

Abstract
Cisplatin is a common metal platinum complex. The platinum atom in the molecule is of great significance to its antitumor effect. Clinically, it can show curative effect on a variety of solid tumors. However, cisplatin has certain adverse effects in treatment, one among which is acute renal injury (AKI). Except for the nuclear DNA damage caused by cisplatin, damage of organelles, and cytoplasm also contribute to side effects. Endoplasmic reticulum stress, mitochondrial apoptosis pathway or cascade reaction caused by complement and caspase protein also play important roles in cisplatin induced renal injury. Therefore, the damage studies of organelles and cytoplasm are also necessary for exploring adverse effects of cisplatin. This paper reviews the damage of endoplasmic reticulum, mitochondria, and indirect DNA apoptosis pathways induced by cisplatin. It also explains in detail why cisplatin is easy to cause kidney damage. Deep understanding of such interactions could be helpful to exploit better drugs which would minimize kidney injury and maximize anti-tumor effects of cisplatin.
Keywords: cisplatin, endoplasmic reticulum stress, mitochondrial OS, apoptosis protein caspase, immune complement, cytotoxicity
Introduction
Cisplatin or (SP-4-2) - dichloroamine platinum (II) is one of the most potential and widely used chemical drugs for the treatment of varieties of solid tumors (Fig. 1).1 However, till now the clinical application of cisplatin is still limitd because of its side effects and drug resistance.
Fig. 1.
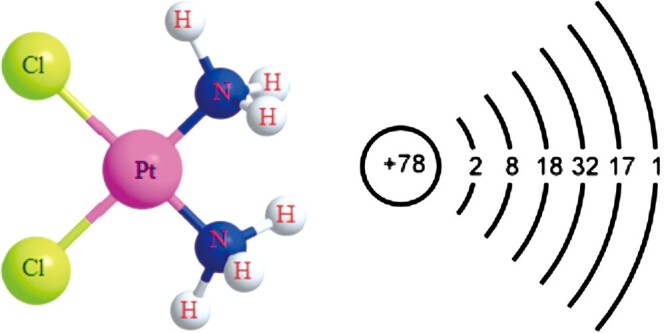
Molecular structure of cisplatin1 and atomic structure of platinum.
The key of anti-tumor effect of cisplatin is damage to DNA of tumor, which also lead to renal injury.2,3 Some reports link cisplatin poisoning with organelle dysfunction, such as endoplasmic reticulum, cytoskeleton, and mitochondria, both in vivo and in vitro. Apoptosis and necrosis of renal tubular cells play a major role in cisplatin induced acute renal injury,4 which is related to infiltration of inflammatory cells,5 regulation of p53 on apoptosis,6 oxidative stress,34 endoplasmic reticulum stress.10 Although cisplatin has obvious nephrotoxicity, so far, the antitumor effect of cisplatin in platinum anticancer drugs cannot be replaced. A correct understanding of its roles is necessary for the clinical therapy and basic experimental research.
Endoplasmic reticulum stress
As an important organelle in cells, endoplasmic reticulum plays an important role in the steady-state processes of protein synthesis, processing, folding, modification and degradation.7 Endoplasmic reticulum stress means that under pathological conditions, cells may lose protease deposition, It will lead to unfolded protein response (UPR), due to the increase of unfolded proteins in endoplasmic reticulum and the accumulation of misfolded proteins.
Some studies have shown that endoplasmic reticulum stress is related to cisplatin induced apoptosis of renal cells and tissues. For instance, It has been found that caspase-12 plays a significant role in cisplatin induced apoptosis of LLC-PK1 renal tubular epithelial cells.8 Caspase 12 mainly exists in the endoplasmic reticulum and mediates endoplasmic reticulum stress-related cell death. Caspase-12 in the activation of innate immune response, pro-inflammatory caspase mediates the maturation of specific cytokines such as pro-IL-1 β And pro-IL-18 to form active IL-1 β and IL-18, which mediates the occurrence of cellular inflammation. But it may also be involved in a form of cell death called pyroptosis. Other studies have shown the blocking effect of kca3. 1 potassium channel protects cisplatin induced AKI through mitochondrial and endoplasmic reticulum stress pathways that attenuate apoptosis9 and cisplatin affects intracellular calcium signaling through the stress on endoplasmic reticulum (ER).10 By increasing intracellular calcium levels, caspase-1 is activated and lysosomes are further activated.11 This mechanism affects the lysosomal degradation of misfolded proteins and further aggravates the unfolded protein response. This effect is not dependent on IL-1 β and IL-18, but is mediated by caspase 7 activation of caspase 1.12 These experiments show that the ion channel plays an important role in causing endoplasmic mesh stress and preventing cisplatin kidney damage.
