

Abstract
Background:
The molecular basis of airway remodeling in chronic obstructive pulmonary disease remains poorly understood. We identified gene expression signatures associated with chest CT scan airway measures to understand molecular pathways associated with airway disease.
Methods:
In 2,396 subjects in the COPDGene Study, we examined the relationship between quantitative CT airway phenotypes and blood transcriptomes to identify airway disease-specific genes and to define an airway wall thickness (AWT) gene set score. Multivariable regression analyses were performed to identify associations of the AWT score with clinical phenotypes, bronchial gene expression and genetic variants.
Results:
Type 1 interferon induced genes were consistently associated with AWT, square root wall area of a hypothetical airway with 10mm internal perimeter (Pi10), and wall area percentage, with the strongest enrichment inAWT. A score derived from 18 genes whose expression was associated with AWT was associated with COPD-related phenotypes including reduced lung function (forced expiratory volume in 1 s percentage predicted β = −3.4, p <0.05) and increased exacerbations (incidence rate ratio 1.7, p <0.05). The AWT score was reproducibly associated with airway wall thickness in bronchial samples from 23 subjects (β = 3.22, p < 0.05). The blood AWT score was associated with genetic variant rs876039, an expression quantitative trait locus (eQTL) for IKZF1, a gene which regulates interferon signaling and is associated with inflammatory diseases.
Conclusion:
A gene expression signature with interferon stimulated genes from peripheral blood and bronchial brushings is associated with CT airway wall thickness, lung function, and exacerbations. Shared genes and genetic associations suggest viral responses and/or autoimmune dysregulation as potential underlying mechanisms of airway disease in COPD.
Keywords: chronic obstructive pulmonary disease, airway disease, innate immunity, blood gene expression
INTRODUCTION
Emphysema and airway disease are two distinct pathological processes that independently lead to airflow obstruction in chronic obstructive pulmonary disease (COPD). However, unlike emphysema which has been extensively studied using animal models, there are considerable differences between species in airway anatomy. Murine models generally do not develop airway remodeling or mucus hypersecretion. Thus, the biological pathways related to airway disease processes remain largely unknown.
Recent advances in chest computed tomography (CT) scan quantification allow distinction between emphysema and airway disease for phenotyping and understanding heterogeneity in the disease process. In addition, CT measures of airways correlate with physiologic and clinical characteristics. Airway wall thickness (AWT), square root wall area of a hypothetical airway with 10mm internal perimeter (Pi10) and airway wall area percentage (WA%) are inversely correlated with forced expiratory volume in 1s (FEV1)[1], and positively associated with exacerbation risk[2], bronchodilator response, and respiratory symptoms such as chronic bronchitis and SGRQ score[3, 4].
The Genetic Epidemiology of COPD Study (COPDGene) is a multicenter study that extensively characterized participants and includes quantitative chest CT scans, genetic analysis and gene expression profiling. Prior studies from COPDGene showed that blood gene expression profiles track with lung gene expression[5] and identified blood gene expression signatures of eosinophilic COPD[6], providing molecular insights.
Therefore, to understand the pathways related to airway disease in COPD, we utilized RNA-sequencing and CT quantification in a large sample size. We hypothesized that blood gene expression profiling would identify molecular signatures associated with CT airway measures. We further hypothesized that a blood airway gene expression score will be associated with specific clinical phenotypes, with bronchial gene expression and with genetic risk.
METHODS
Study population
COPDGene (NCT00608764) is a multicenter observational study which enrolled 10,192 smokers with and without COPD (defined as post-bronchodilator FEV1/FVC<0.7) in Phase 1 [7]. Approximately 5,800 returned for a Phase 2 visit five years later (Supplementary material).
RNA sequencing
The whole blood and bronchial epithelial RNA sequencing were performed from Phase 2 samples. Protocols have been described previously (supplementary material)[5, 8]. Bronchial epithelial cells were obtained by flexible bronchoscopy with brushing of the right mainstem bronchus.
Differential gene expression
Association analyses of CT airway phenotypes and transcript expression were performed with voom transformation in the limma R package[9]. Covariates in the linear models are specified in individual result tables. Differentially expressed genes were defined by false discovery rate (FDR) <0.05 in models with and without surrogate variable adjustment. Pathway analyses were performed using MSigDB[10]. Expression data has been deposited to Database of Genotypes and Phenotypes (dbGaP), accession phs000765.v6.p2.
Interferon response genes
GEO datasets (GSE38351, GSE71634, GSE19491, GSE60244) were analyzed to identify differentially expressed genes associated with IFN treatment, or with disease conditions (supplementary table E1, supplementary material and supplementary material file E2).
Quantification of gene set scores
Gene Set Variation Analysis (GSVA) was performed to calculate airway wall thickness (AWT) and type 1 interferon enrichment scores[11]. Linear or Negative binomial regression were performed to test the associations between scores and continuous variables. The strength of correlation was estimated by the regression coefficient (β), which is the change in the outcome variable associated with a 1 unit increase in AWT score when all other variables were held constant. P values were adjusted using Holm-Bonferroni method.
Genome wide association study (GWAS)
We performed linear regression of the AWT score adjusting for age, sex, smoking status and genetic ancestry in non-Hispanic white subjects using Illumina OmniExpress genotypes, HRC 1.1 imputation[12], and PLINK v2.00a2LM[13]. Functional annotation was conducted using FUMA v1.3.5[14] and Open Targets Genetics platform[15].
RESULTS
CT airway wall thickness is associated with interferon stimulated genes
Peripheral blood RNA sequencing was performed on 2,396 subjects with quantitative CT airway measurements (table 1). COPD subjects were older, more likely to be male non-Hispanic whites and former smokers. All three measures of CT airways -- AWT, WA%, and Pi10 -- were increased in COPD cases compared to controls and inversely associated with post-bronchodilator FEV1 (r =−0.16, −0.38, and −0.55 respectively, p<0.001). We tested blood gene expression levels associated with CT airway measures using linear models adjusted for age, sex, race, pack-years of smoking, current smoking status, white blood cell counts and library construction batch. We found 18 genes whose expression were associated with AWT, 17 genes associated with WA%, and 81 genes associated with Pi10 (supplementary material file E1). Notably, the AWT associated genes were enriched for interferon stimulated genes (ISGs) such as OAS, RSAD2, and IFI44, with overrepresentation of type 1 interferon (IFN) signaling and viral response pathways. These ISGs were positively associated with AWT indicating higher expression was associated with increased airway wall thickness. Similar association of interferon response genes were observed with WA% and Pi10. WA% was associated with ISGs RSAD2 and IFI44L, and Pi10 associated genes included LMO2, GBP2, RABGAP1L induced by IFNα (supplementary file E1) that were all positively associated with CT airway measures. The association of ISGs was strongest with AWT (table 2). WA% associated genes did not show any significant pathways, while Pi10 associated genes were enriched for hemoglobin biosynthetic process and regulation of phosphorus metabolic process. Enrichment of ISGs were unique for airway related phenotypes and was not observed with lung function or emphysema [16]. Unlike interferon stimulated genes, the expression of genes encoding interferons themselves were not associated with CT airway measures.
