

Abstract
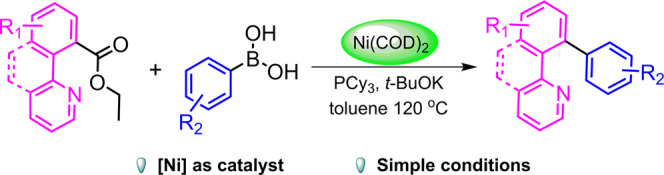
10-Arylbenzo[h]quinolines were synthesized by cross-coupling of ethyl benzo[h]quinoline-10-carboxylate with arylboronic acids via group-directed Ni(0) catalyzation. The catalytic system combining Ni(COD)2 (10 mol %) with PCy3 (20 mol %) and t-BuOK (3 equiv) was optimal for the above transformations. A series of arylboronic acids reacted with ethyl benzo[h]quinoline-10-carboxylates for the production of various substituted 10-phenyl[h]quinolines in moderate and good yields under optimized reaction conditions.
Introduction
Transition-metal-mediated cross-coupling of aryl halides and arylboronic acids, namely, the Suzuki–Miyaura reaction, has been widely applied in the construction of organic materials, pharmaceuticals, and natural products as a powerful and reliable C–C bond formation method.1 Although the use of organic halides as electrophiles dominates for these cross-coupling reactions, in the past few decades, some other compounds have also been found to react with arylboronic acids as alternative electrophiles for the C–C bond formation.2 For example, several advanced methods have been developed to use phenols or anilines as the electrophiles for cross-coupling with arylboronic acids.3 The in situ generated intermediates of aromatic nitriles, tertiary alcohols, aromatic aldehydes, and amides have also been found to react with arylboronic acids to form C–C bonds in the presence of transition metals.4 The transition-metal-mediated decarboxylation intermediates of aromatic acids and anhydrides were found to react with arylboronic acids to give cross-coupling products smoothly.5 The use of these substrates other than aryl halides as electrophiles in coupling reactions has the following advantages: First, these reactions do not produce metal halide wastes that exert a strong corrosive effect on metal reactors and thus are conducive to future industrial production. Second, considering that these reactions use readily available raw materials to construct useful complex molecules through rapid transformation, production costs are considerably reduced.6 Hence, the development of new synthetic strategies and routes that use different electrophiles for cross-coupling with arylboronic acids is a continuous pursuit in synthetic organic chemistry.
Esters are cheap and abundant in nature and are the most commonly used organic substrates. Therefore, there is a growing interest in using esters as electrophiles for coupling with arylboronic acids.7 One such effort involves the cross-coupling of this O-acetylated phenol derivative with phenylboronic acid to selectively oxidize the aryl C–O bond of the O-acetylated phenol in the presence of a metal (Scheme 1, i).8 In addition, some active esters, such as 4-nitrophenyl esters, benzoic acid phenyl esters, and 2-pyridyl esters, when there is a suitably positioned directing group, are used as coupling electrodes to arylboronic acids, which are converted to ketones by the activation of acyl C–O bonds (Scheme 1, ii).9 As far as we know, esters can also form carbon–carbon bonds through direct carbon–carbon bond activation or decarbonylation pathways, which involves the loss of the carbonyl group in the form of carbon monoxide (Scheme 1, iii).10 Cavallo and Rueping found that ligands play an important role in the chemoselective C(acyl)–O bond vs C(aryl)–C bond cleavage of aromatic esters in nickel-catalyzed C(sp2)–C(sp3) cross-couplings with alkyl boron compounds.11 In 2012, our group reported a chelation-assisted rhodium-catalyzed C–C bond activation of ethyl benzo[h]quinoline-10-carboxylate that provided a cross-coupling biaryl compound when reacted with organoboron compounds.12 Although the use of a rhodium catalyst gives high yields in this reaction, there is a lack of methods that are inexpensive and with readily accessible catalysts and can be performed with high cross-coupling selectivity.13 Herein, we report the cross-coupling of ethyl benzo[h]quinoline-10-carboxylate with arylboronic acids in the presence of a Ni(0) catalyst.
Scheme 1. Reaction of Esters with Arylboronic Acids.

Results and Discussion
After screening the decarbonylative cross-coupling reaction of ethyl benzo[h]quinoline-10-carboxylate (1a) and phenylboronic acid (2a), Ni(COD)2 (10 mol %) and PCy3 (20 mol %) as the catalyst in combination with t-BuOK (3.0 equiv) as the base were selected as the optimal catalytic system for the current transformation (Table 1). In the optimal catalytic system, the reaction of 1a and 2a in toluene at 120 °C for 20 h gave the desired decarbonylative cross-coupling biaryl product 3a in 81% yield. Replacing phenylboronic acid with phenylboronic acid pinacol ester (PhBpin) and triphenylboroxine (PhBO)3 resulted in 72 and 75% product yields, respectively, under similar reaction conditions. The use of other nickel compounds, such as NiI2, NiCl2, Ni(OAc)2, and Ni(acac)2, as catalysts in place of Ni(COD)2 provided low yields (0–10%) of the decarbonylative cross-coupling products, indicating that Ni(0) plays a catalytic role in such a reaction. However, the in situ generation of Ni(0) via the reduction of Ni(OAc)2 with Zn powder showed low catalytic activity in this reaction, and the desired product was isolated in 15% yield. Notably, no decarbonylative cross-coupling product was isolated when Ni(PPh3)4, a frequently used Ni(0) complex, was utilized in the reaction. This phenomenon implied that the ligand plays an important role in this nickel-catalyzed reaction. Therefore, different ligands were applied in the coupling reaction. When a bisphosphine ligand, such as 1,2-bis(diphenylphosphino) ethane (dppe), 1,3-bis(diphenylphosphino)propane (dppp), rac-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), or 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene (xantphos), was introduced into the reaction mixture to substitute for PCy3, the desired coupling products were obtained in moderate yields that ranged from 43 to 55%. The substitution of PCy3 with 1,3-bis(2,4,6-trimethylphenyl) imidazolium chloride (IMes·HCl) or 1,3-bis(2,6-diisopropyl phenyl)imidazolium chloride (IPr·HCl), which could generate additional electron-donating NHC ligands in the presence of t-BuOK, did not give positive results and resulted in the isolation of the desired coupling product in 60 and 61% yields, respectively. The type and amount of the base also had a significant effect on the yield of this catalytic coupling reaction. The coupling reactions gave product 3a in 55 and 57% yields when K3PO4 and K2CO3 were used instead of t-BuOK, respectively. When t-BuONa and Cs2CO3 were used as bases in this reaction, the yields of 3a were 71 and 76%, which were lower than the yields when t-BuOK was used as the base. Reducing the amount of the base t-BuOK to 2 equiv caused the product yield to drop to 73%. The coupling reaction yielded no product when the base was completely removed from the reaction mixture. Notably, the self-coupling product of phenylboronic acid significantly increased when the amount of the base applied exceeded 3 equiv. This result was very different from that obtained for a similar reaction catalyzed by rhodium,12 which give high product yields in the absence of a base. This difference may indicate that this coupling reaction may have a different mechanism. Futhermore, copper salt was an effective additive to promote decarbonylation in the rhodium system,12 so we tried to add CuI as an additive and the results suggested that the yield of the product was not ideal. Different solvents also provided different coupling product yields. The reaction furnished 3a in 70% yield in 1,4-dioxane but was inhibited completely in acetonitrile.
Table 1. Optimization of the Reaction Conditions for the Cross-Coupling of Ethyl Benzo[h]quinoline-10-carboxylate with Phenylboronic Acida.

| deviation from “standard conditions” | yield (%)a | |
|---|---|---|
| substrate in place of 2a | none | 81 |
| PhBpin | 72 | |
| (PhBO)3 | 75 | |
| precatalyst in place of Ni(COD)2 | Nil2, NiCI2, Ni(OAc)2, Ni(acac)2 | 0–10 |
| Ni(OAc)2 + Zn (3 equiv) | 15 | |
| Ni(PPh3)4 | 0 | |
| ligands in place of PCy3 | dppe, dppp, BINAP, xantphos | 43–55 |
| IMeS·HCI, IPr·HCI | 60–61 | |
| base in place of t-BuOK | K3PO4 | 55 |
| K2CO3 | 57 | |
| t-BuONa | 71 | |
| Cs2CO3 | 76 | |
| reduce the usage of base | 0 equiv | 0 |
| 2 equiv | 73 | |
| additive Cul | 10 mol % | 61 |
| 1 equiv | 18 | |
| replace toluene with other solvents | acetonitrile | 0 |
| 1,4-dioxane | 70 | |
Reaction conditions: 1a (0.1 mmol), 2a (2.3 equiv), catalyst (10 mol %), ligand (20 mol %), and base (3.0 equiv) in 0.5 mL of toluene, 120 °C, 20 h under N2; isolated yields.