Mitochondrial pathway
Relationship between cisplatin and mitochondria
Another important cytoplasmic target of cisplatin is considered to be mitochondria. Mitochondria supply energy through tricarboxylic acid cyclic oxidation phosphorylation and fatty acid oxidation. Moreover, they can maintain the steady-state level of Ca2+, reduce the equivalent carrier, and participate in the internal apoptotic signal pathway regulating cell death.13,14 And the experiment showed a high correlation between cell sensitivity to cisplatin and cell mitochondrial density. Compared with other tubules, injury of proximal tubule cells which have the highest density of mitochondria are most severe, which is a common feature in cisplatin-induced injury. Cisplatin can enter into cells through passive diffusion, active transport, and promoting diffusion. However, active transport and diffusion can make cisplatin accumulate in specific cells or specific organelles, thus aggravating local damage. Particularly, Copper transporters (Ctr1 and ctr2) can participate in drug uptake by facilitated diffusion and transport cisplatin to cells.15,26 Cisplatin enters the cell through the copper transporter proteins and plays a role in the cytoplasm, organelles and nucleus of the cell. However, cisplatin will reduce the concentration of Ctr1 and the accumulation of cisplatin in cancer cells, which will reduce the efficacy of the drug. In animal models, Ctr1 was found to be highly expressed in both proximal and distal renal tubules, and most of it was located outside the basement membrane.17 Higher cisplatin accumulation can be obtained in cells with higher Ctr1 expression, which makes cells with high Ctr1 expression more sensitive to cisplatin.18 Moreover, copper transporters play an important role in the transport of cell membrane and mitochondria, this mechanism may make cisplatin easy to deposit in mitochondria.
Experimental studies show that negatively charged mitochondria attract positively charged cisplatin. Compared with cisplatin sensitive cells, Ovarian tumor cells have low mitochondrial membrane potential, which limits the entry of cisplatin into mitochondria, so they have certain cisplatin resistance.19 The mitochondrial DNA damage of cisplatin-resistant cells was also small. The renal cells sensitive to cisplatin have higher mitochondrial membrane potential, which makes cisplatin easier to enter their mitochondria and cause more serious damage.
Relationship between cisplatin and mtDNA
Previous studies have shown that only a small amount of cisplatin interacts with nuclear DNA, while the largest amount of cisplatin interacts with protein and mitochondrial DNA.20 The difference between mitochondrial DNA and nuclear DNA is that mitochondrial DNA lacks histone, which makes it more vulnerable to free radicals. Mitochondrial DNA lacks effective DNA repair mechanism, which also makes it more vulnerable to damage. DNA repair mechanism is an important part of cisplatin cytotoxicity. Once cisplatin enters into the cell, one of the two chlorine atoms in cisplatin is hydrated, making it easier to enter nucleus, organelles and react with the DNAs. In addition, In vitro studies have confirmed that monoaquated platinum is more active for DNA binding than diaquated platinum.21 Due to the unsuccessful DNA repair process, this causes DNA damage, which activates DNA protection and repair mechanisms.10,18 The pro apoptotic pathway is triggered, resulting in the activation of caspases (including caspase-3 and -7), which leads to apoptosis.22 Interestingly, They can also induce necrosis through the loss of membrane unity23 (Fig. 2). However, it is worth mentioning that studies have reported that secondary necrosis and focal death caused by cisplatin are related to NLRP1, but not NLRP3.24 The role of other NLR members, including AIM1 and NLRC4, in cisplatin induced AKI is not clear.
Fig. 2.
