatinine; Cys-c, cystatin C; CRP, C-reactive protein; TBA, total bile acid; TSH, thyroid stimulating hormone; FT3, free T3; FT4, free T4; Fe, ferrum; AFP, alpha fetoprotein; CA 19-9, carbohydrate antigen 19-9; WBC, white blood cell; RBC, red blood cell; Hb, hemoglobin; Hct, hematocrit; PLT, platelet; PT, prothrombin time; INR, international normalized ratio; PTA. prothrombin time activity; ANA, antinuclear antibodies; AMA, anti-mitochondrial antibody; AMA-M2, anti-mitochondrial M2 antibody; HAV, hepatitis A virus; HBsAg, hepatitis B surface antigen; HCVAb, hepatitis C virus antibody; HEV, hepatitis E antibody; CMV, cytomegalovirus; EBV, Epstein-Barr virus; DNA, deoxyribonucleic acid.
FIGURE 1.
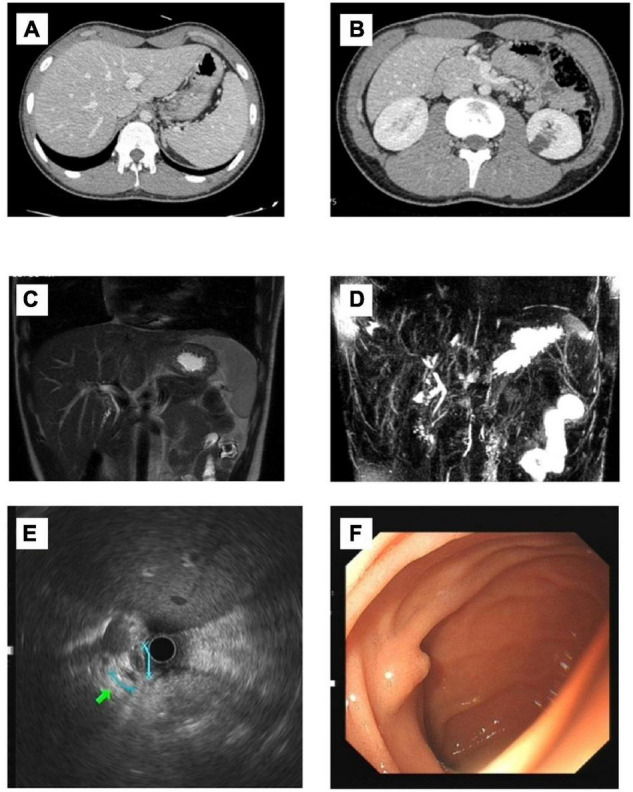
Imaging findings of the patient. (A,B) CT: No pathological findings were found. No dilatation of intrahepatic and external bile ducts was noted. There was no thickening or enhancement of extrahepatic bile ducts. The gallbladder is collapsed without bile filling; (C,D) MRCP: Possible stenosis at the beginning of the common hepatic duct. (E) Endoscopic ultrasonography: The extrahepatic bile duct was fine without dilation, and no definite obstruction was observed. (F) Gastroscopy: The size and morphology of the duodenal papilla were normal.
To clarify the cause of cholestasis, liver biopsy was performed and sections stained with hematoxylin-eosin (HE), Masson, mesh, PAS, DPAS, iron, CK7, CK19, CD10 and bile salt export pump (BSEP). Liver biopsy revealed a clear lobular structure. The main lesions were cholestasis with hepatocytes in central lobules II and III, bile embolism of capillary bile ducts and cholestasis with Kupffer cells, and positive staining for CK7 in some hepatocytes in the lobule (Figure 2). Pathological-clinical diagnosis is simple cholestasis. Genetic testing was performed on the patient and his parents. The patient’s genetic test revealed a rare missense mutation ATP8B1 rs540027832 (chr18-55328450 c.2663C > A p. T888K NM_00560 3.4) (Supplementary Table 1), and genetic tests on his parents showed that the mutation was inherited from his father (Supplementary Figure 1).
FIGURE 2.

