

Abstract
Selective targeting of TNF in inflammatory diseases such as rheumatoid arthritis (RA) has provided great therapeutic benefit to many patients with chronic RA. Although these therapies show initially high response rates, their therapeutic benefit is limited over the lifetime of the patient due to the development of antidrug antibodies that preclude proper therapeutic benefits. As a result, patients often return to more problematic therapies such as methotrexate or hydroxychloroquine, which carry long-term side effects. Thus, there is an unmet medical need to develop alternative treatments enabling patients to regain the benefits of selectively targeting TNF functions in vivo. The protein kinase TAK1 is a critical signaling node in TNF-mediated intracellular signaling, regulating downstream NF-κβ activation, leading to the transcription of inflammatory cytokines. TAK1 inhibitors have been developed but have been limited in their clinical advancement due to the lack of selectivity within the human kinome and, most importantly, lack of oral bioavailability. Using a directed medicinal chemistry approach, driven by the cocrystal structure of the TAK1 inhibitor takinib, we developed HS-276, a potent (Ki = 2.5 nM) and highly selective orally bioavailable TAK1 inhibitor. Following oral administration in normal mice, HS-276 is well tolerated (MTD >100 mg/Kg), displaying >95% bioavailability with μM plasma levels. The in vitro and in vivo efficacy of HS-276 showed significant inhibition of TNF-mediated cytokine profiles, correlating with significant attenuation of arthritic-like symptoms in the CIA mouse model of inflammatory RA. Our studies reinforce the hypothesis that TAK1 can be safely targeted pharmacologically to provide an effective alternative to frontline biologic-based RA therapeutics.
Graphical Abstract
■ INTRODUCTION
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease in which hyperactivated immune cells induce maladaptive and persistent inflammation in the joints, leading to synovial inflammation and bone remodeling.1 While this disease is without a singular cause, there exists a molecular signature exemplified by the increased release of a variety of proinflammatory cytokines. Of these, tumor necrosis factor (TNF) plays an integral role in regulating proinflammatory signaling, especially in the affected joints of RA patients.2 TNF is a potent cytokine involved in the regulation of normal inflammatory and immune system response. However, in RA, the overexpression of TNF has a pleiotropic effect on a variety of immune cells, including macrophages and activated T cells, stimulating the overproduction of other proinflammatory cytokines such as IL-1β, IL-6, and additional TNF.3
Aberrant TNF signaling in the joints of patients leading to downstream activation of NF-κβ and expression of inflammatory cytokines further enhances disease manifestation. Therapies targeting TNF such as humanized anti-TNF antibodies have revolutionized treatment options for patients and provide significant disease mitigation in a majority of patients.4 Unfortunately, the benefits of these biologic-based drugs cannot be sustained for the long term. Up to 34–53% of RA patients develop insufficient therapeutic benefit from frontline biologics such as Remicade and Humira due to the development of neutralizing antidrug antibodies (ADAs) to the anti-TNF antibodies themselves.5–8 The development of ADAs severely limits treatment efficacy and, in some cases, can exacerbate disease in these patients.9 Furthermore, patients who develop immune sensitivity to biologic-based drugs can no longer return to this line of therapy and are left to cycle on older and more problematic drugs such as methotrexate (MTX) or hydroxychloroquine.10 Clearly, controlling TNF signaling has therapeutic utility, and restoring the benefits of managing TNF in patients with chronic RA may provide superior therapeutic benefit. We hypothesize that targeting intracellular signal transduction pathways exclusively mediating TNF receptor signaling in vivo with small-molecule drugs represents a novel therapeutic axis, avoiding issues seen with biologic-based therapies.
The serine/threonine protein kinase TAK1 is a key regulatory signaling node in the TNF pathway, mediating both the prosurvival and inflammatory signaling pathways of TNF. The binding of TNF to the TNF receptor 1 (TNFR1) complex leads to the recruitment of inhibitor of NF-κβ kinase (IKK). TAK1 in complex with TAB1–3 then phosphorylates IKK, leading to the phosphorylation of inhibitor of NF-κβ kinase and allows translocation of NF-κβ to the nucleus and subsequent activation of transcription of prosurvival genes.11 Additionally, TAK1 phosphorylates MKK3/4/6, leading to p38 and cJun activation and subsequent translocation to the nucleus, enhancing the transcription of prosurvival and inflammatory cytokine genes.12 Genetic knockout studies in mice have shown that the loss of TAK1 function significantly attenuates TNF prosurvival/proinflammatory responses.13–16 These observations suggest that selective pharmacological inhibitors of TAK1 will locally block the release of proinflammatory cytokines and cell proliferation specifically at sites of inflammation.
To date, there have been very few examples of inhibitors of protein kinases that specifically target intracellular TNF-mediated signal transduction that have been successfully translated into clinical trials for RA. The closest examples are the JAK/STAT inhibitors such as tofacitinib, baricitinib, and upadacitinib that act by specifically targeting the local release of IL-6 at sites of inflammation.17,18 Recently, we reported on takinib (N-(1-propyl-1,3-dihydro-2H-benzo[d]imidazol-2-ylidene)isophthalamide), a potent (IC50 ≈ 9 nM) and extraordinary selective inhibitor of TAK1.19 In two independent kinome wide screens, the next sensitive protein kinase, IRAK4 (IC50 ≈ 120 nM), is 13-fold less potently inhibited. Prior screens for inhibitors of TAK1 by others identified the fungal metabolite 5(Z)-7-oxozeanol.14 However, this molecule also inhibits a panel of at least 50 other kinases and forms a covalent bond with its target, rendering it inadequate for therapeutic purposes. Medicinal chemistry groups used the chemical scaffold of 5(Z)-7-oxozeanol as a lead molecule to generate more selective inhibitors of TAK1. Their efforts identified additional molecules, namely, LYTAK1, PF-04358168, and AZ-TAK1; however, these remain broadly active across the kinome and have yet to be advanced further.14
Previous work by our group has shown that TAK1 inhibition significantly blocks TNF expression following inflammatory signaling in various immune cells.13 Our first-generation TAK1 inhibitor, takinib, showed poor bioavailability, greatly limiting its use to study the physiological effects of enzymatic inhibition of TAK1 in vivo. Here, we describe an iterative effort to develop an orally bioavailable analogue of TAK1 inhibitor takinib, which retains potency and high selectivity within the human kinome. Based on the cocrystal structure of takinib and TAK1 previously reported as well as the structure of closely related IRAK4, we derived HS-276. Following oral dosing, HS-276 shows >95% bioavailability, is well tolerated (MTD > 100 mg/kg), and exhibits dose-dependent efficacy in the CIA mouse model of human RA. Our findings suggest that TAK1 can be safely targeted, and inhibiting its protein kinase activity in response to TNF can have therapeutic benefit comparable to leading biologic-based anti-TNF drugs.