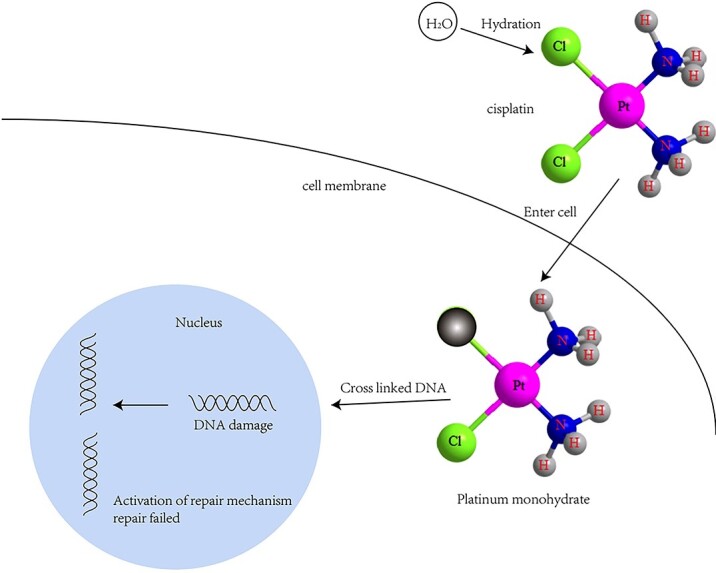
DNA damage pathway after cisplatin administration1 (reproduced with permission, copyright @Elsevier, 2019).
Studies have shown that ROS together with Bax (Bcl2 related x) and Ca2+ lead to mitochondrial DNA damage and repair, and reduce mitochondrial permeability transition.25 This phenomenon promotes mitochondrial rupture.26 In addition, p53 inhibits the anti apoptotic protein on the mitochondrial membrane.27 For example, B-cell lymphoma (Bcl) anti apoptotic protein family. Bcl family proteins are indispensable for keeping mitochondrial function and integrity28 through p53 activation,29 which can aggravate nephrotoxicity.
Results of mitochondrial damage
Mitochondrial dysfunction can lead to a wide range of effects. Dysfunction leads to a decrease in ATP synthesis, which forces stimulated cells to play a role in starvation mode. Mitochondrial rupture releases cytochrome c and pro-caspase-9, which combine with cytoplasmic Apaf-1 and ATP to form an apoptotic complex and activate caspase-9.26 It activates caspase mediated apoptosis by releasing caspase-9 mediators. Peroxidation in the process of lipid metabolism, hypoxia reperfusion injury of cisplatin to local renal vessels and damage in the process of ATP formation lead to the production of reactive oxygen species or free radicals30 (Fig. 3).
Fig. 3.
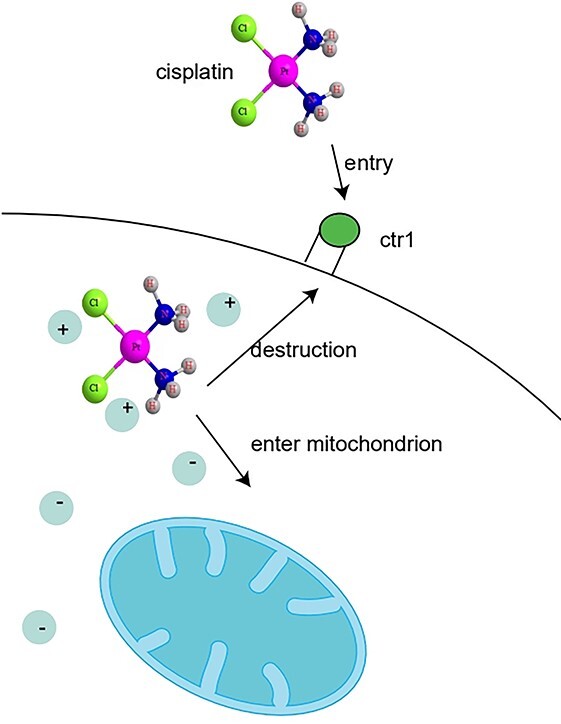
Pathway of kidney injury caused by cisplatin in mitochondria1 (reproduced with permission, copyright @Elsevier, 2019).