Histological findings of liver biopsy in the patient. (A) HE staining at 200× magnification; (B) HE staining at 400× magnification; (C) CK7 staining at 200× magnification; (D) Masson staining at 200× magnification. The lobule structure is clear. The main lesions were cholestasis with hepatocytes in central lobules II and III (red arrow), bile embolism with capillary bile ducts and cholestasis with Kupffer cells. No obvious inflammatory necrosis was observed in the lobules. No enlargement in the sink area or obvious inflammatory cell infiltration was noted. No interfacial inflammation was observed. A small bile duct can be identified. The epithelium of the bile duct is arranged in an orderly manner, and there is no bile duct reaction around the sink area. There was no fibrous tissue proliferation in the interstitium of the portal area, and the portal veins were discernible.
The patient’s clinical manifestations and related examination results conformed to BRIC1. Drug-induced liver injury cannot be excluded. The patient was treated with glycyrrhizin, ursodeoxycholic acid (UDCA), glucocorticoids and haemofiltration. After 5 rounds of haemofiltration, the patient’s symptoms gradually improved, and the bilirubin index gradually decreased. After follow-up, bilirubin gradually decreased to normal within 3 months.
Discussion
We report a patient carrying a rare heterozygous mutation of APT8B1, whose symptoms improved after drug therapy and blood purification. We searched some databases including Clin var, Leiden open variation database, NCBI Gene and gnomAD database. We found only one African male with the same mutation was included in gnomAD database. Besides, there has another variant p.T888M in the same position described, indicates that the mutation has more than one allele. ATP8B1 rs540027832 p.T888M is also a rare mutation, with one European and one East Asian present in gnomAD database. ATP8B1 rs540027832 p.T888K and p.T888M were present at frequencies of 0.000398% (1/251360 alleles) and 0.0007074% (2/282738 alleles), respectively, and never appeared in homozygous status in the gnomAD frequency database. Based on the clinical characteristics, pathological and genetic mutation changes, this patient was ultimately highly suspected of being BRIC1.
ATP8B1 mutations can manifest as a range of diseases with BRIC1 and progressive familial cholestasis (PFIC)1 representing the two extremes of the phenotype. The development of PFIC1 in BRIC1 patients has been reported clinically (4). Intrahepatic cholestasis of pregnancy (ICP) is also associated with ATP8B1 mutations (5). This patient may have an intermediate stages from BRIC1 to PFIC1. Due to its late onset and good results after treatment, we believe that the diagnosis of BRIC is the preferred choice, and the specific diagnosis still needs further follow-up and observation.
Clinical cases of BRIC1 caused by missense mutation of ATP8B1 have been reported. Cases (6, 7) from Korea and Japan with novel heterozygous mutations leading to BRIC1 were reported, showing that BRIC1, as an autosomal dominant disease, may also caused by ATP8B1 heterozygous mutations. Another report has summarized the genetic mutations in the 180 families of BRIC1 or PFIC1 cases (8). The results showed that missense mutations were more common in BRIC, while nonsense, frameshift, and large deletion mutations were more common in PFIC. We have not found any cases of disease caused by this mutation in the past. Our genetic test results can cover 99.9% of the target area. Undetected variation outside the detection range cannot be excluded. We focuses on summarizing the pathophysiology and clinical manifestations of BRIC1 and perspectives for the future development direction.
ATP8B1 is located in the tubular membrane of hepatocytes, and the encoded FIC1 protein has 10 transmembrane domains that participate in the transport of phospholipids in the membrane and turn phosphatidylserine (PS) from the outer lipid leaflets back to the inner lipid leaflets of the tubular membrane to maintain the asymmetry and fluidity of the tubular membrane (9). ATP8B1 and the transmembrane protein CDC50A form a heterodimer complex that promotes its correct transport to the plasma membrane (10). ATP8B1 mutations can affect its stability and its interaction with CDC50A (11, 12). ATP8B1 mutations reduced BSEP activity and impaired bile excretion by affecting PS turnover (13). In ATP8B1 deficient patients, the nuclear translocation of the Farnesoid X receptor (FXR), a transcription factor that controls bile acid homeostasis, is disrupted, and BSEP expression on the hepatic duct membrane is reduced due to its transcriptional inhibition (14, 15) (Figure 3).
FIGURE 3.