■ RESULTS
Development of a Selective Orally Bioavailable TAK1 Inhibitor.
Takinib was originally discovered as a potent and selective inhibitor of TAK1 using a combination of our laboratory’s Fluorescent-Linked Enzyme Chemoproteomic Strategy drug discovery platform coupled with a pan-kinase screen.19,20 Unfortunately, takinib had poor serum surveillance and exhibited some affinity for IRAK4, albeit 13-fold less than for TAK1.19 To improve the selectivity properties of takinib, we began an initial SAR study that showed that the primary amide (Figure 1a, pos. 21) was required and that substitution was not allowed on the isophthalamide ring (Figure 1a, pos. 17–19).19 Subsequent analysis of a cocrystal structure (5v5n) of takinib and TAK1 showed that the isophthalamide is snugly bound and that the benzimidazole (Figure 1b, pos. 1, 2, and N-7) is more solvent-exposed.19 Affinity resins derived from acrylates at positions 1 and 2 were therefore prepared, and interestingly, the resin from position 1 captured both TAK1 and IRAK4, whereas the resin from position 2 only captured TAK1 (Supplemental Figure 1a–c). This suggested that derivatives at position 2 might be selective for TAK1 over IRAK4. An overlap of the takinib-TAK1 crystal structure (5V5N, green) with an IRAK4 structure containing a similar ligand (4RMZ, blue) confirms accommodation of substitutions in the 2 position for TAK1 but not IRAK4 (Figure 1b).
Figure 1.
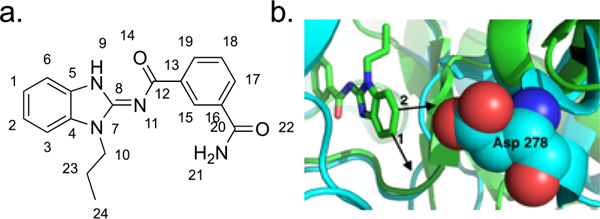
Development of takinib analogues. (a) Takinib atom positions. (b) Overlay of TAK1 (5V5N, blue) and IRAK4 (4RMZ, green) crystal structures with takinib.
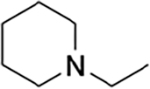
Guided by these observations and informed by previous work on IRAK4 inhibitors, we prepared a number of analogues using the synthetic scheme illustrated in Scheme 1.21 The potencies and selectivities of the analogues prepared are shown in Table 1. Generally, the activity against TAK1 was fairly consistent for all analogues. The hydroxymethyl analogues (HS-268, HS-275, HS-278, and HS-280), intermediates in the synthesis of the more soluble amine analogues, were quite insoluble and therefore not pursued. Interestingly, the substitution of a hydroxylmethyl at R2 is tolerated by IRAK4, suggesting that a larger substitution is necessary to drive selectivity. Addition of the piperidinomethyl substitution at R2, HS-276, showed the greatest selectivity against TAK1 with a 1000-fold preference for TAK1 over IRAK4 (Table 1). HS-277, at 168-fold, was somewhat less selective. Finally, HS-285, a by-product resulting from running the reductive amination in DMF, showed an intermediate level of selectivity. All of the amine products were quite water-soluble (>50 mg/mL) as formate salts (Supplemental Table 1).
Scheme 1. Synthetic Scheme for Takinib Analogue Development.
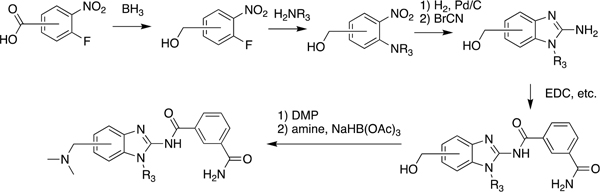
Table 1.
Potency and Selectivity of Takinib Analogues against TAK1 and IRAK4
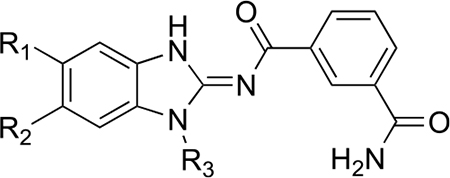
| ||||||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | TAK1 IC50 nM | IRAK4 IC50 nM | Ratio | |
| Takinib | H | H | propyl | 9 | 120 | 13 |
| HS-268 |
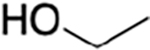
|
H |
|
nt | nt | |
| HS-269 |
|
H |
|
2.8 | 8.5 | 3 |
| HS-275 | H |
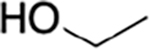
|
propyl | 2.6 | 48 | 18 |
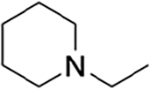
| HS-276 | H |
|
propyl | 2.5 | 2,500 | 1000 |
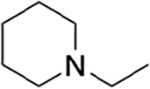
| HS-277 | H |
|
|
2.4 | 402 | 168 |
| HS-278 | H |
|
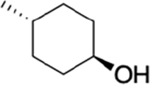
|
1.6 | 8.2 | 5 |
| HS-280 |
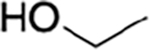
|
H | propyl | 3.1 | 29 | 9 |
| HS-281 |
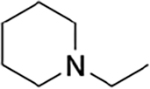
|
H | propyl | 1.7 | 4.2 | 2 |
| HS-285 | H |
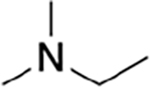
|
propyl | 32 | 1,120 | 35 |
An unbiased assessment of HS-276 kinome selectivity was obtained using the MRC Dundee kinase panel containing 140 human kinases representing all gene family members within the human kinome. At 10 μM, HS-276 showed significant inhibition (<15% enzymatic activity) of TAK1, NUAK1, IRAK1, MAP4K5, CK1γ2, and ULK2 (Figure 2a,b). To further evaluate the inhibitory potential of HS-276 on TAK1 and other targets identified in our initial kinome screen, we performed dose–response experiments of the top 10 kinases inhibited in the initial kinome screen. Of the top 10 kinases screened, TAK1 was most potently inhibited by HS-276 with a half maximal inhibitory concentration (IC50) value of 8 nM, 3.6-fold more potent than the next kinase CLK2 (29 nM) and 4.1-fold more potent than GCK (33 nM). Other kinases such as ULK2 (63 nM), MAP4K5 (124 nM), IRAK1 (264 nM), NUAK (270 nM), CSNK1G2 (809 nM), CAMKKβ−1 (1280 nM), and MLK1 (5585 nM) showed moderate to minimal inhibitory activity (Figure 2c). Due to high homology between the ATP-binding pocket of TAK1 and IRAK4 as well as modest IRAK4 inhibitory activity with parent molecule takinib, we further validated the selectivity of HS-276 toward TAK1 over IRAK4. We found that minimal IRAK4 inhibition was observed in dose–response experiments of HS-276 against IRAK4 (Figure 2d). The selectivity of HS-276 is illustrated with an inhibition dendrogram within the human kinome (Figure 2e).
