

Abstract
Human immune system acts as a pivotal role in the tissue homeostasis and disease progression. Immunomodulatory biomaterials that can manipulate innate immunity and adaptive immunity hold great promise for a broad range of prophylactic and therapeutic purposes. This review is focused on the design strategies and principles of immunomodulatory biomaterials from the standpoint of materials science to regulate macrophage fate, such as activation, polarization, adhesion, migration, proliferation, and secretion. It offers a comprehensive survey and discussion on the tunability of material designs regarding physical, chemical, biological, and dynamic cues for modulating macrophage immune response. The range of such tailorable cues encompasses surface properties, surface topography, materials mechanics, materials composition, and materials dynamics. The representative immunoengineering applications selected herein demonstrate how macrophage-immunomodulating biomaterials are being exploited for cancer immunotherapy, infection immunotherapy, tissue regeneration, inflammation resolution, and vaccination. A perspective on the future research directions of immunoregulatory biomaterials is also provided.
Keywords: immunoengineering, immunomodulatory biomaterials, immunotherapy, macrophages, targeted drug delivery
1. Introduction
Over one century ago, the concept of immunology was made birth officially by the Nobel Prize in Physiology or Medicine 1908; macrophages are the first phagocytes discovered and function as immune effector cells through the pivotal host defense mechanism of phagocytosis.[1,2] Cancer immunotherapy represents a new paradigm for cancer cure and care that can attack and eliminate tumor cells by activating the inherent capacity of the human immune system.[3–5] As its fundamental feature, the human immune system possesses discrimination between self and nonself, so as to attack and clear the invading viruses, bacteria, fungi, parasites, cellular debris, damaged, diseased, or senescent cells, and other foreign matter.[6,7] The immune response can be orchestrated by a sequence of complicated interactions amongst diversified immunocytes. The innate immunity stands at the first line of defense against pathogen exposure, which is implemented by phagocytes including macrophages, dendritic cells (DCs), natural killer (NK) cells and granulocytes (basophils, eosinophils, neutrophils, mast cells). As a pivotal component of innate immune system, macrophages can recruit other immunocytes to infection site, phagocytose and obliterate foreign pathogens, and activate complement system and adaptive immunity.[8–10] The following adaptive immunity encompasses antigen presentation by macrophages and DCs (antigen-presenting cells, APCs) and antigen stimulation on T lymphocytes, B lymphocytes, and macrophages.[11,12]
Among immune cells engaging in defense and homeostasis, macrophages act as a crucial mediator in development, disease (including cancer, infection, and inflammation), and tissue regeneration and remodeling.[13] Macrophages can circulate in bloodstream for immune surveillance and migrate into tissues in response to various dangers; they can also reside in tissues in a steady-state (tissue-resident macrophages).[14,15] The tissue-resident macrophages exist in various organs encompassing skin (Langerhans cells), brain (microglia), eye (intraoccular macrophages), lung (alveolar macrophages), heart (cardiac macrophages), liver (Kupffer cells), spleen (splenic macrophages), kidney (kidney macrophages), small intestine (intestinal macrophages), peritoneum (peritoneal macrophages), bone (bone marrow macrophages) and lymph node (subcapsular sinusoidal macrophages and medullary macrophages).[16,17] Macrophages are derived from the differentiation of the circulating monocytes in peripheral blood,[18] which originate from the hematopoietic stem cells in bone marrow. Monocytes can migrate into diverse tissues from the peripheral blood to supplement and maintain longevous tissue-resident macrophages. In addition, these tissue-resident macrophages can also replenish and renew their populations through rapid local proliferation.[19,20]
Due to the changing states macrophages are in for their varying specific roles, it is of great significance to understand the diversity of macrophage lineage, identity, and regulation, thus to enable macrophages functioning as crucial therapeutic targets for numerous human diseases.[17] Macrophages can migrate into the inflamed tissues and be activated there to represent a full spectrum of polarization phenotypes in a phase-dependent manner correlating with their variational functions. On its one end is the proinflammatory M1 macrophages, and on the other end is the antiinflammatory M2 macrophages.[15] For general detailed information on the macrophage plasticity and polarization, we refer readers to several excellent reviews.[21–25] The M1 macrophages (classically activated macrophages) are produced by the stimulation of proinflammatory signals such as interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), and lipopolysaccharide (LPS), leading to the specific population of macrophage phenotype that can amplify the antimicrobial or antitumoral ability and augment the secretion of proinflammatory cytokines or mediators and the production of reactive oxygen or nitrogen species.[26–28] NK cells can produce IFN-γ in a transient manner while T helper 1 (Th1) cells can produce IFN-γ in a sustained way, thereby maintaining the population of M1 phenotype and affording steady host defense against intracellular microbes (adaptive immunity). Meanwhile, M1 macrophages can efficiently present antigens and promote Th1 lymphocyte differentiation to secrete proinflammatory cytokines (IFN-γ, IL-2, etc.).[15] For example, considering the unique macrophage effector function and capability of penetrating tumor tissues, Gill and co-workers recently genetically engineered the human primary macrophages with chimeric antigen receptor (CAR) to guide their phagocytosis activity against tumor cells.[29] The chimeric adenoviral vector could overcome their intrinsic resistance to genetic manipulation and afford the macrophages with a long-lasting proinflammatory M1 phenotype. The human CAR macrophages were able to express the proinflammatory cytokines and chemokines, transform the bystander macrophages from M2 phenotype to M1 phenotype, upregulate the antigen-presenting machinery, recruit and present antigens to the T cells and resist the impact of the immunosuppressive cytokines, thereby reducing the tumor burden and prolonging overall survival, evidenced with the xenograft mouse solid tumor models. Moreover, the CAR macrophages could also trigger the proinflammatory tumor microenvironment and facilitate the antitumoral T cell activity in the humanized mouse models. Nevertheless, M1 macrophages can also cause damage to surrounding cells or tissues due to their excessive production of proinflammatory cytokines and reactive nitrogen intermediates or reactive oxygen intermediates (RNI/ROI).[30,31] Upon implantation of biomaterials, the presence of M1 macrophages at early phase can create essential inflammatory response whereas their prolonged or unrestrained population will cause excessive inflammation and severe foreign body reaction and fibrotic scar formation around biomaterial implants, which is in particular harmful to biomaterial-mediated tissue repair, replacement, and regeneration and may ultimately cause failure of implants, highlighting the necessity of timely switching of M1 macrophages to M2 macrophages.[32,33]
The M2 macrophages (alternatively activated macrophages) are mediated by the Th2 cytokines such as IL-4 and IL-13 (expressed by eosinophils, basophils, neutrophils, mast cells, and T lymphocytes[34–36]), which is distinct from the IFN-γ induced Th1-type M1 activation.[25] The M2 phenotype encompasses wound-healing macrophages and regulatory macrophages with M2a, M2b, and M2c subtypes.[15,23,37] The M2a and M2b macrophages implement immunomodulatory functions by driving antiinflammatory Th2 immune responses, while the M2c macrophages play an important role in inflammation inhibition and tissue remodeling. Table 1 gives a summary on the characteristics of different macrophage phenotypes including subtypes.[23,30] Consequently, macrophage lineage comprises a noticeable diversity of subset cells that have specialized identity of polarization states and functions by the complicated interactions between microenvironment heterogeneity and transcriptional/chromatin profile.[38–40] Biologic scaffolds from the decellularized tissue extracellular matrix (ECM) can boost a proregenerative (wound-healing) immune phenotype for clinical treatment of tissue loss caused by trauma or tumor resection. Recently, Elisseeff and co-workers investigated how such wound-healing immune responses created by biomaterial microenvironment could influence the tumor formation, development and sensitivity to the immune checkpoint blockade, by implanting the urinary bladder matrix (UBM) scaffold with syngeneic cancer cells in mouse model.[41] The implanted scaffold material could lead to a favorable immune microenvironment that suppressed the tumor formation of B16-F10 melanoma in CD4+ T cell-dependent and macrophage-dependent manners. Further study indicated the activated type 2-like immune response different from classical tumor microenvironment, which included scaffold-associated activated type 2 T helper T cells (Th2 phenotype) and unique macrophage phenotype (with complex M1/M2 polarization and wound-healing phenotype distinct from classical tumor-associated macrophages (TAMs)), as well as eosinophil infiltration, complement and angiogenic factors. Such type 2 wound-healing scaffold immune microenvironment was also capable of potentiating the inhibition effect on tumor growth by the programmed death-1 (PD-1) or programmed deathligand 1 (PD-L1) checkpoint blockade to enhance the immunotherapy potency. Previously, the researchers have validated that, to develop a proregenerative biomaterial scaffold immune microenvironment, T helper 2 cells were required to promote the tissue regeneration of traumatic muscle wounds.[42] The scaffold could induce the proregenerative response featured by mTOR/Rictor-dependent Th2 pathway capable of directing the IL-4-dependent macrophage polarization for fulfilling functional muscle recovery. The same group also defined a specific scaffold-associated CD11b+ macrophages (M2-like) with a high antigen presentation ability.[43] CD3+ T cells were observed surrounding the scaffold implant and colocalized with the CD11b+ macrophages in the cellular aggregates in the scaffold at skin interface, suggesting the communication between the M2 phenotype macrophages and the Th2 T cells in the scaffold immune microenvironment.
Table 1.
| Phenotypes | Inducers | Membrane receptors | Enzymes | Cytokines | Chemokines | Functions |
|---|---|---|---|---|---|---|
|
| ||||||
| M1 | IFN-γ, TNF-α, LPS | CD86, MHC-II, IL-2Ra, IL-15Ra, IL-7R |
iNOS (RNI/ROI), PTGS2 | IL-1, IL-6, IL-10 (low), IL-12 (high), IL-15, IL-23, TNF-α |
CCL8, CCL15, CCL19, CCL20, CXCL9, CXCL10, CXCL11, CXCL13 |
Th1 response; type I inflammation; phagocytosis; intracellular pathogen killing; tumor resistance; proinflammatory |
| M2a | IL-4, IL-13 |
MHC-II, MRC1, SR-A1, DCL-1, DCSIGN, MS4A4A, CLECSF6 |
Arg (Polyamine), PTGS1 | IL-10, Decoy IL-1RII, IL-1ra, FN1, bIG-H3, IGF-1, PDFGC, F13A1, PGL2, TGF-β |
CCL13, CCL14, CCL17, CCL18, CCL23, CCL26 |
Th2 response; type II inflammation; allergy reaction; parasite encapsulation, killing and immunity; antiinflammatory |
| M2b | LPS, IL-1β, antigen–antibody immune complexes (ICs), Toll-like receptor (TLR) agonists, IL-1R ligands |
MHC-II, CD86 | SPHK1 | IL-1, IL-6, IL-10 (high), IL-12 (low), TNF-α |
CCL1, CCL20, CXCL1, CXCL2, CXCL3 |
Th2 activation; immunomodulation; interacting with B lymphocytes and maintaining antibody production; proinflammatory or antiinflammatory |
| M2c | IL-10, TGF-β, glucocorticoids | CD163, TLR-8, TLR-1, IL-21R, SLAM, MR (CD206) |
– | IL-10, TGF-β | CCL18; Matrix (PTX3, versican, α antitrypsin) |
Prohealing; immunomodulation; matrix deposition; tissue remodeling; inflammation termination |
Biomaterials can facilitate the replacement, repair, and/or regeneration of the damaged/diseased human tissues/organs, thereby realizing the rehabilitation or reinforcement of their physiological functions.[44–46] Innate immunocytes are the first to arrive in response to an implanted biomaterial. Proteins, such as fibrinogen, fibronectin, vitronectin, and complement that come from blood or interstitial fluid, can rapidly adsorb to the implant surface and thus form the transient matrix layer releasing chemoattractants and cytokines, which can activate the blood coagulation pathway and the complement system to orchestrate the recruitment of innate immunocytes to the site of injury.[32] The properties of the implanted biomaterials can play a determinant regulatory role in the initiation, severity and outcome of resultant acute or chronic inflammatory reactions. In case of unrestrained/prolonged inflammation or the lack of bioactive cues, the foreign body reaction/response (FBR) can cause the fibrous encapsulation of implants, a cascade involving monocyte recruitment and differentiation, macrophage activation, polarization and fusion into foreign body giant cells (FBGCs), to separate them from surroundings and prevent their direct interactions.[47,48] Nevertheless, there are multiple strategies to manipulate and modulate the host immune response to biomaterials by virtue of the modification and functionalization of their surface or bulk properties.[44] The tailorable/suitable material characteristics, mechanical properties, physical cues, chemical functionalities and biological effects play a critical role in offering regulatory microenvironment cues to direct the fate of immunocytes particularly macrophages in response to biomaterials.
In this review, we will describe the physiological changes of macrophages in response to multiple biomaterial-mediated microenvironment cues, and discuss how the manipulation and modulation of macrophage activation and polarization can be exploited for specific therapeutic functions. We aim to afford a comprehensive overview of biomaterial-mediated immunomodulation of macrophage fate, which can be exploited as a versatile toolbox for researchers in different fields. We anticipate this review will help understand recent research progress of material-mediated modulation of macrophage immune response, guide rational development of advanced immunomodulatory biomaterials specifically for regulating macrophage response, and propel macrophage-associated applications in the immunoengineering field.
In recent years, researchers have made great efforts to this diverse field of immunomodulatory materials and several reviews have been published and served as valuable resources on related particular topics.[32,47,49–55] In this review, we will first summarize and highlight in detail the state-of-the-art design tactics of macrophage-immunomodulating materials from the perspective of materials science and engineering, with the aim at elucidating the tunability of multiple physical, mechanical, chemical, and biological cues for directing macrophage fate (Figure 1). Thereafter, we will introduce and discuss the representative immunoengineering applications relevant to macrophage-modulating biomaterials, encompassing cancer immunotherapy, infection immunotherapy, tissue regeneration immunotherapy, inflammation resolution, and vaccine immunotherapy. In the end, an outlook on future directions of research in this field is also discussed.
Figure 1.

Material-mediated immunomodulation of macrophage fate for immunoengineering applications.
2. Material-Modulated Macrophage Fate
2.1. Surface Properties
Monocytes/macrophages are one of the first-come immune cells that interact with biomaterials after implantation and they act as a key mediator of host foreign body response to the implants.[48] The biomaterial surface triggers sequential foreign body reaction initially including nonspecific protein adsorption, monocyte adhesion, and differentiation into adhering macrophages. Activated macrophages then secrete cytokines and chemokines for recruitment of leukocytes and other related cells to further reconcile inflammation reaction and wound healing on site of biomaterial. In case of unresolved inflammation within 14 to 28 days, fibrotic scar tissues are formed around the implant through macrophage fusion into larger multinucleated foreign body giant cells. The polarization of macrophages in foreign body response is crucial following biomaterial implantation since M2 phenotype/population can be an indicator of constructive tissue remodeling.[56] Therefore, the surface properties of biomaterials particularly surface charge, chirality, and wettability are able to primarily determine the biological response of macrophages when they are approaching to and/or contacting with the biomaterials. Understanding the interactions of macrophages and their microenvironment is of great significance to enable promising strategies for the design and development of immunomodulatory biomaterials and biointerfaces to direct macrophage fate in a precise and selective manner.
2.1.1. Surface Charge
The cellular membranes of macrophages are overall negatively charged. And biomaterials can tune the negative surface charge of macrophage membranes, which holds potential to affect the protein adsorption and conformation and the biological behavior of macrophages. Therefore, the surface charge of a biomaterial can elicit modulatory effects on the macrophage response. In general, positively charged (cationic) particles are more likely to trigger inflammatory response than negatively charged (anionic) and neutral particles.[57] Cationic polymers such as polylysine, polyethylenimine (PEI), cationic gelatin, and dextran could activate M1 macrophages via toll-like receptor-4 (TLR-4) pathway and specifically induce IL-12 secretion to strongly stimulate Th1 response, thereby enabling the reversal of M2-like TAMs toward antitumoral M1 phenotype both in vitro and in vivo, tested on RAW 264.7 macrophages or TAMs isolated from tumors.[58,59] Positively charged particles are generally able to induce greater cellular internalization by macrophages than particles having negative surface charge. Using liposomes comprising 1,2-dioleolyl-sn-glycero-3-phosphatidylcholine (neutral), 1,2-dioleoyl-3-dimethylammonium propanediol (positively charged) and 1,2-dioleolyl-sn-glycero-3-phosphatidylserine (negatively charged), researchers studied how the liposome surface charge affected their binding and endocytosis by J774 macrophages.[60] The in vitro results showed that J774 macrophages endocytosed both the positively charged and negatively charged liposomes to a larger degree than uncharged liposomes. Furthermore, the positively charged liposomes underwent a greater uptake by macrophages compared with the negatively charged liposomes. The surface charge of nanofibrillated cellulose films could be tailored with carboxymethylation (anionic) and hydroxypropyltrimethyl-ammonium groups (cationic).[61] The carboxymethylated films were able to activate monocytes/macrophages toward the proinflammatory phenotype in vitro using THP-1, while unmodified films could boost mild activation. By contrast, the cationic films were not capable of promoting monocyte/macrophage activation, behaving as an inert biomaterial for this purpose. In addition, all these films were not able to directly facilitate the antiinflammatory response.
Modifying the surface charge of poly(ether)urethane biomaterial through introducing the sulfonate ionic groups onto polymer backbone to give different negative charge, Williams and co-workers investigated the influence of biomaterial surface charge on inflammatory response after implantation into a rat model for 2 days to 12 weeks.[62] On day 2, there were significantly more macrophages surrounding all the materials (stained sections) than other later time points. For all samples wherein macrophages were found, cells with staining positive for TNF-α were also seen. These results reveal that the surface charge is able to affect the early stage acute inflammatory response to the implanted biomaterial. By controlling the chemical synthesis, the surface charge of hyperbranched polymer nanoparticles (NPs) were manipulated differently.[63] The cationic NPs showed a more effective and rapid cellular uptake by RAW 264.7 macrophages and cytotoxicity in vitro when compared with their neutral or anionic counterparts. Meanwhile, all the differently charged NPs were able to accumulate within the macrophage cytoplasm; nevertheless, the cationic NPs could also traffic to and accumulate in the cell nucleus. This is due to the fact that the positively charged surfaces of synthetic particles tend to be more easily covered with the opsonin proteins and thereby more visible to the phagocytic cells. Further in vivo pharmacokinetic study showed that, the neutral NPs had longest retention time (≈6 h) in blood, while the cationic NPs had shortest half-life (≈1.8 h), with the anionic NPs cleared at an intermediate rate (≈2.3 h). The distinct plasma half-lives were suggested to reflect their different opsonization rates in bloodstream, which in turn modulates the rates of recognition and clearance by the phagocytes.[64–66] In addition, Au nanorods modified with polyethylene oxide (PEO)–NH2 revealed the antiinflammatory activity, while PEO–COOH modification resulted in the proinflammatory property. Neutrally charged rods only caused minor inflammation in the human monocyte-derived macrophages in vitro.[67] Collectively, the manipulation of surface charge can provide a potent strategy to modulate the immune response of macrophages to an implanted biomaterial for purposes such as immune evasion from phagocytosis, inflammation regulation, and foreign body reaction.
2.1.2. Surface Chirality
Life is a typical chiral system with the high selectivity for chiral molecules such as d-sugar, L-amino acid, helical DNA, and L-phospholipid, which play an important role in maintenance of biological functions of living cells and organisms.[68] The incorporation of chirality into biomaterial surface design will lead to novel strategies to modulate immune cell responses. In 2007, Sun et al. utilized the enantiomers of N-isobutyryl-L(D)-cysteine (L(D)-NIBC) to modify gold sputtered surfaces as the chiral model system to investigate their interactions with macrophages in vitro using the human promyelocytic leukemia cell line HL-60 (Figure 2A).[69] In a typical study, macrophages showed apparent differences on the L-NIBC and D-NIBC surfaces. The L-NIBC surface resulted in a much higher quantity of adhered macrophages than the D-NIBC surface. Furthermore, the majority of macrophages on the L-NIBC surface exhibited deformation, spreading, extruding pseudopods and gathering together, which are indicative of the activated proinflammatory M1 phenotype; however, those macrophages on the D-NIBC surface showed separate distribution and round-shaped morphology, which belong to the polarized antiinflammatory M2 form. This study revealed that surface chirality of biomaterials could serve as a promising regulator to affect the macrophage behavioral responses and provide insights for the design of immunoregulatory biomaterials for intended use.
Figure 2.
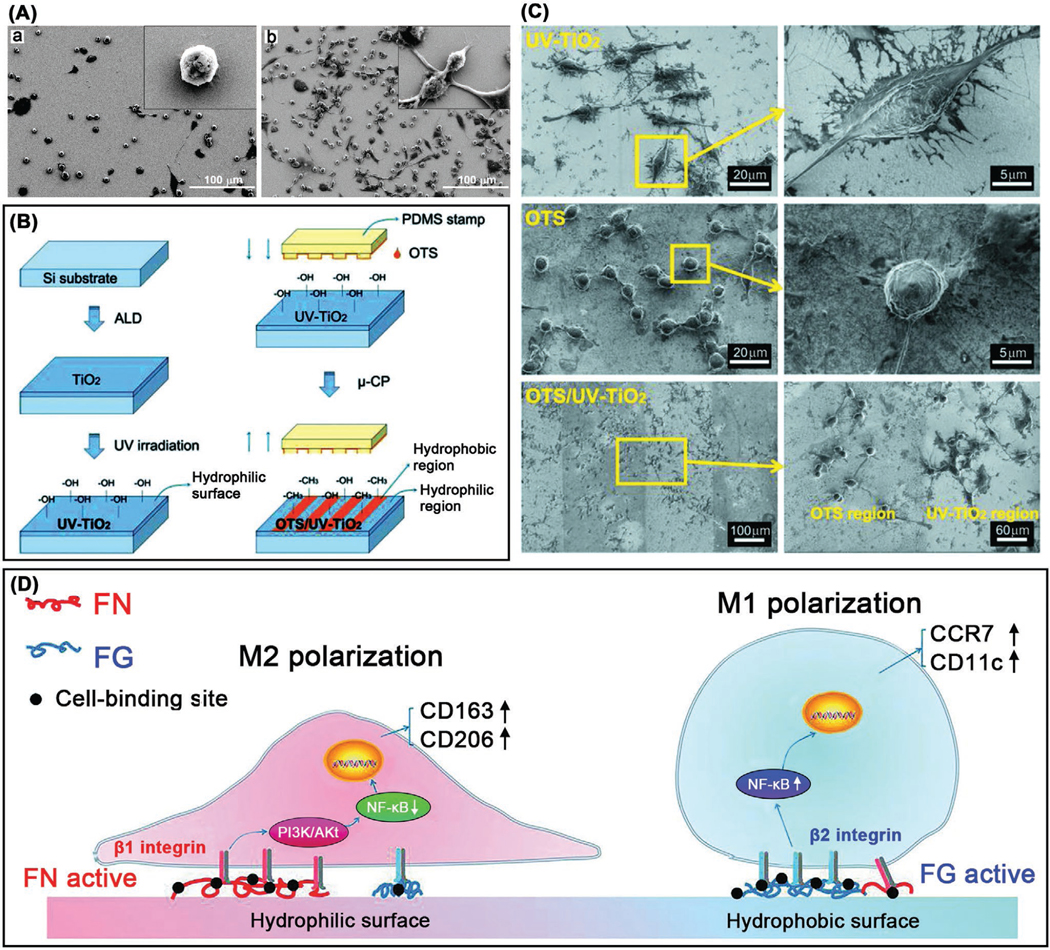
A) Surface chirality affects macrophage adhesion. a) D-NIBC surface; b) L-NIBC surface. Reproduced with permission.[69] Copyright 2007, American Chemical Society. Surface wettability regulates macrophage polarization. B) Fabrication of hydrophilic UV–TiO2 surface (left) and micropatterned hydrophobic/hydrophilic OTS/UV–TiO2 surface (right) through microcontact printing. C) SEM images of RAW 264.7 macrophages cultured on distinct surfaces after 24 h. D) Proposed interactions between surface wettability and macrophage response (adhesion, polarization). B–D) Reproduced with permission.[76]
Kehr and et al. modified the surface chirality of periodic mesoporous organosilicas (PMOs) with the D(L)-mannose (D(L)-MAN) through an enantioselective functionalization method, and then studied how the surface chirality of self-assembled PMO monolayers could have an impact on the adhesion behavior of human primary macrophages in vitro.[70] The number of macrophages adhering on the PMO-D-MAN monolayer was approximately four times larger than on the PMO-L-MAN monolayer or the PMO–NH2 monolayer. This study further demonstrates that upon adhesion, macrophages can not only recognize the surface functionalities, but also distinguish the surface chirality, which is envisioned to be more prominent in the presence of bio-macromolecules such as proteins (serum) and nucleic acids (DNA). In addition, surface chirality also affected the protein adsorption behavior on polymer surface and their interactions,[71] and triggered the surface wettability switching of smart polymers,[72] which could be used to further regulate the macrophage behaviors.
Cyclic azapeptides have revealed the extraordinary binding affinity to the CD36 (cluster of differentiation 36) receptor and capability to alleviate macrophage-driven inflammation via regulating the TLR 2/6 (toll-like receptor 2/6) pathway. To this end, Lubell and co-workers developed a new approach for synthesizing cyclic peptides by A3-macrocyclization to accomplish the controls of their R- and S-configurations and investigate the activity of such CD36 modulators on the RAW 264.7 macrophages in vitro.[73] This study showed the evidence of correlation between dynamic chirality and macrophage-driven inflammation regarding the production of NO, cytokines and chemokines. Immunomodulatory chiral materials are expected to serve as excellent platforms for studying unique chiral phenomena in the immune systems, which is of great significance not only for the development of novel immunomodulatory biomaterials, but also for the understanding of the origin of the marvellous chiral preferences in the nature. One of the future research directions into this field may need to focus on cell biology or molecular biology studies on the intracellular and/or intercellular interaction processes and mechanisms upon stimulation with chiral biomaterials and other external chiral signals.