Molecular mechanisms underlying cholestasis associated with ATP8B1 deficiency. ATP8B1 consists of 10 transmembrane segments, and ATP8B1 and CDC50A assemble to form a heterodimer complex that participates in PS flipping. BESP is a canalicular bile salt transporter. FXR is a nuclear receptor involved in regulating bile acid metabolism. When ATP8B1 is defective, the nuclear translocation of FXR is disrupted, and BSEP expression on the hepatic duct membrane is reduced due to its transcriptional inhibition. *Dotted line indicated a negative effect.
The pathological findings of BRIC1 are non-specific. At the onset, BRIC1 presents as centrilobular cholestasis, in which bile deposits are noted in tubules, hepatocytes and Kupffer cells, and bile embolism may occur in the tubules (16). ATP8B1 detection is particularly important for the diagnosis of BRIC1. In response, resequencing chips have been developed specifically to look for genetic syndromes of cholestasis, which can aid in diagnosis (17). Luketic and Shiffman (16) proposed BRIC diagnostic criteria. For patients with intrahepatic cholestasis of unknown cause, when the known common causes cannot explain the patient’s condition, the possibility of the disease should be considered, and liver biopsy and genetic testing should be pursued.
Although BRIC rarely develops into advanced liver disease, repeated episodes can lead to a significant decline in quality of life. The main aim of treatment is to reduce the frequency of attack and prevent recurrence. Vitamins, UDCA, rifampicin, cholestyramine and corticosteroids (18–22) are currently the main pharmacologic treatments for BRIC. Various new treatment methods are constantly being proposed. Inhibition of ileal bile acid transporter can interrupt hepatoenteric circulation and reduce blood bile acid to relieve pruritus. IBAT inhibitors can be used as a non-invasive method to relieve cholestasis symptoms (23). Endoscopic nasobiliary drainage (ENBD) is a method to improve cholestasis, which can be used in BRIC patients with refractory pruritus during long-term cholestasis attacks. Molecular absorbent recirculating system (MARS) therapy is safe in the treatment of refractory pruritus in BRIC patients and can effectively reduce the biochemical indicators of cholestasis. If MARS is not successful, plasma exchange can be combined (24).
Haploinsufficiency (HI) is defined as insufficient function to maintain a wild-type phenotype in the presence of one wild-type allele and one mutant allele. The relationship between genotype and phenotype is not linear, and the specific function of a gene determines its sensitivity to dose change (25). Previous report (8) showed that different clinical symptoms in FIC patients may be related to the degree of gene mutation and heterozygous mutations are more common in BRIC1, indicates that HI may play a role in FIC, which deserves further investigation and discussion.
Conclusion
The severity of disease caused by the ATP8B1 mutation is related to the severity of the ATP8B1 mutation and the function of the residual ATP8B1. However, the severity is not completely proportional to the reduced expression levels of ATP8B1 protein. In the future, a reliable ATP8B1 protein function detection method is needed to evaluate the severity and prognosis of disease. Research has found that human peripheral blood monocyte-derived macrophages (HMDMs) can be used evaluate ATP8B1 function (15, 26).
New treatments are being proposed continually. ATP8B1 defects lead to cystic fibrosis transmembrane conductance regulator (CFTR) downregulation (27). Targeted damage to the plasma membrane of ATP8B1 caused by I661T, the most common ATP8B1 disease mutation in European patients, was proven to be resolved by the CFTR corrector (28). Protein homeostasis regulators therefore represent a possible therapeutic strategy. Hepatocyte transplantation has been shown to correct pathological changes in PFIC3 model mice (29), but no human experimental studies have been conducted to date. However, 4-phenylbutyrate can be used as a chemical partner of the fold-defect variant BSEP and has been successfully used in BRIC2 (30). In addition, whether regulatory factors are available that can be used evaluate and regulate disease phenotypes still needs to be further explored.
