

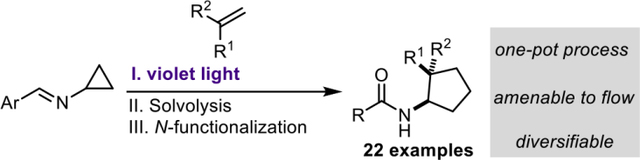
Abstract
Detailed herein is the development of a photochemical intermolecular formal [3+2] cycloaddition between cyclopropylimines and substituted alkenes to generate aminocyclopentane derivatives. The Schiff base of the cyclopropylimine was designed to enable a masked N-centered radical approach in which the requisite open-shell character was achieved upon excitation with visible-light. The cycloaddition products were directly converted to N-functionalized aminocyclopentanes via solvolysis and N-acylation. The photochemical component of this reaction sequence was demonstrated to operate in continuous flow.
Graphical Abstract
The development of milder methods for radical generation over the last 15 years, taking the form of electrochemical and photoredox catalysis approaches, has fueled a resurgence of interest in radical chemistryand has allowed easy access to chemical building blocks via unique reactive intermediates.1,2 Our laboratory recently disclosed a photochemical intramolecular formal [3+2] cycloaddition for the synthesis of 1-aminonorbornanes (1-aminoNBs).3 This methodology exploited visible-light imine photochemistry to generate an excited state diyl, which in turn initiated strain-driven fragmentation of a cyclopropane ring and serial cyclization reactions to generate the final 1-aminoNB product. This strategy offered exceedingly simple entry to N-centered radical reactivity, indicating the potential to enable additional complexity-building methodology by operating through these otherwise difficult-to-access reactive intermediates.
Aminocyclopentanes are common building blocks often found in various pharmaceuticals and agrochemicals.4 Though they are prevalent throughout the literature, they are often difficult to synthesize. Specifically, 2-substituted aminocyclopentanes are known to be synthetically challenging to generate in a modular fashion. Bayer has previously investigated N-cyclopropyl pyrazole carboxamides as potential succinate dehydrogenase inhibitors (SDHIs).5a–b In two patents published in 2010 and 2011, they reported N-cyclopropyl pyrazole carboxamides containing 2-aryl-1-aminocyclopentanes for their SDHI study (Figure 1A). In 2003, Eli Lilly published a patent focused on the synthesis of a number of 2-aryl-1-aminocyclopentanes.6 In 1998, Merck, also disclosed a procedure to generate alkoxy-substituted aminocyclopentanes.7 In all of these cases, a significant number of steps were required to generate the desired aminocyclopentane products. Photochemistry was envisioned as a viable strategy towards accessing these sp3-rich motifs in a manner that matches the interchangeability of sp2-centric cross-coupling techniques. Minimizing the necessary steps required to access these motifs would improve the availability of these unique substructures in drug or agrochemical development programs.
Figure 1.

A) Prior work; B) Masked N-centered radical strategy; C) This work
Previously, our group developed an intramolecular masked N-centered radical strategy that utilized cyclopropylimines with a suitably placed alkene for the synthesis of 1-aminonorbornanes (Figure 1B). Using a similar strategy, the synthesis of various functionalized 1-aminocyclopentane products via a photochemical intermolecular formal [3+2] cycloaddition (Figure 1C) is reported herein. Our lab has also reported a similar strategy towards the synthesis of bicyclo[3.1.1]heptanes.8
Similar to the previously disclosed intramolecular [3+2] cycloaddition, treatment of cyclopropylimine (1) with violet light (390 nm) results in the formation of excited state diyl 11 (Figure 2). The N-centered radical initiates the strain-driven homolysis of the cyclopropane ring, generating a formal β-iminyl radical intermediate 12 (though a non-canonical bonding structure is likely the best representation, given the reactivity observed herein). Intermediate 12 then engages in an intermolecular addition to the alkene substrate, followed by an intramolecular 5-exo-trig cyclization resulting in the formation of cyclopentane 14. Notably, both radicals of the excited state imine are terminated via recombination to regenerate the C-N double bond of the Schiff base and yield the desired Schiff base-protected aminocyclopentane 3. The mild, operationally-simple reaction conditions and saturated architectures it affords make this methodology a uniquely facile route to chemical complexity and 3-D chemical space. Importantly, the formal [3+2] cycloaddition, Schiff base solvolysis, and N-functionalization can all be performed in a one-pot fashion. Furthermore, this methodology was readily translated to continuous flow, allowing for a higher throughput of material in a fraction of the time.
Figure 2.

