

Abstract
Aberrant activation of NF-κB is the most striking oncogenic mechanism in B cell lymphoma; however, its role in anaplastic large cell lymphomas (ALCL) has not been fully established and its activation mechanism(s) remain unclear. Using ALCL cell line models, we revealed the supporting roles for NFKB2 and the NIK pathway in some ALCL lines. To investigate the detailed activation mechanisms for this oncogenic pathway, we performed specifically designed alternative NF-κB reporter CRISPR screens followed by the RNA-seq analysis, which led us to identify STAT3 as the major mediator for NIK-dependent NF-κB activation in ALCL. Consistently, p-STAT3 level was correlated with NFKB2 nuclear accumulation in primary clinical samples. Mechanistically, we found that in NIK positive ALK− ALCL cells, common JAK/STAT3 mutations promote transcriptional activity of STAT3 which directly regulates NFKB2 and CD30 expression. Endogenous expression of CD30 induces constitutive NF-κB activation through binding and degrading of TRAF3. In ALK+ ALCL, the CD30 pathway is blocked by the NPM-ALK oncoprotein, but STAT3 activity and resultant NFKB2 expression can still be induced by NPM-ALK, leading to minimal alternative NF-κB activation. Our data suggest combined NIK and JAK inhibitor therapy could benefit patients with NIK positive ALK− ALCL carrying JAK/STAT3 somatic mutations.
Introduction
Anaplastic large cell lymphoma (ALCL) is a group of mature T-cell neoplasms sharing elevated expression of CD30 and anaplastic cytology1. ALCL subtypes are divided into two classes based on the status of anaplastic lymphoma kinase (ALK)2, 3, ALK positive (ALK+) and ALK negative (ALK−). While ALK+ ALCL systemic is relatively homogeneous, ALK− ALCL represents a heterogeneous group comprising systemic ALK negative and primary cutaneous ALCL (pC-ALCL)4, 5. The recently recognized breast implant-associated (BIA) ALCL, which arises in the seroma cavity surrounding breast implants, was acknowledged as a third subtypes of ALK− ALCL6. Current therapeutic strategies for ALCL are based on aggressive B-cell lymphoma regimens. The ALK+ ALCL systemic has a relatively good prognosis (>70% OS). In ALK− ALCL, although the outcome for pC-ALCL and BIA-ALCL are generally good (90% OS), the systemic ALK− ALCL usually has a poor prognosis (<50% OS)7. Thus, there is an urgent need to develop targeted therapies in these malignancies, especially in systemic ALK− cases.
A recurrent theme in current understanding of the lymphoma biology is that the lymphoid malignancies co-opt signaling proteins and transcription factors that are used in normal immune cell differentiation and activation. When ALCL is compared to B cell lymphoma, several common factors have been established in both diseases, such as JAK/STAT38, 9, IRF410, 11, AP-1 members cJUN and JUNB12, 13, and PI3K/AKT14, 15. However, some gaps in our knowledge of ALCL remain and need to be addressed, for example, the oncogenic role of the transcriptional factor NF-κB. There are five NF-κB family members: RelA (p65), RelB, c-Rel, NFKB1 (p50 and its precursor p105) and NFKB2 (p52 and its precursor p100). Active NF-κB transcription factors are composed of dimeric combinations of these members16, 17, and two major pathways have been identified. The canonical or classical NF-κB pathway is typically activated by the inhibitor of NF-κB (IκB) kinase (IKK) complex, which allows the nuclear translocation of the RelA/p50 heterodimer and subsequent induction of specific gene transcription. In contrast, the noncanonical or alternative NF-κB pathway is mediated by NF-κB-inducing kinase (NIK), a kinase implicated in the phosphorylation and activation of IKKα. IKKα which then targets p100 for phosphorylation and ubiquitination, resulting in the limited proteolysis of its ankyrin-like C-terminus, generating the mature p52 subunit which enters the nucleus to activate the expression of genes.
In B cell lymphoma, BCR induced aberrant activation of the canonical NF-κB pathway is the most striking oncogenic mechanism18. Several studies suggest that NF-κB is activated in primary ALK+ and ALK− ALCLs, which may confer unique clinical outcomes19–21. ALCL express a high level of TNFR superfamily member CD3022. However, this expression alone is not enough to activate NF-κB in ALCL cells23, indicating that mechanisms in addition to CD30 could contribute to NF-κB signaling in ALCL. Thus, the need remains to identify the detailed mechanism of NF-κB pathway activation in both ALK+ and ALK− ALCL.
Materials and Methods
(See Supplemental Experimental Procedures for details)
Depletion CRISPR library screen (see Supplemental Experimental Procedures for details).
In brief, 40 million cells were infected with the pooled lentiviral sgRNA library. Genomic DNA was extracted and sgRNA sequences were amplified by two rounds of PCR. The resulting libraries were sequenced with single end read with dual-index 75 bp.
RNA sequencing.
The bulk RNA-seq libraries were constructed using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB) and sequenced on the Illumina HiSeq 2500 platform with 150 bp paired-end mode. The RNA sequencing data have been submitted to the NCBI Sequence Read Archive (SRA) under accession number: SUB6534613.
Statistical analyses.
All experiments have been repeated and results reproduced. The number of repeats have been clearly described in Figure legends, and in Supplementary Figure legends (for immunoblots and IPs). Where possible, error bars and/or P values are shown to indicate statistical significance. P values were calculated with t-test. P < 0.05 was considered statistically significant.
Result
Expression of nuclear p52 in ALCL primary samples and cell lines
To gain a comprehensive understanding of the unique factors in ALCL, we produced a unique lymphoma signaling-focused sgRNA library as described in24 for use in genetic screens, which includes immune cell signaling components in known oncogenic pathways required for lymphoma pathogenesis25. A depletion (loss-of-function) screen was performed in the ALK+ ALCL line DEL and DLBCL line U2932 (control) (Supplementary Fig. 1A). We then plotted the screen results for all the genes in the library, and analyzed all the positive hits responsible for tumor cell survival in both the DEL and U2932 lines (Supplementary Fig. 1B). In addition to the common general toxic genes identified in both B and T lymphoma lines (UBE2l, CYCS, RPS6, SOD2), the genes identified as strong positive hits in U2932 matched well with previous studies in DLBCL18, including BCL2 and components of B cell receptor pathway (Supplementary Fig. 1B, right), demonstrating the validity of the screen setup. Similarly, the essential genes supporting ALCL survival identified by DEL screening included some of the well-known molecules including STAT3, JUNB, IRF4 and SOS1 (Supplementary Fig. 1B, left). Interestingly, in addition to those established pathways, NFKB2, a family member of NF-κB transcription factors, was identified as a strong positive hit only in DEL, but not in U2932 (Supplementary Fig. 1B). Confirming the CRISPR screen results, depletion of NFKB2 expression by two individual sgRNAs was moderately toxic to ALCL lines DEL and Karpas299 (Supplementary Fig. 1C). Overall, our CRISPR library screen results were consistent with previous findings implicating the essential role of STAT3, AP1, PI3K–AKT and IRF4 in ALCL; however, our screen also identified NFKB2 as a novel gene that supports the viability of ALCL, indicating the potential role of the alternative NF-κB pathway.
We then assessed the clinical relevance of alternative NF-κB activation in primary ALCL cases, by evaluating p100/p52 and RELB level by immunohistochemical (IHC) staining in 35 primary cases diagnosed with ALCL (17 ALK− and 18 ALK+) (Fig. 1A, right, two representative cases are shown). Notably, certain level of p52 nuclear accumulation was detected in 15 out of 17 ALK− cases, and in 14 out of 18 ALK+ cases (Fig. 1A and Supplementary Table 1, 2). Of note, perinuclear staining (p100) was also observed in primary samples, especially in ALK+ cases (Fig. 1A, right). Thus, the alternative NF-κB activity is frequently elevated in both ALK+ and ALK− cases.
Figure 1: Expression of p100/p52 in ALCL primary samples and cell lines.