With the optimal catalytic conditions in hand, the effects of various substituted groups on arylbononic acids and ethyl benzo[h]quinoline-10-carboxylate on this decarbonylative cross-coupling reaction were investigated, and the results are shown in Table 2. When phenylboronic acids with a methyl group at the p-, m-, or o-position were applied in coupling with 1a, the desired products 3b, 3c, and 3d were obtained with 78, 70, and 50% yields, respectively. These results demonstrated that the yields of the corresponding products were decreased by slightly increasing the steric hindrance of phenylboronic acids. The yield of the desired product 3e acquired with phenylboronic acids with the 2,5-Me substitution was 69%, which was lower than that obtained with phenylboronic acids without substitution. Steric hindrance may affect the transmetalation of phenylboronic acids to the metal center, thereby decreasing the product yield. Phenylboronic acids with n-Bu at the p-position reacted with 1a to produce 3f with 76% yield. 1a could also react with phenylboronic acids containing a t-Bu and i-Pr groups to produce the corresponding products 3g and 3h with 75 and 73% yields. In addition, the reaction of 4-Ph-substituted phenylboronic acid with 1a yielded product 3i in 72% yield. These results indicated that the substitution of a bulky alkyl or aryl at the p-position of phenylboronic acid has little effect on the product yield of the reaction. The product 3j was obtained in 81% yield using a phenylboronic acid with an electron-donating p-MeO group. Phenylboronic acid with an electron-withdrawing −CF3O group at the m-position produced the desired product 3k with good yield (79%). These experiments indicated that the lack or enrichment of the electricity of phenylboronic acid has little effect on the coupling reaction. Other boronic acids, such as naphthalen-2-ylboronic acid and naphthalen-1-ylboronic acid, also reacted with 1a smoothly to produce 3l and 3m in yields of 75 and 71%, respectively. Penylboronic acids with fluorine substitutions, such as 4-F, 3,4,5-F, or 3,4-F, produced 3n, 3o, and 3p with 80, 84, and 81% yields, respectively. The F-substituent was shown to have little effect on the yield. However, when phenylboronic acids with 4-Cl and 4-Br substitutions were utilized in the reaction with 1a, the corresponding coupling products 3q and 3r were furnished in 53 and 40% yields. The low yields may be due to the serious self-coupling side reactions of phenylboronic acid with a −Cl or −Br substituent in the presence of a Ni(0) catalyst. Phenylboronic acid with m-NO2, a strong electron-withdrawing group, could also react with 1a smoothly to give the desired product 3s in 72% yield.
Table 2. Cross-Coupling of Ethyl Benzo[h]quinoline-10-carboxylates with Arylboronic Acida,b.

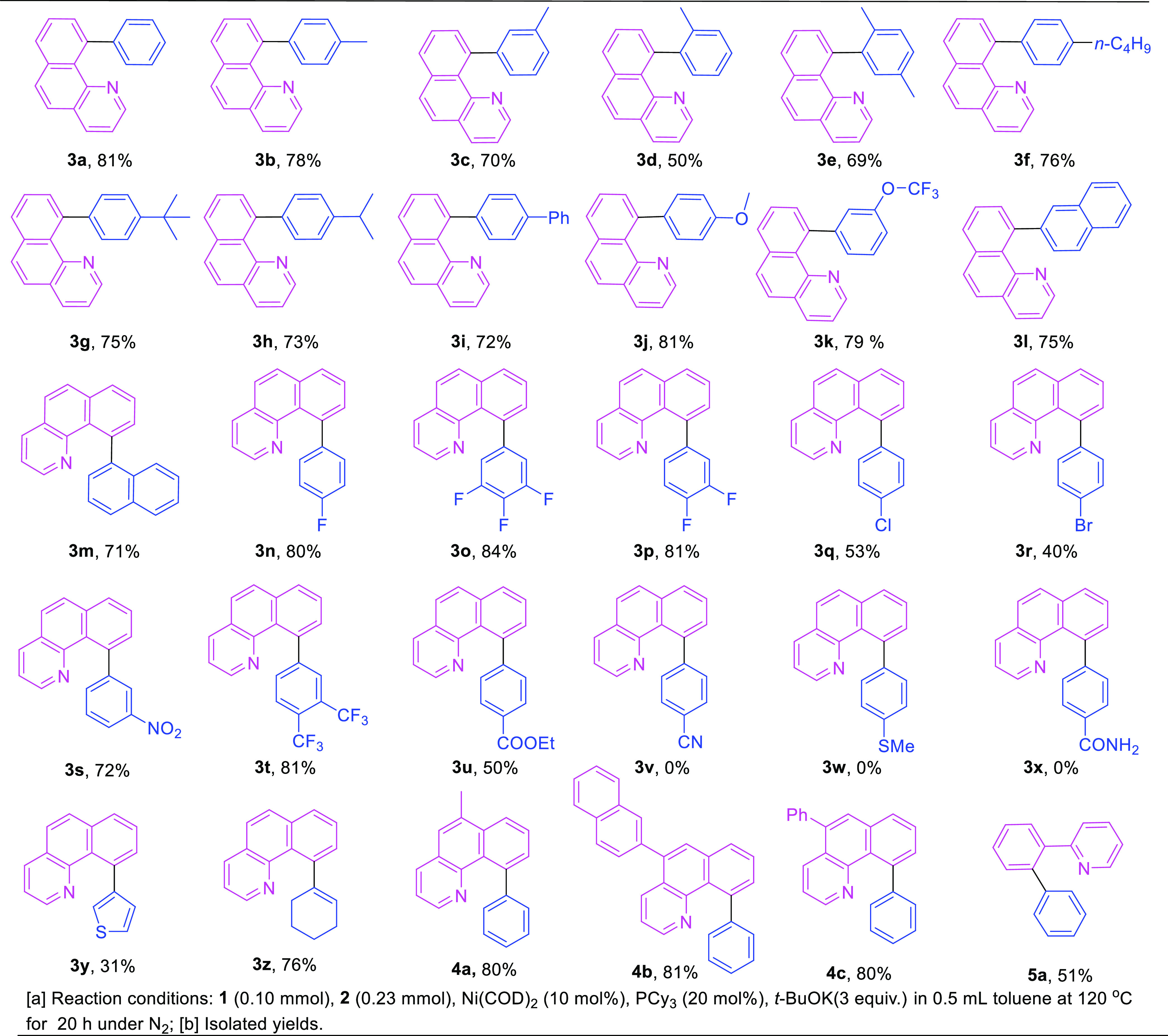
Reaction conditions: 1 (0.10 mmol), 2 (0.23 mmol), Ni(COD)2 (10 mol %), PCy3 (20 mol %), t-BuOK(3 equiv) in 0.5 mL toluene at 120 °C for 20 h under N2.
Isolated yields.
A 3,4-CF3-substituted phenylboronic acid also gave the desired product 3t in 81% yield. The two examples indicated that this decarbonylative cross-coupling reaction was compatible with strongly electron-deficient boronic acids. The 4-COOEt-substituted phenylboronic acid also produced the desired product 3u but with moderate yield (50%). The coordination of the COOEt group may prevent the transmetalization of phenylboronic acid to the metal center, thereby decreasing the product yield. Unfortunately, when the −CN, −SMe, and −CONH2-substituted phenylboronic acids were used as substrates, the desired cross-coupling products were all not observed. Heterocyclic and alkenyl-substituted phenylboronic acids were suitable candidates and gave the desired products 3y and 3z in 31 and 76% yields, respectively. Other ethyl benzo[h]quinoline-10-carboxylate derivatives, such as 5-methyl, 6-(naphthalen-2-yl), or 6-phenyl substitutes, also reacted with phenylboronic acid smoothly to produce 4a, 4b, and 4c in yields of 80, 81, and 80%, respectively, indicating that alkyl or aryl substitutions at the benzo[h]quinoline framework has little effect on the coupling reaction. This result is consistent with the observation for the rhodium (I) catalytic system in our previous report. The reaction was also applied to ethyl 2-(2-pyridinyl)benzoate (5) as a coupling partner to give product 5a in 51% yield.
As a leaving group of the ester group via the C–C bond cleavage of benzoquinoline esters, phenyl benzo[h]quinoline-10-carboxylate (1c) is somewhat less reactive compared to ethyl benzo[h]quinoline-10-carboxylate (1a) and methyl benzo[h]quinoline-10-carboxylate (1b). However, the reaction of ethyl benzoate (6a) with 4-methoxyphenylboronic acid under the same optimal reaction conditions gave only 15% of the coupling product (7a). It is proved that the presence of the guide group in this substrate facilitates carbon–carbon bond activation (Scheme 2).
Scheme 2. Reactivities of 1a, 1b, 1c, and 6a with (4-Methoxyphenyl) Boronic Acid.

A plausible catalytic mechanism is proposed and shown in Figure 1, which is based on the current experiments and previous reports on decarbonylative coupling of ester and boronic acids. The formation mechanism of 3 includes the following steps: First, the highly catalytically active catalyst A is formed through the ligand exchange of Ni(COD)2, the catalytic precursor, with PCy3. Then, the nitrogen of ethyl benzo[h]quinoline-10-carboxylate coordinates with the metal center of catalyst A. In this case, the Ni(0) metal center and the C(acyl)–O bond are brought into proximity. The following oxidative addition between the C(acyl)–O bond and the Ni(0) metal center produces the six-membered cyclic chelated Ni(II) complex B. Subsequently, the carbonyl group of the ester fragment in B migrates to the Ni(II) metal center to give the five-membered cyclic chelated Ni(II) intermediate C. This process is accompanied by the departure of a PCy3 ligand. Next, CO is exchanged and released with the PCy3 ligand to produce the intermediate D. The transmetalation of arylboronic acid into the Ni(II) metal center in the presence of t-BuOK yields the aryl nickel complex E and releases a K[B(OH)2(OEt)(O-t-Bu)] complex at the same time. Finally, the reductive elimination of the aryl and the benzo[h]quinolin-10-yl group from intermediate E yields the desired cross-coupling product 3a, and the regenerated intermediate A can further coordinate with 1a to enter another catalytic cycle. It is worth to note that the use of 3 equiv of the base and an excess amount of arylboronic acid are beneficial to the transmetalation of D into E, thereby providing a high yield of the cross-coupling product. This reaction is significantly different from a similar reaction catalyzed by rhodium without the use of an additional base. This difference implies that the rate-determining steps of these two catalytic reactions may be different. In the proposed reaction mechanism, the slow process from D to E coincides with the reaction that requires a large excess amount of base and arylboronic acids. PCy3 may play an important role in the release of CO by re-cooperating with the Ni (II) metal center of intermediate C.