2.1.3. Surface Wettability
In general, hydrophobic biomaterials can boost monocyte adhesion and induce M1 macrophage activation,[74] and hydrophilic or neutral surfaces tend to inhibit macrophage adhesion and activation, thereby creating an antiinflammatory microenvironment.[75] In a recent study, Zheng and co-workers showed that the hydrophilicity of titanium surface oxide layer was able to regulate the immune response of murine RAW 264.7 macrophages in vitro through polarizing to the antiinflammation and prohealing M2 phenotype (Figure 2B–D).[76] The mechanistic study unveiled that the surface hydrophilicity can govern the adsorption and conformation of fibronectin and fibrinogen and activate the PI3K and NF- κB signaling pathways through the selective expression of integrin β1 or β2, thereby tailoring the macrophage response to create a beneficial immune microenvironment for osteogenesis and bone formation. Surface wetting behavior can undergo reversible switching between hydrophilicity and hydrophobicity in response to diverse external stimuli such as heat,[77] UV,[78] and electrical potential.[79] Such reversible wettability may enable dynamic modulation of macrophage responses to a biomaterial surface. Combining anodic oxidation with hydrogenation, superhydrophilic TiO2 nanotube arrays were created on titanium surface due to the introduction of oxygen vacancies into nanotubes.[80] The hydrogenated surface could lead to the remarkably lower proliferation of RAW 264.7 macrophages in vitro and upregulated secretion of antiinflammatory cytokines (IL-10, bone morphogenetic protein-2 (BMP-2), transforming growth factor-β (TGF-β1)) regardless of LPS stimulation, meanwhile moderate the expression of proinflammatory cytokines (TNF-α, interleukin-6 (IL-6), NO, monocyte chemotactic protein-1 (MCP-1)) triggered by LPS. Furthermore, the superhydrophilic surface was able to upregulate/downregulate the gene expression of M2/M1 surface markers, respectively, thereby implying the potential of using surface wettability control to modulate/direct macrophage immune responses for facilitating inflammation resolution and accelerating tissue repair. Using the hydrophobic (140° water contact angle) and hydrophilic (water entirely adsorbed in 5 s) carbon nanofibers, Webster and co-workers investigated the macrophage response in vitro to wettability upon contact in terms of cytokine expression by and synaptic antigens on the IC-21 macrophages.[81] The hydrophilic carbon nanofibers induced a smallest inflammatory response when compared with their hydrophobic counterparts and titanium, with less secretion of proinflammatory cytokines such as TNF-α and IL-6. Besides, the hydrophobic carbon nanofibers might eventually result in the increased T cell activation compared to the hydrophilic ones.
Tailoring the surface wettability of cellulose microspheres could also influence their phagocytosis by macrophages.[82] Cellulose microspheres with contact angle of 50°–60° were more readily phagocytosed by the macrophages. A recent study by Olivares-Navarrete and co-workers also showed that, how macrophages make response to biomaterial surface properties could lead to the changes in adaptive immune response through regulating the T helper cell population and mesenchymal stem cell recruitment both in vitro and in vivo.[83] It was verified that an increase in the surface wettability and roughness of titanium implants was capable of polarizing adaptive immune response to the prohealing Th2 phenotype, thereby leading to more rapid inflammation resolution and improved MSC recruitment surrounding the implants in the presence of mice primary macrophages. During in vivo studies, the macrophage ablation could decrease the changes in systemic inflammation and populations of T helper cells. Meanwhile, the macrophage ablation was able to cut down the population of stem cells surrounding the implant surface. Taken together, the presence of macrophages and their immune responses to the hydrophilic biomaterials were capable of effectively creating a wound healing microenvironment and modulating/guiding the recruitment of MSCs. Surface wettability control can thus provide an effective approach to modulate the macrophage behavior responses to implanted biomaterials. To this end, surface wettability control can also be combined/coupled with altering other surface properties such as surface roughness and surface topography.
2.2. Surface Topography
2.2.1. Surface Roughness
A key parameter of biomaterials that can direct the macrophage fate is the surface topography, such as surface roughness and ordered/disordered, aligned/unaligned, patterned/unpatterned surface microstructure. For pure titanium with different surface treatments (polished, machined, grit-blasted), the surface adhesion of J774A.1 macrophages increased with time in vitro while their spreading also increased with surface roughness; meanwhile, the adherent macrophages exhibited evident BMP-2 expression, thus having the potential to favor the bone formation on a biomaterial surface.[84] Accompanying the activation, the surface roughness increase also lead to the dramatic secretion of proinflammatory cytokines (interleukin-1β (IL-1β), IL-6, TNF-α) and chemokines (MCP-1, macrophage inflammatory protein-1α (MIP-1α)) from RAW 264.7 macrophages in vitro in a time-dependent way.[85] In addition, macrophage polarization could be jointly modulated by biomaterial surface roughness and hydrophilicity via Wnt signaling regulation in vitro using mice primary bone marrow-derived macrophages (BMDM) and in vivo on a mouse model.[86] It was demonstrated that the loss of the macrophage-derived Wnts could also impair the recruitment of MSCs and T cells toward the titanium implants in vivo. Increasing the surface roughness of titanium material by sandblasting and acid etching treatment, a study showed that RAW 264.7 macrophages cultured on such rough surface in vitro could be activated to the M2-like phenotype, thereby holding the potential to boost wound repair and bone regeneration.[87] Nevertheless, another in vitro study on titanium material with surface roughness from 100 to 400 nm revealed that, the RAW 264.7 macrophages tended to polarize to the M1 phenotype with increasing the surface roughness.[88] With regard to the mineralized collagen material (with different roughness from 0.92 to 12 μm), a rougher surface could lead to the polarization of THP-1-derived macrophages toward M1 phenotype in vitro with high secretion levels of inflammatory factors (TNF-α, IL-6), while a smoother surface was able to promote the M2-phenotype polarization.[89] Corporately, when considering the influence of surface roughness on macrophage immune responses, in addition to the wide range of surface roughness to be designed, other parameters such as surface chemistry, charge and wettability may also need to be analyzed together to obtain a comprehensive understanding for suitable biomaterial surface design.
2.2.2. 2D Topography
Macrophages have an attachment preference to rough substrate surfaces than smooth ones.[74] A regularly microstructured surface of polyvinylidene fluoride could significantly lead to the activation status of human primary macrophages involving both M1 and M2 phenotypes.[90] Leong and co-workers[91] investigated the micro/nanotopography induced behavior changes in RAW 264.7 macrophages (used for in vitro study) through parallel gratings of line width 250 nm to 2 μm imprinted on polycaprolactone (PCL), polylactide (PLA) and polydimethylsiloxane (PDMS) (Figure 3). In comparison with flat control, they observed maximal adhesion and elongation of macrophages on 500 nm grating at 48 h, showing apparently topography-sensitive secretion of TNF-α and vascular endothelial growth factor (VEGF) in a way that greater size of gratings could decrease the secretion levels. In vivo study on a rat model showed that, at day 21, the density of macrophage adhesion and degree of cell fusion on the 2 μm gratings were decreased compared with the planar controls. As a conclusion, surface topographical cues, independent of surface chemical cues, can influence the macrophage behaviors such as morphology change and cytokine secretion during foreign body reaction, though this modulatory role needs further study in longer time points. When considering electrospun polyurethane membranes, nanofiber surface only triggered minimal macrophage inflammatory response and mild foreign body reaction compared with microfiber surface,[92] holding potential application for the development of immunoisolation devices of cell transplantation. To understand how the surface topography could change macrophage morphology and polarization status, Liu and co-workers created titanium surfaces with micro and nanopatterned grooves via a deep etch method.[93] By culturing BMDM in vitro on distinct groove widths, the results showed that micro and nanopatterned grooves were able to affect the macrophage elongation, peaking on the substrates of 400–500 nm wide grooves. Such surface grooves had no influence on the inflammatory activation but promoted the polarization of antiinflammatory, prohealing macrophage phenotype. Macrophages could secrete markedly higher levels of antiinflammatory IL-10 cytokine on the intermediate groove widths, thus highlighting the possibility of exploiting surface topography to modulate/guide macrophage functions and manage biomaterial-mediated wound healing process and tissue repair/regeneration.
Figure 3.
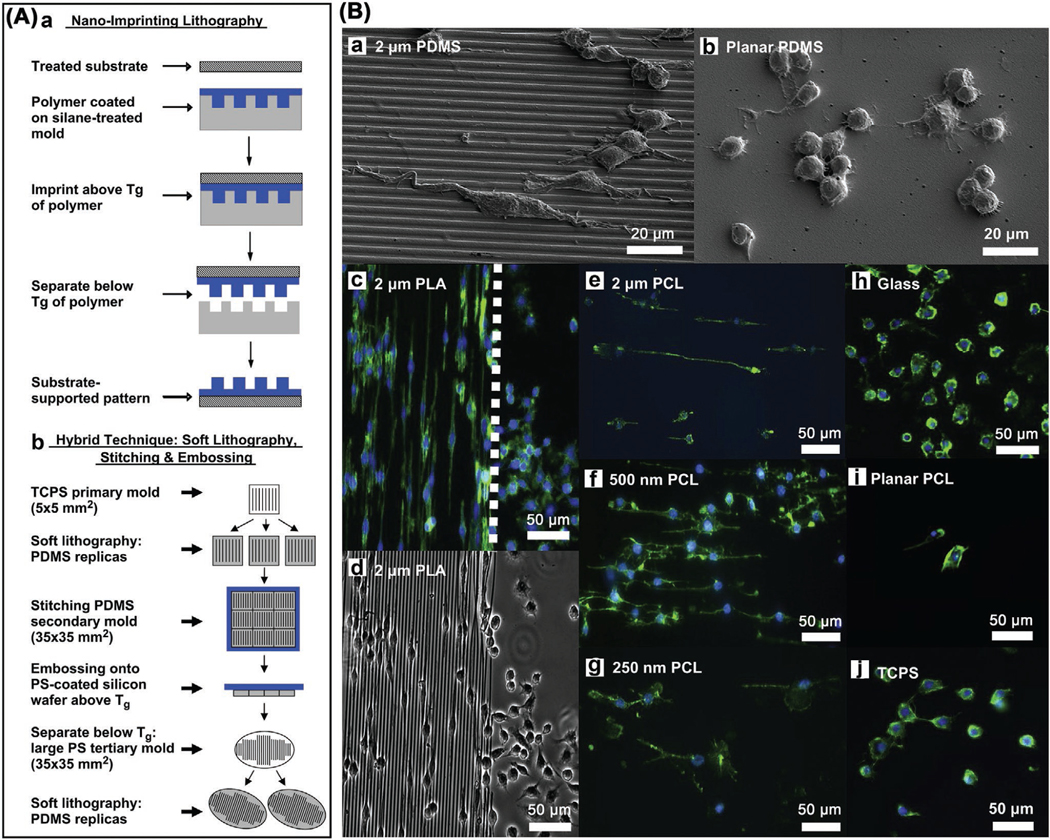
A) Fabrication of topographical substrate. a) Nanoimprint lithography. b) Hybrid technique. B) RAW 264.7 macrophage morphology on topographical gratings at 48 h. a) Elongation in the direction of 2 μm PDMS gratings. b) Natural round shape on planar PDMS. c) Elongation in the direction of 2 μm PLA gratings. d) Macrophage morphology on the border of 2 μm PLA gratings and planar surface. e–g) Macrophage elongation on PCL gratings. h–j) Native round morphology on planar surfaces. A,B) Reproduced with permission.[91]
Chang and co-workers designed well-organized hierarchical micro/nanostructured surfaces on hydroxyapatite bioceramics via combining photolithography and hydrothermal treatment to regulate the macrophage behavior for osteogenesis and angiogenesis, possessing distinct microcircular patterns (4, 12, 36 μm) and nanoscale topographies (nanoneedle, nanosheet, nanorod).[94] It was demonstrated that the designed hierarchical micro/nanostructures with suitable pattern sizes were able to either promote or mitigate RAW 264.7 macrophage polarization in vitro, thereby influencing the outcomes of osteogenesis and angiogenesis through macrophage immunomodulation. Similarly, engineered zinc substrates with microscale surface topography could lead to the reduced inflammatory polarization in vitro of THP-1-derived macrophages for improved biocompatibility and tissue integration.[95] By incorporating gold nanorods into the shape memory PCL film, a dynamic surface topography was acquired with the capability of topography transformation from the flat to the microgrooved through near-infrared irradiation, thus triggering the macrophage elongation and phenotype change in vitro (BMDMs) and in vivo (a rat model), with upregulated arginase-1 and IL-10 expressions.[96] In addition, the PCL fibrous topography could also facilitate the host MSC recruitment by boosting the macrophage phenotype shift from M1 to M2 in vivo.[97] Adopting a high throughput screening method, Alexander and co-workers investigated the relationship between the surface topography of biomaterials and the adhesion and phenotype of human monocyte-derived macrophages with a diversified library of 2176 micropatterns created by an algorithm both in vitro and in vivo (mice model).[98] The micropillars of 5–10 μm diameter could play a predominant role in driving the macrophage adhesion and the combination of micropillar size with density was pivotal to modulate the macrophage phenotype transition from proinflammatory to antiinflammatory status.
2.2.3. 3D Geometry
2D substrate materials are simplified models to elucidate the behavior responses of macrophages toward specific stimulus particularly the surface topography. Nevertheless, cells tend to behave quite differently in a 3D microenvironment and 3D geometrical models can better recapitulate the hierarchical, hybrid and complicated in vivo microenvironments.[99] For example, the expanded electrospun random PCL nanofiber scaffolds, having significantly larger porosity than common 2D nanofiber membranes, could promote evident macrophage infiltration of higher M2/M1 ratios in a subcutaneous rat model within four weeks, together with the formation of new blood vessels inside, while the unexpanded ones only showed surface macrophage adhesion.[100] The interlayer distance, layer thickness and scaffold porosity played major roles in determining macrophage infiltration, neovascularization and host response for in situ tissue regeneration. Besides, the fiber diameter of electrospun poly-L-lactide (PLLA) scaffolds, rather than fiber alignment, affected the in vitro RAW 264.7 macrophage activation by showing minimized inflammation reactions to PLLA nanofiber (≈600 nm) scaffolds compared to the microfiber (≈1.5 μm) ones as well as the 2D flat films that had greater number of FBGCs on surface than 3D scaffolds.[101] By a hydrogel coating strategy to modify the surface chemistry of 2D flat poly(D,L-lactide-co-glycolide) (PLGA) substrate and electrospun 3D nanofiber scaffolds,[102] Bartneck et al. showed that the biomaterial surface topography had much more powerful effects compared with changing surface chemistry on modulating the human primary macrophage immune response in vitro. That is, 2D flat substrate caused the release of a large amount of proinflammatory cytokines (TNF-α, IL-1β), whereas 3D nanofiber scaffolds could dramatically lead to the release of proangiogenesis chemokines (interleukin-8 (IL-8), chemokine ligand 4 (CCL4)/macrophage inflammatory protein-1β (MIP-1β)) and molecules, and meanwhile strongly reduce the proinflammatory cytokine release. In addition, the 3D topography cues could also modulate the crosstalk between macrophages and MSCs.[103] Compared with 2D topographical cues, co-culturing human bone marrow-derived MSCs with THP-1-derived macrophages in vitro on 3D substrate material could markedly reduce the production of IL-6 and MCP-1 related to inflammation and chemotaxis, thereby highlighting the significance of 3D topographical cues in factor-directed communication between macrophages and MSCs. The 3D geometry of 3D-printed chitosan scaffolds with wider angles and larger pores could induce the higher production of proinflammatory cytokines (TNF-α, IL-12/23) from human monocytes/macrophages in vitro.[104] Additionally, performing the plasma electrolytic oxidation treatment on the 3D-printed porous titanium implants could improve the prorepair phenotype polarization of human primary macrophages in vitro from the strong proinflammatory response to the nontreated 3D-printed implants.[105] From the standpoint of extracellular matrix mimetics, 3D topographies of biomaterials can afford better artificial/synthetic extracellular microenvironmental cues for directing the fate and functions of macrophages for clinical applications.
2.3. Material Mechanics
2.3.1. Macrophage Mechanobiology
So far, little knowledge is obtained regarding the macrophage mechanobiology relative to the well-established fibroblast/stem cell mechanobiology.[106,107] Attentions have been increasingly paid to establish the correlation between the mechanical cues of macrophage microenvironment and the macrophage activation and polarization. These efforts are important to deepen the understanding of how macrophages respond to mechanical stimuli, how macrophages correlate with disease progression, and how biomaterials can be designed to direct macrophage fate for numerous applications such as tissue regeneration, infection resolution and cancer treatment.
Podosomes are organelles of high dynamics, constitutively generated in monocytic lineage, including macrophages and osteoclasts.[108] Macrophage podosomes emerge in a typical dot-like shape with tripartite protein substructures of core (such as F-actin, cortactin, and gelsolin), ring (such as vinculin and talin) and cap (such as supervillin and formin), having 0.5–1 μm diameter, 0.2–0.4 μm height, and ≈44 kPa stiffness independent of ECM nature.[109,110] These podosomes can function as i) adhesion structures through integrins or ECM receptors, ii) mechanosensors and mechanotransducers through mechanical signal conversion into chemical cues, and iii) ECM degradation through proteases. Revealed by protrusion force microscopy, human macrophage podosomes could generate an oscillatory protrusion force increasing with substrate stiffness and requiring combination of actin polymerization and actomyosin contraction, which is characteristics of podosome mechanosensing activity.[111] Figure 4A illustrates the macrophage mechanotransduction pathway. For general information of the molecular mechanism of macrophage mechanotransduction, we refer the readers to the review.[112]
Figure 4.
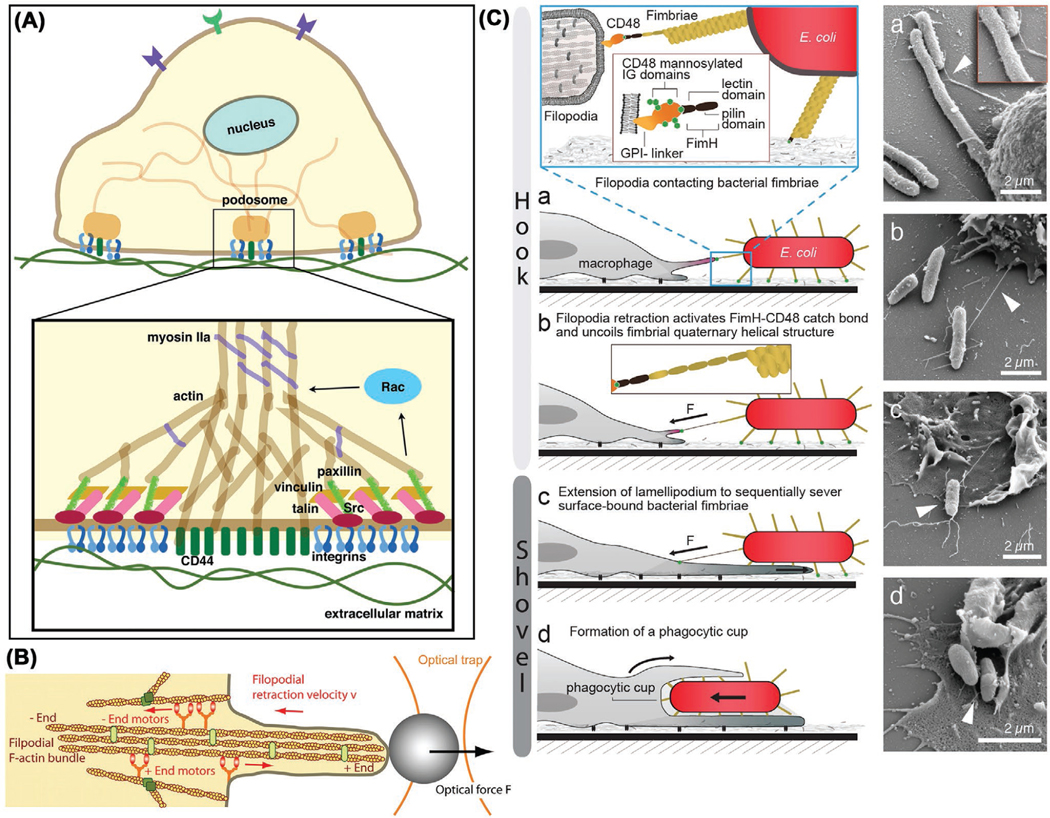
A) Macrophage mechanotransduction pathway. Podosomes mediate macrophage adhesion and connect actin to ECM. Reproduced with permission.[112] Copyright 2014, Springer Basel. B) Filopodial retraction model. Reproduced with permission.[113] Copyright 2007, The National Academy of Sciences of the USA. C) Mechanomodulation of macrophage phagocytosis from prey adhesion to phagocytic cup formation. Reproduced under the terms of the CC-BY 3.0 license.[114]
Macrophages can recognize prey location through chemotaxis navigation and generate stable physical contact for phagocytosis, with the need for producing mechanical forces. Revealed by the optical tweezers, macrophage filopodia exert pico to nanonewton retraction force to pull bound microparticles (Figure 4B).[113] Vogel and co-workers proposed a “Hookand-Shovel” mechanism for macrophage phagocytosis by lifting off and picking up surface-bound bacteria (Figure 4C).[114] Following lift-off, bacteria were engulfed in phagocytic cup, during which the force-activated capture bonds enabled long-term filopodium–fimbrium interplay. The access to prey tip is needed for the phagocytic cup formation and phagocytosis by macrophages.[115]
2.3.2. Substrate Stiffness
Different ECM components, cells, and human tissues have a broad range of mechanical moduli.[116] The stiffness of biomaterials is an important parameter to affect cell fate and function when they interplay with each other. Such mechanical stimulus may provide an efficient way to manipulate the polarization and functions of macrophages.[117,118] Culturing human monocyte-derived macrophages in vitro on fibronectin-coated polyacrylamide (PAAm) hydrogels of varied stiffness, larger macrophage area, and faster proliferation rate were observed on stiffer PAAm (280 kPa–70 GPa) than softer PAAm (1–5 kPa), together with faster migration speed (12.0 μm h−1 for 280 kPa, 5.0 μm h−1 for 3 kPa) and F-actin organization of stress fibers.[119] Stiffer arginine–glycine–aspartic acid (RGD)-poly(ethylene glycol) (PEG) or PEGDA hydrogels induced better RAW 264.7 macrophage adhesion and spreading (flattened rather than rounded on softer) in vitro and in vivo, thereby elevating M1 phenotype and causing more severe foreign body response.[120,121] The migration capacity of macrophages is prerequisite for implementing their tasks and functions, and the mechanosensitive podosomes play a critical role as mechanosensors in macrophage migration and invasion.[109] Besides, macrophages utilize actin-based phagocytosis for clearing intruders and they preferentially phagocytosed stiff PAAm microparticles; for soft particles, the phagocytosis by macrophages could be stimulated by microinjecting the constitutively active Rac1 (small GTP-binding protein) and lysophosphatidic acid (activator of small GTP-binding proteins), implying the Rac1-dependent mechanosensing mechanism for macrophage phagocytosis.[122] To understand the mechanical mechanism for macrophage migration/motility, Hammer and co-workers generated the traction maps of migrating human primary macrophages via observing the in vitro macrophage migration on the compliant PAAm hydrogels.[123] The force produced by the migrating macrophages was concentrated on the cellular leading edge, with a magnitude depending on the underlying substrate stiffness. It was found that the Rac activation by GEF Vav1 is critical for the force generation of macrophages, which also involved the necessary signaling via RhoA kinase ROCK, myosin II and PI3K.
Using THP-1 cultured on the 1%, 4%, and 10% agarose hydrogels as soft substrate or the plastic plate as stiff substrate in vitro, researchers showed that decreasing the stiffness of substrate materials could facilitate the activation of M2-like macrophages, and meanwhile improve the expression of peroxisome proliferator-activated receptor γ (PPARγ), demonstrating that the substrate stiffness serves as a key factor in the balance modulation of proinflammatory M1 and antiinflammatory M2 phenotypes.[124] Altering the collagen scaffold stiffness with different physical or chemical crosslinking methods, O’Brien and co-workers reported the dependence of THP-1-derived macrophage polarization in vitro on both matrix stiffness and crosslinking agents, indicating the coupling effect of scaffold physical and chemical properties on macrophage behavior.[125] Recently, the researchers investigated how the THP-1-derived macrophages adapted their polarization, functions, and migration modes when cultured in vitro on the collagen-coated PAAm hydrogels with different substrate stiffness.[126] The results showed that the stiff PAAm hydrogels (323 kPa) could promote the polarization of proinflammatory macrophage phenotype with an impaired phagocytosis, whereas the soft (11 kPa) or medium-stiffness (88 kPa) hydrogels was able to boost the antiinflammatory and highly phagocytic macrophage phenotype. Moreover, the substrate stiffness could also determine the macrophage migration mode. That is, on the soft or medium-stiffness hydrogels, macrophages exhibited the RhoA kinase ROCK-dependent and podosome-independent rapid amoeboid migration mode, while on the stiff hydrogels, macrophages adopted the ROCK-independent and podosome-dependent slow mesenchymal migration mode. Together, these studies imply that the substrate stiffness of biomaterials is able to guide the macrophage behaviors and functions, independent of the applied biochemical cues to them.
2.3.3. Spatial Confinement
Spatial confinement can regulate macrophage activation and response. As revealed in a recent study by Vogel and co-workers,[127] spatial confinement could block macrophage spreading through micropatterning, microporous substrate, and cell crowding, thereby restraining late LPS-activated gene transcription and epigenetics (Figure 5). The confinement could decrease actin polymerization and downregulate M1 activation and inflammatory response, therefore reducing secretion of proinflammatory cytokines and phagocytizing capacity of macrophages. In vitro polarized macrophages are distinct in cellular morphology.[128] Upon polarization, macrophages have significant cell shape change: M2 phenotype shows an elongated cell shape relative to round M1 phenotype. M2 activation can be promoted by elongation whereas M1 activation is independent of elongation. The macrophage elongation itself could result in M2 marker expression and cut down inflammatory cytokine secretion in the absence of exogenetic cytokines.[129] Elongation could also potentiate the effect of M2-activating cytokines (IL-4, IL-13) and protect macrophages from M1-activating stimuli (LPS, IFN-γ). Jointly, this in vitro study confirmed that altering cell shape through ECM architecture can modulate the phenotype polarization of macrophages (mice BMDMs used here). In addition, the porosity and pore size of 3D scaffolds also have important roles during the interactions between scaffolds and macrophages. An increase in pore size of electrospun 3D polydioxanone scaffolds with irregular pores from 2 to 30 μm could boost M2 marker Arginase 1 (Arg1) expression while inhibit M1 marker inducible nitric oxide synthase (iNOS) expression of mouse BMDMs in vitro, showing higher secretion of angiogenic cytokines VEGF, TGF-β1, and basic fibroblast growth factor (bFGF).[130] For 3D printed PCL fiber scaffolds with box-shaped pores, the pore size decrease from 100 to 40 μm could facilitate the elongation of primary human macrophages and their polarization toward M2 type in vitro.[131] In a study by Ratner and co-workers,[132] cardiac implantation of the acellular hydrogel scaffolds of 30–40 μm pore diameters achieved maximal angiogenesis and minimal fibrotic response, agreeing with the shift/polarization of macrophages toward the prohealing M2 phenotype. Besides, for a retrievable implant made of silicone reservoir and porous polymer membrane from the body after implantation,[133] the membrane of 1 μm pore diameter could permit macrophage migration inside with no loss of encapsulated cells, while the membrane of <0.8 μm pore size could prevent the immunocyte infiltration. Such synthetic polymeric coatings can be optimized to prevent fibrosis and protect transplanted therapeutic cells for long-term survival and function, thus minimizing the chance of graft failure. Besides, creating cone-shaped pores on the surfaces of mesoporous silica rods could regulate the RAW 264.7 macrophage immune response and mitigate the proinflammatory reaction in vitro, thus generating a beneficial immune microenvironment for boosting osteogenesis and new bone regeneration, as evidenced by the improved in vivo bone formation.[134]
Figure 5.