Gene therapy corrects faulty genes that cause diseases to develop. The use of viral vectors for gene therapy has been proposed in PFIC3 (31, 32), and this methodology may be applied in the treatment of patients with clinically specific mutations of PFIC and BRIC in the future. Compensatory modified U1 snRNA is complementary to the mutated donor splicing site and exhibits great therapeutic potential as a new therapeutic strategy for ATP8B1 defects as well as other genetic diseases (33, 34). In the future, how to regulate the expression of pathogenic genes and provide targeted individualized treatment for the functional defects of pathogenic proteins, such as gene therapy with viral vectors and protein homeostasis regulators, are worthy of our efforts.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
HB drafted the script and conducted literature research. BC carried out critical revision of the manuscript for important intellectual content and final approval of the manuscript. Y-LL, DL, and CZ reviewed literature and summarized information. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank our pathology Xuyong Lin for their cooperation in discussing and sharing the pathology pictures.
Abbreviations
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ATP8B1
ATPase class I, type 8B, member 1
- BRIC
benign recurrent intrahepatic cholestasis
- BSEP
bile salt export pump
- CFTR
cystic fibrosis transmembrane conductance regulator
- CT
computed tomography
- DBIL
direct bilirubin
- ENBD
endoscopic nasobiliary drainage
- FIC1
familial intrahepatic cholestasis 1
- FXR
farnesoid X receptor
- GGT
gamma-glutamyltransferase
- HE
hematoxylin-eosin
- HI
haploinsufficiency
- HMDMs
human peripheral blood monocyte-derived macrophages
- ICP
intrahepatic cholestasis of pregnancy
- MARS
molecular absorbent recirculating system
- MRCP
magnetic resonance cholangiopancreatography
- PBC
primary biliary cholangitis
- PS
phosphatidylserine
- PFIC
progressive familial intrahepatic cholestasis
- TBA
total bile acid
- TBIL
total bilirubin
- UDCA
ursodeoxycholic acid.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.897108/full#supplementary-material
References
- 1.Piazzolla M, Castellaneta N, Novelli A, Agolini E, Cocciadiferro D, Resta L, et al. Nonsense variant of ATP8B1 gene in heterozygosis and benign recurrent intrahepatic cholestasis: a case report and review of literature. World J Hepatol. (2020) 12:64–71. 10.4254/wjh.v12.i2.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agarwal S, Lal BB, Rawat D, Rastogi A, Bharathy KGS, Alam S. Progressive familial intrahepatic cholestasis (PFIC) in indian children: clinical spectrum and outcome. J Clin Exp Hepatol. (2016) 6:203–8. 10.1016/j.jceh.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halawi A, Ibrahim N, Bitar R. Triggers of benign recurrent intrahepatic cholestasis and its pathophysiology: a review of literature. Acta Gastro Enterol Belgica. (2021) 84:477–86. 10.51821/84.3.013 [DOI] [PubMed] [Google Scholar]
- 4.van Ooteghem NAM, Klomp LWJ, van Berge-Henegouwen GP, Houwen RHJ. Benign recurrent intrahepatic cholestasis progressing to progressive familial intrahepatic cholestasis: low GGT cholestasis is a clinical continuum. J Hepatol. (2002) 36:439–43. 10.1016/S0168-8278(01)00299-9 [DOI] [PubMed] [Google Scholar]
- 5.Vitale G, Gitto S, Vukotic R, Raimondi F, Andreone P. Familial intrahepatic cholestasis: new and wide perspectives. Digest Liver Dis. (2019) 51:922–33. 10.1016/j.dld.2019.04.013 [DOI] [PubMed] [Google Scholar]
- 6.Lee YS, Kim MJ, Ki CS, Lee YM, Lee Y, Choe YH, et al. Benign recurrent intrahepatic cholestasis with a single heterozygote mutation in the ATP8B1 gene. Pediatr Gastroenterol Hepatol Nutr. (2012) 15:122–6. 10.5223/pghn.2012.15.2.122 [DOI] [Google Scholar]