Table 1.
Study Subjects
| Non-COPD* (n=1368) |
COPD (n=1028) |
P value | |
|---|---|---|---|
| Age, yr (mean(SD)) | 63.11 (8.07) | 68.21 (8.29) | <0.01 |
| Female (%) | 712 (52.9) | 443 (43.1) | <0.01 |
| Non-Hispanic White (%) | 958 (70.0) | 832 (80.9) | <0.01 |
| FEV1% predicted (mean(SD)) | 91.68 (15.95) | 62.17 (22.45) | <0.01 |
| Bronchodilator response (%) | 129 (9.4) | 298 (29) | <0.01 |
| Inhaled corticosteroid use (%)** | 124 (9.1) | 402 (39.1) | <0.01 |
| Airway wall thickness (mean(SD)) | 0.98 (0.20) | 1.10 (0.24) | <0.01 |
| Pi10 (mean(SD)) | 2.04 (0.45) | 2.54 (0.58) | <0.01 |
| Wall area %(mean(SD)) | 47.60 (7.55) | 53.06 (8.30) | <0.01 |
| Current smokers (%) | 533 (39.0) | 331 (32.2) | <0.01 |
| Smoking history (Pack-Years, mean (SD)) | 39.10 (21.06) | 50.61 (26.05) | <0.01 |
| WBC (x103, mean(SD)) | 6.82 (1.96) | 7.37 (1.99) | <0.01 |
| Neutrophil % (mean(SD)) | 57.27 (9.80) | 61.23 (10.24) | <0.01 |
| Lymphocyte % (mean(SD)) | 31.43 (9.38) | 27.11 (9.33) | <0.01 |
| Monocyte % (mean(SD)) | 8.01 (2.36) | 8.29 (2.45) | 0.01 |
| Eosinophil % (mean(SD)) | 2.66 (1.92) | 2.75 (2.11) | 0.28 |
| Basophil % (mean(SD)) | 0.64 (0.60) | 0.61 (0.57) | 0.38 |
Abbreviations: FEV1 = forced expiratory volume in one second, NHW = non-Hispanic white, the remainder was African American. WBC = white blood cell.
including 306 subjects with preserved ratio and impaired spirometry (reduced FEV1 in the setting of a preserved FEV1/FVC[40]),
including inhaled corticosteroid and long acting beta agonist combination inhalers.
Quantitative phenotyping of segmental airways using Thirona software was performed by measuring airway dimensions in 6 bronchial paths: right upper lobe apical bronchus (RB1), right middle lobe lateral bronchus (RB4), right lower lobe posterior basal bronchus (RB10), left upper lobe apicoposterior bronchus (LB1), superior lingular bronchus (LB4) and left lower lobe posterior basal bronchus (LB10)
P values are based on t test or chi-square test for proportions
Table 2.
Genes and pathways associated with CT airway phenotypes
| AWT | WA% | Pi10 | ||
|---|---|---|---|---|
| Number of DE genes | 18 | 17 | 81 | |
| Enrichment Analysis |
GO BP | Regulation of nuclease activity Response to type I interferon Response to virus Viral life cycle Defense response to other organism |
NS | Hemoglobin biosynthetic process Regulation of phosphorus metabolic process |
| MSigDB | Genes upregulated in ovarian cancer progenitor cells in response to IFNα Interferon induced antiviral module in sputum during asthma exacerbations Genes upregulated in SARS-CoV-2 infection |
NS | Genes upregulated in primary fibroblast culture after treatment with IFNα | |
Genes associated with CT airway phenotypes at false discovery rate <0.05. Abbreviations: AWT = segmental airway wall thickness, WA% = wall area percent, DE = differentially expressed, GO BP = GO biologic process, MSigDB = molecular signature database, NS = none significant
Generation of a score associated with CT airway wall thickness
To further understand pathways and phenotypes associated with airway gene expression, we generated a gene signature score with the 18 genes associated with CT airway wall thickness using GSVA which we termed as “blood AWT (gene set) score” (supplementary material file E1). As expected, the blood AWT score was associated with AWT after adjusting for age, sex, race, BMI, smoking status, % emphysema and CT scanner model, and it was also associated with WA%, Pi10, and emphysema % to a lesser degree (Table 3, supplementary table E2, and supplementary figure E1). These correlation between CT airway wall thickness and AWT score was maintained in former and current smokers. When stratified by case-control status, the association between CT airway wall thickness and the blood AWT score was stronger in subjects with COPD (table 3).
Table 3.
Associations between blood AWT score and CT airway wall thickness
| Variables | Beta (95% CI) | Adjusted P value |
|---|---|---|
| All subjects (n= 2396) | 0.25 (0.16–0.34) | <0.001 |
| Control (n=1368) | 0.15 (0.05–0.26) | 0.13 |
CT airway wall thickness standardized using z score. Linear regression adjusted for age, sex, race, body mass index, smoking status, CT scanner model, emphysema (%). Beta denotes regression coefficient, which is the change in the outcome variable associated with a 1 unit increase in blood AWT score when all other variables were held constant.
P value adjusted for multiple testing using Holm-Bonferroni correction.