Figure 1.

Plausible mechanism for the decarbonylative cross-coupling reaction of ethyl benzo[h]quinoline-10-carboxylate and arylboronic acids via chelation-assisted nickel(0)-catalyzed C(acyl)–C bond cleavage
Conclusions
We have developed a new method for the synthesis of biaryl derivatives through decarbonylative cross-coupling of aromatic esters and arylboronic acids via chelation-assisted Ni(0)-catalyzed C(acyl)–C bond cleavage. Ni(COD)2 (10 mol %) and PCy3 (20 mol %) catalysts in combination with t-BuOK (3.0 equiv) were selected as the best catalytic system for the current conversion. Under optimal catalytic conditions, a series of arylbononic acids bearing various substituted groups reacted with ethyl benzo[h]quinoline-10-carboxylate derivatives to produce various 10-aryl-substituted benzo[h]quinolines with moderate-to-good yields. Although these reactions were sensitive to hindrance, arylboronic acids with electron-withdrawing or electron-donating groups were compatible with this reaction. The ligand was observed to play an important role in achieving high yields. A large and more electron-donating phosphine ligand, PCy3, was identified to be the best ligand for this transformation. A reasonable catalytic mechanism, which included C(acyl)–O oxidation, decarbonylation, ligand exchange, transmetalization, and reductive elimination, was proposed to explain the formation of cross-coupling products. Although the yield of the nickel-catalyzed reaction is slightly lower, it is more competitive than the rhodium-catalyzed reaction because nickel catalysts are cheap and readily available. Our laboratory is currently further studying the application of this reaction in the generation of benzoquinoline derivatives with fluorescence properties.
Experimental Section
General Information
Unless otherwise noted, all commercially available reagents were used as received without further purification. Toluene was dried from sodium/benzophenone under a nitrogen (N2) atmosphere and distilled prior to use. All reactions were carried out under an atmosphere of N2 with oven-dried glassware and anhydrous solvents. Reactions were monitored by TLC with silica gel 60 F254. Silica gel (200–300 mesh) was used for column chromatography. 1H NMR, 19F NMR, and 13C NMR were recorded in deuterated solvents on a 400 MHz Bruker DRX-400 spectrometer. Electrospray mass spectra (ESI-MS) of products were recorded on Bruker MiorOTOF-Q II. The starting materials were synthesized and purified according to the corresponding literature procedures, and the 1H NMR and 13C NMR data were consistent with the literature.14−16
Ethyl Benzo[h]quinoline-10-carboxylate (1a)
The product was prepared according to the literature14 as a yellow solid (86% yield). 1H NMR (400 MHz, chloroform-d) δ 8.90 (dd, J = 4.4, 1.8 Hz, 1H), 8.17 (dd, J = 8.0, 1.8 Hz, 1H), 7.97 (dd, J = 7.3, 2.1 Hz, 1H), 7.83 (d, J = 8.8 Hz, 1H), 7.74–7.65 (m, 3H), 7.51 (dd, J = 8.1, 4.3 Hz, 1H), 4.60 (q, J = 7.2 Hz, 2H), 1.43 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, chloroform-d) δ 172.36, 147.73, 145.12, 135.55, 134.02, 132.55, 129.33, 127.76, 127.72, 127.45, 126.97, 126.18, 126.11, 121.99, 61.50, 14.28.
Ethyl 2-(2-Pyridinyl)benzoate (5)
The product was prepared according to the literature17 as a yellow solid (86% yield). 1H NMR (400 MHz, chloroform-d) δ 8.63 (dt, J = 5.0, 1.2 Hz, 1H), 7.84 (d, J = 7.6 Hz, 1H), 7.74 (td, J = 7.7, 1.9 Hz, 1H), 7.59–7.42 (m, 4H), 7.29–7.19 (m, 1H), 4.13 (q, J = 7.1 Hz, 2H), 1.05 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, chloroform-d) δ 168.82, 158.87, 149.00, 140.98, 136.21, 131.79, 131.09, 129.77, 128.31, 122.85, 122.02, 60.96, 13.82.
Experimental Procedure for the Ni-Catalyzed Benzo[h]-quinoline-10-carboxylic Acid Ethyl Ester with Substituted Phenylboronic Acids
Substituted ethyl benzo[h]quinoline-10-carboxylate (0.1 mmol), substituted phenylboronic acid (0.23 mmol), PCy3 (0.02 mmol), t-BuOK (0.3 mmol), and Ni(COD)2 (0.01 mmol) were successively added into a 10 mL sealed tube using anhydrous toluene (0.5 mL) as the solvent. The mixture was vigorously stirred at 120 °C under a nitrogen atmosphere to the end of the reaction. Organic solvents were removed in vacuo, and then the residue was purified by silica gel column chromatography to give the desired product.
10-Phenylbenzo[h]quinolone (3a)
White solid, 20.5 mg (yield: 81%). 1H NMR (400 MHz, chloroform-d) δ 8.45 (dd, J = 4.3, 2.1 Hz, 1H), 8.09 (dd, J = 8.1, 2.3 Hz, 1H), 7.94 (d, J = 7.9 Hz, 1H), 7.87 (dd, J = 8.8, 2.1 Hz, 1H), 7.74–7.64 (m, 2H), 7.58 (d, J = 7.3 Hz, 1H), 7.42 (ddd, J = 11.8, 8.9, 6.7 Hz, 5H), 7.32 (dt, J = 6.1, 3.2 Hz, 1H). 13C NMR (101 MHz, chloroform-d) δ 146.93, 146.86, 146.48, 141.74, 135.25, 135.03, 131.55, 129.06, 128.77, 128.34, 128.01, 127.44, 127.27, 127.09, 125.99, 125.74, 121.13.
10-(p-Tolyl)benzo[h]quinolone (3b)
Colorless oil, 21 mg (yield: 78%). 1H NMR (400 MHz, chloroform-d) δ 8.45 (dd, J = 4.3, 1.9 Hz, 1H), 8.05 (dd, J = 8.0, 1.9 Hz, 1H), 7.88 (dd, J = 7.8, 1.5 Hz, 1H), 7.83 (d, J = 8.8 Hz, 1H), 7.65 (dd, J = 8.3, 5.8 Hz, 2H), 7.53 (dd, J = 7.3, 1.5 Hz, 1H), 7.32–7.18 (m, 5H), 2.45 (s, 3H). 13C NMR (101 MHz, chloroform-d) δ 146.90, 146.85 143.46, 141.73, 135.10, 135.05, 131.68, 129.09, 128.65, 128.33, 128.12, 127.81, 127.23, 127.06, 125.90, 121.04, 21.34.
10-(m-Tolyl)benzo[h]quinolone (3c)
White solid, 18.8 mg (yield: 70%). 1H NMR (400 MHz, chloroform-d) δ 8.49 (dd, J = 4.3, 1.9 Hz, 1H), 8.10 (dd, J = 8.0, 1.9 Hz, 1H), 7.94 (dd, J = 7.9, 1.5 Hz, 1H), 7.88 (d, J = 8.8 Hz, 1H), 7.75–7.66 (m, 2H), 7.59 (dd, J = 7.3, 1.5 Hz, 1H), 7.38–7.29 (m, 2H), 7.26 (t, J = 2.0 Hz, 1H), 7.21 (t, J = 7.2 Hz, 2H), 2.45 (s, 3H). 13C NMR (101 MHz, chloroform-d) δ 146.91, 146.31, 141.87, 136.87, 135.21, 135.05, 131.58, 129.37, 129.07, 128.35, 127.90, 127.25, 127.23, 127.07, 126.47, 126.06, 125.92, 121.10, 21.67.
10-(o-Tolyl)benzo[h]quinolone (3d)
Yellow oil, 13.4 mg (yield: 50%). 1H NMR (400 MHz, chloroform-d) δ 8.40 (dd, J = 4.3, 1.9 Hz, 1H), 8.04 (dd, J = 8.0, 1.9 Hz, 1H), 7.92 (dd, J = 7.9, 1.4 Hz, 1H), 7.84 (d, J = 8.8 Hz, 1H), 7.71–7.61 (m, 2H), 7.45 (dd, J = 7.3, 1.4 Hz, 1H), 7.32–7.21 (m, 4H), 7.17 (dd, J = 7.0, 1.9 Hz, 1H), 1.84 (s, 3H). 13C NMR (101 MHz, chloroform-d) δ 147.50, 147.06, 146.48, 141.06, 135.91, 135.10, 134.67, 130.75, 129.48, 128.76, 128.46, 127.97, 127.93, 127.28, 126.98, 125.96, 125.87, 125.15, 121.00, 20.25.