- 7.Suzuki H, Arinaga-Hino T, Sano T, Mihara Y, Kusano H, Mizuochi T, et al. Case report: a rare case of benign recurrent intrahepatic Cholestasis-Type 1 with a novel heterozygous pathogenic variant of ATP8B1. Front Med. (2022) 9:891659. 10.3389/fmed.2022.891659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klomp LWJ, Vargas JC, van Mil SWC, Pawlikowska L, Strautnieks SS, van Eijk MJT, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. (2004) 40:27–38. 10.1002/hep.20285 [DOI] [PubMed] [Google Scholar]
- 9.Sticova E, Jirsa M, Pawłowska J. New insights in genetic cholestasis: from molecular mechanisms to clinical implications. Can J Gastroenterol. (2018) 2018:1–12. 10.1155/2018/2313675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paulusma CC, Folmer DE, Ho-Mok KS, de Waart DR, Hilarius PM, Verhoeven AJ, et al. ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology. (2008) 47:268–78. 10.1002/hep.21950 [DOI] [PubMed] [Google Scholar]
- 11.Folmer DE, van der Mark VA, Ho-Mok KS, Oude Elferink RPJ, Paulusma CC. Differential effects of progressive familial intrahepatic cholestasis type 1 and benign recurrent intrahepatic cholestasis type 1 mutations on canalicular localization of ATP8B1. Hepatology. (2009) 50:1597–605. 10.1002/hep.23158 [DOI] [PubMed] [Google Scholar]
- 12.Koh S, Takada T, Kukuu I, Suzuki H. FIC1-mediated stimulation of FXR activity is decreased with PFIC1 mutations in HepG2 cells. J Gastroenterol. (2009) 44:592–600. 10.1007/s00535-009-0041-y [DOI] [PubMed] [Google Scholar]
- 13.Andersen JP, Vestergaard AL, Mikkelsen SA, Mogensen LS, Chalat M, Molday RS. P4-ATPases as phospholipid flippases—structure, function, and enigmas. Front Physiol. (2016) 7:275. 10.3389/fphys.2016.00275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen F, Ananthanarayanan M, Emre S, Neimark E, Bull LN, Knisely AS, et al. Progressive familial intrahepatic cholestasis, type 1, is associated with decreased farnesoid X receptor activity. Gastroenterology. (2004) 126:756–64. 10.1053/j.gastro.2003.12.013 [DOI] [PubMed] [Google Scholar]
- 15.Mizutani A, Sabu Y, Naoi S, Ito S, Nakano S, Minowa K, et al. Assessment of adenosine triphosphatase phospholipid transporting 8B1 (ATP8B1) function in patients with cholestasis with ATP8B1 deficiency by using peripheral blood Monocyte-Derived macrophages. Hepatol Commun. (2021) 16:255. 10.1002/hep4.1605/suppinfo [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luketic VA, Shiffman ML. Benign recurrent intrahepatic cholestasis. Clin Liver Dis. (2004) 8:133–49. 10.1016/S1089-3261(03)00133-8 [DOI] [PubMed] [Google Scholar]
- 17.Liu C, Aronow BJ, Jegga AG, Wang N, Miethke Al, Mourya R, et al. Novel resequencing chip customized to diagnose mutations in patients with inherited syndromes of intrahepatic cholestasis. Gastroenterology. (2007) 132:119–26. 10.1053/j.gastro.2006.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beuers U. Drug Insight: mechanisms and sites of action of ursodeoxycholic acid in cholestasis. Nat Clin Pract Gastroenterol Hepatol. (2006) 3:318–28. 10.1038/ncpgasthep0521 [DOI] [PubMed] [Google Scholar]
- 19.Chen H, Wu D, Jiang W, Lei T, Lu C, Zhou T. Case report: a novel homozygous variant identified in a chinese patient with benign recurrent intrahepatic Cholestasis-Type 1. Front Med. (2021) 8:705489. 10.3389/fmed.2021.705489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Dijk R, Kremer AE, Smit W, van den Elzen B, van Gulik T, Gouma D, et al. Characterization and treatment of persistent hepatocellular secretory failure. Liver Int. (2015) 35:1478–88. 10.1111/liv.12603 [DOI] [PubMed] [Google Scholar]
