

Abstract
Purpose:
This study aimed to undertake a multidisciplinary characterization of the phenotype associated with SOX11 variants.
Methods:
Individuals with protein altering variants in SOX11 were identified through exome and genome sequencing and international data sharing. Deep clinical phenotyping was undertaken by referring clinicians. Blood DNA methylation was assessed using Infinium MethylationEPIC array. The expression pattern of SOX11 in developing human brain was defined using RNAscope.
Results:
We reported 38 new patients with SOX11 variants. Idiopathic hypogonadotropic hypogonadism was confirmed as a feature of SOX11 syndrome. A distinctive pattern of blood DNA methylation was identified in SOX11 syndrome, separating SOX11 syndrome from other BAFopathies.
Conclusion:
SOX11 syndrome is a distinct clinical entity with characteristic clinical features and episignature differentiating it from BAFopathies.
Keywords: Exome, Genome sequencing, Hypogonadism, Methylation, Neurodevelopmental disorder, SOX11
Introduction
Coffin-siris syndrome (CSS) is a multiple congenital malformation syndrome, which is associated with variants in genes encoding subunits of the BAF complex (collectively termed BAFopathies).1 The classical CSS phenotype consists of coarse facies, hypoplasia of the nail of the fifth digit, and developmental delay. Variants in ARID1B (OMIM 614556) are the most common cause of CSS, but ARID1B variants are also associated with nonsyndromic intellectual disability (ID).2 ARID1B variants are among the most common causes of neurodevelopmental disorders (NDDs) identified in large scale sequencing studies.3 Variants in other genes can be associated with CSS (eg, ARID1A, DPF2) but are considerably less frequent.4,5
Tsurusaki et al6 reported de novo single nucleotide variants (SNVs) in SOX11 in 2 children with CSS. Hempel et al7 identified microdeletions at 2p25.2 (containing SOX11) and SOX11 SNVs in a series of children with either nonsyndromal ID or CSS. The SOX11 missense variants identified were located in the high-mobility group (HMG) DNA binding domain and were shown to impair activation of SOX11 target genes in vitro. SOX11 is a single exon gene with a single transcript, which is predicted to be haploinsufficient and loss-of-function intolerant.7 The phenotype associated with SOX11 variants was designated as CSS9 (OMIM 615866).
Proteins encoded by SOX genes are a family of transcription factors, which play crucial roles in multiple developmental processes.8 All SOX protein family members contain an HMG box, which is the hallmark of these proteins. The HMG box binds to and regulates target genes. The HMG box also controls protein–protein interactions and trafficking of SOX proteins between cytoplasm and nucleus. Variants in a variety of SOX genes are associated with human developmental disorders (termed SOXopathies); examples include Waardenburg syndrome9 (caused by pathogenic variants in SOX10) and SOX2-anophthalmia syndrome.10 SOXopathies share some common features such as ocular malformations, ID, hypogonadotropic hypogonadism, and genital malformations. SOX11 forms a peripheral component of the SWI/SNF complex, and thus, SOX11-associated syndrome (SOX11 syndrome) may lie in the CSS spectrum. However, given the similarity in protein sequence and function between SOX11 and other SOX gene family members, it is possible that the phenotypes associated with SOX11 variants may be more congruent with SOXopathies.
In this article, we report a large series of individuals with SOX11 variants identified via a genotype first approach in large scale exome and genome sequencing studies. We define the associated phenotypic and molecular genetic spectrum, including detailed developmental milestones. We identify ocular malformations and hypogonadotrophic hypogonadism as being features of SOX11 syndrome. We report a unique peripheral blood DNA methylation signature as a diagnostic biomarker and phenotypic clustering analysis that distinguishes SOX11 syndrome from BAFopathies.
Materials and Methods
Ascertainment of individuals with SOX11 variants
Participants with protein altering SNVs in SOX11 were identified through exome sequencing from the Deciphering Developmental Disorders (DDD) study and genome sequencing undertaken in the 100,000 Genomes Project.11 The sequencing pipelines for DDD3 and the 100,000 Genomes Project have been published elsewhere. Additional participants were identified via GeneMatcher (Paracel, Inc).12 This study complied with the Declaration of Helsinki. Written informed consent was obtained from parents and guardians as appropriate, including for publication of photographs. Clinical and phenotypic data were gathered from medical records by the recruiting clinician. Kaplan-Meier analysis was performed (PASW, http://www.spss.com.hk/statistics/) to summarize acquisition of developmental milestones.
SOX11 variant classification
SOX11 variants (NM_003108.4) were classified using American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology13 criteria applied using VarSome tool.14
In vitro analysis of SOX11 transactivating activity
The SOX11 open reading frame clone was purchased from Promega, and the mutant variants (c.1142_1143insT[p.Gly384Argfs*14], c.527C>A [p.Ala176Glu], and c.882C>G [p.Tyr294*]) of SOX11 were generated through site-directed mutagenesis either with KOD-Plus-Mutagenesis Kit (Toyobo) (for c.1142_1143insT) or with PrimeSTAR Mutagenesis Basal Kit (Takara Bio) (for c.527C>A and c.882C>G). Wild-type and mutant SOX11 complementary DNAs were amplified using polymerase chain reaction and cloned into p3xFLAG-CMV-14 expression vector (Sigma-Aldrich). The GDF5 promoter 5′-flanking sequence (–448/+319, NM_000557.3, GRCh37/hg19) was polymerase chain reaction amplified and cloned into pGL3-basic vector (Promega). All constructs were verified using Sanger sequencing.
HeLa cells were cultured in Dulbecco’s Modified Eagle Medium–high glucose supplemented with penicillin (50 units/mL), streptomycin (50 μg/mL), and 10% fetal bovine serum. Cells were plated in 24-well plates, 24 hours before transfection. Transfections were performed using TransIT-LT1 (Takara) with pGL3 reporter (500 ng per well), effector (250 ng per well), and pRL-SV40 internal control (6 ng per well) vectors. The cells were harvested for 24 hours after transfection, and luciferase activities were measured using the PicaGene Dual Sea Pansy Luminescence Kit (TOYO B-Net Co, Ltd). Wild-type and mutant SOX11 proteins were assessed using immunoblot analyses with monoclonal anti-FLAG M2 HRP antibody (1:3000 dilution; Sigma-Aldrich) following the manufacturer’s instructions. Relative GDF5 promoter activities were evaluated using t test. P < .016 was considered as significant (because 3 statistical comparisons were made and the P value was correct for multiple comparisons [.05/3 =.016]). Relative luciferase activities were compared with that of the empty vector and presented as mean ± SD for 2 independent experiments, with each experiment performed in triplicates.
Study of SOX11 expression in fetal brain
Expression of SOX11 transcripts in fetal structures were evaluated using RNAscope in situ hybridization (ISH) assay and compared with expression pattern of GnRHR. Methods are provided in the online Supplemental Methods.
Phenotype cluster analysis of SOX11 syndrome and CSS
Human Phenotype Ontology (HPO)15 terms were used to describe the phenotypes of individuals with SOX11 or ARID1B variants in a standardized fashion. Individuals with pathogenic ARID1B variants were identified from the open access participants listed on DECIPHER. We selected HPO terms that were well defined and not reliant on subjective clinical evaluation (microcephaly <2 SD [HP 0000252], abnormal eye morphology [HP 0013272], oculomotor apraxia [HP 0000657], hypogonadotrophic hypogonadism [HP 0000044], and coarse facies [HP 0000280]).