10-(2,5-Dimethylphenyl)benzo[h]quinolone (3e)
White solid, 19.5 mg (yield: 69%). 1H NMR (400 MHz, chloroform-d) δ 8.43 (dd, J = 4.3, 1.9 Hz, 1H), 8.06 (dd, J = 8.0, 1.9 Hz, 1H), 7.92 (dd, J = 7.9, 1.5 Hz, 1H), 7.85 (d, J = 8.8 Hz, 1H), 7.71–7.64 (m, 2H), 7.46 (dd, J = 7.2, 1.4 Hz, 1H), 7.29 (dd, J = 8.0, 4.3 Hz, 1H), 7.13–7.07 (m, 2H), 7.02 (d, J = 1.7 Hz, 1H), 2.36 (s, 3H), 1.77 (s, 3H). 13C NMR (101 MHz, chloroform-d) δ 147.50, 147.06, 146.25, 141.20, 135.06, 134.62, 134.35, 132.82, 130.87, 129.48, 128.67, 128.57, 128.47, 127.82, 127.23, 126.93, 126.54, 125.80, 120.97, 21.20, 19.72. ESI-MS [M + H]+ calcd for C21H17N: 284.1439, found: 284.1440.
10-(4-Butylphenyl)benzo[h]quinolone (3f)
White solid, 23.6 mg (yield: 76%). 1H NMR (400 MHz, chloroform-d) δ 8.41 (dd, J = 4.3, 1.9 Hz, 1H), 8.03 (dd, J = 8.0, 1.9 Hz, 1H), 7.87 (dd, J = 7.9, 1.5 Hz, 1H), 7.81 (d, J = 8.8 Hz, 1H), 7.67–7.62 (m, 2H), 7.55 (dd, J = 7.3, 1.5 Hz, 1H), 7.30–7.24 (m, 3H), 7.20 (d, J = 8.0 Hz, 2H), 2.71 (t, J = 7.7 Hz, 2H), 1.77–1.64 (m, 2H), 1.44 (q, J = 7.4 Hz, 2H), 0.98 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, chloroform-d) δ 146.98, 146.84, 143.65, 141.86, 140.19, 135.21, 135.07, 131.61, 129.17, 128.61, 128.36, 127.81, 127.50, 127.24, 127.08, 125.91, 121.06, 35.54, 33.82, 22.48, 14.15. ESI-MS [M + H]+ calcd for C23H21N: 312.1752, found: 312.1756.
10-(4-(tert-Butyl)phenyl)benzo[h]quinolone (3g)
White solid, 23.3 mg (yield: 75%). 1H NMR (400 MHz, chloroform-d) δ 8.44 (dd, J = 4.3, 1.9 Hz, 1H), 8.08 (dd, J = 8.0, 1.9 Hz, 1H), 7.92 (dd, J = 7.8, 1.5 Hz, 1H), 7.86 (d, J = 8.8 Hz, 1H), 7.72–7.66 (m, 2H), 7.60 (dd, J = 7.3, 1.4 Hz, 1H), 7.45 (d, J = 8.1 Hz, 2H), 7.32 (dt, J = 8.2, 2.0 Hz, 3H), 1.46 (d, J = 1.0 Hz, 9H). 13C NMR (101 MHz, chloroform-d) δ 148.47, 146.97, 146.90, 146.88, 143.35, 141.77, 135.21, 135.06, 131.63, 129.19, 128.38, 128.35, 127.20, 127.08, 125.91, 124.28, 121.05, 34.58, 31.62. ESI-MS [M + H]+ calcd for C23H21N: 312.1752, found: 312.1750.
10-(4-Isopropylphenyl)benzo[h]quinolone (3h)
White solid, 21.4 mg (yield: 73%). 1H NMR (400 MHz, chloroform-d) δ 8.40 (dd, J = 4.3, 1.9 Hz, 1H), 7.98 (dd, J = 8.0, 1.9 Hz, 1H), 7.86–7.75 (m, 2H), 7.64–7.51 (m, 3H), 7.30–7.20 (m, 5H), 3.21–2.71 (m, 1H), 1.34 (d, J = 7.0 Hz, 6H). 13C NMR (101 MHz, chloroform-d) δ 147.03, 146.93, 146.24, 143.83, 141.90, 135.25, 135.13, 131.71, 129.23, 128.75, 128.41, 127.87, 127.27, 127.13, 125.95, 125.50, 121.09, 34.00, 24.33.
10-([1,1′-Biphenyl]-4-yl)benzo[h]quinolone (3i)
White solid, 23.8 mg (yield: 72%). 1H NMR (400 MHz, chloroform-d) δ 8.45 (dd, J = 4.3, 1.9 Hz, 1H), 8.10 (dd, J = 8.0, 1.9 Hz, 1H), 7.95 (dd, J = 7.9, 1.5 Hz, 1H), 7.88 (d, J = 8.8 Hz, 1H), 7.77–7.64 (m, 6H), 7.60 (dd, J = 7.2, 1.5 Hz, 1H), 7.51–7.42 (m, 4H), 7.39–7.31 (m, 2H). 13C NMR (101 MHz, chloroform-d) δ 146.89, 146.83, 145.56, 141.44, 141.30, 138.36, 135.25, 135.06, 131.52, 129.22, 129.06, 128.75, 128.32, 128.04, 127.26, 127.10, 127.06, 126.95, 126.08, 125.98, 121.14.
10-(4-Methoxyphenyl)benzo[h]quinolone (3j)
White solid, 23.1 mg (yield: 81%). 1H NMR (400 MHz, chloroform-d) δ 8.46 (dd, J = 4.3, 1.9 Hz, 1H), 8.04 (dd, J = 8.0, 1.9 Hz, 1H), 7.87 (dd, J = 8.0, 1.5 Hz, 1H), 7.81 (d, J = 8.8 Hz, 1H), 7.68–7.60 (m, 2H), 7.53 (dd, J = 7.3, 1.5 Hz, 1H), 7.29 (dd, J = 8.3, 4.8 Hz, 3H), 7.02–6.90 (m, 2H), 3.88 (s, 3H). 13C NMR (101 MHz, chloroform-d) δ 157.98, 147.03, 146.92, 141.41, 138.96, 135.26, 135.13, 131.76, 129.85, 129.17, 128.37, 127.81, 127.25, 127.09, 125.93, 121.07, 112.86, 55.34.
10-(3-(Trifluoromethoxy)phenyl)benzo[h]quinolone (3k)
Yellow solid, 26.8 mg (yield: 79%). 1H NMR (400 MHz, chloroform-d) δ 8.41 (dd, J = 4.3, 1.8 Hz, 1H), 8.08 (dd, J = 8.0, 1.9 Hz, 1H), 7.94 (d, J = 7.9 Hz, 1H), 7.85 (d, J = 8.8 Hz, 1H), 7.68 (t, J = 8.6 Hz, 2H), 7.52 (dd, J = 7.3, 1.4 Hz, 1H), 7.42 (t, J = 8.1 Hz, 1H), 7.32 (dd, J = 7.5, 4.9 Hz, 2H), 7.21 (d, J = 7.7 Hz, 2H). 19F NMR (376 MHz, chloroform-d) δ −57.57. 13C NMR (101 MHz, chloroform-d) δ 148.55, 148.31, 147.00, 146.44, 139.96, 135.31, 134.98, 131.21, 128.86, 128.76, 128.54, 128.21, 127.29, 127.07, 126.85, 126.16, 122.20, 121.97, 121.28, 119.42 (q, J = 254.7 Hz), 118.36.
10-(Naphthalen-2-yl)benzo[h]quinolone (3l)
White solid, 22.9 mg (yield: 75%). 1H NMR (400 MHz, chloroform-d) δ 8.33 (dd, J = 4.3, 1.9 Hz, 1H), 8.10 (dd, J = 8.1, 1.9 Hz, 1H), 8.00–7.86 (m, 5H), 7.78 (d, J = 8.5 Hz, 1H), 7.75–7.70 (m, 2H), 7.65 (dd, J = 7.3, 1.5 Hz, 1H), 7.48 (ddd, J = 23.5, 7.3, 2.5 Hz, 3H), 7.30 (dd, J = 8.0, 4.3 Hz, 1H). 13C NMR (101 MHz, chloroform-d) δ 146.97, 146.72, 144.53, 141.51, 135.22, 135.07, 133.82, 132.20, 131.98, 129.49, 129.09, 128.36, 128.19, 128.13, 127.64, 127.27, 127.24, 126.01, 125.71, 125.57, 125.43, 125.20, 121.14. ESI-MS [M + H]+ calcd for C23H15N: 306.1283, found: 306.1284.
10-(Naphthalen-1-yl)benzo[h]quinolone (3m)
White solid, 21.6 mg (yield: 71%). 1H NMR (400 MHz, chloroform-d) δ 8.07–7.98 (m, 3H), 7.91 (q, J = 8.2 Hz, 3H), 7.80–7.69 (m, 2H), 7.64–7.51 (m, 2H), 7.41–7.33 (m, 2H), 7.22 (d, J = 8.6 Hz, 1H), 7.16–7.04 (m, 2H). 13C NMR (101 MHz, chloroform-d) δ 147.24, 146.48, 144.97, 139.61, 134.96, 134.69, 133.24, 133.08, 131.69, 130.34, 128.36, 128.31, 127.92, 127.29, 127.12, 126.43, 126.22, 126.08, 125.58, 125.10, 125.02, 124.64, 120.85.