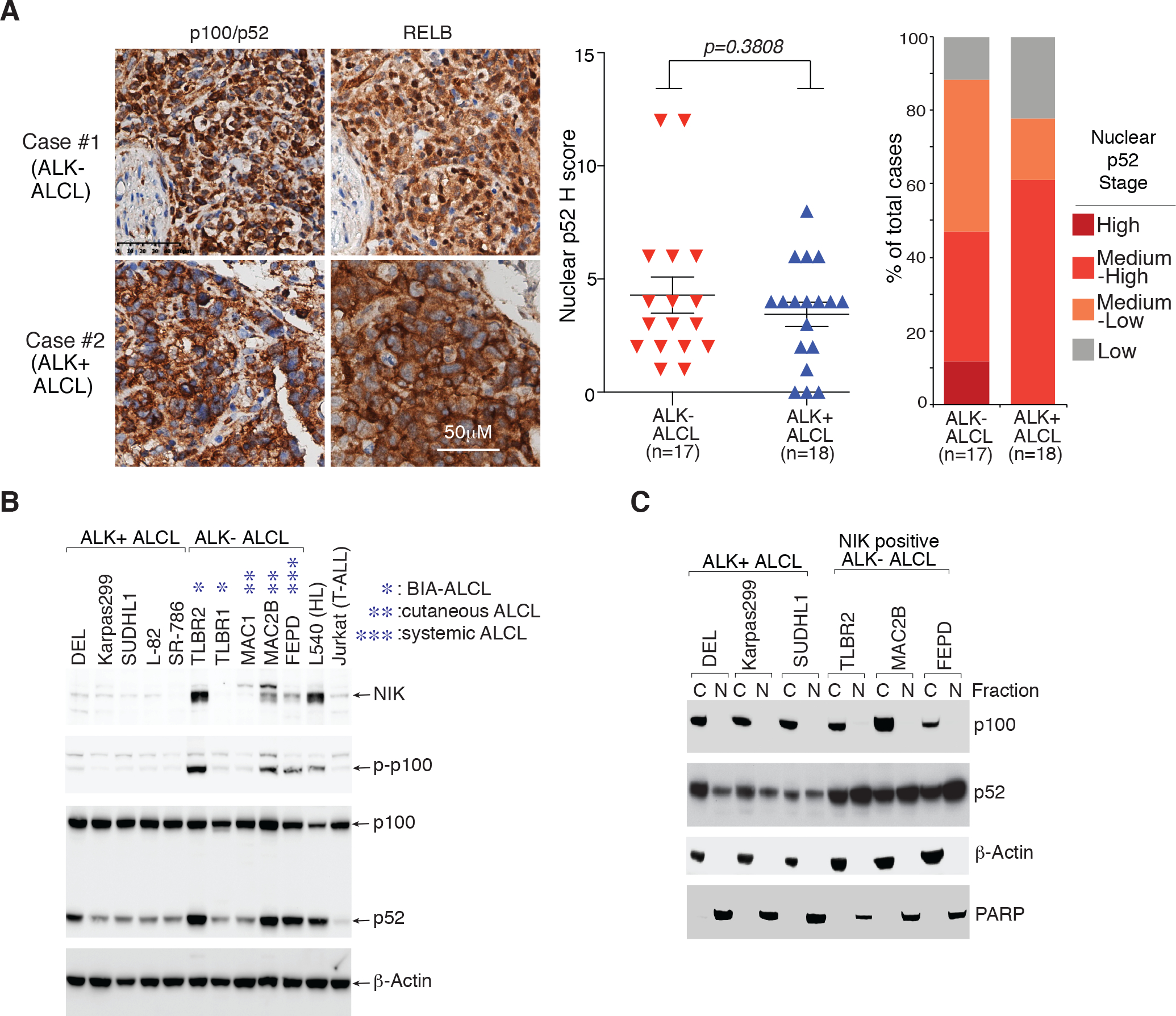
A. Expression of p100/p52 and RELB in ALCL primary cases by immunohistochemistry (IHC). left: Immunohistochemical p100/p52 and RELB staining are shown in two cases. Section of lymph nodes were examined microscopically using a 400X magnification. middle: Nuclear p52 IHC score (detailed at Supplementary Table 1) of the 17 ALK− ALCL and 18 ALK+ ALCL cases examined. p value between ALK− and ALK+ groups is indicated. right: Nuclear p52 stages in primary ALCL cases. IHC score (H Score): <2 (low); >=2, <4 (medium-low); >=4, <9 (medium-low); >=9(high). B. Lysates of indicated cell lines were analyzed by immunoblotting for the indicated proteins. C. Nuclear and cytosol fractions of indicated cell lines were analyzed by immunoblotting for the indicated proteins.
In primary samples, constitutive p100 processing that yields p52 nuclear accumulation was detected, leading us to evaluate the alternative NF-κB activation in a panel of ALCL cell lines, together with a positive control HL line L54026 and a negative control T-ALL line Jurkat. Surprisingly, although constitutive p52 accumulation was observed in the majority of ALCL lines, activated alternative NF-κB signaling, measured by p100 phosphorylation and NIK stabilization, was detected in only a subset of ALK− ALCL lines: TLBR2 (BIA), MAC2B (cutaneous) and FEPD (systemic) (Fig. 1B and Supplementary Fig. 1D), interestingly, in all three subtypes of ALK− ALCL. In line with these results, strong p52 nuclear accumulation was observed in these three NIK positive lines (Fig. 1C). Therefore, ALK+ and ALK− ALCL might have distinct mechanism(s) to maintain alternative NF-κB activation, and only some ALK− ALCLs are likely to have activated NIK-IKKα signaling.
NIK positive ALK− ALCL lines are more dependent on activated NIK-IKKα signaling
Our CRISPR screen revealed a potential role for the alternative NF-κB pathway in ALCL; however, it appears that only some ALK− ALCLs have activated signaling (Fig. 1B, 1C). In alternative NF-κB signaling, p100 is phosphorylated through a NIK-IKKα kinase cascade. We therefore investigated the role of NIK and IKKα in ALCL. In a panel of ALCL cell lines, two individual sgRNAs targeting IKKα were highly toxic to NIK positive ALK− ALCL lines TLBR2 and MAC2B, but not to ALK+ ALCL controls (Fig. 2A, left and Supplementary Fig. 2A). Consistent with these results, depletion or silencing of IKKα largely impaired p52 accumulation in the NIK positive ALK− lines FEPD, TLBR2 and MAC2B, but not so much in the ALK+ ones (Fig. 2A, right, Supplementary Fig. 2B and 2C). Depletion of IKKα also weakly impaired p52 accumulation in the NIK negative ALK− lines MAC1, indicating the partially activated signaling in this line. Similarly, depletion or silencing of NIK significantly inhibited ALCL cell growth and p52 accumulation, preferably in NIK positive ALK− ALCL lines FEPD, TLBR2 and MAC2B (Fig. 2B, Supplementary Fig. 3A, 3B and 3C).
Figure 2: NIK-positive ALK− ALCL lines are more dependent on activated NIK-IKKα signaling.
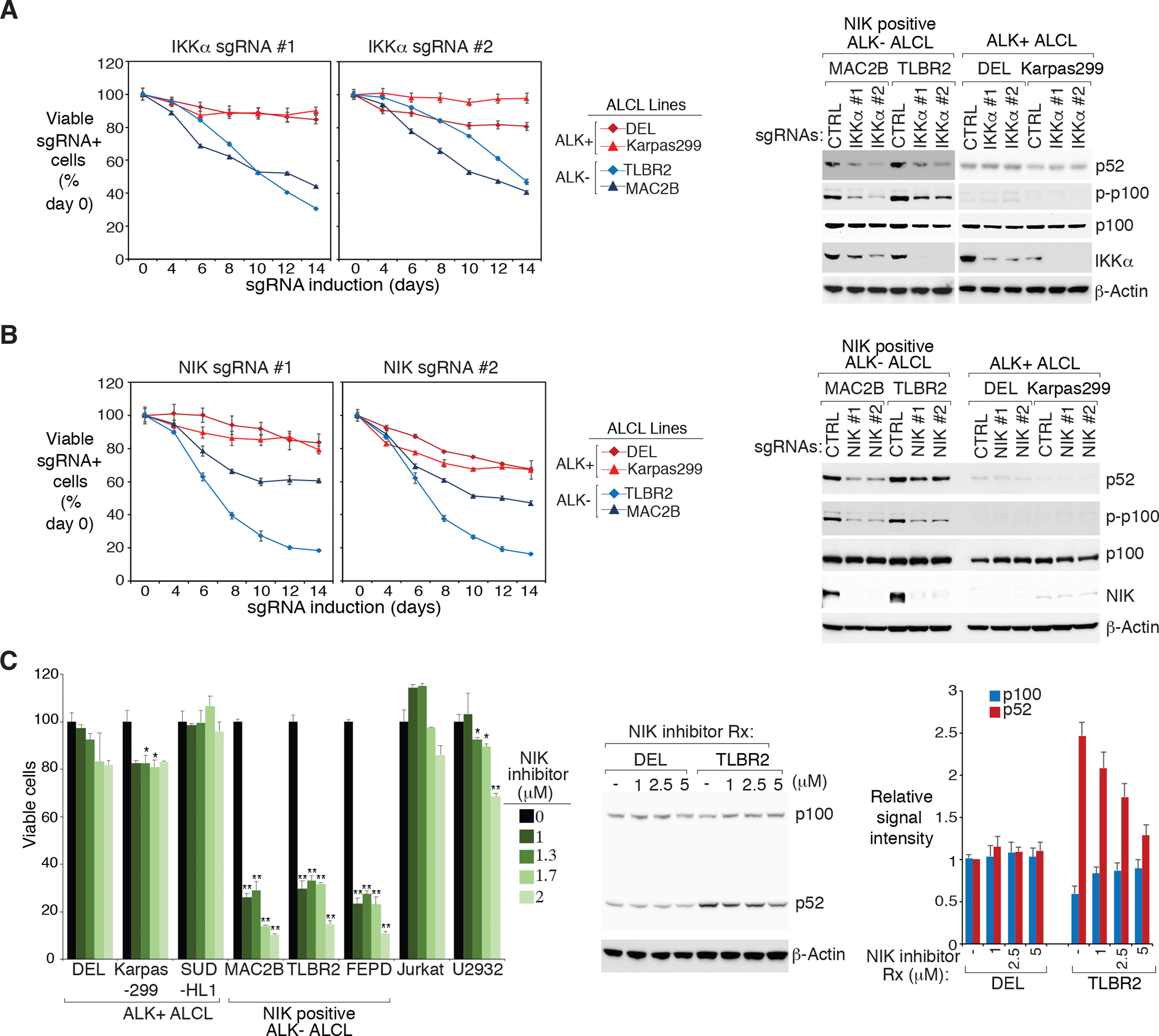
A. left: Indicated ALCL lines were transduced with IKKα or Ctrl sgRNAs along with GFP. The fraction of viable sgRNA-expressing cells relative to the total viable cell fraction at indicated times following induction of the indicated sgRNAs, normalized to day 0 values. Error bars denote SD of triplicates. right: Indicated ALCL lines were transduced with IKKα or Ctrl sgRNAs, selected and expression induced. Lysates were analyzed by immunoblotting for the indicated proteins. B. left: Same as (A). Error bars denote SD of triplicates. right: Same as (A). Lysates were analyzed by immunoblotting for the indicated proteins. C. left: Indicated lines were treated with NIK inhibitor at the indicated concentrations for 4 days. Viability was measured by the MTS assay and normalized to DMSO-treated cells. Error bars denote SEM of triplicates. P were calculated comparing DMSO and treated groups; * indicates P < 0.05; ** indicates P < 0.01. right: ALK+ DEL and ALK− TLBR2 lines were treated with NIK inhibitor at the indicated concentrations overnight. Lysates were analyzed by immunoblotting for the indicated proteins. The relative p100 and p52 signal intensity (compared to β-actin) were determined by densitometric analysis, and normalized to untreated conditions in DEL line (right). Error bars denote SEM of three independent experiments.