Figure 2.
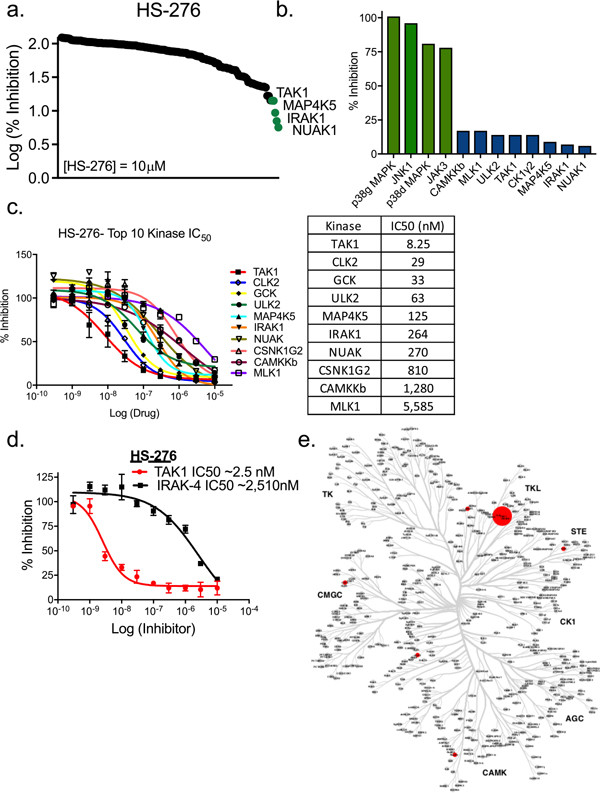
Selectivity of HS-276 in the human kinome. (a) Human kinome wide screening of HS-276 against 140 human kinases. (b) Inhibition of top kinases from kinase screening (% inhibition from control). (c) Kinase inhibition of top 10 kinase hits against HS-276. Data points represent mean ± SEM (n = 2). (d) Inhibition of TAK1 and IRAK4 by HS-276. (e) Dendrogram of human kinase inhibition by HS-276. The sizes of the dots are indicative of kinase activity.
In Vitro Cytokine Inhibition of Lead TAK1 Inhibitors.
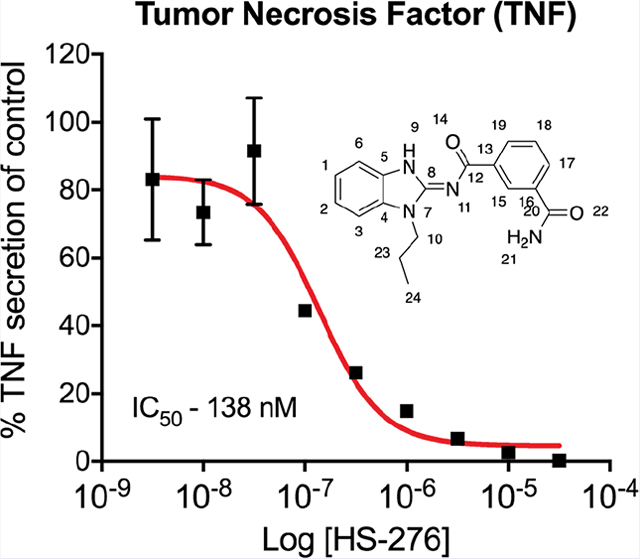
We have previously shown that TAK1 inhibition leads to a significant reduction of TNF expression in proinflammatory stimulated immune cells.13 To evaluate the anti-inflammatory effects of HS-269, HS-276, and HS-277, THP-1 human macrophages were stimulated with LPS and IFNγ in the presence of TAK1 inhibitor (10 μM) or vehicle. Treatment with HS-276 significantly attenuated TNF expression 11-fold, compared to a 2.5-fold reduction with HS-269 and 1.2-fold reduction by HS-277 (Figure 3a,b). Dose–response curves of TNF, IL-6, and IL-1β were generated in response to LPS + HS-276 treatment. Consistent with previous results, HS-276 treatment reduced expression of TNF, IL-6, and IL-1β in a dose-dependent manner with IC50 values of 138, 201, and 234 nM, respectively (Figure 3c).
Figure 3.