AWT score relationship with type 1 IFN signature
We compared the AWT score with type 1 IFN induced genes in blood from previously published studies. The type 1 IFN signature was defined as 110 upregulated genes after treatment of peripheral blood monocytes with IFN-α2 and peripheral blood mononuclear cells (PBMC) with IFN-β, using blood samples from healthy donors[17, 18] (supplementary material file E2). Nine genes (IFI44, IFI44L, RSAD2, CMPK2, DDX60, RIN2, OAS2, OAS3, HERC5) overlapped between AWT genes and type 1 IFN induced genes; the AWT score and type 1 IFN score showed high correlation (Spearman ρ=0.89, p<2.2×10−16), supporting enrichment of ISGs (figure 1). Stratified differential expression analysis of low vs. high AWT score subjects also confirmed enrichment of ISG pathways (supplementary material and supplementary table E3).
Fig 1.
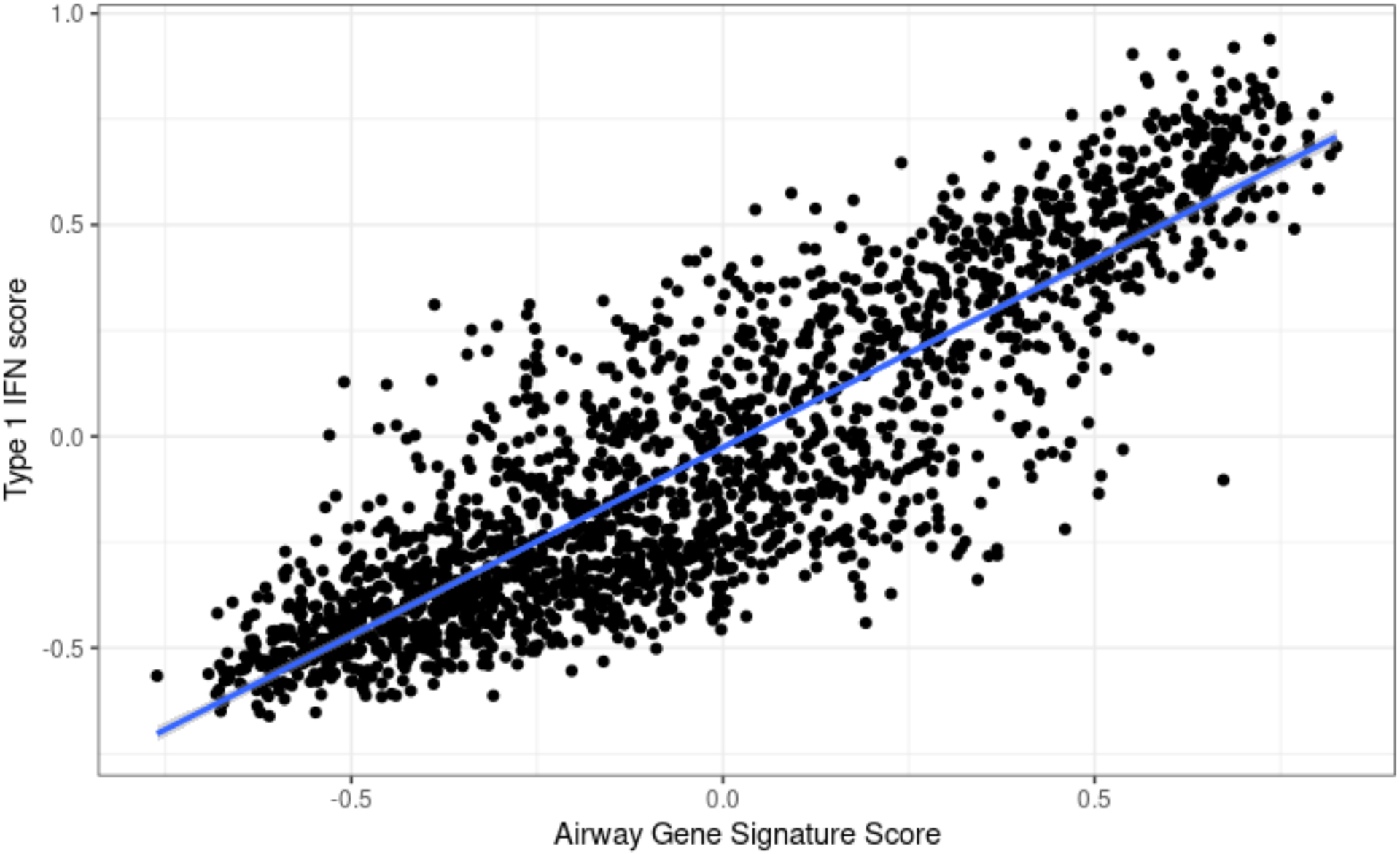
Airway gene signature and Type 1 IFN signature gene set scores are highly correlated.
Gene set enrichment scores are derived from Gene Set Variation Analysis.
Airway gene signature score was derived from 18 genes associated with CT airway wall thickness. Type 1 IFN score was derived from upregulated genes after treatment of peripheral blood with IFN-α and β.
Blood-derived AWT score is recapitulated in bronchial gene expression
Using RNA-seq data from bronchial epithelial brushings from 23 COPDGene subjects (Table E4), we re-created the AWT gene set score with the same genes from the blood signature, except immunoglobin genes IGHG3 and IGLV3–21, which were not expressed in bronchial epithelial cells. The bronchial AWT score was positively associated with AWT and WA% in multivariable linear regression analysis, reproducing the relationship observed in blood. We did not find the AWT score to be associated with Pi10, which showed weakest association with the AWT score in blood (Table 4).
Table 4.
Association between bronchial epithelial AWT score and CT airway measurements
| AWT* | WA%* | Pi10* | ||||
|---|---|---|---|---|---|---|
| beta (95% CI) | adj.p | beta (95% CI) | adj.p | beta (95% CI) | adj.p | |
| Bronchial AWT score(n=23) | 3.22(1.21–5.24) | 0.01 | 2.82(0.76–4.88) | 0.02 | 1.39(−0.91–3.69) | 0.2 |
CT airway measurements are standardized using z-score
Abbreviations: AWT = segmental airway wall thickness, WA% = wall area percent, adj.p = adjusted p value using Holm-Bonferroni correction
Linear regression adjusted for age, sex, race, body mass index, smoking status, and emphysema (%). CT scanner model was identical for all subjects.