Oxidative stress
Oxidative stress also plays a special role in cisplatin induced nephrotoxicity, which is an imbalance between oxidation and antioxidation. Studies have shown that the toxic and side effects caused by cisplatin, such as liver and kidney toxicity, are closely related to the increase of reactive oxygen species.31 Under physiological and pathological conditions, reactive oxygen species are very important to maintain renal function and structure.32,33
Cisplatin induces oxidative stress by forming reactive oxygen species (ROS). NADPH oxidase is the main enzyme source of ROS in cisplatin nephrotoxicity, which is also known as NOXs, plays an important role in maintaining cell homeostasis.35,36 NADPH plays an important role in keeping glutathione (GSH) from being oxidized and is involved in maintaining the content of intracellular reduced GSH. It is worth noting that NOX4 (NADPH Oxidase 4) is mostly expressed in renal cells.37,38 This indicates that NOX4 plays an indispensable role in cisplatin induced renal injury. Therefore, the in-depth study of NOX4 will help to prevent cisplatin induced renal injury. In addition, cisplatin affects the mitochondrial respiratory chain and leads to the excessive production of reactive oxygen species. In kidney, cisplatin can combine with the abundant endogenous antioxidant glutathione in renal tubular cells to form glutathione conjugate. Then pass GGT and APN, which are cleaved into cysteine conjugates in renal tubular cells and further metabolized into active mercaptans in the proximal tubular segment of the kidney.39,40 These highly active mercaptans can combind to some important proteins in proximal renal tubular cells to form more toxic nephrotoxin.41 In addition, high activity mercaptan can produce a large amount of ROS in renal cells at the same time, which further aggravates renal injury. Cisplatin induced ROS accumulation and renal injury were attenuated in CYP2E1 deficient mice.42 It shows that CYP system plays an important role in the formation of ROS induced by cisplatin. In addition, PtII is a kind of soft acid. The cisplatin and molecular species produced by its hydrolysis will preferentially react with soft and easily polarized bases, such as glutathione.43 It leads to the reduction of cellular antioxidants, which leads to oxidative stress and cell death too.
The balance between the generation of active oxygen and the elimination of active oxygen in the human body is very precise. Oxidative stress can initiate autophagy of mitochondria, thus eliminating damaged mitochondria and maintaining homeostasis.44 Therefore, reactive oxygen species are the main mediators of phagocytes and play an important role in the normal survival of cells. On the other hand, excessive oxidative stress may lead to serious cell damage, and even organ failure.45 There may be a cascade amplification reaction in the transport of reactive oxygen species, which leads to the increase and rapid diffusion of reactive oxygen species.
Immune complement and caspase protein
One of the common causes of cisplatin induced renal injury also includes the dysfunction of upstream cell function. Excessive cell death caused by necrosis can trigger the inflammatory process and spread renal tubular injury. It has been reported that complement pathway can amplify the role of caspase pathway. Studies have identified that C3aR, the key mediator of Cpb1 – C3 – C3aR signaling pathway, plays a key role in the early production of proinflammatory mediators in vivo.46 It shows that serine protease and cysteine protease play the same important role in cisplatin induced renal injury.
Cisplatin activates the complement system and has a significant impact on mitochondria, which can be divided into direct impact and indirect impact. Indirect effects of complement activity on mitochondria. Under the stimulation of LPS, the activation of C3aR causes the quick release of cellular ATP to the outside of the cell. Extracellular ATP triggers P2X7 purinergic receptors on monocytes and macrophages in an autocrine manner, activates inflammatory bodies, and supports monocytes to produce pro-inflammatory IL-1.47 This shows that activation of IL-1β may be independent of the NLRP3 inflammasome in cisplatin-treated kidney. Therefore, the role of other inflammatory bodies in cisplatin-induced AKI deserves further study. Direct effects of complement activity on mitochondria, stimulating the expression of C5ar1 on the surface of neutrophils induces its activation, resulting in the production of reactive oxygen species.48 Therefore, intracellular C5 activation and C5aR1 involvement may need to be strictly regulated to maintain the activity of mitochondrial functional effectors supporting immune cells, but inhibit obvious cell damage and death at the same time (Fig. 4).
Fig. 4.