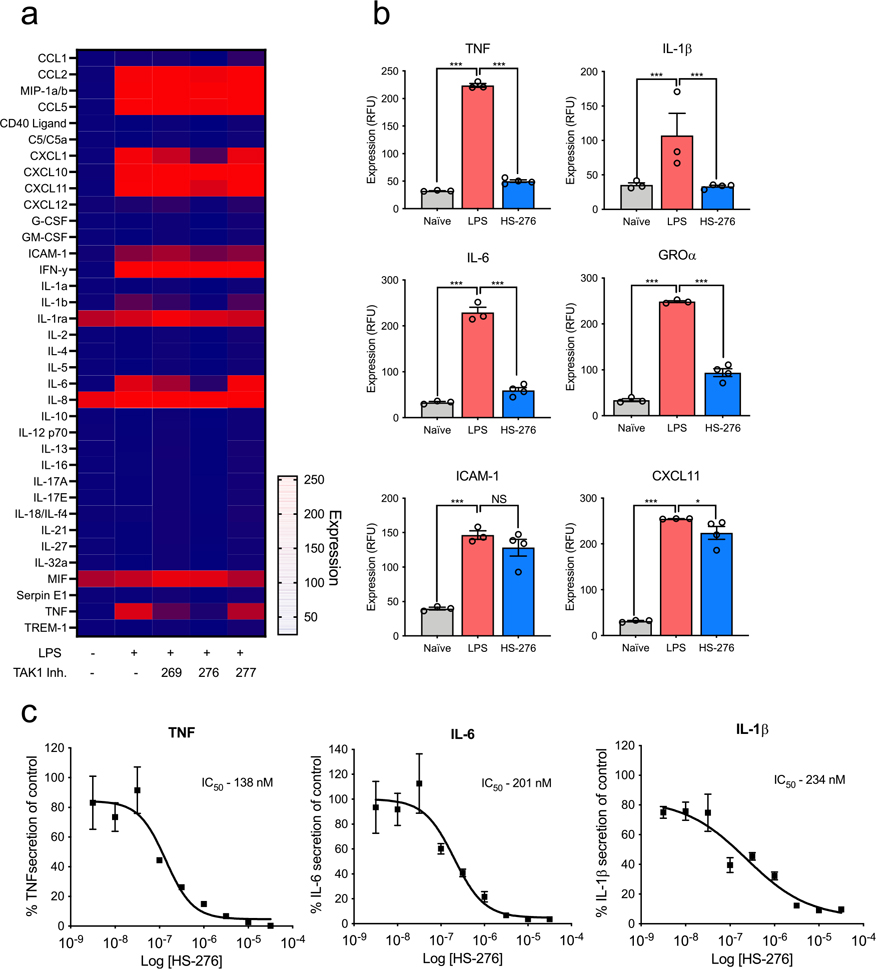
Cellular efficacy of lead TAK1 inhibitors in THP-1 cells. THP-1 human macrophages were differentiated with PMA (10 ng/mL) for 72 h followed by a rest period in PMA-free media. Cells were pretreated with 10 μM of takinib analogues HS-269, HS-276, and HS-277 followed by LPS (10 ng/mL) stimulation. (a) Relative expression of various cytokines compared to vehicle treatment. (b) TNF, IL-1β, IL-6, GROα, ICAM-1, and CXCL11 expression in naive (N =3 ± SEM), LPS (N =3 ± SEM), and HS-276 (N = 4 ± SEM). (c) HS-276 dose-dependent effects of TNF, IL-6, and IL-1β secretion reported as a percent of vehicle control (DMSO) in THP-1 cells, (n = 3 ± SEM).
Oral Bioavailability and Safety.
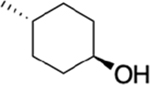
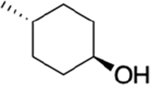
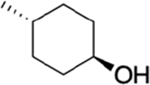
The bioavailability of our three top analogues HS-269, HS-276, and HS-277 were first evaluated in adult Sprague-Dawley rats (male) at 50 mg/kg via oral gavage (n = 3). Sequential blood draws at 1, 2, 4, 8, 12, and 24 h post gavage as well as urine collection at 4, 12, and 24 h post gavage. HS-269 and HS-277 showed low nM plasma levels across all timepoints. Despite low plasma surveillance, both HS-269 and HS-277 showed μM urine levels up to 24 h post gavage. Both HS-269 and HS-277 have the addition of the cyclohexanol to the first nitrogen of the benzimidazole, which may contribute to the lack of bioavailability or metabolism of both these compounds. HS-276, which lacks the cyclohexanol on the first nitrogen of the benzimidazole, exhibited a Cmax of 1.89 μM in plasma 2 h post gavage. HS-276 showed μM plasma concentration up to 8–12 h post gavage. Urine analysis of HS-276 showed a peak in urine concentration at 12 h at 100 μM (Supplemental Figure 2a,b). Further bioavailability analysis of HS-276 in adult CD-1 mice at HS-276 at 30 mg/kg (mice) via oral gavage (PO) or intravenous (IV) was determined. Sequential blood draws at 0.08 (IV only), 0.25, 0.5, 1, 2, 4, 8, 12, and 24 h post gavage were made. Drug concentration was determined by LC–MS as described in the Methods section. HS-276 showed excellent bioavailability in mice with a Cmax of 3.68 μM and %F of 98.1% (Table 2).
Table 2.
Bioavailability of HS-276 in Adult CD-1 Mice and Sprague-Dawley Rat Plasmaa
| species | route | dose (mg/kg) | half-life (h) | Cmax (ng/ml) | Tmax (h) | AUC0-last (h ng/ml) | %F |
|---|---|---|---|---|---|---|---|
| mouse | IV | 30 | 1.24 | 11,162 | 0.08 | 7960.0 | |
| mouse | PO | 30 | 1.41 | 3682 | 1 | 7812 | 98.1 |
| rat | PO | 50 | 5.46 | 1890 | 2 | 6462 |
Values represent means; n = 3 per timepoint.
Maximum tolerated dose studies and toxicology of HS-276 further established the safety and tolerance of HS-276. Here, mice were treated daily with HS-276 (PO, QD) at 5, 30, 50 and 100 mg/kg dosing for 14 days. Daily body weights were taken as well as clinical observations. No significant weight loss was observed between any of the treatment groups (Supplemental Figure 3a). Following termination of the study, relative liver, spleen, kidney, brain, thymus, adrenals, heart, and testes were measured. No significant weight loss was observed between any of the treatment groups in any of the tissues collected (Supplemental Figure 3b).
Efficacy of HS-276 in the CIA Model of Inflammatory Arthritis.
Treatment with HS-276 in the CIA mouse model of inflammatory arthritis significantly delayed disease onset and decreased disease incidence to 92% on day 36 in vehicle treatment compared to 75% disease incidence on day 36 in HS-276 (Figure 4a). Daily paw scores for disease control mice increased over the course of the study, peaking at termination (mean score of 3.50), and were statistically increased as compared to naïve mice (which had no clinical signs of arthritis) on days 27 through 36. Clinical arthritis scores for mice treated with HS-276 (50 mg/kg) were significantly reduced on days 26 through 36 as compared to the disease control group (Figure 4b). Area under the curve (AUC) calculations for clinical arthritis scores were statistically (85%) reduced in mice treated with HS-276 (50 mg/kg) as compared to disease control mice (Figure 4c).