AWT score is associated with reduced lung function and lung function decline
A previous COPDGene publication has shown the CT airway measures to be inversely associated with lung function[1]. We found that the AWT score was inversely associated with FEV1% predicted and FEV1/FVC measured at the Phase 2 visit (concurrent with RNA collection) as well as spirometry measurements from the Phase 1 visit, approximately 5 years prior. Higher AWT score was also associated with greater lung function decline over 5 years (Table 5).
Table 5.
Association of blood AWT score with lung function and exacerbations
| Beta or IRR | 95% CI | Adjusted p value | |
|---|---|---|---|
| FEV1pp* | −3.4 | −5.44, −1.42 | 0.04 |
| FEV1/FVC* | −0.03 | −0.04, −0.02 | <0.0001 |
| ΔFEV1(ml)† | −53.5 | −81.6, −25.4 | <0.01 |
| Exacerbation frequency‡ | 1.7 | 1,24, 2.38 | 0.04 |
Abbreviations: IRR = incidence rate ratio, FEV1pp = FEV1 percent predicted, ΔFEV1/ml= change in FEV1(ml) between over 5 years, adj.p = adjusted p value using Holm-Bonferroni correction
Linear regression adjusted for age, sex, race, body mass index, smoking status, smoking history (PY), emphysema (%). COPD n= 1026, control n = 1367
Linear regression adjusted for age, sex, race, body mass index, smoking status, smoking history (PY), baseline FEV1 percent predicted, emphysema (%). COPD n = 1024, control n = 1360
Acute respiratory disease defined by flare up of chest troubles requiring antibiotic and/or steroid treatment[41]. Negative binomial regression adjusted for age, sex, race, smoking status, baseline FEV1 percent predicted, history of previous exacerbation. COPD n= 937, control n= 1244
AWT score overlap with autoimmune and respiratory viral infection signatures and predicts exacerbations
First discovered as an essential component of antiviral host response, type 1 IFN can also be induced by bacterial pathogens and drive chronic inflammation in autoimmune diseases. To understand whether type 1 IFN signaling in the AWT score may represent active infection or an autoimmune-like process, we compared the AWT gene set with two published blood gene expression studies, first in patients with systemic lupus erythematosus (SLE) or systemic bacterial infection[19] and second with lower respiratory viral or bacterial infections[20] (supplementary material file E2). SLE is an extensively studied autoimmune disease with dysregulation of type 1 IFN response genes. We found the AWT score overlapped with SLE gene expression but not with systemic bacterial infection (p<1.0×10−6). Within lower respiratory infection, the AWT score overlapped with viral respiratory infection (p<2.8×10−14) but not with bacterial infection (supplementary figure E2). The airway genes overlapping with SLE and viral lower respiratory infection genes were 9 identical interferon response genes (OAS1, OAS2, OAS3, RSAD2, IFI44, IFI44L, DDX60, CMPK2, HERC5), supporting the shared molecular pathways between viral infection and autoimmune processes (File E2).
Next we applied the published 396-gene set meta-virus signature (MVS) that could distinguish respiratory viral infections from bacterial infections or healthy controls to our blood gene expression data [21]. A strong association between the AWT score and MVS was found (Spearman ρ=0.43, p<2.2×10−16). As COPD exacerbations are commonly triggered by viral infections[22], we examined whether the AWT score or MVS are associated with exacerbations. The AWT score was correlated with the number of exacerbations in the year prior to enrollment (Spearman ρ=0.059, p<0.005). In a subset of subjects who had prospective follow up (n= 2,181), blood AWT score and MVS score predicted future exacerbations (Table 5, Table E5).
Subgroup and sensitivity analysis
We performed stratified analysis for potential confounders influencing gene expression (sex, smoking, inhaled corticosteroid (ICS) and asthma history, Supplemental Results, Table E6–10). Male sex and lack of inhaled corticosteroid use were associated with stronger association of AWT gene sets with airway wall thickness. Blood AWT score was not significantly associated with CT airway wall thickness in ICS users, suggesting that ICS suppress ISG expression in blood (Table E6).
Genome-wide association of blood AWT score
To understand the genetic factors that contribute to airway signature genes, we performed a GWAS of the airway signature in 1,788 non-Hispanic white subjects from COPDGene, adjusted for age, sex, current smoking status and genetic ancestry. We identified two significant loci -- on chromosome 9 and on chromosome 7 -- associated with AWT score at genome-wide significance (p<5×10−8) (Fig 2, Table 6, File E3). In the chromosome 7p12 locus, one lead SNP rs876039 was identified, which is near the IKZF1 gene and is also an expression quantitative trait locus (eQTL) for IKZF1 in blood (eQTLGen, FDR <0.05)(Fig E3)[23] and more recently described as the lead variant affecting the level of plasmacytoid dendritic cells (pDC)[24]. This signal co-localizes with susceptibility to SLE, and coincides with several eQTLs acting in trans, suggesting other signals that could be mediated by the action of this gene. IKZF1 encodes the transcription factor Ikaros that has previously been suggested as a candidate gene for SLE[25]. Other associations at this locus include monocyte percentage, non-albumin protein, and albumin/globulin ratio [15]. We found the AWT score is positively associated with peripheral blood monocyte percentage in COPDGene, consistent with the GWAS direction of effect (beta=1.39, adj.p<0.001). GWAS mapped genes were enriched for response to type 1 interferon pathway, confirming the association found for the blood AWT score (File E3).
Fig 2.