10-(4-Fluorophenyl)benzo[h]quinolone (3n)
White solid, 21.8 mg (yield: 80%). 1H NMR (400 MHz, chloroform-d) δ 8.46 (dd, J = 4.4, 1.9 Hz, 1H), 8.10 (dd, J = 8.0, 1.9 Hz, 1H), 7.94 (d, J = 7.9 Hz, 1H), 7.86 (d, J = 8.8 Hz, 1H), 7.69 (dd, J = 10.9, 8.2 Hz, 2H), 7.53 (d, J = 7.3 Hz, 1H), 7.33 (ddd, J = 13.5, 8.2, 4.9 Hz, 3H), 7.10 (t, J = 8.7 Hz, 2H). 19F NMR (376 MHz, chloroform-d) δ −118.10. 13C NMR (101 MHz, chloroform-d) 161.59 (d, J = 244.42 Hz), 146.89, 146.76, 142.26 (d, J = 4.0 Hz), 140.67, 135.32, 135.04, 131.52, 130.17 (d, J = 8.0 Hz), 129.06, 128.32, 128.18, 127.29, 127.05, 126.04, 121.17, 114.14 (d, J = 21.2 Hz).
10-(3,4,5-Trifluorophenyl)benzo[h]quinolone (3o)
White solid, 25.9 mg (yield: 84%). 1H NMR (400 MHz, chloroform-d) δ 8.51 (dd, J = 4.3, 1.8 Hz, 1H), 8.12 (dd, J = 8.0, 1.9 Hz, 1H), 7.97 (dd, J = 8.0, 1.4 Hz, 1H), 7.86 (d, J = 8.8 Hz, 1H), 7.75–7.59 (m, 2H), 7.48 (dd, J = 7.2, 1.4 Hz, 1H), 7.38 (dd, J = 8.0, 4.3 Hz, 1H), 6.94 (dd, J = 8.6, 6.6 Hz, 2H). 19F NMR (376 MHz, chloroform-d) δ −137.30 (d, J = 20.5 Hz), −165.78 (t, J = 20.7 Hz). 13C NMR (151 MHz, chloroform-d) δ 151.49 (dd, J = 10.6, 4.5 Hz), 149.85 (dd, J = 10.6, 4.5 Hz), 147.23, 146.26, 142.82–142.09 (m), 139.26 (t, J = 15.5 Hz), 138.59, 137.61 (t, J = 15.5 Hz), 135.52, 135.02, 130.95, 128.98, 128.79, 128.23, 127.46, 127.09, 126.39, 121.51, 112.93 (dd, J = 16.6, 3.0 Hz).
10-(3,4-Difluorophenyl)benzo[h]quinolone (3p)
White solid, 23.5 mg (yield: 81%). 1H NMR (400 MHz, chloroform-d) δ 8.48 (dd, J = 4.3, 1.9 Hz, 1H), 8.11 (dd, J = 8.0, 1.9 Hz, 1H), 7.95 (dd, J = 8.0, 1.4 Hz, 1H), 7.86 (d, J = 8.8 Hz, 1H), 7.75–7.62 (m, 2H), 7.50 (dd, J = 7.3, 1.4 Hz, 1H), 7.36 (dd, J = 8.1, 4.3 Hz, 1H), 7.21–7.10 (m, 2H), 7.04 (dddd, J = 8.3, 4.4, 2.1, 1.4 Hz, 1H). 19F NMR (376 MHz, chloroform-d) δ −140.45 (d, J = 21.7 Hz), −143.07 (d, J = 21.7 Hz). 13C NMR (101 MHz, chloroform-d) δ 149.7 (dd, J = 245.4 Hz, 12.8 Hz), 148.1 (dd, J = 243.6 Hz, 12.8 Hz), 147.03, 146.45, 143.22 (d, J = 10.8 Hz), 139.45, 135.39, 134.97, 131.25, 128.87, 128.56, 128.24, 127.33, 127.03, 126.18, 124.50 (dd, J = 6.0 Hz, 3.5 Hz), 121.32, 117.85 (d, J = 17.2 Hz), 116.03 (d, J = 17.1 Hz).
10-(4-Chlorophenyl)benzo[h]quinolone (3q)
White solid, 15.3 mg (yield: 53%). 1H NMR (400 MHz, chloroform-d) δ 8.46 (dd, J = 4.3, 1.8 Hz, 1H), 8.09 (dd, J = 8.0, 1.9 Hz, 1H), 7.93 (dd, J = 7.9, 1.4 Hz, 1H), 7.85 (d, J = 8.7 Hz, 1H), 7.72–7.64 (m, 2H), 7.53–7.46 (m, 3H), 7.35–7.32 (m, 1H), 7.26–7.19 (m, 2H). 13C NMR (101 MHz, chloroform-d) δ 146.66, 146.30, 145.10, 140.05, 135.07, 134.71, 131.09, 131.02, 130.23, 130.15, 129.84, 128.55, 128.05, 127.99, 127.22, 126.99, 126.81, 125.82, 120.97, 119.40.
10-(4-Bromophenyl)benzo[h]quinolone (3r)
Colorless oil, 13.3 mg (yield: 40%). 1H NMR (400 MHz, chloroform-d) δ 8.46 (dd, J = 4.3, 1.9 Hz, 1H), 8.10 (dd, J = 8.0, 1.9 Hz, 1H), 7.93 (dd, J = 7.9, 1.4 Hz, 1H), 7.86 (d, J = 8.8 Hz, 1H), 7.72–7.64 (m, 2H), 7.50 (td, J = 6.8, 1.7 Hz, 3H), 7.34 (dd, J = 8.0, 4.3 Hz, 1H), 7.26–7.21 (m, 3H). 13C NMR (101 MHz, chloroform-d) δ 146.94, 146.58, 145.37, 140.33, 135.36, 134.99, 131.31, 130.50, 130.43, 128.83, 128.33, 128.27, 127.27, 127.09, 126.10, 121.25, 119.68.
10-(3-Nitrophenyl)benzo[h]quinolone (3s)
White solid, 21.6 mg (yield: 72%). 1H NMR (400 MHz, chloroform-d) δ 8.36 (dd, J = 4.3, 1.9 Hz, 1H), 8.23 (ddq, J = 5.0, 2.4, 1.2 Hz, 2H), 8.12 (dd, J = 8.0, 1.9 Hz, 1H), 8.00 (dd, J = 8.0, 1.4 Hz, 1H), 7.89 (d, J = 8.8 Hz, 1H), 7.76–7.72 (m, 2H), 7.68 (dt, J = 7.6, 1.3 Hz, 1H), 7.57–7.50 (m, 2H), 7.35 (dd, J = 8.0, 4.3 Hz, 1H). 13C NMR (101 MHz, chloroform-d) δ 147.96, 147.73, 146.95, 146.23, 138.95, 135.54, 135.10, 135.01, 131.22, 129.01, 128.72, 128.25, 128.02, 127.38, 127.16, 126.32, 123.98, 121.45, 120.83.
10-(3,4-Bis(trifluoromethyl)phenyl)benzo[h]quinolone (3t)
White solid, 31.6 mg (yield: 81%). 1H NMR (400 MHz, chloroform-d) δ 8.36 (dd, J = 4.3, 1.9 Hz, 1H), 8.12 (dd, J = 8.0, 1.9 Hz, 1H), 8.01 (dd, J = 8.0, 1.4 Hz, 1H), 7.91–7.85 (m, 2H), 7.83 (s, 2H), 7.77–7.70 (m, 2H), 7.52 (dd, J = 7.2, 1.4 Hz, 1H), 7.36 (dd, J = 8.0, 4.3 Hz, 1H). 19F NMR (376 MHz, chloroform-d) δ −62.59. 13C NMR (151 MHz, chloroform-d) δ 148.14, 146.97, 146.11, 138.47, 135.61, 135.11, 131.31, 130.30 (q, J = 32.7 Hz), 129.40, 129.36, 128.70, 128.30, 127.43, 127.26, 126.46, 123.93 (q, J = 272.8 Hz), 121.68, 119.69–119.49 (m).
Ethyl 4-(benzo[h]quinolin-10-yl)benzoate (3u)
White solid, 16.3 mg (yield: 50%). 1H NMR (400 MHz, chloroform-d) δ 8.38 (dd, J = 4.3, 1.9 Hz, 1H), 8.12–8.05 (m, 4H), 7.93 (dd, J = 7.9, 1.4 Hz, 1H), 7.85 (d, J = 8.8 Hz, 1H), 7.71–7.65 (m, 2H), 7.49 (dd, J = 7.2, 1.4 Hz, 1H), 7.43–7.38 (m, 2H), 7.30 (dd, J = 8.0, 4.3 Hz, 1H), 4.43 (q, J = 7.1 Hz, 2H), 1.44 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, chloroform-d) δ 167.18, 151.54, 146.92, 146.46, 140.63, 135.29, 134.94, 131.03, 130.19, 128.85, 128.80, 128.75, 128.45, 128.22, 127.75, 127.28, 127.22, 127.07, 126.12, 121.26, 60.80, 14.48.