Figure 4.
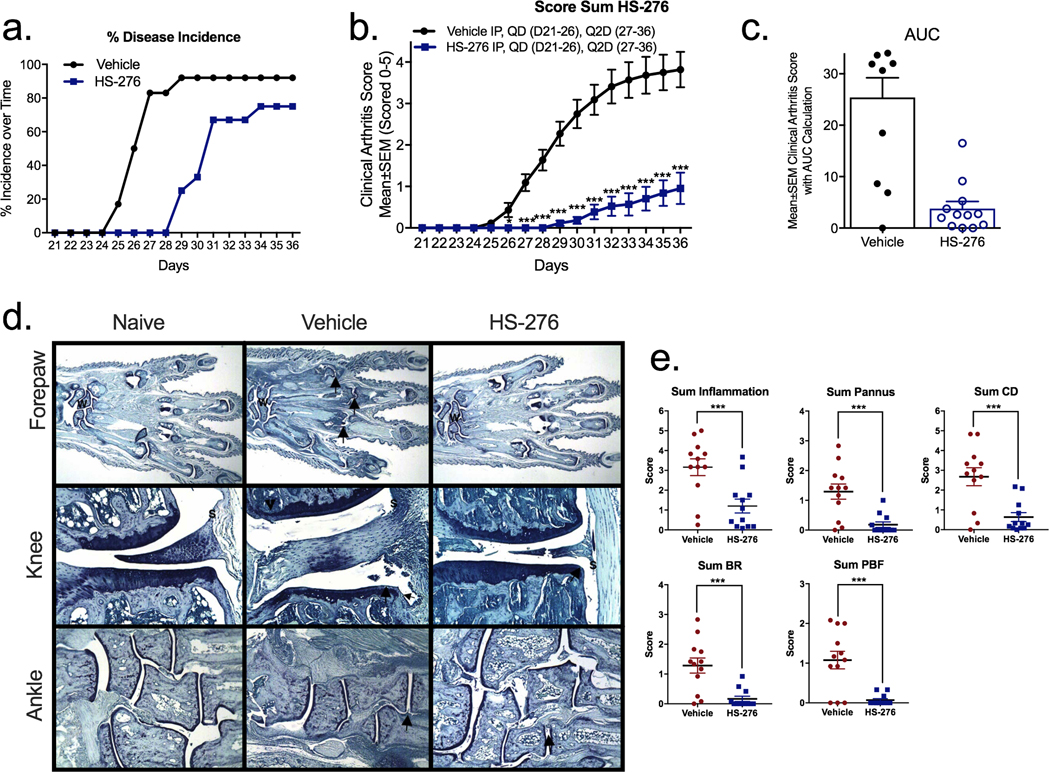
HS-276 reduces the clinical arthritis score. DBA/1 mice induced with CIA demonstrated clinical arthritic scores around day 25, which persisted throughout the study. Mice were initially treated daily (days 21–26) with vehicle control or HS-276 (50 mg/kg IP), followed by Q2D (days 27–36). (a) Disease incidence over time in vehicle (black)- and HS-276 treated (blue) (N =12 ± SEM). (b) HS-276-treated mice show a reduction in clinical arthritic score compared to vehicle control (N =12 ± SEM). (c) Area under the curve calculation of vehicle- and HS-276-treated animals. (d) Representative photomicrographs of the forepaw, knee, and ankle from disease control (naïve)-, diseased vehicle-, and HS-276-treated animals. W identifies the wrist, arrows identify representative affected joints, and S identifies inflammation. (e) HS-276 reduced inflammation, pannus, cartilage damage (CD), bone resorption (BR), and periosteal bone formation (PBF) histological manifestations of CIA 36 days post disease onset. N =12 per group ± SEM. *p < 0.05; ***p < 0.001.
Evaluation of histological changes in showed that vehicle-treated mice had histopathologic changes consistent with those seen in type II collagen-induced arthritis, including microscopic alteration and infiltration of synovium and periarticular tissue with neutrophils and mononuclear inflammatory cells, marginal zone pannus and bone resorption, and cartilage damage (Figure 4d). When considering all joints (paws and knees), mice treated with HS-276 (50 mg/kg IP) had significant reductions in scores: 62% inflammation, 86% pannus, 76% cartilage damage, 87% bone resorption, and 93% periosteal bone formation as compared to the vehicle-treated control group (Figure 4e).
Following the proof of principle using IP injections in Figure 5, we next sought to determine the efficacy of HS-276 delivered via oral gavage in the CIA mouse model. Following disease onset as previously described, mice were treated with HS-276 at 10 and 30 mg/kg QD PO and 25 mg/kg IP for comparison. Treatment with HS-276 did not significantly delay disease onset compared with vehicle-treated mice (Figure 5a); however, daily oral dosing of HS-276 reduced the arthritic clinical score by 18 and 35% when dosed at 10 and 30 mg/kg, respectively. Oral dosing showed similar efficacy compared to IP dosing at 35% (PO) and 36% (IP) reduction in disease clinical score, respectively (Figure 5b,c).
Figure 5.
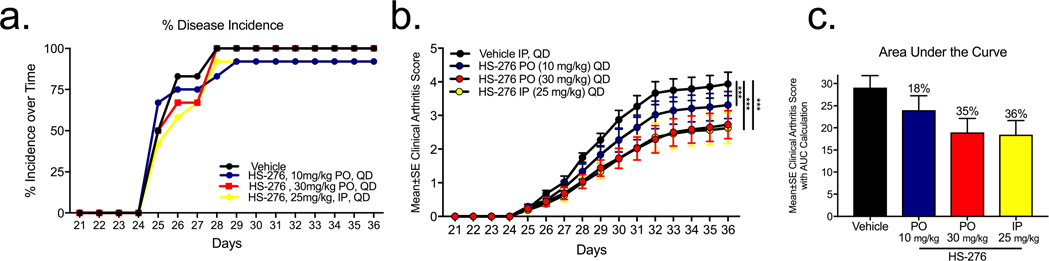
Oral efficacy of HS-276 in the CIA model of RA. (a) Disease incidence over time in vehicle (black)- and HS-276-treated mice at 10 mg/kg PO (blue), 30 mg/kg PO (red), and 25 mg/kg IP (yellow) (N =12 ± SEM). (b) Mean clinical arthritic score of CIA mice treated with HS-276 (10 mg/kg PO, 30 mg/kg PO, and 25 mg/kg IP) (N =12 ± SEM). (c) Area under the curve calculations of HS-276 treated mice. ***p < 0.001.