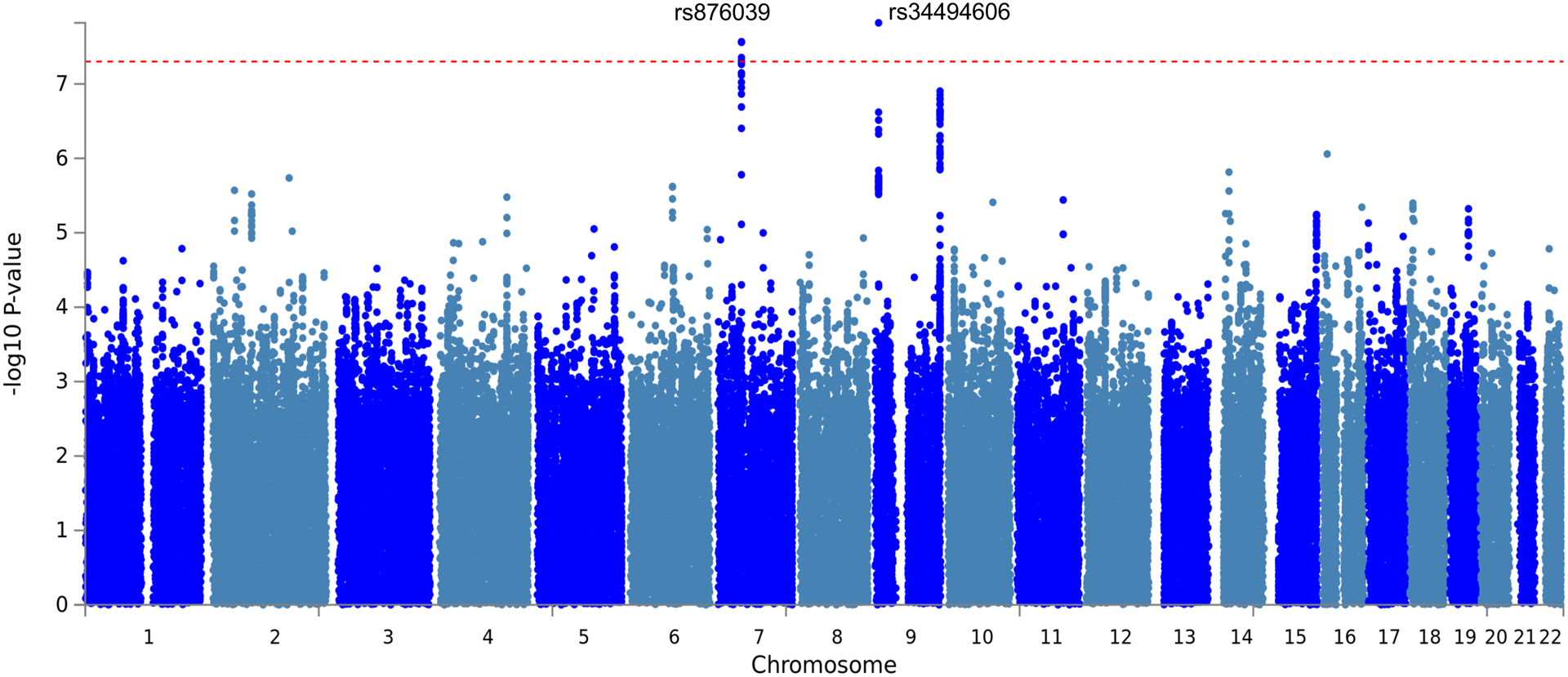
GWAS of airway gene signature score. Manhattan plot showing the association of variants with airway gene signature score in 1,788 non-Hispanic White individuals. The dashed red line marks the threshold for genome-wide significance (p = 5 × 10−8).
Table 6.
Genome-wide significant SNPs associated with blood AWT score from non-Hispanic whites at two loci.
| SNP | Chr | Position | Effect Allele | MAF | Beta | P value |
|---|---|---|---|---|---|---|
| rs34494606 | 9 | 7872062 | C | 0.10 | 0.32 | 1.5 × 10−8 |
| rs876039 | 7 | 50308811 | C | 0.31 | −0.20 | 2.7 × 10−8 |
Abbreviations: SNP = single nucleotide polymorphism, Chr = chromosome, MAF = minor allele frequency
Cellular origin of AWT score
To identify the cell types expressing AWT score genes, we analyzed matched blood and lung single cell RNA-sequencing data[26]. The majority of IFN induced genes were highly expressed in nonclassical monocytes in both blood and lung. In blood, some genes were expressed in neutrophils and pDCs, while in lung AWT genes were also abundantly expressed in endothelial cells and bronchial epithelial cells (club cells and goblet cells) (Fig E4).
DISCUSSION
In a gene expression study of 2,396 individuals with and without COPD, we identified an 18-gene molecular signature associated with CT airway wall thickness that is conserved between blood and bronchial epithelial cells. This AWT score is associated with type 1 IFN signaling. We also identified genetic loci associated with the blood AWT score, providing molecular and genetic insights into further understanding airway disease in COPD.
The CT airway measures were associated with interferon response genes. Among the three complementary CT airway measures, the enrichment was strongest with AWT. The blood AWT gene set was recapitulated in bronchial epithelial gene expression, where the bronchial AWT score was associated with AWT and WA%. Recent studies show dynamic changes in AWT, Pi10 and WA% in COPD, with an increase in AWT in smokers associated with COPD incidence [27]. AWT is a direct measure of wall thickness, compared to Pi10 and WA% which are composite measures of airway lumen and wall thickness. In COPD, the airway lumen is narrowed by inflammatory mucus, whereas wall thickness increases from infiltration of inflammatory cells and remodeling. This suggests that the interferon signaling may represent a specific molecular process involved in airway thickening, early airway inflammation or remodeling in COPD, or a subtype of COPD associated with such processes. Accordingly, the blood AWT score was associated with lung function decline adjusting for emphysema, suggesting that it may potentially serve as a biomarker for lung function decline related to airway disease. Conversely, we also found an association between AWT score and emphysema after adjusting for airway wall thickness, supporting the interconnected relationship between airway disease and emphysema.
As a first line of defense against viral pathogens, interferon response genes encode antiviral effectors and also elicit a wide range of context-specific inflammatory mediators that could be protective or pathogenic. For example, in SARS, early induction of type 1 IFN signaling was associated with reduced inflammation and milder disease, but delayed type 1 IFN signaling and persistent expression of ISGs were associated with sustained inflammation and fatal disease[28]. In COVID-19, a low type 1 IFN transcriptomic score from blood was associated with severe disease[29]. In asthma, impaired IFN-β response in airway epithelial cells upon virus infection was associated with greater viral load and worse symptoms during exacerbations[30]. In COPD, studies on the effect of type 1 IFN response yielded inconsistent results that appear to be context dependent. Some studies demonstrate increased expression of IFN-β in COPD epithelial cells infected with rhinovirus[31] and human metapneumovirus[32]. While others reported deficiency of type 1 IFN upon rhinovirus infection in COPD bronchoalveolar lavage cells [33] and reduced expression of IFN-β in sputum samples from frequent exacerbators, without differential expression of ISGs[34]. Furthermore, in COPD, cigarette smoke and ICS can also impact IFN response. Cigarette smoke suppresses induction of type 1 IFN, but cell death caused by smoking and emphysema can release damage-associated molecular patterns (DAMPs) which lead to type 1 IFN production[35]. We found ICS use is associated with low AWT score in COPD patients, consistent with ICS suppressing ISG expression in mouse viral infection models and primary airway epithelial cells from COPD patients[36].