10-(Thiophen-3-yl)benzo[h]quinolone (3y)
White solid, 8.1 mg (yield: 31%). 1H NMR (400 MHz, chloroform-d) δ 8.60–8.51 (m, 1H), 8.13–8.05 (m, 1H), 7.94–7.89 (m, 1H), 7.84 (dd, J = 8.8, 1.7 Hz, 1H), 7.73–7.59 (m, 3H), 7.40–7.33 (m, 1H), 7.30–7.22 (m, 2H), 7.04 (d, J = 4.9 Hz, 1H). 13C NMR (101 MHz, chloroform-d) δ 147.20, 146.86, 146.50, 136.25, 135.26, 135.00, 131.75, 130.72, 129.44, 128.32, 128.28, 127.21, 127.18, 125.94, 122.74, 121.20, 119.72.
10-(Cyclohex-1-en-1-yl)benzo[h]quinoline (3z)
Colorless oil, 20.4 mg (yield: 76%). 1H NMR (400 MHz, chloroform-d) δ 8.99 (dd, J = 4.3, 1.9 Hz, 1H), 8.10 (dd, J = 8.1, 1.9 Hz, 1H), 7.80 (dd, J = 9.6, 7.7 Hz, 2H), 7.61 (dd, J = 16.4, 8.4 Hz, 2H), 7.54–7.35 (m, 2H), 5.63 (t, J = 3.7 Hz, 1H), 2.58 (d, J = 14.8 Hz, 1H), 2.40–2.21 (m, 2H), 1.89 (dd, J = 79.1, 45.4 Hz, 5H). 13C NMR (101 MHz, chloroform-d) δ 147.70, 146.93, 145.26, 144.25, 135.26, 134.85, 129.98, 129.01, 128.63, 127.55, 127.46, 127.20, 125.55, 120.86, 120.29.
6-Methyl-10-phenylbenzo[h]quinolone (4a)
White solid, 21.5 mg (yield: 80%). 1H NMR (400 MHz, chloroform-d) δ 8.33 (dd, J = 4.3, 1.9 Hz, 1H), 8.11–8.04 (m, 1H), 7.97 (dd, J = 8.0, 1.9 Hz, 1H), 7.76–7.69 (m, 1H), 7.59–7.51 (m, 2H), 7.45–7.29 (m, 7H), 7.28–7.21 (m, 1H), 2.82–2.79 (m, 3H). 13C NMR (101 MHz, chloroform-d) δ 146.91, 146.31, 146.06, 142.07, 134.86, 134.36, 133.68, 131.22, 129.11, 128.69, 128.63, 127.43, 127.09, 126.96, 125.89, 125.54, 123.84, 121.15, 20.62.
5-(Naphthalen-2-yl)-10-phenylbenzo[h]quinolone (4b)
White solid, 30.8 mg (yield: 81%). 1H NMR (400 MHz, chloroform-d) δ 8.41 (dd, J = 4.2, 1.8 Hz, 1H), 8.15 (dd, J = 8.3, 1.9 Hz, 1H), 8.02–7.87 (m, 6H), 7.69 (t, J = 7.6 Hz, 1H), 7.63 (dd, J = 8.4, 1.7 Hz, 1H), 7.55 (td, J = 6.7, 2.4 Hz, 3H), 7.44–7.36 (m, 4H), 7.22 (dd, J = 8.1, 4.4 Hz, 1H). 13C NMR (101 MHz, chloroform-d) δ 147.11, 146.66, 146.55, 141.75, 137.93, 137.04, 134.48, 133.66, 133.50, 132.80, 131.57, 128.84, 128.74, 128.70, 128.16, 128.13, 128.09, 128.02, 127.86, 127.46, 127.43, 126.58, 126.55, 126.36, 125.70, 120.95.
5,10-Diphenylbenzo[h]quinolone (4c)
White solid, 26.5 mg (yield: 80%). 1H NMR (400 MHz, chloroform-d) δ 8.41 (dd, J = 4.2, 1.8 Hz, 1H), 8.14 (dd, J = 8.3, 1.8 Hz, 1H), 7.92 (dd, J = 7.9, 1.4 Hz, 1H), 7.82 (s, 1H), 7.70 (t, J = 7.6 Hz, 1H), 7.58–7.46 (m, 6H), 7.44–7.34 (m, 5H), 7.26 (d, J = 6.0 Hz, 1H). 13C NMR (101 MHz, chloroform-d) δ 147.02, 146.57, 146.52, 141.66, 139.49, 137.96, 134.40, 133.57, 131.48, 130.01, 128.68, 128.59, 128.53, 128.37, 128.02, 127.71, 127.42, 127.36, 126.40, 125.65, 120.88.
2-([1,1′-Biphenyl]-2-yl)pyridine (5a)
Yellow oil, 11.8 mg (yield: 51%). 1H NMR (400 MHz, chloroform-d) δ 8.65–8.60 (m, 1H), 7.70 (dd, J = 5.7, 3.3 Hz, 1H), 7.46 (dq, J = 7.8, 3.6 Hz, 3H), 7.38 (td, J = 7.7, 1.8 Hz, 1H), 7.26–7.20 (m, 3H), 7.16 (dd, J = 6.7, 2.9 Hz, 2H), 7.10 (dd, J = 7.4, 4.9 Hz, 1H), 6.88 (d, J = 7.9 Hz, 1H). 13C NMR (101 MHz, chloroform-d) δ 159.27, 149.44, 141.36, 140.63, 135.22, 130.52, 130.50, 129.73, 128.55, 128.08, 127.67, 126.72, 125.44, 121.37.
4-Methoxy-1,1′-biphenyl (7a)
White solid, 2.8 mg (yield: 15%). 1H NMR (400 MHz, chloroform-d) δ 7.57–7.50 (m, 4H), 7.41 (t, J = 7.7 Hz, 2H), 7.33–7.27 (m, 1H), 7.00–6.95 (m, 2H), 3.85 (s, 3H). 13C NMR (101 MHz, chloroform-d) δ 159.15, 140.85, 133.79, 128.75, 128.19, 126.77, 126.69, 114.21, 55.37.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Nos. 22071177 and 21572155) and the Natural Science Foundation of Tianjin (16JCYBJC19700).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c01105.
NMR spectra for compounds and complexes synthesized (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Schneider N.; Lowe D. M.; Sayle R. A.; Tarselli M. A.; Landrum G. A. Big Data from Pharmaceutical Patents: A Computational Analysis of Medicinal Chemists’ Bread and Butter. J. Med. Chem. 2016, 59, 4385–4402. 10.1021/acs.jmedchem.6b00153. [DOI] [PubMed] [Google Scholar]; b Brown D. G.; Bostrçm J. Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone?. J. Med. Chem. 2016, 59, 4443–4458. 10.1021/acs.jmedchem.5b01409. [DOI] [PubMed] [Google Scholar]; c Molander G. A.; Canturk B. Preparation of Potassium Alkoxymethyltrifluoroborates and Their Cross-Coupling with Aryl Chlorides. Org. Lett. 2008, 10, 2135–2138. 10.1021/ol800532p. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Miyaura N. Organoboron Compounds. Top. Curr. Chem. 2002, 219, 11–59. [Google Scholar]; e Miyaura N.; Yamada K.; Suzuki A. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 20, 3437–3440. 10.1016/S0040-4039(01)95429-2. [DOI] [Google Scholar]; f Miyaura N.; Suzuki A. Stereoselective synthesis of arylated (E)-alkenes by the reaction of alk-1-enylboranes with aryl halides in the presence of palladium catalyst. J. Chem. Soc., Chem. Commun. 1979, 866–867. 10.1039/c39790000866. [DOI] [Google Scholar]
- a Modha S. G.; Mehta V. P.; Van der Eycken E. V. Transition metal-catalyzed C–C bond formation via C–S bond cleavage: an overview. Chem. Soc. Rev. 2013, 42, 5042–5055. 10.1039/c3cs60041f. [DOI] [PubMed] [Google Scholar]; b Rosen B. M.; Quasdorf K. W.; Wilson D. A.; Zhang N.; Resmerita A.-M.; Garg N. K.; Percec V. Nickel-Catalyzed Cross-Couplings Involving Carbon–Oxygen Bonds. Chem. Rev. 2011, 111, 1346–1416. 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sun C.-L.; Li B.-J.; Shi Z.-J. Pd-catalyzed oxidative coupling with organometallic reagents via C–H activation. Chem. Commun. 2010, 46, 677–685. 10.1039/b908581e. [DOI] [PubMed] [Google Scholar]; d Chen X.; Engle K. M.; Wang D.-H.; Yu J.-Q. Palladium(II)-Catalyzed C–H Activation/C–C Cross-Coupling Reactions: Versatility and Practicality. Angew. Chem., Int. Ed. 2009, 48, 5094–5115. 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Yu D.-G.; Yu M.; Guan B.-T.; Li B.-J.; Zheng Y.; Wu Z.-H.; Shi Z.-J. Carbon–Carbon Formation via Ni-Catalyzed Suzuki–Miyaura Coupling through C–CN Bond Cleavage of Aryl Nitrile. Org. Lett. 2009, 11, 3374–3377. 10.1021/ol901217m. [DOI] [PubMed] [Google Scholar]