■ DISCUSSION
Here, we describe the development of HS-276, a selective orally bioavailable TAK1 inhibitor. HS-276 demonstrates relative selectivity within the human kinome as well as μM serum levels up to 8 h post oral gavage. Interestingly, the analysis of our takinib analogues and their respective affinities suggest that TAK1-selective and TAK1/IRAK4 polypharmacological inhibitors can be readily developed based on the positioning of substituents in the 1 and 2 positions of the benzimidazole. The overlaid structures of TAK1 and IRAK4 in Figure 1 show a small eminence comprising several residues in front of the entrance to the ATP-binding pocket for IRAK4 that is not present in TAK1. These residues in IRAK4 are Leu277 and Asp278, which may discriminate TAK1- and IRAK4-specific inhibitors. In fact, we found that the addition of bulky substituents in the 2 position blocked IRAK4 activity and retained TAK1 activity. Previous work by our group has shown the consequences of TAK1 and IRAK4 inhibition on cytokine expression and resulting cellular phenotypes associated with the inhibition of each kinase, respectively.22
Having demonstrated the selectivity of HS-276 against the kinome and related IRAK4, we further evaluated its cellular and in vivo effects on TAK1 signaling. HS-276 shows a potent anti-TNF effect in human macrophages stimulated with LPS, consistent with previous TAK1 inhibitors.23 Previously, our group has explored the therapeutic potential of TAK1 inhibition with our parent molecule takinib in the CIA model of inflammatory arthritis. Due to low bioavailability, takinib showed only a modest effect on CIA arthritis score, reducing the mean clinical score by ~32% when treated at 50 mg/kg IP. Here, we show that HS-276 reduced the mean clinical score by 85% when treated at 50 mg/kg IP. Thus, when compared to takinib, HS-276 shows marked improvement in bioavailability and disease reduction. Furthermore, consistent with disease attenuation by anti-arthritic therapeutics, histological analysis of joints in animals treated with HS-276 compared to vehicle showed reduced inflammatory infiltrates, edema, and bone degeneration by anti-arthritic therapeutics.
The treatment of RA has traditionally progressed from the use of nonsteroidal anti-inflammatory drugs (NSAIDs) for mild symptoms to conventional disease modifying antirheumatic drugs (cDMARDs) such as methotrexate and others as well as corticosteroids. However, a better understanding of the role TNF plays in driving RA pathogenesis led to the development of various anti-TNF biologics. These anti-TNF biologic DMARDs (bDMARDs) work by sequestering circulating TNF to reduce aberrant levels of proinflammatory cytokines, which are driving this disease. While these drugs have revolutionized the treatment options for RA patients across the globe, they are marred by serious safety issues regarding the increased risk of infections, malignancies, and other complications including injection site reactions. Drug noncompliance occurs in 28–66% of patients taking bDMARDs due either to the inconvenient need for self-administration of injectables or routine trips to a healthcare facility for infusions. Also, approximately 40% of patients taking anti-TNF biologics either fail to respond to initial treatment or lose response over time.3 The development of an orally bioavailable inhibitor of the protein kinase TAK1 represents a major advancement in RA treatment strategies by broadening the landscape of therapeutic options for RA patients. Providing the RA treatment market with a novel alternative to current therapies (such as a TAK1 inhibitor), which is safer and equally or more effective than current products, will significantly improve the quality of care for RA patients in the United States and across the globe.
■ METHODS
Macrophage Differentiation.
THP-1 cells were differentiated with phorbol 12-myristate 13-acetate (PMA) (100 nM) for up to 72 h in supplemented RPMI 1640× media. Following PMA differentiation, media was removed and replaced with RPMIX media (without PMA) for 24 h prior to treatments. LPS (10 ng/mL) was used for proinflammatory stimulation.
Cytokine Profile.
THP-1 cells were differentiated as previously described and treated with 10 μM designated drug or vehicle (DMSO). Twenty-four hours after treatment, the supernatant was collected and tested in a Human Cytokine XL proteome array (R&D Systems) in accordance with the manufacturer protocol. Cytokine expression was determined by chemiluminescence.
Dose–Response Curves.
THP-1 cells were differentiated as previously described and treated with HS-276 from 3.2 nM to 32 μM for 24 h, and TNF, IL-6, and IL-1β expression was determined by Luminex as per manufacturer protocol.
Animal Care.
The CIA model was managed in accordance with The Guide for the Care & Use of Laboratory Animals (8th edition) and in accordance with all Bolder BioPATH (Inotiv Inc.) IACUC-approved policies and procedures. The Inotiv Inc. IACUC approved the IACUC protocol for this specific working protocol (BBP-001). All CIA studies were performed in male DBA/1 mice. Clinical evaluation was performed by experienced principal investigators under blinded conditions.
Collagen Type II-Induced Arthritis Induction.
Collagen (4 mg/mL) solution was prepared in 0.01 N acetic acid. Equal volumes of collagen and Freund’s complete adjuvant (5 mg/mL; 1:1) were emulsified by hand mixing for ~5 min, at which point a bead of this solution holds its form when placed in water.
CIA Experimental Design.
On study days 0 and 21, animals were anesthetized with isoflurane and given intradermal injections of a total of 400 μg of Type II collagen in Freund’s complete adjuvant at the base of the tail.24,25 Mice were randomized into study groups based on body weight on day 21. Daily clinical scores and observations were made for each of the paws (right front, left front, right rear, and left rear) throughout study days 21 through 36. At study terminus (day 36), mice were euthanized for necropsy (approximately 3 h after the final dose), and samples were collected.
Immunohistological Staining.
Following 24–48 h in fixative and 4–5 days in 5% formic acid for decalcification, tissues were processed for paraffin embedding. Paws were embedded in paraffin facing the frontal plane, while knees were paraffin embedded with the patella facing down. Ankles were embedded in the frontal plane. Slides were cut and evaluated for inflammatory and histological changes with toluidine blue.