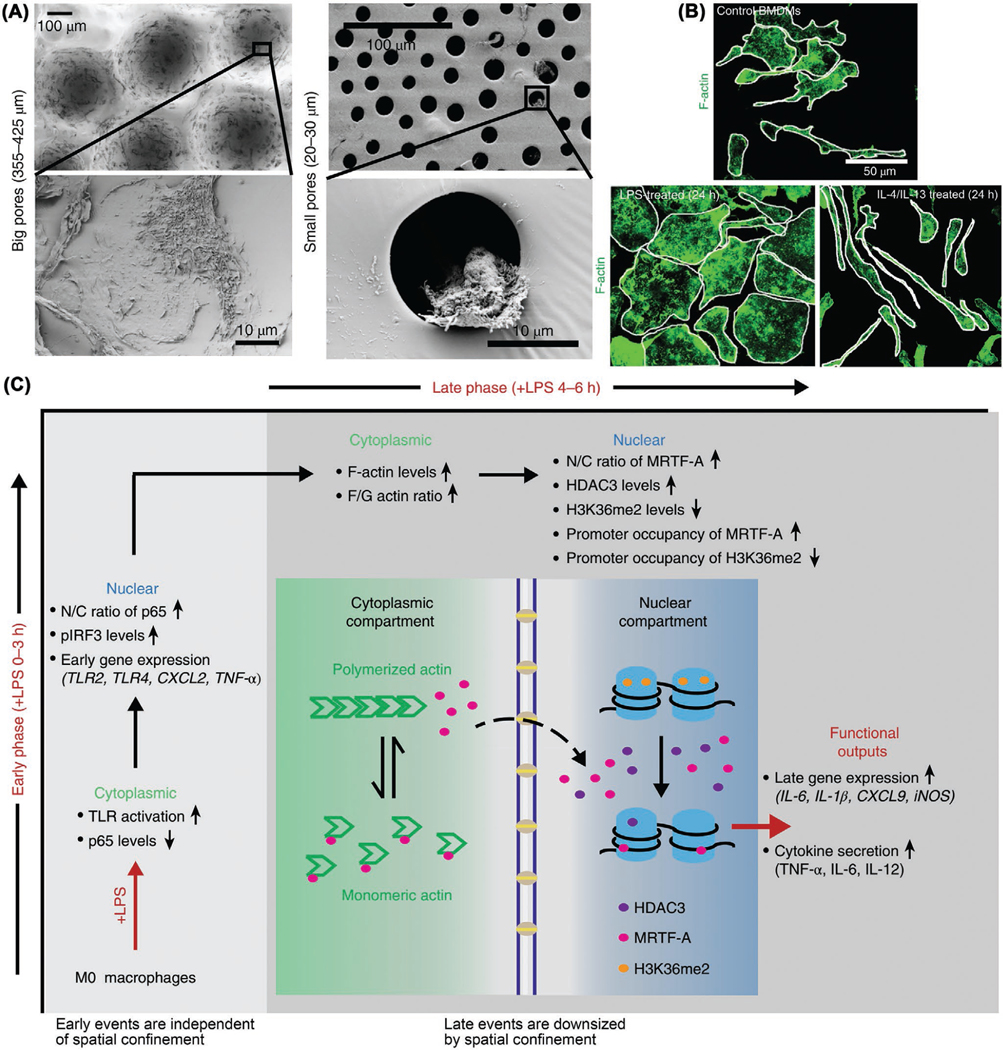
Spatial confinement downregulates macrophage M1 activation and inflammatory response. A) SEM images of cultured bone marrow-derived macrophages (BMDMs) in microwells of big pores (left) and small pores (right). B) F-actin staining (green) images of control (M0, homeostatic status), LPS treated (M1, cell size augment) and IL-4/IL-13 treated (M2, cell shape alteration) BMDMs. C) Schematic of how spatial confinement of macrophages downsizes their late rather than early M1 activation and proinflammatory response. Reproduced with permission.[127]
2.3.4. Phagocytosis Physics
Immunocytes can process particles in size-dependent way: particles of size <0.5 μm are internalized by macropinocytosis; phagocytosis is a process by which phagocytes use plasma membrane to engulf or ingest particulate materials of size >0.5 μm and then digest in phagosome.[57,135,136] The particulate materials can be microorganisms, cellular debris, tumor cells, small biominerals (such as kidney stones) or synthetic particles (such as nanocarriers). Phagocytes consist of the professional phagocytes encompassing diverse types of leukocytes and the nonprofessional phagocytes including fibroblasts and epithelial cells.[137] Macrophages, a key subset of the mononuclear phagocyte system (MPS),[14,38] are able to phagocytose particles as large as 5 μm, but facing larger ones macrophages tend to coalesce to generate multinucleated foreign body giant cells.[52,138] Polystyrene particles with 2–3 μm longest dimension revealed highest recognition and attachment by J774 mouse macrophages in vitro,[139] which correlates with the size range of commonly seen rod-shaped bacteria in nature. An in vitro study by Chan and co-workers showed that the particle size and surface PEG density could determine the serum protein adsorption onto Au nanoparticles and the following phagocytosis by J774A.1 macrophages.[140] In general, large particles (>1 μm) could induce Th1 response, while smaller ones (<500 nm) could trigger Th2 response.[57,141] Nevertheless, it does not always follow this rule and particle size can couple with other parameters to determine Th response and influence Th1/Th2 balance.
Particle shape is also an important physical parameter for designing drug delivery systems and directing cell response to biomaterials.[142,143] Long fibers (>20 μm) such as asbestos and carbon nanotubes cannot be engulfed by macrophages, hence resulting in frustrated phagocytosis.[144] In a study by Mitragotri and co-workers,[145] multilayer polymeric discs (diameter 4–7 μm, thickness <1 μm) were designed with a layer of hyaluronic acid on each disk face to allow tight adhesion to but avoid disk phagocytosis by J774 mouse macrophages in vitro, therefore capable of serving as cellular backpacks for therapeutic drug loading and macrophage-mediated targeted delivery to diseased nidus. Polymeric micelle assemblies (filomicelles) were prepared to research the trafficking and transport behaviors of flexible filaments in comparison with spherical particles of similar chemical identity in vitro and in vivo. The filaments could persist up to one week in circulation post intravenous injection in rat and mouse models, which was around ten times longer than spheres. Under the condition of fluid flow in vitro, THP-1-derived macrophages could more easily phagocytose spherical particles and short filaments than longer filomicelles (≥3 μm), due to the flow-induced extension.[146] Phagocytosis is the main constituent of innate immunity by which macrophages can internalize targets in actin-dependent way. Researchers have investigated the phagocytosis of polystyrene particles with varying sizes and shapes by rat alveolar macrophages in vitro (Figure 6A).[147] Particle shape, rather than particle size, played a predominant role in the phagocytosis. That is, the local particle shape at the initial contact point dictated if macrophages could initiate the phagocytosis or merely spread on the particles without internalization, through determining the complicacy and suitability of actin structure for the phagocytosis initiation and membrane–particle interaction. In the cases that particle volume was larger than macrophage volume, the particle size could mainly affect the implementation of phagocytosis.
Figure 6.
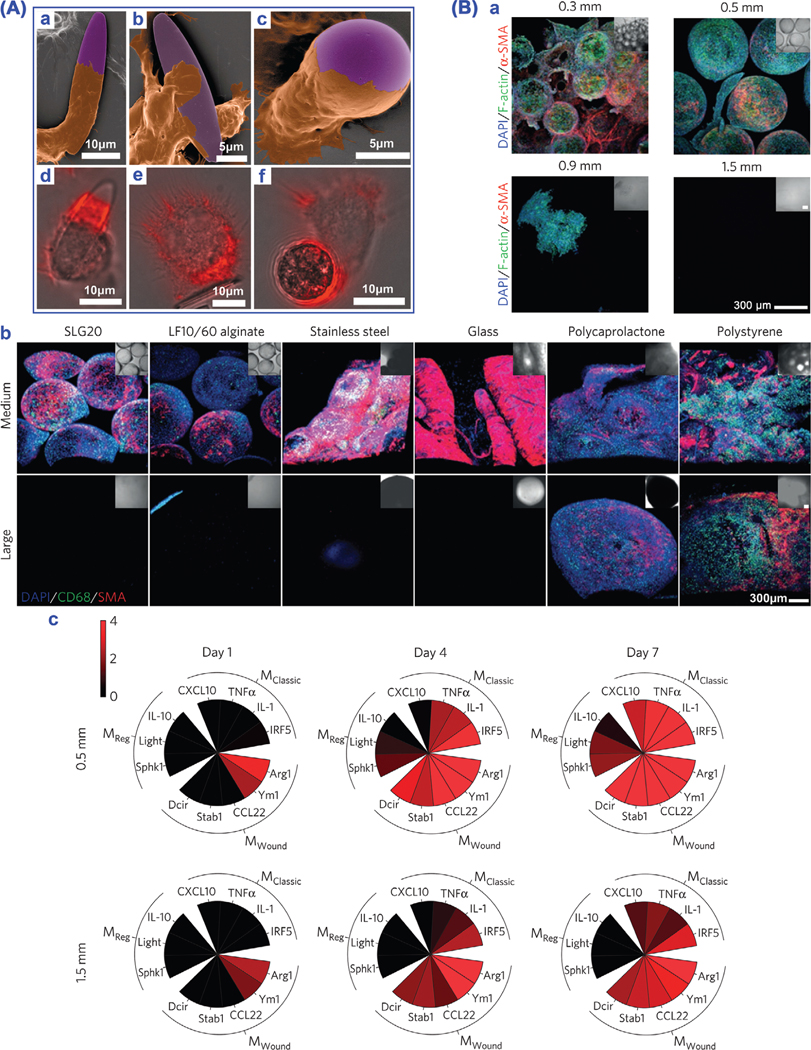
Particle geometry affects macrophage phagocytosis physics. A) Particle shape and size tune phagocytosis by macrophages. a–c) SEM images of macrophages (brown) interacting with particles (purple). d–f) Overlay of fluorescence and bright field images with actin staining (red). Reproduced with permission.[147] Copyright 2006, The National Academy of Sciences of the USA. B) Material size and shape tailor foreign body response. a) Decreased fibrosis on surface with increasing size of alginate spheres. b) Decreased foreign body reaction with increasing sphere diameter of diverse materials. Note: cell nuclei (blue, DAPI), macrophages (green, CD68) and activated myofibroblasts associated with fibrosis (red, α-SMA). c) Analysis of marker expression of macrophage phenotypes. Reproduced with permission.[148]
Anderson and co-workers investigated the role of spherical material geometry on the in vivo biocompatibility. They demonstrated that in the animal models of rodents and nonhuman primates, implanted material spheres (diameter ≥1.5 mm) ranging from hydrogel, plastic, ceramic to metal, could significantly abrogate fibrosis and foreign body response in comparison with their smaller spherical counterparts (Figure 6B).[148] The findings imply that simply tuning the sphere size of biomedical materials/devices can remarkably improve their in vivo biocompatibility. In addition to particle size and shape, particle stiffness is another mechanical parameter that should be considered facing phagocytosis. The stiffness of target can significantly influence the efficacy of phagocytosis. For instance, upon exposure to antibody-coated PAAm beads (1–6 μm) with varied stiffness but the same chemical identity, mice BMDMs strongly preferred to phagocytose rigid opsonized beads sixfold over soft ones in vitro.[122] Similarly, soft (10 kPa) PEGDA hydrogel NPs (200 nm) could dramatically decrease the phagocytosis by J774 macrophages in vitro when compared to rigid (3 MPa) ones, thereby potentially offering an approach to upgrade the in vivo biological fate of NPs with improved blood circulation, decreased immune uptake, and enhanced targeting.[149] A conjunct effect of target size and stiffness on the RAW 264.7 macrophage phagocytosis in vitro was also observed using discoidal polymer NPs of varying shape, size and stiffness.[150] Rigid discocytes showed more efficient uptake by THP-1-derived macrophages than flexible ones through Myosin-II hyperactivation. The shape of rigid erythrocytes could also regulate the “don’t eat me” signal of CD47 and engulfment of macrophages, that is, rigid stomatocytes could signal self-better than rigid discocytes, consistent with the in vivo clearance study on the mice model, thus indicating the shape effect on the CD47 suppression of macrophage phagocytosis.[151] The phagocytosis physics, based on the regulation effects and interactions of particle size, shape, and stiffness (3S), reveal that a synergetic enhancement effect on the phagocytosis efficacy of macrophages can be achieved through tuning the 3S physical parameters. The capacity that macrophages recognize particle 3S has important physiological significance considering the fact that many preys/targets such as pathogens, cellular debris, cancer cells and foreign particles come in diversified 3S parameters. For example, as antigen-presenting cells together with DCs, macrophages can phagocytose, process, and present antigens to T cells, thereby aiding and/or amplifying the adaptive immunity for vaccine immunotherapy against cancer, virus, and bacterial infections.
2.4. Material Composition
Once implanted into human body, bioactive biomaterials, such as silicate/phosphate-based bioceramics, bioglasses, bone cements, and ionically crosslinked hydrogels, in the forms of scaffolds, coatings or films, can undergo natural degradation process in a time-dependent manner, mediated by corrosion, dissolution, hydrolysis, enzymolysis, and phagocytosis.[152] Following the material degradation, bioactive ions can be released to modulate local immune microenvironment. Meanwhile, other strategies to modulate macrophage immune responses to biomaterials include surface chemical modification and incorporating bioactive molecules, which involve inorganic ions, functional groups, cytokines, etc.[32,52] Besides, some glycosaminoglycans including heparin, hyaluronan, and their derivatives also have wide immunomodulatory activities.[153,154] Recently, Elisseeff and co-workers used the single-cell RNA sequencing analysis technique to study the macrophage responses to the biologic UBM and synthetic PCL biomaterials after implantation.[155] The results showed that UBM was capable of boosting the tissue repair via creating a tissue microenvironment featured with the Th2/IL-4 immune profile, while PCL triggered the standard foreign body reaction featured by the Th17/IL-17 and fibrosis. From the UBM implantation, distinct macrophage phenotypes were illustrated and responsible for the chemoattraction, phagocytosis, and antigen presentation. From the PCL tissue microenvironment, a CD9hi+IL-36γ+ macrophage phenotype was identified, expressing the Th17-associated molecules. Taken together, these different macrophage phenotypes can provide potential targets for the therapeutic immunomodulation with elaborately designed biomaterials. The different bioactive ions/molecules incorporated into biomaterial surfaces or matrices are able to produce a wide range of regulating effects on the immune system.
Although many efforts have been devoted to investigating the interactions between biomaterial components and macrophage immune responses and elucidating the possible mechanisms behind, much more work need to be done further to clarify the exact molecular mechanisms. To this end, some issues can be taken into consideration with regard to the material composition. First, bioceramics and bioglasses in general contain multiple bioactive ions. When immersed in cell culture medium or implanted into the body, multiple ion species are released from the material matrices,[156] making it difficult to study and clarify the biological/immunomodulatory effects of a single ion species. Moreover, when releasing ions from surface or inside, bioceramic/bioglass materials may remarkably change the local pH microenvironment,[157–159] which can meanwhile produce important impacts on macrophage behaviors.[160] In addition, to fully illustrate the mechanisms of how specific biomaterial components manipulate macrophage fate, systemic genomics and proteomics researches are quite essential.[161] Similarly, regarding the decellurized biomaterials derived from various natural tissues for regulating macrophage immune responses, one concern for biomaterials scientists and biomedical engineers lies in the complexity and difficulty in clarifying the multiple components of such materials and thus further understanding what are the exact compositions responsible for the observed therapy effects,[49,162,163] highlighting the necessity and importance to make it clear in the future research. Table 2 summarizes the immunoregulation effects of representative material compositions on the monocyte/macrophage behaviors.
Table 2.
Regulatory effects of typical material compositions on monocyte/macrophage immune response.
| Material compositions | Monocytes/macrophages | Immunomodulatory effects | Ref. | |
|---|---|---|---|---|
|
| ||||
| Inorganic ions | Ca | - | Wnt5a/Ca2+ signaling cascade can enhance inflammation | [164] |
| Co | RAW 264.7 | Co-doped TiO2 coating can boost M1 polarization, create inflammatory microenvironment, and promote bacteria phagocytosis and infection eradication | [165] | |
| Cu | RAW 264.7 | Cu-loaded SPEEK can facilitate M1 polarization, bacteria phagocytosis, and infection resolution | [166] | |
| RAW 264.7 | Macrophages phagocytose Cu-doped MSNs and initiate proper inflammation to boost osteogenesis but thwart osteoclastogenesis | [167] | ||
| Fe | Human primary macrophages | Fe overloading will cause M1 activation and inflammatory niche and impair wound healing | [168] | |
| Li | Mouse BMDMs | Li from Li2Ca2Si2O7 bioceramic can suppress in vitro macrophage osteoclastogenesis and in vivo osteolysis | [169] | |
| Mg | Human monocytes | Mg from MgSO4 (2.5 × 10−3 M) decreased maternal IL-6 and TNF-α secretion, showing its broad antiinflammatory function | [170] | |
| RAW 264.7 | Mg from MgSiO3 coating can downregulate inflammatory cytokines and suppress macrophage osteoclastogenesis to boost osseointegration | [171] | ||
| RAW 264.7 | Mg from MgO NPs can boost M2 macrophage switch and inhibit Ti particle-triggered in vivo osteolysis and osteoclastogenesis | [172] | ||
| Se | RAW 264.7 | Se NP-loaded TiO2 nanotubes can show antiinflammatory and antibacterial activities | [173] | |
| Si | RAW 264.7 | Si-doped TiO2 nanotubes can favor prohealing M2 polarization to downregulate inflammation | [174] | |
| Sr | RAW 264.7 | Sr-substituted bioglasses can restrain osteoclast TRAP and resorption activities | [175] | |
| Human primary monocytes | Sr-substituted calcium phosphate can reduce inflammatory cytokine (TNF-α, IL-6) and chemokine (IL-8) production from LPS-stimulated monocytes | [176] | ||
| THP-1 | Sr-loaded titanate coating can inhibit THP-1 osteoclastogenesis and promote osseointegration in an osteoporotic rat model | [177] | ||
| Zn | RAW 264.7 | Zn-loaded SPEEK can facilitate macrophage M2 polarization and osteogenic and antiinflammatory cytokine secretion | [178] | |
| - | Zn deficiency can dysregulate macrophage cytokine secretion, phagocytosis, and intracellular killing. | [179] | ||
| RAW 264.7 | Zn from ZnO films can favor macrophage phagocytosis and inflammatory cytokine production to kill bacteria | [180] | ||
| RAW 264.7 | Zinc silicate from calcium phosphate cement can markedly downregulate inflammatory-related gene expression and restrain osteoclastogenesis | [181] | ||
| Functional groups | Alkene, sulfonic acid | Mouse macrophages | P(NIPAAm-co-AAc) NPs modified with nitro, ether, sulfonic acid and phosphonic acid favor in vivo M1 polarization, while those modified with alkene, amide, epoxide, and ketone boost M2 polarization |
[182] |
| −NH2, guanidinium, –COO[−] |
Mouse macrophages | Carboxylate-based multidomain peptide hydrogels elicit minimal inflammation in vivo; lysine-based ones evoke acute inflammation that fades away; arginine-based ones induce inflammation and fibrous capsule formation. | [183] | |
| –COOH, –NH2 | Human macrophages | COOH− and NH2-modified polystyrene NPs inhibit M2 polarization; −NH2 NPs reduce M1 and M2 phagocytosis; −COOH NPs elevate M1 and M2 protein content, M1 TGF-β1 secretion and M2 ATP level. | [184] | |
| –NH2 | RAW 264.7, BMDMs | NH2-coated MBG can promote M2 polarization and antiinflammatory cytokine (Arg1, IL-10) production, thus creating favorable osteoimmunoregulatory niche | [185] | |
| Cationic polymers | PEI | RAW 264.7, THP-1 | PEI-modified superparamagnetic iron oxide nanoparticles (SPIONs) could induce M1 polarization and inflammatory response |
[186] |
| Integrins | Mac-1 | BMDMs | The absence of Mac-1 or blocking of RGD-binding integrin can mitigate inflammatory response of macrophages and fibrous encapsulation surrounding implanted biomaterials |
[187] |
| Cytokines | IL-4 | Murine primary macrophages | IL-4 from starPEG-heparin hydrogels can have sustained delivery and facilitate prohealing M2 polarization |
[188] |
| IL-4 | - | IL-4/polydopamine coating on Ti-based implants can boost M2 polarization and metal implant–soft tissue integration in vivo |
[189] | |
| IL-13 | Mouse macrophages | IL-13 can skew macrophages toward M2, thereby regulating plaque component and protecting from atherosclerosis | [190] | |
| IL-1 family | - | IL-1 can prolong monocyte/macrophage survival; IL-1 family have diverse immunoregulatory activities | [191] | |
| IFN, IL-4 | Human primary macrophages | The short release of IFN-γ from decellularized bone scaffolds could boost M1-phenotype polarization, and subsequent more sustained IL-4 release was able to favor M2 phenotype, thus enhancing vascularization |
[192] | |
| IL-4 | Rat bone marrow-derived macrophages | IL-4 released from high-stiffness gelatin hydrogels could facilitate the polarization of immunomodulatory M2 macrophages, thereby positively impacting osteogenic differentiation of BMSCs |
[193] | |
| IL4/IL10/TGF-β1 cocktail | Human monocyte-derived macrophages | The cytokine cocktail could trigger stable M2-phenotype macrophages that had markedly reduced proinflammatory cytokine secretion and increased antiinflammatory cytokine production |
[194] | |
| IL-4 | THP-1 | In the presence of IL-4, biomaterials with integrin attachment sites could trigger the antiinflammatory M2-phenotype polarization and propagate the induction effect of IL-4 on M2 macrophages |
[195] | |
| IL-6 | Mouse adipose tissue macrophages | IL-6 could serve as a Th2 cytokine to stimulate the M2-phenotype polarization and local proliferation of macrophages in obesity | [196] | |
| IFN, IL-4 | THP-1 | IFN-γ and IL-4 released from silk biomaterials can accordingly induce THP-1 polarization into M1 and M2, and repolarize macrophages from M2 to M1 and vice versa |
[197] | |
| Cellular components | Cells | - | Acellular ECM scaffolds can elicit dominant M2 response and constructive tissue remodeling, whereas those containing cellular component (even autologous) induced dominant M1 response and dense connective tissue deposition and/or scarring |
[56] |
| Decellularized biomaterials | Porcine tissue-derived scaffolds | Mouse macrophages | Tissue-derived scaffolds could promote the polarization of M2-like macrophages having a high antigen presentation activity | [43] |
| Urinary bladder matrix (UBM) scaffolds | Murine macrophages | The biologic scaffolds could activate type 2-like immune response different from classical tumor microenvironment, with activated Th2 T cells, unique UBM- associated macrophage phenotype, angiogenic factors, eosinophil infiltration and complement, thus collectively suppressing tumor formation and potentiating checkpoint immunotherapy |
[41] | |
| Decellularized bovine pericardium | Human monocyte-derived macrophages | Decellularized matrices may not stimulate macrophages polarizing to the inflammatory phenotype, thus supporting the potential for tissue engineering | [198] | |
Researchers have much focused on how parameters of synthetic particles can affect particle binding and internalization by the macrophages in liver and spleen, thereby promoting particle-mediated systemic therapeutic, diagnostic and/or imaging agent delivery.[199,200] The surface functionalization of synthetic particles with polymer coating/modification may modulate the macrophage fate through tailoring the particle surface properties such as surface charge, wettability, topography and composition.[201–203] Surface chemical modification can provide a method to devise phagocytosis-resistant particles through blocking protein adsorption on and complement interactions with particle surface (opsonization), otherwise the adsorbed serum components can act as molecular handholds for the binding and internalization by phagocytes.[204] The opsonin proteins in blood serum can bind to the nonstealth synthetic particles quickly and favor the macrophages of mononuclear phagocytic system to recognize and clear these particles easily. Anchoring/grafting a dense PEG or PEG-containing layer on particle surface could sterically resist the protein interaction with particles to limit opsonization,[64] in a way to generate a hydrophilic protecting layer around the synthetic particles to repel the opsonin protein absorption through steric repulsion force, which was thus able to block and/or delay the first step of opsonization process. Some zwitterionic polymers such as poly(carboxybetaine) and poly(sulfobetaine) were also able to protect the synthetic particles from opsonization via surface coating and modification to achieve a high resistance to the nonspecific protein adsorption.[205]
Surface modification of polystyrene microparticles with the poloxamer polymer coating could affect the phagocytic uptake by the mouse peritoneal macrophages in vitro.[206] By altering the chain lengths of PEO and polypropylene oxide (PPO), it showed that poloxamer polymers with long chains were able to effectively inhibit the particle phagocytosis by macrophages, which was ascribed to the changed surface features of particles including steric stabilization effect of the coating layer and decreased surface hydrophobicity by the coating layer as well as tunable coating layer thickness. The carboxylated or bovine serum albumin (BSA)-coated polystyrene microparticles with negatively charged surface were less efficiently phagocytosed by macrophages (derived from human peripheral blood monocytes) in vitro; however, the poly-L-lysine (PLL)-coated polystyrene particles with positively charged surface could trigger the high phagocytosis by macrophages.[207] In addition, BSA-coated polystyrene microparticles induced the acidified phagosomal microenvironment with pH 4.6–5.1 after low phagocytosis by the human peripheral blood monocyte-derived macrophages in vitro.[208] By contrast, the cationic polyamine-coated polystyrene microparticles were highly phagocytosed by macrophages (mice BMDMs) in vitro with a diminished acidification in phagosomal microenvironment (pH 6.0–6.8). The surface modification of synthetic particles could also influence the fate of different macrophage phenotypes. Using PEG- or CD47-coated polystyrene nanoparticles as model target, the stimulated macrophages exhibited a higher phagocytic activity than their nonactivated M0 counterpart, and M1 macrophages possessed a stronger phagocytosis ability than M2 macrophages.[209] Furthermore, the PEG coating of surface was able to reduce the clearance of particles by all phenotypes of macrophages and the CD47 coating could preferentially weaken the phagocytic capability of the M1 macrophages.