The NIK stabilization in a subset of ALK− ALCL lines (Fig. 1B) and the NIK sgRNA results (Fig. 2B) led us to test the cytotoxicity of a highly specific NIK inhibitor described in our previous work27. At the dosages used, the NIK inhibitor was highly toxic to ALK− ALCL lines with stabilized NIK, but not to ALK+ ALCL and the control cell lines (Fig. 2C, left). Biochemically, the NIK inhibitor specially inhibited p100 processing in the NIK positive ALK− ALCL line TLBR2 but not in the ALK+ line DEL (Fig. 2C, right). Thus, some ALK− ALCL cell lines with NIK stabilization are specifically more dependent on the activated NIK-IKKα signaling.
STAT3 promotes the alternative NF-κB pathway in both ALK+ and ALK− ALCL
To investigate the mechanisms for alternative NF-κB activation, we generated a CRISPR screen platform to identify the molecules required for p100 processing in ALCL, using a specifically designed p100-EGFP reporter system (Fig. 3A). The sensitivity and efficiency of the p100-EGFP reporter system were verified using the sgRNAs targeting NFKB2 and NIK (Supplementary Fig. 4A). Since NFKB2 is essential for ALCL cell survival, we reasoned that its mediator should as well. We therefore assembled a mini CRISPR library consisting of genes identified as positive hits in our ALCL depletion screen (Supplementary Fig. 1B), as well as multiple positive and negative controls (Supplementary Table 3). The p100-EGFP reporter TLBR2 and DEL lines were infected with this mini library, induced, and p100 accumulation (relative GFP signal vs Ctrl) in each sgRNA transduced cells was calculated and plotted (Fig. 3A and Supplementary Fig. 4B). Remarkably, two individual sgRNAs targeting STAT3, were identified as the top hits only in TLBR2 line (Fig. 3A).
Figure 3: STAT3 supports the alternative NF-κB pathway in ALCL.
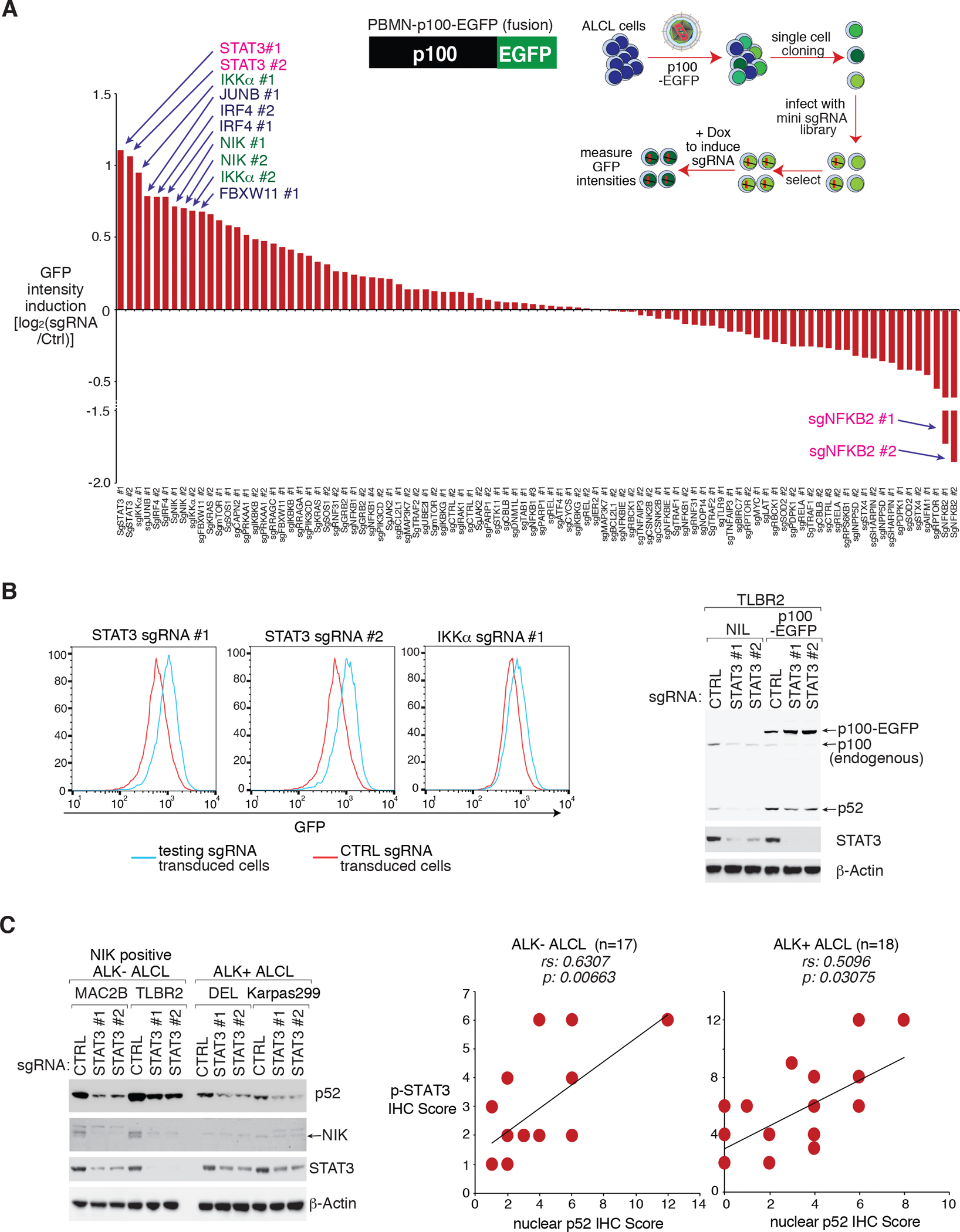
A. Mini CRISPR library screen using the p100-EGFP reporter. The TLBR2 p100-EGFP reporter line was transduced with each individual sgRNAs in the mini CRISPR library, and induced to express for 4 days. The relative GFP intensity in each sgRNA transduced sample was measured by FACS and normalized to Ctrl sgRNA transduced samples. The log2 fold changes of GFP intensity in each sgRNA transduced sample vs Ctrl sgRNA were calculated and plotted. B. Confirmational EXP for (A). left: The TLBR2 p100-EGFP reporter line was transduced with indicated sgRNAs, the relative GFP intensity in each sgRNA transduced sample was measured by FACS. right: The TLBR2 p100-EGFP reporter line or the parental controls were transduced with indicated sgRNAs, selected and expression induced. Lysates were analyzed by immunoblotting for the indicated proteins. C. left: Indicated ALCL lines were transduced with STAT3 or Ctrl sgRNAs, selected and expression induced. Lysates were analyzed by immunoblotting for the indicated proteins. right: Correlation between p-STAT3 IHC scores with nuclear p52 IHC scores, was calculated by Spearman’s rank correlation methods, in 17 ALK− ALCL cases and in 18 ALK+ ALCL cases.
Confirming the screening results, STAT3 deletion inhibited p100 processing, at an extent even greater than IKKα deletion, in our p100-EGFP reporter TLBR2 line (Fig. 3B, left). To confirm the EGFP reporter results, we assessed p100 processing by immunoblotting of the sgRNA transduced cells. Interestingly, while STAT3 sgRNAs impaired ectopically expressed p100 processing, it also inhibited endogenous p100/p52 expression (Fig. 3B, right and Supplementary Fig. 4C). Indeed, STAT3 was required for p52 accumulation in all ALK− and ALK+ ALCL lines tested except SUDHL1 (Fig. 3C, left, Supplementary Fig. 5A and 5B). However, different from ALK+ ALCL, STAT3 is also required for NIK stabilization in NIK-positive ALK− ALCL (MAC2B, TLBR2 and FEPD), indicating it is involved in the activated signaling in these lines.