Given the cross-sectional design, it is not possible to determine whether the enriched IFN response is protective or pathogenic in the disease process, or if pathogenic, whether we observed secondary consequences of viral infections vs. enhanced IFN signaling that is part of the disease pathogenesis. We did not detect the differential expression of IFN itself in our data. Due to the reduced sensitivity of detecting type 1 IFN gene expression, ISG expression is thought to be a more sensitive readout for activation of this pathway than the cytokine itself [37]. Furthermore, we cannot exclude the possibility that the observed ISGs may also be upregulated to some extent by IFN-γ. Alternatively, ISG may be induced independent of IFN as reported in certain viral infections or cell lines[38]. Nonetheless, the finding of genetic variants associated with this signature suggest that future studies may be able to identify potential mechanisms. The blood AWT score overlapped with transcriptomic signatures of respiratory viral infections and was predictive of future exacerbations. In COPDGene, blood was obtained during a stable state, at least 30 days since the last exacerbation, so we do not believe the signature reflects acute viral infection, but may represent sustained, chronic interferon response. Investigations into chronic viral infections and autoimmune diseases have shown that prolonged IFN signaling leads to chronic immune activation and inflammation, leading to immune exhaustion, autoimmunity, and tissue damage. Therefore, we speculate that elevated blood AWT score in the absence of active infection is likely pathologic.
The overlap of the AWT score with the blood transcriptomic signature of SLE patients and the genetic association of the blood AWT score shared with monocyte counts and SLE align with the notion of pathologic chronic immune dysregulation mediated by type 1 IFN response in COPD airway disease. The C allele in rs876039 was associated with reduced blood AWT score, reduced monocyte counts and lower risk of SLE, consistent with our findings. The putative causal gene, IKZF1, a SLE susceptibility gene, not only regulates immune cell development but also interferon pathways. Monocytes are critical producers, effectors, and regulators of type 1 IFN, that showed enrichment of AWT score gene expression from single cell RNA-sequencing data; Type 1 IFN is produced largely by plasmacytoid dendritic cells (pDC) and inflammatory monocytes. In response to type 1 IFN, monocytes can differentiate into antigen presenting dendritic cells that cause tissue damage, produce inflammatory cytokines and regulate pDC production of type 1 IFN. It would be informative to examine how IFN responses are modulated in different monocyte subsets, and other cell types expressing AWT genes such as bronchial epithelial and endothelial cells. Future studies will be required to validate the blood AWT score in independent cohorts. However, through cross-tissue analysis, we showed that the AWT score is conserved between blood and airways.
This study leverages recent advances in quantitative CT imaging analysis and RNA sequencing to dissect molecular processes associated with airway disease in COPD and found IFN signaling genes as possible players in the disease process. Given the importance of ICS, which suppress ISG response, in COPD therapeutics, and a recent study on IFN-β prophylaxis to modulate viral infection in COPD[39], understanding of IFN signaling and airway phenotypes is needed to delineate the protective or pathologic effect of IFN signaling on COPD airway disease. ICS or other modulation of IFN signaling may have potential to treat airway disease. Lastly, how this relates to the airway microbiome, outcomes of viral infections, and prospective changes in CT airway phenotypes and risk of COPD development remains to be demonstrated in future studies.
Supplementary Material
Take home message.
Airway wall thickness in chronic obstructive pulmonary disease is associated with type 1 interferon signaling from peripheral blood gene expression.
Funding
Supported by National Institute of Health (NIH) grants K08HL146972, R01HL130512, R01HL125583, U01HL089856, U01HL089897.
COPDGene® s also supported by the COPD foundation through contributions made to an Industry Advisory Board comprised of AstraZeneca, Boehringer-Ingelheim, Genetech, GlaxoSmithKline, Novartis, Pfizer, Siemens and Sunovion.
Disclosure of potential conflict of interest
J. Yun received personal fees from Bridge Biotherapeutics, M.Cho received personal fees from Genetech, personal fees from AstraZeneca, personal fees from Illumina, grants from Bayer, grants from GlaxoSmithKline outside the submitted work. P.Castaldi received grants from GlaxoSimthKline, personal fees from GlaxoSmithKline, presonal fees from Novartis outside the submitted work. C.Hersh received grants from Bayer, Boehringer-Ingelheim, Novartis and Vertex, and personal fees from Takeda outside the submitted work.
COPDGene Phase 3
Grant Support and Disclaimer
The project described was supported by Award Number U01 HL089897 and Award Number U01 HL089856 from the National Heart, Lung, and Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
COPD Foundation Funding
COPDGene is also supported by the COPD Foundation through contributions made to an Industry Advisory Board comprised of AstraZeneca, Boehringer-Ingelheim, Genentech, GlaxoSmithKline, Novartis, Pfizer, Siemens, and Sunovion.