Tactics that utilize biogenic cell membrane components to cloak synthetic particles can render unique cell-like functions and enrich the concept of surface modification and functionalization of synthetic micro and nanoparticles. Such camouflage strategies can inherit and integrate the merits of those parent components, and create self-signals or serve Trojan Horses for a wide range of biomedical purposes such as eluding opsonization, delaying uptake by phagocytes, prolonging circulation time in blood, targeting inflamed/diseased sites, targeted drug delivery, specific tumor imaging, targeted cancer therapy, and offering antigens for cancer vaccination and immunotherapy. The macrophage membrane camouflage strategy have been widely used for the nanomaterial surface functionalization.[210–217] Furthermore, the combination of macrophage membrane with other types of cell membrane holds potential to enable hybrid cytomembrane components with multiple functionalities for more versatile surface camouflage of synthetic NPs.[218–221]
2.5. Material Dynamics
2.5.1. Material Degradability
The material degradation process, facilitated by physicochemical or cell-mediated dissolution, hydrolysis, or enzymolysis,[152] can lead to the composition dynamics, topography dynamics, and stiffness dynamics of a biomaterial, therefore producing dynamic physical and/or chemical stimulation on macrophages. During degradation, β-tricalcium phosphate substitutes could release Ca2+ ions into local microenvironment to switch RAW 264.7 macrophages to M2 phenotype in vitro via activating the calcium-sensing receptor (CaSR) pathway, and significantly upregulate the expression of BMP-2 for enhancing osteogenesis.[222] Another in vitro work showed that the degradation particles from biphasic calcium phosphate ceramics could initiate the appropriate inflammatory response of RAW 264.7 macrophages at early phase to secrete signaling molecules that recruit MSCs and boost their osteogenic differentiation.[223] Nondegradable biomaterials, such as knitted polypropylene mesh, can frequently cause chronic foreign body reaction and fibrosis. Uncoated polypropylene mesh triggered predominant M1 response on the fiber surface in a rodent model, which could be attenuated by the ECM hydrogel coatings through releasing bioactive ECM fragments during degradation process. The decreased M1 response was also accompanied by reduced number of the foreign body giant cells in vivo.[224] Nondegradable, slowly degradable, or chemically crosslinked ECM scaffolds with limited degradation induced the dominant M1 macrophage response and chronic inflammation post implantation. On the contrary, rapidly degradable ECM scaffolds could elicit the M2 response and constructive tissue remodeling following implantation.[56,225] Harnessing the material degradability via rational design strategies can contribute to the dynamic immunomodulation function on the macrophage fate.
2.5.2. Dynamic Loading
Some human tissues, such as bones, joints, teeth, lungs, and vessels, are in dynamic mechanical loading as the need to fulfill normal functions. Nevertheless, abnormal mechanical stimuli may cause inflammation or other diseases.[226,227] Upon squeezing through subtle capillaries, blood-borne monocytes/macrophages as well as tissue-resident macrophages are quite often under cyclic or constant strain to stretch and elongate shape. Once put into the body, a biomaterial, associated with implant, device, or depot, can also undergo dynamic loading because of apparent reasons, such as joint/tooth motioning, alveoli/airway contracting, vessel pulsing, and muscle constricting, thereby applying dynamic mechanical forces to neighboring cells to affect their behaviors. Baaijens and co-workers investigated the strain modulation of macrophage phenotype polarization of human peripheral blood mononuclear cells (hPBMCs) within electrospun PCL scaffolds subjected to cyclic strain (0%, 7%, and 12%) for one week.[228] The moderate cyclic strain of 7% could increase M2/M1 ratio over time and promote macrophage polarizing to prohealing M2 phenotype in vitro; in contrast, 12% cyclic strain notably decreased the M2/M1 ratio over time and triggered the dominant polarization of M1 macrophages, thereby indicating the double-side effect of cyclic strain for modulating macrophage responses. Human primary alveolar macrophages, monocyte-derived macrophages, and THP-1 leukemic monocytes could sense cyclic pressure-stretching strain and elevate IL-8 secretion in vitro; applying cyclic strain and LPS stimulus together could result in a prominent synergistic effect on TNF-α and IL-6 secretion.[229]
Resorbable scaffolds have been emerging as an attractive strategy for the replacement of diseased blood vessels. Nevertheless, mismatches between the scaffold design and the in vivo hemodynamic loading (cyclic stretch, shear stress) may cause adverse inflammation reaction and tissue remodeling that finally result in the premature graft failure. To understand the underlying mechanisms, an in vitro 3D model was established to mimic the transient inflammation and biomechanics microenvironments.[230] Adopting resorbable supramolecular elastomer as scaffold material, the results showed that the cyclic stretch could initially decrease the production of proinflammatory cytokines using hPBMCs, and stimulate the secretion of IL-10 and downstream matrix deposition. The shear stress was able to mitigate the cyclic stretch-triggered matrix growth through amplifying the collagen remodeling mediated by MMP-1/TIMP-1, thus highlighting the different roles of hemodynamic loading for designing the resorbable vascular grafts.
2.5.3. Physical Fields
Macrophages implement distinct dynamic functions in vivo, dependent on their polarization. Applying external physical fields, such as magnetic, optic, electric, and flow fields, can accordingly exert dynamic stimuli on macrophages and thus regulate their biological behaviors for the intended purpose. The establishment of correlation between macrophage responsiveness and physical field stimulation will allow the remote control of macrophage fate, such as adhesion, migration, polarization, and phagocytosis. Bian and co-workers demonstrated in vitro (RAW 264.7 macrophages) and in vivo (mice subcutaneous model) the remote manipulation of macrophage adhesion and polarization using RGD-superparamagnetic iron oxide nanoparticles (SPIONs) grafted matrix (Figure 7A).[231] The ligand nanooscillation speeds of RGD-bearing SPIONs were tailored through altering the frequencies of applied oscillating magnetic field. Magnetic field oscillation at low frequency could boost macrophage attachment and M2 polarization; by contrast, high frequency oscillation could facilitate M1 polarization but restrain macrophage adhesion. In another work,[232] the researchers conjugated RGD-bearing Au NPs to substrate and magnetic nanocages to Au NPs through flexible linkers. The reversible caging and uncaging of RGD could be fulfilled by regulating nanoscale displacement of magnetic nanocages in magnetic field. Interestingly, the uncaging of RGD could temporally favor macrophage adhesion and M2 polarization but suppress M1 polarization in vitro (RAW 264.7 macrophages) and in vivo (mice model). These studies provide a strategy for remote magnetic control allowing tissue penetration of macrophage behavior response toward the biomaterial implants/devices. In a recent study from Wosik et al.,[233] exerting uneven magnetic field caused extreme elongation of mice peritoneal macrophages in vitro. The magnetic force could realign actin cytoskeleton and modify macrophage marker expression to change their polarization status. Elongated macrophages in magnetic field showed well accordant alignment and position with simulated orientation and distribution of magnetic force lines. The possible interaction mechanisms are depicted in Figure 7B. Nelson and co-workers utilized microrobotic prey (micromagnets) that mimic bacteria to investigate the translational and rotational modes of macrophage attacks preceding phagocytosis in vitro (Figure 7C).[234] J774A.1 and RAW 264.7 macrophages launched push–pull attack mode against translational resistive prey that mimic surface-bound bacteria, whereas they rearranged nonresistive prey mimicking planktonic bacteria with long axis to promote pickup.
Figure 7.

Magnetic field modulates macrophage fate. A) Remotely manipulated ligand nanooscillation controls macrophage attachment and polarization. Reproduced with permission.[231] Copyright 2017, American Chemical Society. B) Magnetic field alters macrophage polarization. a) Magnetic field and RhoA deletion changes actin and relevant structures in M0 macrophages differently. b) Lorentz force acting on membrane calcium channel. c) Susceptibility buoyance acting on diamagnetic macrophage. Reproduced with permission.[233] Copyright 2018, Biophysical Society. C) Dynamic control of translational/rotational resistance of magnetic microprey through magnetic tweezer system (5D-MTS) to investigate macro phage response before phagocytosis. Reproduced with permission.[234]
A recent in vitro study demonstrated the remote control over macrophage polarization through NIR excitation of calcium regulator-loaded upconversion nanoparticles to trigger the intracellular regulator release, in which the increase or decrease of intracellular calcium content could facilitate RAW 264.7 macrophage polarization toward M1 or M2, respectively (Figure 8A).[235] Chen and co-workers reported the photoresponsive RGD release and conjugation for periodically activating RAW 264.7 macrophage ανβ3 integrin to boost the M2 polarization in vitro, enabling dynamic modulation of macrophage immune responses (Figure 8B).[236] Biomimetic 3D in vitro models showed that macrophages (mice BMDMs) could sense and make response to the interstitial fluid flow from tumors to surrounding stroma, inducing M2 polarization through β1 integrin/Src interceded STAT3/6 mechanotransduction pathway (Figure 8C).[237] Interstitial fluid could guide faster macrophage migration against flow, implying its regulatory effect on the polarization and recruitment of M2 macrophages into tumor tissues in favor of tumor invasion and progression. Electrical fields are naturally occurring in the sites of injured tissues to accelerate wound healing. Applying electrical fields (5–300 mV mm−1) guided the migration of macrophages (human monocyte derived) toward anode in vitro, along with phagocytosis enhancement and cytokine production, while monocytes migrated toward cathode, thus being able to modulate macrophage functions.[238] Besides, radiotherapy can trigger M1 macrophage polarization with anticancer function.[239] For instance, in the mouse pancreatic cancer model, low-dose γ ray irradiation could reprogram TAMs to iNOS+ M1 phenotype in vivo, thus orchestrating potent T lymphocyte immunotherapy.[240]
Figure 8.
A) NIR light control over intracellular calcium regulates macrophage polarization. Reproduced with permission.[235] Copyright 2018, Elsevier Ltd. B) Photoresponsive dynamic modulation of macrophage polarization. a) Fabrication of photoresponsive hyaluronan nanocomposite hydrogel. b) Temporal activation of macrophage ανβ3 integrin via UV light for phenotype polarization. Reproduced with permission.[236] Copyright 2018, Wiley-VCH. C) Flow field furthers M2 macrophage polarization. a) Microfluidic system schematic. b) Interaction of macrophages and collagen ECM. c) Flow velocity given by tracking microbead trajectory (green) in collagen gel with macrophages (red). d) Upregulation of M2 marker expression of bone marrow derived macrophages with ≈3 μm s−1 flow for 48 h (left, staining images; right, quantification). e) Transwell flow device for investigating the influence on protein expression of macrophages. f–g) Upregulation of protein expression of macrophage M2 markers (Western blot analysis). Note: Bars represent the mean ± SEM of the data (fold change relative to the no flow control; n = 3, n is the number of independent parallel experiments). Reproduced with permission.[237]
Currently, understanding is still limited with regard to the exact mechanisms for how the physicochemical properties of biomaterials interact with the physiological and/or immune system, thus making it still unclear to optimally design and functionalize the immunomodulatory biomaterials for the in vivo applications. A lot of in vitro studies have been carried out to elucidate the interactions between biomaterials and immune cells in particular macrophages, and how biomaterials create microenvironmental cues to modulate macrophage fate. For in vitro research, various macrophage cell lines and primary macrophages derived from mouse, rat, or human have been widely used for the purpose. Nevertheless, due to the difference in diverse macrophage cell sources, researchers may receive distinct or even contradictory results and conclusions, which should be well taken into consideration when compared with previous studies. When interacting with macrophages, the forms of biomaterials with identical chemistry identity, such as particles, coatings/films, or scaffolds, may also lead to the difference in the ultimate outcomes. In addition, when describing how the specific properties of biomaterials can influence macrophage immune responses, a more quantitative conclusion should be given, for example, the effect of biomaterial surface roughness at a specific value or range on macrophage behavior. The in vitro assessment of macrophage immune responses to biomaterials is crucial to offer insights into the reasonable biomaterial designs, and several studies have demonstrated the good match between in vitro and in vivo results.[241] However, the conclusions from in vitro studies cannot be directly translated to the in vivo, since how the macrophages respond to biomaterials in vitro and in vivo are not always consistent. Furthermore, after entering into the body, the properties of biomaterials can change and differ in what macrophages encounter in vivo. Eventually, it is the in vivo assessment and validation that can approve the feasibility and applicability of biomaterials for the patients in clinic.[242]
3. Immunoengineering Applications
3.1. Materializing Cancer Immunotherapy
3.1.1. Tumor-Associated Macrophages
Macrophages act as the critical driver of tumor-promoting inflammation. The TAMs are dominantly present in tumor microenvironment and can reinforce tumor progression in different levels to boost metastasis, increase genetic instability, tame adaptive immunity, and foster cancer stem cells. Moreover, the TAM phagocytosis activity are thwarted by tumor cells through expressing the transmembrane protein, CD47, a ligand that can bind to the signal regulatory protein α (SIRPα) on TAMs to transmit a “don’t eat me” signal to phagocytes.[243] They can impose a “Yin-Yang” effect on the cytoreductive chemotherapy/radiotherapy, either antagonizing these anticancer therapies or potentiating overall antineoplastic outcome.[244–246] In fact, for the chemotherapy/radiotherapy, antiangiogenic therapy, targeted drug delivery, immunotherapy such as immune checkpoint blockade, they all have profound impact or dependence on TAM function.[246] These TAMs are distinct paradigm of polarized M2 macrophages that can infiltrate the tumor tissues.[247] Therapies that target TAMs can provide promising antitumor strategies to enrich, integrate and synergize the immunotherapy and chemotherapy.[244] TAM-targeting therapeutic tactics mainly involve i) suppression of TAM/monocyte recruitment,[248,249] ii) facilitation of tumor cell phagocytosis,[250] iii) targeted TAM depletion,[251] and iv) TAM reeducation. Among these tactics, promoting M1-like TAM repolarization has acquired particular interest due to its capacity to improve the tumoricidal efficacy of other tumor-infiltrating T cells.[252] One promising strategy is to reverse TAMs toward antitumor M1 phenotype through functional reeducation with micro/nanoformulations. It is anticipated that the TAMs can confer powerful targets to combine immunotherapy and cytoreductive therapies in the manner of precision medicine.
3.1.2. Reeducating Macrophages with Nanomaterials
Nanomedicine-based cancer immunotherapy has been rapidly advancing on the basis of functional nanomaterials.[253–256] TAMs are immunosuppressive effector cells and thus facilitate tumor survival.[257] Therefore, reprogramming the TAMs from antiinflammatory M2-like phenotype toward proinflammatory M1-like phenotype is crucial to elicit tumoricidal immune response.[258] Nanoparticles have been emerging as key tools to target and reeducate M2 TAMs owing to their tailorable physicochemical and biological properties. Daldrup-Link and co-workers reported the intrinsic suppression effect of FDA-approved ferumoxytol (iron oxide NPs) on early breast cancer growth and lung cancer liver/lung metastasis through reversing TAMs to M1 phenotype and triggering proinflammatory response (Figure 9A),[259] implying the capacity of ferumoxytol NPs to amplify macrophage-regulating cancer immunotherapy. In a recent study by Gu and co-workers,[260] an in situ sprayable immunotherapeutic bioresponsive hydrogel was used to restrain postsurgery tumor recurrence and distant tumor development (Figure 9B). The fibrin hydrogel containing CaCO3 NPs loaded with anti-CD47 antibody (denoted as aCD47@CaCO3) could scavenge H+ in site of surgical wound and induce TAMs repolarization toward M1 phenotype. The released aCD47 could augment “don’t eat me” signal blockade in tumor cells and thus their phagocytosis by macrophages. The reeducated macrophages could effectively heighten antigen presentation and activate T cell immunotherapy. The designed fibrin hydrogel has the immunomodulatory capability to “awaken” host innate and adaptive immunity to thwart local and/or metastatic tumor growth. Weissleder and co-workers recently screened and identified R848 (resiquimod) agonist of TLR-7/TLR-8 as the potent driver to reprogram TAMs toward M1 subtype (Figure 9C).[261] R848-laden β-cyclodextrin NPs (denoted as CDNP-R848) could preferentially localize in TAMs and confer efficacious drug delivery for in vivo M1 repolarization. In monotherapy of multiple mouse tumor models, CDNP-R848 administration could bottle tumor growth and secure mice from tumor rechallenge. Combination with T cell directed checkpoint inhibitor antiprogrammed death 1 blocking antibody (aPD-1) could potentiate the antitumoral efficacy of CDNP-R848 including in the aPD-1 resistant tumor model. Therefore, the formulated drug@NPs nanosystem could target and reverse the TAMs to amplify cancer immunotherapy.
Figure 9.
Nanomaterial-based TAM reeducation for cancer immunotherapy. A) Macrophage and ferumoxytol combination suppress tumor growth. a) Sequential injection of ferumoxytol and KP1-GFP-Luc cells into mice. b) Ferumoxytol-treated livers showed no strong bioluminescence on day 21 post injection. Reproduced with permission.[259] Copyright 2016, Springer Nature. B) Schematic of in situ sprayable bioresponsive fibrin hydrogel having aCD47@CaCO3 NPs within postsurgical tumor bed. The aCD47@CaCO3 NPs scavenge H+ in surgical wound and release aCD47, thereby facilitating TAM repolarization to anticancer M1 phenotype and “don’t eat me” signal blockade in tumor cells. Reproduced with permission.[260] Copyright 2018, Springer Nature. C) Schematic of reprogramming M2-like TAMs toward tumoricidal M1-like phenotype by drug treatment. Reproduced with permission.[261]
Liu and co-workers created the core–shell PLGA-R837@Cat NPs using PLGA, catalase (Cat) and imiquimod (R837) for triggering robust cancer immunotherapy.[262] The treatment of PLGA-R837@Cat NPs could lead to the relieved hypoxia in tumor microenvironment, which further resulted in the significant decrease of M2-polarized TAMs and the repolarization of TAMs from M2 phenotype to M1 phenotype. This observation agreed well with their previous studies on PEG-modified hollow MnO2 (loaded with chlorin e6 and doxorubicin (DOX)) nanoplatform,[263] human serum albumin-bound paclitaxel NPs (combined with erlotinib treatment)[264] and liposome NPs (loaded with catalase and H2O2 separately)[265] for clearance of tumor cells by macrophages. Therefore, these designed functional NPs were able to reverse immunosuppressive tumor microenvironment and synergistically amplify cancer treatment outcome. Recently, the same group incorporated chlorin e6-modified catalase and R837-loaded PLGA NPs (as immune adjuvant) into PEGDA hydrogel formed by light-induced in situ gelation for robust photodynamic immunotherapy, with a remarkable reduction of M2-polarized TAMs, thereby boosting the reversal of M2-like immunosuppressive tumor microenvironment for cancer cell clearance.[266] Using the system composed of dextran–hyaluronidase NPs and chlorine e6-loaded liposomes, researchers also acquired similar outcome of tumor treatment with markedly promoted macrophage infiltration.[267]
Zhang and co-workers designed artificial natural killer cells with perfluorohexane and glucose oxidase (cloaked by red blood cell membrane) for specific tumor killing.[268] This artificial system could generate H2O2 that recruited immune cells and reeducated macrophages from M2 to M1 for attacking tumor cells. They also used the hyaluronic acid-modified superparamagnetic iron oxide NPs to artificially reprogram TAMs from protumoral M2 to antitumor M1, which achieved analogous tumor killing outcome.[269] Repolarization of TAMs from M2 to M1 is an effective strategy to facilitate tumor cell clearance by macrophages. Such nanosystems reported also included but not limited to cancer cell membrane camouflaged superparamagnetic iron oxide (loaded with doxorubicin and indocyanine) NPs,[270] PEG–PLGA (enveloped with Gdmetallofullerenol and doxorubicin) NPs[271] and microRNA-125b encapsulated hyaluronic acid-poly(ethylenimine) NPs.[272] Table 3 gives the summaries of representative nanomaterial-based tactics to reeducate TAMs into cancer fighters for immunotherapy. For general information of nanomaterial-based TAM targeting and reprogramming, we refer the readers to the reviews.[273,274]
Table 3.
Representative nanomaterial-based TAM reeducating strategies for cancer immunotherapy.
| Nanomaterials | Drugs | Targets | Functions | Ref. | |
|---|---|---|---|---|---|
|
| |||||
| Inorganic NPs | Ferumoxytol | - | - | Ferumoxytol exposure can induce proinflammatory M1 macrophage polarization and Th1 response and increase M1 presence in tumor tissues to suppress tumor growth |
[259] |
| Au | Macrolide | Macrophage | Macrolide–Au nanorod conjugates can selectively/specifically target TAMs and boost antitumor therapy against breast cancer cells | [275] | |
| ZnO | DOX | DNA | ZnO NPs can protect macrophages from DOX toxicity and induce M1 polarization, thus boosting tumor cell apoptosis | [276] | |
| MoSe2 | - | - | RBC membrane camouflaged MoSe2 nanosheets can reprogram TAMs into M1 phenotype to amplify antitumoral capacity | [277] | |
| CaCO3 | aCD47 | Macrophage | aCD47@CaCO3 NPs can scavenge H+ in surgical wound, release aCD47, and thus boost TAM repolarization toward tumoricidal M1 phenotype and ‘don’t eat me’ signal blockade in tumor cells |
[260] | |
| Polymeric NPs | β-cyclodextrin | R848 | TLR-7/TLR-8 | R848-loaded NPs can efficiently deliver to TAMs and promote M1 polarization to inhibit tumor growth |
[261] |
| PLGA-b-PEG | Pt(IV) prodrug | DNA | Therapeutic NPs can accumulate in TAMs that act as local drug depot to deliver to tumor cells | [278] | |
| PEG-b-PAEMA-PAMAM | Pt(IV) prodrug, BLZ945 | DNA, CSF-1R | BLZ945SCNs/Pt NPs can release BLZ945 to target and deplete TAMs and Pt(IV) prodrug locally in deep tumor tissue to kill cancer cells | [279] | |
| MAN-PLGA-TAA, MAN-PLGA-N-TAA | - | - | NP-based ROS photogeneration can reprogram TAMs to antitumor M1, with amplified antigen presentation for T cell recruitment and tumoricidal response |
[280] | |
| Poly(β-amino ester) | IL-12 | - | Therapeutic NPs can locally release IL-12 in tumor microenvironment and reeducate TAMs from M2 into M1 for cancer treatment | [281] | |
| Lipid-based NPs | PEGylated liposomes | Alendronate | - | NPs can abrogate tumor growth via TAM repolarization into M1 | [282] |
| Supramolecular NPs | BLZ945, anti-CD47–SIRPα | CSF-1R, CD47–SIRPα |
NPs can amplify TAM repolarization to M1, and augment phagocytosis, antitumor and antimetastasis effect | [283] | |
| Biohybrid NPs | Au | siRNA, M2 peptide | - | RNAi-M2pepAuNPs can selectively target, silence and deplete TAMs | [284] |
| sPEG-coated polypeptide | miRNA | - | NPs can effectively favor TAM-targeting miRNA delivery and repolarize TAMs to M1, thus noticeably augmenting activated T cells and natural killer cells in tumor | [285] | |
| Gal-C-dextran, PEG-His-modified alginate |
Oligonucleotides | - | NPs can target and accumulate in TAMs to locally deliver oligonucleotides and efficaciously repolarize TAMs into M1 | [286] | |
| Hyaluronic acid-coated mannan-conjugated MnO2 | - | - | Man-HA-MnO2 can reprogram TAMs into M1 and synergize DOX chemotherapy |
[287] | |
| Cancer cell membrane coated SPION@DOX-ICG |
DOX | DNA | Nanosystems have tumor-homing capacity and can reprogram TAM polarization to M1 |
[270] | |
| Myeloid-derived suppressor cell membrane cloaked Fe3O4 | - | - | NPs can actively target tumor microenvironment to reeducate TAMs into M1 and synergize immunogenic cell death | [288] | |
3.1.3. Reeducated Macrophages as Delivery Systems
MPS and renal clearance pathway can compete with tumor for NPs when administered systemically. A study revealed that only the median 0.7% of administered dose of NPs was able to be delivered into solid tumors.[289] In this context, it is of great significance to develop advanced delivery systems that can target the sites of tumor tissues for precision medication and localized cancer treatment of high efficiency. Tumor-tropic monocytes/macrophages have been pyramidally considered as promising cargo delivery systems to target tumor tissues and surmount biological barriers such as blood brain barrier (BBB), thereby heightening local drug payload and therapy efficacy.[289] After reeducating, these engineered monocytes/macrophages can act as the “Trojan Horses” to deliver anticancer drugs and/or nanomaterials into tumor nidus to maximize the selective eradication and meanwhile minimize or circumvent the toxicity against normal tissues. For the cargo-loaded backpacks carried by monocytes/macrophages, they should circumvent the internalization by these phagocytes otherwise cargos can degrade or fail to deliver.[145] Mitragotri and co-workers recently developed the cellular backpacks for the macrophage immunotherapy (Figure 10).[290] The backpacks were made from biodegradable polymers via microcontact printing, comprising one cell-adhesive PLGA layer, one PVA layer and one more PLGA layer. After 1.5 h incubation, these backpacks could bind to 86.9% of BMDMs from the BALB/c mice. Such robustly adhering backpacks were able to elude the phagocytosis for several days. The IFN-γ was incorporated and stabilized in the PVA layer, the interior of backpacks. Through releasing IFN-γ, the backpacks could continuously guide macrophage polarization toward the durable antitumoral phenotype, even in strongly immunosuppressive microenvironment in murine breast cancer model, which led to the outcomes of the decreased metastasis burden and the slowed tumor growth, highlighting a new way to modulate and maintain the antitumor phenotype of adoptive macrophage immunotherapies.
Figure 10.