Hierarchical cluster analysis was performed in Python using the Scikit-learn library and the AgglomerativeClustering object. The Euclidean distance and Ward parameters were selected to compute the linkage distance and cluster merge strategy. The dendrogram and heatmap were created using the Seaborn library and clustermap object. Input document is provided in Supplemental Methods. The script for the clustering test and plotting functions can be found on https://github.com/Eema-jawahiri/phenotypic-cluster-analysis.git
Identification of SOX11 syndrome episignature
Full methods are provided in the Supplemental Methods. Peripheral blood DNA was extracted using standard techniques. Bisulfite conversion was performed, and samples were analyzed using Illumina Infinium MethylationEPIC BeadChips (Illumina). Details of DNA methylation data analysis and episignature discovery were previously described.16–20 For mapping the episignature (probe and feature selection), MatchIt package21,22 was used to randomly select controls matched for age, sex, and array type from the EpiSign Knowledge Database (EKD), providing a control sample size 5 times larger than that of the cases, resulting in 50 controls.
Methylation levels (β-values) were then transformed into M-values, which were used for linear regression modeling. Using the limma package, linear regression modeling was performed for the purpose of calculating the methylation differences between the case and the control groups, along with the corresponding P value for each probe. A total of 224 differentially methylated probes (DMPs) were identified and considered as the SOX11 episignature.
Using the 224 DMPs, 2 binary support vector machine (SVM) classifiers with a linear kernel were constructed using the e1071 package as described previously.16,17 The first classifier was trained using only the SOX11 samples against the control samples, and then samples from 38 other Mendelian NDDs with an established episignature from the EKD were supplied into the model to assess the specificity of the model. Using the Platt’s scaling method, the classifiers generate a methylation variant pathogenicity (MVP) score ranging from 0 to 1 for each sample, in which a score near 1 is indicative of similarity to the identified SOX11 syndrome episignature, whereas a score near 0 shows that the sample has a methylation profile different from the SOX11 syndrome episignature.
Results
Ascertainment of cohort
We identified 38 new patients with SNVs (n = 34) or deletions (n = 4) of SOX11 (clinical and genomic data are summarized in Supplemental Tables 1, 2, and 3). Participants were identified through exome sequencing in the DDD study or genome sequencing in the 100,000 Genomes Project, with additional cases identified via GeneMatcher. We identified 15 published patients with SOX11 SNVs (Supplemental Table 1).
Spectrum of SOX11 variants in NDDs
We identified 29 distinct SOX11 SNVs from these 38 new patients (clinical information is in Supplemental Tables 1 and 2, and genomic information in Supplemental Table 3). There were 25 unique missense variants (Figure 1), 4 protein truncating variants (PTVs), and 4 microdeletions. One sibling pair shared the same PTV, and 1 sibling pair shared the same missense variant. All PTVs were classified as pathogenic. In total, 5 missense variants were classified as likely pathogenic and 20 as pathogenic. De novo status was confirmed in 30 patients. In addition, 1 variant was inherited from a mosaic parent, and 2 affected sibships (4 participants) had a parent with ID who was presumed to be a SOX11 variant heterozygote (but was not tested). No other likely pathogenic or pathogenic variants that might better explain the phenotype were identified. Case 24 had a BPTF variant (ACMG Class 3). Case 28 had a single variant in IVD (recessive isovaleric acidaemia). Case 36 had a KATB variant (ACMG class 3) inherited from an unaffected mother.
Figure 1. Schematic diagram of reported SOX11 variants.
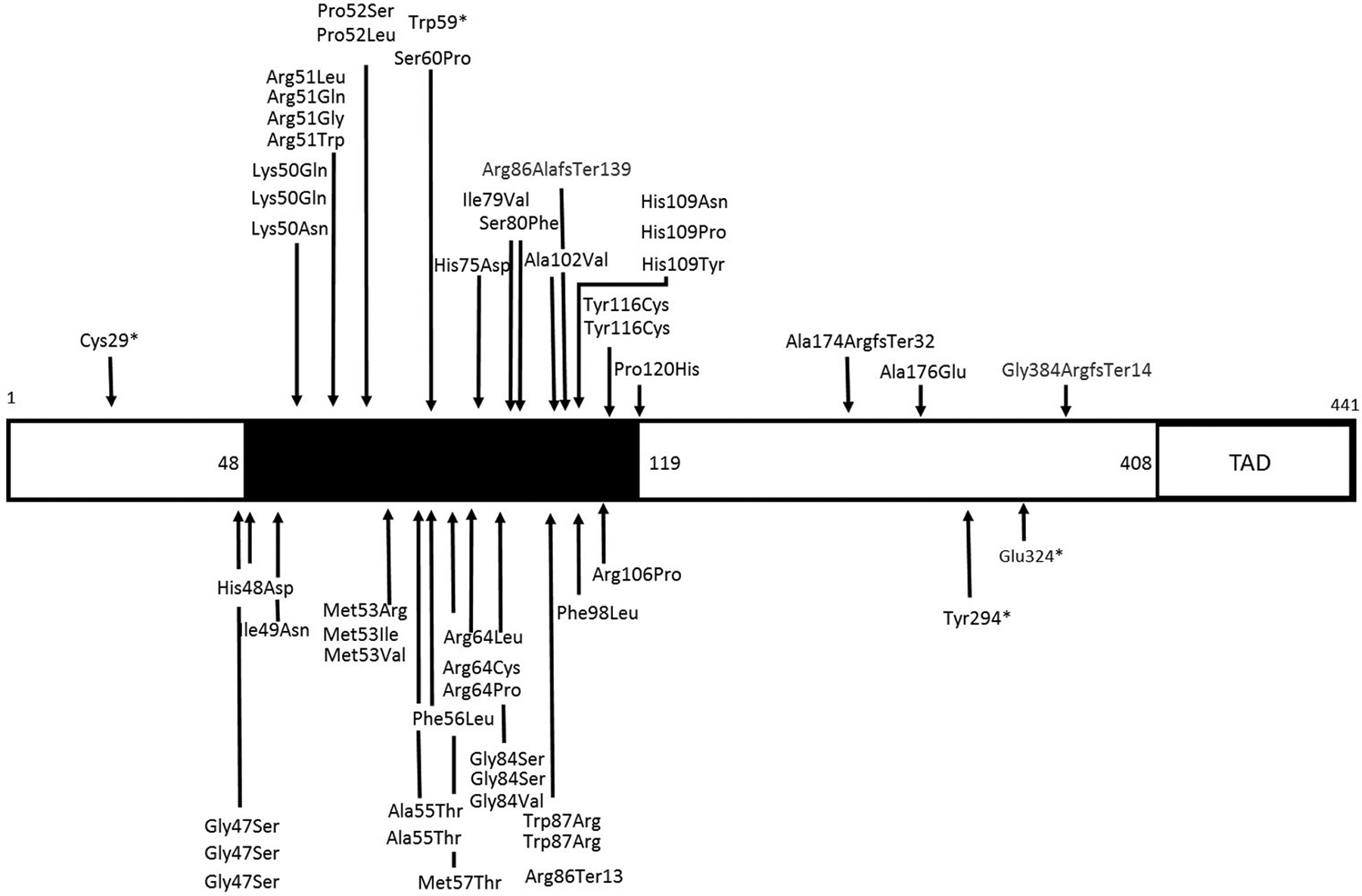
Illustration of missense and protein truncating variants in SOX11 in people with neurodevelopmental disorders. Both novel variants identified in this study and those identified in published patients are shown. The black box indicates the high-mobility group domain. The domain is not drawn to scale. Domain boundaries (amino acid number) as defined by Refseq (NP_003099.1), SwissProt (P35716.2), and International Nucleotide Sequence Database Collaboration (AAB08518.1). TAD, transactivating domain. Refseq, National Center for Biotechnology Information Reference Sequence Database.