We then assessed STAT3 activation (p-STAT3) and p52 nuclear accumulation by immunohistochemistry (IHC), in 35 primary cases described in Fig. 1. Notably, high p52 nuclear accumulation was significantly more common in p-STAT3-high cases (Supplementary Fig. 5C and Supplementary Table 1). p-STAT3 IHC scores correlated well with p52 nuclear IHC scores in both ALK− and ALK+ cases (p=0.00663 and 0.03075 respectively, Fig. 3C, right). As positive controls, strong nuclear p-STAT3 and p52 were observed in TLBR2 and MAC2B cells by IHC (Supplementary Fig. 5D). Thus, STAT3 is likely to regulate the alternative NF-κB pathway in both ALK+ and ALK− ALCL lymphomas.
STAT3 directly regulates NFKB2 expression in ALCL
STAT3 is a transcription factor, thus it likely regulates the alternative NF-κB pathway via mediating expression of certain genes. Therefore, we profiled gene expression changes upon sgRNA depletion of STAT3 in the ALK− line TLBR2 and ALK+ line DEL, and then compared gene expression changes caused by STAT3 sgRNA with a database of gene expression signatures that reflect regulatory processes28. Among STAT3 sgRNA downregulated genes, a signature of NF-κB activated genes was significantly enriched (Fig. 4A and Supplementary Fig. 6A). NF-κB signature genes were downregulated in both TLBR2 and DEL cells (Fig. 4A), and demonstrated significant enrichment in Gene Set Enrichment Analysis (GSEA) (Supplementary Fig. 6A). Thus, in line with our p100-EGFP reporter CRISPR screen, STAT3 supports NF-κB in ALCL. As expected, depletion STAT3 was highly toxic to both ALK− and ALK+ ALCL lines (Supplementary Fig. 6B).
Figure 4: STAT3 regulates NFKB2 and CD30 expression in ALCL.
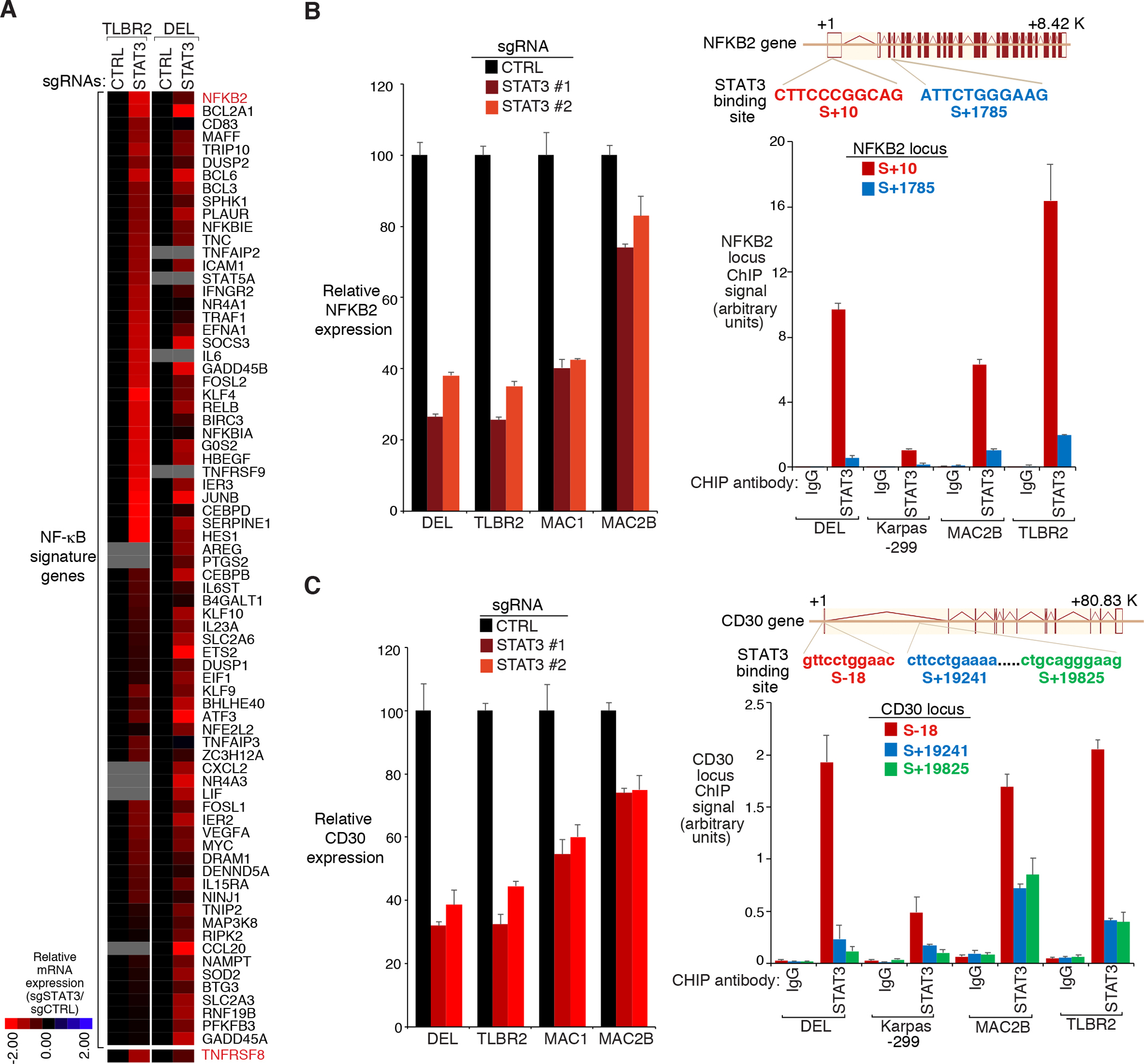
A. Heatmap of mRNA induction of NF-κB signature genes in the STAT3 sgRNA transduced cells relative to the Ctrl sgRNA transduced cells, in TLBR2 and DEL lines. B. left: Indicated ALCL lines were transduced with STAT3 or Ctrl sgRNAs, selected and expression induced, NFKB2 gene expression was measured by real-time PCR. Error bars denote SEM of triplicates. right: Diagrammatic representation of the STAT3 binding sites in NFKB2 locus (top). Chromatin IPs from indicated antibodies were subjected to real-time PCR analysis for candidate STAT3-binding regions in the NFKB2 locus in the indicated ALCL lines (bottom). Error bars denote SEM of triplicates. C. left: Same as (C), CD30 gene expression was measured by real-time PCR. right: Diagrammatic representation of the STAT3 binding sites in CD30 locus (top). Chromatin IPs from indicated antibodies were subjected to real-time PCR analysis for candidate STAT3-binding regions in the CD30 locus in the indicated ALCL lines (bottom). Error bars denote SEM of triplicates.
Interestingly, among NF-κB signature genes, NFKB2 itself was one of the most downregulated genes upon STAT3 deletion, in both ALK+ and ALK− lines (Fig. 4A), which can be confirmed by real-time PCR analysis (Fig. 4B, left). Analyses of the NFKB2 gene revealed a highly-conserved STAT3 binding site near the transcription start site (S+ 10), as well as a weaker binding site in the third intron (S+1785) (Fig. 4B, right). Consistent with this observation, our chromatin immunoprecipitation (ChIP)-coupled real-time PCR experiments demonstrated that STAT3 bound avidly to DNA fragments (~300bp) containing the NFKB2 transcription start site in a panel of ALK+ and ALK− ALCL lines (Fig. 4B, right), supporting that STAT3 directly mediates NFKB2 gene expression. STAT3 also bound to the S+1785 site of NFKB2 gene, although to a much lesser extent (Fig. 4B, right). Therefore, STAT3 supports the alternative NF-κB pathway at least partially through maintaining NFKB2 gene expression in ALCL.
STAT3 mediates NIK-IKKα signaling in NIK positive ALK− ALCL through CD30.
However, STAT3 is also required for upstream NIK-IKKα signal activation in NIK positive ALK− ALCLs (Fig. 3), indicating that other factors in addition to NFKB2 are involved. Indeed, CD30 (TNFRSF8), the main TNF receptor family members expressed in both ALK+ and ALK− ALCL, was also significantly downregulated in STAT3 depleted cells (Fig. 4A). Similar to NFKB2, STAT3 promoted CD30 mRNA transcription in both ALK+ and ALK− ALCL lines Fig. 4C, left), and bound to the transcription start site of CD30 in our ChIP-coupled real-time PCR experiments (Fig. 4C, right). Of note, analyses of the CD30 gene also revealed two highly-conserved STAT3 binding sites in intron 1, ~19kb downstream from the transcription start site (Fig. 4C, right), indicating a candidate enhancer region. STAT3 was found to bind to these two sites in all ALCL lines we tested, in addition to the transcription start site of CD30 (Fig. 4C, right). Confirming the mRNA and ChIP results, STAT3 depletion or silencing largely inhibited CD30 protein accumulation measured by immunoblotting (Fig. 5A, Supplementary Fig. 7A and 7B) and surface FACS analysis (Supplementary Fig. 7C), in all ALCL lines tested.
Figure 5: STAT3 promotes alternative NF-κB signaling in NIK positive ALK− ALCL through CD30.