- a Cope J. D.; Sheridan P. E.; Galloway C. J.; Awoyemi R. F.; Stokes S. L.; Emerson J. P. Synthesis and Characterization of a Tetradentate, N-Heterocyclic Carbene Copper(II) Complex and Its Use as a Chan–Evans–Lam Coupling Catalyst. Organometallics 2020, 39, 4457–4464. 10.1021/acs.organomet.0c00552. [DOI] [Google Scholar]; b Yoo W.-J.; Tsukamoto T.; Kobayashi S. Visible-Light-Mediated Chan–Lam Coupling Reactions of Aryl Boronic Acids and Aniline Derivatives. Angew. Chem., Int. Ed. 2015, 54, 6587–6590. 10.1002/anie.201500074. [DOI] [PubMed] [Google Scholar]; c Deb A.; Agasti S.; Saboo T.; Maiti D. Generation of Arylated Quinones by Iron-Catalyzed Oxidative Arylation of Phenols: Formal Synthesis of Phellodonin, Sarcodonine, Leucomelone and Betulinan A. Adv. Synth. Catal. 2014, 356, 705–710. 10.1002/adsc.201301084. [DOI] [Google Scholar]; d Čermák J. K.; Církva V. Copper-mediated synthesis of mono- and dichlorinated diaryl ethers. Tetrahedron Lett. 2014, 55, 4185–4188. 10.1016/j.tetlet.2014.06.035. [DOI] [Google Scholar]; e Mulla S. A. R.; Inamdar S. M.; Pathana M. Y.; Chavan S. S. Base promoted highly efficient copper fluorapatite catalyzed coupling of phenols with arylboronic acids under mild and ligand-free conditions. RSC Adv. 2012, 2, 12818–12823. 10.1039/c2ra21850j. [DOI] [Google Scholar]; f Roglans A.; Pla-Quintana A.; Moreno-Mañas M. Diazonium Salts as Substrates in Palladium-Catalyzed Cross-Coupling Reactions. Chem. Rev. 2006, 106, 4622–4643. 10.1021/cr0509861. [DOI] [PubMed] [Google Scholar]; g Blakey S. B.; MacMillan D. W. C. The First Suzuki Cross-Couplings of Aryltrimethylammonium Salts. J. Am. Chem. Soc. 2003, 125, 6046–6047. 10.1021/ja034908b. [DOI] [PubMed] [Google Scholar]
- a Castillo-García A. A.; González-Sebastián L.; Lomas-Romero L.; Hernandez-Ortega S.; Toscano R. A.; Morales-Morales D. Novel meta-benzothiazole and benzimidazole functionalised POCOP-Ni(ii) pincer complexes as efficient catalysts in the production of diarylketones. New J. Chem. 2021, 45, 10204–10216. 10.1039/D1NJ01348C. [DOI] [Google Scholar]; b Dutta M. M.; Phukan P. Cu-doped CoFe2O4 nanoparticles as magnetically recoverable catalyst for CeN cross-coupling reaction. Catal. Commun. 2018, 109, 38–42. 10.1016/j.catcom.2018.02.014. [DOI] [Google Scholar]; c Moustafa D.; Sweet C.; Lim H.; Calalpa B.; Kaur P. Mn/Cu catalyzed addition of arylboronic acid to nitriles: Direct synthesis of arylketones. Tetrahedron Lett. 2018, 59, 3816–3820. 10.1016/j.tetlet.2018.09.021. [DOI] [Google Scholar]; d Lei C.; Yip Y. J.; Zhou J. S. Nickel-Catalyzed Direct Synthesis of Aryl Olefins from Ketones and Organoboron Reagents under Neutral Conditions. J. Am. Chem. Soc. 2017, 139, 6086–6089. 10.1021/jacs.7b02742. [DOI] [PubMed] [Google Scholar]; e Huanga H.; Jiang Z.-T.; Wu Y.; Gan C.-Y.; Li J.-M.; Xiang S.-K.; Feng C.; Wang B.-Q.; Yang W.-T. Copper-Catalyzed Amidation of Arylboronic Acids with Nitriles. Synlett 2016, 27, 951–955. 10.1055/s-0035-1561291. [DOI] [Google Scholar]; f Yang J.; Chen X.; Wang Z. The carbonyl group tuned electron-deficient phosphorus ligands and their application in Rhodium catalyzed arylation to aldehydes. Tetrahedron Lett. 2015, 56, 5673–5675. 10.1016/j.tetlet.2015.08.076. [DOI] [Google Scholar]; g Mavila S.; Rozenberg I.; Lemcof N. G. A general approach to mono- and bimetallic organometallic nanoparticles. Chem. Sci. 2014, 5, 4196–4203. 10.1039/C4SC01231C. [DOI] [Google Scholar]; h Raghuvanshi D. S.; Gupta A. K.; Singh K. N. Nickel-Mediated N-Arylation with Arylboronic Acids: An Avenue to Chan-Lam Coupling. Org. Lett. 2012, 14, 4326–4329. 10.1021/ol3021836. [DOI] [PubMed] [Google Scholar]; i Korenaga T.; Ko A.; Uotani K.; Tanaka Y.; Sakai T. Synthesis and Application of 2,6-Bis(trifluoromethyl)-4-pyridylPhosphanes: The Most Electron-Poor Aryl Phosphanes with ModerateBulkiness. Angew. Chem., Int. Ed. 2011, 50, 10703–10707. 10.1002/anie.201104588. [DOI] [PubMed] [Google Scholar]; j Wong Y.-C.; Parthasarathy K.; Cheng C.-H. Direct Synthesis of Arylketones by Nickel-Catalyzed Addition of Arylboronic Acids to Nitriles. Org. Lett. 2010, 12, 1736–1739. 10.1021/ol1003252. [DOI] [PubMed] [Google Scholar]; k Forrat V. J.; Prieto O.; Ramón D. J.; Yus M. trans-1-Sulfonylamino-2-isoborneolsulfonylamino cyclohexane Derivatives: Excellent Chiral Ligands for the Catalytic Enantioselective Addition of Organozinc Reagents to Ketones. Chem. – Eur. J. 2006, 12, 4431–4445. 10.1002/chem.200501397. [DOI] [PubMed] [Google Scholar]
- a Li X.; Hong X. Computational studies on Ni-catalyzed C–O bond activation of esters. J. Organomet. Chem. 2018, 864, 68–80. 10.1016/j.jorganchem.2018.01.019. [DOI] [Google Scholar]; b Shi S.; Nolan S. P.; Szostak M. Well-Defined Palladium(II)–NHC Precatalysts for Cross-Coupling Reactions of Amides and Esters by Selective N–C/O–C Cleavage. Acc. Chem. Res. 2018, 51, 2589–2599. 10.1021/acs.accounts.8b00410. [DOI] [PubMed] [Google Scholar]; c Guo L.; Rueping M. Decarbonylative Cross-Couplings: Nickel Catalyzed Functional Group Interconversion Strategies for the Construction of Complex Organic Molecules. Acc. Chem. Res. 2018, 51, 1185–1195. 10.1021/acs.accounts.8b00023. [DOI] [PubMed] [Google Scholar]; d Guo L.; Rueping M. Transition-Metal-Catalyzed Decarbonylative Coupling Reactions: Concepts, Classifications, and Applications. Chem. – Eur. J. 2018, 24, 7794–7809. 10.1002/chem.201704670. [DOI] [PubMed] [Google Scholar]; e Devarajan N.; Suresh P. Copper-catalyzed oxidative coupling of arylboronic acids with aryl carboxylic acids: Cu3(BTC)2 MOF as a sustainable catalyst to access aryl esters. Org. Chem. Front. 2018, 5, 2322–2331. 10.1039/C8QO00519B. [DOI] [Google Scholar]; f Takise R.; Muto K.; Yamaguchi J. Cross-coupling of aromatic esters and amides. Chem. Soc. Rev. 2017, 46, 5864–5888. 10.1039/C7CS00182G. [DOI] [PubMed] [Google Scholar]; g Gooßen L. J.; Winkel L.; Döhring A.; Ghosh K.; Paetzold J. Pd-Catalyzed Synthesis of Functionalized Arylketones from Boronic Acids and Carboxylic Acids Activated in situ with Dimethyl Dicarbonate. Synlett 2002, 8, 1237–1240. 10.1055/s-2002-32961. [DOI] [Google Scholar]
- Muto K.; Yamaguchi J.; Musaev D. G.; Itami K. Decarbonylative organoboron cross-coupling of esters by nickel catalysis. Nat. Commun. 2015, 6, 7508–7515. 10.1038/ncomms8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yue H.; Guo L.; Liao H.-H.; Cai Y.; Zhu C.; Rueping M. Catalytic Ester and Amide to Amine Interconversion: Nickel-Catalyzed Decarbonylative Amination of Esters and Amides by C–O and C–C Bond Activation. Angew. Chem., Int. Ed. 2017, 56, 4282–4285. 10.1002/anie.201611819. [DOI] [PubMed] [Google Scholar]; b Guo L.; Chatupheeraphat A.; Rueping M. Decarbonylative Silylation of Esters by Combined Nickel and CopperCatalysis for the Synthesis of Arylsilanes and Heteroarylsilanes. Angew. Chem., Int. Ed. 2016, 55, 11810–11813. 10.1002/anie.201604696. [DOI] [PubMed] [Google Scholar]; c Guo L.; Rueping M. Functional Group Interconversion: Decarbonylative Borylation of Esters for the Synthesis of Organoboronates. Chem. – Eur. J. 2016, 22, 16787–16790. 10.1002/chem.201604504. [DOI] [PubMed] [Google Scholar]; d Amaike K.; Muto K.; Yamaguchi J.; Itami K. Decarbonylative C–H Coupling of Azoles and Aryl Esters: Unprecedented Nickel Catalysis and Application to the Synthesis of Muscoride A. J. Am. Chem. Soc. 2012, 134, 13573–13576. 10.1021/ja306062c. [DOI] [PubMed] [Google Scholar]