Evidence that the SOX11 HMG box could be a hotspot for pathogenic variants
The HMG box in SOX11 protein is a domain responsible for SOX11 binding to DNA and regulation of target genes. In addition, the HMG box regulates key protein–protein interactions and trafficking of SOX11 protein between cytoplasm and nucleus. Our previous in vitro studies showed that 4 missense variants in the HMG box impaired SOX11 transactivating activity (p.Lys50Asn, p.Pro120His, p. Ser60Pro, and p. Tyr116Cys). We could not perform functional analyses of all missense variants in the HMG box. It should not be assumed that all HMG box missense variants are pathogenic. However, it is plausible that such variants could interfere with SOX11 transactivating activity and have pathogenic potential.
First, none of the SOX11 missense variants were present in Genome Aggregation Database (gnomAD), and only 8 HMG missense variants in SOX11 were identified in 114,704 individuals in gnomAD v2.1.1 non-neuro data set. This strongly suggests that missense variants in this domain are not compatible with normal neurodevelopment. Second, if the HMG box is a pathogenic variant hotspot, then it should be relatively depleted of missense SNVs in cohorts without NDDs. Using gnomAD (v2.1.1 non-neuro data set) we identified that the percentage of residues with a missense SNV in the HMG box was significantly lower than in the N-terminal, central, or transactivating domains (Supplemental Figure 1). This suggests intolerance to variation in the HMG box. There is significant sequence homology between the HMG box domains of human SOX proteins. We reasoned that if pathogenic variants had been reported at a given residue in a SOX protein, then it could be taken as possible evidence of pathogenicity for the equivalent variant in SOX11. We therefore compared variants from DECIPHER and ClinVar between SOX10 and SOX11. In total, 6 residues in SOX11 had pathogenic variants (DECIPHER and ClinVar) at equivalent residues in SOX10 (Supplemental Table 4). Several of these had identical amino acid change, eg, p. (Arg51Gly) in SOX11 and p. (Arg106Gly) in SOX10.
Missense variants in regions of SOX11 other than the HMG box could also interfere with protein function and have pathogenic potential. The p.(Ala176Glu) variant that we identified enabled us to investigate this. This variant significantly reduced SOX11 transactivating activity in vitro (Figure 2). This supports a potential pathogenic role for the variant.
Figure 2. Assessment of SOX11 variant impact on transactivation of target gene.
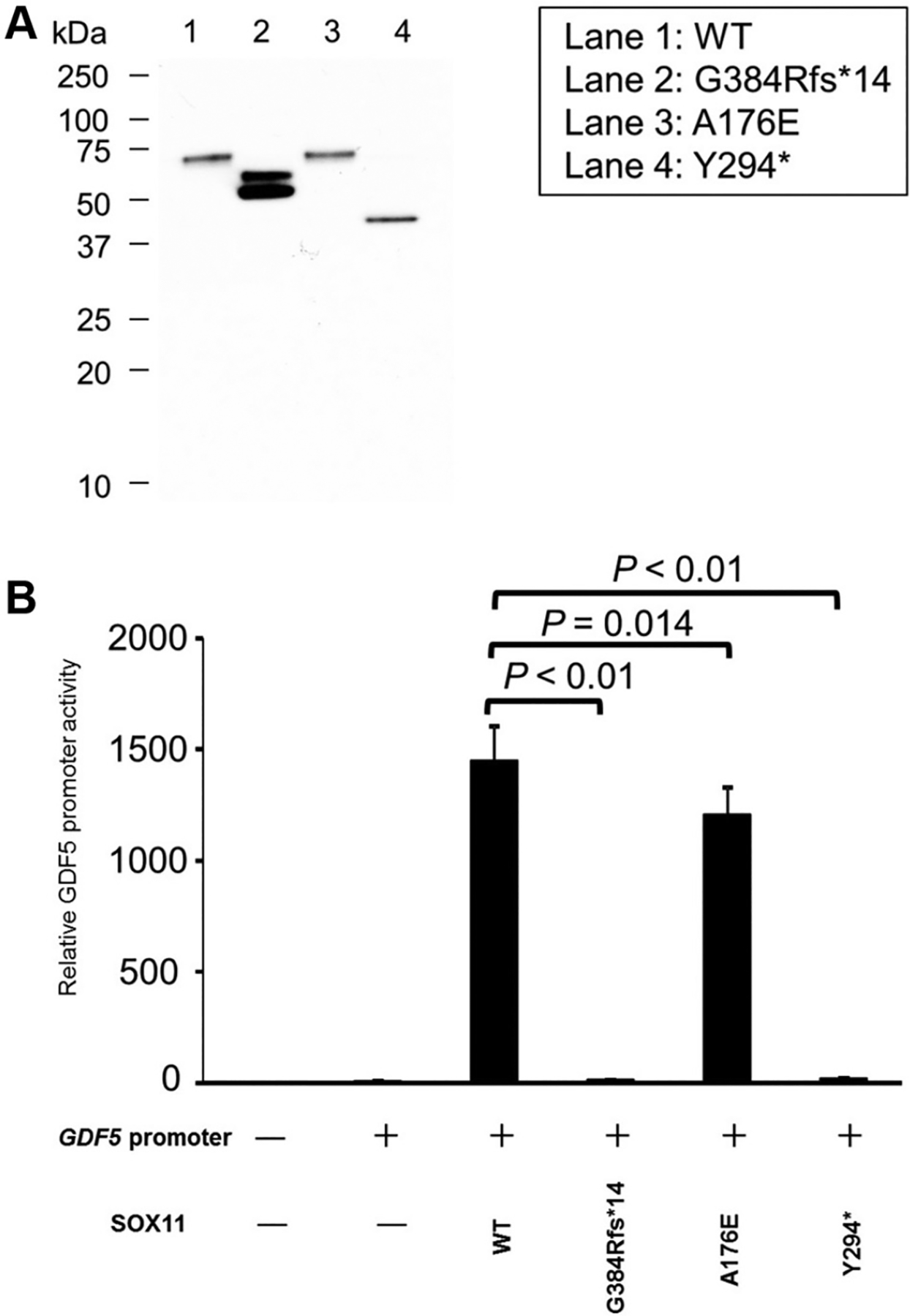
A. Western blot showing lower molecular weight of G384Rfs*14 and Y294* SOX11 variants than that of WT SOX11 protein. An anti-FLAG M2 HRP antibody (1:3000; Sigma-Aldrich) was used. B. Luciferase assay showing impaired activation of GDF5 promotor by G384Rfs*14 and Y294* SOX11 variants. A176E impairs SOX11 activity but to a much lesser extent than Y294* or G384Rfs*14. Data were taken from 2 separate experiments; each experiment was performed in technical triplicate. Activation of GDF5 promotor was compared between WT SOX11 protein and 3 SOX11 variants (G384Rfs*14, A176E, and Y294*) using t test. Correction for multiple comparisons was undertaken with a P value of .016 being taken as significant (0.5/3 = .016). WT, wild type.
We then investigated the effect of PTVs on SOX11 transactivating activity. In vitro analysis of 2 PTVs showed significant impairment of SOX11 transactivating ability (Figure 2). Only 2 PTVs are present in gnomAD (v2.1.1 non-neuro data set), and SOX11 is predicted to be loss-of-function intolerant with probability of loss of function intolerance = 0.86 (observed/expected 0.09 [0.03–0.44]). In vitro evidence of impaired transactivation and depletion of PTV in gnomAD supports a pathogenic role for SOX11 PTV. The mechanism through which PTV leads to reduction in SOX11 transactivating activity may relate to the loss of C-terminal transactivation domain.