Proposed mechanism for intermolecular formal [3+2] cycloaddition.
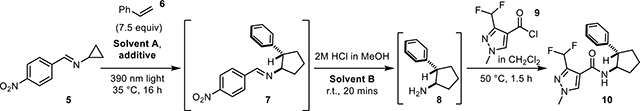
The cyclopropylimine Schiff base starting material 5 was easily prepared on multi-gram scale via condensation of commercially available cyclopropylamine and 4-nitrobenzaldehyde.9 As there was a desire for the [3+2] cycloaddition to be amenable to continuous flow, it was necessary for this reaction to be optimized so that the formation of polymeric materials and other solids were minimized or eliminated.
Several optimization screens were undertaken (Table 1), the first being a screen of polymerization-preventing additives used within the chemical industry (i.e. decolorizing agents; entries 1–8, see SI for structures) 10a–b, the second was a screen of reaction solvent (entries 9–12) and the third a screen of additive loading (entries 5, 12–13). From these studies it was found that the best conditions for the [3+2] component of the sequence utilized ethyl acetate as solvent A, 7.5 mol equivalents of the substituted alkene and 1 mol% of 2,2’-dipyridyl disulfide (aldrithiol) as a polymerization-suppressing additive. This mixture was then irradiated under 390 nm light at 35 °C for 16 hours before being diluted with dichloromethane (solvent B) and subsequently solvolyzed with 2M HCl in MeOH at ambient temperature. The addition of dichloromethane before the solvolysis was found to reduce the amount of precipitate without impacting yield, as compared to the analogous trial with ethyl acetate as the only solvent (of entries 9 and 12); alternative solvents negatively impacted yield. The final phase of the reaction sequence involves the addition of an acid chloride to the crude mixture followed by brief heating, ultimately yielding the amide of the newly-formed aminocyclopentane. It was envisioned that product diversification could be achieved via three different aspects of the reaction: 1) the alkene component; 2) the acyl chlorides; or 3) the cyclopropylimine. Investigations into reaction scope commenced with the exploration of substitution on the alkene component (Figure 3). All of the substituted alkenes explored during this phase of the project were either commercially available or were easily generated via a Wittig olefination of the corresponding ketone.11 Neutral, electron-deficient, and electron-rich alkene systems were all tolerated by the reaction. The best yields were attained from the neutral and electron-deficient alkenes, while electron-donating substituents resulted in decreased yields.
Table 1.
Reaction optimization studies.
| |||||
|---|---|---|---|---|---|
| Trial | Solvent A | Additive | Additive Loading | Solvent B | Isolated Yield |
| 1 | EtOAc | BHT | 1 mol% | CH2Cl2 | 63% |
| 2 | EtOAc | DTDP | 1 mol% | CH2Cl2 | 56% |
| 3 | EtOAc | Irganox | 1 mol% | CH2Cl2 | 64% |
| 4 | EtOAc | 22M46 | 1 mol% | CH2Cl2 | 63% |
| 5 | EtOAc | Aldrithiol | 1 mol% | CH2Cl2 | 66% |
| 6 | EtOAc | TMQ | 1 mol% | CH2Cl2 | 62% |
| 7 | EtOAc | Allopurinol | 1 mol% | CH2Cl2 | 57% |
| 8 | EtOAc | No Additive | No Additive | CH2Cl2 | 55% |
| 9 | EtOAc | Aldrithiol | 10 mol% | EtOAc | 68% |
| 10 | PhMe | Aldrithiol | 10 mol% | PhMe | 38% |
| 11 | CH2Cl2 | Aldrithiol | 10 mol% | CH2Cl2 | 56% |
| 12 | EtOAc | Aldrithiol | 10 mol% | CH2Cl2 | 67% |
| 13 | EtOAc | Aldrithiol | 50 mol% | CH2Cl2 | 66% |
Figure 3.