- a Kuwano R.; Shimizu R. An Improvement of Nickel Catalyst for Cross-coupling Reaction of Arylboronic Acids with Aryl Carbonates by Using a Ferrocenyl Bisphosphine Ligand. Chem. Lett. 2011, 40, 913–915. 10.1246/cl.2011.913. [DOI] [Google Scholar]; b Molander G. A.; Beaumard F. Nickel-Catalyzed C–O Activation of Phenol Derivatives with Potassium Heteroaryltrifluoroborates. Org. Lett. 2010, 12, 4022–4025. 10.1021/ol101592r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Xu L.; Li B.-J.; Wu Z.-H.; Lu X.-Y.; Guan B.-T.; Wang B.-Q.; Zhao K.-Q.; Shi Z.-J. Nickel-Catalyzed Efficient and Practical Suzuki–Miyaura Coupling of Alkenyl and Aryl Carbamates with Aryl Boroxines. Org. Lett. 2010, 12, 884–887. 10.1021/ol9029534. [DOI] [PubMed] [Google Scholar]; d Antoft-Finch A.; Blackburn T.; Snieckus V. N,N-Diethyl O-Carbamate: Directed Metalation Group and Orthogonal Suzuki–Miyaura Cross-Coupling Partner. J. Am. Chem. Soc. 2009, 131, 17750–17752. 10.1021/ja907700e. [DOI] [PubMed] [Google Scholar]; e Quasdorf K. W.; Riener M.; Petrova K. V.; Garg N. K. Suzuki–Miyaura Coupling of Aryl Carbamates, Carbonates, and Sulfamates. J. Am. Chem. Soc. 2009, 131, 17748–17749. 10.1021/ja906477r. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Guan B.-T.; Wang Y.; Li B.-J.; Yu D.-G.; Shi Z.-J. Biaryl Construction via Ni-Catalyzed C–O Activation of Phenolic Carboxylates. J. Am. Chem. Soc. 2008, 130, 14468–14470. 10.1021/ja8056503. [DOI] [PubMed] [Google Scholar]; g Quasdorf K. W.; Tian X.; Garg N. K. Cross-Coupling Reactions of Aryl Pivalates with Boronic Acids. J. Am. Chem. Soc. 2008, 130, 14422–14423. 10.1021/ja806244b. [DOI] [PubMed] [Google Scholar]
- a Ma H.; Bai C.; Bao Y.-S. Heterogeneous Suzuki–Miyaura coupling of heteroaryl ester via chemoselective C (acyl)–O bond activation. RSC Adv. 2019, 9, 17266–17272. 10.1039/C9RA02394A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Xin B.-W. Synthesis of Aryl Ketones by Cross-Coupling Reaction of Arylboronic Acids with Carboxylic Anhydrides in Aqueous Phase. Synth. Commun. 2008, 38, 2826–2837. 10.1080/00397910801979346. [DOI] [Google Scholar]; c Xin B.; Zhang Y.; Cheng K. Phosphine-Free Cross-Coupling Reaction of Arylboronic Acids with Carboxylic Anhydrides or Acyl Chlorides in Aqueous Media. J. Org. Chem. 2006, 71, 5725–5731. 10.1021/jo060749d. [DOI] [PubMed] [Google Scholar]; d Tatamidani H.; Kakiuchi F.; Chatani N. A New Ketone Synthesis by Palladium-Catalyzed Cross-Coupling Reactions of Esters with Organoboron Compounds. Org. Lett. 2004, 6, 3597–35991. 10.1021/ol048513o. [DOI] [PubMed] [Google Scholar]; e Chatani N.; Tatamidani H.; Ie Y.; Kakiuchi F.; Murai S. The Ruthenium-Catalyzed Reductive Decarboxylation of Esters: Catalytic Reactions Involving the Cleavage of Acyl–Oxygen Bonds of Esters. J. Am. Chem. Soc. 2001, 123, 4849–4850. 10.1021/ja0103501. [DOI] [PubMed] [Google Scholar]
- a Yang S.; Zhou T.; Poater A.; Cavallo L.; Nolan S. P.; Szostak M. Suzuki–Miyaura cross-coupling of esters by selective O–C (O) cleavage mediated by air- and moisture-stable [Pd(NHC)(μ-Cl)Cl]2 precatalysts: catalyst evaluation and mechanism. Catal. Sci. Technol. 2021, 11, 3189–3197. 10.1039/D1CY00312G. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lei P.; Ling Y.; An J.; Nolan S. P.; Szostak M. 2-Methyltetrahydrofuran (2-MeTHF): A Green Solvent for Pd-NHC-Catalyzed Amide and Ester Suzuki-Miyaura Cross-Coupling by N-C/O-C Cleavage. Adv. Synth. Catal. 2019, 361, 5654–5660. 10.1002/adsc.201901188. [DOI] [Google Scholar]; c Ben Halima T.; Zhang W.; Yalaoui I.; Hong X.; Yang Y.-F.; Houk K. N.; Newman S. G. Palladium-Catalyzed Suzuki–Miyaura Coupling of Aryl Esters. J. Am. Chem. Soc. 2017, 139, 1311–1318. 10.1021/jacs.6b12329. [DOI] [PubMed] [Google Scholar]; d Sun C.-L.; Wang Y.; Zhou X.; Wu Z.-H.; Li B.-J.; Guan B.-T.; Shi Z.-J. Construction of Polysubstituted Olefins through Ni-Catalyzed Direct Activation of Alkenyl CO of Substituted Alkenyl Acetates. Chem. – Eur. J. 2010, 16, 5844–5847. 10.1002/chem.200902785. [DOI] [PubMed] [Google Scholar]
- Chatupheeraphat A.; Liao H.-H.; Srimontree W.; Guo L.; Minenkov Y.; Poater A.; Cavallo L.; Rueping M. Ligand-Controlled Chemoselective C(acyl)–O Bond vs C(aryl)–C Bond Activation of Aromatic Esters in Nickel Catalyzed C(sp2)–C(sp3) Cross-Couplings. J. Am. Chem. Soc. 2018, 140, 3724–3735. 10.1021/jacs.7b12865. [DOI] [PubMed] [Google Scholar]
- Wang J.; Liu B.; Zhao H.; Wang J. Rhodium-Catalyzed Cross-Coupling Reactions of Carboxylate and Organoboron Compounds via Chelation-Assisted C–C Bond Activation. Organometallics 2012, 31, 8598–8607. 10.1021/om300994j. [DOI] [Google Scholar]
- a Zhou J.-Y.; Liu R.-Q.; Wang C.-Y.; Zhu Y.-M. Synthesis of Biaryls via Decarbonylative Nickel-Catalyzed Suzuki–Miyaura Cross-Coupling of Aryl Anhydrides. J. Org. Chem. 2020, 85, 14149–14157. 10.1021/acs.joc.0c02266. [DOI] [PubMed] [Google Scholar]; b Dindarloo Inaloo I.; Majnooni S.; Eslahi H.; Esmaeilpour M. Nickel(II) Nanoparticles Immobilized on EDTA-Modified Fe3O4@SiO2 Nanospheres as Efficient and Recyclable Catalysts for Ligand-Free Suzuki–Miyaura Coupling of Aryl Carbamates and Sulfamates. ACS Omega 2020, 5, 7406–7417. 10.1021/acsomega.9b04450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhou W.; Li P.; Zhang Y.; Wang L. Palladium-Catalyzed Direct Alkoxycarbonylation of Aromatic C–H Bonds via Selective C–C Cleavage of α-Keto Esters. Adv. Synth. Catal. 2013, 355, 2343–2352. 10.1002/adsc.201300436. [DOI] [Google Scholar]; b Kochi T.; Urano S.; Seki H.; Mizushima E.; Sato M.; Kakiuchi F. Ruthenium-Catalyzed Amino- and Alkoxycarbonylations with Carbamoyl Chlorides and Alkyl Chloroformates via Aromatic C–H Bond Cleavage. J. Am. Chem. Soc. 2009, 131, 2792–2793. 10.1021/ja8097492. [DOI] [PubMed] [Google Scholar]
- a Hoang G. T.; Reddy V. J.; Nguyen H. H. K.; Douglas C. J. Insertion of an Alkene into an Ester: Intramolecular Oxyacylation Reaction of Alkenes through Acyl C–O Bond Activation. Angew. Chem., Int. Ed. 2011, 50, 1882–1884. 10.1002/anie.201005767. [DOI] [PubMed] [Google Scholar]; b Hamada Y.; Takeuchi I. Syntheses of Nitrogen-Containing Heterocyclic Compounds. Reaction of Benzo[f or h]quinolines and Their N-Oxides with Methylsulfinyl Carbanion. J. Org. Chem. 1977, 42, 4209–4213. 10.1021/jo00862a008. [DOI] [Google Scholar]
- Liu C.; Yang W. A fast and oxygen-promoted protocol for the ligand-free Suzuki reaction of 2-halogenated pyridines in aqueous media. Chem. Commun. 2009, 6267–6269. 10.1039/b912364d. [DOI] [PubMed] [Google Scholar]
- Iyori Y.; Takahashi K.; Yamazaki K.; Ano Y.; Chatani N. Nickel-catalyzed reductive defunctionalization of esters in the absence of an external reductant: activation of C–O bonds. Chem. Commun. 2019, 55, 13610–13613. 10.1039/C9CC07710C. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.