Clinical phenotype associated with SOX11 variants
The clinical phenotypes are summarized in Supplemental Table 1. The mean age at examination was 9 years (range: neonate to 23 years). Microcephaly, short stature, and low body weight were common. There was a consistent facial dysmorphology across multiple ethnic groups (Supplemental Figure 2). All but 1 patient was reported to have developmental delay or ID. In total, 80% of patients had begun to sit by age 12 months (Supplemental Figure 3A), 70% were walking independently by age 30 months (Supplemental Figure 3B), and 80% had begun to speak by age 40 months (Supplemental Figure 3C). Internal organ malformations were uncommon, apart from renal anomalies (3 patients). Ocular involvement was infrequent, with oculomotor apraxia (4 patients), coloboma (2 patients), and microphthalmia (1 patient) being reported. Magnetic resonance imaging (MRI) of brain was performed in 20 (42%) patients and was abnormal in 12 (60% of imaged patients). Cerebellar malformations (4 patients), agenesis of the corpus callosum (4 patients), arhinencephaly (1 patient), Rathke’s cleft cyst (1 patient), and small pituitary gland (2 patients) were reported. A total of 8 (21%) patients presented with hypogonadotropic hypogonadism, which was confirmed by endocrine testing (Supplemental Table 2). Investigations were prompted by delayed puberty, cryptorchidism, or genital malformations.
Detection and verification of an episignature for SOX11 syndrome and classification of BAFopathy complex samples
An overall hypomethylation pattern was observed for most probes when comparing 10 SOX11 cases (case details in Supplemental Table 5) and control samples (Supplemental Figure 4). A total of 224 DMPs (Supplemental Figure 5, Supplemental Table 6) were used for the purpose of constructing unsupervised and supervised classification models. To assess the robustness of the episignature in differentiating between cases and controls, hierarchical clustering (Supplemental Figure 6A) and MDS analysis (Supplemental Figure 6B) were performed, resulting in a clear separation between these 2 groups. BAFopathy complex samples were applied to the SOX11 episignature classifier, but none of them were grouped with SOX11 samples (Supplemental Figure 7A and B). Ten rounds of cross-validation on MDS plot were performed using 9 SOX11 samples as training set and a single SOX11 sample as testing set. In all steps, the testing samples were correctly clustered with the training samples, further providing evidence of a robust common DNA methylation signature for SOX11 (Supplemental Figure 8).
Construction of the binary prediction model
Two MVP plots were generated to confirm specificity of the classification model. In the first MVP plot in which the SVM was trained by comparing the 10 SOX11 samples against controls, the classifier showed a high sensitivity for all SOX11 samples, with all samples scoring high on the MVP axis (Figure 3A) further confirming the previous heatmap and MDS results. Some samples from other disorders that are in EKD that are part of the EpiSign V2 clinical assay, including autosomal dominant cerebellar ataxia, deafness, and narcolepsy, HVDAS_T, and Sotos syndrome, plus 1 sample from control (testing), Kabuki syndrome, and mental retardation, autosomal dominanttype 51 cohorts showed an elevated MVP score, suggesting levels of similarity in the DNA methylation profiles between these disorders.
Figure 3. MVP scores plot.
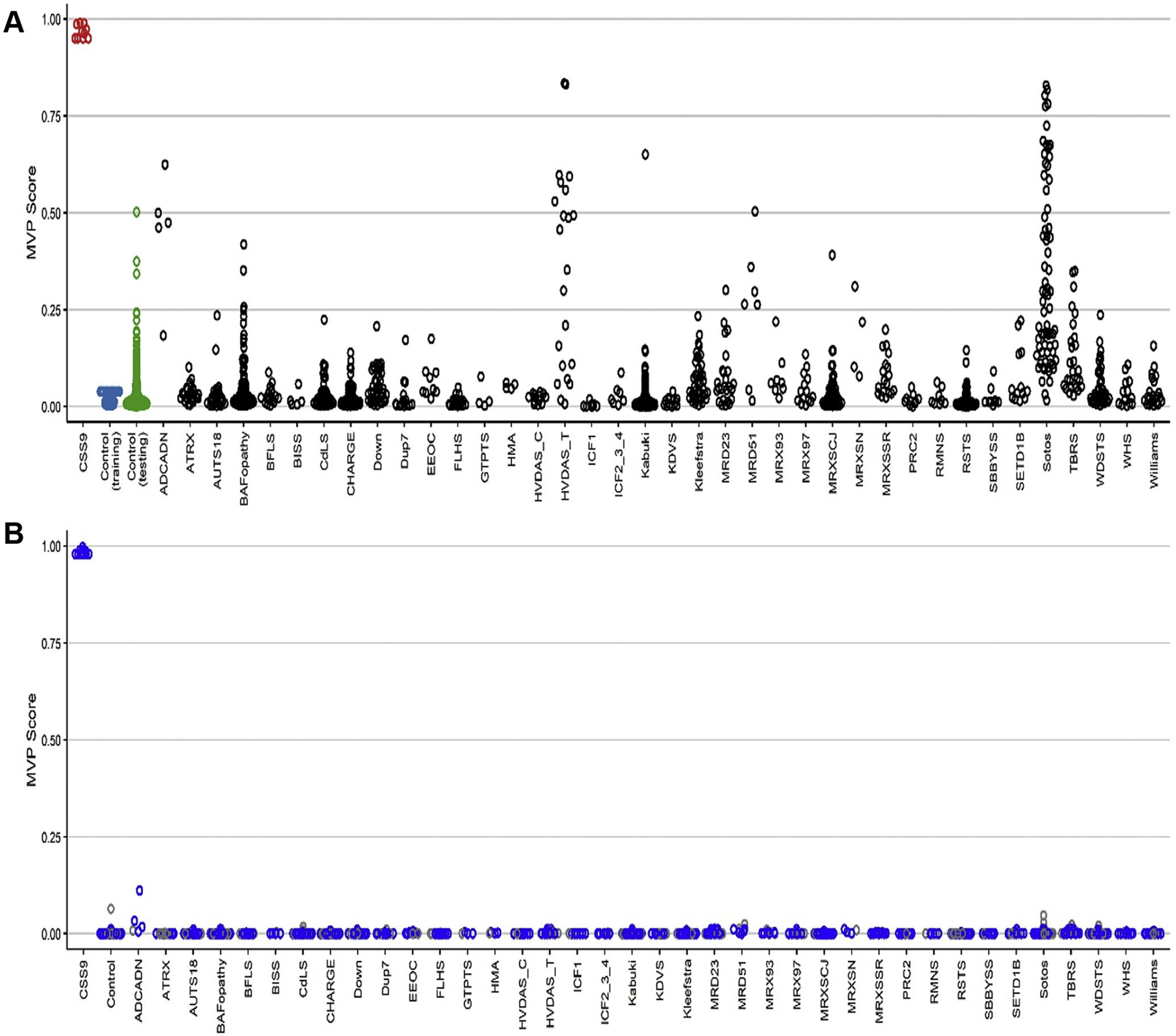
A. MVP scores was created using the support vector machine (SVM) trained by comparing 10 SOX11 samples against controls. B. MVP scores were created using the SVM trained by comparing 10 SOX11 samples against controls and 38 neurodevelopmental disorders and congenital anomalies available in the EpiSign Knowledge Database. The blue circles represent the training samples, and the gray circles represent the testing samples. MVP, methylation variant pathogenicity.
To increase the specificity of the classifier, the SVM was trained by comparing the 10 SOX11 samples against controls as well as 38 NDDs and congenital anomalies with known episignatures present in the EKD. A high MVP score was seen in 10 SOX11 samples with much improved specificity relative to other EpiSign conditions (Figure 3B).