A) Schematic illustrations for i) a backpack and ii) the printing method. B) Confocal images of macrophages (blue, nucleus; green, cytomembrane) displaying backpacks fabricated from PLGA discs (red). C) Schematic illustration of the cellular backpacks for maintaining the proinflammatory phenotype of the adoptive MΦ therapies. a) MΦs stimulated by IFN-γ ex vivo can quickly switch from proinflammatory to antiinflammatory phenotype following penetration through a solid tumor. b) MΦs carrying backpacks loaded with IFN-γ can maintain their proinflammatory phenotype deep within tumor microenvironment, changing the phenotype of the endogenous TAMs. A–C) Reproduced with permission.[290]
The tumor hypoxia is located away from the blood vessels, which makes it difficult for hypoxia-activated prodrugs to accumulate within solid tumors at therapeutically sufficient concentrations. To overcome this barrier, Mitragotri and co-workers recently developed a generalized strategy by adopting macrophages as active drug carriers to heighten the prodrug penetration and accumulation deep into the solid tumors.[291] Macrophages were able to phagocytose and internalize at large amounts the PLGA NPs loading tirapazamine prodrug. By infiltrating the hypoxic regions in the solid tumors, the tirapazamine prodrug could contribute to a potent tumoricidal effect. With the help of macrophages as drug carriers (“Trojan Horse” strategy), light-activated nitric oxide prodrugs (comprising manganese–nitrosyl complex and Nd3+-doped upconverting NPs loaded into PLGA microparticles) could be delivered to the targeted tumor tissues, providing necessary temporal, spatial and dosage control for the treatment of cancer hypoxia.[292] Table 4 gives some representative examples of using reeducated macrophages as tumor-targeting cargo delivery systems.
Table 4.
Representative targeted cargo delivery systems on the basis of macrophages.
| Strategies | Cargos | Functions | Ref. |
|---|---|---|---|
|
| |||
| Trojan Horses | Nanoparticle indinavir | Nanoparticle indinavir packaged BMDMs can effectively decrease the number of HIV-1 infected cells and protect CD4+ T lymphocytes in humanized mouse model | [293] |
| Au–SiO2 nanoshells | Macrophages can deliver therapeutic NPs to tumor hypoxic region for NIR-induced cell killing | [294] | |
| Liposomal DOX, iron oxide | Macrophages can actively deliver DOX to and effectively suppress xenograft tumors in mouse model; iron oxide-laden macrophages can efficaciously penetrate into tumors via MR imaging |
[295] | |
| BSA-coated small Au nanorods | Macrophages can effectively deliver Au agent into entire tumor post intratumoral injection and greatly amplify photothermal therapy efficacy | [296] | |
| Bi2Se3 nanosheets | Bi2Se3-loaded macrophages can efficiently target tumor post intravenous injection and noticeably heighten photothermal therapy efficacy | [297] | |
| Aspirin-laden Au nanocages | Armed monocytes can initiate M1 activation to combat bacterial infection at early stage, and at late stage differentiate into M2 macrophages driven by NIR to restrain excessive inflammation for bone formation and osteomyelitis treatment |
[298] | |
| Polymeric backpacks | Drug-loaded polyelectrolyte backpacks | Monocytes/macrophages can target and deliver cellular backpacks into inflamed tissues to locally release drugs for inflammation treatment | [299] |
| IFN-γ loaded PLGA–PVA–PLGA backpacks | Macrophages can maintain the durable antitumoral phenotype to reduce metastasis burden and slow tumor growth for cancer immunotherapy | [290] | |
3.2. Materializing Infection Immunotherapy
The raise of multidrug-resistance (MDR) bacteria and the famine of new antibiotics have been cumulatively demanding for breakthrough tactics that can surpass classical antibiotics to fight back this oncoming human health calamity.[300,301] In addition, bacterial biofilms, the slimy hydrated matrices of polysaccharides, proteins and extracellular DNA, encasing adherent bacteria on implanted biomaterials or infected tissues, can protect bacteria from antibiotics and contribute to the antibiotic resistance.[302,303] Furthermore, bacteria can evade host immunity by skewing immune response (immune evasion).[304] Mature biofilms of dense polymeric matrices are tough to devour by macrophages and cause frustrated phagocytosis.[305] Staphylococcal biofilms skew host immune response from inflammatory antibacterial status to antiinflammatory profibrotic status.[306] The bacterial biofilms can alter macrophage polarization phenotypes from proinflammatory M1 to antiinflammatory M2, which is immunosuppressive and detrimental against antibacterial immunity (macrophage exhaustion). These macrophages are exhausted and cannot fight back bacteria, pointing out the necessity to reeducate exhausted macrophages as energetic warriors to let M1 phenotype survive longer and fight longer.
Li et al. developed cobalt-doped TiO2 coating to combat biomaterial-associated bacterial infections.[165] The tailored band structure of TiO2 semiconductor by cobalt doping could cause bacteria starvation and subvert biofilm formation, which facilitated innate immunocytes (including macrophages and neutrophils) to efficaciously engulf and exterminate the biofilm-thwarted bacteria in a divide-and-conquer manner. Moreover, the cobalt ions released from coating could skew macrophage polarization toward M1 phenotype, thereby creating a local proinflammatory environment and amplifying methicillin-resistant Staphylococcus aureus (MRSA) phagocytosis and clearance. Such design concept can also apply to other immunomodulatory antibacterial biomaterials. The rational selection of semiconductor dopants can simultaneously result in the potentiation of antibacterial activity and antibacterial immunity.
Zhang and co-workers magnetron-sputtered the copper NPs on the surface of porous sulfonated polyetheretherketone (SPEEK) to fight MRSA infection.[166] The bactericidal action involved dual modes of contact killing and trap killing. Released copper ions at low dosage could trigger activation of M1 macrophages and secretion of inflammatory cytokines (including IL-6 and TNF-α) and MCP-1 chemokine to generate inflammatory microenvironment and potentiate phagocytic activity of macrophages for MRSA killing and resolution. Previously researchers also observed the similar immunoregulatory antibacterial ability of ZnO NP-modified implant surface.[180] Nanomaterial-based surface modification can not only confer direct antibacterial capacity but also create desired immune microenvironment to intensify indirect antibacterial immunity, without significant toxicity against normal tissue cells including immunocytes. Clinical observations proposed that porous Ta implants could attenuate bacterial infection incidence in revision arthroplasty. To delve into the feasibility and mechanism, Shen and co-workers devised TaOx-coated implant surface to mimic the native nanothin oxide layer on Ta materials.[307] Of note, TaOx layer could not afford direct surface antibacterial capacity. Interestingly, TaOx layer could augment bacteria phagocytosis by neutrophils and alleviate neutrophil lysis to let them fight longer. Moreover, TaOx layer could also motivate M1 macrophage polarization and inflammatory cytokine secretion to aid neutrophil migration deeper into biofilms for bacteria elimination since neutrophils can be activated only in the peripheral regions of bacterial biofilms but cannot penetrate into biofilms.[308,309]
In a recent study by Dong and co-workers,[310] researchers demonstrated that the adoptive transfer of macrophages that contained the antimicrobial peptides linked to the cathepsin B in lysosomes (denoted as MACs) could be used for the therapy of sepsis caused by MDR bacteria in immunosuppressive mice (Figure 11). MACs were constructed through transfection of the vitamin C lipid nanoparticles for delivering the antimicrobial peptides and cathepsin B (denoted as AMP-CatB) mRNA. Vitamin C lipid nanoparticles could allow for specific accumulation of the AMP-CatB in the macrophage lysosomes, which are the pivotal site for bactericidal activity. The adoptive MAC transfer could lead to the eradication of MDR bacteria and complete recovery of the immunocompromised septic mice. By incorporating oligomycin, an inhibitor of oxidative phosphorylation, into poly(ethylene glycol)-b-poly(L-glutamic acid) (PEG-b-PGA) NPs as the drug carriers, and then intracellularly delivering oligomycin-loaded NPs into monocytes/macrophages, they could be reprogrammed to the proinflammatory phenotype and exhibit remarkable clearance activity on Staphylococcus aureus biofilms in vivo when combined with systemic antibiotics.[311] Collectively, developing the immunomodulatory antibacterial biomaterials can make contributions to the infection combination therapy that involves direct bacteria/biofilms allopathy and indirect infection immunotherapy.
Figure 11.

Schematic construction of the MACs for the sepsis therapy. AMP-CatB mRNA was encapsulated in VCLNP and delivered to macrophage, in which mRNA was translated in cytoplasm and translocated into lysosomes. Within lysosomes, cleavable linker was cleaved by lysosomal CatB to release AMP-IB367. After the phagosomes carrying the MDR bacteria fused with lysosomes, ingested MDR bacteria were eradicated by prestored AMP-IB367. Reproduced with permission.[310]
3.3. Materializing Tissue Regeneration Immunotherapy
The presence of monocytes/macrophages is indispensable to orchestrate efficacious healing during tissue regeneration process, which comprises the overlapping inflammation, repair and remodeling phases.[312,313] At inflammation phase, one of major roles of monocytes/macrophages is to remove damaged tissues and cells. Moreover, they secrete inflammatory cytokines to orchestrate tissue healing response. Early inflammatory reaction is beneficial while unrestrained inflammation is detrimental to wound healing and tissue regeneration. For example, TNF-α and IL-6 are necessary for efficient tissue healing upon restricted in early days following injuries of murine skins and skeletal muscles,[314,315] though prolonged/excessive cytokine levels do harm to healing. Bone fractures are common traumatic injury; innate and adaptive immunity are the component of fracture healing, which can be amplified by diverse interventions.[316] In repair phase, macrophages switch phenotypes from proinflammatory state to antiinflammatory state, with increased secretion of cytokines (such as IL-10) and growth factors (such as TGF-β), to initiate neotissue growth and maturation. During remodeling phase, the repaired/regenerated new tissues fulfill maturation. In common experimental models, inflammation phase usually occurs in the early few days following injury; repair phase starts within days and culminates in one week following injury; remodeling phase lasts months or longer. A long-lived presence of the M1 macrophages induces chronic inflammation and hampers tissue healing, while a long-lasting existence of the M2 macrophages causes excessive fibrosis.[317,318] Therefore, macrophages are one of the key orchestrators, effectors, and cellular targets to enable the tissue regeneration immunotherapy through reforming their phenotypes and functions.[54,319] Figure 12 illustrates the schematic of inflammation, repair and remodeling phases during bone formation and regeneration with the help of immunomodulatory biomaterials.
Figure 12.

Schematic illustration of the inflammation phase, repair phase and remodeling phase during bone regeneration in the presence of immunomodulatory biomaterials. Note: The changes of cytokine production and cell types involved at different phases refer to previous work.[32,313]
Articular cartilage lesions possess very limited intrinsic self-repair capability and can result in sterile inflammation. To understand how the bioceramics can regulate immune microenvironment and affect cartilage regeneration, Wu and co-workers fabricated the Li2Ca4Si4O13 scaffolds and investigated in vitro whether the scaffolds were capable of facilitating the cartilage maturation through modulating the macrophage polarization.[320] The results showed that, the Li2Ca4Si4O13 scaffolds could promote the antiinflammatory M2-phenotype polarization of RAW 264.7 macrophages, with decreased expression of inflammatory TNF-α, IL-6, and IL-1β genes and increased expression of antiinflammatory IL-10 gene. Both indirect culture of chondrocytes with macrophage-conditioned medium and direct co-culture of chondrocytes with macrophages verified that, the Li2Ca4Si4O13 scaffolds had the immunomodulatory function to promote the M2 macrophage polarization and markedly enhance the chondrocyte maturation, possessing the potential for cartilage repair and regeneration.
Compared with the commercial hydroxyapatite microparticles, researchers validated that the home-made hydroxyapatite nanoparticles could possess effective immunomodulatory capacity to preferentially boost the M2 macrophage polarization both in vitro (human blood monocyte-derived macrophages) and in vivo (rat model by incorporating NPs into ECM scaffolds).[321] The hydroxyapatite nanoparticles were able to specifically elevate the secretion of antiinflammatory IL-10 cytokine. Moreover, nanoparticle-treated macrophages were capable of promoting MSC osteogenesis in vitro with a dependence on IL-10, as well as propelling the proangiogenic response of human macrophages together with HUVECs. A similar profile was further revealed on the rat femoral defect model, ultimately contributing to the enhanced tissue vascularization and bone formation due to the immunomodulatory antiinflammatory potential of the hydroxyapatite nanoparticles.
By incorporating the macrophage recruitment agent, SEW2871, into the gelatin–chitosan multilayer on microstructured titanium implant, the modified surface could effectively recruit macrophages upon releasing the SEW2871 slowly both in vitro (RAW 264.7 macrophages) and in vivo (rat model).[322] The tuned surface roughness and wettability were able to promote the antiinflammatory response of M2 macrophages and mitigate the M1 proinflammatory response. The favorable immune microenvironment created by macrophage–implant interactions further boosted the in vitro osteogenic differentiation of rat mesenchymal stromal cells and the in vivo osteointegration after implantation for 6 weeks. Osteogenic peptide-coated titanium implant demonstrated the potential to attenuate the inflammation response of M1 macrophages in vitro (RAW 264.7) and in vivo (rat model).[323] With osteoimmunomodulatory promotion on M2 macrophage polarization, the bioactive peptide coating could effectively inhibit osteoclastogenesis and ameliorate bone-implant osseointegration in the presence of a chronic inflammation. Herein, we mainly focus on the biomaterial-assisted targeted immunomodulation of monocytes/macrophages in tissue repair and regeneration immunotherapy, and some other representative studies/strategies reported are also summarized in Table 5.
Table 5.
Biomaterial-assisted targeted immunomodulation of monocytes/macrophages for tissue regeneration immunotherapy.
| Tissues | Materials | Functions | Ref. |
|---|---|---|---|
|
| |||
| Bone | CeOx NP-coated Ti implant | Modulating Ce4+/Ce3+ ratio can promote macrophage polarization to prohealing M2 phenotype and create beneficial immune microenvironment for bone regeneration | [324] |
| γ-Fe2O3 and hydroxyapaptite NP-containing PLA scaffold | Superparamagnetic composite scaffold can mechanomodulate macrophage polarization to M2 phenotype upon magnetization, thereby suppressing inflammation and boosting bone formation | [325] | |
| Nanoporous Al2O3 film | Tuning Al2O3 nanopore size can favor M2 macrophage polarization and antiinflammatory response to promote osteogenesis | [326] | |
| Au-loaded MSNs | Au-loaded MSNs can facilitate M2 polarization, antiinflammatory reaction and osteogenic cytokine secretion of macrophages for accelerating bone formation | [327] | |
| PAH/PAA film-coated PEEK implant | Surface functionalization can inhibit macrophage inflammatory response and boost M2 polarization, conferring favorable local immune environment to amplify osseointegration | [328] | |
| Spinal cord | CeOx NPs | CeOx NPs can decrease ROS level and alleviate inflammation, therefore leading to functional recovery of spinal cord injury | [329] |
| Poly(lactide-co-glycolide) NPs | NP administration can reprogram macrophages into proregenerative M2, block inflammation and decrease fibrotic/gliotic scarring to facilitate functional recovery of spinal cord injury | [330] | |
| Cartilage | Squid type II collagen | Squid Col-II can activate M2 phenotype polarization and prochondrogenic gene expression (TGF-β, IGF) of macrophages, and efficiently suppress chondrocyte hypertrophy and apoptosis to favor cartilage repair in osteoarthritis |
[331] |
| Squid type II gelatin–hyaluronic acid hydrogel | Double-network hydrogel can immunomodulate M2 macrophage polarization dynamically and enhance costal cartilage tissue regeneration directly and indirectly stimulated by TGF-β/Smad signaling of M2 macrophages |
[332] | |
| Skeletal muscle | Mammalian ECM-derived bioscaffold | Degradation products of ECM bioscaffold can boost constructive M2 polarization of macrophages for favoring skeletal muscle regeneration | [333] |
| Bone-/myocardium-derived ECM scaffold | ECM bioscaffold can guide IL-4 dependent M2 macrophage polarization and shape local immune milieu in muscle wound to trigger proregenerative immune response and boost muscle tissue repair |
[42] | |
| Skin | StarPEG–heparin hydrogel | Hydrogel can efficiently scavenge inflammatory chemokines (MCP-1, IL-8, MIP-1α, MIP-1β) from patient wound fluids and enhance wound healing and closure in mouse model | [334] |
| Hyaluronan–silk fibroin–PCL scaffold | Hyaluronan can confer hydrophilic nanofibers to inhibit nonspecific protein adsorption, thus decreasing macrophage adhesion and fibrotic tissue thickness | [335] | |
| FTY720-loaded PLGA film | FTY720-loaded PLGA film in site of skin injury can directly recruit nonclassical blood monocytes to perivascular niche for prohealing M2 macrophage activation, thereby boosting skin wound healing |
[336] | |
| Vessel | PCL scaffold | Macroporous PCL scaffold of fiber diameter 5–6 μm and pore size ≈30 μm can promote tissue-remodeling M2 polarization, when compared with small-sized one inducing M1 phenotype, hence favoring arterial regeneration | [337] |
| Nerve | IL-4/IFN-γ laden polysulfone–agarose hydrogel scaffold | Scaffold can locally deliver IL-4/IFN-γ to polarize macrophages to prohealing M2/proinflammatory M1, thereby eliciting favorable peripheral nerve regeneration | [338] |
| Heart | Decellularized pericardium | Decellularized pericardium may not induce inflammatory macrophage phenotype and hold potential for cardiac regeneration | [198] |
3.4. Materializing Inflammation Resolution
An active concerted inflammation resolution program starts in the initial few hours following inflammatory reaction initiation. Granulocytes enter tissues and facilitate prostaglandin and leukotriene switch to initiate termination sequence. Neutrophil recruitment thereby stops and apoptosis wages. Macrophages phagocytize and clear apoptotic neutrophils, and secrete antiinflammatory/reparative cytokines (such as TGF-β1). This antiinflammatory program comes to an end with macrophage emigration through lymphatics.[339] Therefore, macrophages play a critical role in the inflammation resolution and tissue homeostasis restoration.[340] Rheumatoid arthritis (RA) is an inflammatory arthropathy, featured by long-lasting synovitis and joint destruction/disability.[341,342] Normal synovium appears relatively acellular, containing scattered macrophages, yet rheumatoid synovial membrane comprises monocytes/macrophages, dendritic cells and T lymphocytes, orchestrating arthritis progression together.[343,344] The activation of numerous macrophages in inflamed synovium and pannus notably correlates with RA severity, having clear marks of overexpression of proinflammatory cytokines and chemokines. The synovial inflammation can be settled through depletion of proinflammatory M1 phenotype and induction of antiinflammatory M2 phenotype. For example, Hyeon and co-workers developed the ceria and manganese ferrite NP-anchored mesoporous silica nanoparticles (MSNs) to generate O2 and scavenge reactive oxygen species (ROS) in a synergistic manner, hence resulting in efficient shift of M1 to M2 polarization in vitro and in vivo to alleviate RA symptoms. Combining with methotrexate-loaded MSNs could further augment RA therapy effect (Figure 13A).[345] Another work also demonstrated that fumagillin prodrug-based nanotherapy could restrain macrophage inflammation response and drive antiinflammatory phenotype for effective RA treatment.[346] Atherosclerosis is an inflammatory artery wall disease, with accumulation of lipids, ECM and inflammatory cells dominantly macrophages in intima and formation of atherosclerotic plaque (lesion).[347,348] IL-13 administration could decrease monocyte recruitment and plaque macro phage population and induce M2 macrophage polarization to augment lipoprotein clearance, thus regulating plaque composition and protecting against atherosclerosis.[190] Recently, Leeper and co-workers engineered a precision nanotherapy to interrupt the CD47-SIRPα signaling axis in monocytes/macrophages to prevent atherosclerosis.[349] The developed Trojan Horse nanosystem, denoted as SWNT-SHP1i, comprised PEG-modified single-walled carbon nanotubes (SWNTs) loaded with SHP-1 as CD47-SIRPα signaling inhibitor and Cy5.5 as fluorescent probe. Prophagocytic SWNT-SHP1i could specifically accumulate in lesional macrophages to reduce inflammation, reactivate local efferocytosis, and decrease atherosclerotic plaque burden without endangering safety (Figure 13B).
Figure 13.
Inflammation resolution through rational material designs. A) Therapy mechanism of ceria and manganese ferrite NP co-anchored MSNs (MFC-MSNs) against rheumatoid arthritis. Reproduced with permission.[345] Copyright 2019, American Chemical Society. B) 18F-FDG PET/CT imaging validates SWNT-SHP1i could remarkably decrease vascular inflammation. Reproduced with permission.[349] Copyright 2020, The Authors, published by Springer Nature. C) Modulation of fibrous encapsulation on implanted biomaterials via in vivo RGD peptide activation. Reproduced with permission.[355]
Macrophages with the cellular backpacks comprising polymer patches of 7–10 μm diameter and submicron thickness could act as targeted drug delivery systems to travel across blood brain barrier and avoid drug depot phagocytosis.[350] The catalase-loaded backpacks were able to be delivered by autologous macrophages to the inflamed brain and reduce free radical production, hence providing a new strategy for the treatment of neuroinflammatory diseases and neurodegenerative disorders. Covalently immobilized CD47 on polytetrafluoroethylene (PTFE) surface could alleviate monocyte/macrophage inflammatory response and thus favor PTFE-based vascular grafts having long-lasting patency.[351] Besides, inorganic CeO2−x NPs,[352] Ce-doped bioglass NPs,[353] and biogenic lipoaspirate NPs[354] can also be used for antiinflammatory purpose. García and co-workers demonstrated a dynamic, transdermal and noninvasive light activation strategy of cell-adhesive RGD peptide to favor cell adhesion and vascularization but avoid inflammation and fibrous encapsulation.[355] For example, delaying RGD presentation activation could decrease fibrous capsule thickness around implanted biomaterial (Figure 13C). To mitigate the uncontrolled inflammation, Couvreur and co-workers recently developed a formulation of multidrug nanoparticles through conjugating the natural lipid, squalene, to endogenous adenosine immunomodulator and subsequently encapsulating the antioxidant, α-tocopherol.[356] These multidrug NPs could be delivered to the inflammatory cells in vitro (RAW 264.7 macrophages) and the inflamed tissues in vivo (rodent endotoxemia models) in a targeted way, and effectively promote the resolution of inflammation.
3.5. Materializing Vaccination
Synthetic biomaterials are playing cumulatively contributive roles in vaccine development.[357–359] Engineered micro/nanoparticles with packaged antigens or immunostimulators have been increasingly researched as vaccine delivery systems and adjuvants for vaccine immunotherapy.[360–367] APCs, DCs, and monocytes/macrophages, function as the prime particle vaccine targets for initiating/maintaining humoral immunity and cellular immunity. Studies also showed that monocytes could differentiate into DCs; macrophages and/or DCs could internalize the particles at the sites of injection and actively transport them to the draining lymph node (dLN).[368,369] At day 1 and day 4 post injection, the majority of APC population were LN-resident macrophages that localized and accumulated the malaria antigen-laden multilamellar lipid nanoparticle vaccines.[370]
Wang et al. recently showed that hierarchical TiO2 microparticles with nanospikes could activate/amplify immune response for vaccination through imposing mechanical stress on macrophages or dendritic cells, thereafter leading to inflammasome activation and K+ ion efflux during phagocytosis (Figure 14A). In the existence of monophosphoryl lipid A (MPL, TLR-4 agonist), these spiky particles could heighten antigen-specific protective humoral immunity and cellular immunity against EG7 thymoma growth or influenza virus infection.[371] This work is expected to provide new understanding of how to engineer particle physical cues to activate innate immunity and augment adjuvanticity/immunogenicity. Checkpoint inhibition-resistant rodent solid tumors showed low immunogenicity and antigen presentation by CD11b+F4/80+ TAMs, being inactive without antigen-presenting activity, was a major factor for tumor immune resistance. Cholesteryl pullulan nanogel particles could efficaciously target and deliver peptide antigen to the TAMs. In combination with CpG oligo DNA (TLR-9 agonist), this TAM-targeting antigen delivery strategy could effectively elicit the antigen presentation by TAMs, consequently sensitizing resistant tumors to the T cell immunity.[372] Therefore, the status of TAMs plays an important role in the tumor immune resistance. Manipulating TAM functions can amplify tumor immune sensitivity and afford a promising tactic to enhance cancer immunotherapy. In a recent study by Wang and co-workers, researchers developed erythrocyte-derived nanoerythrosome systems for tumor antigen delivery for personalized cancer vaccination and immunotherapy (Figure 14B).[373] Since senescent/impaired erythrocytes are the targets of spleen APCs (macrophages, DCs), the nanoerythrosomes possess intrinsic capability of being captured by the APCs. The tumor antigen-laden nanoerythrosomes (denoted as nanoAg@erythrosomes) could elicit in vivo antigen responses and suppress tumor growth in the 4T1 and B16F10 tumor models when combined with antiprogrammed death ligand 1 blocking antibody (aPDL1). More importantly, “personalized nano-Ag@erythrosomes” could be accomplished through fusing erythrocytes and surgically resected tumors, evidencing the capacity to efficiently decrease tumor relapse and metastasis post surgery in an established tumor model.
Figure 14.