References
- 1.Charbonnier JP, Pompe E, Moore C, Humphries S, van Ginneken B, Make B, Regan E, Crapo JD, van Rikxoort EM, Lynch DA. Airway wall thickening on CT: Relation to smoking status and severity of COPD. Respir. Med Elsevier; 2019; 146: 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jairam PM, Van Graaf Y Der, Lammers JWJ, Th M Mali WP, DeJong PA. Incidental findings on chest CT imaging are associated with increased COPD exacerbations and mortality. Thorax BMJ Publishing Group; 2015; 70: 725–731. [DOI] [PubMed] [Google Scholar]
- 3.Grydeland TB, Dirksen A, Coxson HO, Eagan TML, Thorsen E, Pillai SG, Sharma S, Eide GE, Gulsvik A, Bakke PS. Quantitative computed tomography measures of emphysema and airway wall thickness are related to respiratory symptoms. Am. J. Respir. Crit. Care Med 2010; 181: 353–359. [DOI] [PubMed] [Google Scholar]
- 4.Kim V, Davey A, Comellas AP, Han MK, Washko G, Martinez CH, Lynch D, Lee JH, Silverman EK, Crapo JD, Make BJ, Criner GJ, Farzadegan H, Bragan S, Cayetano S, Curtis J, Kazerooni E, Hanania N, Alapat P, Bandi V, Guntupalli K, Guy E, Mallampalli A, Trinh C, Atik M, Al-Azzawi H, Willis M, Pinero S, Fahr L, Nachiappan A, et al. Clinical and computed tomographic predictors of chronic bronchitis in COPD: A cross sectional analysis of the COPDGene study. Respir. Res 2014; 15: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrow JD, Chase RP, Parker MM, Glass K, Seo M, Divo M, Owen CA, Castaldi P, Demeo DL, Silverman EK, Hersh CP. RNA-sequencing across three matched tissues reveals shared and tissue-specific gene expression and pathway signatures of COPD. Respir. Res BioMed Central Ltd.; 2019; 20: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yun JH, Chase R, Parker MM, Saferali A, Castaldi PJ, Silverman EK, Hersh CP. Peripheral blood gene expression signatures of eosinophilic chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol American Thoracic Society; 2019. p. 398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Regan EA, Hokanson JE, Murphy JR, Make B, Lynch DA, Beaty TH, Curran-Everett D, Silverman EK, Crapo JD. Genetic Epidemiology of COPD (COPDGene) Study Design. COPD J. Chronic Obstr. Pulm. Dis 2011; 7: 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parker MM, Chase RP, Lamb A, Reyes A, Saferali A, Yun JH, Himes BE, Silverman EK, Hersh CP, Castaldi PJ. RNA sequencing identifies novel non-coding RNA and exon-specific effects associated with cigarette smoking. BMC Med. Genomics 2017; 10: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015; 1: 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hänzelmann S, Castelo R, Guinney J, Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013; 14: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, Schlessinger D, Stambolian D, Loh PR, Iacono WG, Swaroop A, Scott LJ, Cucca F, Kronenberg F, Boehnke M, Abecasis GR, Fuchsberger C. Next-generation genotype imputation service and methods. Nat. Genet 2016; 48: 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience BioMed Central Ltd.; 2015; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watanabe K, Taskesen E, Van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun Nature Publishing Group; 2017; 8: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carvalho-Silva D, Pierleoni A, Pignatelli M, Ong CK, Fumis L, Karamanis N, Carmona M, Faulconbridge A, Hercules A, McAuley E, Miranda A, Peat G, Spitzer M, Barrett J, Hulcoop DG, Papa E, Koscielny G, Dunham I. Open Targets Platform: New developments and updates two years on. Nucleic Acids Res. Oxford University Press; 2019; 47: D1056–D1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.AJ G, A S, S L, R C, M M, J M, J Y, PJ C, CP H. Blood RNA sequencing shows overlapping gene expression across COPD phenotype domains. Thorax Thorax; 2021; thoraxjnl-2020–216401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clarelli F, Liberatore G, Sorosina M, Osiceanu AM, Esposito F, Mascia E, Santoro S, Pavan G, Colombo B, Moiola L, Martinelli V, Comi G, Martinelli-Boneschi F. Pharmacogenetic study of long-term response to interferon-β treatment in multiple sclerosis. Pharmacogenomics J Nature Publishing Group; 2017; 17: 84–91. [DOI] [PubMed] [Google Scholar]
- 18.Smiljanovic B, Grün JR, Biesen R, Schulte-Wrede U, Baumgrass R, Stuhlmüller B, Maslinski W, Hiepe F, Burmester GR, Radbruch A, Häupl T, Grützkau A. The multifaceted balance of TNF-α and type I/II interferon responses in SLE and RA: How monocytes manage the impact of cytokines. J. Mol. Med J Mol Med (Berl); 2012; 90: 1295–1309. [DOI] [PubMed] [Google Scholar]
- 19.Berry MPR, Graham CM, McNab FW, Xu Z, Bloch SAA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O’Garra A. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature Nature Publishing Group; 2010; 466: 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suarez NM, Bunsow E, Falsey AR, Walsh EE, Mejias A, Ramilo O. Superiority of transcriptional profiling over procalcitonin for distinguishing bacterial from viral lower respiratory tract infections in hospitalized adults. J. Infect. Dis 2015. p. 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andres-Terre M, McGuire HM, Pouliot Y, Bongen E, Sweeney TE, Tato CM, Khatri P. Integrated, Multi-cohort Analysis Identifies Conserved Transcriptional Signatures across Multiple Respiratory Viruses. Immunity Elsevier Inc.; 2015; 43: 1199–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.2021 GOLD Guidelines [Internet]. [cited 2021 Jul 1].Available from: www.goldcopd.org.