Phenotype clustering separates SOX11 syndrome from ARID1B CSS
We used a phenotype-based clustering analysis to show that SOX11 syndrome and ARID1B CSS can be clinically distinguished (Figure 4). SOX11 variant heterozygotes tended to be microcephalic, have oculomotor apraxia or abnormal eye morphology (cataract, microphthalmia), or have hypogonadotrophic hypogonadism. ARID1B CSS was distinguished by coarse facial features and the absence of the HPO terms prevalent in SOX11 syndrome.
Figure 4. Phenotype-based clustering analysis of SOX11 and ARID1B phenotypes.
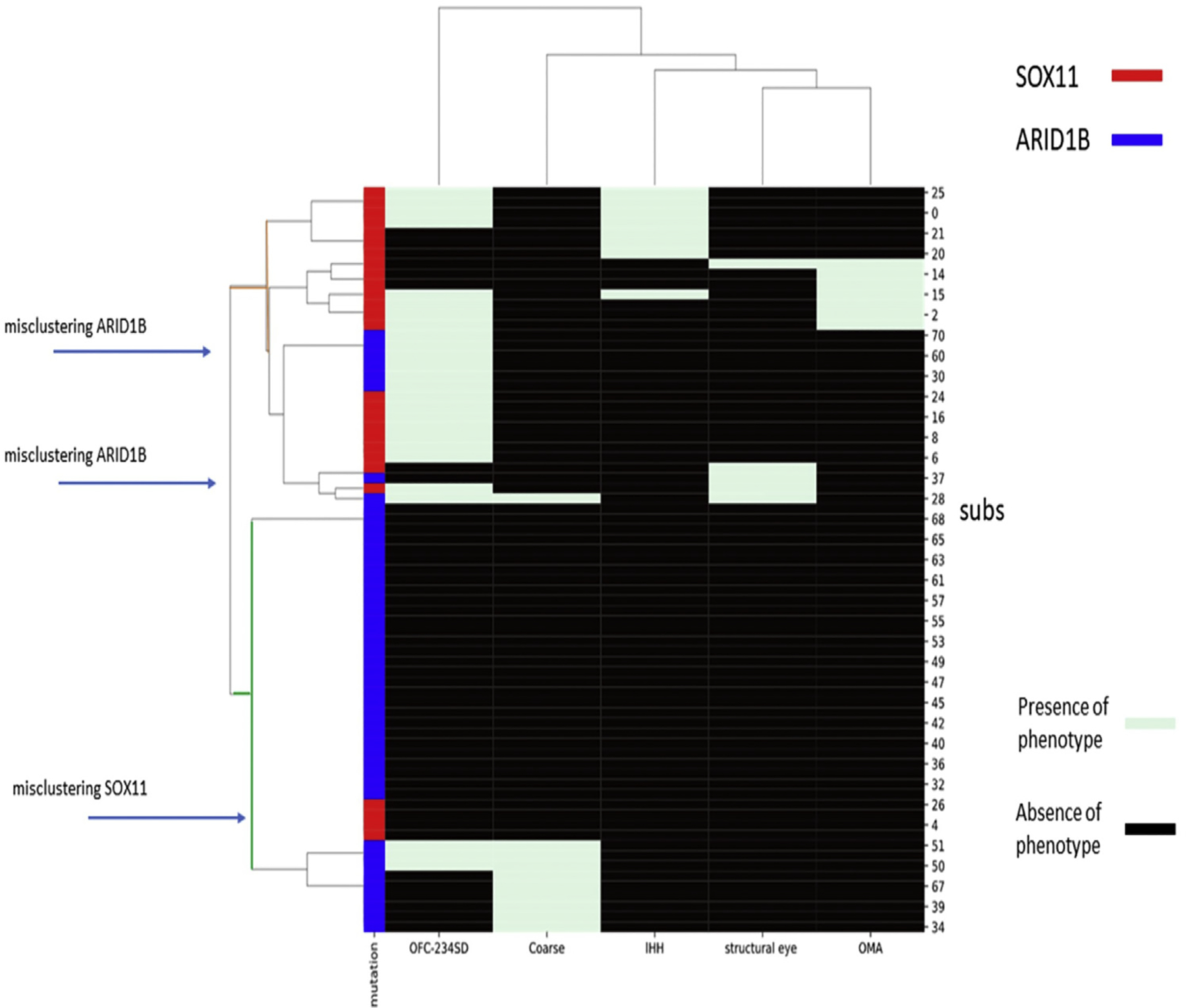
Hierarchical cluster analysis was performed in Python. Euclidean distance and Ward parameters were used to compute linkage distance and cluster merge strategy. SOX11 and ARID1B variant heterozygotes lie in different clusters. OFC indicates OFC < 2 SD. Coarse indicates coarse facial features. Structural eye indicates structural eye disease. Subs indicates individual subject. IHH, idiopathic hypogonadism; OFC, orbitofrontal circumference; OMA, oculomotor apraxia.
SOX11 expression in fetal central nervous system and pituitary gland
ISH showed widespread expression of SOX11 in fetal cranial structures (Figure 5). Use of positive and negative controls confirmed the specificity of this finding (Supplemental Figure 9). At all Carnegie stages examined, SOX11 was strongly expressed in the cerebral cortex and hindbrain (Supplemental Figure 10). Expression within the developing retina and optic nerve was also noted, particularly in Carnegie stage 23. Of interest, SOX11 expression was noted in the developing pituitary, lining the lumen of the adenohypophysis, and also within the neurohypophysis (Figure 5). There was no clear difference observed in spatial localization between SOX11 expression and GnRHR expression (Supplemental Figure 11).
Figure 5. RNAscope images of SOX11 expression using probes to SOX11.