Nanomaterial-based antigen delivery systems and adjuvants for vaccine immunotherapy. A) Spiky particles physically activate innate immunity. a) Activation of immunocytes by spiky particles to augment immune response. b) Activation of inflammasomes by spiky particles. c) Combination of MPL and spiky particles as potent adjuvant. Reproduced with permission.[371] Copyright 2018, The Authors, published by Springer Nature. B) Erythrocyte-derived nanoerythrosome systems deliver tumor antigens to enhance cancer immunotherapy. a) Fabrication of nano-Ag@erythrosomes through fusing tumor cell membrane-associated antigens into nanoerythrosomes. b) Fabrication of personalized nano-Ag@erythrosomes for suppressing tumor relapse and metastasis post surgery. Reproduced with permission.[373]
4. Conclusion and Future Outlook
This review gives a summary of recent advances in the rational designs of immunomodulatory biomaterials for directing macrophage fate for versatile immunoengineering applications. As reviewed and discussed in detail, tailoring the physical, chemical, biological, and dynamic properties of biomaterials can actively regulate the macrophage behavior and immune response, which thus contributes to a variety of immunoengineering purposes. Such material design tactics and principles are also anticipated to apply to other types of immune cells for a wide range of immunotherapy applications. In addition to the surface modification and functionalization of biomaterials/implants regarding surface properties (such as topography, composition, charge, and wettability) to modulate macrophage fate, various scaffold biomaterials/implants with controlled bulk properties are also working to reeducate the macrophage immune responses.[49,54] When developing immunomodulatory biomaterials, particular design strategies need to be exploited specifically according to the targeted applications. For example, to regenerate bone defects, biomaterials with antiinflammatory property are needed to promote M2 macrophage polarization and create prohealing immune microenvironment. To enhance the phagocytosis of bacteria or biofilms by macrophages, the biomaterial-mediated M1-phenotype polarization is required. Similarly, to reprogram the TAMs, biomaterials should be devised to promote TAM repolarization from the immunosuppressive M2 to the immunosupportive M1. Furthermore, the rapidly advancing artificial intelligence (AI) and big data strategies, envisioned to find powerful applications in medicine and healthcare,[374,375] can be integrated with the futuristic design of immunomodulatory biomaterials since vast amounts of information can be acquired from conventional trial-and-error biomaterial design approaches that are continuously contributed by researchers, which can be shared with each other in the future through establishing a Cloud service platform.[376] Such AI and big data approaches can also be used to analyze the responses of immune cells, tissues, organs, and the whole-body reactions to implanted biomaterials. The promising applications include cancer nanomedicine and immunotherapy.[377,378]
Biomaterial-mediated immunotherapy has been rapidly advancing and can efficiently synergize traditional therapies. Materials science holds vast promise to break through some bottlenecks and challenges in the field of immunology and immunotherapy. Leveraging in-depth the roles that biomaterial-associated parameters play in macrophage activation and polarization, including material cues, mechanical cues, physical cues, chemical cues, and biological cues, will largely improve our understanding of material–macrophage interactions, and thus accelerate the development of new macrophage-based therapeutics in combination with immunomodulatory biomaterials. Understanding the interplay between biomaterials and macrophage biology at the molecular, cellular, tissue, organ, immune system, and even whole-organism level can add value in the rational designs of advanced immunomodulatory biomaterials to guide macrophage fate and functions for the future of versatile immunotherapies. Such biomaterial design strategies can also apply to other types of immunocytes in addition to macrophages, such as monocytes,[14] neutrophils,[379] mast cells,[380] and T cells[381] with different phenotypes. It is expected that these design strategies can offer universal guidelines for the research of new immunomodulatory biomaterials to direct immune cell fate and propel the development of novel potent immunotherapeutic strategies mediated by the functional materials. Besides, the material design principles can extend to the crosstalk/interaction between macrophages and other cell types in the material-mediated microenvironment, such as lymphocytes,[382] epithelial cells,[383] cancer cells,[384] marrow stromal cells,[385] and mesenchymal stem cells.[386]
With regard to the prophylaxis and eradication of infectious pathogens (such as viruses, bacteria, fungi, and parasites), NP-based vaccines and vaccine adjuvants can augment the uptake of antigens by APCs, amplify the immune responses of T lymphocytes and B lymphocytes, and induce long-term high-efficiency adaptive immunity.[387] The fast advancement of biohybrid NPs can help expedite the development of patient-specific vaccines. For example, patient-specific tumor antigens and/or adjuvants can be integrated with and delivered by nanovectors to boost cancer vaccine efficacy, thereby fulfilling personalized cancer vaccine immunotherapy. Therefore, the continuous enrichment of immunomodulatory biomaterial designs will greatly advance the rapid development of novel prophylactic/therapeutic vaccines against infectious diseases including the ongoing COVID-19 pandemic[388,389] or HIV pandemic[390,391] or the unknown viruses in the future.
CRISPR–Cas systems[392,393] and synthetic-biology principles[394,395] may provide powerful tools in combination with intelligent nanocarrier delivery systems to edit, educate and engineer (3E) the native immunocytes to manufacture designer immune cells for living therapeutics. For example, genetically engineered macrophages can function as macrophage factory to secrete proinflammatory or antiinflammatory cytokines, chemokines, proteins and growth factors to actively regulate immune microenvironment and innate/adaptive immunity for versatile therapeutic purposes.[396–398] Particulate-laden macrophages can selectively target, localize and deliver drugs/imaging agents to particular sites of tissues or organs in the body; engineered macro phages as Trojan Horses can be optimized through tailoring the nanoparticle parameters and responsiveness for multi ple biomedical applications. These engineered immunomodulatory micro and nanosystems are expected to actively interact with immune cells and immune system to implement tasks and functions from single-cell level to whole-organ level.
3D printing enables the versatile scaffold designs of a wide range of material, mechanical, geometrical, physical, chemical and biological cues in a customized way,[45] thereby providing the great possibility and convenience to investigate macrophage fate in response to various material-mediated stimuli. Hydrogels are an ideal material for this purpose due to their favorable 3D printability, ECM mimics, ease of modification and immunomodulatory potentials.[51] Furthermore, the introduction of 3D printing technology may facilitate the development of novel scaffold vaccines in an individualized manner, which can largely add values to the formation of local vaccination and immunomodulation by creating localized controlled immune microenvironment and regulating on-site immune response.[256,399,400] The customizable scaffold vaccines can further make contributions to precision medicine and patient-specific healthcare. Additionally, dynamic materials have both inherent properties and dynamic cues, which are appealing to generate dynamic stimuli to guide macrophage behavior.
Acknowledgements
J.L. gratefully acknowledges the funding support from the Alexander von Humboldt Stiftung/Foundation. This work was supported by grants from the start-up packages of UCLA, National Institutes of Health (R01 CA234343–01A1), Jonsson Comprehensive Cancer Center at UCLA to Z.G., and National Natural Science Foundation of China (81921002) to X.Q.J.
Biography

Jinhua Li obtained his Ph.D. degree in Materials Science in 2016 from the University of Chinese Academy of Sciences. From 2016 to 2018, he worked at the University of Hong Kong as a postdoctoral researcher. In 2018, he joined the Dresden University of Technology in Germany as the Alexander von Humboldt Fellow. His research interests focus on the functional materials for medical applications.

Xinquan Jiang is a full professor and executive dean of College of Stomatology in Shanghai Jiao Tong University and director of Shanghai Engineering and Research Center for Advanced Dental Technology and Materials. He obtained his Ph.D. in Clinical Dentistry (Oral maxillofacial Surgery) from Shanghai Second Medical University in 2003. Meanwhile, he finished his exchange Ph.D. training at University of Alberta, Canada. His research interests focus on prosthodontics and regenerative medicine, particularly on bone regeneration and oral function restoration, with tissue engineering and regenerative medicine strategies.

Zhen Gu is a Chair Professor and Dean of College of Pharmaceutical Sciences at Zhejiang University. Dr. Gu received his B.S. degree in Chemistry and M.S. degree in Polymer Chemistry and Physics from Nanjing University. In 2010, he obtained his Ph.D. from the Department of Chemical and Biomolecular Engineering at the University of California, Los Angeles (UCLA). He was a Postdoctoral Associate at MIT and Harvard Medical School during 2010 to 2012. Before he moved to Zhejiang University in 2020, he was a Full Professor in the Department of Bioengineering and Director of the NIH Biotechnology Training in Biomedical Sciences and Engineering Program at UCLA. From 2012 to 2018, he was working in the Joint Department of Biomedical Engineering at the University of North Carolina at Chapel Hill and North Carolina State University, where he was appointed as a Jackson Family Distinguished Professor. His group studies controlled drug delivery, biomaterials and cell therapy, especially for cancer and diabetes treatment.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Dr. Jinhua Li, Centre for Translational Bone, Joint and Soft Tissue Research, University Hospital and Faculty of Medicine Carl Gustav Carus, Technische Universität Dresden, Dresden 01307, Germany.
Prof. Xinquan Jiang, Department of Prosthodontics, Shanghai Engineering Research Center of Advanced Dental Technology and Materials, Shanghai Key Laboratory of Stomatology, Shanghai Research Institute of Stomatology, National Clinical Research Center for Oral Diseases, Shanghai Ninth People’s Hospital, College of Stomatology, Shanghai Jiao Tong University School of Medicine, No. 639 Zhizaoju Road, Shanghai 200011, China
Dr. Hongjun Li, Department of Bioengineering, University of California, Los Angeles, Los Angeles, CA 90095, USA Jonsson Comprehensive Cancer Center California NanoSystems Institute and Center for Minimally Invasive Therapeutics, University of California, Los Angeles, Los Angeles, CA 90095, USA; College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, P. R. China.
Prof. Michael Gelinsky, Centre for Translational Bone, Joint and Soft Tissue Research, University Hospital and Faculty of Medicine Carl Gustav Carus, Technische Universität Dresden, Dresden 01307, Germany
Prof. Zhen Gu, Department of Bioengineering, University of California, Los Angeles, Los Angeles, CA 90095, USA Jonsson Comprehensive Cancer Center California NanoSystems Institute and Center for Minimally Invasive Therapeutics, University of California, Los Angeles, Los Angeles, CA 90095, USA; College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, P. R. China.
References
- [1].Kaufmann SHE, Nat. Immunol 2008, 9, 705. [DOI] [PubMed] [Google Scholar]
- [2].Nathan C, Nat. Immunol 2008, 9, 695. [DOI] [PubMed] [Google Scholar]
- [3].Mellman I, Coukos G, Dranoff G, Nature 2011, 480, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pardoll DM, Nat. Rev. Cancer 2012, 12, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ribas A, Wolchok JD, Science 2018, 359, 1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Parkin J, Cohen B, Lancet 2001, 357, 1777. [DOI] [PubMed] [Google Scholar]
- [7].Delves PJ, Roitt IM, N. Engl. J. Med 2000, 343, 37. [DOI] [PubMed] [Google Scholar]
- [8].Akira S, Uematsu S, Takeuchi O, Cell 2006, 124, 783. [DOI] [PubMed] [Google Scholar]
- [9].Janeway CA, Medzhitov R, Annu. Rev. Immunol 2002, 20, 197. [DOI] [PubMed] [Google Scholar]
- [10].Carroll MC, Nat. Immunol 2004, 5, 981. [DOI] [PubMed] [Google Scholar]
- [11].Iwasaki A, Medzhitov R, Nat. Immunol 2015, 16, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Iwasaki A, Medzhitov R, Science 2010, 327, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wynn TA, Chawla A, Pollard JW, Nature 2013, 496, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gordon S, Taylor PR, Nat. Rev. Immunol 2005, 5, 953. [DOI] [PubMed] [Google Scholar]
- [15].Mosser DM, Edwards JP, Nat. Rev. Immunol 2008, 8, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Epelman S, Lavine KJ, Randolph GJ, Immunity 2014, 41, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Murray PJ, Wynn TA, Nat. Rev. Immunol 2011, 11, 723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K, Science 2010, 327, 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S, Immunity 2013, 38, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FMV, Nat. Neurosci 2007, 10, 1538. [DOI] [PubMed] [Google Scholar]
- [21].Sica A, Mantovani A, J. Clin. Invest 2012, 122, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege J-L, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA, Immunity 2014, 41, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Martinez FO, Sica A, Mantovani A, Locati M, Front. Biosci 2008, 13, 453. [DOI] [PubMed] [Google Scholar]
- [24].Martinez FO, Gordon S, F1000Prime Rep. 2014, 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gordon S, Martinez FO, Immunity 2010, 32, 593. [DOI] [PubMed] [Google Scholar]
- [26].O’Shea JJ, Murray PJ, Immunity 2008, 28, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mosser DM, J. Leukocyte Biol 2003, 73, 209. [DOI] [PubMed] [Google Scholar]
- [28].Dale DC, Boxer L, Liles WC, Blood 2008, 112, 935. [DOI] [PubMed] [Google Scholar]
- [29].Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, Schmierer M, Gabrusiewicz K, Anderson NR, Petty NE, Cummins KD, Shen F, Shan X, Veliz K, Blouch K, Yashiro-Ohtani Y, Kenderian SS, Kim MY, O’Connor RS, Wallace SR, Kozlowski MS, Marchione DM, Shestov M, Garcia BA, June CH, Gill S, Nat. Biotechnol 2020, 38, 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M, Trends Immunol. 2004, 25, 677. [DOI] [PubMed] [Google Scholar]
- [31].Ding AH, Nathan CF, Stuehr DJ, J. Immunol 1988, 141, 2407. [PubMed] [Google Scholar]
- [32].Chen Z, Klein T, Murray RZ, Crawford R, Chang J, Wu C, Xiao Y, Mater. Today 2016, 19, 304. [Google Scholar]
- [33].Brown BN, Ratner BD, Goodman SB, Amar S, Badylak SF, Biomaterials 2012, 33, 3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Reese TA, Liang H-E, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM, Nature 2007, 447, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Brandt E, Woerly G, Younes AB, Loiseau S, Capron M, J. Leukocyte Biol 2000, 68, 125. [PubMed] [Google Scholar]
- [36].Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TKA, Bucks C, Kane CM, Fallon PG, Pannell R, Jolin HE, McKenzie ANJ, Nature 2010, 464, 1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gordon S, Nat. Rev. Immunol 2003, 3, 23. [DOI] [PubMed] [Google Scholar]
- [38].Lawrence T, Natoli G, Nat. Rev. Immunol 2011, 11, 750. [DOI] [PubMed] [Google Scholar]
- [39].Amit I, Winter DR, Jung S, Nat. Immunol 2016, 17, 18. [DOI] [PubMed] [Google Scholar]
- [40].Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I, Cell 2014, 159, 1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wolf MT, Ganguly S, Wang TL, Anderson CW, Sadtler K, Narain R, Cherry C, Parrillo AJ, Park BV, Wang G, Pan F, Sukumar S, Pardoll DM, Elisseeff JH, Sci. Transl. Med 2019, 11, eaat7973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sadtler K, Estrellas K, Allen BW, Wolf MT, Fan H, Tam AJ, Patel CH, Luber BS, Wang H, Wagner KR, Powell JD, Housseau F, Pardoll DM, Elisseeff JH, Science 2016, 352, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sadtler K, Allen BW, Estrellas K, Housseau F, Pardoll DM, Elisseeff JH, Tissue Eng., Part A 2017, 23, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Williams DF, Biomaterials 2008, 29, 2941. [DOI] [PubMed] [Google Scholar]
- [45].Li J, Wu C, Chu PK, Gelinsky M, Mater. Sci. Eng., R 2020, 140, 100543. [Google Scholar]
- [46].Lu Y, Aimetti AA, Langer R, Gu Z, Nat. Rev. Mater 2017, 2, 16075. [Google Scholar]
- [47].Vishwakarma A, Bhise NS, Evangelista MB, Rouwkema J, Dokmeci MR, Ghaemmaghami AM, Vrana NE, Khademhosseini A, Trends Biotechnol. 2016, 34, 470. [DOI] [PubMed] [Google Scholar]
- [48].Anderson JM, Rodriguez A, Chang DT, Semin. Immunol 2008, 20, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sadtler K, Singh A, Wolf MT, Wang X, Pardoll DM, Elisseeff JH, Nat. Rev. Mater 2016, 1, 16040. [Google Scholar]
- [50].Sridharan R, Cameron AR, Kelly DJ, Kearney CJ, O’Brien FJ, Mater. Today 2015, 18, 313. [Google Scholar]
- [51].Singh A, Peppas NA, Adv. Mater 2014, 26, 6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Franz S, Rammelt S, Scharnweber D, Simon JC, Biomaterials 2011, 32, 6692. [DOI] [PubMed] [Google Scholar]
- [53].Davenport Huyer L, Pascual-Gil S, Wang Y, Mandla S, Yee B, Radisic M, Adv. Funct. Mater 2020, n/a, 1909331. [Google Scholar]
- [54].Dellacherie MO, Seo BR, Mooney DJ, Nat. Rev. Mater 2019, 4, 379. [Google Scholar]
- [55].Zhou G, Groth T, Macromol. Biosci 2018, 18, 1800112. [DOI] [PubMed] [Google Scholar]
- [56].Brown BN, Valentin JE, Stewart-Akers AM, McCabe GP, Badylak SF, Biomaterials 2009, 30, 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Dobrovolskaia MA, McNeil SE, Nat. Nanotechnol 2007, 2, 469. [DOI] [PubMed] [Google Scholar]
- [58].Chen H, Li P, Yin Y, Cai X, Huang Z, Chen J, Dong L, Zhang J, Biomaterials 2010, 31, 8172. [DOI] [PubMed] [Google Scholar]
- [59].Huang Z, Yang Y, Jiang Y, Shao J, Sun X, Chen J, Dong L, Zhang J, Biomaterials 2013, 34, 746. [DOI] [PubMed] [Google Scholar]
- [60].Miller CR, Bondurant B, McLean SD, McGovern KA, O’Brien DF, Biochemistry 1998, 37, 12875. [DOI] [PubMed] [Google Scholar]
- [61].Hua K, Ålander E, Lindström T, Mihranyan A, Strømme M, Ferraz N, Biomacromolecules 2015, 16, 2787. [DOI] [PubMed] [Google Scholar]
- [62].Hunt JA, Flanagan BF, McLaughlin PJ, Strickland I, Williams DF, J. Biomed. Mater. Res 1996, 31, 139. [DOI] [PubMed] [Google Scholar]
- [63].Chen L, Simpson JD, Fuchs AV, Rolfe BE, Thurecht KJ, Mol. Pharmaceutics 2017, 14, 4485. [DOI] [PubMed] [Google Scholar]
- [64].Owens DE, Peppas NA, Int. J. Pharm 2006, 307, 93. [DOI] [PubMed] [Google Scholar]
- [65].Albanese A, Tang PS, Chan WCW, Annu. Rev. Biomed. Eng 2012, 14, 1. [DOI] [PubMed] [Google Scholar]
- [66].Lynch I, Dawson KA, Nano Today 2008, 3, 40. [Google Scholar]
- [67].Bartneck M, Keul HA, Singh S, Czaja K, Bornemann J, Bockstaller M, Moeller M, Zwadlo-Klarwasser G, Groll J, ACS Nano 2010, 4, 3073. [DOI] [PubMed] [Google Scholar]
- [68].Zhang M, Qing G, Sun T, Chem. Soc. Rev 2012, 41, 1972. [DOI] [PubMed] [Google Scholar]
- [69].Sun T, Han D, Rhemann K, Chi L, Fuchs H, J. Am. Chem. Soc 2007, 129, 1496. [DOI] [PubMed] [Google Scholar]
- [70].Kehr NS, Galla HJ, Riehemann K, Fuchs H, RSC Adv. 2015, 5, 5704. [Google Scholar]
- [71].Wang X, Gan H, Sun T, Adv. Funct. Mater 2011, 21, 3276. [Google Scholar]
- [72].Qing G, Sun T, Adv. Mater 2011, 23, 1615. [DOI] [PubMed] [Google Scholar]
- [73].Danelius E, Ohm RG, Ahsanullah MM, Ong H, Chemtob S, Erdelyi M, Lubell WD, J. Med. Chem 2019, 62, 11071. [DOI] [PubMed] [Google Scholar]
- [74].Rich A, Harris AK, J. Cell Sci 1981, 50, 1. [DOI] [PubMed] [Google Scholar]
- [75].Hotchkiss KM, Reddy GB, Hyzy SL, Schwartz Z, Boyan BD, Olivares-Navarrete R, Acta Biomater. 2016, 31, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Lv L, Xie Y, Li K, Hu T, Lu X, Cao Y, Zheng X, Adv. Healthcare Mater 2018, 7, 1800675. [DOI] [PubMed] [Google Scholar]
- [77].Sun T, Wang G, Feng L, Liu B, Ma Y, Jiang L, Zhu D, Angew. Chem., Int. Ed 2004, 43, 357. [DOI] [PubMed] [Google Scholar]
- [78].Xin B, Hao J, Chem. Soc. Rev 2010, 39, 769. [DOI] [PubMed] [Google Scholar]
- [79].Lahann J, Mitragotri S, Tran T-N, Kaido H, Sundaram J, Choi IS, Hoffer S, Somorjai GA, Langer R, Science 2003, 299, 371. [DOI] [PubMed] [Google Scholar]
- [80].Gao S, Lu R, Wang X, Chou J, Wang N, Huai X, Wang C, Zhao Y, Chen S, J. Biomater. Appl 2020, 34, 1239. [DOI] [PubMed] [Google Scholar]
- [81].Chun YW, Wang W, Choi J, Nam T-H, Lee Y-H, Cho K-K, Im Y-M, Kim M, Gwon Y-H, Kang SS, Lee JD, Lee K, Khang D, Webster TJ, Carbon 2011, 49, 2092. [Google Scholar]
- [82].Tabata Y, Ikada Y, J. Colloid Interface Sci 1989, 127, 132. [Google Scholar]
- [83].Hotchkiss KM, Clark NM, Olivares-Navarrete R, Biomaterials 2018, 182, 202. [DOI] [PubMed] [Google Scholar]
- [84].Takebe J, Champagne CM, Offenbacher S, Ishibashi K, Cooper LF, J. Biomed. Mater. Res., Part A 2003, 64A, 207. [DOI] [PubMed] [Google Scholar]
- [85].Refai AK, Textor M, Brunette DM, Waterfield JD, J. Biomed. Mater. Res., Part A 2004, 70A, 194. [DOI] [PubMed] [Google Scholar]
- [86].Overlin JW, Shah AH, Chaubal M, Hotchkiss KM, Olivares-Navarrete R, Biomaterials 2020, 243, 119920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Barth KA, Waterfield JD, Brunette DM, J. Biomed. Mater. Res., Part A 2013, 101A, 2679. [DOI] [PubMed] [Google Scholar]
- [88].Li X, Huang Q, Elkhooly TA, Liu Y, Wu H, Feng Q, Liu L, Fang Y, Zhu W, Hu T, Biomed. Mater 2018, 13, 045013. [DOI] [PubMed] [Google Scholar]
- [89].Li J, Zhang Y-J, Lv Z-Y, Liu K, Meng C-X, Zou B, Li K-Y, Liu F-Z, Zhang B, Regener. Biomater 2020, 7, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Paul NE, Skazik C, Harwardt M, Bartneck M, Denecke B, Klee D, Salber J, Zwadlo-Klarwasser G, Biomaterials 2008, 29, 4056. [DOI] [PubMed] [Google Scholar]