- 21.Koukoulioti E, Ziagaki A, Weber SN, Lammert F, Berg T. Long-term colestyramine treatment prevents cholestatic attacks in refractory benign recurrent intrahepatic cholestasis type 1 disease. Hepatology. (2021) 74:522–4. 10.1002/hep.31671 [DOI] [PubMed] [Google Scholar]
- 22.Arthur Lorio E, Valadez D, Alkhouri N, Loo N. Cholestasis in Benign Recurrent Intrahepatic Cholestasis 2. ACG Case Rep J. (2020) 7:e412. 10.14309/crj.0000000000000412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamath BM, Stein P, Houwen RHJ, Verkade HJ. Potential of ileal bile acid transporter inhibition as a therapeutic target in Alagille syndrome and progressive familial intrahepatic cholestasis. Liver Int. (2020) 40:1812–22. 10.1111/liv.14553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schoeneich K, Frimmel S, Koball S. Successful treatment of a patient with benign recurrent intrahepatic cholestasis type 1 with albumin dialysis. Artif Organs. (2020) 44:341–2. 10.1111/aor.13572 [DOI] [PubMed] [Google Scholar]
- 25.Johnson AF, Nguyen HT, Veitia RA. Causes and effects of haploinsufficiency. Biol Rev. (2019) 94:1774–85. 10.1111/brv.12527 [DOI] [PubMed] [Google Scholar]
- 26.Hayashi H, Naoi S, Togawa T, Hirose Y, Kondou H, Hasegawa Y, et al. Assessment of ATP8B1 deficiency in pediatric patients with cholestasis using peripheral blood Monocyte-Derived macrophages. Ebiomedicine. (2018) 27:187–99. 10.1016/j.ebiom.2017.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Demeilliers C, Jacquemin E, Barbu V, Mergey M, Paye F, Fouassier L, et al. Altered hepatobiliary gene expressions in PFIC1: ATP8B1 gene defect is associated with CFTR downregulation. Hepatology. (2006) 43:1125–34. 10.1002/hep.21160 [DOI] [PubMed] [Google Scholar]
- 28.van der Woerd WL, Wichers CGK, Vestergaard AL, Andersen JP, Paulusma CC, Houwen RHJ, et al. Rescue of defective ATP8B1 trafficking by CFTR correctors as a therapeutic strategy for familial intrahepatic cholestasis. J Hepatol. (2016) 64:1339–47. 10.1016/j.jhep.2016.02.001 [DOI] [PubMed] [Google Scholar]
- 29.De Vree JML, Ottenhoff R, Bosma PJ, Smith AJ, Aten J, Oude Elferink RPJ. Correction of liver disease by hepatocyte transplantation in a mouse model of progressive familial intrahepatic cholestasis. Gastroenterology. (2000) 119:1720–30. 10.1053/gast.2000.20222 [DOI] [PubMed] [Google Scholar]
- 30.Hayashi H, Naoi S, Hirose Y, Matsuzaka Y, Tanikawa K, Igarashi K, et al. Successful treatment with 4-phenylbutyrate in a patient with benign recurrent intrahepatic cholestasis type 2 refractory to biliary drainage and bilirubin absorption. Hepatol Res. (2015) 46:192–200. 10.1111/hepr.12561 [DOI] [PubMed] [Google Scholar]
- 31.Felzen A, Verkade HJ. The spectrum of progressive familial intrahepatic cholestasis diseases: update on pathophysiology and emerging treatments. Eur J Med Genet. (2021) 64:104317. 10.1016/j.ejmg.2021.104317 [DOI] [PubMed] [Google Scholar]
- 32.Siew SM, Cunningham SC, Zhu E, Tay SS, Venuti E, Bolitho C, et al. Prevention of cholestatic liver disease and reduced tumor igenicity in a murine model of PFIC Type 3 Using Hybrid AAV-piggy Bac gene therapy. Hepatology. (2019) 70:2047–61. 10.1002/hep.30773 [DOI] [PubMed] [Google Scholar]
- 33.van der Woerd WL, Houwen RH, van de Graaf SF. Current and future therapies for inherited cholestatic liver diseases. World J Gastroentero. (2017) 23:763. 10.3748/wjg.v23.i5.763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Woerd WL, Mulder J, Pagani F, Beuers U, Houwen RHJ, van de Graaf SFJ. Analysis of aberrant pre-messenger RNA splicing resulting from mutations in ATP8B1 and efficientin vitro rescue by adapted U1 small nuclear RNA. Hepatology. (2015) 61:1382–91. 10.1002/hep.27620 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.