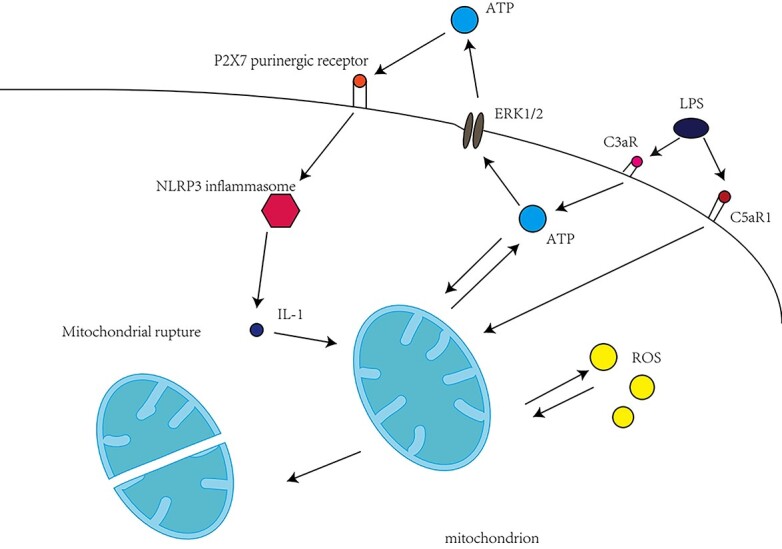
The role of complement in cisplatin induced nephrotoxicity.
The respiratory burst caused by complement will lead to the increase of ROS and the increase of cellular reactive oxygen species. It aggravates the damage of organelles and the nephrotoxicity of cisplatin, resulting in cascade amplification, which will have a serious impact on the whole kidney. Due to the important role of complement in cisplatin induced renal injury, the role of complement in cisplatin induced renal injury may become a research hotspot in the future.
Future trends
In summary, our understanding of how cisplatin causes renal injury has been deepened year by year. However, there are still relatively few studies on the toxic damage of mitochondria, endoplasmic reticulum, and other organelles caused by cisplatin. The complex effect of cisplatin on the organelles of renal cells limits people's understanding of it. Clarifying its toxicological effects on organelles and cytoplasm and the role of complement in damage will attract the attention of experimenters and the public to the rapid development of drug research and development, strategic decision-making, and human clinical experiments. It also lays a solid foundation for the in-depth study of cisplatin induced renal injury, the research and development of other platinum drugs.
Author contributions
Fig. 1 and Fig. 2 were provided by Qian Zhang. Fig. 3 was provided by Chunming Xu and Long Chen. The remaining portion was written by Guochen Huang and Hongxia Zhang.
Funding
This work was supported by the National Natural Science Foundation of China (81873883, 82000525), the Medical and Health Science and Technology Development Plan of Shandong Province (2018WS065), and Youth Innovation Technology in Colleges and Universities of Shandong Province of China (2021KJ106).
Conflict of interest
None declared.
Contributor Information
Guochen Huang, Department of Clinical Pathology, Weifang Medical University, Weifang 261053, Shandong Province, CN; Key Lab for Immunology in Universities of Shandong Province, Weifang Medical University, Weifang 261053, Shandong Province, CN.
Qian Zhang, Department of Clinical Pathology, Weifang Medical University, Weifang 261053, Shandong Province, CN; Key Lab for Immunology in Universities of Shandong Province, Weifang Medical University, Weifang 261053, Shandong Province, CN.
Chunming Xu, Department of Clinical Pathology, Weifang Medical University, Weifang 261053, Shandong Province, CN; Neurologic Disorders and Regenerative Repair Laboratory, Weifang Medical University, Weifang 261053, Shandong Province, CN.
Long Chen, Department of Clinical Pathology, Weifang Medical University, Weifang 261053, Shandong Province, CN; Neurologic Disorders and Regenerative Repair Laboratory, Weifang Medical University, Weifang 261053, Shandong Province, CN.
Hongxia Zhang, Department of Clinical Pathology, Weifang Medical University, Weifang 261053, Shandong Province, CN; Key Lab for Immunology in Universities of Shandong Province, Weifang Medical University, Weifang 261053, Shandong Province, CN; Neurologic Disorders and Regenerative Repair Laboratory, Weifang Medical University, Weifang 261053, Shandong Province, CN.