- [91].Chen S, Jones JA, Xu Y, Low H-Y, Anderson JM, Leong KW, Biomaterials 2010, 31, 3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Wang K, Hou W-D, Wang X, Han C, Vuletic I, Su N, Zhang W-X, Ren Q-S, Chen L, Luo Y, Biomaterials 2016, 102, 249. [DOI] [PubMed] [Google Scholar]
- [93].Luu TU, Gott SC, Woo BWK, Rao MP, Liu WF, ACS Appl. Mater. Interfaces 2015, 7, 28665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Yang C, Zhao C, Wang X, Shi M, Zhu Y, Jing L, Wu C, Chang J, Nanoscale 2019, 11, 17699. [DOI] [PubMed] [Google Scholar]
- [95].Cockerill I, Su Y, Lee JH, Berman D, Young ML, Zheng Y, Zhu D, Nano Lett. 2020, 20, 4594. [DOI] [PubMed] [Google Scholar]
- [96].Zheng X, Xin L, Luo Y, Yang H, Ye X, Mao Z, Zhang S, Ma L, Gao C, ACS Appl. Mater. Interfaces 2019, 11, 43689. [DOI] [PubMed] [Google Scholar]
- [97].Zhang Q, Hwang JW, Oh J-H, Park CH, Chung SH, Lee Y-S, Baek J-H, Ryoo H-M, Woo KM, Biomaterials 2017, 149, 77. [DOI] [PubMed] [Google Scholar]
- [98].Vassey MJ, Figueredo GP, Scurr DJ, Vasilevich AS, Vermeulen S, Carlier A, Luckett J, Beijer NRM, Williams P, Winkler DA, de Boer J, Ghaemmaghami AM, Alexander MR, Adv. Sci 2020, 7, 1903392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Baker BM, Chen CS, J. Cell Sci 2012, 125, 3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Jiang J, Li Z, Wang H, Wang Y, Carlson MA, Teusink MJ, MacEwan MR, Gu L, Xie J, Adv. Healthcare Mater 2016, 5, 2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Saino E, Focarete ML, Gualandi C, Emanuele E, Cornaglia AI, Imbriani M, Visai L, Biomacromolecules 2011, 12, 1900. [DOI] [PubMed] [Google Scholar]
- [102].Bartneck M, Heffels K-H, Pan Y, Bovi M, Zwadlo-Klarwasser G, Groll J, Biomaterials 2012, 33, 4136. [DOI] [PubMed] [Google Scholar]
- [103].Vallés G, Bensiamar F, Crespo L, Arruebo M, Vilaboa N, Saldaña L, Biomaterials 2015, 37, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Almeida CR, Serra T, Oliveira MI, Planell JA, Barbosa MA, Navarro M, Acta Biomater. 2014, 10, 613. [DOI] [PubMed] [Google Scholar]
- [105].Razzi F, Fratila-Apachitei LE, Fahy N, Bastiaansen-Jenniskens YM, Apachitei I, Farrell E, Zadpoor AA, Biomed. Mater 2020, 15, 035017. [DOI] [PubMed] [Google Scholar]
- [106].Jain N, Moeller J, Vogel V, Annu. Rev. Biomed. Eng 2019, 21, 267. [DOI] [PubMed] [Google Scholar]
- [107].Meli VS, Veerasubramanian PK, Atcha H, Reitz Z, Downing TL, Liu WF, J. Leukocyte Biol 2019, 106, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Wiesner C, Le-Cabec V, El Azzouzi K, Maridonneau-Parini I, Linder S, Cell Adhes. Migr 2014, 8, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Linder S, Kopp P, J. Cell Sci 2005, 118, 2079. [DOI] [PubMed] [Google Scholar]
- [110].Labernadie A, Thibault C, Vieu C, Maridonneau-Parini I, Charrière GM, Proc. Natl. Acad. Sci. USA 2010, 107, 21016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Labernadie A, Bouissou A, Delobelle P, Balor S, Voituriez R, Proag A, Fourquaux I, Thibault C, Vieu C, Poincloux R, Charrière GM, Maridonneau-Parini I, Nat. Commun 2014, 5, 5343. [DOI] [PubMed] [Google Scholar]
- [112].McWhorter FY, Davis CT, Liu WF, Cell. Mol. Life Sci 2015, 72, 1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Kress H, Stelzer EHK, Holzer D, Buss F, Griffiths G, Rohrbach A, Proc. Natl. Acad. Sci. USA 2007, 104, 11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Möller J, Lühmann T, Chabria M, Hall H, Vogel V, Sci. Rep 2013, 3, 2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Möller J, Luehmann T, Hall H, Vogel V, Nano Lett. 2012, 12, 2901. [DOI] [PubMed] [Google Scholar]
- [116].Guimarães CF, Gasperini L, Marques AP, Reis RL, Nat. Rev. Mater 2020, 5, 351. [Google Scholar]
- [117].Previtera ML, Sengupta A, PLoS One 2015, 10, e0145813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Smith TD, Nagalla RR, Chen EY, Liu WF, Adv. Drug Delivery Rev 2017, 114, 193. [DOI] [PubMed] [Google Scholar]
- [119].Adlerz KM, Aranda-Espinoza H, Hayenga HN, Eur. Biophys. J 2016, 45, 301. [DOI] [PubMed] [Google Scholar]
- [120].Blakney AK, Swartzlander MD, Bryant SJ, J. Biomed. Mater. Res., Part A 2012, 100A, 1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Nemir S, Hayenga HN, West JL, Biotechnol. Bioeng 2010, 105, 636. [DOI] [PubMed] [Google Scholar]
- [122].Beningo KA, Wang Y.-l., J. Cell Sci 2002, 115, 849. [DOI] [PubMed] [Google Scholar]
- [123].Hind LE, Dembo M, Hammer DA, Integr. Biol 2015, 7, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Okamoto T, Takagi Y, Kawamoto E, Park EJ, Usuda H, Wada K, Shimaoka M, Exp. Cell Res 2018, 367, 264. [DOI] [PubMed] [Google Scholar]
- [125].Sridharan R, Ryan EJ, Kearney CJ, Kelly DJ, O’Brien FJ, ACS Biomater. Sci. Eng 2019, 5, 544. [DOI] [PubMed] [Google Scholar]
- [126].Sridharan R, Cavanagh B, Cameron AR, Kelly DJ, O’Brien FJ, Acta Biomater. 2019, 89, 47. [DOI] [PubMed] [Google Scholar]
- [127].Jain N, Vogel V, Nat. Mater 2018, 17, 1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Donadon M, Torzilli G, Cortese N, Soldani C, Di Tommaso L, Franceschini B, Carriero R, Barbagallo M, Rigamonti A, Anselmo A, Colombo FS, Maggi G, Lleo A, Cibella J, Peano C, Kunderfranco P, Roncalli M, Mantovani A, Marchesi F, J. Exp. Med 2020, 217, e20191847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].McWhorter FY, Wang T, Nguyen P, Chung T, Liu WF, Proc. Natl. Acad. Sci. USA 2013, 110, 17253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Garg K, Pullen NA, Oskeritzian CA, Ryan JJ, Bowlin GL, Biomaterials 2013, 34, 4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Tylek T, Blum C, Hrynevich A, Schlegelmilch K, Schilling T, Dalton PD, Groll J, Biofabrication 2020, 12, 025007. [DOI] [PubMed] [Google Scholar]
- [132].Madden LR, Mortisen DJ, Sussman EM, Dupras SK, Fugate JA, Cuy JL, Hauch KD, Laflamme MA, Murry CE, Ratner BD, Proc. Natl. Acad. Sci. USA 2010, 107, 15211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Bose S, Volpatti LR, Thiono D, Yesilyurt V, McGladrigan C, Tang Y, Facklam A, Wang A, Jhunjhunwala S, Veiseh O, Hollister-Lock J, Bhattacharya C, Weir GC, Greiner DL, Langer R, Anderson DG, Nat. Biomed. Eng 2020, 4, 814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Xu C, Xiao L, Cao Y, He Y, Lei C, Xiao Y, Sun W, Ahadian S, Zhou X, Khademhosseini A, Ye Q, Nano Res. 2020, 13, 2323. [Google Scholar]
- [135].Aderem A, Underhill DM, Annu. Rev. Immunol 1999, 17, 593. [DOI] [PubMed] [Google Scholar]
- [136].Nel AE, Madler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M, Nat. Mater 2009, 8, 543. [DOI] [PubMed] [Google Scholar]
- [137].Rabinovitch M, Trends Cell Biol. 1995, 5, 85. [DOI] [PubMed] [Google Scholar]
- [138].Xia Z, Triffitt JT, Biomed. Mater 2006, 1, R1. [DOI] [PubMed] [Google Scholar]
- [139].Doshi N, Mitragotri S, PLoS One 2010, 5, e10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Walkey CD, Olsen JB, Guo H, Emili A, Chan WCW, J. Am. Chem. Soc 2012, 134, 2139. [DOI] [PubMed] [Google Scholar]
- [141].Oyewumi MO, Kumar A, Cui Z, Expert Rev. Vaccines 2010, 9, 1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Champion JA, Katare YK, Mitragotri S, J. Controlled Release 2007, 121, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Mitragotri S, Lahann J, Nat. Mater 2009, 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Donaldson K, Murphy FA, Duffin R, Poland CA, Part. Fibre Toxicol 2010, 7, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Doshi N, Swiston AJ, Gilbert JB, Alcaraz ML, Cohen RE, Rubner MF, Mitragotri S, Adv. Mater 2011, 23, H105. [DOI] [PubMed] [Google Scholar]
- [146].Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE, Nat. Nanotechnol 2007, 2, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Champion JA, Mitragotri S, Proc. Natl. Acad. Sci. USA 2006, 103, 4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Veiseh O, Doloff JC, Ma M, Vegas AJ, Tam HH, Bader AR, Li J, Langan E, Wyckoff J, Loo WS, Jhunjhunwala S, Chiu A, Siebert S, Tang K, Hollister-Lock J, Aresta-Dasilva S, Bochenek M, Mendoza-Elias J, Wang Y, Qi M, Lavin DM, Chen M, Dholakia N, Thakrar R, Lacík I, Weir GC, Oberholzer J, Greiner DL, Langer R, Anderson DG, Nat. Mater 2015, 14, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Anselmo AC, Zhang M, Kumar S, Vogus DR, Menegatti S, Helgeson ME, Mitragotri S, ACS Nano 2015, 9, 3169. [DOI] [PubMed] [Google Scholar]
- [150].Palomba R, Palange AL, Rizzuti IF, Ferreira M, Cervadoro A, Barbato MG, Canale C, Decuzzi P, ACS Nano 2018, 12, 1433. [DOI] [PubMed] [Google Scholar]
- [151].Sosale NG, Rouhiparkouhi T, Bradshaw AM, Dimova R, Lipowsky R, Discher DE, Blood 2015, 125, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Bohner M, Galea L, Doebelin N, J. Eur. Ceram. Soc 2012, 32, 2663. [Google Scholar]
- [153].Zamboni F, Vieira S, Reis RL, Miguel Oliveira J, Collins MN, Prog. Mater. Sci 2018, 97, 97. [Google Scholar]
- [154].Whitelock JM, Iozzo RV, Chem. Rev 2005, 105, 2745. [DOI] [PubMed] [Google Scholar]
- [155].Sommerfeld SD, Cherry C, Schwab RM, Chung L, Maestas DR, Laffont P, Stein JE, Tam A, Ganguly S, Housseau F, Taube JM, Pardoll DM, Cahan P, Elisseeff JH, Sci. Immunol 2019, 4, eaax4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Zhou Y, Wu C, Chang J, Mater. Today 2019, 24, 41. [Google Scholar]
- [157].Li J, Wang G, Wang D, Wu Q, Jiang X, Liu X, J. Colloid Interface Sci 2014, 436, 160. [DOI] [PubMed] [Google Scholar]
- [158].Shen Y, Liu W, Wen C, Pan H, Wang T, Darvell BW, Lu WW, Huang W, J. Mater. Chem 2012, 22, 8662. [Google Scholar]
- [159].Shen Y, Liu W, Lin K, Pan H, Darvell BW, Peng S, Wen C, Deng L, Lu WW, Chang J, Langmuir 2011, 27, 2701. [DOI] [PubMed] [Google Scholar]
- [160].Wu H, Yin Y, Hu X, Peng C, Liu Y, Li Q, Huang W, Huang Q, ACS Biomater. Sci. Eng 2019, 5, 5548. [DOI] [PubMed] [Google Scholar]
- [161].Gordon S, Plüddemann A, Martinez Estrada F, Immunol. Rev 2014, 262, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Taylor DA, Sampaio LC, Ferdous Z, Gobin AS, Taite LJ, Acta Biomater. 2018, 74, 74. [DOI] [PubMed] [Google Scholar]
- [163].Christman KL, Science 2019, 363, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].De A, Acta Biochim. Biophys. Sin 2011, 43, 745. [DOI] [PubMed] [Google Scholar]
- [165].Li J, Liu W, Kilian D, Zhang X, Gelinsky M, Chu PK, Mater. Horiz 2019, 6, 1271. [Google Scholar]
- [166].Liu W, Li J, Cheng M, Wang Q, Qian Y, Yeung KWK, Chu PK, Zhang X, Biomaterials 2019, 208, 8. [DOI] [PubMed] [Google Scholar]
- [167].Shi M, Chen Z, Farnaghi S, Friis T, Mao X, Xiao Y, Wu C, Acta Biomater. 2016, 30, 334. [DOI] [PubMed] [Google Scholar]
- [168].Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, Hainzl A, Schatz S, Qi Y, Schlecht A, Weiss JM, Wlaschek M, Sunderkötter C, Scharffetter-Kochanek K, J. Clin. Invest 2011, 121, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Pan C, Chen L, Wu R, Shan H, Zhou Z, Lin Y, Yu X, Yan L, Wu C, J. Mater. Chem. B 2018, 6, 8115. [DOI] [PubMed] [Google Scholar]
- [170].Sugimoto J, Romani AM, Valentin-Torres AM, Luciano AA, Ramirez Kitchen CM, Funderburg N, Mesiano S, Bernstein HB, J. Immunol 2012, 188, 6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Wu C, Chen Z, Wu Q, Yi D, Friis T, Zheng X, Chang J, Jiang X, Xiao Y, Biomaterials 2015, 71, 35. [DOI] [PubMed] [Google Scholar]
- [172].Yin Y, Huang Q, Yang M, Xiao J, Wu H, Liu Y, Li Q, Huang W, Lei G, Zhou K, ACS Biomater. Sci. Eng 2020, 6, 3005. [DOI] [PubMed] [Google Scholar]
- [173].Liu W, Golshan NH, Deng X, Hickey DJ, Zeimer K, Li H, Webster TJ, Nanoscale 2016, 8, 15783. [DOI] [PubMed] [Google Scholar]
- [174].Bai L, Liu Y, Zhang X, Huang X, Yao X, Hang R, Tang B, Xiao Y, Nanoscale 2019, 11, 5920. [DOI] [PubMed] [Google Scholar]
- [175].Gentleman E, Fredholm YC, Jell G, Lotfibakhshaiesh N, O’Donnell MD, Hill RG, Stevens MM, Biomaterials 2010, 31, 3949. [DOI] [PubMed] [Google Scholar]
- [176].Buache E, Velard F, Bauden E, Guillaume C, Jallot E, Nedelec JM, Laurent-Maquin D, Laquerriere P, Acta Biomater. 2012, 8, 3113. [DOI] [PubMed] [Google Scholar]
- [177].Wen J, Li J, Pan H, Zhang W, Zeng D, Xu L, Wu Q, Zhang X, Liu X, Jiang X, J. Mater. Chem. B 2015, 3, 4790. [DOI] [PubMed] [Google Scholar]
- [178].Liu W, Li J, Cheng M, Wang Q, Yeung KWK, Chu PK, Zhang X, Adv. Sci 2018, 5, 1800749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Shankar AH, Prasad AS, Am. J. Clin. Nutr 1998, 68, 447S. [DOI] [PubMed] [Google Scholar]
- [180].Wang J, Zhou H, Guo G, Tan J, Wang Q, Tang J, Liu W, Shen H, Li J, Zhang X, ACS Appl. Mater. Interfaces 2017, 9, 33609. [DOI] [PubMed] [Google Scholar]
- [181].Qian G, Lu T, Zhang J, Liu R, Wang Z, Yu B, Li H, Shi H, Ye J, Appl. Mater. Today 2020, 19, 100615. [Google Scholar]
- [182].Bygd HC, Forsmark KD, Bratlie KM, Biomaterials 2015, 56, 187. [DOI] [PubMed] [Google Scholar]
- [183].Lopez-Silva TL, Leach DG, Azares A, Li IC, Woodside DG, Hartgerink JD, Biomaterials 2020, 231, 119667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Fuchs A-K, Syrovets T, Haas KA, Loos C, Musyanovych A, Mailänder V, Landfester K, Simmet T, Biomaterials 2016, 85, 78. [DOI] [PubMed] [Google Scholar]
- [185].Zeng D, Zhang X, Wang X, Huang Q, Wen J, Miao X, Peng L, Li Y, Jiang X, Artif. Cells, Nanomed., Biotechnol 2018, 46, 1425. [DOI] [PubMed] [Google Scholar]
- [186].Mulens-Arias V, Rojas JM, Pérez-Yagüe S, Morales MP, Barber DF, Biomaterials 2015, 52, 494. [DOI] [PubMed] [Google Scholar]
- [187].Zaveri TD, Lewis JS, Dolgova NV, Clare-Salzler MJ, Keselowsky BG, Biomaterials 2014, 35, 3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Schirmer L, Atallah P, Werner C, Freudenberg U, Adv. Healthcare Mater 2016, 5, 3157. [DOI] [PubMed] [Google Scholar]
- [189].Liu R, Chen S, Huang P, Liu G, Luo P, Li Z, Xiao Y, Chen Z, Chen Z, Adv. Funct. Mater 2020, 30, 1910672. [Google Scholar]
- [190].Cardilo-Reis L, Gruber S, Schreier SM, Drechsler M, Papac-Milicevic N, Weber C, Wagner O, Stangl H, Soehnlein O, Binder CJ, EMBO Mol. Med 2012, 4, 1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Mantovani A, Dinarello CA, Molgora M, Garlanda C, Immunity 2019, 50, 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Spiller KL, Nassiri S, Witherel CE, Anfang RR, Ng J, Nakazawa KR, Yu T, Vunjak-Novakovic G, Biomaterials 2015, 37, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].He X-T, Li X, Xia Y, Yin Y, Wu R-X, Sun H-H, Chen F-M, Acta Biomater. 2019, 88, 162. [DOI] [PubMed] [Google Scholar]
- [194].Riabov V, Salazar F, Htwe SS, Gudima A, Schmuttermaier C, Barthes J, Knopf-Marques H, Klüter H, Ghaemmaghami AM, Vrana NE, Kzhyshkowska J, Acta Biomater. 2017, 53, 389. [DOI] [PubMed] [Google Scholar]
- [195].Cha B-H, Shin SR, Leijten J, Li Y-C, Singh S, Liu JC, Annabi N, Abdi R, Dokmeci MR, Vrana NE, Ghaemmaghami AM, Khademhosseini A, Adv. Healthcare Mater 2017, 6, 1700289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Braune J, Weyer U, Hobusch C, Mauer J, Brüning JC, Bechmann I, Gericke M, J. Immunol 2017, 198, 2927. [DOI] [PubMed] [Google Scholar]
- [197].Reeves ARD, Spiller KL, Freytes DO, Vunjak-Novakovic G, Kaplan DL, Biomaterials 2015, 73, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Ariganello MB, Simionescu DT, Labow RS, Michael Lee J, Biomaterials 2011, 32, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Sharma G, Valenta DT, Altman Y, Harvey S, Xie H, Mitragotri S, Smith JW, J. Controlled Release 2010, 147, 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Chellat F, Merhi Y, Moreau A, Yahia LH, Biomaterials 2005, 26, 7260. [DOI] [PubMed] [Google Scholar]
- [201].Ahsan F, Rivas IP, Khan MA, Torres Suárez AI, J. Controlled Release 2002, 79, 29. [DOI] [PubMed] [Google Scholar]
- [202].Muthiah M, Park I-K, Cho C-S, Biotechnol. Adv 2013, 31, 1224. [DOI] [PubMed] [Google Scholar]
- [203].Storm G, Belliot SO, Daemen T, Lasic DD, Adv. Drug Delivery Rev 1995, 17, 31. [Google Scholar]
- [204].Moon JJ, Huang B, Irvine DJ, Adv. Mater 2012, 24, 3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Jiang S, Cao Z, Adv. Mater 2010, 22, 920. [DOI] [PubMed] [Google Scholar]
- [206].Rudt S, Müller RH, J. Controlled Release 1993, 25, 51. [Google Scholar]
- [207].Thiele L, Rothen-Rutishauser B, Jilek S, Wunderli-Allenspach H, Merkle HP, Walter E, J. Controlled Release 2001, 76, 59. [DOI] [PubMed] [Google Scholar]
- [208].Thiele L, Merkle HP, Walter E, Pharm. Res 2003, 20, 221. [DOI] [PubMed] [Google Scholar]
- [209].Qie Y, Yuan H, von Roemeling CA, Chen Y, Liu X, Shih KD, Knight JA, Tun HW, Wharen RE, Jiang W, Kim BYS, Sci. Rep 2016, 6, 26269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Molinaro R, Corbo C, Martinez JO, Taraballi F, Evangelopoulos M, Minardi S, Yazdi IK, Zhao P, De Rosa E, Sherman MB, De Vita A, Toledano Furman NE, Wang X, Parodi A, Tasciotti E, Nat. Mater 2016, 15, 1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Thamphiwatana S, Angsantikul P, Escajadillo T, Zhang Q, Olson J, Luk BT, Zhang S, Fang RH, Gao W, Nizet V, Zhang L, Proc. Natl. Acad. Sci. USA 2017, 114, 11488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Fontana F, Albertini S, Correia A, Kemell M, Lindgren R, Mäkilä E, Salonen J, Hirvonen JT, Ferrari F, Santos HA, Adv. Funct. Mater 2018, 28, 1801355. [Google Scholar]
- [213].Lai J, Deng G, Sun Z, Peng X, Li J, Gong P, Zhang P, Cai L, Biomaterials 2019, 211, 48. [DOI] [PubMed] [Google Scholar]
- [214].Cao H, Dan Z, He X, Zhang Z, Yu H, Yin Q, Li Y, ACS Nano 2016, 10, 7738. [DOI] [PubMed] [Google Scholar]
- [215].Li R, He Y, Zhu Y, Jiang L, Zhang S, Qin J, Wu Q, Dai W, Shen S, Pang Z, Wang J, Nano Lett. 2019, 19, 124. [DOI] [PubMed] [Google Scholar]
- [216].Zhang Y, Cai K, Li C, Guo Q, Chen Q, He X, Liu L, Zhang Y, Lu Y, Chen X, Sun T, Huang Y, Cheng J, Jiang C, Nano Lett. 2018, 18, 1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Zhuang J, Duan Y, Zhang Q, Gao W, Li S, Fang RH, Zhang L, Nano Lett. 2020, 20, 4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Wang D, Dong H, Li M, Cao Y, Yang F, Zhang K, Dai W, Wang C, Zhang X, ACS Nano 2018, 12, 5241. [DOI] [PubMed] [Google Scholar]
- [219].Bu L-L, Rao L, Yu G-T, Chen L, Deng W-W, Liu J-F, Wu H, Meng Q-F, Guo S-S, Zhao X-Z, Zhang W-F, Chen G, Gu Z, Liu W, Sun Z-J, Adv. Funct. Mater 2019, 29, 1807733. [Google Scholar]
- [220].Rao L, Meng Q-F, Huang Q, Wang Z, Yu G-T, Li A, Ma W, Zhang N, Guo S-S, Zhao X-Z, Liu K, Yuan Y, Liu W, Adv. Funct. Mater 2018, 28, 1803531. [Google Scholar]
- [221].Dehaini D, Wei X, Fang RH, Masson S, Angsantikul P, Luk BT, Zhang Y, Ying M, Jiang Y, Kroll AV, Gao W, Zhang L, Adv. Mater 2017, 29, 1606209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Chen Z, Wu C, Gu W, Klein T, Crawford R, Xiao Y, Biomaterials 2014, 35, 1507. [DOI] [PubMed] [Google Scholar]
- [223].Wang J, Liu D, Guo B, Yang X, Chen X, Zhu X, Fan Y, Zhang X, Acta Biomater. 2017, 51, 447. [DOI] [PubMed] [Google Scholar]
- [224].Wolf MT, Dearth CL, Ranallo CA, LoPresti ST, Carey LE, Daly KA, Brown BN, Badylak SF, Biomaterials 2014, 35, 6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Brown BN, Londono R, Tottey S, Zhang L, Kukla KA, Wolf MT, Daly KA, Reing JE, Badylak SF, Acta Biomater. 2012, 8, 978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Goldring MB, Otero M, Curr. Opin. Rheumatol 2011, 23, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Loeser RF, Arthritis Rheum. 2006, 54, 1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Ballotta V, Driessen-Mol A, Bouten CVC, Baaijens FPT, Biomaterials 2014, 35, 4919. [DOI] [PubMed] [Google Scholar]
- [229].Pugin J, Dunn I, Jolliet P, Tassaux D, Magnenat J-L, Nicod LP, Chevrolet J-C, Am. J. Physiol.: Lung Cell. Mol. Physiol 1998, 275, L1040. [DOI] [PubMed] [Google Scholar]
- [230].van Haaften EE, Wissing TB, Kurniawan NA, Smits AIPM, Bouten CVC, Adv. Biosyst 2020, 4, 1900249. [DOI] [PubMed] [Google Scholar]
- [231].Kang H, Kim S, Wong DSH, Jung HJ, Lin S, Zou K, Li R, Li G, Dravid VP, Bian L, Nano Lett. 2017, 17, 6415. [DOI] [PubMed] [Google Scholar]
- [232].Kang H, Jung HJ, Kim SK, Wong DSH, Lin S, Li G, Dravid VP, Bian L, ACS Nano 2018, 12, 5978. [DOI] [PubMed] [Google Scholar]
- [233].Wosik J, Chen W, Qin K, Ghobrial RM, Kubiak JZ, Kloc M, Biophys. J 2018, 114, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Schuerle S, Vizcarra IA, Moeller J, Sakar MS, Özkale B, Lindo AM, Mushtaq F, Schoen I, Pané S, Vogel V, Nelson BJ, Sci. Rob 2017, 2, eaah6094. [DOI] [PubMed] [Google Scholar]
- [235].Kang H, Zhang K, Wong DSH, Han F, Li B, Bian L, Biomaterials 2018, 178, 681. [DOI] [PubMed] [Google Scholar]
- [236].Wang H, Morales R-TT, Cui X, Huang J, Qian W, Tong J, Chen W, Adv. Healthcare Mater 2019, 8, 1801234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Li R, Serrano JC, Xing H, Lee TA, Azizgolshani H, Zaman M, Kamm RD, Mol. Biol. Cell 2018, 29, 1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Hoare JI, Rajnicek AM, McCaig CD, Barker RN, Wilson HM, J. Leukocyte Biol 2016, 99, 1141. [DOI] [PubMed] [Google Scholar]
- [239].Bronte V, Murray PJ, Nat. Med 2015, 21, 117. [DOI] [PubMed] [Google Scholar]
- [240].Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss RH, Timke C, Umansky L, Klapproth K, Schäkel K, Garbi N, Jäger D, Weitz J, Schmitz-Winnenthal H, Hämmerling GJ, Beckhove P, Cancer Cell 2013, 24, 589. [DOI] [PubMed] [Google Scholar]
- [241].Lock A, Cornish J, Musson DS, J. Funct. Biomater 2019, 10, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Hall JB, Dobrovolskaia MA, Patri AK, McNeil SE, Nanomedicine 2007, 2, 789. [DOI] [PubMed] [Google Scholar]
- [243].Willingham SB, Volkmer J-P, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD, Lovelace P, Scheeren FA, Chao MP, Weiskopf K, Tang C, Volkmer AK, Naik TJ, Storm TA, Mosley AR, Edris B, Schmid SM, Sun CK, Chua M-S, Murillo O, Rajendran P, Cha AC, Chin RK, Kim D, Adorno M, Raveh T, Tseng D, Jaiswal S, Enger PØ, Steinberg GK, Li G, So SK, Majeti R, Harsh GR, van de Rijn M, Teng NNH, Sunwoo JB, Alizadeh AA, Clarke MF, Weissman IL, Proc. Natl. Acad. Sci. USA 2012, 109, 6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P, Nat. Rev. Clin. Oncol 2017, 14, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Qian B-Z, Pollard JW, Cell 2010, 141, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Mantovani A, Allavena P, J. Exp. Med 2015, 212, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Mantovani A, Sozzani S, Locati M, Allavena P, Sica A, Trends Immunol. 2002, 23, 549. [DOI] [PubMed] [Google Scholar]