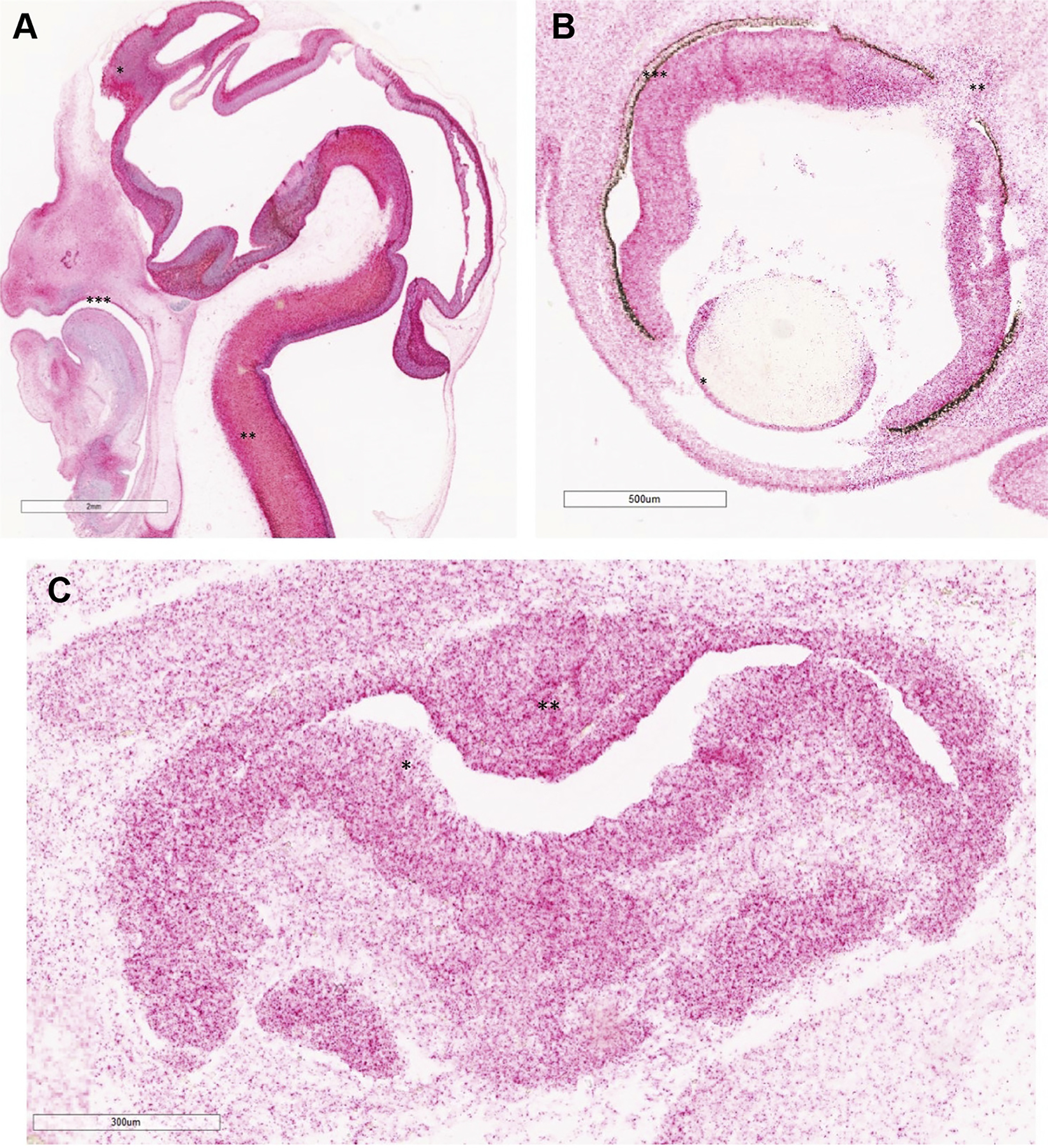
A. Saggital image of Carnegie stage (CS) 21 (CS21) (around 51 days after conception, 2× magnification). Note, diffused staining in central nervous system (red signal). SOX11 expression in frontal cortex (*), spinal cord (**), and palate (***). SOX11 probe is labeled red. No counterstain was used. B. SOX11 expression in developing eye at CS23 (around 56 days after conception, 4×) in lens (*), optic nerve (**), and neuroretina (***). SOX11 probe is labeled red. No counterstain was used. C. SOX11 expression in pituitary at CS20 (around 49 days after conception, 10×) lining lumen of adenohypophysis (*) and also in neurohypophysis (**). SOX11 probe is labeled red. No counterstain was used.
Discussion
In this article, we report a large cohort of people with SOX11 pathogenic variants and define the molecular genetics and clinical spectrum of the disorder. Most pathogenic and likely pathogenic variants were missense. These were all located within or next to the HMG box, a common motif in all SOX proteins that activate target genes via DNA binding.8 The HMG box was significantly depleted of missense variants in samples from gnomAD, suggesting that it does not tolerate variation in healthy individuals. This shows that the HMG box could be classified as a variant “hot spot” for SOX11, further supported by the recurrent variants found at several residues in this domain. Most SOX11 variants in our cohort were de novo, in keeping with a severe syndrome that impairs reproductive fitness. We identified 1 instance of transmission from a mosaic mother. This shows that recurrence could be possible due to mosaicism.
In addition, we identified a single de novo missense variant, p.(Ala176Glu), that was not in the HMG box. This variant reduced SOX11 transactivating ability. We have not identified other affected individuals with missense variants in this region (or other SOX11 protein regions that are not the HMG box). The individual (case 11) died early in the neonatal period. They had cerebellar hypoplasia and microcephaly, which is compatible with SOX11 syndrome. However, it was not possible to ascertain whether they had other features of SOX11 syndrome. It is possible that missense variants in different regions of SOX11 might be pathogenic, as shown by case 11. This warrants further investigation through identification of more cases and functional analyses.
All but 1 of the patients in this cohort had ID or developmental delay. Speech was particularly affected, with Kaplan-Meier analysis suggesting that 20% of the patients may not attain speech. Renal malformations were the only common internal organ malformation. This is in keeping with murine studies indicating that loss of SOX11 results in a spectrum of congenital anomalies of the kidney and urinary tract.23 In mice, these included duplex kidney, mal-positioned kidneys, and hydroureter, which overlaps with the renal anomalies in our cohort. We confirm that ocular malformations—coloboma, lens abnormalities, and microphthalmia—occur in SOX11 syndrome.24–27 These are recapitulated in sox11 null zebrafish, confirming the specificity of the finding.28 In our cohort, 10% of patients had oculomotor apraxia. Diagnosis of oculomotor apraxia requires specialized neuro-ophthalmological evaluation, and therefore, the true prevalence of this feature is likely to be higher. Our study confirms that SOX11 syndrome should be part of the differential diagnosis of oculomotor apraxia.
The extent of brain malformation in SOX11 syndrome is revealed by our work. Of the patients who were imaged, 60% had an abnormality on brain MRI, and the true prevalence is likely to be higher given that children with ID are often not imaged. The most striking findings were those of cerebellar hypoplasia. In Sox11 null mice, there is generalized reduction in size of the cerebrum and cerebellum.29 In this current report, humans with SOX11 variants have microcephaly and cerebellar hypoplasia. The combined human and murine findings indicate that SOX11 has a general role in brain development, rather than a predominant role in the cerebrum or cerebellum. Beyond neurodevelopmental delay, there is little evidence that brain malformations in SOX11 syndrome are associated with neurological disorders. Only 2 participants had epilepsy, and there was no clearly defined ataxia in association with the cerebellar findings on imaging.
In our cohort, 21% of the patients had hypogonadotrophic hypogonadism. A role for SOX11 in hypogonadotrophic hypogonadism is plausible given that other SOX genes (SOX2, SOX10) cause hypogonadotrophic hypogonadism in humans.9,10 In males with SOX11 syndrome presentation, genital malformations at birth was reported, but in both sexes delayed puberty was the principal manifestation. Hypogonadotrophic hypogonadism with anosmia is termed Kallman syndrome30 and is associated with failure of hypothalamic gonadotrophin releasing hormone (GnRH)–releasing neurons to migrate correctly into the hypothalamus. Given the neurodevelopmental delay in SOX11 syndrome, formal assessment of olfaction is not possible. However, 1 participant was reported to have anosmia with hypoplasia of the olfactory nerves on MRI, and the Sox11 null mouse has small olfactory bulbs.29 Based on current evidence, it is not possible to state whether SOX11 syndrome is associated with Kallman syndrome or normosmic hypogonadotrophic hypogonadism.
How SOX11 variants might result in hypogonadotrophic hypogonadism is unclear. GnRH neurons are a central part of the hypothalamic-pituitary-gonad axis.30 GnRH neurons originate in the olfactory neuroepithelium and migrate to the hypothalamus. SOX11 variants could disrupt this process. In support of this, SOX11 has been shown to be expressed in GnRH hypothalamic neurons in mice and has been shown to stimulate expression and secretion of GnRH in vitro.31 However, in an induced human pluripotent cell model of GnRH neurons, SOX11 expression was not enriched.32 A non–mutually exclusive hypothesis is that SOX11 plays a role in pituitary gonadotropes. A single-cell RNA-sequencing study of murine pituitary showed significant enrichment of SOX11 in gonadotropes.33 Our ISH study showed SOX11 expression in developing human pituitary and hypothalamus, compatible with a direct role in the development of both. SOX11 may play multiple roles in development of the hypothalamic-pituitary-gonadal axis, which requires further study.