References
- 1. Ghosh S. Cisplatin: The first metal based anticancer drug. Bioorg Chem. 2019:88:102925. [DOI] [PubMed] [Google Scholar]
- 2. Cullen KJ, Yang Z, Schumaker L, Guo Z. Mitochondria as a critical target of the chemotheraputic agent cisplatin in head and neck cancer. J Bioenerg Biomembr. 2007:39(1):43–50. [DOI] [PubMed] [Google Scholar]
- 3. Mandic A, Hansson J, Linder S, Shoshan MC. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem. 2003:278(11):9100–9106. [DOI] [PubMed] [Google Scholar]
- 4. Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J Am Soc Nephrol. 2015:26(8):1765–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deng J, Kohda Y, Chiao H, et al. Interleukin-10 inhibits ischemic and cisplatin-induced acute renal injury. Kidney Int. 2001:60(6):2118–2128. [DOI] [PubMed] [Google Scholar]
- 6. Kim N, Min WK, Park MH, Lee JK, Jin HK, Bae JS. Neuropeptide Y protects kidney against cisplatin-induced nephrotoxicity by regulating p53-dependent apoptosis pathway. BMB Rep. 2016:49(5):288–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bravo R, Parra V, Gatica D, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol. 2013:301:215–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu H, Baliga R. Endoplasmic reticulum stress-associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. J Am Soc Nephrol. 2005:16(7):1985–1992. [DOI] [PubMed] [Google Scholar]
- 9. Chen CL, Liao JW, Hu OY, Pao LH. Blockade of KCa3.1 potassium channels protects against cisplatin-induced acute kidney injury. Arch Toxicol. 2016:90(9):2249–2260. [DOI] [PubMed] [Google Scholar]
- 10. Florea AM, Büsselberg D. Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel). 2011:3(1):1351–1371 Published 2011 Mar 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bergsbaken T, Fink SL, den Hartigh AB, Loomis WP, Cookson BT. Coordinated host responses during pyroptosis: caspase-1-dependent lysosome exocytosis and inflammatory cytokine maturation. J Immunol. 2011:187(5):2748–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Akhter A, Gavrilin MA, Frantz L, et al. Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog. 2009:5(4):e1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bustos G, Cruz P, Lovy A, Cárdenas C. Endoplasmic reticulum-mitochondria calcium communication and the regulation of mitochondrial metabolism in cancer: a novel potential target. Front Oncol. 2017:7:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zong WX, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell. 2016:61(5):667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ishida S, Lee J, Thiele DJ, Herskowitz I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc Natl Acad Sci U S A. 2002:99(22):14298–14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bodiga S, Vemuri PK, Bodiga VL. Low Ctr1p, due to lack of Sco1p results in lowered cisplatin uptake and mediates insensitivity of rho0 yeast to cisplatin. J Inorg Biochem. 2018:187:14–24. [DOI] [PubMed] [Google Scholar]
- 17. Pabla N, Murphy RF, Liu K, Dong Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am J Physiol Renal Physiol. 2009:296(3):F505–F511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014:740:364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hirama M, Isonishi S, Yasuda M, Ishikawa H. Characterization of mitochondria in cisplatin-resistant human ovarian carcinoma cells. Oncol Rep. 2006:16(5):997–1002. [PubMed] [Google Scholar]
- 20. Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003:22(47):7265–7279. [DOI] [PubMed] [Google Scholar]
- 21. Davies MS, Berners-Price SJ, Hambley TW. Slowing of cisplatin aquation in the presence of DNA but not in the presence of phosphate: improved understanding of sequence selectivity and the roles of monoaquated and diaquated species in the binding of cisplatin to DNA. Inorg Chem. 2000:39(25):5603–5613. [DOI] [PubMed] [Google Scholar]
- 22. Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006:12(9):440–450. [DOI] [PubMed] [Google Scholar]
- 23. Zhang CC, Li CG, Wang YF, et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis. 2019:24(3–4):312–325. [DOI] [PubMed] [Google Scholar]
- 24. Kim HJ, Lee DW, Ravichandran K, et al . NLRP3 inflammasome knockout mice are protected against ischemic but not cisplatin-induced acute kidney injury. J Pharmacol Exp Ther. 2013;346(3):465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997:18(1):44–51. [DOI] [PubMed] [Google Scholar]
- 26. Fuertes MA, Alonso C, Pérez JM. Biochemical modulation of Cisplatin mechanisms of action: enhancement of antitumor activity and circumvention of drug resistance. Chem Rev. 2003:103(3):645–662. [DOI] [PubMed] [Google Scholar]