- 23.Võsa U, Claringbould A, Westra H-J, Bonder MJ, Deelen P, Zeng B, Kirsten H, Saha A, Kreuzhuber R, Kasela S, Pervjakova N, Alvaes I, Fave M-J, Agbessi M, Christiansen M, Jansen R, Seppälä I, Tong L, Teumer A, Schramm K, Hemani G, Verlouw J, Yaghootkar H, Sönmez R, Brown A, Kukushkina V, Kalnapenkis A, Rüeger S, Porcu E, Kronberg-Guzman J, et al. Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv Cold Spring Harbor Laboratory; 2018; 18: 447367. [Google Scholar]
- 24.Orrù V, Steri M, Sidore C, Marongiu M, Serra V, Olla S, Sole G, Lai S, Dei M, Mulas A, Virdis F, Piras MG, Lobina M, Marongiu M, Pitzalis M, Deidda F, Loizedda A, Onano S, Zoledziewska M, Sawcer S, Devoto M, Gorospe M, Abecasis GR, Floris M, Pala M, Schlessinger D, Fiorillo E, Cucca F. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat. Genet Springer US; 2020; 52: 1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bentham J, Morris DL, Cunninghame Graham DS, Pinder CL, Tombleson P, Behrens TW, Martín J, Fairfax BP, Knight JC, Chen L, Replogle J, Syvänen A-C, Rönnblom L, Graham RR, Wither JE, Rioux JD, Alarcón-Riquelme ME, Vyse TJ. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet Nature Publishing Group; 2015; 47: 1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, Chang S, Conley SD, Mori Y, Seita J, Berry GJ, Shrager JB, Metzger RJ, Kuo CS, Neff N, Weissman IL, Quake SR, Krasnow MA. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature Nature Research; 2020; 587: 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oelsner EC, Smith BM, Hoffman EA, Kalhan R, Donohue KM, Kaufman JD, Nguyen JN, Manichaikul AW, Rotter JI, Michos ED, Jacobs DR, Burke GL, Folsom AR, Schwartz JE, Watson K, Graham Barr R. Prognostic significance of large airway dimensions on computed tomography in the general population the multi-ethnic study of atherosclerosis (MESA) lung study. Ann. Am. Thorac. Soc American Thoracic Society; 2018; 15: 718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, Perlman S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe Cell Press; 2016; 19: 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Pere H, Charbit B, Bondet V, Chenevier-Gobeaux C, Breillat P, Carlier N, Gauzit R, Morbieu C, Pene F, Marin N, Roche N, Szwebel T-A, Smith N, Merkling S, Treluyer J-M, Veyer D, Mouthon L, Blanc C, Tharaux P-L, Rozenberg F, Fischer A, Duffy D, Rieux-Laucat F, Kerneis S, Terrier B. Impaired type I interferon activity and exacerbated inflammatory responses in severe Covid-19 patients. Science (80-.) American Association for the Advancement of Science; 2020; 369: 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu J, Message SD, Mallia P, Kebadze T, Contoli M, Ward CK, Barnathan ES, Mascelli MA, Kon OM, Papi A, Stanciu LA, Edwards MR, Jeffery PK, Johnston SL. Bronchial mucosal IFN-α/β and pattern recognition receptor expression in patients with experimental rhinovirus-induced asthma exacerbations. J. Allergy Clin. Immunol Elsevier Inc.; 2019; 143: 114–125.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider D, Ganesan S, Comstock AT, Meldrum CA, Mahidhara R, Goldsmith AM, Curtis JL, Martinez FJ, Hershenson MB, Sajjan U. Increased cytokine response of rhinovirus-infected airway epithelial cells in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med 2010; 182: 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kan-O K, Ramirez R, Macdonald MI, Rolph M, Rudd PA, Spann KM, Mahalingam S, Bardin PG, Thomas BJ. Human Metapneumovirus Infection in Chronic Obstructive Pulmonary Disease: Impact of Glucocorticosteroids and Interferon. J. Infect. Dis 2017; 215: 1536–1545. [DOI] [PubMed] [Google Scholar]
- 33.Mallia P, Message SD, Gielen V, Contoli M, Gray K, Kebadze T, Aniscenko J, Laza-Stanca V, Edwards MR, Slater L, Papi A, Stanciu LA, Kon OM, Johnson M, Johnston SL. Experimental rhinovirus infection as a human model of chronic obstructive pulmonary disease exacerbation. Am. J. Respir. Crit. Care Med 2011; 183: 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singanayagam A, Loo SL, Calderazzo M, Finney LJ, Torralbo MBT, Bakhsoliani E, Girkin J, Veerati P, Pathinayake PS, Nichol KS, Reid A, Footitt J, Wark PAB, Grainge CL, Johnston SL, Bartlett NW, Mallia P. Antiviral immunity is impaired in COPD patients with frequent exacerbations. Am. J. Physiol. - Lung Cell. Mol. Physiol 2019; 317: L893–L903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pouwels SD, Heijink IH, Ten Hacken NHT, Vandenabeele P, Krysko DV, Nawijn MC, Van Oosterhout AJM. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol. 2014; 7: 215–226. [DOI] [PubMed] [Google Scholar]
- 36.Singanayagam A, Glanville N, Girkin JL, Ching YM, Marcellini A, Porter JD, Toussaint M, Walton RP, Finney LJ, Aniscenko J, Zhu J, Trujillo-Torralbo MB, Calderazzo MA, Grainge C, Loo SL, Veerati PC, Pathinayake PS, Nichol KS, Reid AT, James PL, Solari R, Wark PAB, Knight DA, Moffatt MF, Cookson WO, Edwards MR, Mallia P, Bartlett NW, Johnston SL. Corticosteroid suppression of antiviral immunity increases bacterial loads and mucus production in COPD exacerbations. Nat. Commun Springer US; 2018; 9: 2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. U. S. A 2003; 100: 2610–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang W, Yin Y, Xu L, Su J, Huang F, Wang Y, Boor PPC, Chen K, Wang W, Cao W, Zhou X, Liu P, Van Der Laan LJW, Kwekkeboom J, Peppelenbosch MP, Pan Q. Unphosphorylated ISGF3 drives constitutive expression of interferon-stimulated genes to protect against viral infections. Sci. Signal 2017; 10: eaah4248. [DOI] [PubMed] [Google Scholar]
- 39.Watson A, Spalluto CM, McCrae C, Cellura D, Burke H, Cunoosamy D, Freeman A, Hicks A, Hühn M, Ostridge K, Staples KJ, Vaarala O, Wilkinson T. Dynamics of IFN-β responses during respiratory viral infection insights for therapeutic strategies. Am. J. Respir. Crit. Care Med 2020; 201: 83–94. [DOI] [PubMed] [Google Scholar]
- 40.Wan ES, Castaldi PJ, Cho MH, Hokanson JE, Regan EA, Make BJ, Beaty TH, Han MLK, Curtis JL, Curran-Everett D, Lynch DA, DeMeo DL, Crapo JD, Silverman EK, Lantz R, Stepp L, Melanson S, Klanderman B, Laird N, Lange C, Santorico S, McDonald ML, Zhou J, Mattheissen M, Hardin M, Hetmanski J, Parker M, Murray T, Reilly J, Coxson H, et al. Epidemiology, genetics, and subtyping of preserved ratio impaired spirometry (PRISm) in COPDGene. Respir. Res 2014; 15: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bowler RP, Kim V, Regan E, Williams AAA, Santorico SA, Make BJ, Lynch DA, Hokanson JE, Washko GR, Bercz P, Soler X, Marchetti N, Criner GJ, Ramsdell J, Han MK, Demeo D, Anzueto A, Comellas A, Crapo JD, Dransfield M, Wells JM, Hersh CP, MacIntyre N, Martinez F, Nath HP, Niewoehner D, Sciurba F, Sharafkhaneh A, Silverman EK, Beek EJR van, et al. Prediction of Acute Respiratory Disease in Current and Former Smokers With and Without COPD. Chest 2014; 146: 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.