Scores for Synovitis, Pannus Formation, Degradation of Cartilage, and Bone.
Paw Score Criteria.
0 = Normal histology. 0.5 = Very nominal, affects one joint or marginal multifocal periarticular intrusion of inflammatory cells. 1 = Marginal intrusion of inflammatory cells in synovium and periarticular tissue of inflamed joints. 2 = Mild intrusion of inflammatory cells. 3 = Moderate intrusion with modest edema. Generally, three to four joints and the wrist or ankle. 4 = Marked intrusion affecting most areas with noticeable edema, one or two unaltered joints may be present. 5 = Severe diffuse intrusion with stark swelling affecting all joints and periarticular tissues.
Knee Score Criteria.
0 = Normal. 0.5 = Very minimal, affects only one area of the synovium or nominal multifocal periarticular infiltration of inflammatory cells. 1 = Minimal intrusion of inflammatory cells in synovium and periarticular tissue of affected synovial areas. 2 = Mild diffuse intrusion of inflammatory cells. 3 = Moderate diffuse intrusion of inflammatory cells. 4 = Marked diffuse intrusion of inflammatory cells. 5 = Severe diffuse intrusion of inflammatory cells.
Cartilage Damage Score Criteria.
0 = Normal. 0.5 = Very minimal; affects marginal zones only of one to several areas (knees) or joints (paws). 1 = Minimal; mostly nominal loss of toluidine blue staining (proteoglycan) with no noticeable collagen disruption or chondrocyte loss in affected joints/areas. 2 = Mild; mostly mild loss of toluidine blue staining (proteoglycan) with focal areas of chondrocyte loss and/or collagen disruption in some affected joints/areas. Paws may have one or two digit joints with near total to total loss of cartilage. 3 = Moderate; mostly moderate loss of toluidine blue staining (proteoglycan) with collagen disruption and multifocal chondrocyte loss in disturbed joints/areas. 4 = Marked; marked loss of toluidine blue staining (proteoglycan) with multifocal marked collagen disruption or chondrocyte loss in most joints with a few unaltered or slightly affected, knee: one surface with near total cartilage loss. 5 = Severe; severe diffuse loss of toluidine blue staining (proteoglycan) with stark collagen disruption or chondrocyte loss in most/all joints.
Pharmacokinetics.
All serum/urine biological samples were stored at −80 °C prior to analysis. Before quantification in serum or urine, standard curves were established using HS-269, a close structural analogue of HS-276 that serves as an internal standard. LC–MS analysis was performed at the Duke Proteomics and Metabolomics Core Facility or Cyprotex.26 The final concentration of HS-276 in the plasma was calculated by concentration (μM) in serum and urine based on molecular weight.
Statistical Analysis.
GraphPad Prism 8 was used for all statistical analyses. In each analysis, the total sample size (N) and error (SEM) are described in the figure legend. An α of 0.05 was used for all statistical analyses.
Supplementary Material
Funding
This research was supported by the National Institutes of Health (R41AR076772-01 to T.H. and R44AR076772-02A1 to S.S. and T.H.) and the North Carolina Biotechnology Center (2019-BIG-6502L to T.H.).
Footnotes
The authors declare the following competing financial interest(s): The following authors own stock in EydisBio Inc., Scott Scarneo, Philip Hughes, Robert Freeze, and Timothy Haystead. Issued patents US 20180105500A1 and US20210188783A1 and patent application No. 63/141,794 through Duke University.
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.1c00788
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.1c00788.
TAK1 affinity resins; rat pharmacokinetic data; TAK1 inhibitors biochemical data; tolerability of HS-276 in mice; and chemical synthesis of HS-206 analogues (PDF)
Contributor Information
Scott Scarneo, Department of Pharmacology and Cancer Biology, Duke University School of Medicine, Durham, North Carolina 27710, United States; EydiBio Inc., Durham, North Carolina 27710, United States.
Philip Hughes, Department of Pharmacology and Cancer Biology, Duke University School of Medicine, Durham, North Carolina 27710, United States; EydiBio Inc., Durham, North Carolina 27710, United States.
Robert Freeze, EydiBio Inc., Durham, North Carolina 27710, United States.
Kelly Yang, Department of Pharmacology and Cancer Biology, Duke University School of Medicine, Durham, North Carolina 27710, United States.
Juliane Totzke, Department of Pharmacology and Cancer Biology, Duke University School of Medicine, Durham, North Carolina 27710, United States.
Timothy Haystead, Department of Pharmacology and Cancer Biology, Duke University School of Medicine, Durham, North Carolina 27710, United States; EydiBio Inc., Durham, North Carolina 27710, United States.