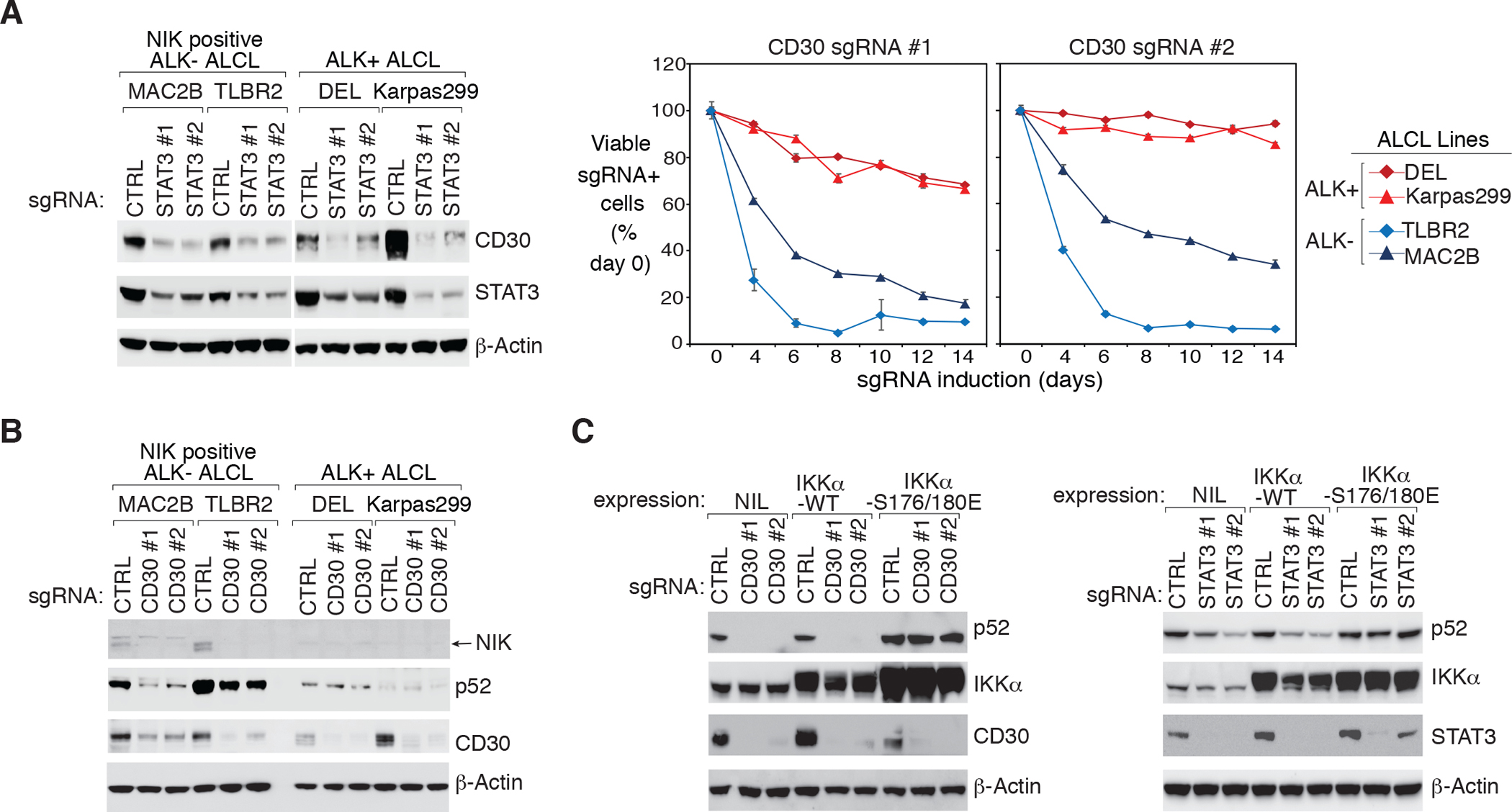
A. left: Indicated ALCL lines were transduced with STAT3 or Ctrl sgRNAs, selected and expression induced. Lysates were analyzed by immunoblotting for the indicated proteins. right: ALCL lines were transduced with CD30 or Ctrl sgRNAs along with GFP. The fraction of viable sgRNA-expressing cells relative to the total viable cell fraction at indicated times following induction of the indicated sgRNAs, normalized to day 0 values. Error bars denote SD of triplicates. B. Indicated ALCL lines were transduced with CD30 or Ctrl sgRNAs, selected and expression induced. Lysates were analyzed by immunoblotting for the indicated proteins. C. ALL- ALCL line TLBR2 was stable engineered with IKKα WT, IKKα S176/180E or empty control, then transduced with CD30 (left), STAT3 (right), or Ctrl sgRNAs, selected and expression induced. Lysates were analyzed by immunoblotting for the indicated proteins.
To examine the role of endogenous CD30 expression in the alternative NF-κB activation of ALCL, we designed two individual sgRNAs against CD30. Using sgRNAs, depletion CD30 was highly toxic to NIK positive ALK− ALCL lines TLBR2 and MAC2B, but had little effect in ALK+ ALCL controls (Fig. 5A, right), suggesting that CD30 plays an important role in transmitting signals only in NIK positive ALK− ALCL. In support, CD30 ablation strongly inhibited NIK stabilization and p52 accumulation only in NIK positive ALK− lines, but not in NIK negative ALK− and ALK+ ones (Fig. 5B, Supplementary Fig. 8A and 8B). Consistently, ectopic expression of a constitutively active IKKα mutant into TLBR2 cells largely mitigated the ability of CD30 or STAT3 (Fig. 5C, Supplementary Fig. 8C and 8D) sgRNAs to inhibit p52 accumulation, but the IKKα WT failed to do so. Together, these data suggest that the STAT3 induced CD30 expression is essential for NIK-IKKα signal activation in NIK positive ALK− ALCL.
CD30 signalosome assembly activates NIK-IKKα signaling in ALK− ALCL
The current model of the alternative NF-κB activation pathway highlights the central role of the NIK-TRAF3 axis29. TRAF3 is targeted for degradation in the receptor complex to allow NIK accumulation. Consistent with this model, depletion of TRAF3 using sgRNAs largely promoted NIK and p52 accumulation in both ALK− and ALK+ ALCL lines (Supplementary Fig. 9A). Therefore, it is likely that CD30 signaling is activated in ALK− ALCL, and a sub pool of TRAF3 is recruited to the CD30 receptor where it is targeted for degradation. In line with this hypothesis, ablation/silencing of either CD30 or STAT3 promoted TRAF3 accumulation in NIK positive ALK− ALCL lines, but not in ALK+ or NIK negative ALK− ALCL lines (Fig. 6A and Supplementary Fig. 9B, 9C).
Figure 6: CD30 signalosome complex formation in ALK− ALCL.
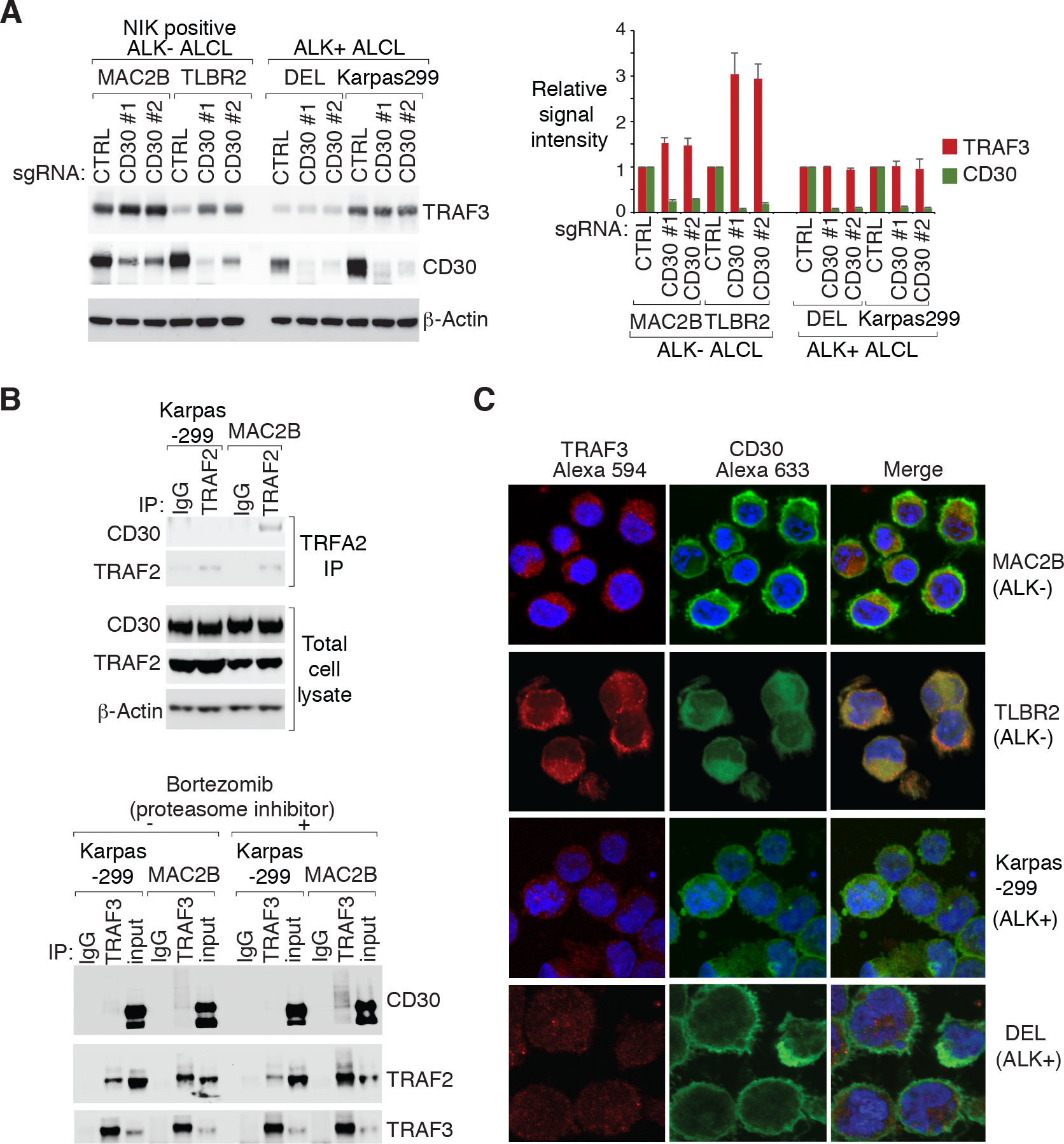
A. Indicated ALCL lines were transduced with CD30 or Ctrl sgRNAs, selected and expression induced. Lysates were analyzed by immunoblotting for the indicated proteins. The relative TRAF3 and CD30 signal intensity (compared to β-actin) were determined by densitometric analysis, and normalized to the Ctrl sgRNA conditions in each cell lines (right). Error bars denote SEM of three independent experiments. D. top: TRAF2 IPs or total lysates from Karpas299 and MAC2B cells were immunoblotted for the indicated proteins. bottom: TRAF3 IPs or total lysates from Karpas299 and MAC2B cells treated with proteasome inhibitor bortezomib (5 nM) or DMSO for overnight were immunoblotted for the indicated proteins. C. Immunofluorescence confocal microscopy analysis of the distribution of endogenous TRAF3 and CD30 in the indicated lines, pretreated with proteasome inhibitor bortezomib (5 nM) overnight. Antibodies used are indicated on the top.