We describe the pattern of SOX11 transcript expression in human development using ISH. As expected, SOX11 is widely expressed in the developing central nervous system including the cerebrum, cerebellum, and brainstem. This confirms previous work using RNA-sequencing and micro-array data from the BrainSpan atlas of the developing brain. SOX11 was also observed in the developing palate. This suggests a direct role of SOX11 in human palatogenesis and SOX11 loss-of-function in the cleft palate observed in some patients of our cohort. Cleft palate has been described in Sox11-null mice.34 This was associated with reduced proliferation of cells in the palatal shelves but also mandibular hypoplasia, with the suggestion that the cleft resulted from a Pierre-Robin sequence. SOX11 was also expressed within the lumen of the developing adenohypophysis and the neurohypophysis. This supports a role for SOX11 in the development of the pituitary gland, as discussed earlier. SOX11 expression in the retina and optic nerve confirms the importance of SOX11 for development of these structures in humans.
Unique genomic DNA methylation patterns, referred to as episignatures, are promising alternatives to diagnose NDDs and overgrowth/ID syndromes. Our group and others have shown the diagnostic utility of genome-wide DNA methylation analysis using peripheral blood samples.14,15,17,35 In this study, we showed a highly sensitive and specific blood-derived episignature with small number of DMPs for SOX11 syndrome, using a relatively small number of patient samples. Many of these DMPs have regulatory roles in neural differentiation and are associated with NDDs (ie, family with sequence similarity 160 member B1 [FAM160B1]36 and FMN237). Some DMPs have regulatory role in the epigenetic machinery, such as DPF138 and AHCTF1.39 This highlights the fact that aberrations in the expression/methylation status of SOX11 affects expression/methylation status of genes involved in neural differentiation and/or epigenetic machinery, in agreement with the observed global hypomethylation seen in Supplemental Figure 5. It also shows the utility of DNA methylation profiling as a useful biomarker for clinical diagnosis of SOX11-related disorders.
DNA methylation is an epigenetic modification affecting molecular mechanisms, including chromatin assembly and gene transcription. Recent advances in sequencing and array technologies, capable of scrutinizing genome-wide DNA methylation patterns, gave unexpected novel insights into the identification of epigenetic biomarkers. This study focuses on DNA methylation as a clinical diagnostic biomarker, enabling interpretation of genetic variation in SOX11. Future studies focusing on integrating epigenomic and gene expression profiles in patients with SOX11-related disorders may provide insights into how epigenetic alterations lead to NDDs. The plastic nature of epigenomic profiles may offer an opportunity to study the use of chromatin and epigenomic targeting agents as a potential therapeutic avenue.
Individuals with SOX11 syndrome were initially reported as having a CSS phenotype. Our phenotype driven clustering analysis shows that, based on HPO terms, SOX11n-syndrome is clinically distinct from ARID1B-related CSS. Particular differentiating features were presence of oculomotor apraxia, ocular malformations, and idiopathic hypogonadotrophic hypogonadism in SOX11 syndrome. This is further supported by methylation analyses of peripheral blood DNA, which shows that SOX11 syndrome and BAFopathies have distinct episignatures (Figure 3). Taken together, this confirms that SOX11 syndrome should be considered as a distinct clinical entity from CSS.
Supplementary Material
Acknowledgments
We thank the families who kindly agreed to participate in this study. This work was supported by a grant from the Baily Thomas Charitable Foundation (TRUST/VC/AC/SG/5399-8436) (atypical neurogenesis) to M.F. and A.M. This work was supported by a grant from the Waterloo Foundation to A.M., AMED grants under grant numbers JP21ek0109486, JP21ek0109549, and JP21ek0109493 to N.M., and grant number JP21ek0109486 to S.I. In addition, funding for this study was provided, in part, by the London Health Sciences Molecular Diagnostics Development Fund and Genome Canada Genomic Applications Partnership Program Grant (Beyond Genomics: Assessing the Improvement in Diagnosis of Rare Diseases using Clinical Epigenomics in Canada, EpiSign-CAN). Sequencing for case 29 was provided by the University of Washington Center for Mendelian Genomics (UW-CMG) and was funded by National Human Genome Research Institute and National Heart, Lung, and Blood Institute grants UM1 HG006493 and U24 HG008956, by the Office of the Director, and National Institutes of Health (NIH) grants under award number S10OD021553 as well as NIH grant number R01EY025718 to E.V.S. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project is funded by the National Institute for Health Research and National Health Service England. The Well-come Trust, Cancer Research UK, and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support. We thank Dr Michela Adamo for help with obtaining clinical data on one of the reproted cases.
Ethics Declaration
A relative or carer gave written consent for publication of clinical details and photographs of the participants in this study. Exome sequencing in the Deciphering Developmental Disorders study has United Kingdom Research Ethics Committee (REC) approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). Genome sequencing undertaken in the 100,000 Genomes Project has approval from the East of England—Cambridge South REC (REC Ref 14/EE/1112). The Human Developmental Brain Resources has approval from the North East—Newcastle and North Tyneside 1 REC (18/NE/0290). The study was approved by the Western University Research Ethics Board (REB 106302 and 116108).
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
Additional Information
The online version of this article (https://doi.org/10.1016/j.gim.2022.02.013) contains supplementary material, which is available to authorized users.
Data Availability
For the episignature work, summarized and anonymized data for each subject are described in the study. The source DNA methylation data are available from the authors upon request. Software used in this study is publicly available, and the detailed analytical methodology is as previously reported (PMID: 34087165). The images from RNAscope (SOX11 and GnRHR probes) experiment are available from Jasmin Turner or Alisdair McNeill. Exome sequencing data from Deciphering Developmental Disorders study are available through application to the Deciphering Developmental Disorders study management committee (https://www.deciphergenomics.org/). Genome sequencing data from the 100,000 Genomes Project is available in the Genomics England Research Environment https://re.extge.co.uk/ovd/
References
- 1.Bögershausen N, Wollnik B. Mutational landscapes and phenotypic spectrum of SWI/SNF-related intellectual disability disorders. Front Mol Neurosci. 2018;11:252. 10.3389/fnmol.2018.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Sluijs PJ, Jansen S, Vergano SA, et al. The ARID1B spectrum in 143 patients: from nonsyndromic intellectual disability to Coffin–Siris syndrome. Genet Med. 2019;21(6):1295–1307. Published correction appears in Genet Med. 2019;21(9):2160–2161. 10.1038/s41436-018-0330-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wright CF, Fitzgerald TW, Jones WD, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;385(9975):1305–1314. 10.1016/S0140-6736(14)61705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vasileiou G, Vergarajauregui S, Endele S, et al. Mutations in the BAF-complex subunit DPF2 are associated with Coffin-Siris syndrome. Am J Hum Genet. 2018;102(3):468–479. 10.1016/j.ajhg.2018.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kosho T, Okamoto N, Coffin-Siris Syndrome International Collaborators. Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am J Med Genet C Semin Med Genet. 2014;166C(3):262–275. 10.1002/ajmg.c.31407. [DOI] [PubMed] [Google Scholar]
- 6.Tsurusaki Y, Koshimizu E, Ohashi H, et al. De novo SOX11 mutations cause Coffin–Siris syndrome. Nat Commun. 2014;5:4011. 10.1038/ncomms5011. [DOI] [PubMed] [Google Scholar]