- [248].Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, Lee KM, Kim JI, Markmann JF, Marinelli B, Panizzi P, Lee WW, Iwamoto Y, Milstein S, Epstein-Barash H, Cantley W, Wong J, Cortez-Retamozo V, Newton A, Love K, Libby P, Pittet MJ, Swirski FK, Koteliansky V, Langer R, Weissleder R, Anderson DG, Nahrendorf M, Nat. Biotechnol 2011, 29, 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, Fowler KJ, Lockhart AC, Suresh R, Tan BR, Lim K-H, Fields RC, Strasberg SM, Hawkins WG, DeNardo DG, Goedegebuure SP, Linehan DC, Lancet Oncol. 2016, 17, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, Jan M, Cha AC, Chan CK, Tan BT, Park CY, Zhao F, Kohrt HE, Malumbres R, Briones J, Gascoyne RD, Lossos IS, Levy R, Weissman IL, Majeti R, Cell 2010, 142, 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, Jones T, Jucknischke U, Scheiblich S, Kaluza K, Gorr IH, Walz A, Abiraj K, Cassier PA, Sica A, Gomez-Roca C, de Visser KE, Italiano A, Le Tourneau C, Delord J-P, Levitsky H, Blay J-Y, Rüttinger D, Cancer Cell 2014, 25, 846. [DOI] [PubMed] [Google Scholar]
- [252].Cassetta L, Pollard JW, Nat. Rev. Drug Discovery 2018, 17, 887. [DOI] [PubMed] [Google Scholar]
- [253].Irvine DJ, Dane EL, Nat. Rev. Immunol 2020, 20, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Wang H, Mooney DJ, Nat. Mater 2018, 17, 761. [DOI] [PubMed] [Google Scholar]
- [255].Martin JD, Cabral H, Stylianopoulos T, Jain RK, Nat. Rev. Clin. Oncol 2020, 17, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Chen Q, Chen M, Liu Z, Chem. Soc. Rev 2019, 48, 5506. [DOI] [PubMed] [Google Scholar]
- [257].Sica A, Bronte V, J. Clin. Invest 2007, 117, 1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Lu X, Miao L, Gao W, Chen Z, McHugh KJ, Sun Y, Tochka Z, Tomasic S, Sadtler K, Hyacinthe A, Huang Y, Graf T, Hu Q, Sarmadi M, Langer R, Anderson DG, Jaklenec A, Sci. Transl. Med 2020, 12, eaaz6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, Pajarinen JS, Nejadnik H, Goodman S, Moseley M, Coussens LM, Daldrup-Link HE, Nat. Nanotechnol 2016, 11, 986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Chen Q, Wang C, Zhang X, Chen G, Hu Q, Li H, Wang J, Wen D, Zhang Y, Lu Y, Yang G, Jiang C, Wang J, Dotti G, Gu Z, Nat. Nanotechnol 2019, 14, 89. [DOI] [PubMed] [Google Scholar]
- [261].Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, Kohler RH, Pittet MJ, Weissleder R, Nat. Biomed. Eng 2018, 2, 578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Chen Q, Chen J, Yang Z, Xu J, Xu L, Liang C, Han X, Liu Z, Adv. Mater 2019, 31, 1802228. [DOI] [PubMed] [Google Scholar]
- [263].Yang G, Xu L, Chao Y, Xu J, Sun X, Wu Y, Peng R, Liu Z, Nat. Commun 2017, 8, 902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Chen Q, Xu L, Chen J, Yang Z, Liang C, Yang Y, Liu Z, Biomaterials 2017, 148, 69. [DOI] [PubMed] [Google Scholar]
- [265].Song X, Xu J, Liang C, Chao Y, Jin Q, Wang C, Chen M, Liu Z, Nano Lett. 2018, 18, 6360. [DOI] [PubMed] [Google Scholar]
- [266].Meng Z, Zhou X, Xu J, Han X, Dong Z, Wang H, Zhang Y, She J, Xu L, Wang C, Liu Z, Adv. Mater 2019, 31, 1900927. [DOI] [PubMed] [Google Scholar]
- [267].Wang H, Han X, Dong Z, Xu J, Wang J, Liu Z, Adv. Funct. Mater 2019, 29, 1902440. [Google Scholar]
- [268].Zou M-Z, Liu W-L, Gao F, Bai X-F, Chen H-S, Zeng X, Zhang X-Z, Adv. Mater 2019, 31, 1904495. [DOI] [PubMed] [Google Scholar]
- [269].Li C-X, Zhang Y, Dong X, Zhang L, Liu M-D, Li B, Zhang M-K, Feng J, Zhang X-Z, Adv. Mater 2019, 31, 1807211. [DOI] [PubMed] [Google Scholar]
- [270].Huang Y, Mei C, Tian Y, Nie T, Liu Z, Chen T, NPG Asia Mater 2018, 10, 1002. [Google Scholar]
- [271].Tang J, Zhang R, Guo M, Zhou H, Zhao Y, Liu Y, Wu Y, Chen C, J. Controlled Release 2020, 320, 293. [DOI] [PubMed] [Google Scholar]
- [272].Parayath NN, Parikh A, Amiji MM, Nano Lett. 2018, 18, 3571. [DOI] [PubMed] [Google Scholar]
- [273].Ovais M, Guo M, Chen C, Adv. Mater 2019, 31, 1808303. [DOI] [PubMed] [Google Scholar]
- [274].Sylvestre M, Crane CA, Pun SH, Adv. Mater 2020, 32, 1902007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Dreaden EC, Mwakwari SC, Austin LA, Kieffer MJ, Oyelere AK, El-Sayed MA, Small 2012, 8, 2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Wang J, Lee JS, Kim D, Zhu L, ACS Appl. Mater. Interfaces 2017, 9, 39971. [DOI] [PubMed] [Google Scholar]
- [277].He L, Nie T, Xia X, Liu T, Huang Y, Wang X, Chen T, Adv. Funct. Mater 2019, 29, 1901240. [Google Scholar]
- [278].Miller MA, Zheng Y-R, Gadde S, Pfirschke C, Zope H, Engblom C, Kohler RH, Iwamoto Y, Yang KS, Askevold B, Kolishetti N, Pittet M, Lippard SJ, Farokhzad OC, Weissleder R, Nat. Commun 2015, 6, 8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Shen S, Li H-J, Chen K-G, Wang Y-C, Yang X-Z, Lian Z-X, Du J-Z, Wang J, Nano Lett. 2017, 17, 3822. [DOI] [PubMed] [Google Scholar]
- [280].Shi C, Liu T, Guo Z, Zhuang R, Zhang X, Chen X, Nano Lett. 2018, 18, 7330. [DOI] [PubMed] [Google Scholar]
- [281].Wang Y, Lin Y-X, Qiao S-L, An H-W, Ma Y, Qiao Z-Y, Rajapaksha RPYJ, Wang H, Biomaterials 2017, 112, 153. [DOI] [PubMed] [Google Scholar]
- [282].Rajan R, Sabnani MK, Mavinkurve V, Shmeeda H, Mansouri H, Bonkoungou S, Le AD, Wood LM, Gabizon AA, La-Beck NM, J. Controlled Release 2018, 271, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Kulkarni A, Chandrasekar V, Natarajan SK, Ramesh A, Pandey P, Nirgud J, Bhatnagar H, Ashok D, Ajay AK, Sengupta S, Nat. Biomed. Eng 2018, 2, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Conde J, Bao C, Tan Y, Cui D, Edelman ER, Azevedo HS, Byrne HJ, Artzi N, Tian F, Adv. Funct. Mater 2015, 25, 4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Liu L, Yi H, He H, Pan H, Cai L, Ma Y, Biomaterials 2017, 134, 166. [DOI] [PubMed] [Google Scholar]
- [286].Huang Z, Zhang Z, Jiang Y, Zhang D, Chen J, Dong L, Zhang J, J. Controlled Release 2012, 158, 286. [DOI] [PubMed] [Google Scholar]
- [287].Song M, Liu T, Shi C, Zhang X, Chen X, ACS Nano 2016, 10, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Yu G-T, Rao L, Wu H, Yang L-L, Bu L-L, Deng W-W, Wu L, Nan X, Zhang W-F, Zhao X-Z, Liu W, Sun Z-J, Adv. Funct. Mater 2018, 28, 1801389. [Google Scholar]
- [289].Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW, Nat. Rev. Mater 2016, 1, 16014. [Google Scholar]
- [290].Shields CW, Evans MA, Wang LL-W, Baugh N, Iyer S, Wu D, Zhao Z, Pusuluri A, Ukidve A, Pan DC, Mitragotri S, Sci. Adv 2020, 6, eaaz6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Evans MA, Shields CW IV, Krishnan V, Wang LL-W, Zhao Z, Ukidve A, Lewandowski M, Gao Y, Mitragotri S, Adv. Ther 2020, 3, 1900162. [Google Scholar]
- [292].Evans MA, Huang P-J, Iwamoto Y, Ibsen KN, Chan EM, Hitomi Y, Ford PC, Mitragotri S, Chem. Sci 2018, 9, 3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Dou H, Destache CJ, Morehead JR, Mosley RL, Boska MD, Kingsley J, Gorantla S, Poluektova L, Nelson JA, Chaubal M, Werling J, Kipp J, Rabinow BE, Gendelman HE, Blood 2006, 108, 2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Choi M-R, Stanton-Maxey KJ, Stanley JK, Levin CS, Bardhan R, Akin D, Badve S, Sturgis J, Robinson JP, Bashir R, Halas NJ, Clare SE, Nano Lett. 2007, 7, 3759. [DOI] [PubMed] [Google Scholar]
- [295].Choi J, Kim H-Y, Ju EJ, Jung J, Park J, Chung H-K, Lee JS, Lee JS, Park HJ, Song SY, Jeong S-Y, Choi EK, Biomaterials 2012, 33, 4195. [DOI] [PubMed] [Google Scholar]
- [296].Li Z, Huang H, Tang S, Li Y, Yu X-F, Wang H, Li P, Sun Z, Zhang H, Liu C, Chu PK, Biomaterials 2016, 74, 144. [DOI] [PubMed] [Google Scholar]
- [297].Li Z, Shao J, Luo Q, Yu X-F, Xie H, Fu H, Tang S, Wang H, Han G, Chu PK, Biomaterials 2017, 133, 37. [DOI] [PubMed] [Google Scholar]
- [298].Shi M, Zhang P, Zhao Q, Shen K, Qiu Y, Xiao Y, Yuan Q, Zhang Y, Small 2020, 16, 1905185. [DOI] [PubMed] [Google Scholar]
- [299].Anselmo AC, Gilbert JB, Kumar S, Gupta V, Cohen RE, Rubner MF, Mitragotri S, J. Controlled Release 2015, 199, 29. [DOI] [PubMed] [Google Scholar]
- [300].Levy SB, Marshall B, Nat. Med 2004, 10, S122. [DOI] [PubMed] [Google Scholar]
- [301].Spellberg B, Bartlett JG, Gilbert DN, N. Engl. J. Med 2013, 368, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Stewart PS, William Costerton J, Lancet 2001, 358, 135. [DOI] [PubMed] [Google Scholar]
- [303].Davies D, Nat. Rev. Drug Discovery 2003, 2, 114. [DOI] [PubMed] [Google Scholar]
- [304].Arciola CR, Campoccia D, Montanaro L, Nat. Rev. Microbiol 2018, 16, 397. [DOI] [PubMed] [Google Scholar]
- [305].Thurlow LR, Hanke ML, Fritz T, Angle A, Aldrich A, Williams SH, Engebretsen IL, Bayles KW, Horswill AR, Kielian T, J. Immunol 2011, 186, 6585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].Foster TJ, Nat. Rev. Microbiol 2005, 3, 948. [DOI] [PubMed] [Google Scholar]
- [307].Yang C, Li J, Zhu C, Zhang Q, Yu J, Wang J, Wang Q, Tang J, Zhou H, Shen H, Acta Biomater. 2019, 89, 403. [DOI] [PubMed] [Google Scholar]
- [308].Leid JG, Shirtliff ME, Costerton JW, Stoodley P, Infect. Immun 2002, 70, 6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Arciola CR, Campoccia D, Speziale P, Montanaro L, Costerton JW, Biomaterials 2012, 33, 5967. [DOI] [PubMed] [Google Scholar]
- [310].Hou X, Zhang X, Zhao W, Zeng C, Deng B, McComb DW, Du S, Zhang C, Li W, Dong Y, Nat. Nanotechnol 2020, 15, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Yamada KJ, Heim CE, Xi X, Attri KS, Wang D, Zhang W, Singh PK, Bronich TK, Kielian T, PLoS Pathog. 2020, 16, e1008354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Novak ML, Koh TJ, Am. J. Pathol 2013, 183, 1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Claes L, Recknagel S, Ignatius A, Nat. Rev. Rheumatol 2012, 8, 133. [DOI] [PubMed] [Google Scholar]
- [314].Zhang C, Li Y, Wu Y, Wang L, Wang X, Du J, J. Biol. Chem 2013, 288, 1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [315].Warren GL, Hulderman T, Jensen N, McKinstry M, Mishra M, Luster MI, Simeonova PP, FASEB J. 2002, 16, 1630. [DOI] [PubMed] [Google Scholar]
- [316].Einhorn TA, Gerstenfeld LC, Nat. Rev. Rheumatol 2015, 11, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Wynn TA, Vannella KM, Immunity 2016, 44, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [318].Braga TT, Agudelo JSH, Camara NOS, Front. Immunol 2015, 6, 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [319].Novak ML, Koh TJ, J. Leukocyte Biol 2013, 93, 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Zhai D, Chen L, Chen Y, Zhu Y, Xiao Y, Wu C, Biomater. Sci 2020, 8, 4521. [DOI] [PubMed] [Google Scholar]
- [321].Mahon OR, Browe DC, Gonzalez-Fernandez T, Pitacco P, Whelan IT, Von Euw S, Hobbs C, Nicolosi V, Cunningham KT, Mills KHG, Kelly DJ, Dunne A, Biomaterials 2020, 239, 119833. [DOI] [PubMed] [Google Scholar]
- [322].He Y, Xu K, Li K, Yuan Z, Ding Y, Chen M, Lin C, Tao B, Li X, Zhang G, Liu P, Cai K, Appl. Mater. Today 2020, 20, 100673. [Google Scholar]
- [323].Bai J, Wang H, Chen H, Ge G, Wang M, Gao A, Tong L, Xu Y, Yang H, Pan G, Chu PK, Geng D, Biomaterials 2020, 255, 120197. [DOI] [PubMed] [Google Scholar]
- [324].Li J, Wen J, Li B, Li W, Qiao W, Shen J, Jin W, Jiang X, Yeung KWK, Chu PK, Adv. Sci 2018, 5, 1700678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [325].Hao S, Meng J, Zhang Y, Liu J, Nie X, Wu F, Yang Y, Wang C, Gu N, Xu H, Biomaterials 2017, 140, 16. [DOI] [PubMed] [Google Scholar]
- [326].Chen Z, Ni S, Han S, Crawford R, Lu S, Wei F, Chang J, Wu C, Xiao Y, Nanoscale 2017, 9, 706. [DOI] [PubMed] [Google Scholar]
- [327].Liang H, Jin C, Ma L, Feng X, Deng X, Wu S, Liu X, Yang C, ACS Appl. Mater. Interfaces 2019, 11, 41758. [DOI] [PubMed] [Google Scholar]
- [328].Gao A, Liao Q, Xie L, Wang G, Zhang W, Wu Y, Li P, Guan M, Pan H, Tong L, Chu PK, Wang H, Biomaterials 2020, 230, 119642. [DOI] [PubMed] [Google Scholar]
- [329].Kim J-W, Mahapatra C, Hong J-Y, Kim MS, Leong KW, Kim H-W, Hyun JK, Adv. Sci 2017, 4, 1700034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [330].Park J, Zhang Y, Saito E, Gurczynski SJ, Moore BB, Cummings BJ, Anderson AJ, Shea LD, Proc. Natl. Acad. Sci. USA 2019, 116, 14947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [331].Dai M, Sui B, Xue Y, Liu X, Sun J, Biomaterials 2018, 180, 91. [DOI] [PubMed] [Google Scholar]
- [332].Dai M, Sui B, Hua Y, Zhang Y, Bao B, Lin Q, Liu X, Zhu L, Sun J, Biomaterials 2020, 240, 119841. [DOI] [PubMed] [Google Scholar]
- [333].Sicari BM, Dziki JL, Siu BF, Medberry CJ, Dearth CL, Badylak SF, Biomaterials 2014, 35, 8605. [DOI] [PubMed] [Google Scholar]
- [334].Lohmann N, Schirmer L, Atallah P, Wandel E, Ferrer RA, Werner C, Simon JC, Franz S, Freudenberg U, Sci. Transl. Med 2017, 9, eaai9044. [DOI] [PubMed] [Google Scholar]
- [335].Li L, Qian Y, Jiang C, Lv Y, Liu W, Zhong L, Cai K, Li S, Yang L, Biomaterials 2012, 33, 3428. [DOI] [PubMed] [Google Scholar]
- [336].Olingy CE, San Emeterio CL, Ogle ME, Krieger JR, Bruce AC, Pfau DD, Jordan BT, Peirce SM, Botchwey EA, Sci. Rep 2017, 7, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [337].Wang Z, Cui Y, Wang J, Yang X, Wu Y, Wang K, Gao X, Li D, Li Y, Zheng X-L, Zhu Y, Kong D, Zhao Q, Biomaterials 2014, 35, 5700. [DOI] [PubMed] [Google Scholar]
- [338].Mokarram N, Merchant A, Mukhatyar V, Patel G, Bellamkonda RV, Biomaterials 2012, 33, 8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [339].Serhan CN, Savill J, Nat. Immunol 2005, 6, 1191. [DOI] [PubMed] [Google Scholar]
- [340].Watanabe S, Alexander M, Misharin AV, Budinger GRS, J. Clin. Invest 2019, 129, 2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [341].Firestein GS, Nature 2003, 423, 356. [DOI] [PubMed] [Google Scholar]
- [342].Smolen JS, Aletaha D, Nat. Rev. Rheumatol 2015, 11, 276. [DOI] [PubMed] [Google Scholar]
- [343].Firestein GS, McInnes IB, Immunity 2017, 46, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [344].McInnes IB, Schett G, Nat. Rev. Immunol 2007, 7, 429. [DOI] [PubMed] [Google Scholar]
- [345].Kim J, Kim HY, Song SY, Go S.-h., Sohn HS, Baik S, Soh M, Kim K, Kim D, Kim H-C, Lee N, Kim B-S, Hyeon T, ACS Nano 2019, 13, 3206. [DOI] [PubMed] [Google Scholar]
- [346].Zhou H.-f., Yan H, Hu Y, Springer LE, Yang X, Wickline SA, Pan D, Lanza GM, Pham CTN, ACS Nano 2014, 8, 7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [347].Tabas I, Nat. Rev. Immunol 2010, 10, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [348].Glass CK, Witztum JL, Cell 2001, 104, 503. [DOI] [PubMed] [Google Scholar]
- [349].Flores AM, Hosseini-Nassab N, Jarr K-U, Ye J, Zhu X, Wirka R, Koh AL, Tsantilas P, Wang Y, Nanda V, Kojima Y, Zeng Y, Lotfi M, Sinclair R, Weissman IL, Ingelsson E, Smith BR, Leeper NJ, Nat. Nanotechnol 2020, 15, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [350].Klyachko NL, Polak R, Haney MJ, Zhao Y, Gomes Neto RJ, Hill MC, Kabanov AV, Cohen RE, Rubner MF, Batrakova EV, Biomaterials 2017, 140, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [351].Gao A, Hang R, Li W, Zhang W, Li P, Wang G, Bai L, Yu X-F, Wang H, Tong L, Chu PK, Biomaterials 2017, 140, 201. [DOI] [PubMed] [Google Scholar]
- [352].Hirst SM, Karakoti AS, Tyler RD, Sriranganathan N, Seal S, Reilly CM, Small 2009, 5, 2848. [DOI] [PubMed] [Google Scholar]
- [353].Zheng K, Torre E, Bari A, Taccardi N, Cassinelli C, Morra M, Fiorilli S, Vitale-Brovarone C, Iviglia G, Boccaccini AR, Mater. Today Bio 2020, 5, 100041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [354].Tian M, Ticer T, Wang Q, Walker S, Pham A, Suh A, Busatto S, Davidovich I, Al-Kharboosh R, Lewis-Tuffin L, Ji B, Quinones-Hinojosa A, Talmon Y, Shapiro S, Rückert F, Wolfram J, Small 2020, 16, 1904064. [DOI] [PubMed] [Google Scholar]
- [355].Lee TT, García JR, Paez JI, Singh A, Phelps EA, Weis S, Shafiq Z, Shekaran A, del Campo A, García AJ, Nat. Mater 2015, 14, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [356].Dormont F, Brusini R, Cailleau C, Reynaud F, Peramo A, Gendron A, Mougin J, Gaudin F, Varna M, Couvreur P, Sci. Adv 2020, 6, eaaz5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [357].Irvine DJ, Nat. Rev. Mater 2016, 1, 15008. [Google Scholar]
- [358].Hubbell JA, Thomas SN, Swartz MA, Nature 2009, 462, 449. [DOI] [PubMed] [Google Scholar]
- [359].Pulendran B, Ahmed R, Nat. Immunol 2011, 12, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [360].Roy K, Mao H-Q, Huang SK, Leong KW, Nat. Med 1999, 5, 387. [DOI] [PubMed] [Google Scholar]
- [361].Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu H, Huang B, Sohail M, Luo S, Ho Um S, Khant H, Goodwin JT, Ramos J, Chiu W, Irvine DJ, Nat. Mater 2011, 10, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [362].Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan RJ, García-Sastre A, Compans R, Pulendran B, Nature 2011, 470, 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [363].Leleux J, Roy K, Adv. Healthcare Mater 2013, 2, 72. [DOI] [PubMed] [Google Scholar]
- [364].Li AW, Sobral MC, Badrinath S, Choi Y, Graveline A, Stafford AG, Weaver JC, Dellacherie MO, Shih T-Y, Ali OA, Kim J, Wucherpfennig KW, Mooney DJ, Nat. Mater 2018, 17, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [365].Moyer TJ, Kato Y, Abraham W, Chang JYH, Kulp DW, Watson N, Turner HL, Menis S, Abbott RK, Bhiman JN, Melo MB, Simon HA, Herrera-De la Mata S, Liang S, Seumois G, Agarwal Y, Li N, Burton DR, Ward AB, Schief WR, Crotty S, Irvine DJ, Nat. Med 2020, 26, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [366].Miao L, Li L, Huang Y, Delcassian D, Chahal J, Han J, Shi Y, Sadtler K, Gao W, Lin J, Doloff JC, Langer R, Anderson DG, Nat. Biotechnol 2019, 37, 1174. [DOI] [PubMed] [Google Scholar]
- [367].Li X, Wang X, Ito A, Chem. Soc. Rev 2018, 47, 4954. [DOI] [PubMed] [Google Scholar]
- [368].Randolph GJ, Beaulieu S, Lebecque S, Steinman RM, Muller WA, Science 1998, 282, 480. [PubMed] [Google Scholar]
- [369].Randolph GJ, Inaba K, Robbiani DF, Steinman RM, Muller WA, Immunity 1999, 11, 753. [DOI] [PubMed] [Google Scholar]
- [370].Moon JJ, Suh H, Li AV, Ockenhouse CF, Yadava A, Irvine DJ, Proc. Natl. Acad. Sci. USA 2012, 109, 1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [371].Wang J, Chen H-J, Hang T, Yu Y, Liu G, He G, Xiao S, Yang B.-r., Yang C, Liu F, Tao J, Wu MX, Xie X, Nat. Nanotechnol 2018, 13, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [372].Muraoka D, Seo N, Hayashi T, Tahara Y, Fujii K, Tawara I, Miyahara Y, Okamori K, Yagita H, Imoto S, Yamaguchi R, Komura M, Miyano S, Goto M, Sawada S.-i., Asai A, Ikeda H, Akiyoshi K, Harada N, Shiku H, J. Clin. Invest 2019, 129, 1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [373].Han X, Shen S, Fan Q, Chen G, Archibong E, Dotti G, Liu Z, Gu Z, Wang C, Sci. Adv 2019, 5, eaaw6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [374].Obermeyer Z, Emanuel EJ, Engl N. J. Med 2016, 375, 1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [375].Murdoch TB, Detsky AS, JAMA J. Am. Med. Assoc 2013, 309, 1351. [DOI] [PubMed] [Google Scholar]
- [376].Zhang L, Luo Y, Tao F, Li BH, Ren L, Zhang X, Guo H, Cheng Y, Hu A, Liu Y, Enterp. Inf. Syst 2014, 8, 167. [Google Scholar]
- [377].Adir O, Poley M, Chen G, Froim S, Krinsky N, Shklover J, Shainsky-Roitman J, Lammers T, Schroeder A, Adv. Mater 2020, 32, 1901989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [378].Zhou X, Qu M, Tebon P, Jiang X, Wang C, Xue Y, Zhu J, Zhang S, Oklu R, Sengupta S, Sun W, Khademhosseini A, Adv. Sci 2020, n/a, 2001447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [379].Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, Catz SD, Sci. Immunol 2018, 3, eaat4579. [DOI] [PubMed] [Google Scholar]
- [380].Galli SJ, Borregaard N, Wynn TA, Nat. Immunol 2011, 12, 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [381].Appay V, van Lier RAW, Sallusto F, Roederer M, Cytometry, Part A 2008, 73A, 975. [DOI] [PubMed] [Google Scholar]
- [382].Biswas SK, Mantovani A, Nat. Immunol 2010, 11, 889. [DOI] [PubMed] [Google Scholar]
- [383].Han CZ, Juncadella IJ, Kinchen JM, Buckley MW, Klibanov AL, Dryden K, Onengut-Gumuscu S, Erdbrügger U, Turner SD, Shim YM, Tung KS, Ravichandran KS, Nature 2016, 539, 570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [384].Georgouli M, Herraiz C, Crosas-Molist E, Fanshawe B, Maiques O, Perdrix A, Pandya P, Rodriguez-Hernandez I, Ilieva KM, Cantelli G, Karagiannis P, Mele S, Lam H, Josephs DH, Matias-Guiu X, Marti RM, Nestle FO, Orgaz JL, Malanchi I, Fruhwirth GO, Karagiannis SN, Sanz-Moreno V, Cell 2019, 176, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [385].Németh K, Leelahavanichkul A, Yuen PST, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X, Jelinek I, Star RA, Mezey É, Nat. Med 2009, 15, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [386].Kim J, Hematti P, Exp. Hematol 2009, 37, 1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [387].Moyer TJ, Zmolek AC, Irvine DJ, J. Clin. Invest 2016, 126, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [388].Cao X, Nat. Rev. Immunol 2020, 20, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [389].Moore BJB, June CH, Science 2020, 368, 473. [DOI] [PubMed] [Google Scholar]
- [390].Bekker L-G, Tatoud R, Dabis F, Feinberg M, Kaleebu P, Marovich M, Ndung’u T, Russell N, Johnson J, Luba M, Fauci AS, Morris L, Pantaleo G, Buchbinder S, Gray G, Vekemans J, Kim JH, Levy Y, Corey L, Shattock R, Makanga M, Williamson C, Dieffenbach C, Goodenow MM, Shao Y, Staprans S, Warren M, Johnston MI, Lancet 2020, 395, 384. [DOI] [PubMed] [Google Scholar]
- [391].Burton DR, Nat. Rev. Immunol 2019, 19, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [392].Wang H-X, Li M, Lee CM, Chakraborty S, Kim H-W, Bao G, Leong KW, Chem. Rev 2017, 117, 9874. [DOI] [PubMed] [Google Scholar]
- [393].Xu C-F, Chen G-J, Luo Y-L, Zhang Y, Zhao G, Lu Z-D, Czarna A, Gu Z, Wang J, Adv. Drug Delivery Rev 2019, S0169, 10.1016/j.addr.2019.11.005. [DOI] [PubMed] [Google Scholar]
- [394].Xie M, Fussenegger M, Nat. Rev. Mol. Cell Biol 2018, 19, 507. [DOI] [PubMed] [Google Scholar]
- [395].Sedlmayer F, Aubel D, Fussenegger M, Nat. Biomed. Eng 2018, 2, 399. [DOI] [PubMed] [Google Scholar]
- [396].Moyes KW, Lieberman NAP, Kreuser SA, Chinn H, Winter C, Deutsch G, Hoglund V, Watson R, Crane CA, Hum. Gene Ther 2017, 28, 200. [DOI] [PubMed] [Google Scholar]
- [397].Wu M, Hussain S, He Y-H, Pasula R, Smith PA, Martin WJ, Proc. Natl. Acad. Sci. USA 2001, 98, 14589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [398].Bailey SR, Maus MV, Nat. Biotechnol 2019, 37, 1425. [DOI] [PubMed] [Google Scholar]
- [399].Shah NJ, Najibi AJ, Shih T-Y, Mao AS, Sharda A, Scadden DT, Mooney DJ, Nat. Biomed. Eng 2020, 4, 40. [DOI] [PubMed] [Google Scholar]
- [400].Adu-Berchie K, Mooney DJ, Acc. Chem. Res 2020, 53, 1749. [DOI] [PubMed] [Google Scholar]