■ REFERENCES
- (1).Firestein GS Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [DOI] [PubMed] [Google Scholar]
- (2).Feldmann M Development of anti-TNF therapy for rheumatoid arthritis. Nat. Rev. Immunol. 2002, 2, 364–371. [DOI] [PubMed] [Google Scholar]
- (3).Vasanthi P; Nalini G; Rajasekhar G Role of tumor necrosis factor-alpha in rheumatoid arthritis: a review. APLAR J. Rheumatol. 2007, 10, 270–274. [Google Scholar]
- (4).Taylor PC; Feldmann M Anti-TNF biologic agents: still the therapy of choice for rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 578–582. [DOI] [PubMed] [Google Scholar]
- (5).Radstake TRDJ; Svenson M; Eijsbouts AM; van den Hoogen FHJ; Enevold C; van Riel PLCM; Bendtzen K Formation of antibodies against infliximab and adalimumab strongly correlates with functional drug levels and clinical responses in rheumatoid arthritis. Ann. Rheum. Dis. 2009, 68, 1739–1745. [DOI] [PubMed] [Google Scholar]
- (6).Bartelds GM; Wijbrandts CA; Nurmohamed MT; Stapel S; Lems WF; Aarden L; Dijkmans BAC; Tak PP; Wolbink GJ Clinical response to adalimumab: relationship to anti-adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Ann. Rheum. Dis. 2007, 66, 921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cohen S; Genovese MC; Choy E; Perez-Ruiz F; Matsumoto A; Pavelka K; Pablos JL; Rizzo W; Hrycaj P; Zhang N; Shergy W; Kaur P Efficacy and safety of the biosimilar ABP 501 compared with adalimumab in patients with moderate to severe rheumatoid arthritis: a randomised, double-blind, phase III equivalence study. Ann. Rheum. Dis. 2017, 76, 1679–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cohen SB; Alonso-Ruiz A; Klimiuk PA; Lee EC; Peter N; Sonderegger I; Assudani D Similar efficacy, safety and immunogenicity of adalimumab biosimilar BI 695501 and Humira reference product in patients with moderately to severely active rheumatoid arthritis: results from the phase III randomised VOLTAIRE-RA equivalence study. Ann. Rheum. Dis. 2018, 77, 914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Laine J; Jokiranta TS; Eklund KK; Vakevainen M; Puolakka K Cost-effectiveness of routine measuring of serum drug concentrations and anti-drug antibodies in treatment of rheumatoid arthritis patients with TNF-a blockers. Biologics 2016, 10, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).O’Dell JR; Leff R; Paulsen G; Haire C; Mallek J; Eckhoff PJ; Fernandez A; Blakely K; Wees S; Stoner J; Hadley S; Felt J; Palmer W; Waytz P; Churchill M; Klassen L; Moore G Treatment of rheumatoid arthritis with methotrexate and hydroxychloroquine, methotrexate and sulfasalazine, or a combination of the three medications: results of a two-year, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002, 46, 1164–1170. [DOI] [PubMed] [Google Scholar]
- (11).Vaiopoulos AG; Athanasoula KC; Papavassiliou AG NF-κB in colorectal cancer. J. Mol. Med. 2013, 91, 1029–1037. [DOI] [PubMed] [Google Scholar]
- (12).Wang C; Deng L; Hong M; Akkaraju GR; Inoue J; Chen ZJ TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [DOI] [PubMed] [Google Scholar]
- (13).Scarneo SA; Mansourati A; Eibschutz LS; Totzke J; Roques JR; Loiselle D; Carlson D; Hughes P; Haystead TAJ Genetic and pharmacological validation of TAK1 inhibition in macrophages as a therapeutic strategy to effectively inhibit TNF secretion. Sci Rep. 2018, 8, 17058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Totzke J; Scarneo SA; Yang KW; Haystead TAJ TAK1: a potent tumour necrosis factor inhibitor for the treatment of inflammatory diseases. Open Biol. 2020, 10, 200099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Scarneo SA; Yang KW; Roques JR; Dai A; Eibschutz LS; Hughes P; Haystead TAJ TAK1 regulates the tumor microenvironment through inflammatory, angiogenetic and apoptotic signaling cascades. Oncotarget 2020, 11, 1961–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Morioka S; Broglie P; Omori E; Ikeda Y; Takaesu G; Matsumoto K; Ninomiya-Tsuji J TAK1 kinase switches cell fate from apoptosis to necrosis following TNF stimulation. J. Cell Biol. 2014, 204, 607–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li Y; Yuan L; Yang J; Lei Y; Zhang H; Xia L; Shen H; Lu J Changes in Serum Cytokines May Predict Therapeutic Efficacy of Tofacitinib in Rheumatoid Arthritis. Mediators Inflammation 2019, 2019, 5617431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Boyle DL; Soma K; Hodge J; Kavanaugh A; Mandel D; Mease P; Shurmur R; Singhal AK; Wei N; Rosengren S; Kaplan I; Krishnaswami S; Luo Z; Bradley J; Firestein GS The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1311–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Totzke J; Gurbani D; Raphemot R; Hughes PF; Bodoor K; Carlson DA; Loiselle DR; Bera AK; Eibschutz LS; Perkins MM; Eubanks AL; Campbell PL; Fox DA; Westover KD; Haystead TAJ; Derbyshire ER Takinib, a Selective TAK1 Inhibitor, Broadens the Therapeutic Efficacy of TNF-α Inhibition for Cancer and Autoimmune Disease. Cell Chem. Biol. 2017, 24, 1029–1039.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Haystead TAJ Fluorescent-Linked Enzyme Chemoproteomic Strategy (FLECS) for Identifying HSP70 Inhibitors. Methods Mol. Biol. 2018, 2018, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wang Z; Sun D; Johnstone S; Cao Z; Gao X; Jaen JC; Liu J; Lively S; Miao S; Sudom A; Tomooka C; Walker NPC; Wright M; Yan X; Ye Q; Powers JP Discovery of potent, selective, and orally bioavailable inhibitors of interleukin-1 receptor-associate kinase-4. Bioorg. Med. Chem. Lett. 2015, 25, 5546–5550. [DOI] [PubMed] [Google Scholar]
- (22).Scarneo SA; Hughes PF; Yang KW; Carlson DA; Gurbani D; Westover KD; Haystead TAJ A highly selective inhibitor of interleukin-1 receptor-associated kinases 1/4 (IRAK-1/4) delineates the distinct signaling roles of IRAK-1/4 and the TAK1 kinase. J. Biol. Chem. 2020, 295, 1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Scarneo SA; Eibschutz LS; Bendele PJ; Yang KW; Totzke J; Hughes P; Fox DA; Haystead TAJ Pharmacological inhibition of TAK1, with the selective inhibitor takinib, alleviates clinical manifestation of arthritis in CIA mice. Arthritis Res. Ther. 2019, 21, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bendele AM Animal models of rheumatoid arthritis. J. Musculoskeletal Neuronal Interact. 2001, 1, 377–385. [PubMed] [Google Scholar]
- (25).Jain A; Olsen HS; Vyzasatya R; Burch E; Sakoda Y; Mérigeon EY; Cai L; Lu C; Tan M; Tamada K; Schulze D; Block DS; Strome SE Fully recombinant IgG2a Fc multimers (stradomers) effectively treat collagen-induced arthritis and prevent idiopathic thrombocytopenic purpura in mice. Arthritis Res. Ther. 2012, 14, R192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Howe MK; Bodoor K; Carlson DA; Hughes PF; Alwarawrah Y; Loiselle DR; Jaeger AM; Darr DB; Jordan JL; Hunter LM; Molzberger ET; Gobillot TA; Thiele DJ; Brodsky JL; Spector NL; Haystead TAJ Identification of an allosteric small-molecule inhibitor selective for the inducible form of heat shock protein 70. Chem. Biol. 2014, 21, 1648–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.