- 27. Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochim Biophys Acta. 2009:1787(5):414–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sadhukhan P, Saha S, Dutta S, Sil PC. Mangiferin ameliorates cisplatin induced acute kidney injury by upregulating Nrf-2 via the activation of PI3K and exhibits synergistic anticancer activity with cisplatin. Front Pharmacol. 2018:9:638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang YC, Tsai MS, Hsieh PC, et al. Galangin ameliorates cisplatin-induced nephrotoxicity by attenuating oxidative stress, inflammation and cell death in mice through inhibition of ERK and NF-kappaB signaling. Toxicol Appl Pharmacol. 2017:329:128–139. [DOI] [PubMed] [Google Scholar]
- 30. Isnard-Bagnis C, Moulin B, Launay-Vacher V, Izzedine H, Tostivint I, Deray G. Toxicité rénale des anticancéreux [Anticancer drug-induced nephrotoxicity]. Nephrol Ther. 2005:1(2):101–114. [DOI] [PubMed] [Google Scholar]
- 31. Kaur T, Borse V, Sheth S, et al. Adenosine A1 receptor protects against cisplatin ototoxicity by suppressing the NOX3/STAT1 inflammatory pathway in the Cochlea. J Neurosci. 2016:36(14):3962–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holterman CE, Read NC, Kennedy CR. Nox and renal disease. Clin Sci (Lond). 2015:128(8):465–481. [DOI] [PubMed] [Google Scholar]
- 33. Nlandu-Khodo S, Dissard R, Hasler U, et al. NADPH oxidase 4 deficiency increases tubular cell death during acute ischemic reperfusion injury. Sci Rep. 2016:6:38598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chirino YI, Pedraza-Chaverri J. Role of oxidative and nitrosative stress in cisplatin-induced nephrotoxicity. Exp Toxicol Pathol. 2009:61(3):223–242. [DOI] [PubMed] [Google Scholar]
- 35. Wang Y, Luo X, Pan H, et al. Pharmacological inhibition of NADPH oxidase protects against cisplatin induced nephrotoxicity in mice by two step mechanism. Food Chem Toxicol. 2015:83:251–260. [DOI] [PubMed] [Google Scholar]
- 36. Schreck C, O'Connor PM. NAD(P)H oxidase and renal epithelial ion transport. Am J Physiol Regul Integr Comp Physiol. 2011:300(5): R1023–R1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sedeek M, Nasrallah R, Touyz RM, Hébert RL. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol. 2013:24(10):1512–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Geiszt M, Kopp JB, Várnai P, Leto TL. Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci U S A. 2000:97(14):8010–8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Townsend DM, Tew KD, He L, King JB, Hanigan MH. Role of glutathione S-transferase Pi in cisplatin-induced nephrotoxicity. Biomed Pharmacother. 2009:63(2):79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Townsend DM, Deng M, Zhang L, Lapus MG, Hanigan MH. Metabolism of cisplatin to a nephrotoxin in proximal tubule cells. J Am Soc Nephrol. 2003:14(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Qi L, Luo Q, Zhang Y, Jia F, Zhao Y, Wang F. Advances in toxicological research of the anticancer drug cisplatin. Chem Res Toxicol. 2019:32(8):1469–1486. [DOI] [PubMed] [Google Scholar]
- 42. Liu H, Baliga R. Cytochrome P450 2E1 null mice provide novel protection against cisplatin-induced nephrotoxicity and apoptosis. Kidney Int. 2003:63(5):1687–1696. [DOI] [PubMed] [Google Scholar]
- 43. Mezencev R. Interactions of cisplatin with non-DNA targets and their influence on anticancer activity and drug toxicity: the complex world of the platinum complex. Curr Cancer Drug Targets. 2015:14(9):794–816. [DOI] [PubMed] [Google Scholar]
- 44. Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002:82(1):47–95. [DOI] [PubMed] [Google Scholar]
- 45. Zorov DB, Plotnikov EY, Jankauskas SS, et al. The phenoptosis problem: what is causing the death of an organism? Lessons from acute kidney injury Biochemistry (Mosc). 2012:77(7):742–753. [DOI] [PubMed] [Google Scholar]
- 46. Napier BA, Brubaker SW, Sweeney TE, et al. Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity. J Exp Med. 2016:213(11):2365–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Asgari E, Le Friec G, Yamamoto H, et al. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. 2013:122(20):3473–3481. [DOI] [PubMed] [Google Scholar]
- 48. Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009:20(2):289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Howell SH. Endoplasmic reticulum stress responses in plants. Annu Rev Plant Biol. 2013:64:477–499. [DOI] [PubMed] [Google Scholar]