Substituted alkene scope.
Monosubstituted styrenyl compounds (18a and 18b), α-alkyl styrenes (18c and 18d) as well as symmetrically (17e) and unsymmetrically (18f) substituted 1,1-diaryl alkenes were all tolerated by the reaction. However, lower yields were observed for the disubstituted styrenyl substrates, which is suspected to be due to steric hinderance. Both mono-substituted and unsymmetrically 1,1’-disubstituted alkenes led to ~1:1 mixtures of diastereomers. Fortunately, the diastereomers were separable through chromatography, HPLC, and/or re-crystallization (trans-substituted compounds typically crystallized preferentially; see X-ray structure of compound 18d in Figure 3). Unfortunately, solely alkyl substituted alkenes, tetra and tri-substituted alkenes, and 1,2-disubstituted alkenes were not tolerated by the reaction, either reacting negligibly or not at all (see Figure S5 for selected failed alkenes). This lack of reactivity is likely a result of steric hinderance in the cases of the tetra and tri-substituted alkenes, while in the case of the the 1,2-disubstituted alkenes and terminal olefins, a lack of electronic polarization is the assumed explanation for the loss of reactivity that is observed. Acrylonitriles (18g-18i) were also successful substrate, reinforcing the electronic polarization observations.
The scope for the acid chlorides was then explored (Figure 4). Using 1,1-diphenyl ethylene as the alkene component of the reaction, a range of aromatic (22a-d) and alkyl (22e and 22f) acyl chlorides were successful. With the aromatic acyl chlorides, neutral, electron-deficient, and electron-rich variations were all successful, with electron-deficient systems performing best. For alkyl substituted acyl chlorides, the material was concentrated under reduced pressure in order to remove excess HCl and methanol after solvolysis.
Figure 4.

Substituted acyl chloride scope. Conditions: EtOAc, Aldrithiol, 390 nm light, 35 °C, 16 h then 2M HCl in MeOH, CH2Cl2, r.t. 20 mins. aAfter solvolysis the solvent is removed under reduced pressure.
The final facet of substrate scope exploration focused on substitution on the cyclopropyl ring of the cyclopropylimine starting material (Scheme 1). A methyl (23) substituent at the C1 position of the cyclopropylimine starting material resulted in the formation of the corresponding C1 substituted aminocyclopentane product 25, albeit in modest yields. The reaction is also tolerant of substitution at the C2 position of the cyclopropyl ring as is evidenced by dimethyl substituted compounds 28a and 28b. Unfortunately, a phenyl (29) substituent at the C2 position -of the cyclopropylimine prohibited progression to product and returned only starting material, presumably due to the increased stability of the generated benzylic radical (31) (Scheme 1).12
Scheme 1.

Substituted cyclopropylimine scope. Conditions: EtOAc, Aldrithiol, 390 nm light, 35 °C, 16 h then 2M HCl in MeOH, CH2Cl2, r.t., 20 mins.
To further expand upon the utility of this reactivity, efforts to optimize the photochemical [3+2] reaction in flow were pursued. Upon optimization (see Supplementary Information), it was found that the reaction mixture could be prepared similarly to that of the optimized batch conditions, as the only modifications were the addition of an extra 1.5 equivalents of styrene (9.0 equivalents total) and a constant temperature of 50 °C. After the mixture was degassed, a peristaltic pump was used to flow the mixture into the flow reactor (14 mL coil, 1.0mm ID, 1/16” OD HPFA tubing) and a commercial photoreactor was used to irradiate the mixture at 385 nm. The higher surface-area-to-volume ratio of the tubing compared to that of the batch reactor allowed for a significant reduction in reaction time for the [3+2] cycloaddition, with the reaction time reduced from 16 hours to between 55 minutes and 1 hour and 35 minutes (40 minutes residence time to 1 hour and 15 minute residence time).13–17 As a result of this reduction in time, the entire reaction sequence was ran in a single day rather than over the course of two days. The subsequent solvolysis and acylation were performed in batch. For comparison purposes, products 18a-18f were run using this flow system, with yields similar to that of the batch reactions (see Figures 3 and 5). Other aryl substituted alkenes (18j-178) and diphenyl ethylene (18m) were also well tolerated under continuous flow conditions (Figure 5).
Figure 5.