The differences of CD30 signaling activation between ALK+ and NIK positive ALK− ALCL lead us to further investigate the CD30 signalosome assembling in ALCL cells. In our co-immunoprecipitation experiments, TRAF2 was found to constitutively bind to CD30 in NIK positive ALK− ALCL lines but not in ALK+ ones (Fig. 6B, top, Supplementary Fig. 9D and 9E), in line with previous findings30. Following these results, we co-immunoprecipitated TRAF3 with or without proteasome inhibitor (bortezomib) treatment in ALCL cells. Again, TRAF3 constitutively bound to CD30 more strongly in MAC2B cells than in Karpas299 controls. Of note, bortezomib treatment significantly increased TRAF3-CD30 binding in MAC2B cells, suggesting that TRAF3 was degraded in the CD30 receptor (Fig. 6B, bottom, and Supplementary Fig. 9F). Lastly, the distribution of TRAF3 and CD30 in ALCL cells was studied by laser confocal immunofluorescence microscopy with bortezomib treatment. A co-localization of CD30 and TRAF3 on the plasma membrane and the Golgi region of the cytoplasm was detected more significantly in ALK− than in ALK+ ALCL cells (Fig. 6C and Supplementary Fig. 9G). Taken together, it is likely that CD30 signalosome assembling activates NIK-IKKα signaling in NIK positive ALK− ALCL.
NPM-ALK oncoprotein and JAK/STAT3 somatic mutations are required for alternative NF-κB activation in ALCL
It is still a puzzle that only a subset of ALK− ALCL lines engage in alternative NF-κB activation (Fig. 1B), indicating unique features of these lines. Different from other hematological malignancies31–33, there were no TRAF3 or NIK genetic alterations reported in ALCL so far. Therefore, it is likely that there are other oncogenic genetic lesions in these lines that contribute to NF-κB activation.
In ALK+ ALCL cells, the NPM-ALK oncoprotein mediates the activation of multiple oncogenic signaling pathways including STAT334, we therefore examined the role of NPM-ALK oncoprotein on alternative NF-κB activation in ALK+ ALCL. In our studies, treatment of a panel of ALK+ ALCL lines with the ALK kinase inhibitor crizotinib significantly downregulated STAT3 phosphorylation, p100 expression and p52 accumulation, but not in ALK− ALCL controls (Fig. 7A and Supplementary Fig. 10A). Consistently, two specific ALK shRNAs delivered very similar effects in DEL and Karpas299 cells (Fig. 7B and Supplementary Fig. 10B). Together, these data suggest that the NPM-ALK oncoprotein is required for alternative NF-κB activation in ALK+ ALCL.
Figure 7: Genetic alternations in ALCL are required for alternative NF-κB activation.
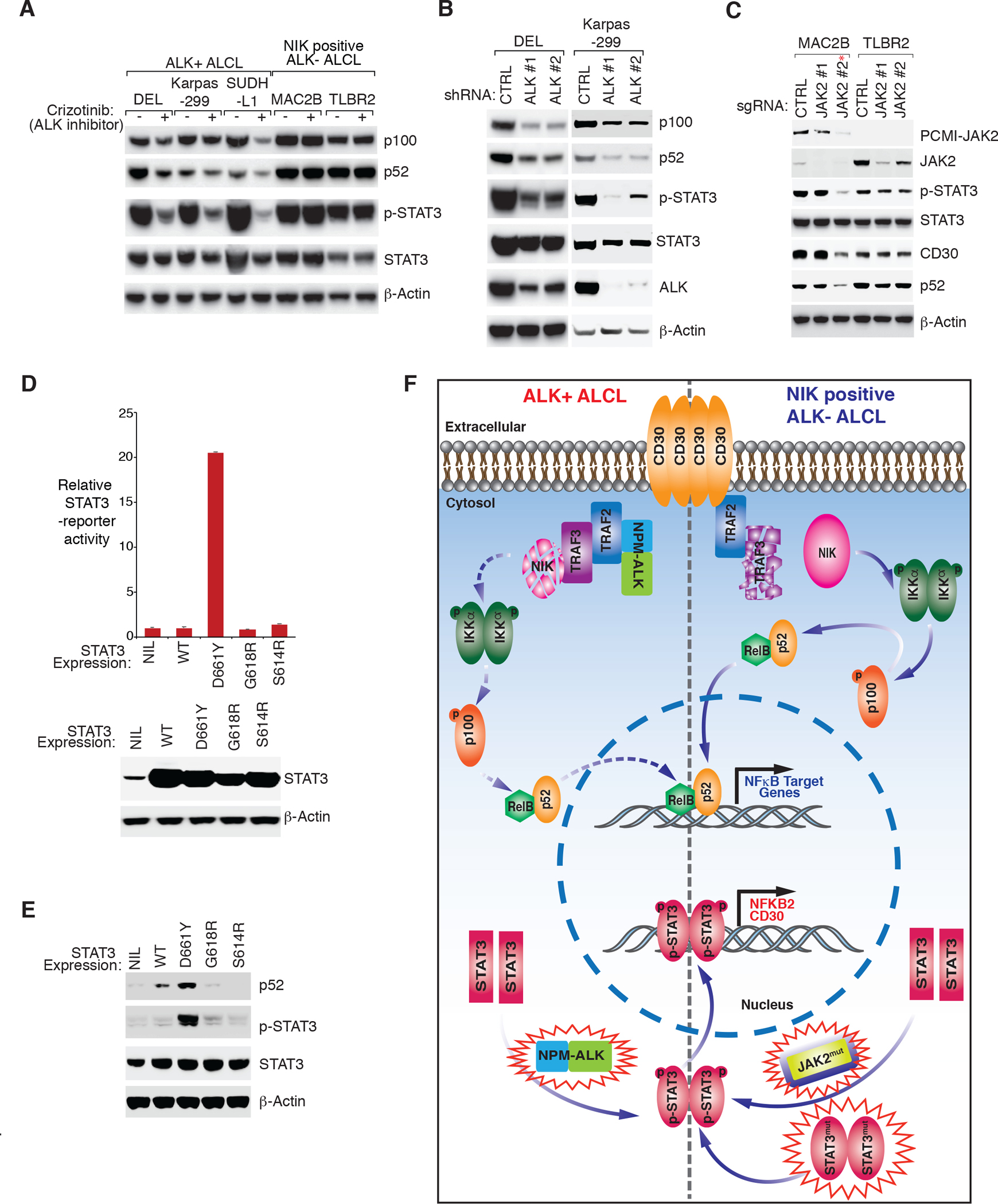
A. Indicated ALCL lines were treated with ALK inhibitor crizotinib (300 nM) or DMSO for 16 hours. Lysates were analyzed by immunoblotting for the indicated proteins. B. ALK+ ALCL line DEL and Karpas299 were transduced with ALK or Ctrl shRNAs, selected and expression induced. Lysates were analyzed by immunoblotting for the indicated proteins. C. ALK− ALCL lines MAC2B and TLBR2 were transduced with two JAK2 or Ctrl sgRNAs, selected and expression induced. Lysates were analyzed by immunoblotting for the indicated proteins. *indicates that this sgRNA targets both JAK2 WT and PCMI-JAK2 fusion. D. HEK293 cells were co-transfected of a STAT3 reporter and a Renilla luciferase control, together with an empty vector, STAT3 WT or indicated STAT3 mutant plasmids for 24 hours. The relative STAT3 reporter activity was measured and normalized to empty vector controls. The expression of STAT3 WT and mutants were analyzed by immunoblotting for the indicated proteins (lower). E. Lymphoma cell line MedB1 was transduced with a control vector, WT STAT3 or indicated STAT3 mutants, selected and expression induced. Lysates were analyzed by immunoblotting for the indicated proteins. F. Model of the alternative NF-κB activation in ALCL. The cartoon depicts a working model of how recurrent genetic alternations in ALK− and ALK+ ALCL regulate STAT3 activity, thereby amplifying NFKB2 and CD30 expression. NFKB2 induction promotes alternative NF-κB activation in both ALK+ and ALK− ALCL, while CD30 expression triggers alternative NF-κB through TRAF3-NIK-IKKα signaling cassette only in ALK− ALCL.