- 7.Hempel A, Pagnamenta AT, Blyth M, et al. Deletions and de novo mutations of SOX11 are associated with a neurodevelopmental disorder with features of Coffin–Siris syndrome. J Med Genet. 2016;53(3):152–162. 10.1136/jmedgenet-2015-103393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Angelozzi M, Lefebvre V. SOXopathies: growing family of developmental disorders due to SOX mutations. Trends Genet. 2019;35(9):658–671. 10.1016/j.tig.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pingault V, Bondurand N, Kuhlbrodt K, et al. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat Genet. 1998;18(2):171–173. 10.1038/ng0298-171. [DOI] [PubMed] [Google Scholar]
- 10.Kelberman D, de Castro SCP, Huang S, et al. SOX2 plays a critical role in the pituitary, forebrain, and eye during human embryonic development. J Clin Endocrinol Metab. 2008;93(5):1865–1873. 10.1210/jc.2007-2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turro E, Astle WJ, Megy K, et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature. 2020;583(7814):96–102. 10.1038/s41586-020-2434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36(10):928–930. 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richards S, Aziz N, Bale S. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kopanos C, Tsiolkas V, Kouris A, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019;35(11):1978–1980. 10.1093/bioinformatics/bty897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Notaro M, Schubach M, Robinson PN, Valentini G. Prediction of Human Phenotype Ontology terms by means of hierarchical ensemble methods. BMC Bioinformatics. 2017;18(1):449. 10.1186/s12859-017-1854-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aref-Eshghi E, Kerkhof J, Pedro VP, et al. Evaluation of DNA methylation episignatures for diagnosis and phenotype correlations in 42 Mendelian neurodevelopmental disorders. Am J Hum Genet. 2020;106(3):356–370. Published correction appears in Am J Hum Genet. 2021;108(6):1161–1163. 10.1016/j.ajhg.2020.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sadikovic B, Levy MA, Kerkhof J, et al. Clinical epigenomics: genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet Med. 2021;23(6):1065–1074. Published correction appears in Genet Med. 2021;23(11):2228. 10.1038/s41436-020-01096-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aref-Eshghi E, Bend EG, Colaiacovo S, et al. Diagnostic utility of genome-wide DNA methylation testing in genetically unsolved individuals with suspected hereditary conditions. Am J Hum Genet. 2019;104(4):685–700. 10.1016/j.ajhg.2019.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pidsley RY Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14(1):293. 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiewel MA, Huson MA, van Vught LA, et al. Impact of HIV infection on the presentation, outcome and host response in patients admitted to the intensive care unit with sepsis; a case control study. Crit Care. 2016;20(1):322. 10.1186/s13054-016-1469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neirijnck Y, Reginensi A, Renkema KY, et al. Sox11 gene disruption causes congenital anomalies of the kidney and urinary tract (CAKUT). Kidney Int. 2018;93(5):1142–1153. 10.1016/j.kint.2017.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho CY, Tsai WY, Lee CT, et al. Clinical and molecular features of idiopathic hypogonadotropic hypogonadism in Taiwan: a single center experience. J Formos Med Assoc. 2022;121(1 Pt 1):218–226. 10.1016/j.jfma.2021.03.010. [DOI] [PubMed] [Google Scholar]
- 25.Wakim V, Nair P, Delague V, et al. SOX11-related syndrome: report on a new case and review. Clin Dysmorphol. 2021;30(1):44–49. 10.1097/MCD.0000000000000348. [DOI] [PubMed] [Google Scholar]
- 26.Diel H, Ding C, Grehn F, Chronopoulos P, Bartsch O, Hoffmann EM. First observation of secondary childhood glaucoma in Coffin-Siris syndrome: a case report and literature review. BMC Ophthalmol. 2021;21(1):28. Published correction appears in BMC Ophthalmol. 2021;21(1):57. 10.1186/s12886-020-01788-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanker B, Gillessen-Kaesbach G, Hüning I, Lüdecke HJ, Wieczorek D. Maternal transmission of a mild Coffin–Siris syndrome phenotype caused by a SOX11 missense variant. Eur J Hum Genet. 2022;30(1):126–132. 10.1038/s41431-021-00865-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pillai-Kastoori L, Wen W, Wilson SG, et al. Sox11 is required to maintain proper levels of Hedgehog signaling during vertebrate ocular morphogenesis. PLoS Genet. 2014;10(7):e1004491. 10.1371/journal.pgen.1004491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Lin L, Lai H, Parada LF, Lei L. Transcription factor Sox11 is essential for both embryonic and adult neurogenesis. Dev Dyn. 2013;242(6):638–653. 10.1002/dvdy.23962. [DOI] [PubMed] [Google Scholar]
- 30.Acién P, Acién M. Disorders of sex development: classification, review, and impact on fertility [review]. J Clin Med. 2020;9(11):3555. 10.3390/jcm9113555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim HD, Choe HK, Chung S, et al. Class-C Sox transcription factors control GnRH gene expression via the intronic transcriptional enhancer. Mol Endocrinol. 2011;25(7):1184–1196. 10.1210/me.2010-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lund C, Yellapragada V, Vuoristo S, et al. Characterization of the human GnRH neuron developmental transcriptome using a GNRH1-TdTomato reporter line in human pluripotent stem cells. Dis Model Mech. 2020;13(3):dmm040105. 10.1242/dmm.040105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ho Y, Hu P, Peel MT, et al. Single-cell transcriptomic analysis of adult mouse pituitary reveals sexual dimorphism and physiologic demand-induced cellular plasticity. Protein Cell. 2020;11(8):565–583. 10.1007/s13238-020-00705-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang H, Yang X, Bao M, et al. Ablation of the Sox11 gene results in clefting of the secondary palate resembling the pierre robin sequence. J Biol Chem. 2016;291(13):7107–7118. 10.1074/jbc.M115.690875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butcher DT, Cytrynbaum C, Turinsky AL, et al. CHARGE and Kabuki syndromes: gene-specific DNA methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. Am J Hum Genet. 2017;100(5):773–788. 10.1016/j.ajhg.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anazi S, Maddirevula S, Faqeih E, et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol Psychiatry. 2017;22(4):615–624. 10.1038/mp.2016.113. [DOI] [PubMed] [Google Scholar]
- 37.Law R, Dixon-Salazar T, Jerber J, et al. Biallelic truncating mutations in FMN2, encoding the actin-regulatory protein formin 2, cause nonsyndromic autosomal-recessive intellectual disability. Am J Hum Genet. 2014;95(6):721–728. 10.1016/j.ajhg.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villanueva-Chimal E, Salinas LS, Fernández-Cardenas LP, Huelgas-Morales G, Cabrera-Wrooman A, Navarro RE. DPFF-1 transcription factor deficiency causes the aberrant activation of MPK-1 and meiotic defects in the Caenorhabditis elegans germline. Genesis. 2017;55(11). 10.1002/dvg.23072. [DOI] [PubMed] [Google Scholar]
- 39.Scholz BA, Sumida N, de Lima CDM, et al. WNT signaling and AHCTF1 promote oncogenic MYC expression through super-enhancer-mediated gene gating. Nat Genet. 2019;51(12):1723–1731. Published correction appears in Nat Genet. 2020;52(11):1265. 10.1038/s41588-019-0535-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For the episignature work, summarized and anonymized data for each subject are described in the study. The source DNA methylation data are available from the authors upon request. Software used in this study is publicly available, and the detailed analytical methodology is as previously reported (PMID: 34087165). The images from RNAscope (SOX11 and GnRHR probes) experiment are available from Jasmin Turner or Alisdair McNeill. Exome sequencing data from Deciphering Developmental Disorders study are available through application to the Deciphering Developmental Disorders study management committee (https://www.deciphergenomics.org/). Genome sequencing data from the 100,000 Genomes Project is available in the Genomics England Research Environment https://re.extge.co.uk/ovd/