Substrates accessed using flow conditions. Conditions: 2M HCl in MeOH, CH2Cl2, r.t. 20 mins then benzoyl chloride 50 °C, 1.5 hours.
Importantly, running the photochemical step in flow allowed for more facile scale-up, evidenced by the production of 18k at a rate of 0.79 mmol/hr on gram-scale. It is also worth noting that continuous flow processing enabled aminocyclopentene 18n to be generated, implementing an alkyne – phenyl acetylene – as the two-carbon fragment. This is significant because this particular reaction showed no conversion to the cyclopentene product when run in batch. 4-Vinylpyridine derived product 32 could not be solvolyzed under standard conditions or at elevated temperatures. As such this compound was isolated as the Schiff base intermediate.
This work expands the use of a masked N-centered radical strategy to generate highly functionalized and saturated amino-substituted carbocycles. In this case, a wide variety of aminocyclopentanes were accessed via an intermolecular formal [3+2] cycloaddition. This modular three phase reaction sequence allows for rapid one-pot synthesis of complex cyclopentyl building blocks, readily scaled in continuous flow. This methodology lends itself to the rapid generation of biologically relevant molecules.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge the financial support for this research from the NIH NIGMS (R35-GM144286) and the University of Michigan. JLC was supported by the University of Michigan’s Rackham Merit Fellowship and a NIH F31 Pre-doctoral Fellowship (5F31GM140677-02). DS was supported by a Postdoctoral Fellowship, PF-16-236-01-CDD, from the American Cancer Society. MJS was supported by the Australian Governments Endeavour Leadership Fellowship. The authors also acknowledge Dr. Jeffrey Kampf for his help obtaining the crystals structures presented herein.
Footnotes
Notes
The authors declare no competing financial interests.
ASSOCIATED CONTENT
Supporting Information
For detailed experimental procedures and characterization data for all compounds and precursors presented above, see the Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, images of reaction setups, and characterization data along with copies of the spectra (PDF)
REFERENCES
- 1.Douglas JJ; Sevrin MJ; Stephenson CRJ Visible Light Photocatalysis: Applications and New Disconnections in the Synthesis of Pharmaceutical Agents. Org. Process Res. Dev. 2016, 20(7), 1134–1147. [Google Scholar]
- 2.Karkas M; Porco J; Stephenson CRJ Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis. Chem. Rev. 2016, 116, 9683–9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Staveness D, Collins JL, McAtee RC, Stephenson CRJ Exploiting Imine Photochemistry for Masked N-Centered Radical Reactivity. Angew. Chem. Int. Ed. 2019, 58, 19000–19006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mcgrath NA; Brichacek M; Njardarson JTA Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ. 2010, 87(12), 1348–1349. [Google Scholar]
- 5.a) Benting J; Dahmen P, Bayer International Publication No. WO 2010/094666, August 26, 2010, [Google Scholar]; b) Benting J; Braun C, Bayer International Publication No. WO 2011/151369, December 08, 2011 [Google Scholar]
- 6.Aikins JA; Smith ECR; Shepherd TA; Zimmerman DM, Eli Lilly US Patent No. US 6,639,107 B1, May 14, 2002
- 7.Caldwell CG; Chen P; Durette PL; Finke P; Hale J; Holson E; Kopka I; MacCoss M; Meurer L; Mills SG; Robichaud A, Merck US Patent No. 5,750,549, May 12, 1998
- 8.Harmata S, A.; Spiller E, T.; Sowden J, M.; Stephenson RJ, C. Photochemical Formal (4 + 2)-Cycloaddition of Imine-Substituted Bicyclo[1.1.1]Pentanes and Alkenes. J. Am. Chem. Soc. 2021, 143 (50), 21223–21228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. See Supporting Information.
- 10.a) Allen NS; Edge M; Norman Allen CS Perspectives on Additives for Polymers. 1. Aspects of Stabilization. J Vinyl Addit. Technol. 2021, 27, 5–27. [Google Scholar]; b) Allen NS; Edge M; Norman Allen CS Perspectives on Additives for Polymers. Part 2. Aspects of Photostabilization and Role of Fillers and Pigments. J Vinyl Addit. Technol. 2021, 27, 211–239. [Google Scholar]
- 11.Schollkopf U; Wittig G About Triphenyl Phosphine Methylene as Olefin-Forming Reagents. Chemische Berichte. 1954, 87, 1318–1330. [Google Scholar]
- 12.Walter RI, Substituent Effects on the Properties of Stable Aromatic Free Radicals. The Criterion for Non-Hammett Behavior. J. Am. Chem. Soc. 1966. 88: 1923–1930. [Google Scholar]
- 13.Cambié D; Bottecchia C; Straathof JW, N.; Hessel V; Noël T Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev. 2016, 116 (17), 10276–10341. [DOI] [PubMed] [Google Scholar]
- 14.Plutschack MB; Us Pieber B; Gilmore K; Seeberger PH The Hitchhiker’s Guide to Flow Chemistry ∥. Chem. Rev. 2017, 117 (18), 11796–11893. [DOI] [PubMed] [Google Scholar]
- 15.Sambiagio C; Noë T Flow Photochemistry: Shine Some Light on Those Tubes! Trends Chem. 2020, 2 (2), 92–106. [Google Scholar]
- 16.Garlets ZJ; Nguyen JD; Stephenson CRJ The Development of Visible-Light Photoredox Catalysis in Flow. Isr. J. Chem. 2014, 54 (4), 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bou-Hamdan FR; Seeberger PH Visible-Light-Mediated Photochemistry: Accelerating Ru(Bpy) 3 2+-Catalyzed Reactions in Continuous Flow †. Chem. Sci. 2012, No. 3, 1612–1616. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.