In ALK− ALCL, recurrent JAK and STAT3 mutations have been identified8, 9, 35. In line with previous reports9, 35, we detected a JAK2 translocation in MAC1/2A/2B cells, and MAC2B appears to have the strongest PCMI-JAK2 expression (Supplementary Fig. 10C), consistent with the result that MAC2B is one of the lines with the strongest NIK-IKKα signaling (Fig. 1B). To understand the role of PCMI-JAK2 fusion in alternative NF-κB activation, we designed two JAK2 sgRNAs, one targeting both JAK2 WT and PCMI-JAK2, and the other one only targeting JAK2 WT. In MAC2B cells, the sgRNA targeting PCMI-JAK2 significantly inhibited STAT3 phosphorylation, CD30 expression, and p52 accumulation, while the sgRNA targeting JAK2 WT only had very little effect (Fig. 7C and Supplementary Fig. 10D). JAK2 depletion had minor effect in the TLBR2 cells as well. These data suggest that the JAK2 translocation or PCMI-JAK2 fusion specially activates STAT3 and alternative NF-κB. In addition to JAK2, STAT3 mutations were found in the other three ALK− lines: TLBR2 (D661Y), TLBR1 (S614R) and FEPD (G618R). In line with previous results9, the TLBR2 D661Y mutation is the only one with high activity in the STAT3 reporter assay without any further stimulation (Fig. 7D). Lastly, ectopic expression of TLBR2 D661Y into a B cell lymphoma cell line MedB1 and NIK negative ALCL line MAC1 strongly stimulated p52 accumulation, while ectopic expression of STAT3 WT or the other two mutants S614R and G618R had very little effect (Fig. 7E, Supplementary Fig. 10E and 10F). Similarly, TLBR2 D661Y was the only one with high STAT3 phosphorylation (Fig. 7E), consistent with our STAT3 reporter results (Fig. 7D). Of note, FEPD line also carries a JAK1 mutation (G1097V), which was shown to be able to strongly activate STAT39, which can explain the strong alternative NF-κB activation in this line (Fig. 1B). Therefore, the high expression of PCMI-JAK2 fusion or the highly activated STAT3/JAK1 mutation are responsible for NIK-dependent NF-κB activation.
Discussion
Constitutive activation of the NF-κB pathway is perhaps the most common mechanism by which B cell lymphoid malignancies avoid cell death36, 37. Early studies reported elevated NF-κB activity in primary ALCL samples19, 20. A recent study confirmed these findings in both ALK+ and ALK− ALCL21. Our results in a large cohort of primary ALCL samples are consistent with previous findings that some level of p52 nuclear accumulation occurs in the majority of ALK+ and ALK− cases, indicating a constitutive activation of the alternative NF-κB pathway. Importantly, using ALCL cell line models and a CRISPR library screen, we uncovered a role for the alternative NF-κB pathway in supporting ALCL, in line with our results in primary cases. It is noteworthy that our CRISPR library screens were only performed in ALK+ ALCL line and DLBCL control, but not in NIK positive ALK− ALCL lines. Therefore, a comprehensive understanding of the unique factors, including classical and alternative NF-κB pathways, in ALK− ALCL is still missing. This limitation will certainly need to be addressed in future studies.
Stabilization of NIK is the key step in alternative NF-κB activation, which has been implied to play an oncogenic role in human T-cell lymphoma cells38. Using CRISPR deletion and a specific pharmacological inhibitor, we established a role of the NIK-IKKα signaling cascade in supporting NF-κB and tumor survival, preferably in NIK positive ALK− ALCL lines. To understand the difference between ALK− and ALK+ ALCL, an p100-EGFP reporter mini CRISPR library screen followed by the RNA-seq analysis were performed, which led us to identify STAT3 as the major mediator of alternative NF-κB activation in ALCL. In some ALK− ALCL lines, specific JAK/STAT3 mutations promote transcriptional activity of STAT3 which directly regulates NFKB2 and CD30 expression. Endogenous expression of CD30 induces constitutive NF-κB activation through binding and degrading of TRAF3, the E3 ligase of NIK. In ALK+ ALCL, the CD30 pathway is blocked by the NPM-ALK oncoprotein, but STAT3 activity and resultant NFKB2 expression can still be induced by NPM-ALK, leading to minimal alternative NF-κB activation (Fig. 7F). Taken together, our findings provide a comprehensive understanding of how the alternative NF-κB pathway is regulated in ALCL.
Similar to other TNF superfamily members, CD30 robustly activates NF-κB upon ligand stimulation39–43. In tumor cells without ligand stimulation, high expression of CD30 to drive NF-κB activation in a ligand-independent manner was reported in HL44, but not in ALK+ ALCL30. Our study demonstrated that ligand-independent CD30 signaling essential for tumor cell survival can also occur in some ALK− ALCL lines, though NIK-dependent NF-κB activation. Importantly, NIK and CD30 are known to be able to activate both the classical and alternative NF-κB pathways45, therefore, it is very likely that the elevated expression of CD30 in ALCL can also lead to classic NF-κB activation in the NIK positive ALCL, which will be examined in future.
Abnormal alternative NF-κB activation has been shown in several hematological malignancies, with TRAF3 inactivation being the most frequent alteration. In multiple myeloma, various mutations and deletions that inactivate TRAF3 occur in more than 10% of cases32, 33. In a recent study, recurrent (roughly 15%) TRAF3 genetic alterations were observed in a cohort of DLBCL cases46. Genetic lesions of the TRAF3 and NIK genes have also been reported in cHL31. Until now, no TRAF3 and NIK genetic alterations have been reported in ALCL cases or cell lines, although elevated alternative NF-κB activity was detected in primary ALCL cases. Our model (Fig. 7F) presents a novel mechanism showing that the NPM-ALK oncoprotein or recurrent JAK/STAT3 mutations adequately drive alternative NF-κB activation in absence of common TRAF3 and NIK genetic abnormalities. Activating mutations of JAK/STAT3 genes were observed in ∼20% ALK− ALCL cases8. Therefore, our study provides support for therapeutic approaches using highly specific and potent NIK inhibitors, alone or in combination with JAK/STAT inhibitors for patients with NIK positive ALK− ALCL who carrying recurrent JAK/STAT3 genetic abnormalities.
Supplementary Material
Acknowledgements.
This research was supported by American Cancer Society (ASC) IRG-92–027-21 (Y.Y), and the Medical Research Grant from the W. W. SMITH Charitable Trust (Y.Y). The study was supported in part by the Intramural Research Program of the National Cancer Institute, NIH.
The authors thank the patients for their participation. We thank D. L. Wiest (FCCC) and K.S. Campbell (FCCC) for discussions and comments. We also thank Dr. Alan L. Epstein (USC Keck School of Medicine) for the TLBR1 and TLBR2 cell lines and Dr. Annarosa Del Mistro (The Veneto Institute of Oncology) for the FE-PD cell line.
Footnotes
Conflicts of Interest.
The authors declare no competing financial interests.
References
- 1.Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016. May 19; 127(20): 2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994. Mar 4; 263(5151): 1281–1284. [DOI] [PubMed] [Google Scholar]
- 3.Shiota M, Fujimoto J, Semba T, Satoh H, Yamamoto T, Mori S. Hyperphosphorylation of a novel 80 kDa protein-tyrosine kinase similar to Ltk in a human Ki-1 lymphoma cell line, AMS3. Oncogene 1994. Jun; 9(6): 1567–1574. [PubMed] [Google Scholar]
- 4.Kempf W CD30+ lymphoproliferative disorders: histopathology, differential diagnosis, new variants, and simulators. J Cutan Pathol 2006. Feb; 33 Suppl 1: 58–70. [DOI] [PubMed] [Google Scholar]
- 5.Falini B, Martelli MP. Anaplastic large cell lymphoma: changes in the World Health Organization classification and perspectives for targeted therapy. Haematologica 2009. Jul; 94(7): 897–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lazzeri D, Agostini T, Bocci G, Giannotti G, Fanelli G, Naccarato AG, et al. ALK-1-negative anaplastic large cell lymphoma associated with breast implants: a new clinical entity. Clin Breast Cancer 2011. Oct; 11(5): 283–296. [DOI] [PubMed] [Google Scholar]
- 7.Tsuyama N, Sakamoto K, Sakata S, Dobashi A, Takeuchi K. Anaplastic large cell lymphoma: pathology, genetics, and clinical aspects. J Clin Exp Hematop 2017; 57(3): 120–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crescenzo R, Abate F, Lasorsa E, Tabbo F, Gaudiano M, Chiesa N, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015. Apr 13; 27(4): 516–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Zhang Y, Petrus MN, Xiao W, Nicolae A, Raffeld M, et al. Cytokine receptor signaling is required for the survival of ALK− anaplastic large cell lymphoma, even in the presence of JAK1/STAT3 mutations. Proc Natl Acad Sci U S A 2017. Apr 11; 114(15): 3975–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weilemann A, Grau M, Erdmann T, Merkel O, Sobhiafshar U, Anagnostopoulos I, et al. Essential role of IRF4 and MYC signaling for survival of anaplastic large cell lymphoma. Blood 2015. Jan 1; 125(1): 124–132. [DOI] [PubMed] [Google Scholar]
- 11.Boddicker RL, Kip NS, Xing X, Zeng Y, Yang ZZ, Lee JH, et al. The oncogenic transcription factor IRF4 is regulated by a novel CD30/NF-kappaB positive feedback loop in peripheral T-cell lymphoma. Blood 2015. May 14; 125(20): 3118–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atsaves V, Lekakis L, Drakos E, Leventaki V, Ghaderi M, Baltatzis GE, et al. The oncogenic JUNB/CD30 axis contributes to cell cycle deregulation in ALK+ anaplastic large cell lymphoma. Br J Haematol 2014. Nov; 167(4): 514–523. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe M, Sasaki M, Itoh K, Higashihara M, Umezawa K, Kadin ME, et al. JunB induced by constitutive CD30-extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase signaling activates the CD30 promoter in anaplastic large cell lymphoma and reed-sternberg cells of Hodgkin lymphoma. Cancer Res 2005. Sep 1; 65(17): 7628–7634. [DOI] [PubMed] [Google Scholar]
- 14.Slupianek A, Nieborowska-Skorska M, Hoser G, Morrione A, Majewski M, Xue L, et al. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res 2001. Mar 1; 61(5): 2194–2199. [PubMed] [Google Scholar]
- 15.Marzec M, Kasprzycka M, Liu X, El-Salem M, Halasa K, Raghunath PN, et al. Oncogenic tyrosine kinase NPM/ALK induces activation of the rapamycin-sensitive mTOR signaling pathway. Oncogene 2007. Aug 16; 26(38): 5606–5614. [DOI] [PubMed] [Google Scholar]
- 16.Rothwarf DM, Karin M. The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE 1999. Oct 26; 1999(5): RE1. [DOI] [PubMed] [Google Scholar]
- 17.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev 2004. Sep 15; 18(18): 2195–2224. [DOI] [PubMed] [Google Scholar]
- 18.Shaffer AL 3rd, Young RM, Staudt LM. Pathogenesis of human B cell lymphomas. Annu Rev Immunol 2012; 30: 565–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathas S, Johrens K, Joos S, Lietz A, Hummel F, Janz M, et al. Elevated NF-kappaB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood 2005. Dec 15; 106(13): 4287–4293. [DOI] [PubMed] [Google Scholar]
- 20.Eckerle S, Brune V, Doring C, Tiacci E, Bohle V, Sundstrom C, et al. Gene expression profiling of isolated tumour cells from anaplastic large cell lymphomas: insights into its cellular origin, pathogenesis and relation to Hodgkin lymphoma. Leukemia 2009. Nov; 23(11): 2129–2138. [DOI] [PubMed] [Google Scholar]
- 21.Abate F, Todaro M, van der Krogt JA, Boi M, Landra I, Machiorlatti R, et al. A novel patient-derived tumorgraft model with TRAF1-ALK anaplastic large-cell lymphoma translocation. Leukemia 2015. Jun; 29(6): 1390–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kadin ME. Ki-1/CD30+ (anaplastic) large-cell lymphoma: maturation of a clinicopathologic entity with prospects of effective therapy. J Clin Oncol 1994. May; 12(5): 884–887. [DOI] [PubMed] [Google Scholar]
- 23.Bargou RC, Leng C, Krappmann D, Emmerich F, Mapara MY, Bommert K, et al. High-level nuclear NF-kappa B and Oct-2 is a common feature of cultured Hodgkin/Reed-Sternberg cells. Blood 1996; 87(10): 4340–4347. [PubMed] [Google Scholar]
- 24.Zhang JP, Song Z, Wang HB, Lang L, Yang YZ, Xiao W, et al. A novel model of controlling PD-L1 expression in ALK(+) anaplastic large cell lymphoma revealed by CRISPR screening. Blood 2019. Jul 11; 134(2): 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 2006. May 4; 441(7089): 106–110. [DOI] [PubMed] [Google Scholar]
- 26.Ranuncolo SM, Pittaluga S, Evbuomwan MO, Jaffe ES, Lewis BA. Hodgkin lymphoma requires stabilized NIK and constitutive RelB expression for survival. Blood 2012. Nov 1; 120(18): 3756–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang Y, Kelly P, Shaffer AL 3rd, Schmitz R, Yoo HM, Liu X, et al. Targeting Non-proteolytic Protein Ubiquitination for the Treatment of Diffuse Large B Cell Lymphoma. Cancer Cell 2016. Apr 11; 29(4): 494–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaffer AL, Wright G, Yang L, Powell J, Ngo V, Lamy L, et al. A library of gene expression signatures to illuminate normal and pathological lymphoid biology. Immunol Rev 2006. Apr; 210: 67–85. [DOI] [PubMed] [Google Scholar]
- 29.Sun SC. The noncanonical NF-kappaB pathway. Immunol Rev 2012. Mar; 246(1): 125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horie R, Watanabe M, Ishida T, Koiwa T, Aizawa S, Itoh K, et al. The NPM-ALK oncoprotein abrogates CD30 signaling and constitutive NF-kappaB activation in anaplastic large cell lymphoma. Cancer Cell 2004. Apr; 5(4): 353–364. [DOI] [PubMed] [Google Scholar]
- 31.Otto C, Giefing M, Massow A, Vater I, Gesk S, Schlesner M, et al. Genetic lesions of the TRAF3 and MAP3K14 genes in classical Hodgkin lymphoma. Br J Haematol 2012. Jun; 157(6): 702–708. [DOI] [PubMed] [Google Scholar]
- 32.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007. Aug; 12(2): 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007. Aug; 12(2): 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Q, Raghunath PN, Xue L, Majewski M, Carpentieri DF, Odum N, et al. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J Immunol 2002. Jan 1; 168(1): 466–474. [DOI] [PubMed] [Google Scholar]
- 35.Ehrentraut S, Nagel S, Scherr ME, Schneider B, Quentmeier H, Geffers R, et al. t(8;9)(p22;p24)/PCM1-JAK2 activates SOCS2 and SOCS3 via STAT5. PLoS One 2013; 8(1): e53767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol 2010. Jun 1; 2(6): a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, Staudt LM. Protein ubiquitination in lymphoid malignancies. Immunol Rev 2015. Jan; 263(1): 240–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Odqvist L, Sanchez-Beato M, Montes-Moreno S, Martin-Sanchez E, Pajares R, Sanchez-Verde L, et al. NIK controls classical and alternative NF-kappaB activation and is necessary for the survival of human T-cell lymphoma cells. Clin Cancer Res 2013. May 1; 19(9): 2319–2330. [DOI] [PubMed] [Google Scholar]
- 39.Aizawa S, Nakano H, Ishida T, Horie R, Nagai M, Ito K, et al. Tumor necrosis factor receptor-associated factor (TRAF) 5 and TRAF2 are involved in CD30-mediated NFkappaB activation. J Biol Chem 1997. Jan 24; 272(4): 2042–2045. [DOI] [PubMed] [Google Scholar]
- 40.Duckett CS, Gedrich RW, Gilfillan MC, Thompson CB. Induction of nuclear factor kappaB by the CD30 receptor is mediated by TRAF1 and TRAF2. Mol Cell Biol 1997. Mar; 17(3): 1535–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gedrich RW, Gilfillan MC, Duckett CS, Van Dongen JL, Thompson CB. CD30 contains two binding sites with different specificities for members of the tumor necrosis factor receptor-associated factor family of signal transducing proteins. J Biol Chem 1996. May 31; 271(22): 12852–12858. [DOI] [PubMed] [Google Scholar]
- 42.Horie R, Aizawa S, Nagai M, Ito K, Higashihara M, Ishida T, et al. A novel domain in the CD30 cytoplasmic tail mediates NFkappaB activation. Int Immunol 1998. Feb; 10(2): 203–210. [DOI] [PubMed] [Google Scholar]
- 43.Lee SY, Lee SY, Kandala G, Liou ML, Liou HC, Choi Y. CD30/TNF receptor-associated factor interaction: NF-kappa B activation and binding specificity. Proc Natl Acad Sci U S A 1996. Sep 3; 93(18): 9699–9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horie R, Watanabe T, Morishita Y, Ito K, Ishida T, Kanegae Y, et al. Ligand-independent signaling by overexpressed CD30 drives NF-kappaB activation in Hodgkin-Reed-Sternberg cells. Oncogene 2002. Apr 11; 21(16): 2493–2503. [DOI] [PubMed] [Google Scholar]
- 45.Buchan SL, Al-Shamkhani A. Distinct motifs in the intracellular domain of human CD30 differentially activate canonical and alternative transcription factor NF-kappaB signaling. PLoS One 2012; 7(9): e45244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang B, Calado DP, Wang Z, Frohler S, Kochert K, Qian Y, et al. An oncogenic role for alternative NF-kappaB signaling in DLBCL revealed upon deregulated BCL6 expression. Cell Rep 2015. May 5; 11(5): 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.